fingerprinting the australian rhizobial inoculant mother cultures using refined pcr protocols yields...
TRANSCRIPT

IntroductionInoculation with the soil bacteria rhizobia has been used
for decades to improve nitrogen (N) input to farmingsystems. In association with members of the plant family,Leguminosae, rhizobia are responsible for most of the N2fixed biologically in agricultural systems and contributesignificantly to their sustainability. By reducing thedependence on N-based fertilisers, the use of rhizobialinoculants also minimises environmental problems, such aswater pollution (Giller 2001). Exploiting this symbiosis inAustralian farming systems has allowed millions of hectaresof infertile land to be brought into production (Brockwellet al. 1995).
Legumes are typically inoculated at sowing. To benefitfrom inoculation, inoculant strains must first survive tonodulate their host legumes. Thereafter, inoculation responseis primarily determined by soil N availability in relation tocrop N demand and the degree to which an effective inoculantstrain establishes itself and fixes N in the root nodules in
competition with indigenous rhizobia (Thies et al. 1991). Forperennial legumes, farmers rely on populations of rhizobia,which are generally competent saprophytes, to establishthemselves after the first year and persist in soil to nodulatelegume roots in subsequent years. Both short-term survivaland longer-term persistence of introduced rhizobia depend toa large degree on the interplay between climate and soilfactors and the continued presence of compatible legumespecies (Brockwell et al. 1995). For example, high soiltemperatures, low rainfall and acid soil conditions lead topoorer persistence than more moderate conditions (Howiesonand Ballard 2004). Monitoring the fate of the inoculant in thefield and in nodules over time is thus important for thesuccessful management of this symbiosis in farming systems.
In Australia, mother cultures of the rhizobial strains usedin the manufacture of commercial inoculants are maintainedand monitored by the Australian Legume Inoculant ResearchUnit (ALIRU, NSW Agriculture, Gosford). Annually, afterstandard quality assurance tests have been performed,
Australian Journal of Experimental Agriculture, 2005, 45, 141–150
0816-1089/05/03014110.1071/EA04061© CSIRO 2005
A-M. Vachot-GriffinA and J. E. ThiesA,B,C,D
ACentre for Farming Systems Research, University of Western Sydney — Hawkesbury, Locked Bag 1797, Penrith South DC, NSW 1797, Australia.
BCentre for Biostructural and Biomolecular Research, University of Western Sydney — Hawkesbury, Locked Bag 1797, Penrith South DC, NSW 1797, Australia.
CDepartment of Crop and Soil Sciences, Cornell University, Ithaca, NY 14853, USA.DCorresponding author. Email: [email protected]
Abstract. Monitoring the success of rhizobial inoculation requires reliable identification of the introduced strainsin nodules and when recovered from field soil. The polymerase chain reaction (PCR) coupled with the use of eitherrandom or directed primers has increasingly become the molecular method of choice for characterising bacteria atthe strain level. We have investigated the use of 5 markers (REP, ERIC, BOXA1R, RPO1 and IGS) commonly usedfor PCR fingerprinting to characterise rhizobia bacteria used in the manufacture of rhizobial inoculants in Australia.PCR with random primers often yields inconsistent results because most protocols do not specify stringent cyclingand non-cycling parameters. We have increased the stringency and improved the specificity of reaction conditionsfor 4 of the 5 markers tested. Optimised protocols were then used to fingerprint the 39 strains of rhizobia bacteriaheld in the 1998 mother culture collection of the Australian Legume Inoculant Research Unit (ALIRU). Results for34 strains using at least one marker are presented. Although the mother cultures of these inoculant strains undergonumerous quality assurance tests annually, it was not until PCR fingerprinting was applied that 2 strains, believedto be unique, were found to be identical. In the subsequent investigation, we determined that the 2 strains wereoriginally unique but that a mix-up in the cultures had occurred at least 3 years before our analysis. Use of serology,plant infection tests and field tests were not sufficient to detect this problem. The use of PCR fingerprinting withoptimised protocols has now been incorporated into the annual quality assurance regime used by the ALIRU whomonitor strain quality for the Australian rhizobial inoculant industry. Higher quality rhizobial inoculant for use byAustralian farmers is a beneficial outcome of this work.
Fingerprinting the Australian rhizobial inoculant mother culturesusing refined PCR protocols yields beneficial
inoculant management applications
www.publish.csiro.au/journals/ajea
CSIRO PUBLISHING

A-M. Vachot-Griffin and J. E. Thies142 Australian Journal of Experimental Agriculture
subcultures of these strains are provided to the Australianinoculant manufacturers who then produce the rhizobialinoculants sold to farmers. Quality assurance requires thatinoculant strains of rhizobia can be identified at a taxonomiclevel as specific as the biovar or strain variant and that theirgenetic stability can be monitored reliably in the laboratoryand the field.
Since the late 1980s, attention has focused on molecular-based methods for characterising bacterial isolates,especially methods that use polymerase chain reaction(PCR) to genetically fingerprint rhizobial taxa (for reviewsee Thies et al. 2001). Until 1998, no molecular techniqueshad been used to characterise or monitor the inoculant strainmother cultures in terms of their identity or genetic andphenotypic stability over time. Serological techniques andhost nodulation tests were the main monitoring methodsemployed. Lack of genetic benchmarking was stronglyunderlined in recent years when controversy arose over(i) variations in strain effectiveness and strain performancein the field, and (ii) observed variations in strain phenotypeover time. In 1998, such a case involving the inoculant strainfor lucerne (WSM688) gave us the opportunity to test theutility and the reliability of characterising strains by PCRfingerprinting (see McInnes et al. 2005). The work presentedhere employs this molecular tool to genetically benchmarkthe Australian commercial inoculant strains to ensure strainidentity and genetic stability on an annual basis and to allowresearchers to follow strain persistence in the field over time.To achieve this aim, optimal DNA extraction methodsneeded to be defined and the sensitivity of these PCR-basedmethods to reaction conditions needed to be increased. Thisdemanded that the protocols be stringently optimised so thatthey could be employed reliably for strain characterisation.Once conditions were optimized, we benchmarked theALIRU mother culture strains in the 1998 collection byPCR-fingerprinting with 5 independent primers: 3 directedtowards repeated sequences, REP, ERIC and BOXA1R(Versalovic et al. 1991); 1 directed towards a conservedsequence in the nifH promoter region, RPO1 (Richardsonet al. 1995); and 1 aimed at amplifying the 16S intergenicspacer sequences, IGS (Laguerre et al. 1996). Severaloligonucleotide primers were used to enhance thediscrimination between strains and the robustness of thisdiscrimination. The ALIRU mother culture collectionconsists of strains that differ from each other at thesubspecies level (e.g. several strains of Rhizobiumleguminosarum bv. trifolii) to the inter-generic taxonomiclevel (e.g. strains of Bradyrhizobium, Rhizobium,Mesorhizobium and Sinorhizobium). Hence, we choseprimers that would provide strain discrimination at thesedifferent levels of taxonomic resolution. Increasing thenumber of markers used for PCR-fingerprinting alsoincreases the chance of detecting genetic variation in aparticular isolate from 1 year to the next.
Materials and methodsRhizobial strains and growth media
The rhizobial strains used in this study and their respective hosts arelisted in Table 1. Single colonies of the bacteria were streaked on yeastextract mannitol agar [YMA (Vincent 1970)] plates and/or inoculatedinto YM broth and incubated at 26°C for 3–5 days for Rhizobium andSinorhizobium strains and 6–10 days for Mesorhizobium andBradyrhizobium strains, before DNA extraction.
Isolation of genomic DNAThree different DNA extraction methods were used: (i) the classic
phenol–chloroform extraction method (Sambrook et al. 1989) usingrhizobia cultured on YMA; (ii) an intermediate method of crude celllysis by freezing, then heating at 96°C, followed by ethanolprecipitation using cells cultured in YM broth; and (iii) crude cell lysisby freezing, then heating at 96°C for 10 min without any furtherpurification step using cells cultured on YMA then placed into 50 µLMilliQ water [Millipore Corp. (Richardson et al. 1995)]. DNAextracted by the classic phenol–chloroform method and by crude lysisfollowed by ethanol precipitation was stored in Tris–EDTA at –20°C,aliquoted, thawed immediately before use and used undiluted. Crudecell lysates were stored at –20°C, thawed immediately before use andused undiluted after precipitation of the cell debris by mildcentrifugation (6000 rpm, 1 min). The crude extracts were renewedevery few weeks, as the DNA extracted by this method degradedrapidly.
Oligonucleotides and PCR reaction conditionsThe oligonucleotides used were synthesised by Life Technology
Inc. (GIBCO BRL Custom Primers, Mulgrave, Vic.). The DNAsequences of the 8 primers used are given in Table 2. PCR reactionswere first performed following published protocols (referenced inTable 2 and referred to as pre-optimisation conditions in Table 3). Theconcentration of each reagent used in the PCR reaction was then testedmethodically against that of all other reagents until the most robust,repeatable profiles for each marker were obtained. Pre- and post-optimisation concentrations for all reagents for each marker are givenin Table 3. PCR reactions (25 µL final volume) were carried out in200 µL Dome Cap PCR tubes (Life Technology Inc., Mulgrave, Vic.)in a PC-960 cooled or gradient model thermal cycler (CorbettResearch, Sydney, NSW). Taq DNA polymerase with Buffer A(Promega Corp., Madison, WI) was used with all primer sets. The IGSprimer set was also tested with TAQ3 DNA polymerase andgelatin-free buffer (Fisher Biotech Inc., Belmont, WA) and AmpliTaqpolymerase (Roche Molecular Systems Inc., Castle Hill, NSW). Forthe RPO1 primer, cycling conditions were 1 cycle at 94°C for 1 min;5 cycles at 92°C for 30 s, 50°C for 2 min, and 72°C for 1 min 30 s;35 cycles at 92°C for 10 s, 55°C for 30 s, and 72°C for 1 min 30 s;1 cycle at 72°C for 10 min; then held briefly at 4°C. For the ERICprimers, cycling conditions were 1 cycle at 94°C for 4 min; 35 cyclesat 94°C for 1 min, 52°C for 1 min, and 65°C for 8 min; 1 cycle at 68°Cfor 16 min; then held briefly at 4°C. For the REP primers, cyclingconditions were 1 cycle at 95°C for 3 min; 35 cycles at 94°C for 1 min,40°C for 1 min, and 65°C for 8 min; 1 cycle at 65°C for 16 min; thenheld briefly at 4°C. For the BOXA1R primer, cycling conditions were1 cycle at 95°C for 3 min; 35 cycles at 90°C for 30 s, 53°C for 1 min,and 65°C for 8 min; 1 cycle at 65°C for 16 min; then held briefly at4°C. For the 16S-23S rRNA intergenic spacer (IGS) primers, cyclingconditions were 1 cycle at 94°C for 3 min; 35 cycles at 94°C for 1 min,55°C for 45 s, and 72°C for 1 min; 1 cycle at 72°C for 10 min; thenheld briefly at 4°C. PCR amplification products (4–7 µL) wereseparated on 2% agarose gels, stained with ethidium bromide,visualised under UV light and photographed using Polaroid Type 667film.

Australian Journal of Experimental Agriculture 143PCR fingerprinting Australian rhizobial inoculant strains
Table 1. List of the 39 mother cultures held in the Australian Legume Inoculant Research Unit 1998 collection that were analysed in this study
StrainA SpeciesB Inoculant strain for:
CB1015 Bradyrhizobium sp. Vigna aconitifolia, V. mungo, V. radiata, V. umbellata, V. unguiculataCB1024 Bradyrhizobium sp. Cajanus cajan, Lablab purpureus, Macrotyloma axillare, M. uniflorumCB1650C Bradyrhizobium sp. Stylosanthes hamataCB1809 B. japonicum Glycine maxCB1923 Bradyrhizobium sp. Centrosema pubescens, C. pascuorumCB2312 Bradyrhizobium sp. Aeschynomene americana, A. falcate, A. villosaCB3035D Bradyrhizobium sp. Cyamopsis tetragonolobaCB3060 Sinorhizobium Leucaena leucocephalaCB3090 Rhizobium sp. Gliricidia sepiaCB3126 Bradyrhizobium sp. Desmanthus spp.CB3481 Bradyrhizobium sp. Stylosanthes seabranaCB376 Bradyrhizobium sp. Lotononis bainesiiCB627 Bradyrhizobium sp. Desmodium intortum, D. uncinatumCB756 Bradyrhizobium sp. Calopogonium mucunoides, Canavalia ensiformis (Brenan ex R. Wilczek) Verdc., Clitoria
ternatea, Macroptilium atropurpureum, M. lathyroides, Mucuna deeringiana, Neonotonia wightii, Psophocarpus tetragonolobus, Pueraria lobata, P. phaseoloides, and some others ofminor economic importance
CB782 R. leguminosarum bv. trifolii Trifolium semipilosumCB82 Bradyrhizobium sp. Stylosanthes erecta, S. fruticosa, S. guianensis var. intermedia (J. Vogel) Hassler, S. humilis,
S. scabra, S. viscosaCC1099 Rhizobium sp. Onobrychis viciifolia (Onobrychis)CC1192 Mesorhizobium ciceri Cicer arietinum, C. ciceriCC1253 Rhizobium sp. Astragalus hamosusCC1335 Rhizobium sp. Hedysarum coronariumCC1502 Mesorhizobium loti Chamaecytisus palmensisCC2483g R. leguminosarum bv. trifolii Trifolium resupinatum (Katzn. et Morley) Zohary & HellerCC283b R. leguminosarum bv. trifolii Trifolium ambiguumCC829 Mesorhizobium loti Lotus pedunculatusCIAT3101 Bradyrhizobium sp. Arachis pintoiNC92 Bradyrhizobium sp. Arachis hypogeae, A. pintoiRCR3644 R. leguminosarum bv. phaseoli Phaseolus lunatus, P. vulgarisSU277 Sinorhizobium meliloti Trigonella foenum-graecumSU303 R. leguminosarum bv. viciae Pisum sativum, Vicia benghalensis, V. ervilia, V. sativa, V. villosa subsp. dasycarpaSU343 Bradyrhizobium sp. Lotus corniculatusTA1 R. leguminosarum bv. trifolii Trifolium alexandrinum, T. dubium, T. fragiferum, T. glomeratum, T. hybridum, T. pratense,
T. purpureum, T. repensWSM1274 R. leguminosarum bv. viciae Lathyrus cicera, L. odoratus, L. sativus, Lens culinaris, Vicia fabaWSM1497 Rhizobium sp. Biserrula pelecinusWSM409 R. leguminsarum bv. trifolii Trifolium cherleri, T. clypeatum, T. glanduliferum, T. hirtum, T. incarnatum, T. michelianum,
T. purpureum, T. subterraneum, T. vesiculosumWSM471 Bradyrhizobium sp. Ornithopus compressus, O. perpusillus, O. pinnatus, O. sativusWSM688 Sinorhizobium meliloti Medicago murex, M. polymorpha, M. rotata, M. rugosa, M. scutellate, M. sphaerocarpus,
M. truncatulaWSM826 Sinorhizobium meliloti Medicago littoralis, M. sativa, M. tornataWU425E Bradyrhizobium sp. Lupinus albus, L. angustifolius, L. cosentinii, L. luteusWU95 R. leguminosarum bv. trifolii Trifolium subterraneum
AStrain designations for original collections: CC, CSIRO Canberra; CB, CSIRO Brisbane; NC, North Carolina; RCR, Rothamsted collection ofRhizobium; SU, Sydney University; TA, Tasmanian Department of Agriculture; WSM, CLIMA/Agriculture Western Australia; WU, University ofWestern Australia.
BThe taxonomy of rhizobia is in a state of flux (Young 1996; Sprent 2001). To the best of our knowledge, the identities given for the 39 strains arecorrect; however, taxonomic authorities may disagree with some of them.
CAmplification profile shown for the RPO1 primer only (Fig. 1c).DAmplification profiles for this strain are not shown in Figures 1 or 2.EAmplification profile shown for the ERIC primer only (Fig. 2c).
A-M. Vachot-Griffin and J. E. Thies144 Australian Journal of Experimental Agriculture
Results and discussionDNA extractions
We tested 3 DNA extraction methods: (i) phenolchloroform; (ii) an intermediate method using freeze–thawlysis with ethanol precipitation; and (iii) crude lysis. Theintermediate and crude lysis DNA extraction methods weretested to see if it was possible to reduce DNA extraction costand time without lowering the quality of the subsequent PCRamplifications. Genomic DNA extracted by thephenol–chloroform method was successfully amplifiedusing the 3 rep-primers (BOXA1R, REP and ERIC) and thedirected primer RPO1 for most of the strains (Table 4). Weexperienced some difficulties with a few strains that produceexcess exopolysaccharide (EPS), such as SU303 (Rhizobiumleguminosarum bv. viciae), CC283b (Rhizobium trifoliibv. trifolii) and CB782 (Rhizobium leguminosarumbv. trifolii), making the protein removal step difficult toperform. However, this didn’t seem to jeopardise the PCRamplification success. Genomic DNA for some strains, inparticular Bradyrhizobium strains displaying dry-looking,hard colonies, was generally not successfully amplified byPCR. Those that proved particularly challenging were theBradyrhizobium strains CB376, CB627, CB1015, CB1024,CB1650, CB1923, CB3481, CIAT3101, WSM471 andWSM425. The intermediate DNA extraction method usingliquid cultures overcame the difficulties encountered during
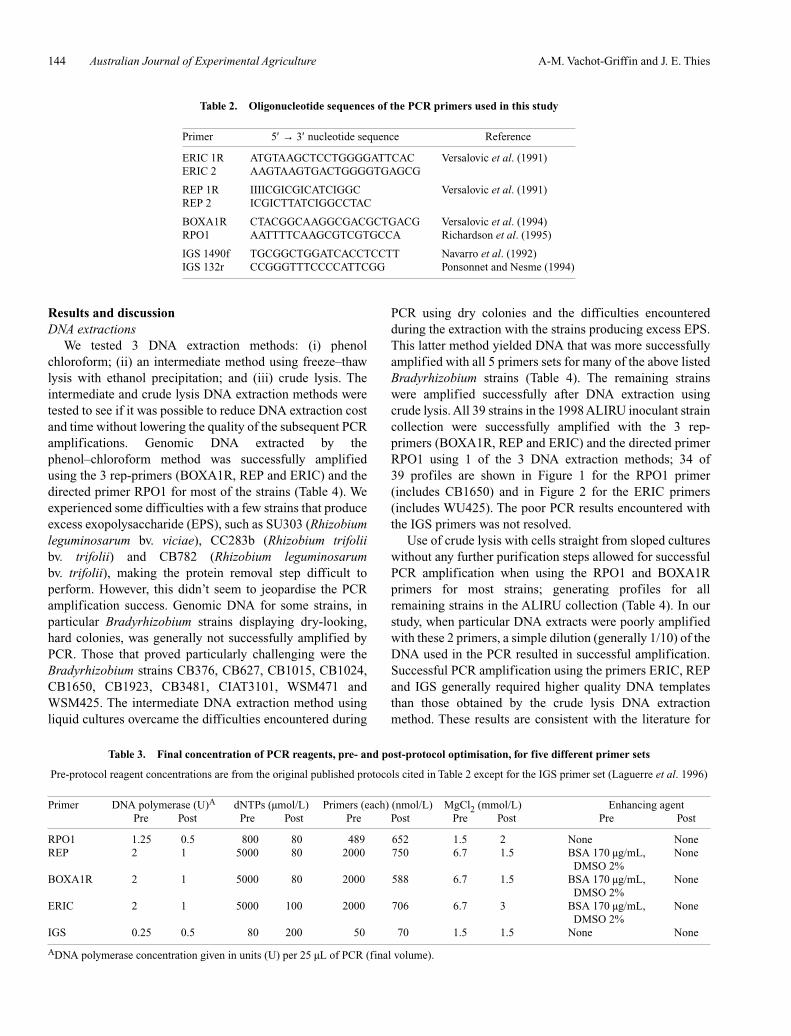
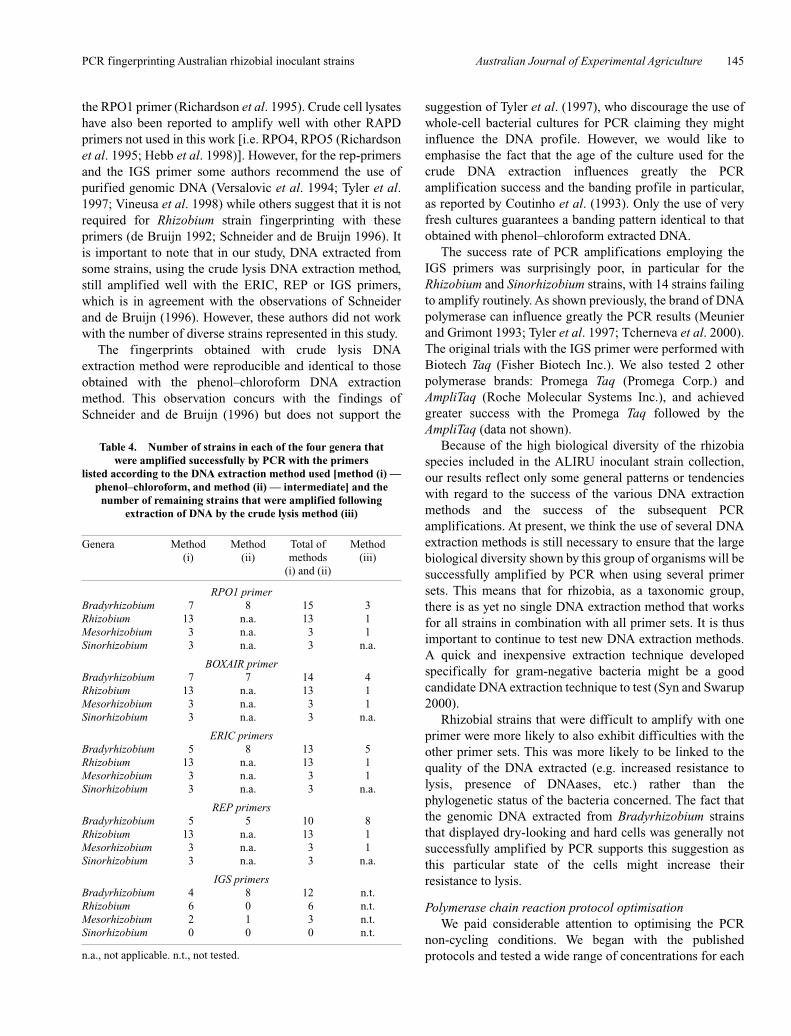
PCR using dry colonies and the difficulties encounteredduring the extraction with the strains producing excess EPS.This latter method yielded DNA that was more successfullyamplified with all 5 primers sets for many of the above listedBradyrhizobium strains (Table 4). The remaining strainswere amplified successfully after DNA extraction usingcrude lysis. All 39 strains in the 1998 ALIRU inoculant straincollection were successfully amplified with the 3 rep-primers (BOXA1R, REP and ERIC) and the directed primerRPO1 using 1 of the 3 DNA extraction methods; 34 of39 profiles are shown in Figure 1 for the RPO1 primer(includes CB1650) and in Figure 2 for the ERIC primers(includes WU425). The poor PCR results encountered withthe IGS primers was not resolved.
Use of crude lysis with cells straight from sloped cultureswithout any further purification steps allowed for successfulPCR amplification when using the RPO1 and BOXA1Rprimers for most strains; generating profiles for allremaining strains in the ALIRU collection (Table 4). In ourstudy, when particular DNA extracts were poorly amplifiedwith these 2 primers, a simple dilution (generally 1/10) of theDNA used in the PCR resulted in successful amplification.Successful PCR amplification using the primers ERIC, REPand IGS generally required higher quality DNA templatesthan those obtained by the crude lysis DNA extractionmethod. These results are consistent with the literature for
Table 2. Oligonucleotide sequences of the PCR primers used in this study
Primer 5′ → 3′ nucleotide sequence Reference
ERIC 1R ATGTAAGCTCCTGGGGATTCAC Versalovic et al. (1991)ERIC 2 AAGTAAGTGACTGGGGTGAGCG
REP 1R IIIICGICGICATCIGGC Versalovic et al. (1991)REP 2 ICGICTTATCIGGCCTAC
BOXA1R CTACGGCAAGGCGACGCTGACG Versalovic et al. (1994)RPO1 AATTTTCAAGCGTCGTGCCA Richardson et al. (1995)
IGS 1490f TGCGGCTGGATCACCTCCTT Navarro et al. (1992)IGS 132r CCGGGTTTCCCCATTCGG Ponsonnet and Nesme (1994)
Table 3. Final concentration of PCR reagents, pre- and post-protocol optimisation, for five different primer sets
Pre-protocol reagent concentrations are from the original published protocols cited in Table 2 except for the IGS primer set (Laguerre et al. 1996)
Primer DNA polymerase (U)A dNTPs (µmol/L) Primers (each) (nmol/L) MgCl2 (mmol/L) Enhancing agentPre Post Pre Post Pre Post Pre Post Pre Post
RPO1 1.25 0.5 800 80 489 652 1.5 2 None NoneREP 2 1 5000 80 2000 750 6.7 1.5 BSA 170 µg/mL, None
DMSO 2%BOXA1R 2 1 5000 80 2000 588 6.7 1.5 BSA 170 µg/mL, None
DMSO 2%ERIC 2 1 5000 100 2000 706 6.7 3 BSA 170 µg/mL, None
DMSO 2%IGS 0.25 0.5 80 200 50 70 1.5 1.5 None None
ADNA polymerase concentration given in units (U) per 25 µL of PCR (final volume).
Australian Journal of Experimental Agriculture 145
the RPO1 primer (Richardson et al. 1995). Crude cell lysateshave also been reported to amplify well with other RAPDprimers not used in this work [i.e. RPO4, RPO5 (Richardsonet al. 1995; Hebb et al. 1998)]. However, for the rep-primersand the IGS primer some authors recommend the use ofpurified genomic DNA (Versalovic et al. 1994; Tyler et al.1997; Vineusa et al. 1998) while others suggest that it is notrequired for Rhizobium strain fingerprinting with theseprimers (de Bruijn 1992; Schneider and de Bruijn 1996). Itis important to note that in our study, DNA extracted fromsome strains, using the crude lysis DNA extraction method,still amplified well with the ERIC, REP or IGS primers,which is in agreement with the observations of Schneiderand de Bruijn (1996). However, these authors did not workwith the number of diverse strains represented in this study.
The fingerprints obtained with crude lysis DNAextraction method were reproducible and identical to thoseobtained with the phenol–chloroform DNA extractionmethod. This observation concurs with the findings ofSchneider and de Bruijn (1996) but does not support the
suggestion of Tyler et al. (1997), who discourage the use ofwhole-cell bacterial cultures for PCR claiming they mightinfluence the DNA profile. However, we would like toemphasise the fact that the age of the culture used for thecrude DNA extraction influences greatly the PCRamplification success and the banding profile in particular,as reported by Coutinho et al. (1993). Only the use of veryfresh cultures guarantees a banding pattern identical to thatobtained with phenol–chloroform extracted DNA.
The success rate of PCR amplifications employing theIGS primers was surprisingly poor, in particular for theRhizobium and Sinorhizobium strains, with 14 strains failingto amplify routinely. As shown previously, the brand of DNApolymerase can influence greatly the PCR results (Meunierand Grimont 1993; Tyler et al. 1997; Tcherneva et al. 2000).The original trials with the IGS primer were performed withBiotech Taq (Fisher Biotech Inc.). We also tested 2 otherpolymerase brands: Promega Taq (Promega Corp.) andAmpliTaq (Roche Molecular Systems Inc.), and achievedgreater success with the Promega Taq followed by theAmpliTaq (data not shown).
Because of the high biological diversity of the rhizobiaspecies included in the ALIRU inoculant strain collection,our results reflect only some general patterns or tendencieswith regard to the success of the various DNA extractionmethods and the success of the subsequent PCRamplifications. At present, we think the use of several DNAextraction methods is still necessary to ensure that the largebiological diversity shown by this group of organisms will besuccessfully amplified by PCR when using several primersets. This means that for rhizobia, as a taxonomic group,there is as yet no single DNA extraction method that worksfor all strains in combination with all primer sets. It is thusimportant to continue to test new DNA extraction methods.A quick and inexpensive extraction technique developedspecifically for gram-negative bacteria might be a goodcandidate DNA extraction technique to test (Syn and Swarup2000).
Rhizobial strains that were difficult to amplify with oneprimer were more likely to also exhibit difficulties with theother primer sets. This was more likely to be linked to thequality of the DNA extracted (e.g. increased resistance tolysis, presence of DNAases, etc.) rather than thephylogenetic status of the bacteria concerned. The fact thatthe genomic DNA extracted from Bradyrhizobium strainsthat displayed dry-looking and hard cells was generally notsuccessfully amplified by PCR supports this suggestion asthis particular state of the cells might increase theirresistance to lysis.
Polymerase chain reaction protocol optimisationWe paid considerable attention to optimising the PCR
non-cycling conditions. We began with the publishedprotocols and tested a wide range of concentrations for each
PCR fingerprinting Australian rhizobial inoculant strains
Table 4. Number of strains in each of the four genera thatwere amplified successfully by PCR with the primers
listed according to the DNA extraction method used [method (i) —phenol–chloroform, and method (ii) — intermediate] and the
number of remaining strains that were amplified followingextraction of DNA by the crude lysis method (iii)
Genera Method Method Total of Method (i) (ii) methods (iii)
(i) and (ii)
RPO1 primerBradyrhizobium 7 8 15 3Rhizobium 13 n.a. 13 1Mesorhizobium 3 n.a. 3 1Sinorhizobium 3 n.a. 3 n.a.
BOXAIR primerBradyrhizobium 7 7 14 4Rhizobium 13 n.a. 13 1Mesorhizobium 3 n.a. 3 1Sinorhizobium 3 n.a. 3 n.a.
ERIC primersBradyrhizobium 5 8 13 5Rhizobium 13 n.a. 13 1Mesorhizobium 3 n.a. 3 1Sinorhizobium 3 n.a. 3 n.a.
REP primersBradyrhizobium 5 5 10 8Rhizobium 13 n.a. 13 1Mesorhizobium 3 n.a. 3 1Sinorhizobium 3 n.a. 3 n.a.
IGS primersBradyrhizobium 4 8 12 n.t.Rhizobium 6 0 6 n.t.Mesorhizobium 2 1 3 n.t.Sinorhizobium 0 0 0 n.t.
n.a., not applicable. n.t., not tested.
A-M. Vachot-Griffin and J. E. Thies146 Australian Journal of Experimental Agriculture
reagent to define those concentrations, in combination, thatyielded the most robust, highly repeatable DNA profiles. Thenecessity to further optimise previously published PCRprotocols arose because, in our experience and that of others(A. McInnes pers. comm.; J. Howieson pers. comm.), theprotocols employing directed (RPO1) or repeated sequence(REP, ERIC, BOXA1R) primers are prone to variation inamplification profiles. This is likely due to the potential formis-priming when using low stringency PCR reactionconditions. This point is exemplified in Table 3, whichdetails the non-cycling parameters for several PCR
fingerprinting protocols. In most cases, the PCR reactionmix contains high concentrations of MgCl2 (Versalovic et al.1991, 1994) and/or a high concentration of DNApolymerase, oligonucleotides or dNTPs (Richardson et al.1995; Versalovic et al. 1991, 1994). Other often citedpublications using rep-primers (de Bruijn 1992; Laguerreet al. 1996; Niemann et al. 1997; Vineusa et al. 1998) alsoemploy an excess of reagents in the PCR reaction mix. Useof these reagents in excess of reaction needs is known todecrease the PCR product specificity (Roux 1995; Tyleret al. 1997). As a consequence, the fingerprint patterns
(a)
MW
M
MW
M
WS
M47
1
CC
283b
CC
2483
g
MW
M
SU
277
WS
M68
8
CB
3126
CB
3090
CB
3060
SU
303
WS
M82
6
CB
1809
800 bp
MW
M
MW
M
NC
92
CC
829
CC
1335
MW
M
CC
1192
CC
1099
CB
3481
CB
627
CB
756
CB
782
CB
82
RC
R36
44800 bp
(b)
MW
M
MW
M
CB
1923
CB
1650
CB
1024
MW
M
CB
1015
WU
95
SU
343
TA
1W
SM
1274
WS
M14
97
WS
M40
9
CB
2312
800 bp
(c)
Figure 1. RPO1 fingerprints of 32 strains from the 1998ALIRU collection (a, b and c). Strain CB82 shown in (b)was from the CSIRO Brisbane collection. MWM, 100 bpmolecular weight marker. DNA used in these PCRs wasextracted primarily using the crude lysis method.

Australian Journal of Experimental Agriculture 147
obtained with these protocols generally have a considerablenumber of poorly resolved bands (de Bruijn 1992; Versalovicet al. 1994), which are not present consistently reaction toreaction (Richardson et al. 1995; Tyler et al. 1997;Tcherneva et al. 2000). Contamination in the no-templateDNA PCR control that varies from a smear to distinctbanding may also become a recurrent problem (Richardsonet al. 1995; Meunier and Grimont 1993; Tyler et al. 1997;Tcherneva et al. 2000). These observations lead us toquestion the reaction specificity, and make the interpretationof patterns obtained difficult and data analysis unreliable.Non-cycling parameters are not the only parametersaffecting PCR reaction specificity (Roux 1995); however, we
chose to concentrate principally on optimising non-cyclingparameters in this study. Standardisation of all other aspectsof PCR-fingerprinting, including number of thermal cyclesand denaturation, annealing and extension times andtemperatures, is also critical for comparing the integrity ofstrains held in collections from year-to-year and forevaluating variation in inoculant strains re-isolated fromdifferent field sites over time.
A comparison between the post-optimisation PCRprotocols for the primers RPO1, REP, ERIC, BOXA1R andIGS and the original published protocols in terms of PCRreagent final concentrations is given in Table 3. The finalconcentration of Taq polymerase and dNTPs in the PCR
PCR fingerprinting Australian rhizobial inoculant strains
MW
M
MW
M
CB
3090
CB
3060
SU
303
WS
M68
8W
SM
826
MW
M
CB
1809
WS
M47
1C
C28
3bC
C24
83b
SU
277
CB
3126
800 bp
(a)
800 bp
(b)
(c)
MW
M
MW
M
NC
92
CC
829
CC
133
5
MW
M
CC
119
2C
C10
99
CB
3481
CB
627
CB
756
CB
782
CB
82
RC
R3
644
MW
M
MW
M
CB
1923
WU
425
CB
1024
MW
M
CB
1015
WU
95
SU
343
TA
1
WS
M12
74W
SM
1497
WS
M40
9
CB
2312
800 bp
Figure 2. ERIC fingerprints of 32 strains from the 1998ALIRU collection (a, b and c). Strain CB82 shown in (b) wasfrom the CSIRO Brisbane collection. MWM, 100 bpmolecular weight marker. DNA used in these PCRs wasextracted primarily using the crude lysis method.

A-M. Vachot-Griffin and J. E. Thies148 Australian Journal of Experimental Agriculture
reaction has been significantly reduced for 4 of the 5 primers(RPO1, REP, ERIC, BOXA1R) and the final concentrationof primers and MgCl2 have also been significantly reducedfor 3 of the 5 primers (REP, ERIC, BOXA1R), comparedwith the originally published protocols (Versalovic et al.1991). All of these combined changes are known tosignificantly increase the PCR product specificity (Roux1995). These changes also allow a great savings in terms ofcost and the time required for stock management. No extraenhancing agents (e.g. BSA, DMSO) were used in any of thepost-optimisation PCR protocols as their use did not improvethe success rate and/or the quality of the amplification. Byeliminating these agents, we also eliminated importantpotential sources of PCR contamination. Modifications toreported protocols, for the rep-primers in particular, are notuncommon [ERIC primer (Di Giovanni et al. 1999), REPprimer (Di Meo et al. 2000) and BOXA1R primer (Jeong andMyrold 1999)]. However, to our knowledge, this study is theonly study that has focused on a large panel of commercialrhizobial inoculant strains and that has modified the originalPCR non-cycling protocols of RPOI, REP, ERIC andBOXA1R to such an extent.
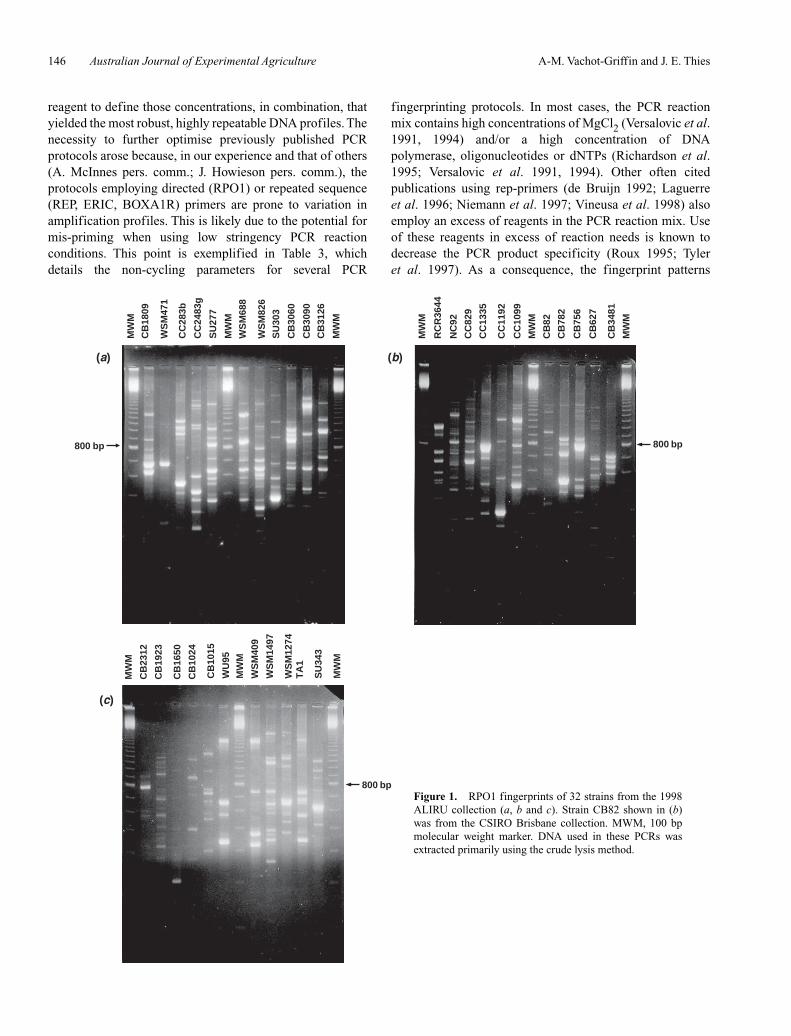
A comparison of the fingerprints obtained using theoriginal and optimised protocols with the ERIC primer setfor 2 strains is given in Figure 3. As expected, the morestringent post-optimisation protocol reduced the number ofDNA bands generated and in particular, secondary DNAbands, which are not always consistent between PCRs from
the same DNA aliquot. The post-optimisation protocol alsoeliminated hazing observed in the no-template DNA PCRcontrol and reduced that observed in the profiles for the2 strains shown. This protocol not only gave morereproducible results at a reduced cost, but also improved theinterpretation of the banding patterns and thus made the dataanalysis more robust. To illustrate this, 34 of the 39 ALIRUmother culture strain fingerprints obtained with the post-optimisation PCR protocols are shown for the RPO1 primerin Figure 1 and for the ERIC primer in Figure 2 (with strainWU425 substituted for strain CB1650 in panel 3c). In theinterest of space, 5 strains analysed infrequently are notpictured (CB376, CB3035, CC1253, CC1502, CIAT3101).
Unlike the fingerprints generated with the rep-primer setsor the directed primers, the unique band generated by theIGS primer set showed very limited diversity in terms offragment size, with a range from 900 to 1500 base pairs [bp(data not shown)]. This result agrees with previousobservations that amplification using the IGS primer alone isunable to resolve differences between closely related species(Young and Haukka 1996). After amplification, treatment ofthe PCR product by sequencing or by restriction fragmentlength polymorphism (RFLP) analysis would overcome thislimitation and further discriminate between closely relatedrhizobia strains, as shown previously (Laguerre et al. 1996;Segundo et al. 1999; Vineusa et al. 1998).
An important issue that had to be addressed during theoptimisation process was the sensitivity of the PCR reactionto concentrations of reaction mix components as expressedby the reproducibility of resulting PCR fingerprints. Asreported before in the literature (Richardson et al. 1995), weobserved that some of the DNA fragments generated,particularly those of minor intensity (secondary bands), werenot always consistently amplified on each occasion. With thestringency of our post-optimisation protocols, theseobservations were very rare in our study and when observed,were closely linked to either the age of the extracted DNA orthe freshness of the cell culture (Coutinho et al. 1993). Insuch instances, some variation in the intensity of thesecondary bands generated was observed. This, however, didnot compromise the ability to recognise the identity of thefingerprints particularly because the polymorphismsgenerated between the strains remained very high for the4 primer sets (RPO1, REP, ERIC, BOXA1R) and were verydiscriminative (Figs 1 and 2).
We never observed contamination or non-specificpriming in the no-template DNA PCR control for the5 different primer sets, which gives us confidence in both thereproducibility and the specificity of the fingerprintsobtained with our post-optimisation protocol.
Beneficial inoculant management applicationsIn Australia, mother cultures of strains used in
commercial inoculants are maintained and monitored by
| CB3090 | NC92 | Control | MWM | P/O P | P/O P | P/O P | MWM
Figure 3. Comparison between ERIC fingerprints obtained with thepublished [P (Versalovic et al. 1991)] and the optimised (P/O) PCRprotocols for strains CB3090 and NC92. Control, no template control.MWM, 100 bp molecular weight marker, bright band = 800 bp. DNA used in these PCRs was extracted using thephenol–chloroform method.

Australian Journal of Experimental Agriculture 149
ALIRU. Each year, strain performance against selectioncriteria are reviewed by the ALIRU Steering Committee.Characteristics evaluated are symbiotic capability on thetarget legume, performance under field conditions, capacityto fix N and selected cultural and biochemical andserological characteristics. Once quality assurance tests havebeen met, mother cultures are subcultured and distributed toinoculant manufacturers for the annual production ofrhizobial inoculants (E. Hartley and G. Gemell pers. comm.).
Lack of genetic benchmarking of these rhizobialinoculants was strongly underlined in recent years whencontroversy arose due to observed variations in strainphenotype over time and variations in strain effectivenessand performance in the field (McInnes et al. 2005; Hartmannet al. 1998).
The genetic fingerprinting of the ALIRU mother culturestrains not only provided an additional robust tool for straincharacterisation, but also led to a significant outcome interms of product quality assurance. Results of the PCRfingerprinting confirmed that 2 strains in the 1998 collectionwere identical for all 5 markers. These strains are CB82, theinoculant strain for fine-stemmed Stylosanthes, and CB756,the inoculant for Siratro. With the help of CSIRO PlantIndustry (Brisbane, Qld), Bio-Care Technology Pty Ltd(Sommersby, NSW) and ALIRU, we determined that this
outcome resulted from a culture mix-up (see Fig. 4). StrainCB82 was replaced accidentally by strain CB756 in the 1998ALIRU collection and, consequently, in the 1999 collectionas well. By typing a sample of the CB82 inoculantmanufactured by Bio-Care Technology in 1997, we were alsoable to show that this mix-up occurred before 1997. Thisdiscovery led to the recall of all faulty inoculant from themarket in 1999 and the supply of the correct inoculant to thefarmers. Another significant outcome was the reconstitutionof the ALIRU culture collection with the correct CB82 strainstock culture, held by the CSIRO in Brisbane, that was usedfor producing the inoculant in 2000. This result illustrates theneed for ensuring strain identity and clearly demonstrates thebenefit of using molecular markers to benchmark inoculantstrains on an annual basis. It is important to underline thefact that without PCR-fingerprinting, this culture confusionmay never have been uncovered. Annual serological testingusing polyclonal antisera prepared more than 10 years agofailed to identify the problem as antisera to these strainscross-reacted (E. Hartley pers. comm.). This situation is inagreement with other such reports in the literature (Hebbet al. 1998; Moawad et al. 1998). It is clear that thecontribution of this work directly benefits the inoculantmanufacturers, the quality assurance governing body(Vachot et al. 1999) and the Australian farming community.
AcknowledgmentsThe authors are grateful to Elizabeth Hartley and Greg
Gemell of ALIRU, Gosford, NSW, for supplying the 1998 and1999 AIRCS rhizobial mother cultures for our analysis. Wethank Alison McInnes, formerly of CSIRO Plant Industry,Brisbane, for revitalising strain CB82 from the CSIROcollection to reinstate it in the ALIRU mother culturecollection, and Gary Bullard formerly of Bio-CareTechnology Pty Ltd (Sommersby, NSW) for supplyingcommercial inoculant for our examination. We thank JudyGray for her technical assistance, and John Brockwell andJohn Howieson for their assistance with chronicling strainhistory and application. This work was supported by theGrains Research and Development Corporation grantUWS20.
ReferencesBrockwell J, Bottomley PJ, Thies JE (1995) Manipulation of rhizobia
microflora for improving legume productivity and soil fertility: acritical assessment. Plant and Soil 174, 143–180. doi:10.1007/BF00032245
de Bruijn FJ (1992) Use of repetitive (Repetitive ExtragenicPalindromic and Enterobacterial Repetitive IntergenericConsensus) sequences and the polymerase chain reaction tofingerprint the genomes of Rhizobium meliloti isolates and othersoil bacteria. Applied and Environmental Microbiology 58,2180–2187.
Coutinho H, Handley B, Kay H, Stevenson L, Beringer J (1993) Theeffect of colony age on PCR fingerprinting. Letters in AppliedMicrobiology 17, 282–284.
PCR fingerprinting Australian rhizobial inoculant strains
1 2 3 4 5 6 7 8 9 10 11
800 bp
| ----- ‘CB756’ ----- | | ------- ‘CB82’ -------- |
MWM MWMMWM
Figure 4. RPO1 amplification profiles of strains labelled CB756(lanes 1–5) and CB82 (lanes 6–11). The profiles illustrate the accidentalsubstitution of strain CB82 with strain CB756 in the 1998 and 1999ALIRU culture collection. Source and year of CB756 strains are lane 1:Bio-Care, 1997; lane 2: ALIRU, 1998 (strain used to generate profilesshown in Figs 2 and 3); lane 3: CSIRO Brisbane, 1991; lane 4: CSIROBrisbane, 1995; lane 5: CSIRO Brisbane (labelled old). Source and yearof CB82 strains are lane 6: Bio-Care, 1997; lane 7: ALIRU, 1999; lane8: ALIRU, 1998; lane 9: CSIRO Brisbane, 1984 (note: this sample wasa contaminant later removed from the CSIRO collection); lane 10:CSIRO Brisbane, 1995 (strain used to reconstitute the ALIRUcollection and to generate the profiles shown in Figs 2 and 3); lane 11:CSIRO Brisbane, 1980. MWM, 100 bp molecular weight marker. DNAused in these PCRs was extracted by the crude lysis method.

A-M. Vachot-Griffin and J. E. Thies150 Australian Journal of Experimental Agriculture
Di Giovanni GD, Watrud LS, Seidler RJ, Widmer F (1999) Comparisonof parental and transgenic alfalfa rhizosphere bacterial communitiesusing Biolog GN metabolic fingerprinting and enterobacterialrepetitive intergenic consensus sequence-PCR (ERIC-PCR).Microbial Ecology 37, 129–139. doi:10.1007/s002489900137
Di Meo C, Wilbur A, Holben W, Feldman R, Vrijenhoek R, Cary S(2000) Genetic variation among endosymbionts of widelydistributed vestimentiferan tubeworms. Applied and EnvironmentalMicrobiology 66, 651–658. doi:10.1128/AEM.66.2.651-658.2000
Giller KE (2001) ‘Nitrogen fixation in tropical cropping systems.’ 2ndedn. (CABI Publishing: Wallingford, UK)
Hartmann A, Giraud JJ, Catroux G (1998) Genotypic diversity ofSinorhizobium (formerly Rhizobium) meliloti strains isolateddirectly from a soil and from nodules of alfalfa (Medicago sativa)grown in the same soil. FEMS Microbiology Ecology 25, 107–116.doi:10.1016/S0168-6496(97)00087-1
Hebb DM, Richardson AE, Reid R, Brockwell J (1998) PCR as anecological tool to determine the establishment and persistence ofRhizobium strains introduced into the field as seed inoculant.Australian Journal of Agricultural Research 49, 923–934.doi:10.1071/A98007
Howieson J, Ballard R (2004) Optimising the legume symbiosis instressful and competitive environments within southern Australia —some contemporary thoughts. Soil Biology and Biochemistry 36,1261–1273.
Jeong S, Myrold D (1999) Genomic fingerprinting of Frankiamicrosymbionts from Ceanothus copopulations using repetitivesequences and polymerase chain reactions. Canadian Journal ofBotany-Revue Canadienne de Botanique 77, 1220–1230.doi:10.1139/cjb-77-9-1220
Laguerre G, Mavingui P, Allard MR, Charnay MP, Louvrier P,Mazurier SI, Rigottier-Gois L, Amarger N (1996) Typing ofrhizobia by PCR DNA fingerprinting and PCR-restriction fragmentlength polymorphism analysis of chromosomal and symbiotic generegions: application to Rhizobium leguminosarum and its differentbiovars. Applied and Environmental Microbiology 62, 2029–2036.
McInnes A, Holford P, Thies JE (2005) Characterisation of dry andmucoid colonies isolated from Australian rhizobial inoculant strainsfor Medicago species. Australian Journal of ExperimentalAgriculture 45, 151–159.
Meunier J, Grimont P (1993) Factors affecting reproducibility ofrandom amplified polymorphic DNA fingerprinting. Research inMicrobiology 144, 373–379. doi:10.1016/0923-2508(93)90194-7
Moawad H, El-Din SM, Abdel-Aziz RA (1998) Improvement ofbiological nitrogen fixation in Egyptian winter legumes throughbetter management of Rhizobium. Plant and Soil 204, 95–106.doi:10.1023/A:1004335112402
Navarro E, Simonet P, Normand P, Bardin R (1992) Characterization ofnatural populations of Nitrobacter spp. using PCR/RFLP analysis ofthe ribosomal intergenic spacer. Archives of Microbiology 157,107–115.
Niemann S, Pühler A, Tichy HV, Simon R, Selbitschka W (1997)Evaluation of the resolving power of three different DNAfingerprinting methods to discriminate among isolates of a naturalRhizobium meliloti population. Journal of Applied Microbiology 82,477–484. doi:10.1046/j.1365-2672.1997.00141.x
Ponsonnet C, Nesme X (1994) Identification of Agrobacterium strainsby PCR-RFLP analysis of PTI and chromosomal regions. Archivesof Microbiology 161, 300–309.
Richardson AE, Viccars LA, Watson JM, Gibson AH (1995)Differentiation of Rhizobium strains using the polymerase chainreaction with random and directed primers. Soil Biology andBiochemistry 27, 515–524. doi:10.1016/0038-0717(95)98626-Y
Roux K (1995) Optimization and troubleshooting in PCR. PCRMethods and Application 4, S185–S194.
Sambrook J, Fritsch EF, Maniatis TA (Eds) (1989) ‘Molecular cloning:a laboratory manual.’ 2nd edn. (Cold Spring Harbor LaboratoryPress: New York)
Schneider M, de Bruijn FJ (1996) Rep-PCR mediated genomicfingerprinting of rhizobia and computer-assisted phylogeneticpattern analysis. World Journal of Microbiology and Biotechnology12, 163–174. doi:10.1007/BF00364681
Segundo E, Martinez-Abarca F, van Dillewijn P, Fernández-López M,Lagares A, Martinez-Drets G, Niehaus K, Pühler A, Toro N (1999)Characterisation of symbiotically efficient alfafa-nodulatingrhizobia isolated from acid soils of Argentina and Uruguay. FEMSMicrobiology Ecology 28, 169–176. doi:10.1016/S0168-6496(98)00102-0
Sprent JI (2001) ‘Nodulation in legumes.’ (Royal Botanic Gardens:Kew, UK)
Syn C, Swarup S (2000) A scalable protocol for the isolation of large-sized genomic DNA within an hour from several bacteria.Analytical Biochemistry 278, 86–90. doi:10.1006/abio.1999.4410
Tcherneva E, Rijpens N, Jersek B, Herman LMF (2000) Differentiationof Brucella species by random amplified polymorphic DNAanalysis. Journal of Applied Microbiology 88, 69–80. doi:10.1046/j.1365-2672.2000.00945.x
Thies JE, Holmes E, Vachot AM (2001) Application of moleculartechniques to studies in Rhizobium ecology: a review. AustralianJournal of Experimental Agriculture 41, 299–319. doi:10.1071/EA99171
Thies JE, Singleton PW, Bohlool BB (1991) Influence of the size ofindigenous rhizobial populations on establishment and symbioticperformance of introduced rhizobia on field-grown legumes.Applied and Environmental Microbiology 57, 19–28.
Tyler KD, Wang G, Tyler SD, Johnson WM (1997) Factors affectingreliability and reproducibility of amplification-based DNAfingerprinting of representative bacterial pathogens. Journal ofClinical Microbiology 35, 339–346.
Vachot AM, Holmes EM, Thies JE (1999) Fingerprinting of the AIRCSmother culture isolates: PCR optimisation and strain specificity. In‘The 12th Australian nitrogen fixation conference’. (Eds J Slattery,E Curran) pp. 20–21. (The Australian Society for Nitrogen Fixation:Wagga Wagga, NSW)
Versalovic J, Koeuth T, Lupski JR (1991) Distribution of repetitiveDNA sequences in eubacteria and application to fingerprinting ofbacterial genomes. Nucleic Acids Research 19, 6823–6831.
Versalovic J, Schneider M, de Bruijn FJ, Lupski JR (1994) Genomicfingerprinting of bacteria using repetitive sequence-basedpolymerase chain reaction. Methods in Molecular and CellularBiology 5, 25–40.
Vincent JM (1970) ‘Manual for the practical study of root-nodulebacteria.’ (Blackwell Scientific: Oxford)
Vineusa P, Rademaker JLW, de Bruijn FJ, Werner D (1998) Genotypiccharacterisation of Bradyrhizobium strains nodulating endemicwoody legumes of the Canary Islands by PCR-restriction fragmentlength polymorphism analysis of genes encoding 16S rRNA (16SrDNA) and 16S-23S rDNA intergenic spacers, repetitive extragenicpalindromic PCR genomic fingerprinting, and partial 16S rDNAsequencing. Applied and Environmental Microbiology 64,2096–2104.
Young JPW (1996) Phylogeny and taxonomy of rhizobia. Plant and Soil186, 45–52. doi:10.1007/BF00035054
Young JPW, Haukka KE (1996) Diversity and phylogeny of rhizobia.The New Phytologist 133, 87–94.
Received 28 March 2004, accepted 9 February 2005
http://www.publish.csiro.au/journals/ajea