erk activity facilitates activation of the s-phase dna damage checkpoint by modulating atr function
TRANSCRIPT

ORIGINAL ARTICLE
ERK activity facilitates activation of the S-phase DNA damage checkpoint
by modulating ATR function
D Wu1,4, B Chen2,4, K Parihar1,2,4, L He2, C Fan1,2, J Zhang,1,2, L Liu1, A Gillis1, A Bruce2,A Kapoor3 and D Tang1,2
1Father Sean O’Sullivan Research Institute, St Joseph’s Hospital, Hamilton, Ontario, Canada; 2Division of Nephrology, Departmentof Medicine, McMaster University, Hamilton, Ontario, Canada and 3Department of Surgery, McMaster University, Hamilton,Ontario, Canada
Although Erk kinase has been recently reported tofunction in the DNA damage response, the mechanismgoverning this process is unknown. We report here thathydroxyurea (HU) activates Erk via MEK1, a processthat is sensitized by a constitutively active MEK1(MEK1Q56P) and attenuated by a dominant-negativeMEK1 (MEK1K97M). While ectopic MEK1Q56P sensi-tized HU-induced S-phase arrest, inhibition of Erkactivation via U0126, PD98059, and MEK1K97Mattenuated the arrest, and thereby enhanced cells toHU-induced toxicity. Taken together, we demonstrate animportant contribution of Erk to the activation of theS-phase DNA damage checkpoint. This can be attributedto Erk’s regulatory role in modulating ATR function.Inhibition of Erk activation with U0126/PD98059 andMEK1K97M substantially reduced HU-induced ATRnuclear foci, leading to a dramatic reduction of cH2AXand its nuclear foci. Reduction of MEK1 function by asmall interference RNA (siRNA) MEK1 and ectopicMEK1K97M significantly decreased HU-inducedcH2AX. Conversely, ectopic MEK1Q56P enhancedcH2AX foci. Furthermore, immunofluorescent and cellfractioning experiments revealed cytosolic and nuclearlocalization of ATR. HU treatment caused the redistri-bution of ATR from the cytosol to the nucleus, a processthat is inhibited by U0126. Collectively, we show that Erkkinase modulates HU-initiated DNA damage response byregulating ATR function.Oncogene (2006) 25, 1153–1164. doi:10.1038/sj.onc.1209148;published online 26 September 2005
Keywords: hydroxyurea; MEK1; Erk; ATR; S-phasecheckpoint
Introduction
Eukaryotic cells employ multiple mechanisms to ensureaccurate transmission of genetic information between
generations. Critical surveillance of this transmission isprovided by the DNA damage response (Zhou andElledge, 2000). Disruption or attenuation of thisresponse plays an essential role in promoting tumorigen-esis (Lengauer et al., 1998; Hoeijmakers, 2001; Schar,2001; Rouse and Jackson, 2002). Stalled replicationforks are sensed by replication protein A (RPA)-mediatedATR (ATM- and Rad3-related kinase) recruitment toDNA damage-induced nuclear foci (Zou and Elledge,2003). ATR and its related ATM kinases play centralroles in the amplification of double-stranded break(DSB) signals, coordination of DNA damage repair andactivation of cell cycle checkpoints (Zhou and Elledge,2000; Shiloh, 2003).One of the first events initiated by DSBs is the
phosphorylation of serine 139 in the SQE motif locatedon the tail of histone H2AX (gH2AX) by ATM/ATRkinases, with subsequent rapid formation of gH2AX fociat the DSB sites (Redon et al., 2002). These foci areessential in facilitating the assembly of repair factors,including Brca1 and the MRE11–RAD50–NBS1 com-plex, on damaged DNA (Paull et al., 2000; Celeste et al.,2002), and also aid in the transduction of DNA damagesignals by binding to 53BP1 and MDC1 (Fernandez-Capetillo et al., 2002; Stewart et al., 2003). gH2AX focialso form at sites of physiological DSBs in lymphocytesand germ cells (Chen et al., 2000; Mahadevaiah et al.,2001; Petersen et al., 2001; Fernandez-Capetillo et al.,2003). Knocking out H2AX produces mice with immunedeficiency and male infertility (Celeste et al., 2002), whileloss or reduction of H2AX compromises genomestability and facilitates tumorigenesis (Celeste et al.,2002; Bassing et al., 2003; Celeste et al., 2003). Takentogether, gH2AX foci play an essential role in thecellular DNA damage response.Cell cycle progression is regulated by Erk (extra-
cellular signal regulated) kinase, a member of themitogen-activated protein kinase family (Kolch, 2000;Johnson and Lapadat, 2002). Erk is activated by itsupstream kinases, MEK1 and MEK2 (Krepinsky et al.,2002). Activation of Erk is sufficient to transformNIH3T3 cells or mouse embryonic fibroblast (MEF)lacking either p53 or p16 (Cowley et al., 1994; Lin et al.,1998). However, Erk may also play a role in the cellularDNA damage response, which is a tumour suppressionprocess. Erk activation was observed in response to
Received 20 May 2005; revised 10 August 2005; accepted 23 August2005; published online 26 September 2005
Correspondence: Dr D Tang, T3310, St Joseph’s Hospital, 50 CharltonAve East, Hamilton, Ontario, Canada L8N 4A6.E-mail: [email protected] authors contributed equally to this work
Oncogene (2006) 25, 1153–1164& 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00
www.nature.com/onc
multiple DNA damage stimuli (Lee et al., 2000; Personset al., 2000; Wang et al., 2000; Tang et al., 2002; Pippinet al., 2003) in either a p53-dependent (Lee et al., 2000)or independent manner (Tang et al., 2002). However,the mechanisms responsible for Erk modulation of theDNA damage response are unknown. While Erk activityfacilitates cisplatin- and etoposide-induced apoptosisand cell cycle arrest (Wang et al., 2000; Tang et al.,2002), it antagonizes C5b-9-induced DNA damage inpodocytes (Pippin et al., 2003).We report here that Erk kinase contributes to
hydroxyurea (HU)-induced S-phase DNA checkpointactivation by regulating ATR function. HU induces Erkactivation in several cell lines. Inhibition of Erkactivation by specific MEK inhibitors U0126 andPD98059 reduces HU-induced ATR nuclear foci,leading to the reduction of gH2AX nuclear foci.Furthermore, we demonstrate for the first time that asmall portion of ATR resides in the cytosol and that HUtreatment leads to complete nuclear localization ofATR. Inhibition of Erk activation prevents this ATRredistribution in response to HU. Thus, Erk kinase playsan important role in modulation of the DNA damageresponse.
Results
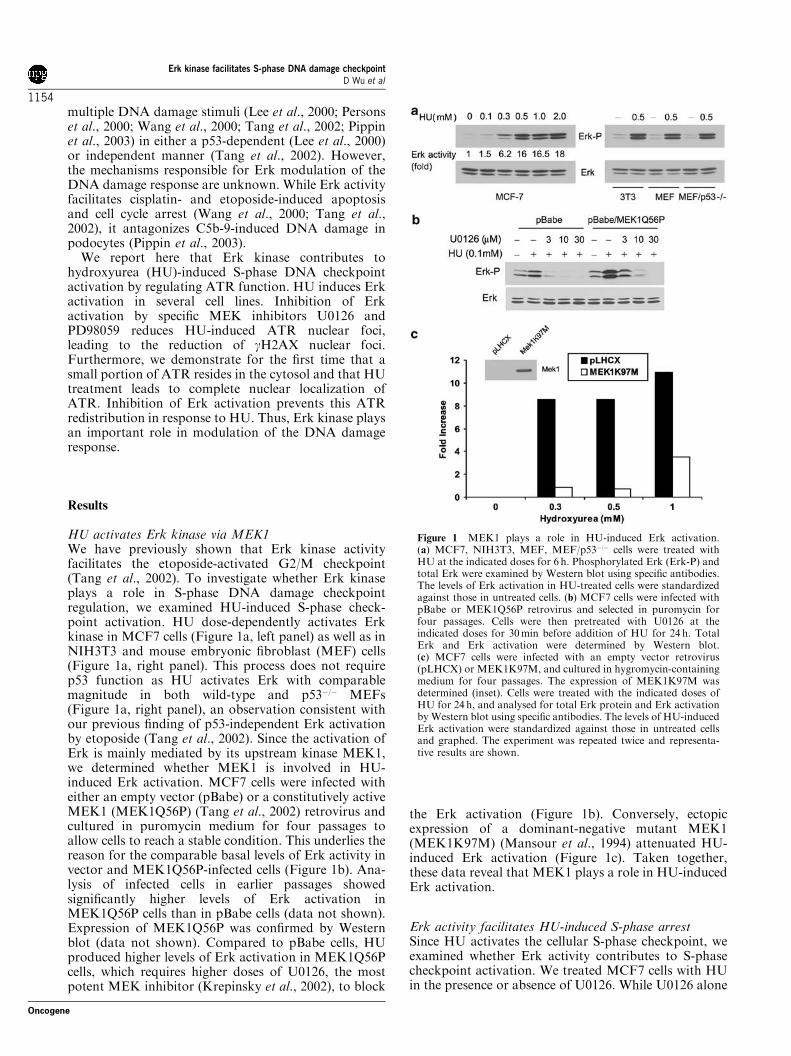
HU activates Erk kinase via MEK1We have previously shown that Erk kinase activityfacilitates the etoposide-activated G2/M checkpoint(Tang et al., 2002). To investigate whether Erk kinaseplays a role in S-phase DNA damage checkpointregulation, we examined HU-induced S-phase check-point activation. HU dose-dependently activates Erkkinase in MCF7 cells (Figure 1a, left panel) as well as inNIH3T3 and mouse embryonic fibroblast (MEF) cells(Figure 1a, right panel). This process does not requirep53 function as HU activates Erk with comparablemagnitude in both wild-type and p53�/� MEFs(Figure 1a, right panel), an observation consistent withour previous finding of p53-independent Erk activationby etoposide (Tang et al., 2002). Since the activation ofErk is mainly mediated by its upstream kinase MEK1,we determined whether MEK1 is involved in HU-induced Erk activation. MCF7 cells were infected witheither an empty vector (pBabe) or a constitutively activeMEK1 (MEK1Q56P) (Tang et al., 2002) retrovirus andcultured in puromycin medium for four passages toallow cells to reach a stable condition. This underlies thereason for the comparable basal levels of Erk activity invector and MEK1Q56P-infected cells (Figure 1b). Ana-lysis of infected cells in earlier passages showedsignificantly higher levels of Erk activation inMEK1Q56P cells than in pBabe cells (data not shown).Expression of MEK1Q56P was confirmed by Westernblot (data not shown). Compared to pBabe cells, HUproduced higher levels of Erk activation in MEK1Q56Pcells, which requires higher doses of U0126, the mostpotent MEK inhibitor (Krepinsky et al., 2002), to block
the Erk activation (Figure 1b). Conversely, ectopicexpression of a dominant-negative mutant MEK1(MEK1K97M) (Mansour et al., 1994) attenuated HU-induced Erk activation (Figure 1c). Taken together,these data reveal that MEK1 plays a role in HU-inducedErk activation.
Erk activity facilitates HU-induced S-phase arrestSince HU activates the cellular S-phase checkpoint, weexamined whether Erk activity contributes to S-phasecheckpoint activation. We treated MCF7 cells with HUin the presence or absence of U0126. While U0126 alone
Figure 1 MEK1 plays a role in HU-induced Erk activation.(a) MCF7, NIH3T3, MEF, MEF/p53�/� cells were treated withHU at the indicated doses for 6 h. Phosphorylated Erk (Erk-P) andtotal Erk were examined by Western blot using specific antibodies.The levels of Erk activation in HU-treated cells were standardizedagainst those in untreated cells. (b) MCF7 cells were infected withpBabe or MEK1Q56P retrovirus and selected in puromycin forfour passages. Cells were then pretreated with U0126 at theindicated doses for 30min before addition of HU for 24 h. TotalErk and Erk activation were determined by Western blot.(c) MCF7 cells were infected with an empty vector retrovirus(pLHCX) or MEK1K97M, and cultured in hygromycin-containingmedium for four passages. The expression of MEK1K97M wasdetermined (inset). Cells were treated with the indicated doses ofHU for 24 h, and analysed for total Erk protein and Erk activationby Western blot using specific antibodies. The levels of HU-inducedErk activation were standardized against those in untreated cellsand graphed. The experiment was repeated twice and representa-tive results are shown.
Erk kinase facilitates S-phase DNA damage checkpointD Wu et al
1154
Oncogene
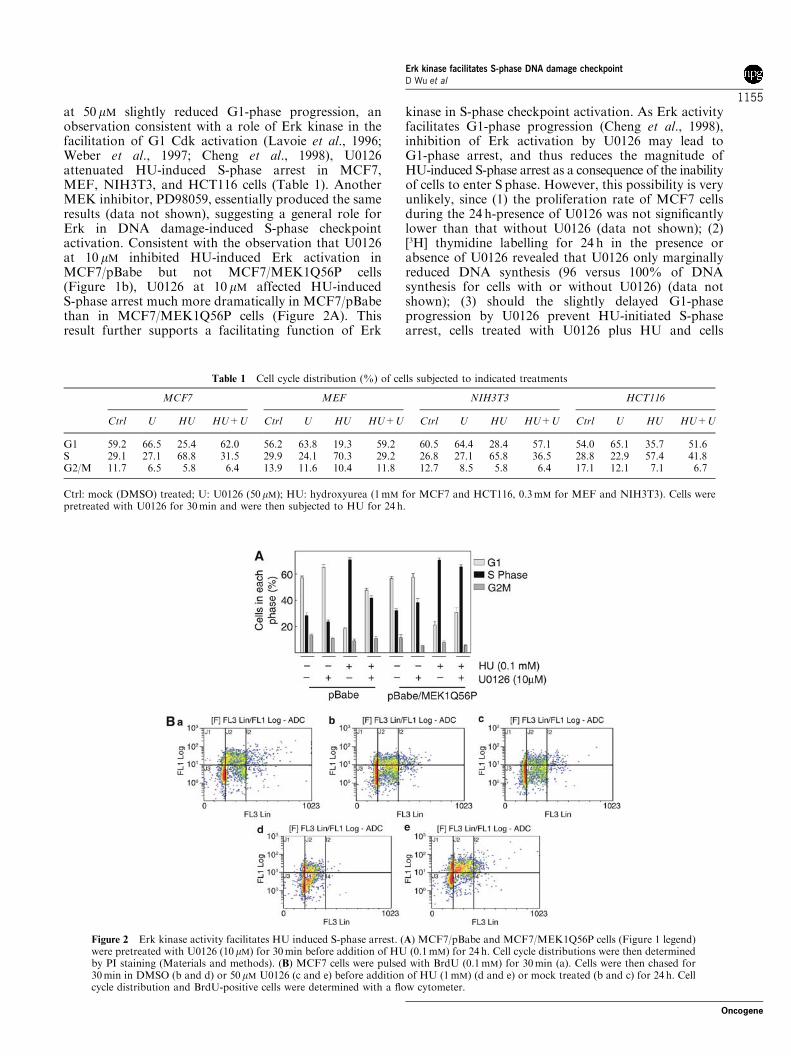
at 50 mM slightly reduced G1-phase progression, anobservation consistent with a role of Erk kinase in thefacilitation of G1 Cdk activation (Lavoie et al., 1996;Weber et al., 1997; Cheng et al., 1998), U0126attenuated HU-induced S-phase arrest in MCF7,MEF, NIH3T3, and HCT116 cells (Table 1). AnotherMEK inhibitor, PD98059, essentially produced the sameresults (data not shown), suggesting a general role forErk in DNA damage-induced S-phase checkpointactivation. Consistent with the observation that U0126at 10 mM inhibited HU-induced Erk activation inMCF7/pBabe but not MCF7/MEK1Q56P cells(Figure 1b), U0126 at 10 mM affected HU-inducedS-phase arrest much more dramatically in MCF7/pBabethan in MCF7/MEK1Q56P cells (Figure 2A). Thisresult further supports a facilitating function of Erk
kinase in S-phase checkpoint activation. As Erk activityfacilitates G1-phase progression (Cheng et al., 1998),inhibition of Erk activation by U0126 may lead toG1-phase arrest, and thus reduces the magnitude ofHU-induced S-phase arrest as a consequence of the inabilityof cells to enter S phase. However, this possibility is veryunlikely, since (1) the proliferation rate of MCF7 cellsduring the 24 h-presence of U0126 was not significantlylower than that without U0126 (data not shown); (2)[3H] thymidine labelling for 24 h in the presence orabsence of U0126 revealed that U0126 only marginallyreduced DNA synthesis (96 versus 100% of DNAsynthesis for cells with or without U0126) (data notshown); (3) should the slightly delayed G1-phaseprogression by U0126 prevent HU-initiated S-phasearrest, cells treated with U0126 plus HU and cells
Table 1 Cell cycle distribution (%) of cells subjected to indicated treatments
MCF7 MEF NIH3T3 HCT116
Ctrl U HU HU+U Ctrl U HU HU+U Ctrl U HU HU+U Ctrl U HU HU+U
G1 59.2 66.5 25.4 62.0 56.2 63.8 19.3 59.2 60.5 64.4 28.4 57.1 54.0 65.1 35.7 51.6S 29.1 27.1 68.8 31.5 29.9 24.1 70.3 29.2 26.8 27.1 65.8 36.5 28.8 22.9 57.4 41.8G2/M 11.7 6.5 5.8 6.4 13.9 11.6 10.4 11.8 12.7 8.5 5.8 6.4 17.1 12.1 7.1 6.7
Ctrl: mock (DMSO) treated; U: U0126 (50 mM); HU: hydroxyurea (1mM for MCF7 and HCT116, 0.3mM for MEF and NIH3T3). Cells werepretreated with U0126 for 30min and were then subjected to HU for 24 h.
Figure 2 Erk kinase activity facilitates HU induced S-phase arrest. (A) MCF7/pBabe and MCF7/MEK1Q56P cells (Figure 1 legend)were pretreated with U0126 (10 mM) for 30min before addition of HU (0.1mM) for 24 h. Cell cycle distributions were then determinedby PI staining (Materials and methods). (B) MCF7 cells were pulsed with BrdU (0.1mM) for 30min (a). Cells were then chased for30min in DMSO (b and d) or 50 mM U0126 (c and e) before addition of HU (1mM) (d and e) or mock treated (b and c) for 24 h. Cellcycle distribution and BrdU-positive cells were determined with a flow cytometer.
Erk kinase facilitates S-phase DNA damage checkpointD Wu et al
1155
Oncogene
treated with U0126 only should display comparableportions of G1-phase cells. However, the G1-phase cellsfor the U0126þHU population is significantly less thanthe U0126 population and the control population(Table 1, Figure 2A). This indicates that the delayedS-phase entry associated with U0126 is not thepredominant event that reduces HU-induced S-phasearrest by preventing cells entry into S phase.To further exclude such a possibility, S-phase
progression in response to HU with or without U0126was determined. MCF7 cells were briefly pulsed with50-bromo-2-deoxyuridine (BrdU) for 30min to label theS-phase cells (Figure 2Ba). A 24-h chase resulted inprogression of the majority of BrdU-positive cells intoG1 (Figure 2Bb, red BrdU-positive population), whichwas not obviously affected by U0126 at 50 mM (Figure2Bc). As expected, HU prevented S-phase progression(Figure 2Bd). Addition of U0126 partially releasedthe S-phase arrest, as cells advanced further intoS phase with U0126 (Figure 2Be) than those without(Figure 2Bd). Similar results were also obtained inNIH3T3 cells (data not shown). Taken together, thesedata show that Erk activity facilitates HU-inducedS-phase arrest.
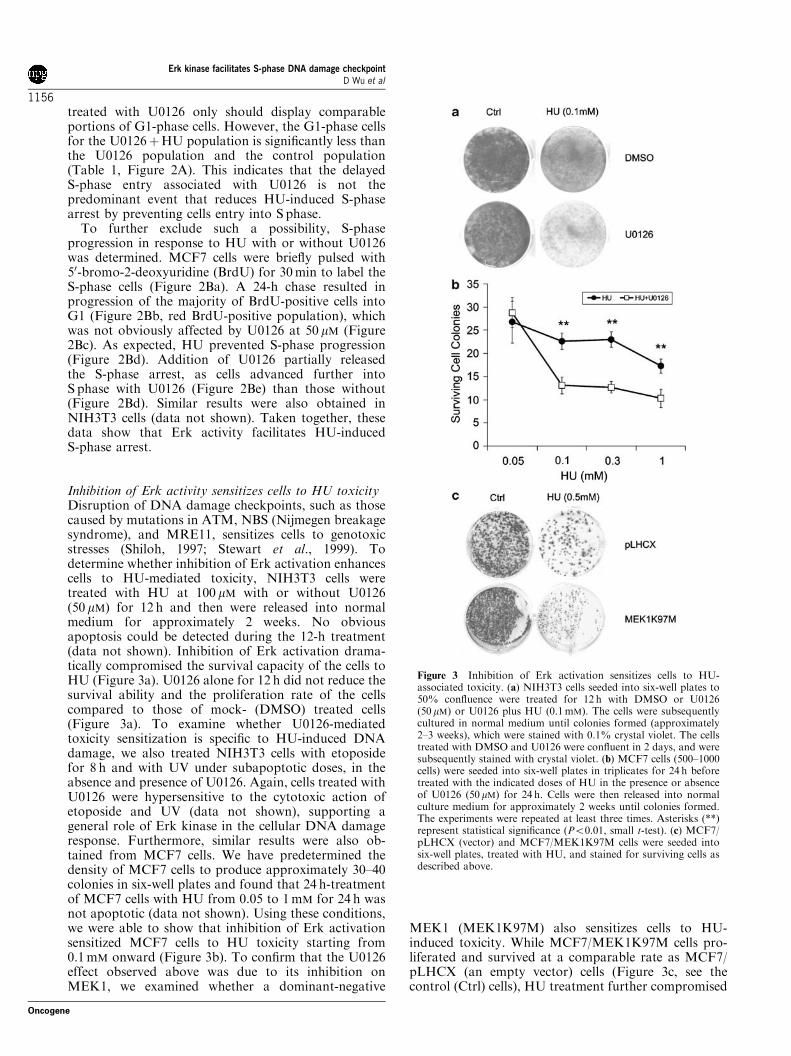
Inhibition of Erk activity sensitizes cells to HU toxicityDisruption of DNA damage checkpoints, such as thosecaused by mutations in ATM, NBS (Nijmegen breakagesyndrome), and MRE11, sensitizes cells to genotoxicstresses (Shiloh, 1997; Stewart et al., 1999). Todetermine whether inhibition of Erk activation enhancescells to HU-mediated toxicity, NIH3T3 cells weretreated with HU at 100 mM with or without U0126(50 mM) for 12 h and then were released into normalmedium for approximately 2 weeks. No obviousapoptosis could be detected during the 12-h treatment(data not shown). Inhibition of Erk activation drama-tically compromised the survival capacity of the cells toHU (Figure 3a). U0126 alone for 12 h did not reduce thesurvival ability and the proliferation rate of the cellscompared to those of mock- (DMSO) treated cells(Figure 3a). To examine whether U0126-mediatedtoxicity sensitization is specific to HU-induced DNAdamage, we also treated NIH3T3 cells with etoposidefor 8 h and with UV under subapoptotic doses, in theabsence and presence of U0126. Again, cells treated withU0126 were hypersensitive to the cytotoxic action ofetoposide and UV (data not shown), supporting ageneral role of Erk kinase in the cellular DNA damageresponse. Furthermore, similar results were also ob-tained from MCF7 cells. We have predetermined thedensity of MCF7 cells to produce approximately 30–40colonies in six-well plates and found that 24 h-treatmentof MCF7 cells with HU from 0.05 to 1mM for 24 h wasnot apoptotic (data not shown). Using these conditions,we were able to show that inhibition of Erk activationsensitized MCF7 cells to HU toxicity starting from0.1mM onward (Figure 3b). To confirm that the U0126effect observed above was due to its inhibition onMEK1, we examined whether a dominant-negative
MEK1 (MEK1K97M) also sensitizes cells to HU-induced toxicity. While MCF7/MEK1K97M cells pro-liferated and survived at a comparable rate as MCF7/pLHCX (an empty vector) cells (Figure 3c, see thecontrol (Ctrl) cells), HU treatment further compromised
Figure 3 Inhibition of Erk activation sensitizes cells to HU-associated toxicity. (a) NIH3T3 cells seeded into six-well plates to50% confluence were treated for 12 h with DMSO or U0126(50 mM) or U0126 plus HU (0.1mM). The cells were subsequentlycultured in normal medium until colonies formed (approximately2–3 weeks), which were stained with 0.1% crystal violet. The cellstreated with DMSO and U0126 were confluent in 2 days, and weresubsequently stained with crystal violet. (b) MCF7 cells (500–1000cells) were seeded into six-well plates in triplicates for 24 h beforetreated with the indicated doses of HU in the presence or absenceof U0126 (50mM) for 24 h. Cells were then released into normalculture medium for approximately 2 weeks until colonies formed.The experiments were repeated at least three times. Asterisks (**)represent statistical significance (Po0.01, small t-test). (c) MCF7/pLHCX (vector) and MCF7/MEK1K97M cells were seeded intosix-well plates, treated with HU, and stained for surviving cells asdescribed above.
Erk kinase facilitates S-phase DNA damage checkpointD Wu et al
1156
Oncogene

MEK1K97M cell survival when compared to pLHCXcells (comparing the sizes of pLHCX-surviving cellcolonies to the sizes of MEK1K97M-surviving cellcolonies upon HU treatment) (Figure 3c). The decreasedpotency of MEK1K97M compared to U0126 in thesensitization of MCF7 cells to HU toxicity may be dueto the observation that U0126 inhibits Erk activationmore efficiently than ectopic expression of MEK1K97M(data not shown). Taken together, these results supportthe concept that the MEK1-Erk pathway functionallycontributes to S-phase checkpoint activation in responseto HU treatment.
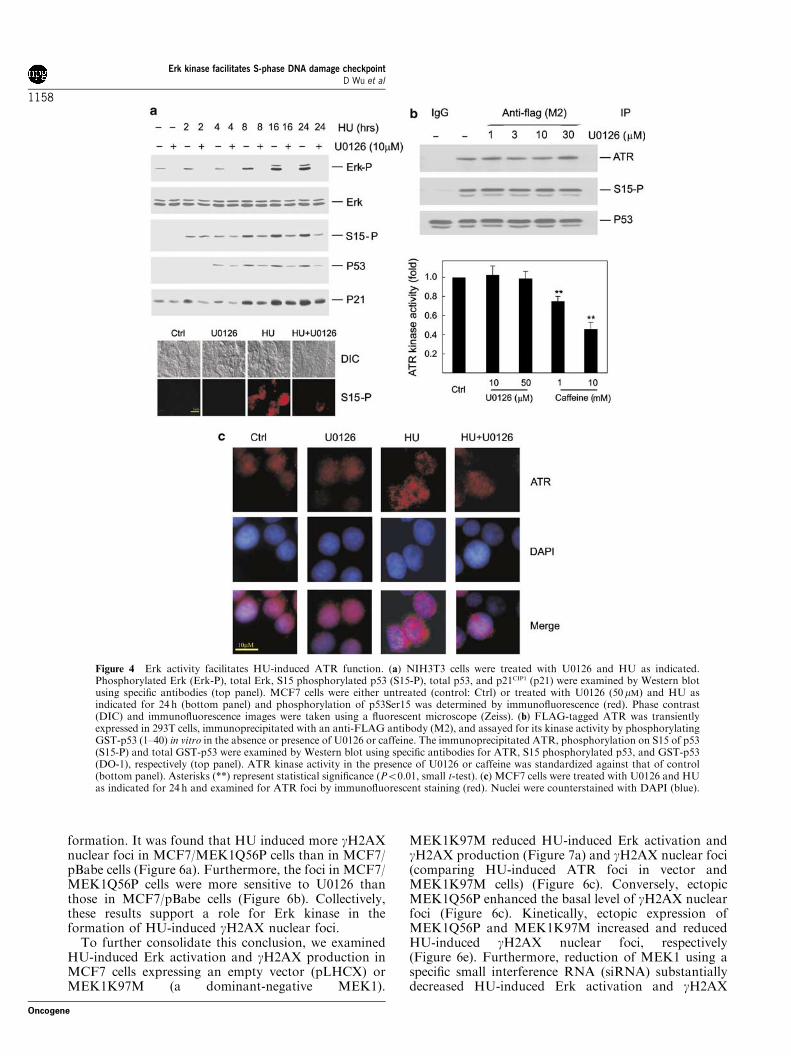
Erk activity regulates ATR function in response to HUTo examine the possible mechanism responsible for Erkfunction in S-phase checkpoint activation, we deter-mined HU-induced p53 serine 15 (p53Ser15) phosphor-ylation and p21CIP1 upregulation. NIH3T3 cells weretreated with HU with or without U0126 for a 24-h timecourse before analyzing for p53Ser15 phosphorylation,p53 stabilization, and p21CIP1 upregulation. WhileU0126 had no effect on HU-induced p53Ser15 phospho-rylation and p53 stabilization in the early phase (up to4 h), it attenuated these events after 8 h of HUtreatment. Kinetically, this matches the profile of HU-induced Erk activation (Figure 4a, top panel). U0126consistently decreased HU-induced p21CIP1 upregulation(Figure 4a, top panel). As p53 stabilization and p21CIP1
upregulation contribute to the DNA damage response,the attenuation of these events by U0126 thus supportsthe notion that Erk functions in S-phase checkpointregulation. Similar results were also obtained in MCF7cells (Figure 4a, bottom panel), consistent with U0126attenuating HU-induced S-phase arrest in both MCF7and NIH3T3 cells (Table 1). Taken together, the aboveresults reveal that Erk kinase activity functionally con-tributes to HU-initiated S-phase checkpoint activation.Phosphorylation of p53Ser15 is mediated by ATM
and ATR in the DNA damage response, and ATR playsan essential role in HU-induced DNA damage response.The fact that U0126 reduces p53Ser15 phosphorylationindicates that Erk kinase may facilitate HU-inducedATR function. To test this hypothesis, we first examinedwhether ATR kinase activity is modulated by Erkkinase. FLAG-tagged ATR was expressed in 293T cells,and immunoprecipitated for an in vitro kinase assayusing recombinant GST-p53 (1–40) as a substrate. Whilecaffeine, as expected, inhibited ATR kinase activity,U0126 exhibited no effects on ATR kinase activity(Figure 4b). This is in accordance with the theme ofATR activation, in that DNA damage does not affectATR kinase activity but induces ATR nuclear foci.Formation of ATR nuclear foci in response to DNAdamage contributes to ATR function (Tibbetts et al.,2000).To determine whether Erk activity regulates ATR
nuclear foci, MCF7 cells were treated with 1mM HUwith or without 50 mM U0126 for 24 h. Consistent with aprevious report (Tibbetts et al., 2000), HU inducednuclear ATR foci (Figure 4c). Interestingly, U0126
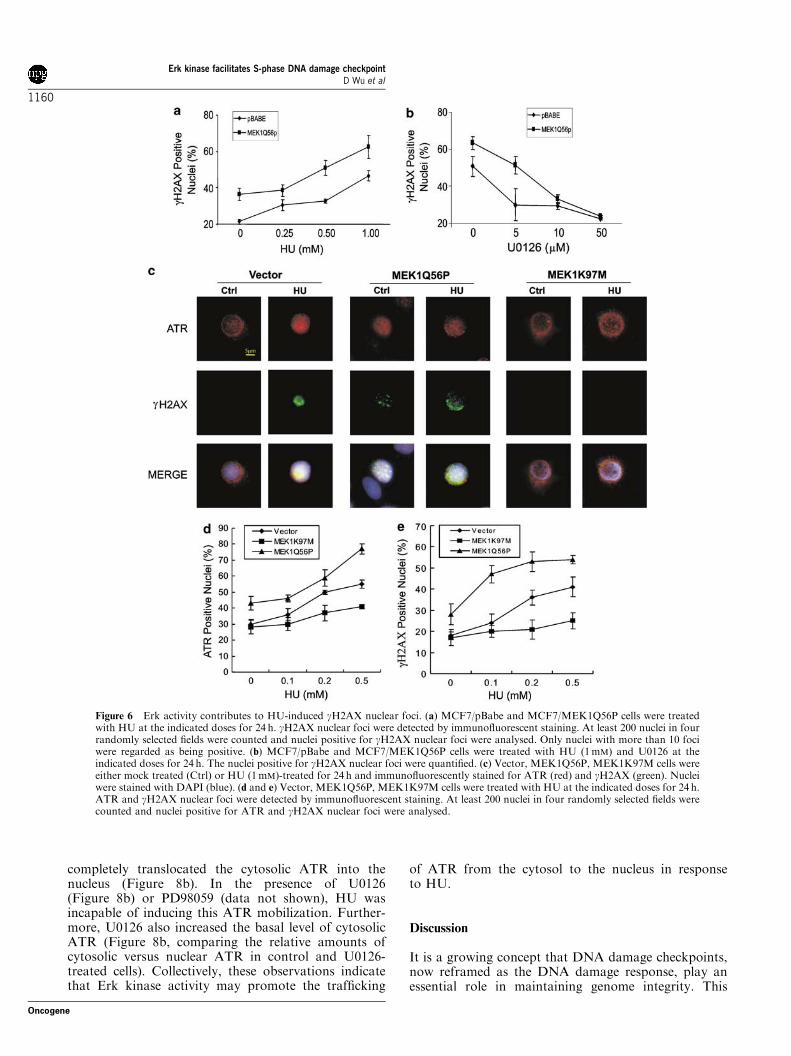
substantially reduced the magnitude of these foci(Figure 4c). Consistent with both U0126 and PD98059being among the most specific protein kinase inhibitors,PD98059 produced essentially the same results as thoseobtained using U0126 (data not shown). The samephenomenon was also observed in NIH3T3 cells (datanot shown). To exclude the possibility that U0126-mediated reduction of ATR nuclear foci was due topossible nonspecific actions instead of its inhibition ofErk activation, we showed that a large portion of ATRlocalizes in the perinuclear region in untreated (control)vector cells, while enforced activation of Erk via ectopicexpression of a constitutively active MEK1Q56P resultsin the redistribution of ATR throughout the nucleus(Figure 6c). Furthermore, HU induces extensive ATRnuclear foci in MEK1Q56P cells (Figure 6c). While HUinduces ATR nuclear foci in vector cells, a significantamount of ATR still resides in the perinuclear region inthe MEK1K197M cells upon HU treatment, whichresembles what was observed in untreated vector cells(Figure 6c). Downregulation of MEK1 via smallinterference RNA (siRNA) also reduced HU-inducedATR nuclear foci (data not shown). By countingapproximately 200 nuclei, we further demonstrated thatectopic expression of MEK1Q56P and MEK1K97Msensitizes and attenuates ATR nuclear focus formation,respectively, in a HU-dose-dependent manner(Figure 6d). Taken together, the above results demon-strate that activities modulating Erk activation plays arole in HU-induced formation of ATR nuclear foci.
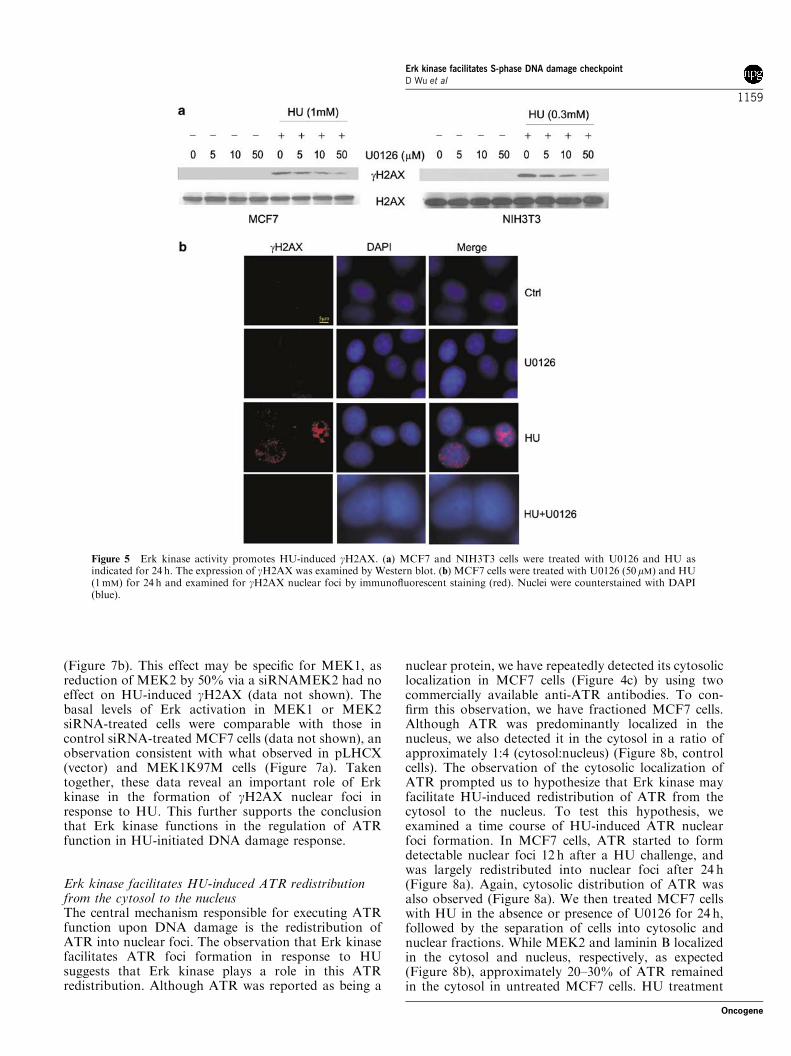
Erk activity plays a role in HU-induced formationof gH2AX nuclear fociH2AX is phosphorylated by ATR on Ser139 (gH2AX)in response to HU (Ward and Chen, 2001). NucleargH2AX foci are formed on DNA DSBs and function inrecruiting DNA repair factors (Celeste et al., 2002),suggesting that HU-induced production of gH2AX isfacilitated by Erk kinase activity. To test this possibility,MCF7 and NIH3T3 cells were treated with HU with orwithout U0126 for 24 h before Western blot analysis forgH2AX. While HU treatment led to the production ofgH2AX, U0126 reduced the levels of gH2AX in MCF7and NIH3T3 cells in a dose-dependent manner(Figure 5a). PD98059 produced the same results in bothcell lines (data not shown). Since gH2AX is recruitedonto DNA breaks and functions in DNA damage repair(Zhou and Elledge, 2000), we have examined whetherinhibition of Erk activation would affect HU-inducedgH2AX nuclear foci. Indeed, both U0126 and PD98059(data not shown) significantly attenuated HU-inducedgH2AX nuclear foci in MCF7 (Figure 5b) and NIH3T3cells (data not shown).To consolidate the notion that Erk activity promotes
the formation of gH2AX nuclear foci due to HUtreatment, we examined whether elevated levels of Erkactivation results in enhanced HU-induced gH2AXnuclear foci. MCF7/pBabe and MCF7/MEK1Q56Pcells were treated with increasing doses of HU todetermine the magnitude of gH2AX nuclear focus
Erk kinase facilitates S-phase DNA damage checkpointD Wu et al
1157
Oncogene
formation. It was found that HU induced more gH2AXnuclear foci in MCF7/MEK1Q56P cells than in MCF7/pBabe cells (Figure 6a). Furthermore, the foci in MCF7/MEK1Q56P cells were more sensitive to U0126 thanthose in MCF7/pBabe cells (Figure 6b). Collectively,these results support a role for Erk kinase in theformation of HU-induced gH2AX nuclear foci.To further consolidate this conclusion, we examined
HU-induced Erk activation and gH2AX production inMCF7 cells expressing an empty vector (pLHCX) orMEK1K97M (a dominant-negative MEK1).
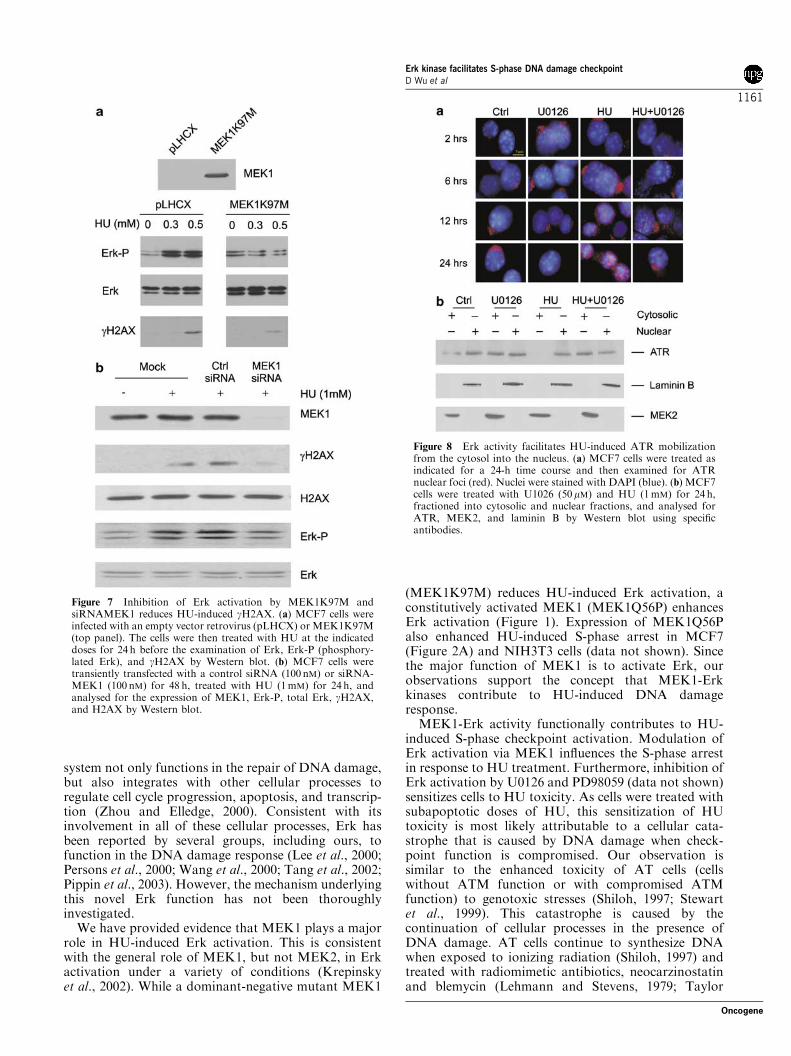
MEK1K97M reduced HU-induced Erk activation andgH2AX production (Figure 7a) and gH2AX nuclear foci(comparing HU-induced ATR foci in vector andMEK1K97M cells) (Figure 6c). Conversely, ectopicMEK1Q56P enhanced the basal level of gH2AX nuclearfoci (Figure 6c). Kinetically, ectopic expression ofMEK1Q56P and MEK1K97M increased and reducedHU-induced gH2AX nuclear foci, respectively(Figure 6e). Furthermore, reduction of MEK1 using aspecific small interference RNA (siRNA) substantiallydecreased HU-induced Erk activation and gH2AX
Figure 4 Erk activity facilitates HU-induced ATR function. (a) NIH3T3 cells were treated with U0126 and HU as indicated.Phosphorylated Erk (Erk-P), total Erk, S15 phosphorylated p53 (S15-P), total p53, and p21CIP1 (p21) were examined by Western blotusing specific antibodies (top panel). MCF7 cells were either untreated (control: Ctrl) or treated with U0126 (50mM) and HU asindicated for 24 h (bottom panel) and phosphorylation of p53Ser15 was determined by immunofluorescence (red). Phase contrast(DIC) and immunofluorescence images were taken using a fluorescent microscope (Zeiss). (b) FLAG-tagged ATR was transientlyexpressed in 293T cells, immunoprecipitated with an anti-FLAG antibody (M2), and assayed for its kinase activity by phosphorylatingGST-p53 (1–40) in vitro in the absence or presence of U0126 or caffeine. The immunoprecipitated ATR, phosphorylation on S15 of p53(S15-P) and total GST-p53 were examined by Western blot using specific antibodies for ATR, S15 phosphorylated p53, and GST-p53(DO-1), respectively (top panel). ATR kinase activity in the presence of U0126 or caffeine was standardized against that of control(bottom panel). Asterisks (**) represent statistical significance (Po0.01, small t-test). (c) MCF7 cells were treated with U0126 and HUas indicated for 24 h and examined for ATR foci by immunofluorescent staining (red). Nuclei were counterstained with DAPI (blue).
Erk kinase facilitates S-phase DNA damage checkpointD Wu et al
1158
Oncogene
(Figure 7b). This effect may be specific for MEK1, asreduction of MEK2 by 50% via a siRNAMEK2 had noeffect on HU-induced gH2AX (data not shown). Thebasal levels of Erk activation in MEK1 or MEK2siRNA-treated cells were comparable with those incontrol siRNA-treated MCF7 cells (data not shown), anobservation consistent with what observed in pLHCX(vector) and MEK1K97M cells (Figure 7a). Takentogether, these data reveal an important role of Erkkinase in the formation of gH2AX nuclear foci inresponse to HU. This further supports the conclusionthat Erk kinase functions in the regulation of ATRfunction in HU-initiated DNA damage response.
Erk kinase facilitates HU-induced ATR redistributionfrom the cytosol to the nucleusThe central mechanism responsible for executing ATRfunction upon DNA damage is the redistribution ofATR into nuclear foci. The observation that Erk kinasefacilitates ATR foci formation in response to HUsuggests that Erk kinase plays a role in this ATRredistribution. Although ATR was reported as being a
nuclear protein, we have repeatedly detected its cytosoliclocalization in MCF7 cells (Figure 4c) by using twocommercially available anti-ATR antibodies. To con-firm this observation, we have fractioned MCF7 cells.Although ATR was predominantly localized in thenucleus, we also detected it in the cytosol in a ratio ofapproximately 1:4 (cytosol:nucleus) (Figure 8b, controlcells). The observation of the cytosolic localization ofATR prompted us to hypothesize that Erk kinase mayfacilitate HU-induced redistribution of ATR from thecytosol to the nucleus. To test this hypothesis, weexamined a time course of HU-induced ATR nuclearfoci formation. In MCF7 cells, ATR started to formdetectable nuclear foci 12 h after a HU challenge, andwas largely redistributed into nuclear foci after 24 h(Figure 8a). Again, cytosolic distribution of ATR wasalso observed (Figure 8a). We then treated MCF7 cellswith HU in the absence or presence of U0126 for 24 h,followed by the separation of cells into cytosolic andnuclear fractions. While MEK2 and laminin B localizedin the cytosol and nucleus, respectively, as expected(Figure 8b), approximately 20–30% of ATR remainedin the cytosol in untreated MCF7 cells. HU treatment
Figure 5 Erk kinase activity promotes HU-induced gH2AX. (a) MCF7 and NIH3T3 cells were treated with U0126 and HU asindicated for 24 h. The expression of gH2AX was examined by Western blot. (b) MCF7 cells were treated with U0126 (50mM) and HU(1mM) for 24 h and examined for gH2AX nuclear foci by immunofluorescent staining (red). Nuclei were counterstained with DAPI(blue).
Erk kinase facilitates S-phase DNA damage checkpointD Wu et al
1159
Oncogene
completely translocated the cytosolic ATR into thenucleus (Figure 8b). In the presence of U0126(Figure 8b) or PD98059 (data not shown), HU wasincapable of inducing this ATR mobilization. Further-more, U0126 also increased the basal level of cytosolicATR (Figure 8b, comparing the relative amounts ofcytosolic versus nuclear ATR in control and U0126-treated cells). Collectively, these observations indicatethat Erk kinase activity may promote the trafficking
of ATR from the cytosol to the nucleus in responseto HU.
Discussion
It is a growing concept that DNA damage checkpoints,now reframed as the DNA damage response, play anessential role in maintaining genome integrity. This
Figure 6 Erk activity contributes to HU-induced gH2AX nuclear foci. (a) MCF7/pBabe and MCF7/MEK1Q56P cells were treatedwith HU at the indicated doses for 24 h. gH2AX nuclear foci were detected by immunofluorescent staining. At least 200 nuclei in fourrandomly selected fields were counted and nuclei positive for gH2AX nuclear foci were analysed. Only nuclei with more than 10 fociwere regarded as being positive. (b) MCF7/pBabe and MCF7/MEK1Q56P cells were treated with HU (1mM) and U0126 at theindicated doses for 24 h. The nuclei positive for gH2AX nuclear foci were quantified. (c) Vector, MEK1Q56P, MEK1K97M cells wereeither mock treated (Ctrl) or HU (1mM)-treated for 24 h and immunofluorescently stained for ATR (red) and gH2AX (green). Nucleiwere stained with DAPI (blue). (d and e) Vector, MEK1Q56P, MEK1K97M cells were treated with HU at the indicated doses for 24 h.ATR and gH2AX nuclear foci were detected by immunofluorescent staining. At least 200 nuclei in four randomly selected fields werecounted and nuclei positive for ATR and gH2AX nuclear foci were analysed.
Erk kinase facilitates S-phase DNA damage checkpointD Wu et al
1160
Oncogene
system not only functions in the repair of DNA damage,but also integrates with other cellular processes toregulate cell cycle progression, apoptosis, and transcrip-tion (Zhou and Elledge, 2000). Consistent with itsinvolvement in all of these cellular processes, Erk hasbeen reported by several groups, including ours, tofunction in the DNA damage response (Lee et al., 2000;Persons et al., 2000; Wang et al., 2000; Tang et al., 2002;Pippin et al., 2003). However, the mechanism underlyingthis novel Erk function has not been thoroughlyinvestigated.We have provided evidence that MEK1 plays a major
role in HU-induced Erk activation. This is consistentwith the general role of MEK1, but not MEK2, in Erkactivation under a variety of conditions (Krepinskyet al., 2002). While a dominant-negative mutant MEK1
(MEK1K97M) reduces HU-induced Erk activation, aconstitutively activated MEK1 (MEK1Q56P) enhancesErk activation (Figure 1). Expression of MEK1Q56Palso enhanced HU-induced S-phase arrest in MCF7(Figure 2A) and NIH3T3 cells (data not shown). Sincethe major function of MEK1 is to activate Erk, ourobservations support the concept that MEK1-Erkkinases contribute to HU-induced DNA damageresponse.MEK1-Erk activity functionally contributes to HU-
induced S-phase checkpoint activation. Modulation ofErk activation via MEK1 influences the S-phase arrestin response to HU treatment. Furthermore, inhibition ofErk activation by U0126 and PD98059 (data not shown)sensitizes cells to HU toxicity. As cells were treated withsubapoptotic doses of HU, this sensitization of HUtoxicity is most likely attributable to a cellular cata-strophe that is caused by DNA damage when check-point function is compromised. Our observation issimilar to the enhanced toxicity of AT cells (cellswithout ATM function or with compromised ATMfunction) to genotoxic stresses (Shiloh, 1997; Stewartet al., 1999). This catastrophe is caused by thecontinuation of cellular processes in the presence ofDNA damage. AT cells continue to synthesize DNAwhen exposed to ionizing radiation (Shiloh, 1997) andtreated with radiomimetic antibiotics, neocarzinostatinand blemycin (Lehmann and Stevens, 1979; Taylor
Figure 7 Inhibition of Erk activation by MEK1K97M andsiRNAMEK1 reduces HU-induced gH2AX. (a) MCF7 cells wereinfected with an empty vector retrovirus (pLHCX) or MEK1K97M(top panel). The cells were then treated with HU at the indicateddoses for 24 h before the examination of Erk, Erk-P (phosphory-lated Erk), and gH2AX by Western blot. (b) MCF7 cells weretransiently transfected with a control siRNA (100 nM) or siRNA-MEK1 (100 nM) for 48 h, treated with HU (1mM) for 24 h, andanalysed for the expression of MEK1, Erk-P, total Erk, gH2AX,and H2AX by Western blot.
Figure 8 Erk activity facilitates HU-induced ATR mobilizationfrom the cytosol into the nucleus. (a) MCF7 cells were treated asindicated for a 24-h time course and then examined for ATRnuclear foci (red). Nuclei were stained with DAPI (blue). (b) MCF7cells were treated with U1026 (50mM) and HU (1mM) for 24 h,fractioned into cytosolic and nuclear fractions, and analysed forATR, MEK2, and laminin B by Western blot using specificantibodies.
Erk kinase facilitates S-phase DNA damage checkpointD Wu et al
1161
Oncogene

et al., 1979; Shiloh et al., 1982). A similar situation wasalso observed for MCF7 cells treated with HU in thepresence of U0126, in that these cells progressed furtherinto S phase than the cells treated with HU alone(Figure 2B). HU causes depletion of dNTP pools byinhibiting ribonucleotide reductase (Atkin et al., 1973;Johns and Gao, 1998), and thereby activates the S-phasecheckpoints. HU was commonly used at 2mM toachieve a high degree of DNA synthesis inhibition. Toinvestigate the function of S-phase checkpoint activationin HU-mediated S-phase arrest, we intentionally usedsuboptimal HU doses to avoid complete DNA synthesisinhibition. At the highest dose used (1mM), HU reducedDNA synthesis in MCF7 cells by approximately 50%when assayed by the standard [3H]:[14C] methodology(data not shown) (Stewart et al., 1999; Falck et al.,2002). Interestingly, addition of U0126 to HU-treatedcells increased DNA synthesis by 15–20% (data notshown). Taken together, we provide compellingevidence that MEK1-Erk functionally contributes toHU-activated S-phase checkpoint activation.Mechanistically, this is mediated by modulating ATR
function. U0126 and PD98059, which are MEK1 andMEK2 inhibitors, effectively block HU-induced ATRnuclear foci. Both inhibitors are well characterized andare among the most specific protein kinase inhibitors(Favata et al., 1998; Davies et al., 2000). U0126 at 50 mMis widely used to prevent Erk activation in vivo withouttoxic effects on cells (Ahn et al., 2001). In a panel ofmore than 29 kinases tested, U0126 has little effect onkinases (including those of the most closely relatedMKK3, MKK4, MKK6, and MKK7) other thanMEK1 and MEK2 (Favata et al., 1998; Davies et al.,2000). We show here that these inhibitors also block thedownstream events of ATR in response to HU. HU-induced generation of gH2AX and its nuclear foci weresubstantially reduced by U0126 (Figures 5 and 6).Inhibition of MEK1 by ectopic expression ofMEK1K97M or using a siRNAMEK1 also led to thereduction of HU-induced gH2AX (Figure 7), whileenhancing MEK1 activation using MEK1Q56P-promoted gH2AX nuclear foci upon HU exposure(Figure 6). These results provide additional supportfor Erk-mediated ATR function.Furthermore, we show that Erk facilitates HU-
induced ATR nuclear foci by promoting, at least inpart, the redistribution of ATR from the cytosol to thenucleus upon HU treatment. Our finding that someATR is also localized in the cytosol is intriguing, as it isgenerally believed that ATR is a nuclear protein(Tibbetts et al., 2000). However, our findings were notartifacts, as (1) we have used two specific anti-ATRantibodies, (2) both immunofluorescent as well as cellfractioning experiments confirmed ATR cytosolic loca-lization, (3) the cytosolic localization of ATR wasobserved in MCF7 (Figures 4c and 8) and NIH3T3 cells(data not shown), and (4) HU exposure abolished thepresence of ATR in the cytosol (Figure 8b). Our findingof the cytosolic localization of ATR is consistent withthe cytosolic and nuclear localization of ATM (Yangand Kastan, 2000), as ATR and ATM are regarded as
functional equivalents in mediating a variety of DNAdamage responses (Zhou and Elledge, 2000).How MEK1-Erk regulates ATR trafficking in the
DNA damage response is not clear. Erk may directlyphosphorylate ATR, resulting in ATR translocationfrom the cytosol into the nucleus. Erk may alsophosphorylate other factors, which subsequently pro-mote this ATR movement. However, these two mechan-isms are not mutually exclusive. Regardless of thepossible mechanisms, our data reveal an interestingaspect of Erk function, especially considering the veryupstream nature of ATR function in regulating theDNA damage response.
Materials and methods
Materials, cell lines, plasmids, and cell cycle determinationPropidium iodide (PI) and HU were purchased from Sigma.MEK1 inhibitors U0126 and PD98059 were purchased fromPromega and Calbiochem, respectively. U0126 and PD98059were prepared in DMSO and used at the concentrationsindicated.NIH3T3 and MCF7 cells were cultured in DMEM
supplemented with 10% fetal calf serum (Gibco BRL) at371C in a tissue culture incubator. MEF and MEF/p53�/� weregifts from Dr Tak Mak and cultured in DMEM, 10% FCS,55 mM b-mercaptoethanol and 10 mg/ml gentamycin. IMR-90was purchased from ATCC and cultured in MEM and 10%FCS. The AT fibroblast line, GM05823, was purchased fromNIGMS Human Genetic Cell Repository and cultured inMEM, 10% FCS, with 2� concentrated essential and non-essential amino acids and vitamins.The constitutively activated MEK1Q56P cloned in the
retroviral vector, pBabe, was kindly provided by Dr ScottLowe of Cold Spring Harbor Laboratories. The dominant-negative MEK1 (MEK1K97M) was kindly provided by DrNatalie G Ahn of University of Colorado. We have subse-quently subcloned MEK1K97M into a retroviral vectorpLHCX. Cell cycle determination was carried out by PIstaining following our published procedure (Tang et al., 2002).
Retroviral infectionRetroviral infection was performed following our previouslypublished procedure (Tang et al., 2001; Li et al., 2004). Briefly,a gag–pol-expressing vector and an envelope-expressing vector(VSV-G) (Stratagene) were transiently cotransfected with adesigned retroviral plasmid into 293T cells. After 48 h, thevirus-containing medium was harvested, filtered through a0.45mM filter, and centrifuged at 50 000 g for 90min toconcentrate the retrovirus. Following the addition of 10mg/ml of polybrene (Sigma), the medium was used to infect cells.Infection was selected in puromycin (1 mg/ml).
Cell lysis and Western blotAfter exposure to the DNA damage agents as indicated, cellswere lysed in a buffer containing 20mM Tris (pH 7.4), 150mM
NaCl, 1mM EDTA, 1mM EGTA, 1% Triton X-100, 25mM
sodium pyrophosphate, 1mM NaF, 1mM b-glycerophosphate,0.1mM sodium orthovanadate, 1mM PMSF, 2mg/ml leupeptinand 10 mg/ml aprotinin. In all, 50mg of total cell lysate wasseparated on SDS–PAGE gel and transferred onto Immobi-lon-P membranes (Millipore). Membranes were blocked with5% skim milk and then incubated with the indicatedantibodies at room temperature for 1 h. Signals were detected
Erk kinase facilitates S-phase DNA damage checkpointD Wu et al
1162
Oncogene

using an ECL Western Blotting Kit (Amersham). Primaryantibodies and concentrations used were: anti-p53 (FL-353 at1mg/ml, Santa Cruz); anti-phospho-p53(S15) (Cell Signaling,1:1000); anti-Erk (1:500, New England Biolabs); anti-phospho-Erk (1:500 New England Biolabs); anti-H2AX (Upstate,1:1000); anti-p21CIP1 (1 mg/ml, Santa Cruz), anti-gH2AX(Upstate, 1:1000), anti-MEK1 (H-8, Santa Cruz, 1:200), anti-MEK2 (N-20, Santa Cruz, 1:200), and anti-laminin B (M-20,Santa Cruz, 1:200).
ImmunofluorescenceCells were treated as defined in the figure legends. Doubleimmunofluorescent staining was carried out by fixing cells withprechilled (�201C) acetone–methanol for 15min. The primaryantibodies, anti-gH2AX (Upstate, 0.5mg/ml), anti-ATR (Ab-2from Oncogenet Research Products and N-19 from SantaCruz; 0.5 mg/ml), anti-S15 phosphorylated p53 (Cell Signalling,1mg/ml) were then added to the slides at 41C overnight. Afterwashing, secondary antibodies, FITC-Donkey anti-mouse IgG(1:200, Jackson Immuno Research Lab) and Rhodamine-Donkey anti-rabbit IgG (1:200, Jackson Immuno ResearchLab), were then applied for 1 h in room temperature. The slidewas then covered with VECTASHIELD-mounting mediumwith DAPI (VECTOR Lab Inc., Burlingame, CA, USA).Images were taken with a fluorescent microscope (Carl Zeiss,Axiovert 200).
ATR kinase assaypcDNA3-ATR (FLAG-tagged) was transiently expressed in293T cells. ATR was immunoprecipitated with an anti-FLAGantibody (M2) and used to phosphorylate recombinant GST-p53 (containing N-terminal 1–40 residues of the p53 protein)(2 mg) in vitro at 301C for 30min according to publishedprocedures (Sarkaria et al., 1999; Chen et al., 2000) in thepresence or absence of different doses of U0126 or caffeine asindicated in Figure 4b. The reactions were terminated byaddition of SDS-protein sample buffer and analysed byWestern blot.
Isolation of cytosolic and nuclear fractionsMCF7 cells (107) were resuspended in 1ml of ice-cold buffer A(10mM Hepes, pH 7.5, 10mM KCl, 2mM MgCl2, 1mM DTT,
1mM PMSF, 2mg/ml leupeptin, 10mg/ml aprotinin, 200mMNaF, and 1mM b-glycerophosphate) on ice for 5min,centrifuged at 1200 rpm (41C) for 5min, resuspended in 1volume of buffer A, and incubated on ice for 20min. The cellswere then homogenized. The cytosolic fraction (supernatant)was harvested after a centrifugation at 15 000 g for 30min.Nuclei were obtained by overlaying the homogenized cellsolution on a 50% sucrose cushion, followed by a centrifuga-tion at 15 000 g for 1min, and resuspension of the nucleuspellet in 2� SDS protein sample buffer.
Downregulation of MEK1 and MEK2 by siRNAsiRNAMEK1 and siRNAMEK2 were synthesized by Dhar-mocon. The target sequence for human MEK1 was 50-AAGCAACTCATGGTTCATGCTTT-30 and for humanMEK2 was 50-AAGAAGGAGAGCCTCACAGCA-30. MCF7cells were seeded at 60–70% confluence overnight beforetransfection with individual siRNA at 100 nM usingLipofectAMINE2000 (Invitrogen) according to the manufac-turer’s procedure. A nonspecific siRNA was also purchasedfrom Dharmocon.
Radio-resistant DNA synthesis (RDS) assayRDS assay was carried out following published methodology(Stewart et al., 1999; Falck et al., 2002). Cells were labelledwith [14C] thymidine (20 nCi/ml) for 24 h and released intonormal medium for additional 24 h. Cells were then pretreatedwith U0126 for 30min before addition of HU for 24 h in[3H] thymidine (2.5 mCi/ml) medium. Cells were washedthree times with PBS, lysed in 0.25M NaOH and countedin a liquid scintillation counter. DNA synthesis was convertedto the ratio of [3H]:[14C] and expressed as a percentage ofcontrols.
Acknowledgements
This work was supported by a CIHR grant to D Tang. Theauthors are very grateful to Drs Alistair Ingram and RichardAustin for thoughtful discussions, and acknowledge thecontributions of MEK1Q56P by Dr Scott Lowe, MEK1K97Mby Dr Natalie G Ahn, ATR by Dr Shlomo Handeli of FredHutchinson Cancer Research Center, and MEF (wild type andp53�/�) by Dr Tak Mak.
References
Ahn NG, Nahreini TS, Tolwinski NT, Resing KR. (2001).Methods Enzymol 332: 417–431.
Atkin CL, Thelander L, Reichard P, Lang G. (1973). J BiolChem 248: 7464–7472.
Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J,Eckersdorff M et al. (2003). Cell 114: 359–370.
Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA et al. (2003). Cell114: 371–383.
Celeste A, Petersen S, Romanienko PJ, Fernandez-CapetilloO, Chen HT, Sedelnikova OA et al. (2002). Science 296:922–927.
Chen HT, Bhandoola A, Difilippantonio MJ, Zhu J, BrownMJ, Tai X et al. (2000). Science 290: 1962–1965.
Cheng M, Sexl V, Sherr CJ, Roussel MF. (1998). Proc NatlAcad Sci USA 95: 1091–1096.
Cowley S, Paterson H, Kemp P, Marshall CJ. (1994). Cell 77:841–852.
Davies SP, Reddy H, Caivano M, Cohen P. (2000). Biochem J351: 95–105.
Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. (2002).Nat Genet 30: 290–294.
Favata MF, Yoriuchi KY, Manos EJ, Daulerio AJ,Stradley DA, Feeser WS et al. (1998). J Biol Chem 273:18623–18632.
Fernandez-Capetillo O, Chen HT, Celeste A, Ward I,Romanienko PJ, Morales JC et al. (2002). Nat Cell Biol 4:993–997.
Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, Roma-nienko PJ, Camerini-Otero RD, Bonner WM et al. (2003).Dev Cell 4: 497–508.
Hoeijmakers JH. (2001). Nature 411: 366–374.Johnson GL, Lapadat R. (2002). Science 298: 1911–1912.Johns DG, Gao WY. (1998). Biochem Pharmacol 55:1551–1556.
Kolch W. (2000). Biochem J 351: 289–305.Krepinsky J, Wu D, Ingram A, Scholey J, Tang D. (2002).
Expert Opin Ther Patents 12: 1795–1811.Lavoie JN, L’Allemain G, Brunet A, Muller R, Pouyssegur J.(1996). J Biol Chem 271: 20608–20616.
Erk kinase facilitates S-phase DNA damage checkpointD Wu et al
1163
Oncogene

Lee SW, Fang L, Igarashi M, Ouchi T, Lu KP, Aaronson SA.(2000). Proc Natl Acad Sci USA 97: 8302–8305.
Lehmann AR, Stevens S. (1979). Nucl Acids Res 6: 1953–1960.Lengauer C, Kinzler KW, Vogelstein B. (1998). Nature 396:643–649.
Li Y, Wu D, Chen B, Ingram A, Iie L, Liu L et al. (2004).Oncogene 23: 7355–7365.
Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M,Lowe SW. (1998). Genes Dev 12: 3008–3019.
Mahadevaiah SK, Turner JM, Baudat F, Rogakou EP, deBoer P, Blanco-Rodriguez J et al. (2001). Nat Genet 27:271–276.
Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S,Fukasawa K et al. (1994). Science 265: 966–970.
Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU,Gellert M, Bonner WM. (2000). Curr Biol 10: 886–895.
Persons DL, Yazlovitskaya EM, Pelling JC. (2000). J BiolChem 275: 35778–35785.
Petersen S, Casellas R, Reina-San-Martin B, Chen HT,Difilippantonio MJ, Wilson PC et al. (2001). Nature 414:660–665.
Pippin JW, Durvasula R, Petermann A, Hiromura K,Couser WG, Shankland SJ. (2003). J Clin Invest 111:877–885.
Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K,Bonner W. (2002). Curr Opin Genet Dev 12: 162–169.
Rouse J, Jackson SP. (2002). Science 297: 547–551.
Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, KarnitzLM et al. (1999). Cancer Res 59: 4375–4382.
Schar P. (2001). Cell 104: 329–332.Shiloh Y. (1997). Annu Rev Genet 31: 635–662.Shiloh Y. (2003). Nat Rev Cancer 3: 155–168.Shiloh Y, Tabor E, Becker Y. (1982). Carcinogenesis 3:815–820.
Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI,Jaspers NG et al. (1999). Cell 99: 577–587.
Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ.(2003). Nature 421: 961–966.
Tang D, Okada H, Ruland J, Liu L, Stambolic V, Mak TWet al. (2001). J Biol Chem 276: 30461–30466.
Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B et al.(2002). J Biol Chem 277: 12710–12717.
Taylor AM, Rosney CM, Campbell JB. (1979). Cancer Res 39:1046–1050.
Tibbetts RS, Cortez D, Brumbaugh KM, Scully R,Livingston D, Elledge SJ et al. (2000). Genes Dev 14: 2989–3002.
Wang X, Martindale JL, Holbrook NJ. (2000). J Biol Chem275: 39435–39443.
Ward IM, Chen J. (2001). J Biol Chem 276: 47759–47762.Weber JD, Raben DM, Phillips PJ, Baldassare JJ. (1997).
Biochem J 326: 61–68.Yang DQ, Kastan MB. (2000). Nat Cell Biol 2: 893–898.Zhou BB, Elledge SJ. (2000). Nature 408: 433–439.Zou L, Elledge SJ. (2003). Science 300: 1542–1548.
Erk kinase facilitates S-phase DNA damage checkpointD Wu et al
1164
Oncogene