electrical contact of redox enzyme layers associated with electrodes: routes to amperometric...
TRANSCRIPT

965
Review
Electrical Contact of Redox Enzyme Layers Associated with Electrodes: Routes to Amperometric Biosensors ltamar Willner,* Eugenii Katz and Bilha Willner
Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 9 1904, Israel
Received: April 24, 1997 Final version: June 9, 1997
Abstract Tailoring of electrically contacted enzyme electrodes provides the grounds for bioelectronic and biosensor systems. Redox-enzymes organized onto electrodes as monolayer assemblies, and chemically functionalized by redox-relay groups, yield electrically contacted enzyme electrodes exhibiting bioelectrocatalytic features. The sensitivity of the enzyme electrode can be enhanced, or tuned, by the organization of multilayer enzyme electrodes and the application of rough metal supports. Enzyme electrodes of extremely efficient electrical communication with the electrode are generated by the reconstitution of apo-flavoenzymes onto relay-FAD monolayers associated with electrodes. The reconstitution process results in an aligned enzyme on the surface, and its effective electrical contact with the electrode yields selective enzyme electrodes of unprecedented high current responses. Integrated electrodes consisting of relay-NAD(P)+-cofactor and enzyme units are generated by the reconstitution of NAD(P)+- dependent enzymes onto a relay-NAD(P)+ monolayer assembly followed by lateral crosslinking of the enzyme network.
Keywords: Bioelectronics, Biosensors, Enzyme electrodes, Enzyme monolayers, Enzyme multilayers, Reconstituted enzymes, Electrically contacted enzyme electrodes, Bioelectrocatalysis, Amperometric biosensors, NAD(P)*-dependent enzyme electrodes
1. Introduction
The lack of electrical contact of enzyme redox-centers and electrode surfaces is well recognized in bioelectrochemistry [I]. Electron transfer theory [Z] indicates that the electron transfer rate constant between a donor-acceptor pair is given by Equation 1, where AGO and X correspond to the free energy and reorganization energy accompanying electron transfer and do and dare the Van der Waals distance and actual distance separating the donor-acceptor centers, respectively.
ket 0~ exp[-P(d - do)]. exp[-(AGO + X)*/4RTX] ( I )
The electrode and redox-center embedded in the enzyme can be viewed as a donor-acceptor pair. Thus, the thick protein layer surrounding the active-center introduces a spatial separation of the donor-acceptor pair and a kinetic bamer for electron transfer. This leads to the electrical insulation of the active sites of most redox proteins. Some enzymes that include their redox-active site at the protein periphery [3] or that their active center is structurally aligned in respect to the electrode [4], exhibit direct electrical contact with the electrode surface. Redox enzymes mediate the most fundamental electron transfer reactions in nature, and one might ask how do these reactions proceed despite these intrinsic kinetic barriers, and why did Mother Nature select complex routes for electron transfer. Several biological mechanisms stimulate electron transfer: Oxygenases use molecular oxygen as their electron acceptor. Molecular diffusion of oxygen into the protein results in sufficiently short electron transfer distances that facilitate electron transfer with the active site. Dehydrogenases are often NAD(P+)/NAD(P)H cofactor-dependent enzymes. These low molecular weight cofactors act as electron carriers that diffusionally attain appropriate distances in respect to the protein-embedded redox centers that facilitate electron transfer. With other redox enzymes, the formation of labile interprotein supramolecular complexes results in appropriate spatial alignment that permits electron transfer, i.e., cytochrome c and cytochrome oxidase or lactate dehydrogenase. These complex mechanisms of biological electron transfer, seem to be, however, the secret of life. The electrical insulation of redox-enzymes yields vectorial and
controlled electron transfer reactions. The kinetic barriers for electron transfer permit the slow assimilation of oxygen, the sequence of biosynthetic transformations, and the synthesis of energy-rich substances, despite the thermodynamic driving force for the conversion of oxygen to water.
The use of redox-enzymes as bioactive matrices in the development of amperometric biosensors [5, 61 is an obvious application (Fig. 1). Provided that the enzyme is brought into electrical contact with the electrode surface, the transduced current is controlled by the substrate concentration in the system. The intrinsic barriers for direct electron transfer between the enzyme redox center and the electrode resulted in the development of various methods to establish electrical communication between redox enzymes and the electrode interfaces. Diffusional electron mediators, such as ferricyanide, ferrocene derivatives, N , N’- bipyridinium salts, quinones, or transition metal complexes, were employed to electrically contact biocatalysts and the electrode surface [7] (Fig. 2a). In these systems, the electron mediator acts as an electron carrier that accepts or donates the electron to the active center. The architecture of integrated biocatalytic assemblies lacking diffusional components has important advantages in bioelectronics and biosensor technology. For example, tailoring of invasive sensors requires active implantable electrodes which lack any diffusional components. Several methods to electrically contact enzymes with electrodes by a nondiffusional route, were
Transduced
P (product)
Fig. 1. A bioelectronic device (biosensor) based on a redox-enzyme electrode.
electrode
Electroanalysis 1997, 9, No. 13 0 WILEY-VCH Verlag GmbH, D-69469 Weinheim, 1997 1040-0397/97/1309-0965 $ 17.50+.50/0

966 I. Willner et al.
C
c product
ubstrate
4 product
Fig. 2. Electrical contact of a redox-enzyme with electrode surfaces: a) by a diffusional electron mediator; b) by tethering redox relay units to the protein; c) by immobilization of the biocatalyst in a redox-polymer.
reported. These include the chemical modification of enzymes with redox-active electron mediators [8-121 (Fig. 2b), and the immobilization of the enzymes in redox-active polymers [ 13- 161 (Fig. 2c). Tethering of redox-relay groups to the protein, i.e., ferrocene [8, 91 or N,N'-bipyridinium units [11, 121, provides a pathway for electron hopping (or tunneling) through the protein. Immobilization of the enzymes in redox co-polymers exhibiting structural flexibility, such as Os'U"'-polypyridine/poly-pyridinium [ 131 or N , N'-dialkyL4,4'-bipyridiniurn polythiophene [ 161, allows dynamic interactions between the electron-relay sites and the protein active-center that yield appropriate distances for mutual electron transfer.
Two basic aspects must be addressed in the application of redox- enzymes as active interfaces in bioelectronic devices. One issue relates to the method of assembly of the enzyme electrode, while the second aspect involves the electrical contact of the bioelec- trocatalyst within the enzyme electrode assembly. It is the aim of this article to address the molecular architecture elements required to assemble monolayer and multilayer enzyme electrodes exhibit- ing effective electrical communication and bioelectrocatalytic features. Our discussion will address the interrelation between the two issues, and the importance of developing biomolecular architecture to attain effective electrical communication in the enzyme electrode system. Our research group is involved in recent years, in tailoring enzyme electrodes by the assembly of mono- layers and multilayers of bioelectrocatalysts on electrode supports.
We will summarize our accomplishments and highlight the future implications of biomolecular architecture in bioelectronics.
2. Electrically Contacted Monolayer Enzyme Electrodes
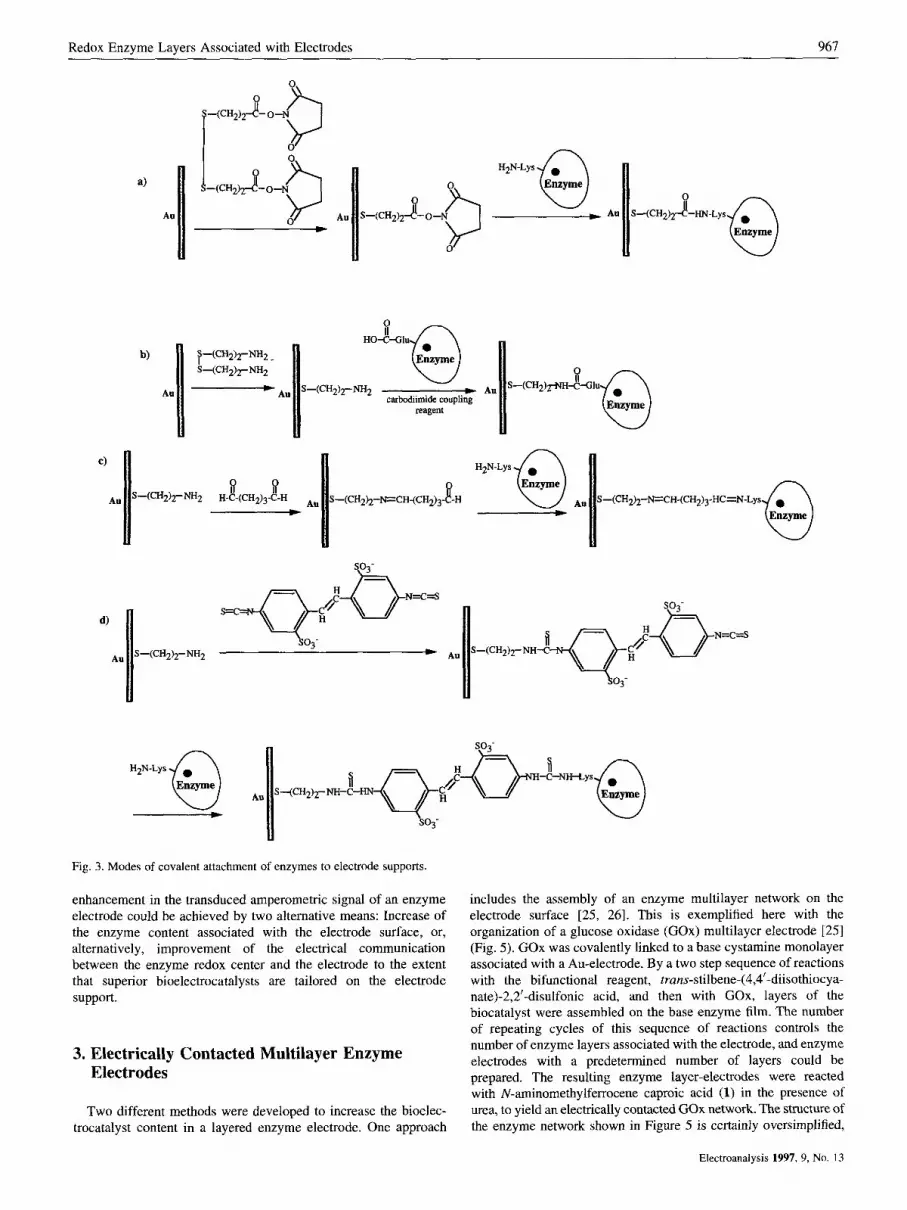
The self-assembly of thiols or disulfides on metal surfaces such as Au, Ag or Pt, has been the subject of extensive research efforts in the last decade [ 17,181. Structural characterization of the monolayer assemblies by various physical means [ 171, chemical functionaliza- tion of solid supports [17, 191 and patterning of surfaces by molecular functionalities [20] represent some of the fascinating research activities in the molecular architecture of surfaces. We used functionalized thiols or disulfides to assemble redox-enzyme electrodes. Figure 3 outlines a few of the functionalized thiolated monolayers that were employed as base interfaces to covalently link the enzyme to the electrode support. Coupling of monolayer carboxylic functions or their active ester to lysine residues of the protein [ I I , 211, direct coupling of monolayer amino functions to glutamic/aspartic protein residues [22], or stepwise coupling of the enzyme lysine groups to the monolayer by bifunctional reagents, i.e., glutaric dialdehyde [23] or di(isothiocyanat0) trans-stilbene disulfate [24, 251, represent a few methods to assemble the enzyme electrodes. The effectiveness of tailoring the enzyme monolayer is controlled by the coupling method. For example, glucose oxidase (Gox), linked to the monolayer by trans-~tiIbene-(4,4'-diisothiocya- nate)-2,2'-disulfonic acid and glutathione reductase (GR) assembled as a monolayer via lysine residues linked to surface carboxylic groups yield surface coverages corresponding to 7.5 x molcm-2 [25] and 2 x lo-" molcm-* [12], respectively.
The enzyme electrode consisting of glutathione reductase coupled to a carboxylic acid-functionalized monolayer via lysine residues, was electrically contacted with the electrode by covalent attachment of N-methyl-N'-carboxyalkyl-4,4'-bipyridinium elec- tron relay units [ l 1, 121 (Fig. 4A). These electron relay groups were tethered to the protein with variable chain lengths (n = 2, 5, 11). The resulting enzyme electrodes revealed bioelectrocatalytic features, and upon the application of a potential more negative than the formal potential of the bipyridinium units, ( Z f = -0.58 V vs. SCE), bioelectrocatalyzed reduction of oxidized glutathione (GSSG) was stimulated (Fig. 4B). The modification of the GR- electrode by the bipyridinium units was performed in the presence of urea, a protocol that turned to be an essential step to generate electrical contact in the resulting enzyme electrode. This was attributed to the partial unfolding of the protein by urea that enabled to implant redox-relay sites in inner protein positions, close to the enzyme redox center. Positioning of these relay groups at inner protein sites and at the enzyme periphery, provides the pathway for electrical communication and activation of the enzyme. The effectiveness of the bioelectrocatalyzed reduction of GSSG is controlled by the chain length tethering the bipyridinium site to the protein (Fig. 4B). The longer the tether, the more efficient is the bioelectrocatalytic reduction of oxidized glutathione. This was attributed to improved electrical contact between the redox-enzyme and the electrode surface with longer tethers. The flexibility of the chains, linking the redox-relay to the protein, enables, with the longer tethers, the formation of shorter relay-redox-site distances that enhance the electrical communication.
The use of electrically contacted enzyme monolayer as sensing interfaces in amperometric biosensors, suffers from the limitation that the amount of biocatalyst in a monolayer is very low. This results in very low amperometric responses upon bioelectrocata- lyzed oxidation (or reduction) of the analyte-substrate. An
Electroandlysis 1997, 9, No, 13
Redox Enzyme Layers Associated with Electrodes 967
a)
Au
H 2 N - L y s o * Enzyme
S-(CH2)2-N=CH-(CH2)3- -H _____)
F
so - \3
Enzyme S-(CH2b-N=CH-(CH2),-HC=N-Lys
-C-NH-Lys 0 n 2 N - L y s o Enzyme
____) W
Fig. 3. Modes of covalent attachment of enzymes to electrode supports.
enhancement in the transduced amperometric signal of an enzyme electrode could be achieved by two alternative means: Increase of the enzyme content associated with the electrode surface, or, alternatively, improvement of the electrical communication between the enzyme redox center and the electrode to the extent that superior bioelectrocatalysts are tailored on the electrode support.
3. Electrically Contacted Multilayer Enzyme Electrodes
Two different methods were developed to increase the bioelec- trocatalyst content in a layered enzyme electrode. One approach
includes the assembly of an enzyme multilayer network on the electrode surface [25, 261. This is exemplified here with the organization of a glucose oxidase (GOx) multilayer electrode [25] (Fig. 5). GOx was covalently linked to a base cystamine monolayer associated with a Au-electrode. By a two step sequence of reactions with the bifunctional reagent, trans-~tilbene-(4,4’-diisothiocya- nate)-2,2’-disulfonic acid, and then with GOx, layers of the biocatalyst were assembled on the base enzyme film. The number of repeating cycles of this sequence of reactions controls the number of enzyme layers associated with the electrode, and enzyme electrodes with a predetermined number of layers could be prepared. The resulting enzyme layer-electrodes were reacted with N-aminomethylferrocene caproic acid (1) in the presence of urea, to yield an electrically contacted GOx network. The structure of the enzyme network shown in Figure 5 is certainly oversimplified,
Electroanalysis 1997, 9, No. 13

968 I. Willner et al.
B
time I min
GSH = GIu-CYS-GIY SH Fig. 4. A) Schematic configuration of an electrically contacted glutathione
reductase monolayer electrode. B) Rate of reduced glutathione (GSH) formation using GR-monolayer electrodes with bipyridinium tethers consisting of different chain lengths: a) n = 2, b) n = 5, c) n = 11. For all systems [GSSG] = 1 x lo-* M and the applied potential corresponded to E = -0.72 (VS. SCE).
GSSG =
and enzyme units within a layer or between layers, can be variable numbers of enzyme layers in the presence of glucose. The crosslinked by the coupling reagent. Analysis of the enzyme content catalytic anodic currents occur at the oxidation potential of in the multilayer assembly revealed, however, an almost identical ferrocene (I? = 0.32V vs. SCE) implying that the enzyme biocatalyst coverage per layer (7.5 x molcm-2) up to a networks are electrically contacted with the electrodes by the network that consists of eight GOx layers. Figure 6A shows the covalently linked ferrocene relay groups. This electrical commu- amperometric responses of layered GOx-electrodes consisting of nication activates the bioelectrocatalyzed oxidation of glucose. It is
n f i - I , .
n
n
303
Fig. 5. Assembly of an electrically contacted glucose oxidase multilayer array on a base cystamine monolayer associated with a Au-electrode.
Electroanalysis 1997, 9, No. 13
Redox Enzyme Layers Associated with Electrodes 969
0 5 10 15 20
[Glucose] / mM 0 0.2 0.4 0.6 0.8
E I V vs. SCE
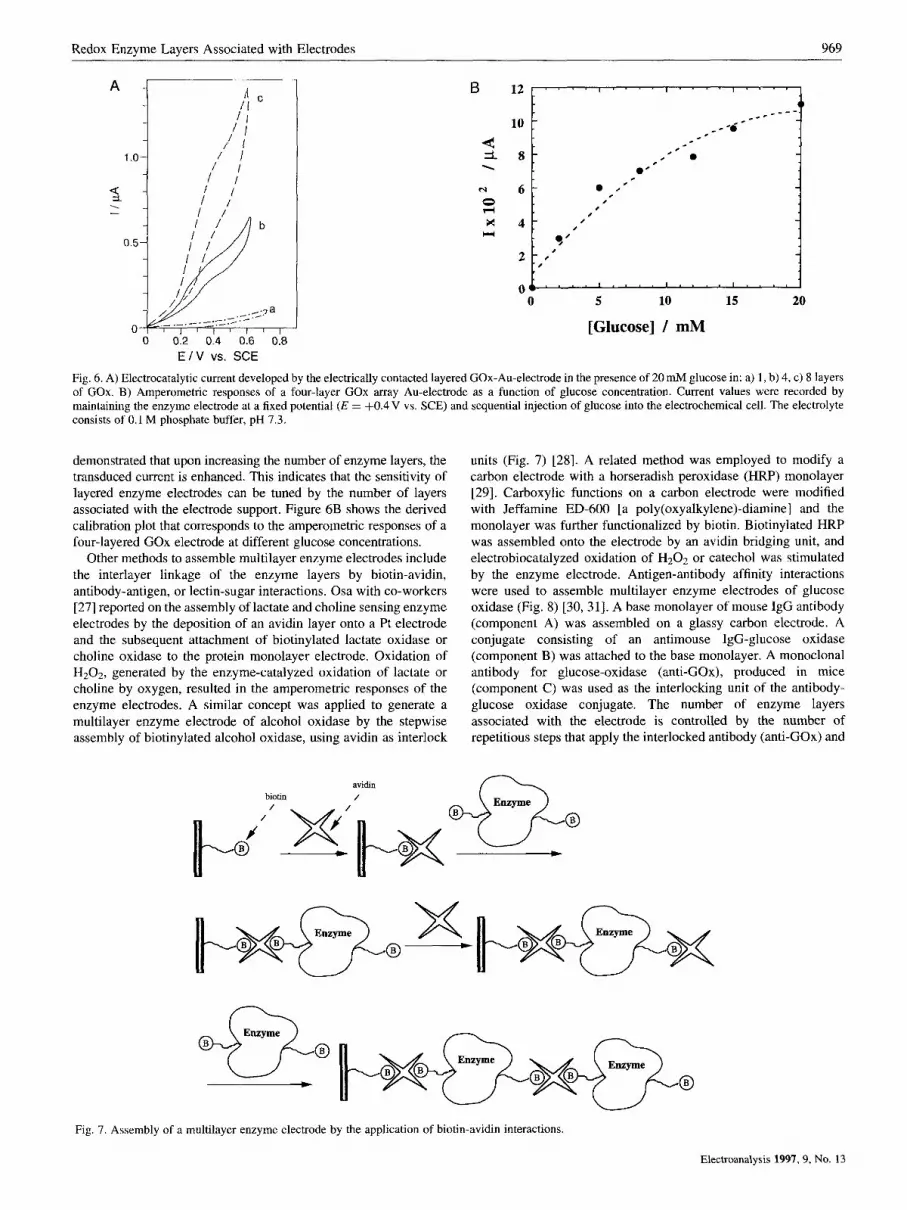
Fig. 6. A) Electrocatalytic current developed by the electrically contacted layered GOx-Au-electrode in the presence of 20 mM glucose in: a) 1 , b) 4, c) 8 layers of GOx. B) Amperometric responses of a four-layer COX array Au-electrode as a function of glucose concentration. Current values were recorded by maintaining the enzyme electrode at a fixed potential ( E = f0.4 V vs. SCE) and sequential injection of glucose into the electrochemical cell. The electrolyte consists of 0.1 M phosphate buffer, pH 7.3.
demonstrated that upon increasing the number of enzyme layers, the transduced current is enhanced. This indicates that the sensitivity of layered enzyme electrodes can be tuned by the number of layers associated with the electrode support. Figure 6B shows the derived calibration plot that corresponds to the amperometric responses of a four-layered GOx electrode at different glucose concentrations.
Other methods to assemble multilayer enzyme electrodes include the interlayer linkage of the enzyme layers by biotin-avidin, antibody-antigen, or lectin-sugar interactions. Osa with co-workers [27] reported on the assembly of lactate and choline sensing enzyme electrodes by the deposition of an avidin layer onto a Pt electrode and the subsequent attachment of biotinylated lactate oxidase or choline oxidase to the protein monolayer electrode. Oxidation of H202, generated by the enzyme-catalyzed oxidation of lactate o r choline by oxygen, resulted in the amperometric responses of the enzyme electrodes. A similar concept was applied to generate a multilayer enzyme electrode of alcohol oxidase by the stepwise assembly of biotinylated alcohol oxidase, using avidin as interlock
units (Fig. 7) [28]. A related method was employed to modify a carbon electrode with a horseradish peroxidase (HRP) monolayer [29]. Carboxylic functions on a carbon electrode were modified with Jeffamine ED-600 [a poly(oxyalky1ene)-diamine] and the monolayer was further functionalized by biotin. Biotinylated HRP was assembled onto the electrode by an avidin bridging unit, and electrobiocatalyzed oxidation of H202 or catechol was stimulated by the enzyme electrode. Antigen-antibody affinity interactions were used to assemble multilayer enzyme electrodes of glucose oxidase (Fig. 8) [30, 311. A base monolayer of mouse IgG antibody (component A) was assembled on a glassy carbon electrode. A conjugate consisting of an antimouse IgG-glucose oxidase (component B) was attached to the base monolayer. A monoclonal antibody for glucose-oxidase (anti-GOx), produced in mice (component C) was used as the interlocking unit of the antibody- glucose oxidase conjugate. The number of enzyme layers associated with the electrode is controlled by the number of repetitious steps that apply the interlocked antibody (anti-GOx) and
avidin biotin /
Fig. 7. Assembly of a multilayer enzyme electrode by the application of biotin-avidin interactions.
Electroanalysis 1997,9, No. 13

910 I. Willner et al.
Fig. 8. Assembly of a multilayer enzyme electrode by the application of antigen/antibody/anti
2
-antibody
- (9)
interactions.
choline J
Fig. 9. Assembly of a bifunctional multilayer network consisting of choline oxidase (ChO) and acetylcholine esterase (AChE) for the amperometric detection of acetylcholine.
the antibody-GOx conjugate. The magnitude of the amperometric responses of the multilayer enzyme electrodes resulting from the bioelectrocatalyzed oxidation of glucose in the presence of the diffusional electron mediator, ferrocene methanol, are controlled by the number of biocatalyst layers associated with the electrode.
The one-step synthesis of a crosslinked electrically contacted enzyme array on a Au-surface was reported by Yoneyama and co- workers [32]. Crosslinking of glucose oxidase and ferrocene ethylamine in the presence of glutaric dialdehyde leads to crosslinking between the ferrocene units and the protein, and parallel bridging of enzyme units. This method of assembling the enzyme electrode is less-ordered in respect to the number of biocatalyst layers associated with the electrode. The simplicity of generating this electrically contacted electrode, and the fact that it exhibits almost linear amperometric responses at glucose concen- trations corresponding to 0-10 mM, contribute to an effective glucose sensing interface.
The stepwise assembly of enzyme multilayers on electrode surfaces and the possibility to tune the enzyme electrode sensitivity by the number of layers associated with the electrode, opens the way to assemble complex enzyme networks for biosensor applications. That is, two enzymes where only one protein is a redox-active biocatalyst, can be assembled in an ordered array on the electrode surface. The nonredox-active enzyme transforms the analyte-substrate to a product that is electrochemically sensed by the redox-active biocatalyst. This approach was applied for the electrochemical detection of acetylcholine [33]. Two layers of
choline oxidase (ChO) and two layers of acetylcholine esterase (AChE) were stepwise assembled on a Au-electrode (Fig. 9). Hydrolysis of acetylcholine leads to the formation of choline. The latter product is electrochemically detected by the ChO-electrode via the electrocatalyzed oxidation of choline in the presence of 2,6- dichlorophenolindophenol (DCPIP) (h? = -0.01 V vs. SCE, pH 7.0) as diffusional electron mediator. The resulting currents are proportional to the hydrolytically generated choline and therefore relate to the acetylcholine concentration.
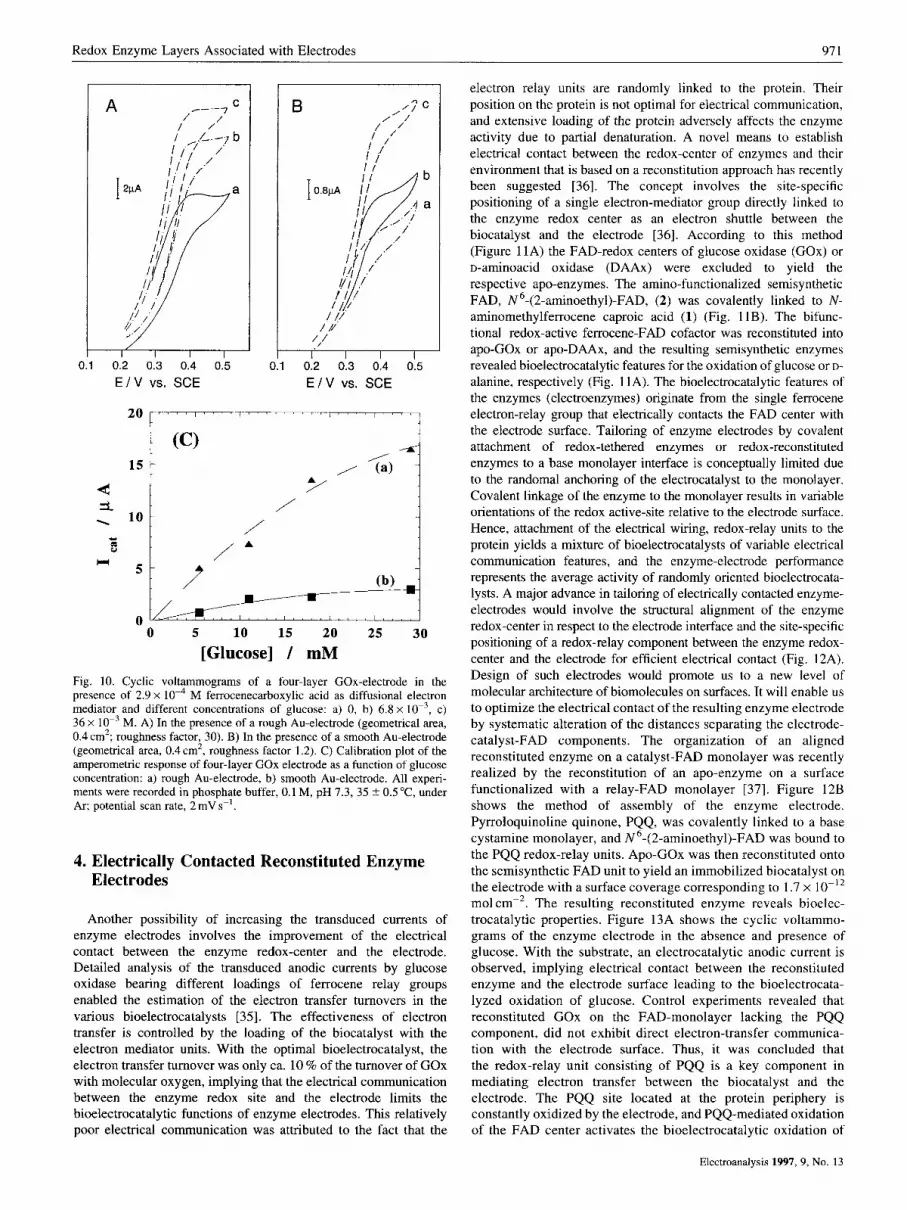
A further method to increase the enzyme content on the electrode support, involves the pretreatment of the electrode itself. The systems described up to now were assembled on commercial Au- surfaces (wires or foils). The typical roughness factors of these surfaces (real surface aredgeometric surface area) corresponds to 1.2-1.3. Treatment of the Au-electrode with mercury and further dissolution of the resulting amalgam yields surfaces with a typical roughness factor of 15-20 [34]. These roughened electrodes allow the immobilization of substantially higher amounts of the biocatalyst [33]. Figures 10A and B show the amperometric responses of enzyme electrodes consisting of an assembly of four layers of GOx, immobilized as described in (Fig. 5) , on a rough and a smooth Au-electrode, respectively. In these systems, ferrocene carboxylic acid is used as diffusional electron mediators. The amperometric responses of the rough electrode are ca. 7-fold higher than that of the smooth electrodes. The derived calibration plots, Figure lOC, reveal a significantly enhanced sensitivity with the rough electrode support.
Electroanalysis 1997, 9, No. 13
Redox Enzyme Layers Associated with Electrodes 97 1
0.1 0.2 0.3 0.4 0.5 E I V vs. SCE E I V vs. SCE
/
0 5 10 15 20 25 30 [Glucose] / mM
Fig. 10. Cyclic voltammograms of a four-layer GOx-electrode in the presence of 2.9 x M ferrocenecarboxylic acid as diffusional electron mediator and different concentrations of glucose: a) 0, b) 6.8 x lo-', c) 36 x lo-' M. A) In the presence of a rough Au-electrode (geometrical area, 0.4 cm2; roughness factor, 30). B) In the presence of a smooth Au-electrode (geometrical area, 0.4 cm', roughness factor 1.2). C) Calibration plot of the amperometric response of four-layer GOx electrode as a function of glucose concentration: a) rough Au-electrode, b) smooth Au-electrode. All experi- ments were recorded in phosphate buffer, 0.1 M, pH 7.3, 35 ? 0.5"C, under Ar; potential scan rate, 2 mV s-'.
4. Electrically Contacted Reconstituted Enzyme Electrodes
Another possibility of increasing the transduced currents of enzyme electrodes involves the improvement of the electrical contact between the enzyme redox-center and the electrode. Detailed analysis of the transduced anodic currents by glucose oxidase bearing different loadings of ferrocene relay groups enabled the estimation of the electron transfer turnovers in the various bioelectrocatalysts [35]. The effectiveness of electron transfer is controlled by the loading of the biocatalyst with the electron mediator units. With the optimal bioelectrocatalyst, the electron transfer turnover was only ca. 10 % of the turnover of GOx with molecular oxygen, implying that the electrical communication between the enzyme redox site and the electrode limits the bioelectrocatalytic functions of enzyme electrodes. This relatively poor electrical communication was attributed to the fact that the
electron relay units are randomly linked to the protein. Their position on the protein is not optimal for electrical communication, and extensive loading of the protein adversely affects the enzyme activity due to partial denaturation. A novel means to establish electrical contact between the redox-center of enzymes and their environment that is based on a reconstitution approach has recently been suggested [36]. The concept involves the site-specific positioning of a single electron-mediator group directly linked to the enzyme redox center as an electron shuttle between the biocatalyst and the electrode [36]. According to this method (Figure 11A) the FAD-redox centers of glucose oxidase (GOx) or o-aminoacid oxidase (DAAx) were excluded to yield the respective apo-enzymes. The amino-functionalized semisynthetic FAD, N6-(2-aminoethyl)-FAD, (2) was covalently linked to N- aminomethylferrocene caproic acid (1) (Fig. 11B). The bifunc- tional redox-active ferrocene-FAD cofactor was reconstituted into apo-GOx or apo-DAAx, and the resulting semisynthetic enzymes revealed bioelectrocatalytic features for the oxidation of glucose or D-
alanine, respectively (Fig. 1 1A). The bioelectrocatalytic features of the enzymes (electroenzymes) originate from the single ferrocene electron-relay group that electrically contacts the FAD center with the electrode surface. Tailoring of enzyme electrodes by covalent attachment of redox-tethered enzymes or redox-reconstituted enzymes to a base monolayer interface is conceptually limited due to the randomal anchoring of the electrocatalyst to the monolayer. Covalent linkage of the enzyme to the monolayer results in variable orientations of the redox active-site relative to the electrode surface. Hence, attachment of the electrical wiring, redox-relay units to the protein yields a mixture of bioelectrocatalysts of variable electrical communication features, and the enzyme-electrode performance represents the average activity of randomly oriented bioelectrocata- lysts. A major advance in tailoring of electrically contacted enzyme- electrodes would involve the structural alignment of the enzyme redox-center in respect to the electrode interface and the site-specific positioning of a redox-relay component between the enzyme redox- center and the electrode for efficient electrical contact (Fig. 12A). Design of such electrodes would promote us to a new level of molecular architecture of biomolecules on surfaces. It will enable us to optimize the electrical contact of the resulting enzyme electrode by systematic alteration of the distances separating the electrode- catalyst-FAD components. The organization of an aligned reconstituted enzyme on a catalyst-FAD monolayer was recently realized by the reconstitution of an apo-enzyme on a surface functionalized with a relay-FAD monolayer [37]. Figure 12B shows the method of assembly of the enzyme electrode. Pyrroloquinoline quinone, PQQ, was covalently linked to a base cystamine monolayer, and N6-(2-aminoethyl)-FAD was bound to the PQQ redox-relay units. Apo-GOx was then reconstituted onto the semisynthetic FAD unit to yield an immobilized biocatalyst on the electrode with a surface coverage corresponding to 1.7 x mol cm-2. The resulting reconstituted enzyme reveals bioelec- trocatalytic properties. Figure 13A shows the cyclic voltammo- grams of the enzyme electrode in the absence and presence of glucose. With the substrate, an electrocatalytic anodic current is observed, implying electrical contact between the reconstituted enzyme and the electrode surface leading to the bioelectrocata- lyzed oxidation of glucose. Control experiments revealed that reconstituted GOx on the FAD-monolayer lacking the PQQ component, did not exhibit direct electron-transfer communica- tion with the electrode surface. Thus, it was concluded that the redox-relay unit consisting of PQQ is a key component in mediating electron transfer between the biocatalyst and the electrode. The PQQ site located at the protein periphery is constantly oxidized by the electrode, and PQQ-mediated oxidation of the FAD center activates the bioelectrocatalytic oxidation of
Electroanalysis 1997, 9, No. 13

972 I. Willner et al.
A
GOx-holoenzyme u GOx-apoenzyme u \gluconic acid
H- C- OH H- c- on n- c- on I
I H - F - H
O = P - 0 - P - 9 9 I ,
0' 0'
GOx
. glucose
ocdn Fy;q H
on OH I EDC
H H-C-H - I - 0
NH-CHz-CHpNH -C-(CH&- HN- CH2 n- c- on n- c- OH I Fe
I H- c- on H-F-H nc, y 2 ';CH
N N
FAD-FC
0- 0
OH OH
Fig. 11. A) Electrical contacting of a flavoenzyme by its reconstitution with a relay-FAD semisynthetic cofactor. B) Synthesis of a ferrocene-FAD semisynthetic cofactor for the reconstitution of apo-flavoenzymes, EDC = 1 -(3-dimethylamino-propyl)-3-ethylcarbodiimide hydrochloride.
glucose. The resulting electrical current is controlled by the recycling rate of the reduced FAD by the substrate, or the concentration of glucose. Figure 13B shows the derived calibration plot for the amperometric responses of the reconstituted enzyme electrode at different concentrations of glucose. The resulting current densities are unprecedentally high in the area of biosensors and bioelectronic devices (300 pA cm-' at 8 O m M of glucose).
The electron-transfer turnover rate of COX with molecular
oxygen as electron acceptor corresponds to ca. 600 s-l at 25 "C. Using the activation energy of 7.2 kcal mol-', the electron-transfer turnover rate of COX at 35 "C is estimated to be ca. 900 s-' [37]. A densely packed monolayer of COX (2.9 x lo-'' mol cm-*) that exhibits the theoretical electron-transfer turnover rate, is expected to yield an amperometric response of ca. 300pAcrn-*. This indicates that reconstituted COX on the PQQ-FAD monolayer exhibits an electron-transfer turnover with the electrode of similar
Electroanalysis 1997, 9, No. 13

Redox Enzyme Layers Associated with Electrodes 973
A
57 Apoenzyme
Au
PQQ = H B C W H
HOOC 0
PQQ - Au
Apo - GOx - Au
-substrate
Gluconic acid
u Glucose 2e-
2e-
H Z N - ~ ~ ~
. . OH OH
Fig. 12. A) Reconstitution of an apo-flavoenzyme on a relay-FAD monolayer linked to an electrode support. B) Reconstitution of GOx-apoenzyrne on PQQ- FAD monolayer linked to a Au-electrode.
effectiveness observed with the enzyme and oxygen as mediator. Besides the high sensitivity of the resulting enzyme electrode, the unprecendented efficiency of electrical contact has important consequences in tailoring future enzyme electrodes. Amperometric glucose sensing electrodes suffer from nonspecific oxidation of various interferants, such as ascorbic acid or uric acid. Also, oxygen
interferes with the current transduction as a result of non- electrochemical oxidation of the enzyme redox-site. The efficient electrical contact of the biocatalyst and the electrode and the resulting effective electrocatalyzed oxidation of glucose, suggest that the nonspecific oxidation of the interfering substrates or the accompanying reaction of the biocatalyst with oxygen, should have
Electroanalysis 1997, 9, No. 13

974 I. Willner et al.
I I 400 ' I I I ' I I I I I I I I
A
. 4 zoo{
- 1 100
I I"1
ii 04 1- -f
-0.6 -0.4 -0.2 0 0.2 0.4 E I v vs.SCE
. . . 1.5
. .
0:' I I b.bi I I b.od I I b.bt: I I b.bd I I ':I
. C glucose I
Fig. 13. A) Cyclic voltammograms of the PQQ-FAD-surface reconstituted GOx electrode in the absence of glucose (curve a) and in the presence of glucose, 80mM (curve b). Data recorded under argon, 3 5 T , 0.01 M phosphate buffer and 0.1M sodium sulfate, pH 7.0, scan rate 5mVs- ' . (Geometrical electrode area, 0.4cm2, roughness factor 15 2 5) . B) Ampero- metric responses of the reconstituted enzyme electrode at different glucose concentrations. Data recorded by chronoamperometric measurements at a final potential +0.2 V (vs. SCE), 35 "C.
product
Fig. 14. Electrochemical contact of an NAD(P)+-dependent enzyme with the electrode in the presence of diffusional NAD(P)+ cofactor.
little effect on the resulting current. Indeed, it was found that the transduced amperometric response at a glucose concentration of 5 m M was almost unaffected in the presence of oxygen or the interfering compounds [37]. The resulting selectivity of the reconstituted enzyme electrode and the high current densities achieved have further importance in the application of these electrodes as invasive glucose sensors. The pain accompanying the penetration of a needle-electrode is proportional to its diameter, and microelectrodes are anticipated to be applied as invasive sensing electrodes. The high electron-transfer turnover rates of the
Electroanalysis 1997, 9, No. 13
reconstituted biocatalyst suggests that even with the low surface- area of the microelectrode, physically detectable current responses will be obtained.
5. Integrated NAD(P)'-Dependent Enzyme Electrodes
The development of amperometric enzyme electrodes concen- trated, so far, on the application of flavoenzymes, as bioactive sensing interfaces. With these enzymes the FAD-redox cofactor is a part of the protein and synthetic redox-relays are capable of generating the desirable electrical contact. Most of the redox enzymes are, however, cofactor-dependent biocatalysts, particu- larly nicotinamide adenine dinucleotide, NAD+, and nicotinamide adenine dinucleotide phosphate, NADP+-dependent enzymes. In this series of enzymes the NAD(P)+-cofactors stimulate the oxidation of the enzyme redox-site by a diffusional pathway and the resulting reduced cofactor, NAD(P)H, that diffuses out of the protein, can be oxidized by another biocatalytic reductive transformation, or, alternatively, oxidized by an electrode. Electro- chemical oxidation of NAD(P)H cofactors was the subject of extensive research activities [38]. Electrochemical oxidation of NAD(P)H is accompanied by kinetic barriers and requires a substantial overpotential. Thus, electrochemical activation of NAD(P)+-dependent enzymes requires the participation of an electrocatalyst capable of regenerating the NAD(P)H/NAD(P)+ couple close to the thermodynamic redox potential (Fig. 14). Different redox relays, such as o-quinones [39], p-quinones [40], phenazine, phenoxazine and phenothiazine derivatives [41], ferrocenes [42] and 0s-complexes [43], were applied as electro- catalysts for the regeneration of NAD(P)+ cofactors. For the practical application of NAD(P)+-dependent enzymes as bioactive sensing matrices, integration of the electrocatalyst, the cofactor and the enzyme, as a united assembly that allows the electrochemical activation of the biocatalyst, is essential. This is particularly difficult since the NAD(P)+-cofactors participate in the electron transfer chain by a diffusional route. Different configurations that employ polymeric NAD(P)+ cofactors entrapped with the respec- tive biocatalysts in crosslinked polymers, or by means of a membrane, were used to confine the cofactor and enzyme at the electrode surface [44-461.
Recent research activities demonstrated, however, possible means to tailor NAD(P)+-dependent enzyme electrodes and the potential of using this important class of biocatalysts as active sensing interfaces in biosensor devices. Pyrroloquinoline quinone, PQQ, was assembled as a monolayer on Au-electrodes by its covalent coupling to a cystamine monolayer [34, 47, 481. The quinone monolayer revealed electrocatalytic properties for NAD(P)H oxidation [47, 481. The PQQ monolayer-electrode revealed enhanced electrocatalyzed oxidation of glucose in the presence of added Ca2+-ions [47]. The electrocatalytic features of the PQQ-monolayer were used to develop an integrated PQQ enzyme monolayer as depicted in (Figure 15). The NADP+- dependent malic enzyme was covalently linked to the PQQ monolayer assembled on the Au-electrode [48]. In the presence of NADP+ as diffusional cofactor and malic acid as substrate, biocatalyzed oxidation of malic acid yields the reduced cofactor, that is electrocatalytically oxidized by PQQ. The resulting current is controlled by the concentration of NADPH, while the concentration of the reduced cofactor is determined by the analyte (malic acid). This system is still not electrically integrated as the cofactor is in a diffusional configuration. Tethering of NAD(P)+ units to the enzyme monolayer could be a future extension of generated,

Redox Enzyme Layers Associated with Electrodes 975
pyruvic acid + C02 jmJ Malic
v EDC + HEPES-buffer. pH=7 3
malic acid
PQQ = n~ jXjcJrnH 0
NADP+ =
Fig. 15. Assembly of a PQQ-malic enzyme electrode.
integrated, catalyst-cofactor-enzyme electrodes. Recent reports applied polymeric NAD(P)+ analogs and enzymes as bioactive components entrapped in membrane assemblies for continuous biosynthetic application [44-461. This suggests that entrapment of polymeric NAD(P)+ at the PQQ-enzyme electrode, by means of an appropriate membrane, could also lead in the future to reusable cofactor-dependent enzyme electrode.
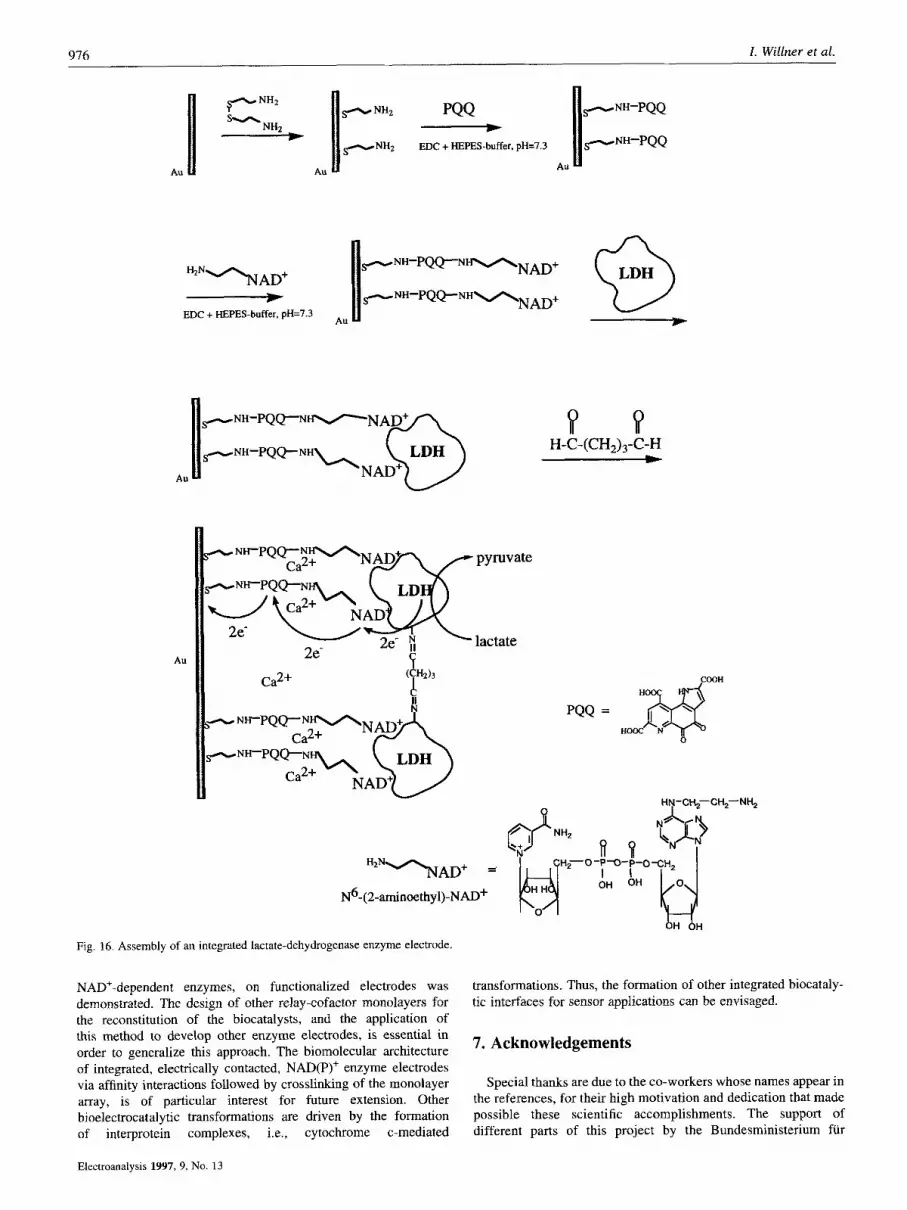
A major advance in the architecture of electrically integrated NAD(P)+-dependent enzyme electrodes was recently accomplished by the application of the reconstitution method (Fig. 16) [49]. NAD(P)+-cofactors exhibit substrate affinities for the respective biocatalysts, and the mutual recognition and associative interactions between the cofactor and the enzymes can be used to generate a cofactor-enzyme complex [50]. The fact that PQQ acts as an electrocatalyst for the oxidation of NAD(P)H, led to the assembly of a PQQ-NAD+ monolayer by covalent linkage of N6-(2- aminoethy1)-NAD+ (3) to the PQQ monolayer interface. Interaction of the PQQ-NAD+ monolayer electrode with the NAD+-dependent enzyme lactate dehydrogenase (LDH), led to the selective association of the biocatalyst to the NAD+-cofactor units. Micro- gravimetric, quartz-crystal-microbalance, experiments revealed that the coverage of the surface-associated LDH by the respective affinity interactions is 3.5 x lo-'' mol cm-'. The PQQ-NAD+- bound LDH exhibited bioelectrocatalytic oxidation of lactate. The surface-bound LDH revealed temporary stability on the electrode surface, and only ca. 10 % of the biocatalyst were dissociated from the electrode within 30 min. The surface-bound LDH was, however, immediately detached from the surface upon addition of diffusional NAD+ to the electrolyte solution implying the labile association of LDH to the monolayer electrode. The temporary stability of the monolayer of LDH associated with the PQQ-NAD' interface enabled the crosslinking of the enzyme layer with glutaric- dialdehyde. The resulting enzyme-monolayer-electrode represents an electrically integrated NAD+-dependent enzyme electrode. The
enzyme is neither dissociated from the electrode by itself, nor upon the addition of diffusional NAD+. The resulting PQQ-NAD+- crosslinked enzyme assembly is electroactive for the oxidation of lactate. Figure 17 shows the calibration plot that corresponds to the amperometric response of the electrode at different lactate concentrations. This approach appears to be general, and a similar pathway was used to assemble an alcohol sensing electrode using the NAD+-dependent alcohol dehydrogenase.
6. Conclusions and Future Perspectives
This review article has addressed recent advances in tailoring monolayer and multilayer enzyme electrodes for biosensor applica- tions. The conceptual method of designing the enzyme electrodes involved the assembly of covalently linked protein assemblies on base thiolate monolayers associated with Au-electrodes. Methods to electrically contact the enzyme layers with the electrode surface, and means to increase the biocatalyst content on the electrode supports by the construction of protein multilayer arrays, and using rough Au-supports, were discussed. This enabled enhancement and tuning of the enzyme electrode sensitivities and the combination of two (or more) enzymes as biocatalytic sensing interfaces.
The methods to chemically functionalize Au-electrodes by redox-active monolayers and to construct complex redox-relay/ cofactor assemblies, opened the venue to the novel development of surface reconstituted enzyme electrodes. Beyond the practical implications of these electrodes, the reconstitution of biocatalysts on their cofactors, has important basic scientific elements. Structural alignment and covalent association of proteins on the electrodes, and the stimulation of electron transfer chains in the resulting assemblies, highlight new means of tailoring bioelectronic systems. At present, the reconstitution of flavoenzymes and
Electroanalysis 1997, 9, No. 13
976 1. Willner et al.
PQQ p NH2 u
NHZ I P N H z EDC + HEPES-buffer. pH=7.3
All Au Au
Au
P P H-C-(CH&-C-H I
Au
Fig. 16. Assembly of an integrated lactate-dehydrogenase enzyme electrode
NAD+-dependent enzymes, on functionalized electrodes was demonstrated. The design of other relay-cofactor monolayers for the reconstitution of the biocatalysts, and the application of this method to develop other enzyme electrodes, is essential in order to generalize this approach. The biomolecular architecture of integrated, electrically contacted, NAD(P)+ enzyme electrodes via affinity interactions followed by crosslinking of the monolayer array, is of particular interest for future extension. Other bioelectrocatalytic transformations are driven by the formation of interprotein complexes, i.e., cytochrome c-mediated
transformations. Thus, the formation of other integrated biocataly- tic interfaces for sensor applications can be envisaged.
7. Acknowledgements
Special thanks are due to the co-workers whose names appear in the references, for their high motivation and dedication that made possible these scientific accomplishments. The support of different parts of this project by the Bundesministerium fur
Electroanalysis 1997, 9, No. 13

Redox Enzyme Layers Associated with Electrodes 977
1 , , , , , , , , , , , , , , , I ' 0
0 0 5 10 15 20
[lactate] / mM Fig. 17. Amperometric responses of an integrated lactate dehydrogenase enzyme electrode at different concentrations of lactate. Data recorded under argon, 25"C, 0.05M Tris-HC1 buffer, pH 8.0, +10mM CaC12; electrode geometrical area, 0.2 cm'; roughness factor, 1.2; applied potential +0.1 V (vs. S C E l
Bildung, Wissenschaft, Forschung und Technologie (BMBF, Germany), The Israel Ministry of Science, The Szold Foundation of The Hebrew University of Jerusalem and Savyon Diagnostics Ltd., Ashdod, Israel, is gratefully acknowledged.
8. References
[I] A. Heller, Acc. Chem. Res. 1990, 23, 128. [2] R.A. Marcus, N. Sutin, Biochim. Biophys. Acta 1985,811,265. [3] J. Kazlauskaite, A.C.G. Westlake, L.-L. Wong, H.A.O. Hill, Chem. Commun.
1996, 2189. [4] B.A. Kuznetsov, N.A. Byzova, G.P. Shumakovich, J. Electroanal. Chem.
1994, 371, 85. [5] H.-L. Schmidt, W. Schuhmann, F.W. Scheller, F. Schubert, in Sensors, Vol.
3: Chemical and Biochemical Sensors, Part 2 (Eds: W. Gopel, T.A. Jones, M. Kleitz, I. Lundstrom, T. Seiyama), VCH, Weinheim 1992, p. 719.
161 P.N. Bartlett, in Biosensor Technology, Fundamentals and Applications (Eds: R.P. Buck, W.E. Hatfield, M. Umana, E.F. Bowden), Marcel Dekker,
[71 P.N. Bartlett, P. Tebbutt, R.G. Whitaker, Prog. React. Kinetics 1991,16,55. [81 Y. Degani, A. Heller, J. Phys. Chem. 1987, 91, 1285. [91 Y. Degani, A. Heller, J. Am. Chem. Soc. 1988, 110, 2615.
[lo] W. Schuhmann, Biosens. Bioelectron. 1995, 10, 181. Llll I. Willner, E. Katz, A. Riklin, R. Kasher, J . Am. Chem. Soc. 1992, 114,
[I21 I. Willner, N. Lapidot, A. Riklin, R. Kasher, E. Zahavy, E. Katz, J. Am.
New York 1990, p, 95.
10965.
Chem. Soc. 1994,116, 1428.
[I31 A. Heller, J. Phys. Chem. 1992, 96, 3579. [14] P.D. Hale, T. Inagaki, H. I. Karan, Y. Okamoto, T.A. Skotheim, J. Am. Chem.
[I51 T. Kaku, H.I. Karan, Y. Okamoto, Anal. Chem. 1994, 66, 1231. [I61 1. Willner, E. Katz, N. Lapidot, Bioelectrochem. Bioenerg. 1992, 29, 29. [17] H.O. Finklea, in Electroanalylical Chemistry, Vol. 19 (Eds: A.J. Bard, 1.
Rubinstein), Marcel Dekker, New York 1996, p. 109. [I81 J. Xu. H:L. Li, J. Colloid Inteflace Sci. 1995, 176, 138. [191 D. Mandler, I. Turyan, Electroanalysis 1996, 8, 207. [20] A. Kumar, N.L. Abbott, E. Kim, H.A. Biebuyck, G.M. Whitesides, Acc.
121) M. Lion-Dagan, E. Katz, I. Willner, J. Am. Chem. Soc. 1994, 116, 7913. [22] B.A. Kuznetsov, N.A. Byzova, G.P. Shumakovich, J. Electroanal. Chem.
1231 I. Willner, M. Lion-Dagan, S. Marx-Tibbon, E. Katz, J. Am. Chem. SOC.
[24] E. Katz, A. Riklin, I. Willner, J. Electroanal. Chem. 1993, 354, 129. [25] I. Willner, A. Riklin, B. Shoham, D. Rivenzon, E. Katz, Adv. Mater. 1993,5,
[26] B. Shoham, Y. Migron, A. Riklin, 1. Willner, B. Tartakovsky, Biosens.
[27] T. Hoshi, H. Takeshita, J. Anzai, T. Osa, Anal. Sci. 1995, 11, 3 1 1 . [28J X-yan Du, J. Anzai, T. Osa, R. Motohashi, Electroanalysis, 1996, 8, 813. [29] P. Pantano, T.H. Morton, W.G. Kuhr, J. Am. Chem. Soc. 1991, 113, 1832. [30] C. Bourdillon, C. Demaille, J. Moiroux, J.-M. SavCant, J. Am. Chem. Soc.
1311 C. Boudillon, C. Demaille, J. Moiroux, J.-M. Saviant, Ace. Chem. Re.s. 1996,
[32] S. Kuwabata, T. Okamoto, Y. Kajiya, H. Yoneyarna, Anal. Chem. 1995, 67,
[33] A. Riklin, 1. Willner, Anal. Chem. 1995, 67, 4118. [34] E. Katz, D.D. Schlereth, H.-L. Schmidt, J. Electroanal. Chem. 1994,367,59. [35] A. Badia, R. Carlini, A. Fernandez, F. Battaglini, S.R. Mikkelsen, A.M.
English, J. Am. Chem. Soc. 1993, 115, 7053. [36] A. Riklin, E. Katz, I. Willner, A. Stocker, A.F. Buckmann, Nature 1995,376,
672. [37] I. Willner, V. Heleg-Shabtai, R. Blonder, E. Katz, G. Tao, A.F. Buckmann,
A. Heller, J. Am. Chem. Soc. 1996, 118, 10321. [38] L. Gorton, B. Persson, P.D. Hale, L.I. Boguslavsky, H.I. Karan, H.S. Lee,
T.A. Skotheim, H.L. Lan, Y. Okamoto, in Biosensors and Chemical Sensors (Eds: P.G. Edelman, J . Wang), ACS Symp. Ser. 487, ACS, Washington, DC,1992, p. 56.
[39] H. Jaegfeldt, T. Kuwana, G. Johansson, J. Am. Chem. Soc. 1983, 105, 1805. 1401 H. Huck, H:L. Schmidt, Angew. Chem. Int. Ed. Engl. 1981, 93,421. [41] L. Gorton, J. Chem. Soc., Faraday Trans. I 1986, 82, 1245. [42] B.W. Carlson, L.L. Miller, J. Am. Chem. Soc. 1983, 105, 7345. [43] T. Vering, W. Schuhmann, D. Seiwald, H.-L. Schmidt, B. Speiser, L. Ye,
[44] M.-0. Mansson, P:O. Larsson, K. Mosbach, Eur. J. Biochem. 1978,86,455. [45] Y. Yamazaki, H. Maeda, Agricult. Biol. Chem. 1982, 46, 1571. [46] M. MontagnC, J.-L. Marty, Anal. Chim. Acta, 1995, 315, 297. [47] E. Katz, T. Lotzbeyer, D.D. Schlereth, W. Schuhmann, H.-L. Schmidt, J.
[48] I. Willner, A. Riklin, Anal. Chem. 1994, 66, 1535. [49] A. Bardea, E. Katz, A.F. Biickmann, I. Willner, J. Am. Chem. Soc. 1997, in
press. [50] C.R. Lowe, I.P. Trayer, H.R. Trayer, in Methods in Enzymology, Vol. 66, Part
E (Eds: D.B. McCormick, L.D. Wright), Academic Press, New York 1980, p. 192.
Soc. 1989, 1 1 I, 3482.
Chem. Res. 1995, 28,219.
1994, 371, 85.
1995, 117, 6581.
912.
Bioelectron, 1995, 10, 341.
1994,116, 10328.
29, 529.
1684.
J. Electroanal. Chem. 1994, 364, 275.
Electroanal. Chem. 1994, 373, 189.
Electroanalysis 1997, 9, No. 13