drug permeation across intestinal epithelial cells using porous silicon nanoparticles
TRANSCRIPT

lable at ScienceDirect
Biomaterials 32 (2011) 2625e2633
Contents lists avai
Biomaterials
journal homepage: www.elsevier .com/locate/biomater ia ls
Drug permeation across intestinal epithelial cells using porous siliconnanoparticles
Luis M. Bimbo a,*, Ermei Mäkilä b, Timo Laaksonen a, Vesa-Pekka Lehto c, Jarno Salonen b, Jouni Hirvonen a,Hélder A. Santos a,*
aDivision of Pharmaceutical Technology, Faculty of Pharmacy, University of Helsinki, FI-00014, Finlandb Laboratory of Industrial Physics, Department of Physics and Astronomy, University of Turku, FI-20014, FinlandcDepartment of Physics and Mathematics, University of Eastern Finland, FI-70211 Kuopio, Finland
a r t i c l e i n f o
Article history:Received 8 October 2010Accepted 4 December 2010Available online 30 December 2010
Keywords:SiliconNanoparticleDegradationCytotoxicityMacrophageEpithelial cell
* Corresponding authors. Tel.: þ358 9 19159160; faE-mail addresses: [email protected] (L.M. Bim
(H.A. Santos).
0142-9612/$ e see front matter � 2010 Elsevier Ltd.doi:10.1016/j.biomaterials.2010.12.011
a b s t r a c t
Mesoporous silicon particles hold great potential in improving the solubility of otherwise poorly solubledrugs. To effectively translate this feature into the clinic, especially via oral or parenteral administration,a thorough understanding of the interactions of the micro- and nanosized material with the physio-logical environment during the delivery process is required. In the present study, the behaviour ofthermally oxidized porous silicon particles of different sizes interacting with Caco-2 cells (both non-differentiated and polarized monolayers) was investigated in order to establish their fate in a model ofintestinal epithelial cell barrier. Particle interactions and TNF-a were measured in RAW 264.7 macro-phages, while cell viabilities, reactive oxygen species and nitric oxide levels, together with transmissionelectron microscope images of the polarized monolayers, were assessed with both the Caco-2 cells andRAW 264.7 macrophages. The results showed a concentration and size dependent influence on cellviability and ROS-, NO- and TNF-a levels. There was no evidence of the porous nanoparticles crossing theCaco-2 cell monolayers, yet increased permeation of the loaded poorly soluble drug, griseofulvin, wasshown.
� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
The oral delivery of drug molecules remains the preferred routeof administration due to its simplicity and patient compliance[1]. However, the poor pharmacokinetics of many of the newlydeveloped, lipophilic/hydrophobicmolecules hinders their effectivedelivery [2]. The dissolution and solubility of the drug in the intes-tinal lumen, poor permeation properties in the gastrointestinal (GI)tract, as well as high intestinal or hepatic first pass metabolism aresome common aspects that hamper the drug’s oral bioavailability[3]. Several newapproaches andmaterials havebeen actively soughtand investigated in order to improve the pharmacokinetic proper-ties and to overcome the inherent difficulties of oral delivery [4].These new materials face the challenge to prove their efficacy, aswell as safety and biocompatibility issues.
Porous silicon (PSi) possesses several properties that make it anattractive material for controlled release and drug delivery applica-tions [5e7]. It has been envisioned as a promising oral drug delivery
x: þ358 9 19159144.bo), [email protected]
All rights reserved.
carrierdue to its apparentbiocompatibility [8,9] andexcellent abilityin increasing the solubility of otherwise poorly soluble drugs [10,11].Several reports have alreadybeenpublishedabout theeffectivedrug,peptide and even small interfering RNA delivery using mesoporoussilicon materials [10,12e15]. Furthermore, these materials can betailored to withstand the conditions of the stomach and gastroin-testinal lumen, and improving the drug dissolution and fast releasekinetics at the appropriate site of action [6].
PSi shows distinctive advantages when compared with othermaterials, such as its top-down production method [6], the possi-bility to stabilize loaded drugs or peptides within its pores and easysurface modification for imaging purposes [16]. Several studies havealready been published about the biological interaction of meso-porous silicon particles at a micro- and nanoscale [16e18] whichfurther support the claim of its biocompatibility. The size of theseparticulate systems should be confined to a range adequate for drugdelivery applications, and that can exhibit optimal stability. Amongthe several stabilization treatmentsof the silicon surface, the thermaloxidation is one of the simplest [19]. This method, depending ontreatment temperature, forms a thin layer of silicon oxide on thesurfaces of PSi, resulting in a slight decrease in pore diameter. Inaddition to the increased stability, the oxidation changes the surface

L.M. Bimbo et al. / Biomaterials 32 (2011) 2625e26332626
from hydrophobic to hydrophilic, which is useful for drug deliveryapplications under physiological conditions [6]. An ideal oral drugdelivery agent requires biocompatibility, improved solubility ofa loaded drug or peptide, releasing of the payload at the absorptionsite and, at the same time, leaving undisturbed cell structure andfunction and maintaining the physiological milieu.
To our knowledge, the therapeutic applications of this nano-scaled thermally oxidized-PSi (TOPSi) material, alongside with theassessment of its compatibility and drug permeation propertieshave not been tested in human cell models. Here we show that thenanoparticulate mesoporous silicon system effectively enhancesthe permeability of a poorly soluble model drug across the mono-layers of differentiated Caco-2 cells. Thorough evaluation is alsodone on the TOPSi particles’ compatibility profile and their inter-actions with the Caco-2 epithelial cells and RAW 264.7 macro-phages are also investigated.
2. Material and methods
2.1. Preparation of TOPSi particles
2.1.1. MicroparticlesFree standing PSi films were fabricated from monocrystalline h100i silicon
wafers (Cemat Silicon S.A.) by electrochemical anodization in a 1:1 (v/v) hydrofluoricacid (38%) e ethanol solution with a constant current density of 50 mA/cm2. Thewafers were boron-doped pþ-type with a resistivity of 0.01e0.02 U cm. After theetching, the porous films were separated from the substrate by abruptly increasingthe current density to the electropolishing region. The films were milled in a highenergy ball mill (Pulverisette 7, Fritsch GmbH) in an agate grinding jar and dry sievedrepeatedly in test sieves until the desired size fractions were obtained. Finally, eachfraction of microparticles was wet sieved with ethanol to separate the largeraggregates and dried at 65 �C under low pressure. The microparticles were thenstabilized by thermal oxidation for 2 h at 300 �C in ambient air.
2.1.2. NanoparticlesFree standing multilayer PSi films were fabricated from monocrystalline h100i
pþ silicon wafers (Cemat Silicon S.A.) by electrochemical anodization in a 1:1 (v/v)hydrofluoric acid (38%) e ethanol solution. The multilayer structure was producedby pulse etching with a low current density followed by a short burst of electro-polishing current, which functions as a fracture plane in the PSi film [16]. The highcurrent burst was followed by a short pause, in order to compensate for possibleconcentration gradients in the electrolyte. The size of the nanoparticles can beaffected through the duration of the low current density pulse. The multilayer filmwas finally separated from the substrate by an abrupt increase of current to theelectropolishing region.
The multilayer films were stabilized by thermal oxidation for 2 h at 300 �C inambient air. The nanoparticles were produced by wet milling the TOPSi multilayerfilms in ethanol with a high energy ball mill (Pulverisette 7, Fritsch GmbH) ina zirconia grinding jar. The nanoparticles were separated to different particle sizesby centrifugation.
2.1.3. Nanoparticle loadingThe low-solubility model drug studied was griseofulvin (Orion Pharma). Initially,
the TOPSi particles were stored in ethanol dispersion. The storage dispersant wasremoved after centrifugation by collecting the supernatant and replaced withdichloromethane (DCM). After redispersion of the particles into the DCM, thedispersant was again removed after centrifugation and replaced with the loadingsolution (griseofulvineDCM solution with a concentration of 75 mg/ml), with a ratioof approximately 0.5 ml of solution for 15 mg of nanoparticles and redispersed. Theloading time was 3 h after which the particles were again centrifuged and thesupernatant (loading solution) removed. The particles were dried at 65 �C overnight.In every case, the dispersing was done by ultrasonication. The average loading of thegriseofulvin in the nanoparticles was determined by immersing the loaded nano-particles in a 50/50 (v/v) ethanol/water solution for 1 h under vigorous stirring andthen determining the drug concentration in the solution by HPLC (Agilent 1100series). The average loading degree of the drug obtained from four differentmeasurements was 17.3 � 0.4% in weight.
2.2. Particle characterization
The pore volume, average pore diameter, and specific surface area of the TOPSimicro- and nanoparticles were calculated from desorption branch of nitrogensorption (TriStar 3000, Micromeritics Inc.) measurements. Although the surface areaand pore volume were reduced in the nanoparticles, they were still sufficientconsidering drug delivery applications. The average nanoparticle sizes were
measured from transmission electron microscope (TEM, FEI Tecnai F12, PhilipsElectron Optics) images and with dynamic light scattering (Zetasizer Nano ZS,Malvern Instruments Ltd.). The zeta potential of the nanoparticles was calculatedfrom the measured electrophoretic mobility using the Schmolukovski equation(Zetasizer Nano ZS, Malvern Instruments Ltd.). The samples for TEM were preparedby adding droplets of the TOPSi nanoparticle suspensions on the copper-carbongrids and allowing them to dry in a dry box for ca. 24 h. The size distribution wasthen estimated using public domain ImageJ image processing software. Themicroparticle size was determined by automated optical microscopy analysis(Morphologi G3, Malvern Instruments Ltd.).
2.3. Cell line and culture
The in vitro studies with the TOPSi particles were performed with human coloncarcinoma (Caco-2) and RAW 264.7 murine macrophage cells. Both cell lines wereobtained from American Type Culture Collection (ATCC). Non-differentiated Caco-2cells were cultured in 75 cm2 culture flasks (Corning Inc. Life Sciences) using Dul-becco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovineserum, 1% non-essential amino acids, 1% L-glutamine, penicillin (100 IU/ml), andstreptomycin (100 mg/ml) (all from Euroclone). The culture was maintained at 37 �C(BB 16 gas incubator, Heraeus Instruments GmbH) in an atmosphere of 5% CO2 and95% relative humidity. The growth medium was changed every other day until thetime of use. Caco-2 cells from passage numbers 31e40 and RAW 264.7 macrophagesfrom passages 17e20 were used in the experiments. Prior to each test, the RAW264.7 macrophages were harvested using phosphate buffered saline (PBS)-ethyl-enediamine tetraacetic acid (EDTA) and the Caco-2 cells were harvested using 0.25%(v/v) trypsineEDTAePBS. For the toxicity assessment, 2 � 104 cells in DMEM wereseeded in 96-well plates and allowed to attach overnight.
2.4. Fluorescent cell viability assay
This testwas performed as described in detail elsewhere [20,21]. The fluorescenttest measures the relative number of living cells in the cell population based on onemarker for cell viability (CellTiter-Fluor�Cell, Promega). The generated fluorescentsignal is proportional to the number of living cells. After the cells were attached tothe 96-well plates, the wells were washed with 100 ml of 10 mM HBSS (Hank’s buffersalt solution) (pH 7.4). After washing, suspensions of 250, 100, 50 and 15 mg/ml ofTOPSi particles were added to the 96-well plates. The cells were then incubated at37 �C for 24 h at a stirrer speed of 25 rpm. After incubation, 100 ml of the reagentassay was added to each well. The cells were further incubated for at least 45 min. AHBSS buffer solutionwas used as a negative control. The fluorescence was measuredat an excitation and emission wavelength of 400 and 505 nm, respectively, withVarioskan Flash (Thermo Fisher Scientific Inc). The cell viability as a percentage ofthe negative control was calculated from the fluorescent values.
2.5. Luminescent cell viability assay
The assay quantifies the number of viable cells in culture based on the amount ofATP produced by metabolically active cells (CellTiter-Glo Luminescent Cell ViabilityAssay, Promega) [20,21]. The test is based on the luciferase reaction in the presenceof Mg2þ, ATP, and oxygen. The amount of ATP is directly proportional to the numberof living cells present in the culture. An identical procedure to the fluorescent assaywas also used here, with the difference that after the 96-well plates (containing thecells and 250e15 mg/ml of the TOPSi particles) were incubated at 37 �C for 24 h, theplates were equilibrated at room temperature for about 30 min. Thereafter, 100 ml ofthe reagent assay was added to each well. Negative (HBSS buffer solution) controlwells were also used and treated similarly as described earlier. The luminescencewas measured using Varioskan Flash (Thermo Fisher Scientific Inc). The cell viabilityas a percentage of the negative control was calculated from the luminescent values.
2.6. TNF-a assay
In order to assess the TNF-a production by the macrophages, an enzyme-linkedimmunosorbent assay (ELISA) was performed using a commercially available kit(Mouse TNF-a ELISA, Bender MedSystems GmbH). The experiments were performedas described elsewhere [16]. First, a serial dilution of murine TNF-a was preparedand pipetted into the 96-well plates pre-coated with immobilized antibodies. Pre-collected supernatants from all the treated and control cells were then added to thewells. After 2 h the wells were washed and biotin-conjugate specific for mouseTNF-a was added to the wells. The wells were then completely washed to removeany unbound reactants after another 2 h. The amount of TNF-a present in each wellwas determined by measuring the absorbance for each sample with Varioskan Flash(Thermo Fisher Scientific Inc) at 450 nm. The amount of TNF-a produced by the RAW264.7 macrophage cells was then calculated from the standard curve of knownamounts of murine TNF-a.

L.M. Bimbo et al. / Biomaterials 32 (2011) 2625e2633 2627
2.7. Reactive oxygen species (ROS) determination
The experiments were performed as described elsewhere [16]. In 96-well plates,2 � 104 cells were seeded and allowed to attach overnight. The medium was thenaspirated and 100 ml of 10 mM 20 ,70-dichlorofluorescein diacetate (DCF-DA) solutionwas added and allowed to incubate 1 h at 37 �C. The solution was subsequentlywashed with HBSS and TOPSi suspension solutions of concentrations of 250, 100, 50and 15 mg/ml were added to thewells. Hydrogen peroxide treated cells (H2O2, 0.09%)were used as a positive control. After treatment for 24 h, the plate wells werewashed and DCF fluorescence was measured with Varioskan Flash (Thermo FisherScientific). Excitation and emissionwavelengths were 498 and 522 nm, respectively.
2.8. Nitric oxide (NO) determination
In 96-well plates, 2� 104 cells were seeded and allowed to attach overnight. Themediumwas then aspirated and 100 ml of TOPSi solutionwith concentrations of 250,100, 50 and 15 mg/ml were added to the wells and incubated for 24 h. The super-natants were collected by aspirating half the volume of the particle solutions in thewells. In new 96-well plates, 20 ml of the collected supernatants or nitrite standardswere added to each well and the volume completed with 80 ml of ultrapure water.Next, 10 ml of 100 mg/ml 2,3-Diaminonaphtalene (DAN) solution prepared in 0.25 M
HCl was added to each well. The plate was then incubated for 10 min in the dark and20 ml of 2.8 M NaOH was added in order to stop the reaction and to enhance fluo-rescence. The fluorescence was measured with Varioskan Flash (Thermo FisherScientific), at an excitation wavelength of 360 and emission wavelength of 430 nm.
2.9. Flat embedding TEM
For the TEM experiments, and for the TOPSi association studies with the RAWmacrophage cells, the next protocol was followed. In 12-well plates (Corning Inc. LifeSciences), 13 mm round coverslips were placed at the bottom of each well. Next,3 � 104 cells were seeded in DMEM and allowed to attach overnight. The mediumwas then aspirated and 100 ml of TOPSi particle suspensions with concentrations of50 and 15 mg/ml were added to each well and incubated for 24 h. After incubation,the particle suspension was carefully removed and the cells were fixed with 2%glutaraldehyde in 0.1 M PBS solution (pH 7.4) for 30 min at room temperature. Thewells were then washed twice with sodium cacodylate buffer (NaCac) for 3 min.Afterwards, the cells were post-fixedwith 1% osmium tetroxide in 0.1 M NaCac buffer(pH 7.4). The cells were then dehydrated and embedded in epoxy resin. Ultrathinsections (60 nm) were cut parallel to the coverslip, post-stained with uranyl acetateand lead citrate, and examined with a TEM FEI Tecnai 10 (Philips) at 120 kV.
In the polarized Caco-2 cell permeation experiments, 7.5�104 cells were seededon 12-Transwell cell culture inserts (polyester membranes, pore size 3 mm, growtharea 1.12 cm2, Corning Inc. Life Sciences) and kept under the same conditions aspreviously described. The cells were used in the permeation experiments 21e28days after the seeding, with the medium changed every other day. The Eponembedding procedure was similar to that described above, except that the ultrathinsections were cut perpendicular to the insert.
2.10. Caco-2 permeability experiments
The permeability across the Caco-2 cell monolayers was performed in the apical-to-basolateral direction for griseofulvin. After seeding the cells in 12-Transwell cellculture inserts, the cells were allowed to attach overnight. Themediumwas replacedevery other day and the cells were used in the permeation experiments 21e28 daysafter the seeding. The transport experiments were performed in HBSS at pH 5.5 and7.4 (apical compartment) at 37 �C using an orbital shaker (50 rpm). The solution inthe basolateral compartment was kept at pH 7.4 in all the experiments. 1 mg ofgriseofulvin loaded (17.3% w/w) TOPSi particles with an average size of 170 nm andthe corresponding amount (173 mg) of pure bulk griseofulvin (Sigma) were pipettedin the apical side of the inserts in 500 ml of HBSS pH 5.5 and 7.4. 100 ml samples weretaken from the basolateral side of the inserts at different time points, and 100 ml offresh HBSS buffer was added to replace withdrawn volume. Sample concentrationswere quantified by HPLC (Agilent 1100 series) and the amount of griseofulvinreleased from the particles was calculated related from the original drug amountpresent in the particles.
Fig. 1. TEM images of 164 nm TOPSi nanoparticles. Scale bars are 1 mm, and 100 nm forthe inset.
2.11. Degradation of TOPSi particles in fasted state simulated intestinal fluid (FaSSIF)
The Si degradation of the TOPSi particles was assessed by incubating 1 mg ofTOPSi particles with 3 ml of blank (without the enzymes) FaSSIF [22], under agita-tion at 37 �C in an orbital incubator (Stuart SI50) for several days. At different timepoints, 300 ml samples were collected and replaced by fresh medium. The sampleswere diluted 10 times and the results were analyzed by ICP Spectrometry (ICAP 6000Series with Gas Cryo Systems and Autosampler ASX-260, Thermo Scientific) andnormalized for the total amount of Si.
2.12. Statistical analyses
Results from the several tests are expressed as mean � SD from of at least threeindependent experiments. The level of significance was set at a probability ofp < 0.05 for *, p < 0.01 for **, and p < 0.001 for ***. A one-way analysis of variance(ANOVA), followed by a Dunnett’s multiple comparison test was used to analyze thedata. The analysis was carried out using GraphPad Prism v. 5.01 (GraphPadSoftware).
3. Results and discussion
3.1. Characterization of the TOPSi particles
The PSi films were produced by electrochemical etching the Siwafers with HF and ethanol. TOPSi particles with several sizes,ranging from micro- to nanosize were prepared. The two micro-sized TOPSi fractions were: 1e10 and 10e25 mm, with a specificsurface area of 202 m2/g. The average pore diameter was 9.2 nmand the pore volume was 0.638 cm3/g. In the case of TOPSi nano-particles, the fractions prepared had an average size of 97, 125 and164 nm. The specific surface area was 177 m2/g. The average porediameter was 15.7 nm and the pore volume was 0.636 cm3/g. Thedecreased surface area and increased average pore diameter area result of the different etching current profile used in theproduction of the nanoparticles. The current modulates the poremorphology enlarging the pore openings in the nanoparticles,leading to the increase in the average pore diameter and decreasein surface area. Due to the same reason, the pore volume of thenanoparticles remained almost invariable and rather similar to thepore volume of the microparticles.
The average zeta (z)-potential measured for all the nanoparticleswas �33.67 � 4.40 mV.
Fig.1 shows the TEMpictures of the 164 nmnanoparticles (Fig.1)prepared by the abovementioned method. These nanoparticlesshow a very spherical-round shape and stable dispersion, with noevidence of aggregates. In theDLSmeasurements, the polydispersityindex of all nanoparticles was below 0.15 (range 0.05e0.15),
L.M. Bimbo et al. / Biomaterials 32 (2011) 2625e26332628
indicating highly monodisperse systems. The measured z-potentialalso indicates a rather negatively charged surface, which preventsaggregation and stabilizes the particles in suspension.
3.2. ROS production, inflammatory reaction and cytotoxicity ofRAW 264.7 and Caco-2 cells
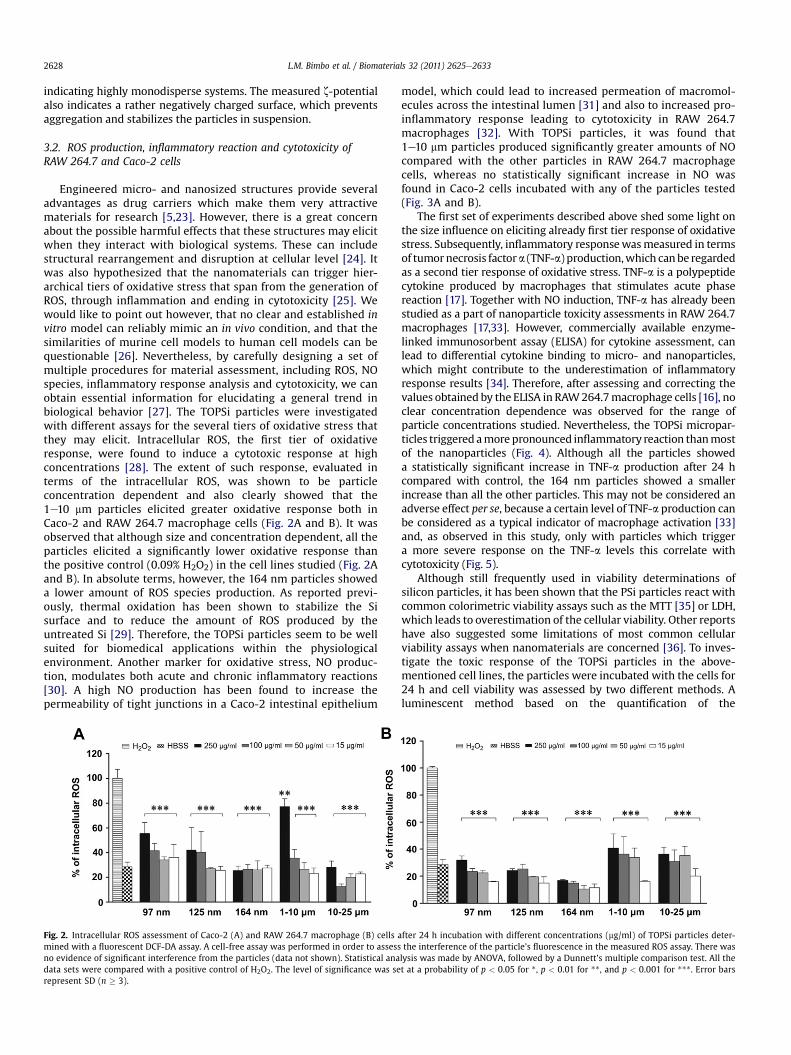
Engineered micro- and nanosized structures provide severaladvantages as drug carriers which make them very attractivematerials for research [5,23]. However, there is a great concernabout the possible harmful effects that these structures may elicitwhen they interact with biological systems. These can includestructural rearrangement and disruption at cellular level [24]. Itwas also hypothesized that the nanomaterials can trigger hier-archical tiers of oxidative stress that span from the generation ofROS, through inflammation and ending in cytotoxicity [25]. Wewould like to point out however, that no clear and established invitro model can reliably mimic an in vivo condition, and that thesimilarities of murine cell models to human cell models can bequestionable [26]. Nevertheless, by carefully designing a set ofmultiple procedures for material assessment, including ROS, NOspecies, inflammatory response analysis and cytotoxicity, we canobtain essential information for elucidating a general trend inbiological behavior [27]. The TOPSi particles were investigatedwith different assays for the several tiers of oxidative stress thatthey may elicit. Intracellular ROS, the first tier of oxidativeresponse, were found to induce a cytotoxic response at highconcentrations [28]. The extent of such response, evaluated interms of the intracellular ROS, was shown to be particleconcentration dependent and also clearly showed that the1e10 mm particles elicited greater oxidative response both inCaco-2 and RAW 264.7 macrophage cells (Fig. 2A and B). It wasobserved that although size and concentration dependent, all theparticles elicited a significantly lower oxidative response thanthe positive control (0.09% H2O2) in the cell lines studied (Fig. 2Aand B). In absolute terms, however, the 164 nm particles showeda lower amount of ROS species production. As reported previ-ously, thermal oxidation has been shown to stabilize the Sisurface and to reduce the amount of ROS produced by theuntreated Si [29]. Therefore, the TOPSi particles seem to be wellsuited for biomedical applications within the physiologicalenvironment. Another marker for oxidative stress, NO produc-tion, modulates both acute and chronic inflammatory reactions[30]. A high NO production has been found to increase thepermeability of tight junctions in a Caco-2 intestinal epithelium
Fig. 2. Intracellular ROS assessment of Caco-2 (A) and RAW 264.7 macrophage (B) cells amined with a fluorescent DCF-DA assay. A cell-free assay was performed in order to assessno evidence of significant interference from the particles (data not shown). Statistical anadata sets were compared with a positive control of H2O2. The level of significance was serepresent SD (n � 3).
model, which could lead to increased permeation of macromol-ecules across the intestinal lumen [31] and also to increased pro-inflammatory response leading to cytotoxicity in RAW 264.7macrophages [32]. With TOPSi particles, it was found that1e10 mm particles produced significantly greater amounts of NOcompared with the other particles in RAW 264.7 macrophagecells, whereas no statistically significant increase in NO wasfound in Caco-2 cells incubated with any of the particles tested(Fig. 3A and B).
The first set of experiments described above shed some light onthe size influence on eliciting already first tier response of oxidativestress. Subsequently, inflammatory responsewasmeasured in termsof tumornecrosis factora (TNF-a) production,which canbe regardedas a second tier response of oxidative stress. TNF-a is a polypeptidecytokine produced by macrophages that stimulates acute phasereaction [17]. Together with NO induction, TNF-a has already beenstudied as a part of nanoparticle toxicity assessments in RAW 264.7macrophages [17,33]. However, commercially available enzyme-linked immunosorbent assay (ELISA) for cytokine assessment, canlead to differential cytokine binding to micro- and nanoparticles,which might contribute to the underestimation of inflammatoryresponse results [34]. Therefore, after assessing and correcting thevalues obtained by the ELISA in RAW264.7macrophage cells [16], noclear concentration dependence was observed for the range ofparticle concentrations studied. Nevertheless, the TOPSi micropar-ticles triggered amore pronounced inflammatory reaction thanmostof the nanoparticles (Fig. 4). Although all the particles showeda statistically significant increase in TNF-a production after 24 hcompared with control, the 164 nm particles showed a smallerincrease than all the other particles. This may not be considered anadverse effect per se, because a certain level of TNF-a production canbe considered as a typical indicator of macrophage activation [33]and, as observed in this study, only with particles which triggera more severe response on the TNF-a levels this correlate withcytotoxicity (Fig. 5).
Although still frequently used in viability determinations ofsilicon particles, it has been shown that the PSi particles react withcommon colorimetric viability assays such as the MTT [35] or LDH,which leads to overestimation of the cellular viability. Other reportshave also suggested some limitations of most common cellularviability assays when nanomaterials are concerned [36]. To inves-tigate the toxic response of the TOPSi particles in the above-mentioned cell lines, the particles were incubated with the cells for24 h and cell viability was assessed by two different methods. Aluminescent method based on the quantification of the
fter 24 h incubation with different concentrations (mg/ml) of TOPSi particles deter-the interference of the particle’s fluorescence in the measured ROS assay. There waslysis was made by ANOVA, followed by a Dunnett’s multiple comparison test. All thet at a probability of p < 0.05 for *, p < 0.01 for **, and p < 0.001 for ***. Error bars
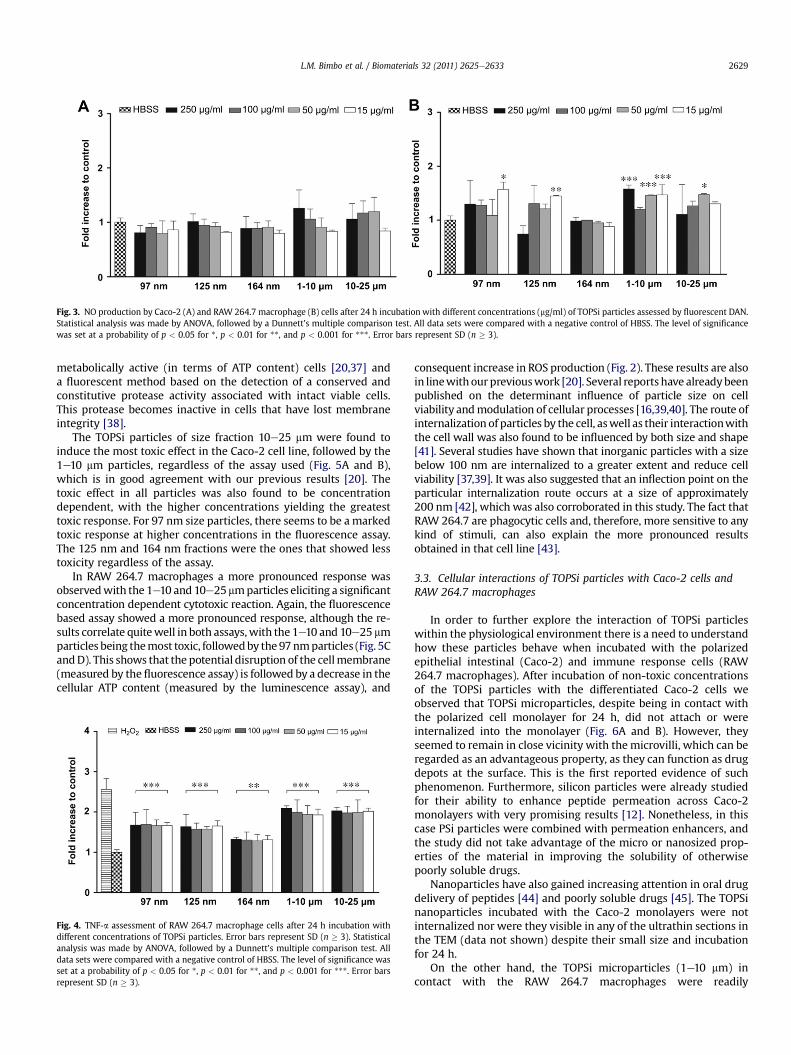
Fig. 3. NO production by Caco-2 (A) and RAW 264.7 macrophage (B) cells after 24 h incubation with different concentrations (mg/ml) of TOPSi particles assessed by fluorescent DAN.Statistical analysis was made by ANOVA, followed by a Dunnett’s multiple comparison test. All data sets were compared with a negative control of HBSS. The level of significancewas set at a probability of p < 0.05 for *, p < 0.01 for **, and p < 0.001 for ***. Error bars represent SD (n � 3).
L.M. Bimbo et al. / Biomaterials 32 (2011) 2625e2633 2629
metabolically active (in terms of ATP content) cells [20,37] anda fluorescent method based on the detection of a conserved andconstitutive protease activity associated with intact viable cells.This protease becomes inactive in cells that have lost membraneintegrity [38].
The TOPSi particles of size fraction 10e25 mm were found toinduce the most toxic effect in the Caco-2 cell line, followed by the1e10 mm particles, regardless of the assay used (Fig. 5A and B),which is in good agreement with our previous results [20]. Thetoxic effect in all particles was also found to be concentrationdependent, with the higher concentrations yielding the greatesttoxic response. For 97 nm size particles, there seems to be amarkedtoxic response at higher concentrations in the fluorescence assay.The 125 nm and 164 nm fractions were the ones that showed lesstoxicity regardless of the assay.
In RAW 264.7 macrophages a more pronounced response wasobservedwith the 1e10 and 10e25mmparticles eliciting a significantconcentration dependent cytotoxic reaction. Again, the fluorescencebased assay showed a more pronounced response, although the re-sults correlate quitewell in both assays,with the 1e10 and 10e25 mmparticles being themost toxic, followedby the97nmparticles (Fig. 5CandD). This shows that the potential disruption of the cellmembrane(measured by the fluorescence assay) is followed by a decrease in thecellular ATP content (measured by the luminescence assay), and
Fig. 4. TNF-a assessment of RAW 264.7 macrophage cells after 24 h incubation withdifferent concentrations of TOPSi particles. Error bars represent SD (n � 3). Statisticalanalysis was made by ANOVA, followed by a Dunnett’s multiple comparison test. Alldata sets were compared with a negative control of HBSS. The level of significance wasset at a probability of p < 0.05 for *, p < 0.01 for **, and p < 0.001 for ***. Error barsrepresent SD (n � 3).
consequent increase in ROS production (Fig. 2). These results are alsoin linewithour previouswork [20]. Several reports have alreadybeenpublished on the determinant influence of particle size on cellviability andmodulation of cellular processes [16,39,40]. The route ofinternalization of particles by the cell, aswell as their interactionwiththe cell wall was also found to be influenced by both size and shape[41]. Several studies have shown that inorganic particles with a sizebelow 100 nm are internalized to a greater extent and reduce cellviability [37,39]. It was also suggested that an inflection point on theparticular internalization route occurs at a size of approximately200 nm [42], which was also corroborated in this study. The fact thatRAW 264.7 are phagocytic cells and, therefore, more sensitive to anykind of stimuli, can also explain the more pronounced resultsobtained in that cell line [43].
3.3. Cellular interactions of TOPSi particles with Caco-2 cells andRAW 264.7 macrophages
In order to further explore the interaction of TOPSi particleswithin the physiological environment there is a need to understandhow these particles behave when incubated with the polarizedepithelial intestinal (Caco-2) and immune response cells (RAW264.7 macrophages). After incubation of non-toxic concentrationsof the TOPSi particles with the differentiated Caco-2 cells weobserved that TOPSi microparticles, despite being in contact withthe polarized cell monolayer for 24 h, did not attach or wereinternalized into the monolayer (Fig. 6A and B). However, theyseemed to remain in close vicinity with themicrovilli, which can beregarded as an advantageous property, as they can function as drugdepots at the surface. This is the first reported evidence of suchphenomenon. Furthermore, silicon particles were already studiedfor their ability to enhance peptide permeation across Caco-2monolayers with very promising results [12]. Nonetheless, in thiscase PSi particles were combined with permeation enhancers, andthe study did not take advantage of the micro or nanosized prop-erties of the material in improving the solubility of otherwisepoorly soluble drugs.
Nanoparticles have also gained increasing attention in oral drugdelivery of peptides [44] and poorly soluble drugs [45]. The TOPSinanoparticles incubated with the Caco-2 monolayers were notinternalized nor were they visible in any of the ultrathin sections inthe TEM (data not shown) despite their small size and incubationfor 24 h.
On the other hand, the TOPSi microparticles (1e10 mm) incontact with the RAW 264.7 macrophages were readily
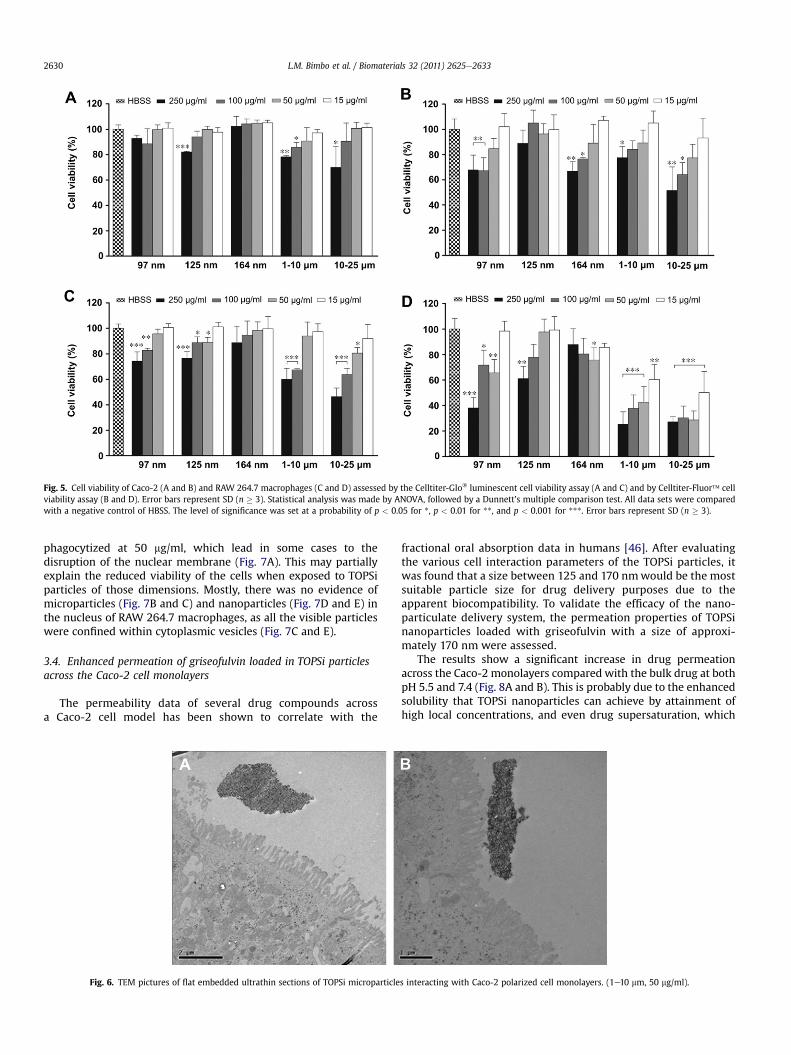
Fig. 5. Cell viability of Caco-2 (A and B) and RAW 264.7 macrophages (C and D) assessed by the Celltiter-Glo� luminescent cell viability assay (A and C) and by Celltiter-Fluor� cellviability assay (B and D). Error bars represent SD (n � 3). Statistical analysis was made by ANOVA, followed by a Dunnett’s multiple comparison test. All data sets were comparedwith a negative control of HBSS. The level of significance was set at a probability of p < 0.05 for *, p < 0.01 for **, and p < 0.001 for ***. Error bars represent SD (n � 3).
L.M. Bimbo et al. / Biomaterials 32 (2011) 2625e26332630
phagocytized at 50 mg/ml, which lead in some cases to thedisruption of the nuclear membrane (Fig. 7A). This may partiallyexplain the reduced viability of the cells when exposed to TOPSiparticles of those dimensions. Mostly, there was no evidence ofmicroparticles (Fig. 7B and C) and nanoparticles (Fig. 7D and E) inthe nucleus of RAW 264.7 macrophages, as all the visible particleswere confined within cytoplasmic vesicles (Fig. 7C and E).
3.4. Enhanced permeation of griseofulvin loaded in TOPSi particlesacross the Caco-2 cell monolayers
The permeability data of several drug compounds acrossa Caco-2 cell model has been shown to correlate with the
Fig. 6. TEM pictures of flat embedded ultrathin sections of TOPSi microparticle
fractional oral absorption data in humans [46]. After evaluatingthe various cell interaction parameters of the TOPSi particles, itwas found that a size between 125 and 170 nmwould be the mostsuitable particle size for drug delivery purposes due to theapparent biocompatibility. To validate the efficacy of the nano-particulate delivery system, the permeation properties of TOPSinanoparticles loaded with griseofulvin with a size of approxi-mately 170 nm were assessed.
The results show a significant increase in drug permeationacross the Caco-2 monolayers compared with the bulk drug at bothpH 5.5 and 7.4 (Fig. 8A and B). This is probably due to the enhancedsolubility that TOPSi nanoparticles can achieve by attainment ofhigh local concentrations, and even drug supersaturation, which
s interacting with Caco-2 polarized cell monolayers. (1e10 mm, 50 mg/ml).
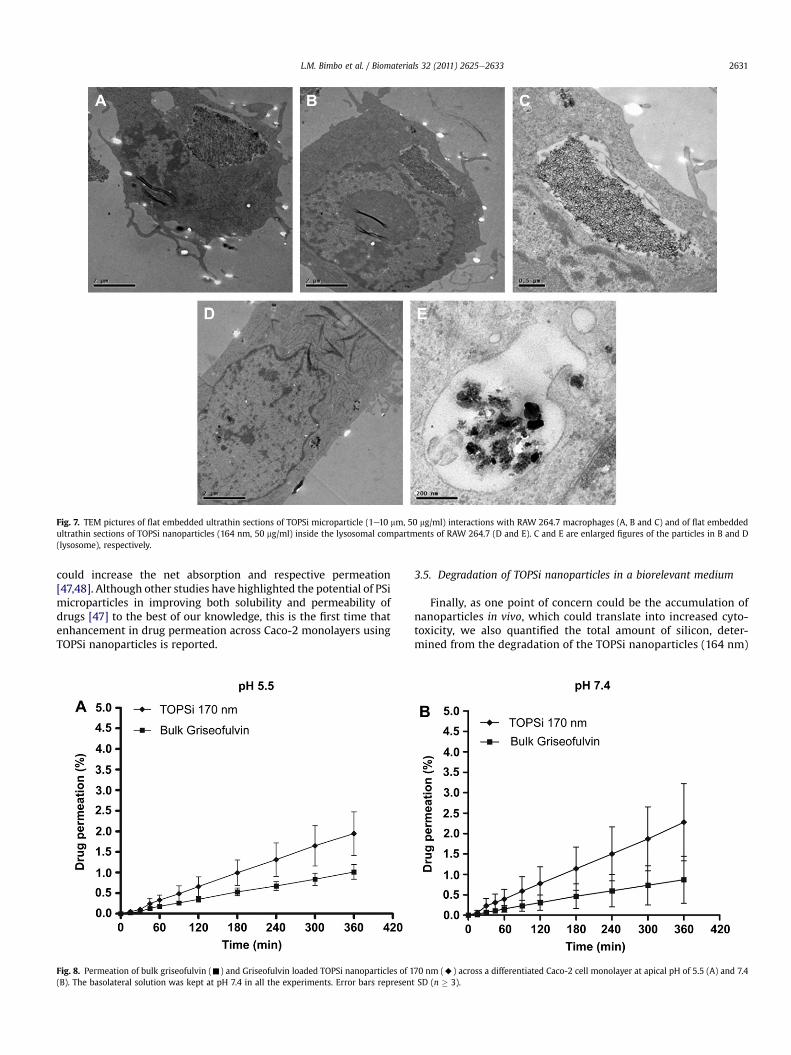
Fig. 7. TEM pictures of flat embedded ultrathin sections of TOPSi microparticle (1e10 mm, 50 mg/ml) interactions with RAW 264.7 macrophages (A, B and C) and of flat embeddedultrathin sections of TOPSi nanoparticles (164 nm, 50 mg/ml) inside the lysosomal compartments of RAW 264.7 (D and E). C and E are enlarged figures of the particles in B and D(lysosome), respectively.
L.M. Bimbo et al. / Biomaterials 32 (2011) 2625e2633 2631
could increase the net absorption and respective permeation[47,48]. Although other studies have highlighted the potential of PSimicroparticles in improving both solubility and permeability ofdrugs [47] to the best of our knowledge, this is the first time thatenhancement in drug permeation across Caco-2 monolayers usingTOPSi nanoparticles is reported.
Fig. 8. Permeation of bulk griseofulvin (-) and Griseofulvin loaded TOPSi nanoparticles of 1(B). The basolateral solution was kept at pH 7.4 in all the experiments. Error bars represen
3.5. Degradation of TOPSi nanoparticles in a biorelevant medium
Finally, as one point of concern could be the accumulation ofnanoparticles in vivo, which could translate into increased cyto-toxicity, we also quantified the total amount of silicon, deter-mined from the degradation of the TOPSi nanoparticles (164 nm)
70 nm (A) across a differentiated Caco-2 cell monolayer at apical pH of 5.5 (A) and 7.4t SD (n � 3).
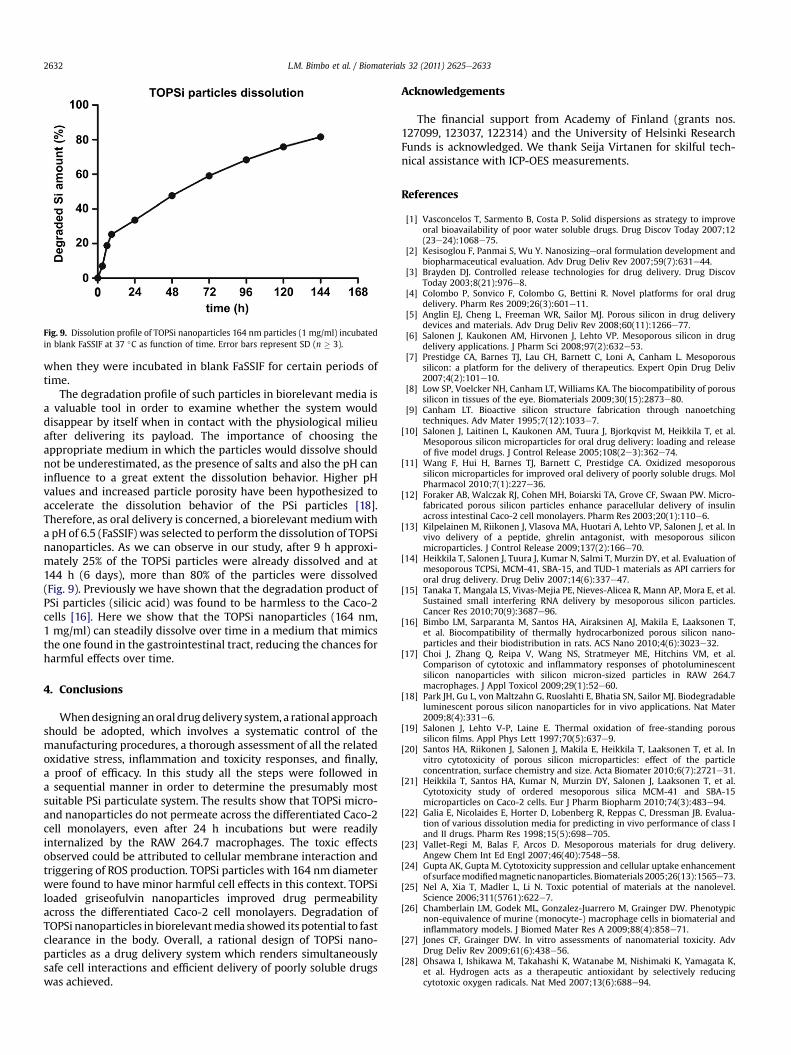
Fig. 9. Dissolution profile of TOPSi nanoparticles 164 nm particles (1 mg/ml) incubatedin blank FaSSIF at 37 �C as function of time. Error bars represent SD (n � 3).
L.M. Bimbo et al. / Biomaterials 32 (2011) 2625e26332632
when they were incubated in blank FaSSIF for certain periods oftime.
The degradation profile of such particles in biorelevant media isa valuable tool in order to examine whether the system woulddisappear by itself when in contact with the physiological milieuafter delivering its payload. The importance of choosing theappropriate medium in which the particles would dissolve shouldnot be underestimated, as the presence of salts and also the pH caninfluence to a great extent the dissolution behavior. Higher pHvalues and increased particle porosity have been hypothesized toaccelerate the dissolution behavior of the PSi particles [18].Therefore, as oral delivery is concerned, a biorelevant mediumwitha pH of 6.5 (FaSSIF) was selected to perform the dissolution of TOPSinanoparticles. As we can observe in our study, after 9 h approxi-mately 25% of the TOPSi particles were already dissolved and at144 h (6 days), more than 80% of the particles were dissolved(Fig. 9). Previously we have shown that the degradation product ofPSi particles (silicic acid) was found to be harmless to the Caco-2cells [16]. Here we show that the TOPSi nanoparticles (164 nm,1 mg/ml) can steadily dissolve over time in a medium that mimicsthe one found in the gastrointestinal tract, reducing the chances forharmful effects over time.
4. Conclusions
Whendesigning anoral drugdelivery system, a rational approachshould be adopted, which involves a systematic control of themanufacturing procedures, a thorough assessment of all the relatedoxidative stress, inflammation and toxicity responses, and finally,a proof of efficacy. In this study all the steps were followed ina sequential manner in order to determine the presumably mostsuitable PSi particulate system. The results show that TOPSi micro-and nanoparticles do not permeate across the differentiated Caco-2cell monolayers, even after 24 h incubations but were readilyinternalized by the RAW 264.7 macrophages. The toxic effectsobserved could be attributed to cellular membrane interaction andtriggering of ROS production. TOPSi particles with 164 nm diameterwere found to have minor harmful cell effects in this context. TOPSiloaded griseofulvin nanoparticles improved drug permeabilityacross the differentiated Caco-2 cell monolayers. Degradation ofTOPSi nanoparticles in biorelevantmedia showed its potential to fastclearance in the body. Overall, a rational design of TOPSi nano-particles as a drug delivery system which renders simultaneouslysafe cell interactions and efficient delivery of poorly soluble drugswas achieved.
Acknowledgements
The financial support from Academy of Finland (grants nos.127099, 123037, 122314) and the University of Helsinki ResearchFunds is acknowledged. We thank Seija Virtanen for skilful tech-nical assistance with ICP-OES measurements.
References
[1] Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improveoral bioavailability of poor water soluble drugs. Drug Discov Today 2007;12(23e24):1068e75.
[2] Kesisoglou F, Panmai S, Wu Y. Nanosizingeoral formulation development andbiopharmaceutical evaluation. Adv Drug Deliv Rev 2007;59(7):631e44.
[3] Brayden DJ. Controlled release technologies for drug delivery. Drug DiscovToday 2003;8(21):976e8.
[4] Colombo P, Sonvico F, Colombo G, Bettini R. Novel platforms for oral drugdelivery. Pharm Res 2009;26(3):601e11.
[5] Anglin EJ, Cheng L, Freeman WR, Sailor MJ. Porous silicon in drug deliverydevices and materials. Adv Drug Deliv Rev 2008;60(11):1266e77.
[6] Salonen J, Kaukonen AM, Hirvonen J, Lehto VP. Mesoporous silicon in drugdelivery applications. J Pharm Sci 2008;97(2):632e53.
[7] Prestidge CA, Barnes TJ, Lau CH, Barnett C, Loni A, Canham L. Mesoporoussilicon: a platform for the delivery of therapeutics. Expert Opin Drug Deliv2007;4(2):101e10.
[8] Low SP, Voelcker NH, Canham LT, Williams KA. The biocompatibility of poroussilicon in tissues of the eye. Biomaterials 2009;30(15):2873e80.
[9] Canham LT. Bioactive silicon structure fabrication through nanoetchingtechniques. Adv Mater 1995;7(12):1033e7.
[10] Salonen J, Laitinen L, Kaukonen AM, Tuura J, Bjorkqvist M, Heikkila T, et al.Mesoporous silicon microparticles for oral drug delivery: loading and releaseof five model drugs. J Control Release 2005;108(2e3):362e74.
[11] Wang F, Hui H, Barnes TJ, Barnett C, Prestidge CA. Oxidized mesoporoussilicon microparticles for improved oral delivery of poorly soluble drugs. MolPharmacol 2010;7(1):227e36.
[12] Foraker AB, Walczak RJ, Cohen MH, Boiarski TA, Grove CF, Swaan PW. Micro-fabricated porous silicon particles enhance paracellular delivery of insulinacross intestinal Caco-2 cell monolayers. Pharm Res 2003;20(1):110e6.
[13] Kilpelainen M, Riikonen J, Vlasova MA, Huotari A, Lehto VP, Salonen J, et al. Invivo delivery of a peptide, ghrelin antagonist, with mesoporous siliconmicroparticles. J Control Release 2009;137(2):166e70.
[14] Heikkila T, Salonen J, Tuura J, Kumar N, Salmi T, Murzin DY, et al. Evaluation ofmesoporous TCPSi, MCM-41, SBA-15, and TUD-1 materials as API carriers fororal drug delivery. Drug Deliv 2007;14(6):337e47.
[15] Tanaka T, Mangala LS, Vivas-Mejia PE, Nieves-Alicea R, Mann AP, Mora E, et al.Sustained small interfering RNA delivery by mesoporous silicon particles.Cancer Res 2010;70(9):3687e96.
[16] Bimbo LM, Sarparanta M, Santos HA, Airaksinen AJ, Makila E, Laaksonen T,et al. Biocompatibility of thermally hydrocarbonized porous silicon nano-particles and their biodistribution in rats. ACS Nano 2010;4(6):3023e32.
[17] Choi J, Zhang Q, Reipa V, Wang NS, Stratmeyer ME, Hitchins VM, et al.Comparison of cytotoxic and inflammatory responses of photoluminescentsilicon nanoparticles with silicon micron-sized particles in RAW 264.7macrophages. J Appl Toxicol 2009;29(1):52e60.
[18] Park JH, Gu L, von Maltzahn G, Ruoslahti E, Bhatia SN, Sailor MJ. Biodegradableluminescent porous silicon nanoparticles for in vivo applications. Nat Mater2009;8(4):331e6.
[19] Salonen J, Lehto V-P, Laine E. Thermal oxidation of free-standing poroussilicon films. Appl Phys Lett 1997;70(5):637e9.
[20] Santos HA, Riikonen J, Salonen J, Makila E, Heikkila T, Laaksonen T, et al. Invitro cytotoxicity of porous silicon microparticles: effect of the particleconcentration, surface chemistry and size. Acta Biomater 2010;6(7):2721e31.
[21] Heikkila T, Santos HA, Kumar N, Murzin DY, Salonen J, Laaksonen T, et al.Cytotoxicity study of ordered mesoporous silica MCM-41 and SBA-15microparticles on Caco-2 cells. Eur J Pharm Biopharm 2010;74(3):483e94.
[22] Galia E, Nicolaides E, Horter D, Lobenberg R, Reppas C, Dressman JB. Evalua-tion of various dissolution media for predicting in vivo performance of class Iand II drugs. Pharm Res 1998;15(5):698e705.
[23] Vallet-Regi M, Balas F, Arcos D. Mesoporous materials for drug delivery.Angew Chem Int Ed Engl 2007;46(40):7548e58.
[24] Gupta AK, Gupta M. Cytotoxicity suppression and cellular uptake enhancementof surfacemodifiedmagneticnanoparticles. Biomaterials 2005;26(13):1565e73.
[25] Nel A, Xia T, Madler L, Li N. Toxic potential of materials at the nanolevel.Science 2006;311(5761):622e7.
[26] Chamberlain LM, Godek ML, Gonzalez-Juarrero M, Grainger DW. Phenotypicnon-equivalence of murine (monocyte-) macrophage cells in biomaterial andinflammatory models. J Biomed Mater Res A 2009;88(4):858e71.
[27] Jones CF, Grainger DW. In vitro assessments of nanomaterial toxicity. AdvDrug Deliv Rev 2009;61(6):438e56.
[28] Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K,et al. Hydrogen acts as a therapeutic antioxidant by selectively reducingcytotoxic oxygen radicals. Nat Med 2007;13(6):688e94.

L.M. Bimbo et al. / Biomaterials 32 (2011) 2625e2633 2633
[29] Low SP, Williams KA, Canham LT, Voelcker NH. Generation of reactive oxygenspecies from porous silicon microparticles in cell culture medium. J BiomedMater Res A 2010;93(3):1124e31.
[30] Vignoli AL, Srivastava RC, Stammati A, Turco L, Tanori M, Zucco F. Nitric oxideproduction in Caco-2 cells exposed to different inducers, inhibitors andnatural toxins. Toxicol In Vitro 2001;15(4e5):289e95.
[31] Salzman AL, Menconi MJ, Unno N, Ezzell RM, Casey DM, Gonzalez PK, et al.Nitric oxide dilates tight junctions and depletes ATP in cultured Caco-2BBeintestinal epithelial monolayers. Am J Phys 1995;268(2 Pt 1):G361e73.
[32] Park EJ, Park K. Oxidative stress and pro-inflammatory responses induced bysilica nanoparticles in vivo and in vitro. Toxicol Lett 2009;184(1):18e25.
[33] Scheel J, Weimans S, Thiemann A, Heisler E, Hermann M. Exposure of themurine RAW 264.7 macrophage cell line to hydroxyapatite dispersions ofvarious composition and morphology: assessment of cytotoxicity, activationand stress response. Toxicol In Vitro 2009;23(3):531e8.
[34] Kocbach A, Totlandsdal AI, Lag M, Refsnes M, Schwarze PE. Differentialbinding of cytokines to environmentally relevant particles: a possible sourcefor misinterpretation of in vitro results? Toxicol Lett 2008;176(2):131e7.
[35] Laaksonen T, Santos H, Vihola H, Salonen J, Riikonen J, Heikkila T, et al. Failureof MTT as a toxicity testing agent for mesoporous silicon microparticles. ChemRes Toxicol 2007;20(12):1913e8.
[36] Monteiro-Riviere NA, Inman AO, Zhang LW. Limitations and relative utility ofscreening assays to assess engineered nanoparticle toxicity in a human cellline. Toxicol Appl Pharmacol 2009;234(2):222e35.
[37] AshaRani PV, Low Kah Mun G, Hande MP, Valiyaveettil S. Cytotoxicity andgenotoxicity of silver nanoparticles in human cells. ACS Nano 2009;3(2):279e90.
[38] Niles AL, Moravec RA, Eric Hesselberth P, Scurria MA, Daily WJ, Riss TL.A homogeneous assay to measure live and dead cells in the same sampleby detecting different protease markers. Anal Biochem 2007;366(2):197e206.
[39] Jiang W, Kim BY, Rutka JT, Chan WC. Nanoparticle-mediated cellular responseis size-dependent. Nat Nanotechnol 2008;3(3):145e50.
[40] Chithrani BD, Ghazani AA, Chan WC. Determining the size and shapedependence of gold nanoparticle uptake into mammalian cells. Nano Lett2006;6(4):662e8.
[41] Gratton SE, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, et al. Theeffect of particle design on cellular internalization pathways. Proc Natl AcadSci U S A 2008;105(33):11613e8.
[42] Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization ofparticles via the pathways of clathrin- and caveolae-mediated endocytosis.Biochem J 2004;377(Pt 1):159e69.
[43] Chapekar MS, Zaremba TG, Kuester RK, Hitchins VM. Synergistic induction oftumor necrosis factor alpha by bacterial lipopolysaccharide and lipoteichoicacid in combination with polytetrafluoroethylene particles in a murinemacrophage cell line RAW 264.7. J Biomed Mater Res 1996;31(2):251e6.
[44] Yin L, Ding J, He C, Cui L, Tang C, Yin C. Drug permeability and mucoadhesionproperties of thiolated trimethyl chitosan nanoparticles in oral insulindelivery. Biomaterials 2009;30(29):5691e700.
[45] Zhang Y, Zhi Z, Jiang T, Zhang J, Wang Z, Wang S. Spherical mesoporous silicananoparticles for loading and release of the poorly water-soluble drug tel-misartan. J Control Release 2010;145(3):257e63.
[46] Artursson P, Karlsson J. Correlation between oral drug absorption in humansand apparent drug permeability coefficients in human intestinal epithelial(Caco-2) cells. Biochem Biophys Res Commun 1991;175(3):880e5.
[47] Kaukonen AM, Laitinen L, Salonen J, Tuura J, Heikkila T, Limnell T, et al.Enhanced in vitro permeation of furosemide loaded into thermally carbonizedmesoporous silicon (TCPSi) microparticles. Eur J Pharm Biopharm 2007;66(3):348e56.
[48] Brouwers J, Brewster ME, Augustijns P. Supersaturating drug deliverysystems: the answer to solubility-limited oral bioavailability? J Pharm Sci2009;98(8):2549e72.