electronic, energetic, and geometric properties of methylene-functionalized c60
TRANSCRIPT

1 23
Journal of Cluster ScienceIncluding Nanoclusters andNanoparticles ISSN 1040-7278Volume 24Number 3 J Clust Sci (2013) 24:669-678DOI 10.1007/s10876-013-0563-6
Electronic, Energetic, and GeometricProperties of Methylene-Functionalized C60
Mohammad T. Baei, Ali AhmadiPeyghan & Zargham Bagheri

1 23
Your article is protected by copyright and all
rights are held exclusively by Springer Science
+Business Media New York. This e-offprint is
for personal use only and shall not be self-
archived in electronic repositories. If you wish
to self-archive your article, please use the
accepted manuscript version for posting on
your own website. You may further deposit
the accepted manuscript version in any
repository, provided it is only made publicly
available 12 months after official publication
or later and provided acknowledgement is
given to the original source of publication
and a link is inserted to the published article
on Springer's website. The link must be
accompanied by the following text: "The final
publication is available at link.springer.com”.

ORI GIN AL PA PER
Electronic, Energetic, and Geometric Propertiesof Methylene-Functionalized C60
Mohammad T. Baei • Ali Ahmadi Peyghan • Zargham Bagheri
Received: 10 January 2013 / Published online: 2 February 2013
� Springer Science+Business Media New York 2013
Abstract Chemical functionalization of C60 fullerene with one to six carbene
(CH2) molecule(s) has been investigated using density functional theory. We have
found that the reaction is regioselective so that a CH2 molecule prefers to be
adsorbed atop a C–C bond which is shared between two hexagonal rings of the C60,
releasing energy of -3.95 eV. Singly occupied molecular orbital (SOMO) of the
CH2 interacts with LUMO of the C60 via a [2 ? 1] cycloaddition reaction. Energy
of the reaction and work function of the system are decreased by increasing the
number of adsorbed CH2 molecules. The HOMO/LUMO energy gap of C60 is
slightly changed and the electron emission from its surface is facilitated upon the
functionalization.
Keywords Fullerene � DFT � C60 � Adsorption
Introduction
Nanomaterials have attracted great interest in recent years due to their excellent
mechanical, electrical, optical, magnetic, and surface properties [1–6]. Since the
discovery of C60 fullerene [7], several studies on different nanostructures such as
fullerene-like clusters, nanotubes, nanocapsules, nanopolyhedra, cones, cubes, and
M. T. Baei
Department of Chemistry, Azadshahr Branch, Islamic Azad University, Azadshahr, Golestan, Iran
A. A. Peyghan (&)
Young Researchers and Elite Club, Central Tehran Branch, Islamic Azad University, Tehran, Iran
e-mail: [email protected]
Z. Bagheri
Physics Group, Science Department, Islamic Azad University, Islamshahr Branch,
P.O. Box: 33135-369, Islamshahr, Tehran, Iran
123
J Clust Sci (2013) 24:669–678
DOI 10.1007/s10876-013-0563-6
Author's personal copy

onions have been reported [8–10]. Being interesting candidates for constructing
units of new materials, increasing variety of species have been utilized for
functionalization of the fullerenes [11]. These derivatives have also enhanced
stability of the endohedral fullerenes [12], which might be very interesting for
applications in medicinal chemistry, materials science, and nanotechnology [13].
Several types of functionalized fullerenes have been recently synthesized for
possible applications in electronics and optical devices as well as for developing
possible applications in biology and medicine [14]. Exploration of the reactivity in
addition reactions of the fullerenes has motivated special interest for preparation of
specific fullerene derivatives by exohedral functionalization [15]. Synthesis of
uniformly exohedrally monofunctionalized fullerene derivatives in preparative
quantities has been explored with success by a variety of addition reactions [16, 17],
most notably by a range of cycloaddition [18, 19] and organometallic addition
reactions [20]. The preparation of specific multiply functionalized derivatives of C60
with organic addends has been taken up in several research groups [21], exploiting
the regioselectivity of the cycloaddition step.
In the present work, [2 ? 1] cycloaddition reactions of one to six carbene(s) with
a C60 molecule have been theoretically investigated in terms of geometry, energies,
electronic structures, stability, etc. using density functional theory (DFT). Carbenes
have played an important role as transient intermediates over the past five decades
[22]. Introduced by Doering into organic chemistry in the 1950s [23] and by Fischer
into organometallic chemistry in 1964 [24], these fascinating species are involved in
many reactions of high synthetic interest. The [2 ? 1] cycloaddition of carbene to
nanotubes was reported for the first time by Haddon et al. and coworkers in 1998
[25]. It was proposed initially that the [2 ? 1] cycloaddition on the nanotube
sidewall gave rise to the formation of three-membered-ring species [26]. However,
our results may be useful for further studies in functionalization of C60 and
construction of nanodevices.
Computational Methods
Geometry optimizations and density of states (DOS) analysis have been performed
on a C60 fullerene and different n CH2/C60 complexes using B3LYP functional with
6–31G (d) basis sets as implemented in GAMESS suite of program [27]. GaussSum
program [28] has been used to obtain DOS results. The B3LYP has been
demonstrated to be a reliable and commonly used functional for study of different
nanostructures [29–33]. Vibrational frequencies were also calculated at the same
level to confirm that all the stationary points correspond to true minima on the
potential energy surface. All frequency calculations were performed using
numerical second derivatives and verified that all of the structures are true minima
by frequency analysis and obtained positive Hessian eigenvalues. We used B3PW91
functional to study the electronic properties of the systems. Xiao et al. [34] have
shown that for several studied semiconductors, B3PW91 predicts gaps substantially
better than all modern hybrid functionals. Reaction energy (Er) of the CH2 molecule
with the C60 has been calculated using the following equation:
670 M. T. Baei et al.
123
Author's personal copy
Er ¼ H n CH2=C60ð Þ � H C60ð Þ � nH CH2ð Þ; n ¼ 1� 6 ð1Þ
where H (n CH2/C60) is the enthalpy of the complex of n CH2 adsorbed on the C60
and H (CH2) refers to the enthalpies of an isolated CH2 molecule at 298 K and 1
atom. The H (C60) is the enthalpy of the pristine C60. The negative value of Er
indicates the exothermic character of the adsorption process.
Results and Discussion
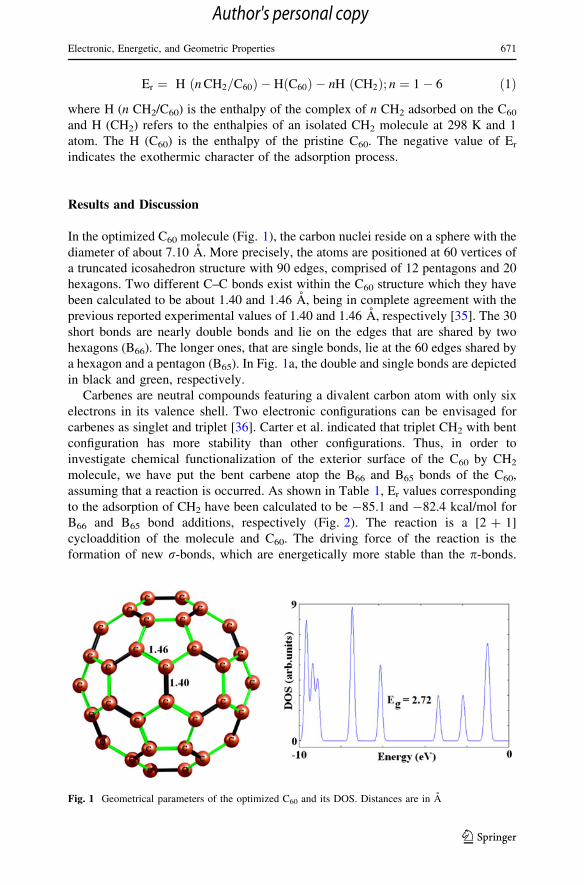
In the optimized C60 molecule (Fig. 1), the carbon nuclei reside on a sphere with the
diameter of about 7.10 A. More precisely, the atoms are positioned at 60 vertices of
a truncated icosahedron structure with 90 edges, comprised of 12 pentagons and 20
hexagons. Two different C–C bonds exist within the C60 structure which they have
been calculated to be about 1.40 and 1.46 A, being in complete agreement with the
previous reported experimental values of 1.40 and 1.46 A, respectively [35]. The 30
short bonds are nearly double bonds and lie on the edges that are shared by two
hexagons (B66). The longer ones, that are single bonds, lie at the 60 edges shared by
a hexagon and a pentagon (B65). In Fig. 1a, the double and single bonds are depicted
in black and green, respectively.
Carbenes are neutral compounds featuring a divalent carbon atom with only six
electrons in its valence shell. Two electronic configurations can be envisaged for
carbenes as singlet and triplet [36]. Carter et al. indicated that triplet CH2 with bent
configuration has more stability than other configurations. Thus, in order to
investigate chemical functionalization of the exterior surface of the C60 by CH2
molecule, we have put the bent carbene atop the B66 and B65 bonds of the C60,
assuming that a reaction is occurred. As shown in Table 1, Er values corresponding
to the adsorption of CH2 have been calculated to be -85.1 and -82.4 kcal/mol for
B66 and B65 bond additions, respectively (Fig. 2). The reaction is a [2 ? 1]
cycloaddition of the molecule and C60. The driving force of the reaction is the
formation of new r-bonds, which are energetically more stable than the p-bonds.
Fig. 1 Geometrical parameters of the optimized C60 and its DOS. Distances are in A
Electronic, Energetic, and Geometric Properties 671
123
Author's personal copy
Experimental work has indicated that in cycloadditions the molecule prefers to
attack the B66 bond (double bond) of the fullerene over the B65 bond [37].
Therefore, it can be concluded that the Er of the CH2 interaction with the B66 bond is
higher than that with the B65 which is in good agreement with the experimental
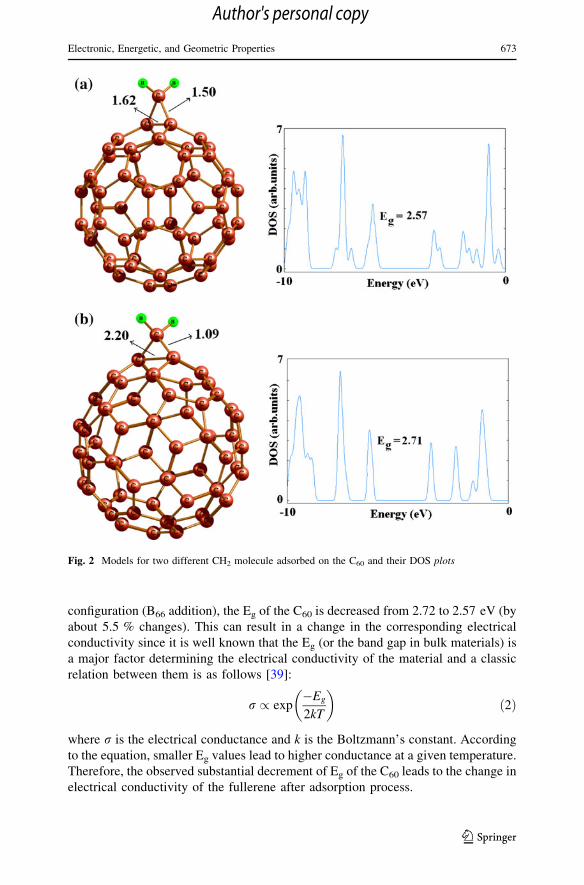
results. Subsequently, kinetic favorability of reactions was explored. As shown in
Fig. 3, barrier energy must be overcome to get into a final configuration in each
reaction. The barrier energies for the configurations B66 and B65 are about 10.3 and
56.7 kcal/mol, respectively. It can be inferred that barrier energy for configuration
B65 is large that the reaction process may not overcome that at the room
temperature. Thus, the CH2 addition on C60 through C66 bond is both kinetically and
energetically more favorable than that on the B65 one.
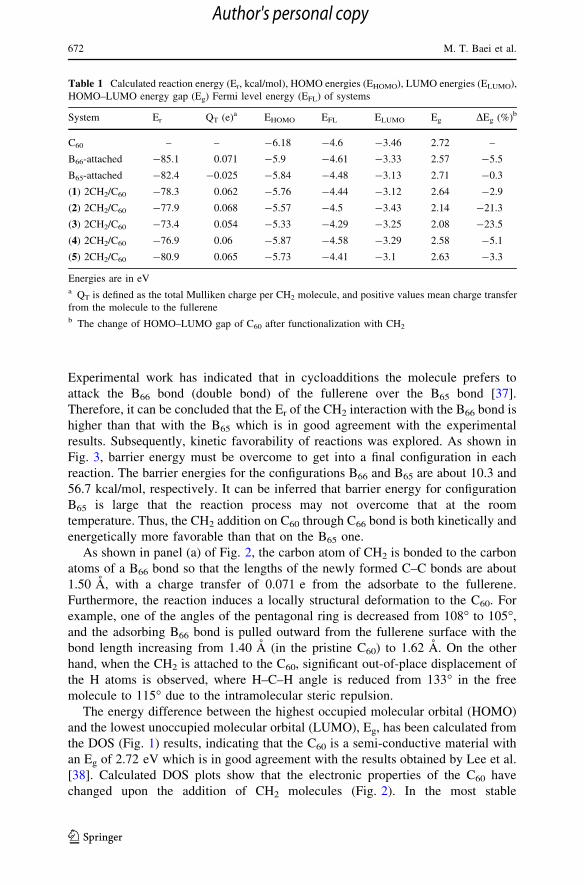
As shown in panel (a) of Fig. 2, the carbon atom of CH2 is bonded to the carbon
atoms of a B66 bond so that the lengths of the newly formed C–C bonds are about
1.50 A, with a charge transfer of 0.071 e from the adsorbate to the fullerene.
Furthermore, the reaction induces a locally structural deformation to the C60. For
example, one of the angles of the pentagonal ring is decreased from 108� to 105�,
and the adsorbing B66 bond is pulled outward from the fullerene surface with the
bond length increasing from 1.40 A (in the pristine C60) to 1.62 A. On the other
hand, when the CH2 is attached to the C60, significant out-of-place displacement of
the H atoms is observed, where H–C–H angle is reduced from 133� in the free
molecule to 115� due to the intramolecular steric repulsion.
The energy difference between the highest occupied molecular orbital (HOMO)
and the lowest unoccupied molecular orbital (LUMO), Eg, has been calculated from
the DOS (Fig. 1) results, indicating that the C60 is a semi-conductive material with
an Eg of 2.72 eV which is in good agreement with the results obtained by Lee et al.
[38]. Calculated DOS plots show that the electronic properties of the C60 have
changed upon the addition of CH2 molecules (Fig. 2). In the most stable
Table 1 Calculated reaction energy (Er, kcal/mol), HOMO energies (EHOMO), LUMO energies (ELUMO),
HOMO–LUMO energy gap (Eg) Fermi level energy (EFL) of systems
System Er QT (e)a EHOMO EFL ELUMO Eg DEg (%)b
C60 – – -6.18 -4.6 -3.46 2.72 –
B66-attached -85.1 0.071 -5.9 -4.61 -3.33 2.57 -5.5
B65-attached -82.4 -0.025 -5.84 -4.48 -3.13 2.71 -0.3
(1) 2CH2/C60 -78.3 0.062 -5.76 -4.44 -3.12 2.64 -2.9
(2) 2CH2/C60 -77.9 0.068 -5.57 -4.5 -3.43 2.14 -21.3
(3) 2CH2/C60 -73.4 0.054 -5.33 -4.29 -3.25 2.08 -23.5
(4) 2CH2/C60 -76.9 0.06 -5.87 -4.58 -3.29 2.58 -5.1
(5) 2CH2/C60 -80.9 0.065 -5.73 -4.41 -3.1 2.63 -3.3
Energies are in eVa QT is defined as the total Mulliken charge per CH2 molecule, and positive values mean charge transfer
from the molecule to the fullereneb The change of HOMO–LUMO gap of C60 after functionalization with CH2
672 M. T. Baei et al.
123
Author's personal copy
configuration (B66 addition), the Eg of the C60 is decreased from 2.72 to 2.57 eV (by
about 5.5 % changes). This can result in a change in the corresponding electrical
conductivity since it is well known that the Eg (or the band gap in bulk materials) is
a major factor determining the electrical conductivity of the material and a classic
relation between them is as follows [39]:
r / exp�Eg
2kT
� �ð2Þ
where r is the electrical conductance and k is the Boltzmann’s constant. According
to the equation, smaller Eg values lead to higher conductance at a given temperature.
Therefore, the observed substantial decrement of Eg of the C60 leads to the change in
electrical conductivity of the fullerene after adsorption process.
Fig. 2 Models for two different CH2 molecule adsorbed on the C60 and their DOS plots
Electronic, Energetic, and Geometric Properties 673
123
Author's personal copy
With each B66 bond on the C60 being a potential chemisorption site, the
possibility of multiple adsorption is interesting to evaluate CH2 concentration on the
C60. Thus, it seems evident that the formation of a symmetric hexakisadduct will
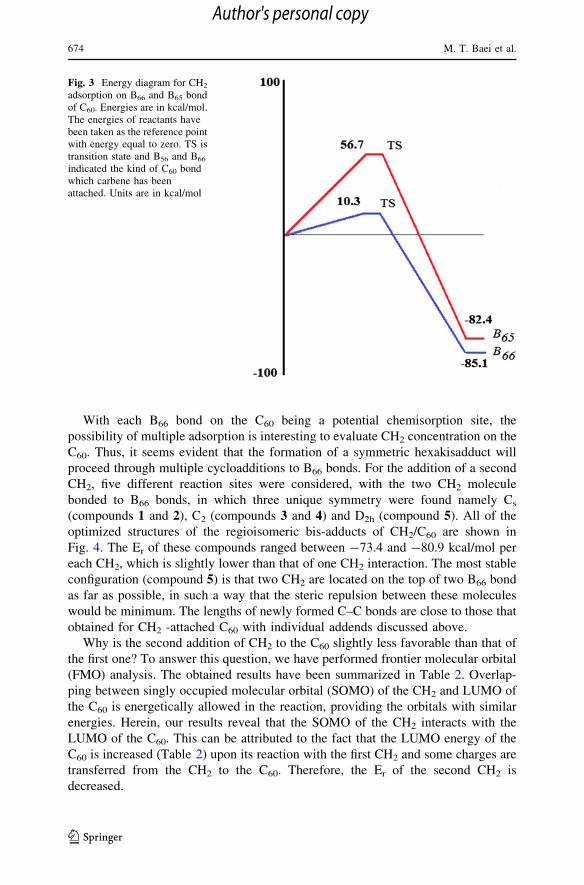
proceed through multiple cycloadditions to B66 bonds. For the addition of a second
CH2, five different reaction sites were considered, with the two CH2 molecule
bonded to B66 bonds, in which three unique symmetry were found namely Cs
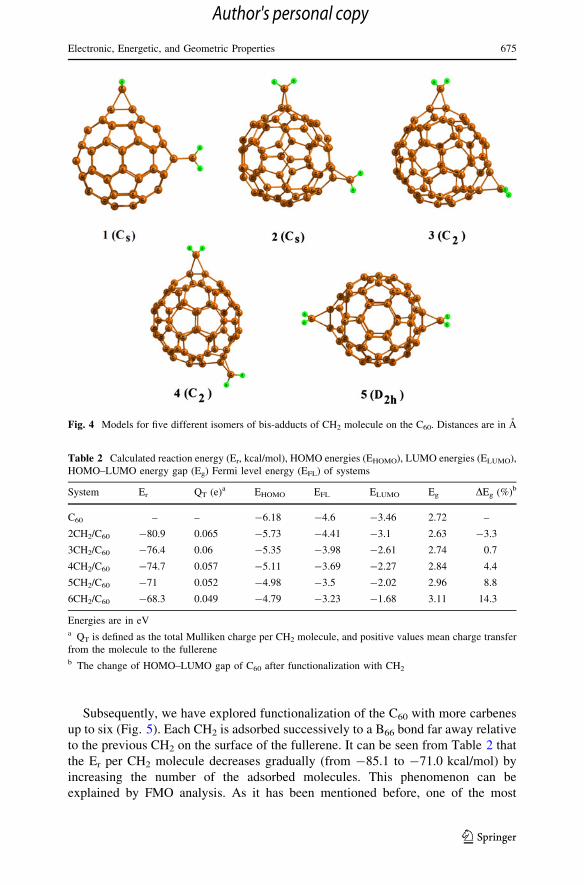
(compounds 1 and 2), C2 (compounds 3 and 4) and D2h (compound 5). All of the
optimized structures of the regioisomeric bis-adducts of CH2/C60 are shown in
Fig. 4. The Er of these compounds ranged between -73.4 and -80.9 kcal/mol per
each CH2, which is slightly lower than that of one CH2 interaction. The most stable
configuration (compound 5) is that two CH2 are located on the top of two B66 bond
as far as possible, in such a way that the steric repulsion between these molecules
would be minimum. The lengths of newly formed C–C bonds are close to those that
obtained for CH2 -attached C60 with individual addends discussed above.
Why is the second addition of CH2 to the C60 slightly less favorable than that of
the first one? To answer this question, we have performed frontier molecular orbital
(FMO) analysis. The obtained results have been summarized in Table 2. Overlap-
ping between singly occupied molecular orbital (SOMO) of the CH2 and LUMO of
the C60 is energetically allowed in the reaction, providing the orbitals with similar
energies. Herein, our results reveal that the SOMO of the CH2 interacts with the
LUMO of the C60. This can be attributed to the fact that the LUMO energy of the
C60 is increased (Table 2) upon its reaction with the first CH2 and some charges are
transferred from the CH2 to the C60. Therefore, the Er of the second CH2 is
decreased.
Fig. 3 Energy diagram for CH2
adsorption on B66 and B65 bondof C60. Energies are in kcal/mol.The energies of reactants havebeen taken as the reference pointwith energy equal to zero. TS istransition state and B56 and B66
indicated the kind of C60 bondwhich carbene has beenattached. Units are in kcal/mol
674 M. T. Baei et al.
123
Author's personal copy
Subsequently, we have explored functionalization of the C60 with more carbenes
up to six (Fig. 5). Each CH2 is adsorbed successively to a B66 bond far away relative
to the previous CH2 on the surface of the fullerene. It can be seen from Table 2 that
the Er per CH2 molecule decreases gradually (from -85.1 to -71.0 kcal/mol) by
increasing the number of the adsorbed molecules. This phenomenon can be
explained by FMO analysis. As it has been mentioned before, one of the most
Fig. 4 Models for five different isomers of bis-adducts of CH2 molecule on the C60. Distances are in A
Table 2 Calculated reaction energy (Er, kcal/mol), HOMO energies (EHOMO), LUMO energies (ELUMO),
HOMO–LUMO energy gap (Eg) Fermi level energy (EFL) of systems
System Er QT (e)a EHOMO EFL ELUMO Eg DEg (%)b
C60 – – -6.18 -4.6 -3.46 2.72 –
2CH2/C60 -80.9 0.065 -5.73 -4.41 -3.1 2.63 -3.3
3CH2/C60 -76.4 0.06 -5.35 -3.98 -2.61 2.74 0.7
4CH2/C60 -74.7 0.057 -5.11 -3.69 -2.27 2.84 4.4
5CH2/C60 -71 0.052 -4.98 -3.5 -2.02 2.96 8.8
6CH2/C60 -68.3 0.049 -4.79 -3.23 -1.68 3.11 14.3
Energies are in eVa QT is defined as the total Mulliken charge per CH2 molecule, and positive values mean charge transfer
from the molecule to the fullereneb The change of HOMO–LUMO gap of C60 after functionalization with CH2
Electronic, Energetic, and Geometric Properties 675
123
Author's personal copy

important factors in SOMO/LUMO interactions is the energy difference between
the SOMO and LUMO of the reactants. The FMO analysis shows that the SOMO
energy of the nucleophile agent (CH2) is about -6.61 eV and that of the LUMO of
the pristine C60 is about -3.46 eV which is gradually increased by increasing the
number of attached CH2 (Table 1) species. It suggests that the Er decrement by
increasing the number of carbenes is due to the larger difference between the SOMO
energy of the CH2 and the LUMO energy of the adsorbent.
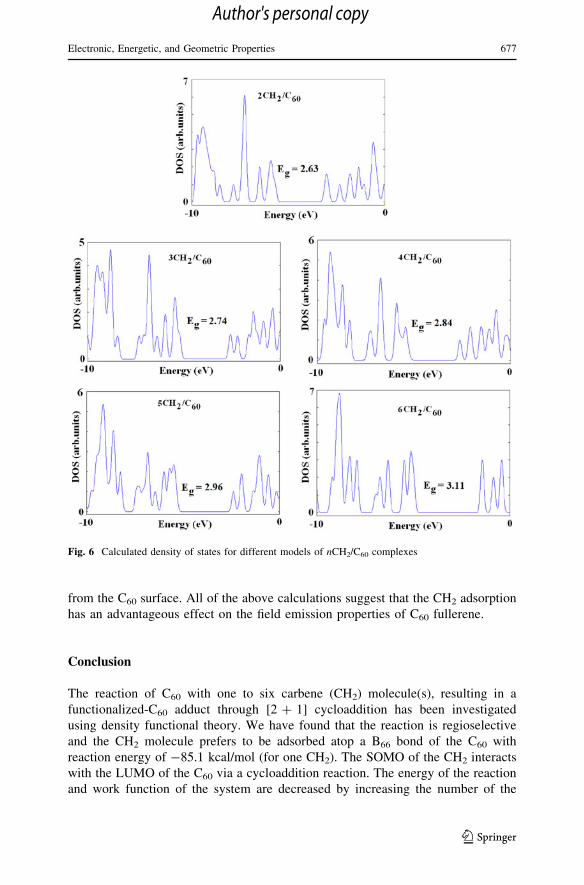
Total electronic DOSs for the CH2/C60 complexes are shown in Fig. 6, indicating
that their Eg have changed about 0.7–14.3 % after the functionalization with two or
more carbenes. It is noteworthy to say that as shown in Table 2, not also the Eg of
the C60 is changed but also the Fermi level energy (EFL) is gradually increased by
increasing the number of the adsorbed CH2. The canonical assumption for Fermi
level is that in a molecule (at T = 0 K) it lies approximately in the middle of the Eg.
In fact, what lies in the middle of the Eg is the chemical potential, and since the
chemical potential of a free gas of electrons is equal to its Fermi level as
traditionally defined, herein, the Fermi level of the considered systems is at the
middle of the Eg.
When one or two CH2 molecules are adsorbed on the fullerene, the EFL obviously
increases by 0.21–0.38 eV. When six CH2 molecules are adsorbed on the C60, the
EFL is increased from -4.82 eV in the pristine fullerene to -3.23 eV in the
adsorbed form. This phenomenon leads to a decrement in the work function that is
important in field emission applications. The work function can be found using the
standard procedure by calculating the potential energy difference between the
vacuum level and the Fermi level, which is the minimum energy required for one
electron to be removed from the Fermi level to the vacuum. The decrement in the
work function indicates that the field emission properties of the C60 are facilitated
upon the adsorption of CH2 molecules. Furthermore, this results in reduced potential
barrier of the electron emission for the fullerene, facilitating the electron emission
Fig. 5 Model for a 4CH2 and b 6CH2 fucntionalized-C60
676 M. T. Baei et al.
123
Author's personal copy
from the C60 surface. All of the above calculations suggest that the CH2 adsorption
has an advantageous effect on the field emission properties of C60 fullerene.
Conclusion
The reaction of C60 with one to six carbene (CH2) molecule(s), resulting in a
functionalized-C60 adduct through [2 ? 1] cycloaddition has been investigated
using density functional theory. We have found that the reaction is regioselective
and the CH2 molecule prefers to be adsorbed atop a B66 bond of the C60 with
reaction energy of -85.1 kcal/mol (for one CH2). The SOMO of the CH2 interacts
with the LUMO of the C60 via a cycloaddition reaction. The energy of the reaction
and work function of the system are decreased by increasing the number of the
Fig. 6 Calculated density of states for different models of nCH2/C60 complexes
Electronic, Energetic, and Geometric Properties 677
123
Author's personal copy

adsorbed CH2 molecules. The Eg of the C60 is slightly changed and the electron
emission from its surface is facilitated upon the reaction.
References
1. S. Iijima (1991). Nature 354, 56.
2. M. Moradi, A. A. Peyghan, Z. Bagheri, and M. Kamfiroozi (2012). J. Mol. Model 18, 3535.
3. A. Ma, J. Lu, S. Yang, and K. M. Ng (2006). J. Cluster Sci. 17, 599.
4. G. Ge, Q. Jing, H. Cao, and H. Yan (2012). J. Cluster Sci. 23, 189.
5. J. Beheshtian, Z. Bagheri, M. Kamfiroozi, and A. Ahmadi (2012). J. Mol. Model 18, 2653.
6. A.A. Peyghan, M.T. Baei, M. Moghimi, S. Hashemian (2012) J. Cluster Sci. doi:10.1007/
s10876-012-0512-9.
7. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl, and R. E. Smalley (1985). Nature 318, 162.
8. J. Beheshtian, Z. Bagheri, M. Kamfiroozi, and A. Ahmadi (2012). Struct. Chem. 23, 653.
9. B. Hong, Y.-F. Chang, L.-L. Sun, X.-M. Pan, and R.-S. Wang (2011). J. Cluster Sci. 22, 1.
10. J. Beheshtian, M. Kamfiroozi, Z. Bagheri, and A. Ahmadi (2012). Comput. Mater. Sci. 54, 115.
11. G. Torres-Garcıa and J. Mattaym (1996). Tetrahedron 52, 5421.
12. X. Y. Ren, C. Y. Jiang, J. Wang, and Z. Y. Liu (2008). J. Mol. Graph. Model 27, 558.
13. H. Prinzbach, A. Weiler, P. Landenberger, et al. (2000). Nature 407, 60.
14. G. L. Marcorin, T. Da Ros, S. Castellano, G. Stefancich, I. Bonin, S. Miertus, and M. Prato (2000).
Org. Lett. 2, 3955.
15. J. Averdung and J. Mattay (1996). Tetrahedron 52, 5407.
16. P. R. Birkett, P. B. Hitchcock, H. W. Kroto, R. Taylor, and D. R. M. Walton (1992). Nature 357, 479.
17. P. J. Krusic, E. Wasserman, P. N. Keizer, J. R. Morton, and K. F. Preston (1991). Science 254, 1183.
18. S. Kotha and A. K. Ghosh (2004). Tetrahedron Lett. 45, 293.
19. H. Yang, X. J. Ruan, C. Miao, H. Xi, Y. Jiang, Q. Meng, and X. Sun (2009). Tetrahedron Lett. 50,
7337.
20. Y. Matsuo and E. Nakamura (2006). Inorg. Chim. Acta 359, 1979.
21. A. Duarte-Ruiz, T. Muller, K. Wurst, and B. Krautler (2001). Tetrahedron 57, 3709.
22. H. Li, C. Risko, J. H. Seo, C. Campbell, G. Wu, J. L. Bredas, and G. C. Bazan (2011). J. Am. Chem.
Soc. 133, 12410.
23. W. V. E. Doering and A. K. Hoffmann (1954). J. Am. Chem. Soc. 76, 6162.
24. E. O. Fischer and A. Maasbol (1964). Angew. Chem. 3, 580.
25. J. Chen, M. A. Hamon, H. Hu, Y. Chen, A. M. Rao, P. C. Eklund, and R. C. Haddon (1998). Science
282, 95.
26. X. Lu, F. Tian, and Q. Zhang (2003). J. Phys. Chem. B 107, 8388.
27. M. Schmidt, et al. (1993). J. Comput. Chem. 14, 1347.
28. N. M. O’Boyle, A. L. Tenderholt, and K. M. Langner (2008). J. Comp. Chem. 29, 839.
29. J. Beheshtian, A. A. Peyghan, and Z. Bagheri (2012). Comput. Mater. Sci. 62, 71.
30. L. Chen, C. Xu, X. F. Zhang, and T. Zhou (2009). Phys. E 41, 852.
31. J. Beheshtian, A. A. Peyghan, Z. Bagheri, and M. Kamfiroozi (2012). Struct. Chem. 23, 1567.
32. M. T. Baei, A. A. Peyghan, M. Moghimi, and S. Hashemian (2012). J. Cluster Sci. 23, 1119.
33. M. Breza (2006). Chem. Phys. 330, 224.
34. H. Xiao, J. Tahir-Kheli, and W. A. Goddard (2011). J. Phys. Chem. Lett. 2, 212.
35. K. Hedberg, L. Hedberg, D. S. Bethune, C. A. Brown, H. C. Dorn, R. D. Johnson, and M. Devries
(1992). Science 254, 410.
36. E. A. Carter and W. A. Goddard (1987). J. Chem. Phys. 86, 862.
37. B. Krautler and M. Puchberger (1993). Helv. Chim. Acta 76, 1626.
38. S. M. Lee, R. J. Nicholls, D. Nguyen-Manh, D. G. Pettifor, G. A. D. Briggs, S. Lazar, D. A. Pankurst,
and D. J. H. Cockayne (2005). Chem. Phys. Lett. 404, 206.
39. S. Li Semiconductor physical electronics, 2nd ed (Springer, USA, 2006).
678 M. T. Baei et al.
123
Author's personal copy