dna hypermethylation as a chemotherapy target
TRANSCRIPT

Cellular Signalling 23 (2011) 1082–1093
Contents lists available at ScienceDirect
Cellular Signalling
j ourna l homepage: www.e lsev ie r.com/ locate /ce l l s ig
Review
DNA hypermethylation as a chemotherapy target
Juan Ren a,⁎,1, Brahma N. Singh b,1, Qiang Huang b,c, Zongfang Li c, Ya Gao c, Prachi Mishra d, Yi L. Hwa e,Jinping Li b,f, Sean C. Dowdy f, Shi-Wen Jiang b,f,⁎⁎a Cancer Center, First Hospital of Xi'an Jiaotong University, Xi'an, Shaanxi Province, 710061, Chinab Department of Biological Science, Mercer University School of Medicine, 4700 Waters Avenue, Savannah, GA 31405, USAc Second Hospital of Xi'an Jiaotong University, Xi'an 710061, Shaanxi Province, 710004, Chinad School of Biotechnology, Devi Ahilya University, Khandwa Road, Indore, M.P., Indiae Department of Internal Medicine, Mayo College of Medicine, 2nd Street, Rochester, MN 55902, USAf Department of Obstetrics and Gynecology, Mayo College of Medicine, 2nd Street, Rochester, MN 55902, USA
⁎ Correspondence to: J. Ren, First Hospital of Xi'an Ji⁎⁎ Correspondence to: S.W. Jiang, Department of Biol
E-mail address: [email protected] (S.-W. Jiang)1 These authors contributed equally.
0898-6568/$ – see front matter © 2011 Elsevier Inc. Aldoi:10.1016/j.cellsig.2011.02.003
a b s t r a c t
a r t i c l e i n f oArticle history:Received 1 February 2011Accepted 10 February 2011Available online 21 February 2011
Keywords:DNA methylationDNA methyltranferasesHypermethylationCancerDNMT inhibitors
Epigenetics refers to partially reversible, somatically inheritable, but DNA sequence-independent traits thatmodulate gene expression, chromatin structure, and cell functions such as cell cycle and apoptosis. DNAmethylation is an example of a crucial epigenetic event; aberrant DNA methylation patterns are frequentlyfound in human malignancies. DNA hypermethylation and the associated expression silencing of tumorsuppressor genes represent a hallmark of neoplastic cells. The cancer methylome is highly disrupted, makingDNA methylation an excellent target for anti-cancer therapies. Several small synthetic and natural molecules,are able to reverse the DNA hypermethylation through inhibition of DNAmethyltransferase (DNMT). DNMT isthe enzyme catalyzing the transfer of methyl groups to cytosines in genomic DNA. These reagents are studiedintensively in cell cultures, animal models, and clinical trials for potential anti-cancer activities. It was foundthat accompanying DNA demethylation is a dramatic reactivation of the silenced genes and inhibition ofcancer cell proliferation, promotion of cell apoptosis, or sensitization of cells to other chemotherapeuticreagents. During the last few decades, an increasing number of DNMT inhibitors (DNMTi) targeting DNAmethylation have been developed to increase efficacy with reduced toxicity. This review provides an updateon new findings on cancer epigenetic mechanisms, the development of new DNMTi, and their application inthe clinical setting. Current challenges, potential solutions, and future directions concerning the developmentof DNMTi are also discussed in this review.
aotong University, Xi'an, Shaanxi Province, 710061, Chinogical Science, Mercer University School of Medicine, 47.
l rights reserved.
© 2011 Elsevier Inc. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10832. Transcriptional down regulation due to DNA methylation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1085
2.1. DNA methylation and gene silencing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10852.2. Cancer epigenetics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1085
3. DNMT inhibitors (DNMTi). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10853.1. Nucleoside DNMTi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1085
3.1.1. Azacytidine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10853.1.2. Zebularine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10863.1.3. 5-Fluoro-2′-deoxycytidine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1086
3.2. Non-nucleoside DNMTi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10873.2.1. Procaine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10873.2.2. Procainamide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10873.2.3. Hydralazine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10873.2.4. RG108 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10873.2.5. Miscellaneous agents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1087
a.00 Waters Avenue, Savannah, GA 31405, USA.

1083J. Ren et al. / Cellular Signalling 23 (2011) 1082–1093
4. Clinical studies on DNMTi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10894.1. 5-Azacytidine (Vidaza, Azacytidine) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10894.2. 5-Aza-CdR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1089
5. Combination therapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10905.1. In vitro studies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10905.2. Clinical trial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1090
6. Conclusion and future directions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1091References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1091
1. Introduction
The sequences of genomic DNA carry genetic codes that in thesemiconservational replication of DNA accounts for the biologicalinheritance from generation to generation. An increasing body ofevidence indicates thatbesidesDNAsequences, covalentmodification ofthe nucleotides and histone tails also regulate gene expression and cellfunction [1]. The "epigenetic" traits,with their characteristic inheritancepattern along the somatic lineages, effectively complement the geneticinformation encoded by DNA sequences, and play a critical role in theembryogenesis, tissue/organ development, maintenance of physiolog-ical function, aging and pathogenesis of diseases [1,2]. The cytosinemethylation-mediated pathway is the best studied subject and itsinvolvement in various cell processes including imprinting [3], X-chromosome inactivation [4], suppression of retrotransposon andrepetitive elements, and tissue-specific genes [5]. Moreover, aberrantDNAmethylation profiles have been found to be associatedwith a broadspectrum of disorders, including cancer, atherosclerosis, obesity, insulinresistance, kidney disease, and autoimmunity [6–8].
DNA methylation is a covalent biochemical modification, resultingin the addition of a methyl group to the 5th carbon position in thepyrimidine ring of cytosine located in the context of cytosine–phosphate–guanine (CpG) dinucleotides. Hypermethylation oftenlimits the accessibility of transcription factors to promoters, promotesmethyl-CpG binding domain (MBD) binding, which results inrecruitment of additional silencing-associated proteins, and ulti-mately, gene silencing [9,10]. The human genome is not methylateduniformly, but contains unmethylated segments interspersed by meth-ylated regions [11]. An estimated 60% of mammalian gene promoterscontain CpG islands, many of these being housekeeping genes thatare commonly unmethylated and transcriptionally active. It has beenwell known that CpG islands in certain genes, especially tumor sup-pressor genes, often become aberrantly hypermethylated during thedevelopment of cancer (Fig. 1a & b). This may give the cell a growthadvantage by increased proliferation or increased resistance to apo-ptotic factors. Some hypermethylation events, such as the methylationof glutathione S-transferase Pi in prostate cancer, are readily detectedin precancerous or early stages of cancer development [12]. Theseepigenetic alterations may cause or improve conditions for malignanttransformation. Herman and Baylin [13] found that genes such as O6-methylguanine methyltransferase (MGMT) retinoic acid receptor β(RAR-β), the tumor suppressor p16INK4a, and the DNA repair genehMLH1 were frequently inactivated by hypermethylation in earlylesions of esophageal basal cell hyperplasia.
Epigenetic changes are connected to genetic aberrations by severalpathways. Epigenetic silencing of the DNA repair gene MLH1 in thehyperplastic stage can lead to genetic hypermutations that are found incancers [14]. Global hypomethylation, as detected inmany tumor types,is associated with genomic instability and may contribute to thedevelopment of aneuploidy, LOH and genetic amplifications [15]. It hasbeen shown that the leukemia-promoting PML–RAR fusion protein canrecruit DNMTs to target the genes and thereby induce epigeneticsilencing [10]. It has also been found that in some tumor suppressorgenes, one allele is genetically mutated and another allele is epigenet-ically silenced [16], providing an example of a cooperative mechanismby which epigenetic and genetic lesions promote tumorigenesis.
Patterns of DNA methylation are established by the coordinatedaction of the DNA methyltransferases (DNMTs) and associated fac-tors, such as the polycomb proteins, in the presence of S-adenosyl-methionine (SAM) that serves as a methyl donor [8]. The mammalianDNMT family includes four active members: DNMT1, DNMT3A,DNMT3B, and DNMT3L [17]. Mammalian DNMTs are responsible formethylation pattern acquisition during gametogenesis, embryogene-sis and somatic tissue development [18]. The DNMT1 is the majorenzyme responsible for maintenance of the DNAmethylation pattern.DNMT1 is located at the replication fork and methylates newlybiosynthesized DNA [19]. The mammalian DNMTs are comprised oftwo parts: a C-terminal catalytic part and a large multi-domain N-terminal part of variable size, which has regulatory functions. The C-terminal part is composed of 500 amino acids, which is conservedbetween C5 DNMTs of eukaryotics and prokaryotics that harbor theactive center of the enzyme and contains amino acids motifscharacteristic of the cytosine-C5 methyltransferases. The N-terminalpart contains 621 amino acids that are not essential for DNMT1activity [20], but are required for the discrimination between hemi-methylated and unmethylated DNA. The catalytic domains of allthe DNMTs share a common core structure, known as "AdoMet-dependent methyltransferase" [21]. This domain is involved, both, incofactor binding (motifs I and X) and catalysis (motifs IV, VI and VIII)of substrate to DNMT. The non-conserved region between motifs VIIIand IX, the so called target recognition domain, is involved in DNArecognition and specificity [22]. DNMT2 is the smallest mammalianDNA methyltransferase. It is composed solely of the C-terminaldomain, and does not possess the regulatory N-terminal region. Thestructure of DNMT2 suggests that this enzyme participates in therecognition of DNA damage, DNA recombination and mutation repair[23]. However, recent studies suggest that an interaction betweenDNMT1 and DNMT3B may be vital for the maintenance of patterns ofDNA methylation in human colon-cancer cells, particularly in repeatregions and imprinted genes [24,25]. During early embryogenesis, denovo DNA methylation is mediated by DNMT3A and DNMT3Bassociated with DNMT3L. The DNMT3L lacks conserved motifs of thecatalytic domain but is otherwise closely related to the C-terminaldomain of DNMT3A and DNMT3B [23,26]. DNMT3A and DNMT3Bexhibit a high degree of primary structural homology. Upondifferentiation of embryonic stem cells the activities of both enzymesare reduced and remain low in adult somatic tissues. The expressionof DNMT3A is ubiquitous, while DNMT3B is expressed at very lowlevels in most tissues except the testis, thyroid and bone marrow [27].It has recently been reported that DNMT3A and DNMT3B in the cellare tightly associated with nucleosomes containing methylated DNA[25]. Both the direct interaction of these proteins with the histone tailsand the polymerization of DNMT3A could contribute to the stableassociation of these enzymes with chromatin. The levels of DNMTs,especially those of DNMT3B and DNMT3A, are often increased invarious cancer tissues and cell lines, which may partially account forthe hypermethylation of promoter CpG-rich regions of TSGs in avariety of malignancies [28,29]. For these reasons, DNMTs are con-sidered valuable targets for the design of specific anti-cancerstrategies.
The earliest andmost successful epigenetic drug to date, DAC (alsoknown as 5-Aza-2′-deoxycytidine zebularine), is currently

Fig. 1. Different DNA methylation patterns, histone modifications, and chromatin structures between normal and tumor cells. Mechanism of DNMT inhibitors. Unmethylated,transcriptionally active genes are packaged with acetylated histone proteins, and constitute an ‘open’ chromatin structure which favors transcription. During tumerigenesis, thesame gene may become hypermethylated (b), and the methylated CpG sites are recognized by the methyl-binding proteins (MBDs), which are coupled with repressor (R) andhistone deacetyltransferase (HDAC) proteins to remove the acetyl group from the histones, generating a tightly closed chromatin status to shut down gene expression. During DNAreplication, Aza-deoxycytidine (DAC) is incorporated into DNA and trap DNMT molecule attempting to methylate the CpG sites (c). DNMT molecules are subsequently targeted forproteosomal degradation. DNA-containing DAC are hemi-methylated after the first round of DNA replication and become progressively more demethylated after several rounds ofreplication due to the dilution effect. Using DNMT inhibitors, the silenced epigenetic modifications could be switched to an active status.
1084 J. Ren et al. / Cellular Signalling 23 (2011) 1082–1093
recommended as the first-line treatment of high-risk myelodysplasticsyndromes. During the last few decades, an increasing number ofdrugs targeting DNA methylation have been developed for increasedefficacy and stability, as well as reduced toxicity. These drugs have
been shown to inhibit cancer cell growth, induce cancer cell apoptosis,and reduce tumor volumes in mice [30–33]. Encouraging results frompreclinical and clinical studies have prompted further efforts toelucidate epigenetic alterations in cancer, and to subsequently

1085J. Ren et al. / Cellular Signalling 23 (2011) 1082–1093
develop new epigenetic therapies. In this review, we will focus on therecent findings from the in vitro experiments, animal models, andclinical trials on DNMT inhibitory agents.
2. Transcriptional down regulation due to DNA methylation
2.1. DNA methylation and gene silencing
The role of DNAmethylation in cancer has become one of the mostextensively investigated areas. Recent genome-wide studies havedemonstrated distinct patterns of DNA methylation in canceroustissues, contrary to their normal counterparts [34–36]. Theseepigenetic alterations, specifically changes in the DNA methylationpattern, can be classified under two major categories: 1) Genome-wide hypomethylation which usually involves repeated DNAsequences such as long interspersed nuclear elements (LINE) [37],and 2) hypermethylation of CpG islands [38] within specific genes.Early studies that measured the global content of 5-methylcytosineof tumors showed that hypomethylation was a common feature inseveral malignancies such as metastatic hepatocellular carcinoma[39], cervical cancer [40], and prostate tumors [41]. This epigeneticchange leads to the relaxation of the DNA methylation-mediatedtranscriptional suppression of LINE-1 and endogenous retroviralsequences, which has been shown to play an oncogenic role [14].Hypomethylation caused chromosomal destabilization and maycontribute to cancer progression [42]. However, most of the studiesconcentrated on focal CpG island hypermethylation, often associatedwith tumor suppressor genes, including those for steroid receptor, celladhesion molecules, and inhibitors of matrix metalloproteinases.[43,44]. One of the most detailed studies was conducted on tissuecultures from a patient with lung cancer. More than 40 genes werefound to contain some degree of alteration in DNA methylation. Themost commonly hypermethylated genes include RAR, RASSF1A,CDNK2A, CHD13, and APC [45].
While there is a general correlation between increased DNMTexpression levels and rates of gene hypermethylation in cancercompared to normal controls, regression analysis based on individualsamples does not support a one-to-one correlation between DNMTlevels and gene hypermethylation, reflecting the complex control ofDNA methylation in vivo. Nevertheless, in acute leukemia over-expression of DNMTs appears to be well correlated with hypermethy-lation of p15 and p16. At the time of diagnosis, it was also found thatp15 hypermethylation was associated with lower survival, andtransformation of myelodysplastic syndromes to acute myeloidleukemia. Based on this data the authors proposed to use p15 as amarker of leukemic transformation [46,47]. Gene methylation andsilencing has also been linked to clinical manifestations. In apopulation-based study on human bladder carcinoma, the epigeneticsilencing of three tumor suppressor genes, p16INK4A, RASSF1A andPRSS3, were examined. RASSF1A and PRSS3 promoter methylation wasfound to be associated with advanced tumor stage [46].
2.2. Cancer epigenetics
The detailed mechanism by which a gene undergoes hyper- orhypomethylation are still unclear. Early evidence suggested thatelevated DNMT levels might trigger DNA hypermethylation, whichmay afford a growth advantage [48]. Indeed, the most epigeneticallysusceptible genes are those involved in cell cycle regulation (p16INK4a,p15INK4a, Rb, p14ARF), DNA repair (BRCA1, MGMT), apoptosis (DAPK,TMS1), drug resistance, detoxification, differentiation, angiogenesis,and metastasis. Considering the new findings from the stem cells,Cedar and Bergman [15] proposed an alternative mechanism bywhich cancer cells undergo a process similar to active epigeneticreprogramming during development. This ‘epigenetic switch’ may beregulated by DNMTs and other proteins (Fig. 1b).
Accompanying DNA methylation are the posttranscriptionalmodifications of histones. Upon binding to methylated DNA, MBDscan recruit histone deacetylases (HDACs) to local chromatin [49].Indeed, HDACs are often found to be over-expressed in various typesof cancerous cells (Fig. 1b), resulting in histone deacetylation aroundthe transcription start site and the formation of a more compactstructure in the region of the silenced genes [50,51]. A close linkbetween DNA methylation and histone methylation has also beenobserved. H3 Lys9 methylation in Neurospora and Arabidopsis appearsto be a prerequisite for DNA methylation [52]. Lehnertz et al. [53]observed that Suv39H1/2 knockout also alters the DNA methylationpattern in the pericentric heterochromatin. Recently, Lim et al. [54]pointed out that H3K4me is selectively demethylated by histonelysine demethylase (LSD1), which is upregulated in cancer. Thus, DNAand histone methylation are likely to form a positive feedback loopresulting in long term gene silencing. From this point of view, someeffects of DNMT inhibitors may be achieved through the disruptioncaused by the interactions between DNMTs- and HDACs-mediatedpathways. This partially explains why the simultaneous targeting ofthe two pathways can often achieve synergistic effects.
3. DNMT inhibitors (DNMTi)
For most of the currently available DNMT inhibitors, blocking ofDNAmethylationwas originally identified as a demethylating activity;which raises questions about their specificity as well as cytotoxicity.Nevertheless, optimization of treatment regimens combined withadditional drugs have much improved their profile and inspiredexpectation for their clinical application. Approval of 5-Azacytidine bythe FDA as an anti-tumor agent for the treatment of myelodysplasticsyndrome, has invigorated efforts for the development of novelstrategies to inhibit DNMTs. Despite the recent reports of several novelDNMT inhibitors, the number of available DNMTi compounds remainslimited. Targeted design of DNMTi and expended screening, using aneffective reporter are considered feasible approaches that willfacilitate the identification of more specific molecules [55].
3.1. Nucleoside DNMTi
3.1.1. AzacytidineThe two best studied DNMT inhibitors are Azacytidine (5-
Azacytidine; 5-Aza-CR), a simple derivative of the nucleoside cytidine,and deoxycytidine (5-Aza-2′-deoxycytidine; 5-Aza-CdR), the deoxy-ribose analogue of 5-Azacytidine. The two pyrimidine analogues,which were first synthesized as cytotoxic agents, have been approvedby the FDA in theUSA for the treatment ofmyeloidmalignancies. In the1980s, these potential drugs were found to show demethylatingactivity after incorporation into the DNA of actively replicating tumorcells by forming a covalent protein–DNA complex [56]. Their anti-cancer activity is mediated by two main mechanisms: 1) cytotoxicityresulting from incorporation into the RNA and/or genomicDNA, and2)restoring normal growth and differentiation by demethylation oftumor suppressor genes [10]. Upon uptake by the concentrativenucleoside transporter 1 (hCNT1) [57], 5-Aza-CR and 5-Aza-CdR arephosphorylated sequentially by uridine–cytidine and diphosphatekinases which convert them to the active triphosphate forms 5-Aza-CTP and 5-Aza-dCTP, respectively (Fig. 1c) [58,59]. 5-Aza-CdR ingenomic DNA can prevent the resolution of the covalent reactionintermediate of DNMT and DNA [60] which leads to the trapping andinactivation of DNMT. 5-Aza-CTP is incorporated into RNA whichaffects nuclear and cytoplasmic RNA metabolism including ribosomebiogenesis and protein synthesis [61]. Therefore, 5-Aza-CR can exertcellular effects independent of demethylation [62]. These drugs appearto be most toxic during the S-phase of the cell cycle; however, themechanism of cytotoxicity is not fully understood.

1086 J. Ren et al. / Cellular Signalling 23 (2011) 1082–1093
Recently, Stresemann et al. [63] pointed out that the 5-Aza-CR canalso be incorporated into DNA after intracellular conversion to 5-Aza-dCTP by phosphorylation and reduction. Thus, both drugs are able toinhibit the methylation of replicating DNA by the stoichiometricbinding and trapping of DNMTs for proteosomal degradation. Thedepletion of DNMT in combination with continued DNA replicationlead to genomic DNA hypomethylation through the passive dilutionof methylated cytosine [64,65] rather than active demethylation. Indaughter cells the decreased levels of DNAmethylation are associatedwith reduced H3K9me3 levels and increased H3ac and H3K4me3modifications around gene promoter regions. Lin et al. [66] observedthe formation of compact nucleosomes in themethylated and silencedMLH1 promoter. A recent genome-wide study of modifications in theepigenetic landscape after 5-Aza-CR treatment further validated thisconcept [67]. Interestingly, studies from Momparler et al. [62]indicated that 5-Aza-CdR may be more specific and less toxic than5-Aza-CR. Indeed, the molecule shows a greater inhibitory effect onDNA methylation as well as higher anti-tumor activity againstmyelodysplastic syndrome, acute myelogenous leukemia (AML),and chronic myelogenous leukemia (CML) [68]. More recently, Qinet al. [69] observed that the demethylation functions of 5-Aza-CR and5-Aza-CdR are potent inhibitors for DNA methylation even at lowconcentrations. This observation bears strong clinical implicationssince these reagents at higher concentrations exhibit cytotoxic effectssuch as myelosuppression with neutropenic fever [70] due to DNAdamage and/or interference with DNA synthesis [71]. These cytotoxiceffects highlight the necessity for cautious interpretation of laboratoryand clinical data derived from DNMTi studies. Based on the action ofthese reagents, S-phase is required for the effective incorporation ofthese two drugs into DNA. Therefore, rapidly proliferating cells suchas malignant cells may be more sensitive, than normal cells,undergoing regular cell cycle. Further exploration of this specificfeature may provide opportunity for achieving an improved efficacyand at the same time, reduced side effects by these drugs in theclinical setting.
3.1.2. ZebularineSeveral cytidine analogues have been developed in order to
improve the stability and efficacy of 5-Aza-nucleosides. Zebularine (1-(β-D-ribofuranosyl)-2(1H)- pyrimidinone), a cytidine lacking 4-amino group of the pyrimidine ring, is the most recent addition tothe list of demethylating agents in the family of nucleoside analogues[72]. This agent is described as a potent inhibitor that acts by forminga covalent complex with DNMT and cytidine deaminase whenincorporated into DNA. Synthesized in 1961, it was first characterizedas a potent inhibitor of cytidine deaminasewith anti-tumor properties[40]. It was later described as a DNMTi [73] leading to DNAdemethylation [59] and reactivation of methylation-silenced TSGs[74,75] in cancer cell lines. Zebularine appears to be less cytotoxic andmore stable than 5-Aza-CTP and 5-Aza-dCTP [73,76]. Zebularine's lowtoxicity allowed it to be given continuously, for long period of time, tomaintain the reversal of aberrant hypermethylation [77]. Moreover,zebularine enhances tumor cell chemo and radiation sensitivity [78]and has angiostatic and antimitogenic activities [79,80]. Additionally,zebularine is stable in vivo and suitable for oral administration. It hasbeen well known that when 5-Aza-CR or 5-Aza-CdR is withdrawn,demethylated DNA targets become remethylated. However, whencancer cells are transiently treatedwith 5-Aza-CdR and then subjectedto a continuous treatment with zebularine, remethylation is hinderedand gene expression is maintained, suggesting that this treatmentmodality is able to achieve a sustained response [77,81,82]. However,zebularine is not without drawbacks and therefore further clinicalinvestigations are required for better understanding of its efficiency,as well as side effects in cancer patients. For example, it is not clearwhy higher concentrations of zebularine are needed to obtain similarlevels of demethylation in cells in comparisonwith 5-Aza-CdR [75,83].
Moreover, Suzuki and their colleagues [84] reported that zebularinesuppresses the apoptotic potential of 5-fluorouracil against humanoral squamous cell carcinoma cells, indicating that combinationtherapies with this drug must be carefully investigated. It has alsobeen observed that zebularine can act as a potent mutagen inEscherichia coli [85]. Its impact on genomic DNA stability inmammalian cells remains to be examined.
3.1.3. 5-Fluoro-2′-deoxycytidineThe cytidine analogue 5-fluoro-2′-deoxycytidine (FdCyd, NSC
48006) was previously investigated as a tumor type-selective prodrugof the more potent thymidylate synthase inhibitor 5-fluoro-2′-dUMP(FdUMP) [68,86]. FdUMP can be produced from FdCyd throughdeamination of FdCyd to 5-fluoro-2′-deoxyuridine (FdUrd) bycytidine deaminase followed by phosphorylation to FdUMP [87].This compound was later found to have demethylating effect in themouse cells as well as in the human breast and lung carcinoma cells[87]. Newman and Santi [88] concluded that FdCyd is not only aprodrug for FdUMP, but also has antineoplastic effects by inhibition ofcytidine deaminase within the FdUrd resistant S-49 mutant cell lines[89]. In FdCyd, a fluorine atom replaced one hydrogen at C5, whichduring themethylation reaction receives themethyl group. The FdCydcan be incorporated into DNA which then exerts an inhibitory effecton the action of DNMT at the β-elimination step of themethyl transferreaction. The presence of the fluorine atom makes it difficult for theDNMTmolecule to leave the moiety [90]. The DNMT enzyme becomestrapped in an abortive covalent complex at targeted 5-fluorocytosineresidues in DNA in a similar manner as with 5-Aza-CdR [91]. This drughas already entered phase-I clinical trials for the treatment of breastcancer and other solid tumors [92]. A number of clinical studiesshowed that combination therapy of FdCyd with tetrahydrouridine(THU), a potent cytidine deaminase inhibitor, improves the stability ofFdCyd [89]. More recently Kratzke et al. [93] tested the combinedtherapy with FdCyd and another stable analogue, dihydro-5-Azacytidine (DHAC), for the treatment of malignant mesothelioma.DHAC was later found to significantly inhibit DNMT1 activity, and tohave demethylating activity in human breast cancer cells [94]. DHACalso competes with cytidine triphosphate for incorporation into RNA,leading to ribosomal degradation and defective protein synthesis [95].FdCyd failed to showclear clinical benefit in this study. The clinical useofFdCyd is also limited by its in vivo generation of the potentially toxic5-fluorodeoxyuridine as a metabolites [96].
Efforts to develop new nucleoside analogue DNMTi with improvedstability are underway [12]. One preclinical study showed thatNPEOC–DAC (a prodrug of 5-Aza-CdR containing a 2-(p-nitrophenyl)ethoxycarbonyl (NPEOC) group at the 4 position of the pyrimidinering) can be incorporated into DNA that inhibits DNMTs after itsactivation by human carboxylesterase in a liver cancer cell line [97].Byun et al. [97] reported that the NPEOC–DAC, compared todecitabine, was 23-fold less potent at low doses (b10 μM) forinhibiting DNA methylation; and was also associated with a 3-daydelay in its effect. However, at higher doses (N10 μM) NPEOC–DACwas more effective in inhibiting DNMT. NPEOC–DAC is chemicallydistinct from decitabine with differing metabolism. Thus, changingthe N4 NPEOC group of NPEOC–DAC to a smaller carbon chain maylead to amoleculemuchmore efficient at inhibiting DNAmethylation.Moreover, the activity of NPEOC–DAC is dependent upon carbox-ylesterases, which are not expressed in every tissue. Further studiesare required to explore the use of this compound in combinationtherapy [97]. S110 (a 5′-AzapG-3′ dinucleotide, containing the 5-azacytosine ring) has also been shown to improve the efficacy of 5-Aza-CdR by protecting it from deamination by blocking the activity ofdeaminase enzyme. The compound is well-tolerated and has beenshown to reduce the level of DNA methylation in the CDKN2Apromoter region in xenografts [98].

1087J. Ren et al. / Cellular Signalling 23 (2011) 1082–1093
3.2. Non-nucleoside DNMTi
Nucleoside analogues, though effective in inducing DNA demethy-lation and reactivation of hypermethylated genes, carry a consider-able concern on cytotoxicity [99], which is probably associated withthe drugs' incorporation into DNA. This concern prompted the searchfor non-nucleoside DNMT inhibitors. Some compounds assessed fortheir potential to induce hypomethylation in solid tumors includeprocaine, L-tryptophan derivative RG108, hydralazine, MG98, procai-namide, and (−)-epigallocatechin-3-gallate (EGCG), which is themain polyphenol compound in green tea [100]. Several non-nucleoside compounds are documented for anti-DNMT activity [68]:dietary polyphenols like EGCG, the bisulfide bromotyrosine deriva-tives psammaplins, the L-tryptophan derivative RG108, and procaineand procainamide, which were originally approved by the U.S. Foodand Drug Administration as local anesthetics and for the treatment ofcardiac arrhythmias, respectively. Chuang et al. [101] observed thatthe non-nucleoside compounds, however, induce limited epigeneticchanges in living cells.
3.2.1. ProcaineThe local anesthetic drug procaine acts as an inhibitor of DNMT in
breast cancer cells, causing global genomic DNA demethylation andreactivation of TSGs with hypermethylated CpG islands [55]. Procainealso exerts demethylating activity in the mouse tumor xenograft[39,102]. Procaine appears to bind CpG-rich sequences and therebyblock the binding of DNMTs to DNA [39]. According to Vilar-Gareaet al. [102] procaine acts as an effective DNMTi at high concentrations(100–500 μM) in a cell type-dependentmanner. Castellano et al. [103]prepared a series of procaine analogues and tested their inhibitoryactivity against DNMT1. Among those tested, the procaine derivativepyrrolidine exhibited the highest inhibiting potency on demethyla-tion of chromosomal satellite repeats in HL60 human myeloidleukemia cells. Derivative pyrrolidine is considered a lead compoundfor further studies in this field. In an experiment using humanhepatoma cell lines, the cell viability was significantly decreased byprocaine treatment. Many genes transcriptionally suppressed by DNAhypermethylation were demethylated and reactivated followingprocaine treatment. Moreover, the combined treatment with TSAand procaine produced a stronger reduction in cell viability [104].These data indicated that procaine had DNA demethylating andgrowth-inhibitory effects on human hepatoma cells. Recently,procaine was shown to be effective for induction of DNA demethyla-tion and growth inhibition in the human breast cancer cell line MCF-7[102]. These findings point to the possible application of procaine andits derivatives for epigenetic therapies.
3.2.2. ProcainamideThe antiarrhythmic drug procainamide has been known as an
inhibitor of DNAmethylation in humanT cells. Its demethylating effecton T cells led to the over-expression of lymphocyte function-associated antigen 1 thatmakes T cells autoreactive [105,106]. Initiallyproposed as a perturbative of the interactions between DNMTs andCpG-rich sequences, procainamide was reported to specifically inhibitthe maintenance methyltransferase activity of DNMT1 and todemethylate hypermethylated genes [107]. Procainamide causesglobal DNA hypomethylation and restores the expression of thedetoxifier gene glutathione S-transferase P1 (GSTP1) [105]. Themechanism of action is thought to be mediated by its binding to GC-rich DNA sequences [108]. The nitrogen atoms of procainamide mayinteract with the Lys-162 and Arg-240 residues of DNMT and block theenzyme's catalytic activity [109,110]. Procainamide was also reportedto be capable of restoring the expression of tumor suppressor genesRAR-β, GSTP1, and p16INK4a, which were frequently silenced bypromoter hypermethylation in cancer cells. In one xenograft study,the drug showed a mild inhibitory effect on tumor growth [111]. Gao
et al. [112] demonstrated that WIF-1 is silenced due to promoterhypermethylation in lung cancer cell lines, and both procaine andprocainamide can reactivate WIF-1 and down-regulate the Wntpathway. These results suggest that procaine and procainamide mayhave a potential use for preventing the development of lung cancer[112].
3.2.3. HydralazineHydralazine is a small-molecule non-nucleoside compoundwidely
used as a vasodilator or antiarrhythmic agent [111]. Described as arelatively weak DNMTi [113], hydralazine has been shown todemethylate and reactivate the expression of several tumor suppres-sor genes in cervical cell lines [110]. This activity is enhanced whenhydralazine was applied in combination with valproic acid (VAP)[114]. Hydralazine uses the nitrogen atoms to interact with the Lys-162 andArg-240 residues of DNMT1and inhibit its activity through thesame mechanism as procainamide [115]. Clinical studies havedemonstrated that this combination of epigenetic agents were well-tolerated in treating a number of solid tumors. The combinedtreatment also led to re-expression of methylation-silenced genes inthe primary tumor tissues of cancer patients [116]. The DNAdemethylating and gene reactivating activity of hydralazine in tumorshave also been demonstrated in cervical cancer within an oral doserange of 50–150 mg/day [117].
3.2.4. RG108RG108 was the first DNMT inhibitor developed by rational drug
design. RG108, a non-nucleoside drug and small-molecule inhibitor ofDNMT, was selected for its ability to inhibit free DNMTs without anydetectable cytotoxicity [118]. RG108 appears to directly block thecatalytic pocket of free DNMT proteins, without the formation ofcovalent reaction intermediates [63]. RG108 is capable of inhibitinghuman DNMT activities and reactivate TSGs in human colon-cancercells [119]. However, the action of RG108 is not fully understood.Direct interactionwith DNMT (rather than forming a covalent enzymetrap) and/or the binding to CG-rich DNA sequences [102] wereconsidered possible mechanisms. Brueckner et al. [118] concludedthat RG108 treatment induced demethylation and reactivation ofTSGs in a human colon-cancer cell line, but did not affect themethylation of centromeric satellite sequences [119]. The preferentialaction of RG108 for euchromatic regions might suggest thateuchromatin and heterochromatic sequences are methylated bydistinct pools of DNMTs. This gene- or genomic domain-specificaction, if proven to be true, may be used for the development of morespecific epigenetic therapies. We can take advantage of the selectiveactivity of this drug to reactivate silenced TSG, yet at the same timeconserve the methylation status of satellite DNA and therefore, thechromosome stability in treated cells [38,120]. Thus, RG108 isconsidered as an attractive candidate for further evaluation.
3.2.5. Miscellaneous agentsRecently, the lipophilic, quinoline-based compound SGI-1027
containing a 5-Aza-CdR moiety was demonstrated to be a novelDNMTi in vitro. Treatment of RKO cells with SGI-1027 resulted in thedegradation of DNMT1, demethylation of the CDKN2A gene promoter,and reactivation of methylation-silenced genes [121]. A uniqueproperty of SGI-1027 and probably other compounds in this class isthat, unlike the nucleoside compounds, it is not incorporated intoDNA. Alternative strategies to inhibit DNMT1 include the use of shortanti-sense oligodeoxynucleotides or microRNAs. MG98 is a 20-bpanti-sense oligonucleotide that specifically binds to the 3′ UTR ofhuman DNMT1mRNA thereby interfering with its translation. Despitepromising results in preclinical studies, the clinical use of MG98 hasnot been validated [122]. Recently, Garzon et al. [123] reported thatthe microRNA specifically, miR29a can act as a potential DNMTi.MiR29a targets DNMT3A/B directly and DNMT1 indirectly by binding
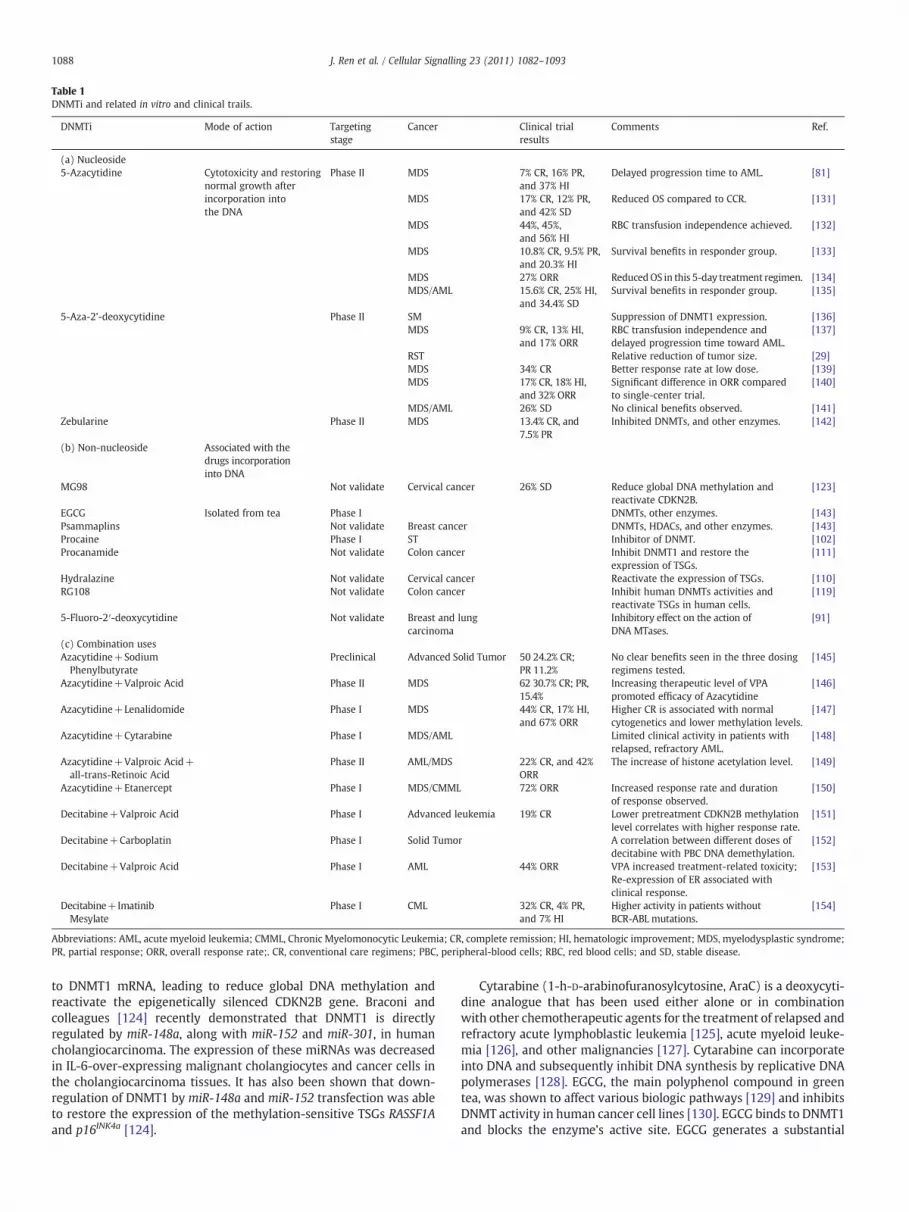
Table 1DNMTi and related in vitro and clinical trails.
DNMTi Mode of action Targetingstage
Cancer Clinical trialresults
Comments Ref.
(a) Nucleoside5-Azacytidine Cytotoxicity and restoring
normal growth afterincorporation intothe DNA
Phase II MDS 7% CR, 16% PR,and 37% HI
Delayed progression time to AML. [81]
MDS 17% CR, 12% PR,and 42% SD
Reduced OS compared to CCR. [131]
MDS 44%, 45%,and 56% HI
RBC transfusion independence achieved. [132]
MDS 10.8% CR, 9.5% PR,and 20.3% HI
Survival benefits in responder group. [133]
MDS 27% ORR ReducedOS in this 5-day treatment regimen. [134]MDS/AML 15.6% CR, 25% HI,
and 34.4% SDSurvival benefits in responder group. [135]
5-Aza-2’-deoxycytidine Phase II SM Suppression of DNMT1 expression. [136]MDS 9% CR, 13% HI,
and 17% ORRRBC transfusion independence anddelayed progression time toward AML.
[137]
RST Relative reduction of tumor size. [29]MDS 34% CR Better response rate at low dose. [139]MDS 17% CR, 18% HI,
and 32% ORRSignificant difference in ORR comparedto single-center trial.
[140]
MDS/AML 26% SD No clinical benefits observed. [141]Zebularine Phase II MDS 13.4% CR, and
7.5% PRInhibited DNMTs, and other enzymes. [142]
(b) Non-nucleoside Associated with thedrugs incorporationinto DNA
MG98 Not validate Cervical cancer 26% SD Reduce global DNA methylation andreactivate CDKN2B.
[123]
EGCG Isolated from tea Phase I DNMTs, other enzymes. [143]Psammaplins Not validate Breast cancer DNMTs, HDACs, and other enzymes. [143]Procaine Phase I ST Inhibitor of DNMT. [102]Procanamide Not validate Colon cancer Inhibit DNMT1 and restore the
expression of TSGs.[111]
Hydralazine Not validate Cervical cancer Reactivate the expression of TSGs. [110]RG108 Not validate Colon cancer Inhibit human DNMTs activities and
reactivate TSGs in human cells.[119]
5-Fluoro-2′-deoxycytidine Not validate Breast and lungcarcinoma
Inhibitory effect on the action ofDNA MTases.
[91]
(c) Combination usesAzacytidine+SodiumPhenylbutyrate
Preclinical Advanced Solid Tumor 50 24.2% CR;PR 11.2%
No clear benefits seen in the three dosingregimens tested.
[145]
Azacytidine+Valproic Acid Phase II MDS 62 30.7% CR; PR,15.4%
Increasing therapeutic level of VPApromoted efficacy of Azacytidine
[146]
Azacytidine+Lenalidomide Phase I MDS 44% CR, 17% HI,and 67% ORR
Higher CR is associated with normalcytogenetics and lower methylation levels.
[147]
Azacytidine+Cytarabine Phase I MDS/AML Limited clinical activity in patients withrelapsed, refractory AML.
[148]
Azacytidine+Valproic Acid+all-trans-Retinoic Acid
Phase II AML/MDS 22% CR, and 42%ORR
The increase of histone acetylation level. [149]
Azacytidine+Etanercept Phase I MDS/CMML 72% ORR Increased response rate and durationof response observed.
[150]
Decitabine+Valproic Acid Phase I Advanced leukemia 19% CR Lower pretreatment CDKN2B methylationlevel correlates with higher response rate.
[151]
Decitabine+Carboplatin Phase I Solid Tumor A correlation between different doses ofdecitabine with PBC DNA demethylation.
[152]
Decitabine+Valproic Acid Phase I AML 44% ORR VPA increased treatment-related toxicity;Re-expression of ER associated withclinical response.
[153]
Decitabine+ImatinibMesylate
Phase I CML 32% CR, 4% PR,and 7% HI
Higher activity in patients withoutBCR-ABL mutations.
[154]
Abbreviations: AML, acute myeloid leukemia; CMML, Chronic Myelomonocytic Leukemia; CR, complete remission; HI, hematologic improvement; MDS, myelodysplastic syndrome;PR, partial response; ORR, overall response rate;. CR, conventional care regimens; PBC, peripheral-blood cells; RBC, red blood cells; and SD, stable disease.
1088 J. Ren et al. / Cellular Signalling 23 (2011) 1082–1093
to DNMT1 mRNA, leading to reduce global DNA methylation andreactivate the epigenetically silenced CDKN2B gene. Braconi andcolleagues [124] recently demonstrated that DNMT1 is directlyregulated by miR-148a, along with miR-152 and miR-301, in humancholangiocarcinoma. The expression of these miRNAs was decreasedin IL-6-over-expressing malignant cholangiocytes and cancer cells inthe cholangiocarcinoma tissues. It has also been shown that down-regulation of DNMT1 by miR-148a and miR-152 transfection was ableto restore the expression of the methylation-sensitive TSGs RASSF1Aand p16INK4a [124].
Cytarabine (1-h-D-arabinofuranosylcytosine, AraC) is a deoxycyti-dine analogue that has been used either alone or in combinationwith other chemotherapeutic agents for the treatment of relapsed andrefractory acute lymphoblastic leukemia [125], acute myeloid leuke-mia [126], and other malignancies [127]. Cytarabine can incorporateinto DNA and subsequently inhibit DNA synthesis by replicative DNApolymerases [128]. EGCG, the main polyphenol compound in greentea, was shown to affect various biologic pathways [129] and inhibitsDNMT activity in human cancer cell lines [130]. EGCG binds to DNMT1and blocks the enzyme's active site. EGCG generates a substantial

1089J. Ren et al. / Cellular Signalling 23 (2011) 1082–1093
amount of the oxidizing agent H2O2 [130], and the oxidation ofDNMTs and other proteins might contribute to its inhibition of DNAmethylation as well. The same action, however, may also causeoxidative damage and substantially increase its cytotoxicity.
4. Clinical studies on DNMTi
Accumulated in vitro data indicates that DNMTi can reversehypermethylation DNA and restore normal gene expression profilesin the different cancer cell lines. Promising in vitro results haveencouraged in vivo evaluation of these drug efficacy, adverse effects,and the best regimens and/or combination strategies that fulfill theirfull therapeutic potential. Some important clinical studies are summa-rized in Table 1.
4.1. 5-Azacytidine (Vidaza, Azacytidine)
5-Aza-CR was the first drug approved by the FDA for the treatmentof myelodysplastic syndromes (MDS). The approval was based onpositive results from three clinical studies conducted by the Cancerand Leukemia Group B (CALGB) [131]. Before approval of 5-Aza-CR,no specific drug was available and the mainstay therapy wassupportive care, including RBC/platelet transfusions and treatmentwith hematopoietic growth factors. In the FDA clinical trials,treatment with 5-Aza-CR resulted in consistent responses in about16% (11.8–18.8%) of patients. The response rate was reproducibleamong the three trials and is consistent with other published reports[155]. Clinical benefits regarding the decreased incidence of bleedingor infections could not be established because of the low incidences ofevents during the trial period [156]. The response rates in the 5-Aza-CR-treated group, and the control group receiving supportive carewere 60% and 5% (Pb0.0001) respectively, with a median responseduration of 14 months in the 5-Aza-CR treated group. These datapresented the first evidence in support of Azacytidine's efficacy inMDS patients with low-risk disease [156]. 5-Aza-CR treatment alsoextended the overall survival time in patients diagnosed withRefractory Anemia with Excess Blasts (RAEB) or Refractory Anemiawith Excess of Blasts in Transformation (RAEB-T), by delaying theprogression to AML [131], and improved the quality of life [157].
A European Aza-001 trial was conducted to further validate theefficacy of 5-Aza-CR inMDS patients, and to test the cytarabine effects.It was found that the 5-Aza-CR treated patients exhibited a significantimprovement in the median survival over the patients that receivedconventional care regimens (CCR) (24.5 months and 15.0 months,respectively) [158]. This drug also benefited older AML patients byprolonging the overall survival (OS) from 16.0 months to 24.5 monthswhile reducing the adverse effects on the hemoglobin levels andneutrophil count. The same trial also indicated that low-dosecytarabine was less effective but more toxic as compared with 5-Aza-CR in MDS patients [158]. Based on the outcome of the Aza-001trial, the National Comprehensive Cancer Network (NCCN) recom-mended 5-Aza-CR as the preferred therapy for patients with high-riskMDS. In addition to high-risk MDS, 5-Aza-CR can also be used fortreating patients with low-risk MDS [12]. In 2009, the FDA approved a7-day regimen that requires weekend treatment, which is inconve-nient for some patients and care providers. To overcome this problem,three alternative regimens that avoid weekend treatment weredesigned by Lyons et al. [134]. Patients who received any one of thethree regimens showed similar hematological improvement as the 7-day regimen, as well as a higher transfusion independent rate. Aphase-II trial on a 5-day alternative 5-Aza-CR intravenous schedulereported partial (PR) and complete remission (CR) rates comparablewith the 7-day subcutaneous regimen [135]. Further studies arerequired to demonstrate the survival benefits of these modifiedregimens. According to the International Working Group, 4 treatmentcycles of treatment with 5-Aza, valproic acid and all-trans retinoic acid
can achieve anoverall response rate of 50%.However,most clinical trialsapplied longer treatment (7–10 treatment cycles). Results from thesetrials provided consistent results on 5-Aza-CR's efficacy in clinicalsettings [137,159]. Interestingly, it was reported that the in patientswith myeloid malignancies, epigenetic changes and DNA damage werenot accurate indicators for clinical response in an overlapping scheduleof 5-azacytidine and HDAC inhibitor entinostat [144].
Safety evaluation of 5-Aza-CR was confounded by the pathophys-iology of MDS which overlaps to a great extent with the commontoxicities of this drug. Serious adverse events (SAEs) occurred in about60% of Azacytidine-treated patients and in about 36% of observationarm patients. The most common SAEs resulting in hospitalization inboth arms were thrombocytopenia, febrile neutropenia, fever andpneumonia. Virtually all (99%) 5-Aza-CR-treated patients and over96% of the observation arm patients reported adverse events.Gastrointestinal events, hematologic events, injection site events,arthralgia cough, dyspnea, headache, weakness, dizziness, andinsomnia were more commonly reported by patients treated with5-Aza-CR than by patients in the observation arm [136,137]. MostMDS patients die from bleeding or infection and from progression toAML. A recent clinical trial observed that 5-Aza-CR, when adminis-tered at high doses to patients with osteogenic sarcoma or othercancers, caused side effects such as nausea, fatigue, neutropenia,thrombocytopenia, vomiting and fevers [28]. Interestingly, the studyalso indicated that 5-Aza-CR could act as DNMTi with minimal impacton DNA synthesis if applied at lower dosages.
4.2. 5-Aza-CdR
5-Aza-CdR was also approved by the FDA for treatment of MDS.However, there is no clear evidence indicating that 5-Aza-CdRimproves OS as observed in a recent CTCL phase-II trial. While only30% responded, thosewho did not respond still benefited from relief ofpruritus early in the trial [140]. Two large phase-II trials of 5-Aza-CdRin MDS were carried out in Europe. In the first of two phase-II studies,patients with intermediate and high-risk MDS received 5-Aza-CdR(135 mg/m2 total dose per course) that resulted in response rates of51% with the high-risk MDS and 46% with the intermediate-1 disease.In the second phase-II trial on 5-Aza-CdR, both, dose intensity andsubcutaneous route of administration were tested in secondary MDS[160,161]. Overall, 32 patients (34%) achieved a CR and 69 (73%) hadan objective response (OR), or hematologic improvement (HI) [161].The authors concluded that 5-Aza-CdR produces durable clinicalresponses and delays the time to MDS transformation with amanageable toxicity profile.
A European phase-III study was conducted in 2007. 170 patientswith MDS were randomized to receive 5-Aza-CdR dose of 15 mg/m2
intravenously over 3 h, every 8 h, for 3 days and the regimens wererepeated every 6 weeks for 6 cycles. 5-Aza-CdR resulted in a higherobjective response rate (17%, CR in 9% and PR in 8%) compared to asupportive care response rate (0%). In another trial, patients treatedwith 5-Aza-CdR had a 17% ORR, which was significantly higher thanthat in the best supportive care (BSC) group (0%). However, nosignificant improvement (14.0 vs 14.9 months) was observed whencomparing the OS between the 5-Aza-CdR and control arms, eventhough clinical benefits (e.g. elongation of median time to AMLprogression) were seen after 5-Aza-CdR treatment [162]. A similarnegative outcome of OS was observed in older patients with MDS orChronic Myelomonocytic Leukemia (CMML) [139]. Grades 3 to 4nonhematologic toxicities, possibly related to 5-Aza-CdR includedhyperbilirubinemia (12%), pneumonia (10%) and constipation (1%)[162]. Based on these data, 5-Aza-CdR received approval from the FDAfor therapy ofMDS and chronicmyelomenocytic leukemia. To increasethe CR rate of patients with these diseases, taking 5-Aza-CdR in theoutpatient setting, several clinical trials explored alternative sche-dules. Kantarjian et al. [140] showed that a 5-day intravenous schedule

1090 J. Ren et al. / Cellular Signalling 23 (2011) 1082–1093
with the highest dose-intensity yielded the highest CR rate (39%). Inanother study, the ADOPT trial reported anORR of 32%, suggesting thatthis 5-day schedule was as effective as the in-patient regimen [138].
It is often difficult to determine whether the effects on geneexpression and cellular function (or even on anti-tumor activity)associated with the inhibitor treatment are due to non-specificcytotoxicity, or to the demethylation of genomic DNA. Intriguingly,data from a recent phase-II study of decitabine in patients withchronic myelogenous leukemia showed that drug-induced hypo-methylation of DNA in peripheral-blood cells was less pronounced inresponders than in nonresponders [70]. 5-Aza-CdR may be applied totreat AML patients by several ways: 1) as a single agent in elderlypatients not fit for intensive chemotherapy; 2) in combination withother agents such as HDAC inhibitors; and 3) as a maintenancetherapy after the completion of consolidation therapy to reduce thepotential of disease recurrence through the prevention of DNA re-hypermethylation. Decitabine alone or in combination with tyrosinekinase inhibitors should be further explored in CML patientswho havedeveloped resistance to tyrosine kinase inhibitors. Decitabine mayalso be beneficial for ALL patients, or patients with other hematologicdisorders. Based on its clear demethylating effects in virtually all thecells examined, there is a compelling scientific rationale to explore thetherapeutic application of 5-Aza-CdR for treating different types ofsolid tumors.
5. Combination therapy
5.1. In vitro studies
Combination therapies are expected to achieve the followinggoals: (1) to enhance or extend the molecular effects of the inhibitors;(2) to counter the molecular effects of inhibitors that abrogate theirefficacy; or (3) to reduce the side effects through applying lowerdosages of one or both drugs. Combination therapies employingDNMTi and other agents are being pursued clinically. Given the invitro evidence for the synergy, this approach remains a topic of activestudy, with initial trials focusing on hematologic malignancies [163].As discussed above, restoration of the aberrantly silenced TSGexpression is coordinated by multiple epigenetic events such aspromoter histone acetylation and DNA demethylation. As listed inTable 1, numerous clinical trials have been conducted to assesscombined treatment with DNMTi and other reagents.
HDAC is the enzyme responsible for the removal of the acetylgroup from histones as well as non-histone proteins [164]. Just likeDNMT inhibitors, HDAC inhibitors such as VPA, vorinistat, MS-275,FK228, sodium phenylbutyrate, and others are small hydrophobiccompounds that can easily penetrate cell membranes and reach theirnuclear targets. HDAC inhibitors induce dramatic changes in chroma-tin structure, which is accompanied by significant alterations in geneexpression patterns and cell functions such as proliferation rate andapoptosis. Since DNAmethylation and histone modification representthe two most important epigenetic pathways involved in TSGsilencing, combined treatment with the two drugs often generatesynergistic effects. Numerous reports have demonstrated the benefitof combined use of the two drugs on both the gene reactivation andchemotherapy levels [165,166]. For example, the combination of 5-Aza-dC with HDAC inhibitors synergistically activated the silencedTSGs and an enhanced antineoplastic effects on breast and lungcarcinomas [167], while DNMTi decitabine (5-Aza-dC) and the HDACinhibitors (LBH589 or MGCD0103) synergistically reduced theproliferation in SCLC cell lines by inducing DNA damage [16].Similarly, sequential treatment of malignant cell lines, first with 5-azacytosine nucleosides and subsequently with an HDAC inhibitorresulted in a more robust re-expression of methylated tumorsuppressor genes such as MLH1, TIMP3 (TIMP3), CDKN2B (INK4B,p15) and CDKN2A in vitro than either agent alone [168]; NY-ESO-1,
one of the most immunogenic cancer/testis antigens, was demon-strated to be expressed in a range of solid tumors via DNAdemethylation and/or histone modification. Oi et al. [169] reportedthat VPA, acting as an HDACi, enhanced induction of NY-ESO-1 in asynergistic action with DNMTi by eliciting DNA demethylation,histone H3 Lys9 demethylation, and acetylation. HDAC inhibitors(FK228, TSA) and DNA hypomethylating agent (5-Aza) exhibitedvarious growth-inhibitory effects on transitional cell carcinoma celllines in a dose- and time-dependentmanner [170]. In addition to G2/M cell cycle arrest, FK228 is more potent in inducting apoptosis thanthe two other single agents, and combination of both FK228 and 5-Azafurther enhances this effect. The p21 induction is closely associatedwith FK228 or TSA but not 5-Aza, which is mediated via p53-independent pathway [170].
Functional interactions between histone acetylation and DNAmethylation have been observed for more than a decade. InNeurospora it was observed that TSA was able to greatly reduceDNAmethylation in several specific regions [171]. Hu et al. [172] haveshown that TSA treatment induces a partial relaxation of imprinting aswell as demethylation of the IGF2R gene. Cosgrove and Cox [173]found that sodium butyrate administration to several human cell linesresulted in global hypomethylation. This mechanism may contributeto the synergy observed between HDAC and DNMT inhibitors withregard to gene reactivation and anti-tumor activity [174]. Studiesfrom this laboratory showed that TSA down-regulates DNMT3BmRNA and protein expression in human endometrial cancer cellsthrough destabilization of DNMT3B mRNA [166]. This finding, asproved later in independent investigations by other laboratories usingdifferent cell lines [172], provided a pathway through which theremarkable synergism is achieved by DNMT and HDAC inhibitors, indifferent tissue/cell types.
5.2. Clinical trial
In a phase-I study, Lin and their colleagues [151] co-administered5-Aza-CR and sodium phenylbutyrate to patients with advanced solidtumors. The three dose schedule administered in that study showedmild toxicity such as neutropenia and anemia; however, theantineoplastic effects were not significant. Braiteh et al. [175]observed promising outcomes in a combination therapy of 5-Aza-CRand VPA for the therapy of breast cancer, colon cancer and othercancers at advanced stages. Recently, Soriano et al. [146] conducted aphase-I/II clinical trial using 5-Aza-CR and VPA in MDS patients. AnORR of 42% indicated that this combination strategy was clinicallyeffective. Another phase-II study with sequential subcutaneousadministration of VPA and 5-Aza-CR reported a CR+PR of 30.7%.VPA concentrations were confirmed to be increased in the plasma(N50 mg/mL vs b50 mg/mL) and better median survival was observed(18.7 vs 10 months) by Voso et al. [176], indicating that HDACi has thepotential to increase the efficacy of DNMTi. Patients with MDS or AMLwho received 5-Aza-CR supplemented with HDACi vorinostat showedpromising outcomes. Among them, 7 patients had hematologicimprovement, 3 patients showed CR (24.2%) and 4 patients showedPR (11.2%) [145]. Reversal of promoter methylation (p15INK4B, CDH-1,DAPK-1, and SOCS-1) after therapy was observed. 5-Aza-CdR+VPAappeared to achieve a better OR (22% vs 9.9%) in patients withleukemia although, in this case, the higher level of VPA did notcorrelate with clinical activity [153]. A later study conducted by Blumet al. [147] confirmed these findings, demonstrating that 25 mg/kg/dof VPA with 5-Aza-CdR neither enhanced the efficacy of 5-Aza-CdRnor did it increase the re-expression of CDKN2B. A very recent studycombining 5-Aza-CR with the FDA-approved MDS chemotherapydrug lenalidomide revealed an impressive 67% overall response rate,32% CR, and 4% PR [154]. In addition, combination of 5-Aza-CdR withother agents such as Imatinib, Carboplatin, or with gemtuzumabozogamicin were examined in various clinical settings [152,172,177].

1091J. Ren et al. / Cellular Signalling 23 (2011) 1082–1093
The encouraging clinical outcomes could broaden the application ofDNMTi to various tumor types, but further randomized clinical trialswith control arms are needed to demonstrate the feasibility of thesemultidrug transporters.
6. Conclusion and future directions
Since DNA methylation is established and maintained by enzy-matic reactions, aberrant DNA methylation patterns are potentiallyreversible by small DNMT inhibitors. The use of these compoundsrepresents a promising and novel approach to cancer prevention andtherapy. Following decades of development, with the approval ofthree epigenetic agents and additional ones in the pipeline, we havecreated a toolbox for manipulating the epigenome in vivo. DNMTinhibitors for chemotherapy are still in its early stage and far fromreaching its full potential. Progress made in epigenetic research willlead to a better understanding of the actions of DNMTi, which willpromote the translation from "bench to the bedside".
While the stability of the DNA methylation pattern is wellrecognized, recent studies have demonstrated the dynamic natureof epigenetic regulation. The dynamic and flexible feature of DNAmethylation is best demonstrated by its reversibility through pharma-cological interference. Despite their significant power in changing theDNA methylation pattern, currently available DNMT inhibitors are allnonselective, which account for most of the side effects. An idealepigenetic therapy should be able to distinguish aberrantly methyl-ated genes (such as TSG) from normally methylated genes. UsingDNMTi as a study probe, in-depth investigation on the interactionsamong DNA methylation, chromatin structure and gene expressionregulation will be performed in the near future. Insights on DNMTtargeting and DNMTi interaction with DNMT molecules will facilitatethe rational design of selective inhibitors. Also, development of moreefficient screening assays will speed up the identification andvalidation of specific DNMT inhibitors.
TSG silencing is mediated by multiple mechanisms including themodification of histones. Consequently, the use of a single demethy-lating agent might not be sufficient to achieve the full reversal ofepigenetic alterations in native cancer tissues. In addition, concernsabout side effects are the driving force for experiments on combineddrug therapy. So far most studies on combinational therapy areperformed in cell culture and animal models. Although the availabledata appears to be optimistic, more clinical trials are required tovalidate its benefit. Another new development would be individual-ized therapy based on laboratory tests for the individual patient'ssensitivity to DNMTi treatment. For example, it was reported that AMLpatients who best respond to demethylation therapy are those withlower methylation levels in the CDKN2B promoter or higher levels ofmiR29 by 10-day schedule of 5-Aza-CdR [147]. Developments in thisnew area have the potential for substantial improvements for treatingcancer patients.
References
[1] M. Kulis, M. Esteller, Advance Genetics 70 (2010) 56.[2] I.A. Qureshi, M.F. Mehler, Archives of Neurology 67 (2010) 1441.[3] T.A. Hore, R.W. Rapkins, J.A. Graves, Trends in Genetics 23 (2007) 448.[4] Z.C. Yen, I.M. Meyer, S. Karalic, C.J. Brown, Genomics 90 (2007) 463.[5] L. Lande-Diner, J. Zhang, I. Ben-Porath, N. Amariglio, I. Keshet, M. Hecht, V.
Azuara, A.G. Fisher, G. Rechavi, H. Cedar, The Journal of Biological Chemistry 282(2007) 12200.
[6] P.D. Gluckman, M.A. Hanson, T. Buklijas, F.M. Low, A.S. Beedle, Nature ReviewsEndocrinology 5 (2009) 408.
[7] A. Portela, M. Esteller, Nature Biotechnology 8 (2010) 1068.[8] K.D. Robertson, Nature Reviews. Genetics 6 (2005) 606.[9] Y. Kondo, L. Shen, A.S. Cheng, S. Ahmed, Y. Boumber, C. Charo, T. Yamochi, T.
Urano, K. Furukawa, B. Kwabi-Addo, D.L. Gold, Y. Sekido, T.H. Huang, J.P. Issa,Nature Genetics 40 (2008) 750.
[10] P.A. Jones, G. Liang, Nature Reviews. Genetics 10 (2009) 881.[11] M.M. Suzuki, A. Bird, Nature Reviews. Genetics 9 (2008) 465.
[12] X. Yang, F. Lay, H. Han, P.A. Jones, Trends in Pharmacological Sciences 31 (2010)546.
[13] J.G. Herman, S.B. Baylin, The New England Journal of Medicine 349 (2003) 2054.[14] K. Saito, K. Kawakami, I. Matsumoto, M. Oda, G. Watanabe, T. Minamoto, Clinical
Cancer Research 16 (2010) 2426.[15] H. Cedar, Y. Bergman, Nature Reviews. Genetics 10 (2009) 304.[16] W. Luszczek, V. Cheriyath, T.M. Mekhail, E.C. Borden, Molecular Cancer Therapy
(2010) 2321.[17] R.Z. Jurkowska, T.P. Jurkowski, A. Jeltsch, Chembiochem 54 (2010) 18.[18] J. Turek-Plewa, P.P. Jagodziński, Cellular & Molecular Biology Letters 10 (2005)
647.[19] A. Hermann, R. Goyal, A. Jeltsch, The Journal of Biological Chemistry 279 (2004)
48359.[20] S. Pradhan, P.O. Esteve, Clinical Immunology 109 (2003) 16.[21] X. Cheng, R.J. Roberts, Nucleic Acids Research 29 (2001) 3795.[22] A. Jeltsch, Chembiochem 3 (2002) 293.[23] M. Okano, D.W. Bell, D.A. Haber, E. Li, Cell 99 (1999) 257.[24] I. Rhee, K.E. Bachman, B.H. Park, K.W. Jair, R.W. Yen, K.E. Schuebel, H. Cui, A.P.
Feinberg, C. Lengauer, K.W. Kinzler, S.B. Baylin, B. Vogelstein, Nature 416 (2002) 556.[25] S. Jeong, G. Liang, S. Sharma, J.C. Lin, S.H. Choi, H. Han, C.B. Yoo, G. Egger, A.S.
Yang, P.A. Jones, Molecular Cell Biology 29 (2009) 5376.[26] D. Bourc'his, G.L. Xu, C.S. Lin, B. Bollman, T.H. Bestor, Science 294 (2001) 2539.[27] S. Xie, Z. Wang, M. Okano, M. Nogami, Y. Li, W.W. He, K. Okumura, E. Li, Gene 236
(1999) 95.[28] C.B. Yoo, P.A. Jones, Nature Reviews. Drug Discovery 5 (2006) 50.[29] F. Jin, S.C. Dowdy, Y. Xiong, N.L. Eberhardt, K.C. Podratz, S.W. Jiang, Gynecologic
Oncology 96 (2005) 538.[30] S.C. Dowdy, S. Jiang, X.C. Zhou, X. Hou, F. Jin, K.C. Podratz, S.W. Jiang, Molecular
Cancer Therapy 5 (2006) 2776.[31] N. Xiang, R. Zhao, G. Song, W. Zhong, Carcinogenesis 29 (2008) 2181.[32] E. Jang, S. Lim, E. Lee, G. Jeong, T. Kim, Y. Bang, J. Lee, Oncogene 23 (2004) 1736.[33] X. Yang, D. Phillips, A. Ferguson, W. Nelson, J. Herman, N. Davidson, Cancer
Research 61 (2001) 7029.[34] J.M. Ordway, M.A. Budiman, Y. Korshunova, R.K. Maloney, J.A. Bedell, R.W. Citek,
B. Bacher, S. Peterson, T. Rohlfing, PLoS ONE 2 (2007) 1314.[35] T.A. Rauch, X. Zhong, X. Wu, M. Wang, K.H. Kernstine, Z. Wang, A.D. Riggs, G.P.
Pfeifer, The Proceedings of National Academy of Sciences U.S.A. 105 (2008) 257.[36] M.E. Figueroa, L. Skrabanek, Y. Li, A. Jiemjit, T.E. Fandy, E. Paietta, H. Fernandez,
M.S. Tallman, J.M. Greally, H. Carraway, J.D. Licht, S.D. Gore, A. Melnick, Blood 114(2009) 3458.
[37] L. Di Croce, V.A. Raker, M. Corsaro, F. Fazi, M. Fanelli, M. Faretta, F. Fuks, F. LoCoco, T. Kouzarides, C. Nervi, S. Minucci, P.G. Pelicci, Science 295 (2002) 1082.
[38] M. Ehrlich, In: M. Ehrlich, (Editor), BioTechniques Book (1999).[39] X. Lin, M. Tascilar,W.-H. Lee, W.J. Vles, B.H. Lee, R. Veeraswamy, K. Asgari, D. Freije,
B. van Rees, W.R. Gage, G.S. Bova, W.B. Isaacs, J.D. Brooks, T.L. DeWeese, A.M. DeMarzo, W.G. Nelson, Cancer Research 61 (2001) 8617.
[40] C.H. Kim, V.E. Marquez, D.T. Mao, D.R. Haines, J.J. McCormack, Journal of MedicalChemistry 29 (1986) 1380.
[41] M.T. Bedford, P.D. van Helden, Cancer Research 47 (1987) 5276.[42] I.P. Pogribny, F.A. Beland, Life Sciences 66 (2009) 2261.[43] A. Lujambio, G.A. Calin, A. Villanueva, S. Ropero, M. Sánchez-Céspedes, D. Blanco,
L.M.Montuenga, S. Rossi,M.S. Nicoloso,W.J. Faller,W.M. Gallagher, S.A. Eccles, C.M.Croce, M. Esteller, The Proceedings of National Academy of Sciences U.S.A. 105(2008) 13561.
[44] M. Toyota, H. Suzuki, Y. Sasaki, R. Maruyama, K. Imai, Y. Shinomura, T. Tokino,Cancer Research 68 (2008) 4132.
[45] J.A. Tsou, J.A. Hagen, C.L. Carpenter, I.A. Laird-Offringa, Oncogene 21 (2002) 5461.[46] C.J. Marsit, M.R. Karagas, H. Danaee, M. Liu, A. Andrew, A. Schned, H.H. Nelson, K.T.
Kelsey, Carcinogenesis 27 (2006) 116.[47] T. Uchida, T. Kinoshita, H. Nagai, Y. Nakahara, H. Saito, T. Hotta, T. Murate, Blood
90 (1997) 1409.[48] T.L. Kautiainen, P.A. Jones, The Journal of Biological Chemistry 261 (1986) 1598.[49] X. Nan, H.H. Ng, C.A. Johnson, C.D. Laherty, B.M. Turner, R.N. Eisenman, A. Bird,
Nature 393 (1998) 396.[50] K. Halkidou, L. Gaughan, S. Cook, H.Y. Leung, D.E. Neal, C.N. Robson, The Prostate
59 (2004) 189.[51] J. Song, J.H. Noh, J.H. Lee, J.W. Eun, Y.M. Ahn, S.Y. Kim, S.H. Lee, W.S. Park, N.J. Yoo,
J.Y. Lee, S.W. Nam, APMIS 113 (2005) 268.[52] J.P. Jackson, A.M. Lindroth, X. Cao, S.E. Jacobsen, Nature 416 (2002) 560.[53] B. Lehnertz, Y. Ueda, A.A. Derijck, U. Braunschweig, L. Perez-Burgos, S. Kubicek, T.
Chen, E. Li, T. Jenuwein, A.H. Peters, Current Biology 13 (2003) 1200.[54] S. Lim, A. Janzer, A. Becker, A. Zimmer, R. Schüle, R. Buettner, J. Kirfel,
Carcinogenesis 31 (2010) 520.[55] L. Jin, E.M. Lee, H.S. Ramshaw, S.J. Busfield, A.G. Peoppl, L. Wilkinson, M.A.
Guthridge, D. Thomas, E.F. Barry, A. Boyd, D.P. Gearing, G. Vairo, A.F. Lopez, J.E.Dick, R.B. Lock, Cell Stem Cell 5 (2009) 42.
[56] J. Vesely´, A. Cihak, Pharmacological Therapy A 2 (1978) 840.[57] M. Rius, C. Stresemann, D. Keller, M. Brom, E. Schirrmacher, D. Keppler, F. Lyko,
Molecular Cancer Therapy 8 (2009) 231.[58] J.P. Issa, H.M. Kantarjian, Clinical Cancer Research 15 (2009) 3946.[59] C. Stresemann, F. Lyko, International Journal of Cancer 123 (2008) 13.[60] D.V. Santi, A. Norment, C.E. Garrett, The Proceedings of National Academy of
Sciences U.S.A. 81 (1984) 6997.[61] A. Cihak, Oncology 30 (1974) 422.[62] R.L. Momparler, L.F. Momparler, J. Samson, Leukemia Research 8 (1984)
1049.

1092 J. Ren et al. / Cellular Signalling 23 (2011) 1082–1093
[63] C. Stresemann, B. Brueckner, T. Musch, H. Stopper, F. Lyko, Cancer Research 66(2006) 2800.
[64] K. Ghoshal, J. Datta, S. Majumder, S. Bai, H. Kutay, T. Motiwala, S.T. Jacob,Molecular Cell Biology 25 (2005) 4741.
[65] H.K. Kuo, J.D. Griffith, K.N. Kreuzer, Cancer Research 67 (2007) 8254.[66] J.C. Lin, S. Jeong, G. Liang, D. Takai, M. Fatemi, Y.C. Tsai, G. Egger, E.N. Gal-Yam, P.A.
Jones, Cancer Cell 12 (2007) 444.[67] V.M. Komashko, P.J. Farnham, Epigenetics 5 (2010) 240.[68] F. Lyko, R. Brown, Journal of The National Cancer Institute 97 (2005) 1506.[69] T. Qin, J. Jelinek, J. Si, J. Shu, J.P. Issa, Blood 113 (2009) 667.[70] J.P. Issa, V. Gharibyan, J. Cortes, J. Jelinek, G. Morris, S. Verstovsek, M. Talpaz, G.
Garcia-Manero, H.M. Kantarjian, Journal of Clinical Oncology 23 (2005) 3956.[71] R. Juttermann, E. Li, R. Jaenisch, The Proceedings of National Academy of Sciences
U.S.A. 91 (1994) 11801.[72] C. Champion, D. Guianvarc'h, C. Se'namaud-Beaufort, R.Z. Jurkowska, A. Jeltsch, L.
Ponger, P.B. Arimondo, A.L. Guieysse-peugeot, PLoS ONE 5 (2010) 12388.[73] L. Zhou, X. Cheng, B.A. Connolly, M.J. Dickman, P.J. Hurd, D.P. Hornby, Journal of
Molecular Biology 321 (2002) 599.[74] M. Billam, M.D. Sobolewski, N.E. Davidson, Breast Cancer Research and
Treatment 120 (2010) 592.[75] C. Flotho, R. Claus, C. Batz, M. Schneider, I. Sandrock, S. Ihde, C. Plass, C.M.
Niemeyer, M. Lübbert, Leukemia 23 (2009) 1028.[76] J.C. Cheng, C.B. Matsen, F.A. Gonzales, W. Ye, S. Greer, V.E. Marquez, P.A. Jones, E.U.
Selker, Journal of The National Cancer Institute 95 (2003) 409.[77] J.C. Cheng, C.B. Yoo, D.J. Weisenberger, J. Chuang, C. Wozniak, G. Liang, V.E.
Marquez, S. Greer, T.F. Orntoft, T. Thykjaer, P.A. Jones, Molecular Cell Biology 24(2004) 1278.
[78] H. Dote, D. Cerna, W.E. Burgan, D.J. Carter, M.A. Cerra, M.G. Hollingshead, K.Camphausen, P.J. Tofilon, Clinical Cancer Research 11 (2005) 4579.
[79] C. Balch, P. Yan, T. Craft, S. Young, D.G. Skalnik, T. H-M, K.P. Nephew Huang,Molecular Cancer Therapy 4 (2005) 1514.
[80] D.M. Hellebrekers, K.W. Jair, E. Vire, S. Eguchi, N.T. Hoebers, M.F. Fraga, M.Esteller, F. Fuks, S.B. Baylin, M. van Engeland, A.W. Griffioen, Molecular CancerTherapy 5 (2006) 475.
[81] C.B. Yoo, J.C. Chuang, H.M. Byun, G. Egger, A.S. Yang, L. Dubeau, T. Long, P.W.Laird, V.E. Marquez, P.A. Jones, Cancer Prevention Research (Phila Pa) 1 (2008)240.
[82] M. Lemaire, L.F. Momparler, N.J. Raynal, M.L. Bernstein, R.L. Momparler, CancerChemotherapy and Pharmacology 63 (2009) 416.
[83] C.B. Yoo, J.C. Cheng, P.A. Jones, Biochemical Society Transactions 32 (2004)912.
[84] M. Suzuki, F. Shinohara, M. Endo, M. Sugazaki, S. Echigo, H. Rikiishi, CancerChemotherapy and Pharmacology 64 (2009) 232.
[85] G. Lee, E. Wolff, J.H. Miller, DNA Repair (Amst) 3 (2004) 161.[86] B. Van Triest, H.M. Pinedo, G. Giaccone, G.J. Peters, Annals of Oncology 11 (2000)
391.[87] J.H. Beumer, J.L. Eiseman, R.A. Parise, E. Joseph, J.L. Holleran, J.M. Covey, M.J.
Egorin, Clinical Cancer Research 12 (2006) 7483.[88] E.M. Newman, D.V. Santi, The Proceedings of National Academy of Sciences U.S.A.
79 (1982) 6423.[89] J.H. Beumer, R.A. Parise, E.M. Newman, J.H. Doroshow, T.W. Synold, H.J. Lenz, M.J.
Egorin, Cancer Chemotherapy and Pharmacology 62 (2008) 368.[90] P.A. Jones, S.M. Taylor, Cell 20 (1980) 93.[91] S. Reither, F. Li, H. Gowher, A. Jeltsch, Journal of Molecular Biology 329 (2003)
684.[92] H. Gowher, A. Jeltsch, Cancer Biology & Therapy 3 (2004) 1068.[93] R.A. Kratzke, X. Wang, L. Wong, M.G. Kratzke, M.R. Green, E.E. Vokes, N.J.
Vogelzang, H.L. Kindler, J.A. Kern, Journal of Thoracic Oncology 3 (2008) 421.[94] B.L. Samuels, J.N. Herndon, D.C. Harmon, R. Carey, J. Aisner, J.M. Corson, Y. Suzuki,
M.R. Green, N.J. Vogelzang, Cancer 82 (1998) 1584.[95] N.J. Vogelzang, J.N. Herndon, C. Cirrincione, D.C. Harmon, K.H. Antman, J.M.
Corson, Y. Suzuki, M.R. Green, N.J. Vogelzang, Cancer 79 (1997) 2242.[96] D.A. Boothman, T.V. Briggle, S. Greer, Molecular Pharmacology 27 (1985) 594.[97] H.M. Byun, S.H. Choi, P.W. Laird, B. Trinh, M.A. Siddiqui, V.E. Marquez, A.S. Yang,
Cancer Letter 266 (2008) 2488 2’-.[98] J.C. Chuang, S.L. Warner, D. Vollmer, H. Vankayalapati, S. Redkar, D.J. Bearss, X.
Qiu, C.B. Yoo, P.A. Jones, Molecular Cancer Therapy 9 (2010) 1450.[99] B. Brueckner, D. Kuck, F. Lyko, Cancer Journal 13 (2007) 22.
[100] B. Segura-Pacheco, E. Perez-Cardenas, L. Taja-Chayeb, A. Chavez-Blanco, A.Revilla-Vazquez, L. Benitez-Bribiesca, A. Duenas-González, Journal of Transla-tional Medicine 4 (2006) 32.
[101] J.C. Chuang, C.B. Yoo, J.M. Kwan, T.W. Li, G. Liang, A.S. Yang, P.A. Jones, MolecularCancer Therapy 4 (2005) 1520.
[102] A. Villar-Garea, M.F. Fraga, J. Espada, M. Esteller, Cancer Research 63 (2003)4993.
[103] S. Castellano, D. Kuck, M. Sala, E. Novellino, F. Lyko, G. Sbardella, Journal ofMedicinal Chemistry 51 (2008) 2325.
[104] M. Tada, F. Imazeki, K. Fukai, A. Sakamoto, M. Arai, R. Mikata, T. Tokuhisa, O.Yokosuka, Hepatology International 1 (2007) 364.
[105] L.S. Scheinbart, M.A. Johnson, L.A. Gross, S.R. Edelstein, B.C. Richardson, TheJournal of Rheumatology 18 (1991) 534.
[106] R. Yung, S. Chang, N. Hemati, K. Johnson, B. Richardson, Arthritis andRheumatism 40 (1997) 1443.
[107] B.H. Lee, S. Yegnasubramanian, X. Lin, W.G. Nelson, The Journal of BiologicalChemistry 280 (2005) 40756.
[108] W. Zacharias, W.J. Koopman, Arthritis and Rheumatism 33 (1990) 374.
[109] N. Singh, A. Dueñas-González, F. Lyko, J.L. Medina-Franco, ChemMedChem 4(2009) 799.
[110] Y. Song, C. Zhang, Cancer Chemotherapy and Pharmacology 63 (2009) 613.[111] B. Segura-Pacheco, C. Trejo-Becerril, E. Perez-Cardenas, L. Taja-Chayeb, I.
Mariscal, A. Chavez, C. Acuña, A.M. Salazar, M. Lizano, A. Dueñas-Gonzalez,Clinical Cancer Research 9 (2003) 1603.
[112] Z. Gao, Z. Xu, M.-S. Hung, Y.-C. Lin, T. Wang, M. Gong, X. Zhi, D.M. Jablons, L. You,Oncology Report 22 (2009) 1484.
[113] M. Candelaria, D. Gallardo-Rinco´ n, C. Arce, L. Cetina, J.L. Aguilar-Ponce, O.Arrieta, A. González-Fierro, A. Chávez-Blanco, E. de la Cruz-Hernández, M.F.Camargo, C. Trejo-Becerril, E. Pérez-Cárdenas, C. Pérez-Plasencia, L. Taja-Chayeb,T. Wegman-Ostrosky, A. Revilla-Vazquez, A. Dueñas-González, Annals ofOncology 18 (2007) 1538.
[114] H. Li, S. Chen, Y. Shu, Y. Chen, Y. Su, X. Wang, S. Zou, Frontiers of Medicine inChina 3 (2009) 157.
[115] C. Mund, B. Brueckner, F. Lyko, Epigenetics 1 (2006) 13.[116] A. Duenas-Gonzalez, M. Candelaria, C. Perez-Plascencia, Cancer Treatment
Reviews 34 (2008) 222.[117] P. Zambrano, B. Segura-Pacheco, E. Perez-Cardenas, L. Cetina, A. Revilla-Vazquez,
L. Taja-Chayeb, A. Chavez-Blanco, E. Angeles, G. Cabrera, K. Sandoval, C. Trejo-Becerril, J. Chanona-Vilchis, A. Duenas-González, BMC Cancer 5 (2005) 44.
[118] B. Brueckner, R. Garcia Boy, P. Siedlecki, T. Musch, H.C. Kliem, P. Zielenkiewicz, S.Suhai, M. Wiessler, F. Lyko, Cancer Research 65 (2005) 6311.
[119] T. Suzuki, R. Tanaka, S. Hamada, H. Nakagawa, N. Miyata, Bioorganic & MedicinalChemistry Letters 20 (2010) 1127.
[120] F. Gaudet, J.G. Hodgson, A. Eden, L. Jackson-Grusby, J. Dausman, J.W. Gray, H.Leonhardt, R. Jaenisch, Science 300 (2003) 492.
[121] J. Datta, K. Ghoshal, W.A. Denny, S.A. Gamage, D.G. Brooke, P. Phiasivongsa, S.Redkar, S.T. Jacob, Cancer Research 69 (2009) 4285.
[122] R.J. Amato, Clinical Genitourinary Cancer 5 (2007) 426.[123] R. Garzon, S. Liu, M. Fabbri, Z. Liu, C.E. Heaphy, E. Callegari, S. Schwind, J. Pang, J.
Yu, N. Muthusamy, V. Havelange, S. Volinia, W. Blum, L.J. Rush, D. Perrotti, M.Andreeff, C.D. Bloomfield, J.C. Byrd, K. Chan, L.C. Wu, C.M. Croce, G. Marcucci,Blood 113 (2009) 6418.
[124] C. Braconi, N. Huang, T. Patel, Hepatology 51 (2010) 890.[125] A. Candoni, A. Michelutti, E. Simeone, D. Damiani, M. Baccarani, R. Fanin,
European Journal of Hematology 77 (2006) 299.[126] H. Tsurumi, N. Kanemura, T. Hara, S. Kasahara, T. Yamada, M. Sawada, M.
Oyama, H. Moriwaki, Journal of Cancer Research and Clinical Oncology 133(2007) 553.
[127] S. Giebel, M. Krawczyk-Kulis, M. Adamczyk-Cioch, B. Jakubas, G. Palynyczko, K.Lewandowski, A. Dmoszynska, A. Skotnicki, K. Nowak, J. Holowiecki, Annals ofHematology 85 (2006) 722.
[128] H. Iwasaki, P. Huang, M.J. Keating, W. Plunkett, Blood 90 (1997) 278.[129] S.B. Moyers, N.B. Kumar, Nutrition Review 62 (2004) 211.[130] M.Z. Fang, Y. Wang, N. Ai, Z. Hou, Y. Sun, H. Lu, W. Welsh, C.S. Yang, Cancer
Research 63 (2003) 7570.[131] L.R. Silverman, D.R. McKenzie, B.L. Peterson, J.F. Holland, J.T. Backstrom, C.L.
Beach, R.A. Larson, Journal of Clinical Oncology 24 (2006) 3903.[132] P. Musto, L. Maurillo, A. Spagnoli, A. Gozzini, F. Rivellini, M. Lunghi, O. Villani, M.A.
Aloe-Spiriti, A. Venditti, V. Santini, Cancer 116 (2010) 1494.[133] V. Santini, Leukemia Research 33 (2009) 26.[134] R.M. Lyons, T.M. Cosgriff, S.S. Modi, R.H. Gersh, J.D. Hainsworth, A.L. Cohn, H.J.
McIntyre, I.J. Fernando, J.T. Backstrom, C.L. Beach, Journal of Clinical Oncology 27(2009) 1856.
[135] M.G. Martin, R.A. Walgren, E. Procknow, G.L. Uy, K. Stockerl-Goldstein, A.F.Cashen, P. Westervelt, C.N. Abboud, F. Kreisel, K. Augustin, J.F. Dipersio, R. Vij,American Journal of Hematology 84 (2009) 564.
[136] E. Winquist, J. Knox, J.P. Ayoub, L. Wood, N. Wainman, G.K. Reid, L. Pearce, A.Shah, E. Eisenhauer, Investigation of New Drugs 24 (2006) 167.
[137] C. Müller-Thomas, T. Schuster, C. Peschel, K.S. Götze, Annals of Hematology 88(2009) 219.
[138] D.P. Steensma, M.R. Baer, J.L. Slack, R. Buckstein, L.A. Godley, G. Garcia-Manero,M. Albitar, J.S. Larsen, S. Arora, M.T. Cullen, H. Kantarjian, Journal of ClinicalOncology 27 (2009) 3848.
[139] P. Wijermans, S. Suciu, L. Baila, U. Platzbecker, A. Giagounidis, D. Selleslag, et al.,Blood 112 (2008) 226.
[140] H. Kantarjian, Y. Oki, G. Garcia-Manero, X. Huang, S. O'Brien, J. Cortes, S. Faderl, C.Bueso-Ramos, F. Ravandi, Z. Estrov, A. Ferrajoli, W. Wierda, J. Shan, J. Davis, F.Giles, H.I. Saba, J.P. Issa, Blood 109 (2007) 57.
[141] D.S. Schrump, M.R. Fischette, D.M. Nguyen, M. Zhao, X. Li, T.F. Kunst, A. Hancox, J.A.Hong, G.A. Chen, V. Pishchik, W.D. Figg, A.J. Murgo, S.M. Steinberg, Clinical CancerResearch 12 (2006) 5785.
[142] S.D. Gore, C. Jones, P. Kirkpatrick, Nature Reviews. Drug Discovery 5 (2006) 892.[143] B. Brueckner, F. Lyko, Trends in Pharmacological Sciences 25 (2004) 553.[144] T.E. Fandy, J.G. Herman, P. Kerns, A. Jiemjit, E.A. Sugar, S.H. Choi, A.S. Yang, T.
Aucott, T. Dauses, R. Odchimar-Reissig, J. Licht, M.J. McConnell, C. Nasrallah, M.K.Kim, W. Zhang, Y. Sun, A. Murgo, I. Espinoza-Delgado, K. Oteiza, I. Owoeye, L.R.Silverman, S.D. Gore, H.E. Carraway, Blood 114 (2009) 2764.
[145] A.O. Soriano, H. Yang, S. Faderl, Z. Estrov, F. Giles, F. Ravandi, J. Cortes, W.G.Wierda, S. Ouzounian, A. Quezada, S. Pierce, E.H. Estey, J.P. Issa, H.M. Kantarjian,G. Garcia-Manero, Blood 110 (2007) 2308.
[146] J.L. Blum, J. Kohles, E. McKenna, N. Scotto, S. Hu, D. Odom, J.A. Kaye, S. Glück,Breast Cancer Research and Treatment 125 (2011) 439.
[147] E. Jabbour, S. Giralt, H. Kantarjian, G. Garcia-Manero, M. Jagasia, P. Kebriaei, L. dePadua, E.J. Shpall, R. Champlin, M. de Lima, Cancer 115 (2009) 1905.

1093J. Ren et al. / Cellular Signalling 23 (2011) 1082–1093
[148] R. Plummer, L. Vidal, M. Griffin, M. Lesley, J. de Bono, S. Coulthard, J. Sludden, L.L.Siu, E.X. Chen, A.M. Oza, G.K. Reid, A.R. McLeod, J.M. Besterman, C. Lee, I. Judson,H. Calvert, A.V. Boddy, Clinical Cancer Research 15 (2009) 3183.
[149] G. Borthakur, X. Huang, H. Kantarjian, S. Faderl, F. Ravandi, A. Ferrajoli, R. Torma,G. Morris, D. Berry, J.P. Issa, Leukemia & Lymphoma 51 (2010) 78.
[150] J. Lin, J. Gilbert, M.A. Rudek, J.A. Zwiebel, S. Gore, A. Jiemjit, M. Zhao, S.D. Baker, R.F.Ambinder, J.G. Herman, R.C. Donehower, M.A. Carducci, Clinical Cancer Research15 (2009) 6249.
[151] S. Chowdhury, S. Seropian, P.W. Marks, American Journal of Hematology 84(2009) 600.
[152] G. Garcia-Manero, H.M. Kantarjian, B. Sanchez-Gonzalez, H. Yang, G. Rosner, S.Verstovsek, M. Rytting, W.G. Wierda, F. Ravandi, C. Koller, L. Xiao, S. Faderl, Z.Estrov, J. Cortes, S. O'brien, E. Estey, C. Bueso-Ramos, J. Fiorentino, E. Jabbour, J.P.Issa, Blood 108 (2006) 3279.
[153] M.A. Sekeres, A.F. List, D. Cuthbertson, R. Paquette, R. Ganetzky, D. Latham, K.Paulic, M. Afable, H.I. Saba, T.P. Loughran Jr., J.P. Maciejewski, Journal of ClinicalOncology 28 (2010) 2258.
[154] J. Gryn, Z.R. Zeigler, R.K. Shadduck, J. Lister, J.M. Raymond, I. Sbeitan, C.S.D.Meisner, C. Evans, Leukemia Research 26 (2002) 897.
[155] R.K. Jain, Nature Medicine 7 (2001) 989.[156] A.B. Kornblith, J.E. Herndon, L.R. Silverman, E.P. Demakos, R. Odchimar-Reissig, J.F.
Holland, B.L. Powell, C. De Castro, J. Ellerton, R.A. Larson, C.A. Schiffer, J.C. Holland,Journal of Clinical Oncology 20 (2002) 2452.
[157] P. Fenaux, N. Gattermann, J.F. Seymour, E. Hellström-Lindberg, G.J. Mufti, U.Duehrsen, S.D. Gore, F. Ramos, O. Beyne-Rauzy, A. List, D. McKenzie, J. Backstrom,C.L. Beach, British Journal of Hematology 149 (2010) 249.
[158] X. Qiu, C. Hother, U.M. Ralfkiær, A. Søgaard, Q. Lu, C.T. Workman, G. Liang, P.A.Jones, K. Grønbæk, PLoS ONE 5 (2010) 2994.
[159] E.A. Olsen, Y.H. Kim, T.M. Kuzel, T.R. Pacheco, F.M. Foss, S. Parker, S.R. Frankel, C. Chen,J.L. Ricker, J.M. Arduino, M. Duvic, Journal of Clinical Oncology 25 (2007) 3115.
[160] B.D. Cheson, P.L. Greenber, J.M. Bennett, B. Lowenberg, P.W. Wijermans, S.D.Nimer, A. Pinto, M. Beran, T.M. deWitte, R.M. Stone, M. Mittelman, G.F. Sanz, S.D.Gore, C.A. Schiffer, H. Kantarjian, Blood 108 (2006) 425.
[161] H. Kantarjian, J.P. Issa, C.S. Rosenfeld, J.M. Bennett, M. Albitar, J. DiPersio, V.Klimek, J. Slack, C. de Castro, F. Ravandi, R. Helmer, L. Shen, S.D. Nimer, R. Leavitt,A. Raza, H. Saba, Cancer 106 (2006) 1803.
[162] M. Bots, R. Johnstone, Clinical Cancer Research 15 (2009) 3977.
[163] B.N. Singh, G. Zhang, Y.L. Hwa, J. Li, S.C. Dowdy, S.W. Jiang, Expert Review ofAnticancer Therapy 10 (2010) 954.
[164] F.A. Steiner, J.A. Hong, M.R. Fischette, D.G. Beer, Z.S. Guo, G.A. Chen, T.S. Weiser,E.S. Kassis, D.M. Nguyen, S. Lee, J.B. Trepel, D.S. Schrump, Oncogene 24 (2005)2397.
[165] Y. Xiong, S.C. Dowdy, K.C. Podratz, F. Jin, J.R. Attewell, N.L. Eberhardt, S.-W. Jiang,Cancer Research 65 (2005) 2689.
[166] A. Raza, M. Mehdi, M. Mumtaz, F. Ali, S. Lascher, N. Galili, Cancer 113 (2008)1604.
[167] E.E. Cameron, K.E. Bachman, S. Myohanen, J.G. Herman, S.B. Baylin, NatureGenetics 21 (1999) 107.
[168] S. Oi, A. Natsume, M. Ito, Y. Kondo, S. Shimato, Y. Maeda, K. Saito, T. Wakabayashi,Journal of Neurology 92 (2008) 22.
[169] J.A. Karam, J. Fan, J. Stanfield, E. Richer, A. Elie Benaim, E. Frenkel, P. Antich, A.I.Sagalowsky, R.P.J.-T. Mason Hsieh, International Journal of Cancer 120 (2007)1802.
[170] E.U. Selker, The Proceedings of National Academy of Sciences U.S.A. 95 (1998)9435.
[171] J.F. Hu, J. Pham, I. Dey, T. Li, T.H. Vu, A.R. Hoffman, Endocrinology 141 (2000)4435.
[172] D.E. Cosgrove, G.S. Cox, Biochimica et Biophysica Acta 1087 (1990) 86.[173] M. Primeau, J. Gagnon, R.L. Momparler, International Journal of Cancer 103
(2003) 184.[174] F. Braiteh, A.O. Soriano, G. Garcia-Manero, D. Hong, M.M. Johnson, P. Silva Lde, H.
Yang, S. Alexander, J. Wolff, R. Kurzrock, Clinical Cancer Research 14 (2008)6301.
[175] M.T. Voso, V. Santini, C. Finelli, P. Musto, E. Pogliani, E. Angelucci, G. Fioritoni, G.Alimena, L. Maurillo, A. Cortelezzi, F. Buccisano, M. Gobbi, L. Borin, A. Di Tucci, G.Zini, M.C. Petti, G. Martinelli, E. Fabiani, P. Fazi, M. Vignetti, A. Piciocchi, V. Liso, S.Amadori, G. Leone, Clinical Cancer Research 15 (2009) 5007.
[176] Y. Oki, H.M. Kantarjian, V. Gharibyan, D. Jones, S. O'brien, S. Verstovsek, J. Cortes,G.M. Morris, G. Garcia-Manero, J.P. Issa, Cancer 109 (2007) 906.
[177] K. Appleton, H.J. Mackay, I. Judson, J.A. Plumb, C. McCormick, G. Strathdee, C. Lee,S. Barrett, S. Reade, D. Jadayel, A. Tang, K. Bellenger, L. Mackay, A. Setanoians, A.Schätzlein, C. Twelves, S.B. Kaye, R. Brown, Journal of Clinical Oncology 25 (2007)4609.