distinct evolutionary pressures underlie diversity in simian immunodeficiency virus and human...
TRANSCRIPT

Published Ahead of Print 10 October 2012. 2012, 86(24):13217. DOI: 10.1128/JVI.01862-12. J. Virol.
T. KorberBeatrice H. Hahn, Norman L. Letvin, Gary J. Nabel and BetteLi, Rajeev Gautam, Ivona Pandrea, George M. Shaw, Will Fischer, Cristian Apetrei, Mario L. Santiago, Yingying Lineagesand Human Immunodeficiency Virus
VirusDiversity in Simian Immunodeficiency Distinct Evolutionary Pressures Underlie
http://jvi.asm.org/content/86/24/13217Updated information and services can be found at:
These include:
SUPPLEMENTAL MATERIAL Supplemental material
REFERENCEShttp://jvi.asm.org/content/86/24/13217#ref-list-1at:
This article cites 91 articles, 52 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from

Distinct Evolutionary Pressures Underlie Diversity in SimianImmunodeficiency Virus and Human Immunodeficiency VirusLineages
Will Fischer,a Cristian Apetrei,b,c Mario L. Santiago,d Yingying Li,e Rajeev Gautam,c Ivona Pandrea,b,c George M. Shaw,e,f
Beatrice H. Hahn,e,f Norman L. Letvin,g† Gary J. Nabel,h and Bette T. Korbera,i
Theoretical Biology, Los Alamos National Laboratory, Los Alamos, New Mexico, USAa; University of Pittsburgh Center for Vaccine Research, Pittsburgh, Pennsylvania, USAb;Tulane National Primate Research Center, Covington, Louisiana, USAc; Division of Infectious Diseases, University of Colorado School of Medicine, Aurora, Colorado, USAd;Departments of Microbiologye and Medicine,f Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA; Beth Israel Deaconess MedicalCenter, Boston, Massachusetts, USAg; Vaccine Research Center, Bethesda, Maryland, USAh; and The Santa Fe Institute, Santa Fe, New Mexico, USAi
Simian immunodeficiency virus (SIV) infection of rhesus macaques causes immune depletion and disease closely resemblinghuman AIDS and is well recognized as the most relevant animal model for the human disease. Experimental investigations ofviral pathogenesis and vaccine protection primarily involve a limited set of related viruses originating in sooty mangabeys(SIVsmm). The diversity of human immunodeficiency virus type 1 (HIV-1) has evolved in humans in about a century; in con-trast, SIV isolates used in the macaque model evolved in sooty mangabeys over millennia. To investigate the possible conse-quences of such different evolutionary histories for selection pressures and observed diversity in SIVsmm and HIV-1, we iso-lated, sequenced, and analyzed 20 independent isolates of SIVsmm, including representatives of 7 distinct clades of virusesisolated from natural infection. We found SIVsmm diversity to be lower overall than HIV-1 M group diversity. Reduced positiveselection (i.e., less diversifying evolution) was evident in extended regions of SIVsmm proteins, most notably in Gag p27 and Envgp120. In addition, the relative diversities of proteins in the two lineages were distinct: SIVsmm Env and Gag were much lessdiverse than their HIV-1 counterparts. This may be explained by lower SIV-directed immune activity in mangabeys relative toHIV-1-directed immunity in humans. These findings add an additional layer of complexity to the interpretation and, potentially,to the predictive utility of the SIV/macaque model, and they highlight the unique features of human and simian lentiviral evolu-tion that inform studies of pathogenesis and strategies for AIDS vaccine design.
The potential efficacy of human immunodeficiency virus type 1(HIV-1) interventions, including vaccines, can be investigated
by using the macaque/simian immunodeficiency virus (SIV)model of infection, wherein rhesus macaques (Macaca mulatta)are exposed to a macaque-adapted simian immunodeficiency vi-rus (SIVmac). The SIV isolates in common use as vaccine andchallenge strains originated in U.S. primate centers (2, 29) viaaccidental, incidental, or experimental transmission of retrovi-ruses from captive sooty mangabeys (Cercocebus atys) to rhesusmacaques, often followed by additional macaque passages. Theendemic sooty mangabey virus (SIVsmm) is not overtly patho-genic in its natural host, but many SIVsmm-derived SIVmacstrains cause AIDS in Asian macaques, as can native SIVsmm,SIVagm from African green monkeys, SIVmnd from mandrills,and SIVlho from L’Hoest monkeys, although adaptation may berequired for full virulence (reviewed in reference 41). Natural SIVinfections are often considered to be nonpathogenic in general,and indeed, there is little overt disease in the best-studied naturalhost/SIV pairings. We note, however, that the natural histories ofmost of the 40-some SIV strains have not been studied and areessentially unknown, that SIVcpz infection can cause AIDS-likedisease in free-ranging chimpanzees (39), and that AIDS has, infact, been reported to have occurred in a few “natural-host” Afri-can primates (reviewed in references 65 and 82), including a sootymangabey (54). SIV-induced pathogenesis is therefore possible,albeit rare, in putatively well-adapted hosts.
The similar pathologies of HIV infection of humans and SIVinfection of macaques (reviewed in references 9 and 41) are char-
acterized by an initial high peak viremia, massive depletion ofCD4� T cells in the gastrointestinal tract during acute infection(10, 58, 83), lowering of viremia by 2 to 4 logs during chronicinfection, progressive loss of CD4� T cells, and eventual progres-sion to AIDS. Furthermore, and critical to the relevance of vaccinestudies in the macaque model, the dynamics of infection in low-dose SIV vaccine challenge models are similar to those of naturalHIV infection of humans (40). These striking parallels, in bothinfection and disease, make SIV infection of macaques a powerfuland informative animal model for HIV infection in humans. An-imal models are extremely useful for vaccine studies, allowingcontrol of the mode, dose, and timing of viral exposure, permit-ting any desired tissue sampling, and enabling efficacy testing viaviral challenge to compare vaccine design strategies. Recent het-
Received 18 July 2012 Accepted 28 September 2012
Published ahead of print 10 October 2012
Address correspondence to Will Fischer, [email protected], or Bette T. Korber,[email protected].
† Deceased.
This article is dedicated to the memory of Norm Letvin, whose many questionsoriginally stimulated our efforts on this project.
Supplemental material for this article may be found at http://jvi.asm.org/.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01862-12
The authors have paid a fee to allow immediate free access to this article.
December 2012 Volume 86 Number 24 Journal of Virology p. 13217–13231 jvi.asm.org 13217
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from

erologous-challenge studies with macaques have provided newhope and direction for the HIV vaccine field: vaccine-elicited an-tibodies correlated with protection from infection (4), and persis-tent CD8� effector memory cells localized in lymph nodes canprovide protection from infection, in some cases, and stringentcontrol of viremia and protection from disease in challenged an-imals that do become infected (20, 26). Nevertheless, despite themany similarities in the biology noted above, and the clear value ofnew insights the macaque vaccine model provides, simian virusesand simian immune responses differ significantly from their hu-man counterparts (64), and the direct applicability of SIV/ma-caque challenge models to human vaccine studies is a matter oflong-standing debate (29, 85).
In this study, we examined a potential complicating factor ofthe SIVmac/macaque model that could affect the interpretation ofheterologous-challenge results as they relate to cross-reactive pro-tection against the extraordinary population diversity of HIV(44). It has been noted that the genetic diversity of naturally cir-culating SIVsmm strains is roughly comparable to the diversity ofthe HIV-1 M group (1), and the predictive utility of SIV heterol-ogous-challenge models rests in part on this observation. How-ever, SIVsmm in sooty mangabeys represents a far more ancientlineage than HIV-1 in humans: the global diversity of the currentHIV-1 epidemic harks back to a common ancestor about a cen-tury ago (43, 89), while SIVsmm origins in sooty mangabey goback many millennia (90). Additionally, the immunological envi-ronments of SIVsmm and HIV-1 are quite different: in sootymangabeys, anti-SIV antibody titers, cytotoxic T lymphocyte(CTL) responses, and immune activation are lower than in HIV-1-infected humans or SIVmac-infected macaques (21, 35, 77; seealso Discussion). Thus, although the levels of protein diversityobserved in the two lineages are roughly comparable, they have
evolved under different selective forces over different timescales,which could in principle give rise to patterns of amino acid diver-sity that reflect these distinct biological histories. We thereforeinvestigated patterns of natural selection in HIV-1 and SIVsmm,finding distinct evolutionary patterns that could potentially affectvaccine-induced immune responses. Here we present new nat-ural-isolate SIVsmm sequences and compare signatures of nat-ural selection (both diversifying and stabilizing selection) to acomparable set of viral sequences drawn from the global HIV-1pandemic.
MATERIALS AND METHODSViral cDNA isolation and sequencing. Near-complete genomes wereamplified by limiting dilution, cloned, and sequenced for 20 new SIVsmmisolates (Table 1), representing seven of the nine SIVsmm lineages previ-ously recovered from U.S. primate centers (2), and for 4 previously iso-lated viruses (Table 2). Of the new isolates, 17 SIVsmm strains (represent-ing lineages 1 to 6) were amplified from tissue culture supernatants (TC)of short-term sooty mangabey peripheral blood mononuclear cell(PBMC) cultures, in which primary SIVsmm isolates were propagated(23), while the remaining 3 (representing lineage 7) were amplified fromuncultured sera (see Table S1 in the supplemental material). The latter
TABLE 1 Origins of newly reported SIVsmm isolatesa
IsolateID Source facility
Host age(yr) Sex
Virallineage
Infectiondurationb Viral load
CD4� Tcell count GenBank accession no.(s)
G078 TNPRC 19 M 1 12 47,363 899 JX860415M919 TNPRC 13 F 1 7 181,553 880 JX860417M922 TNPRC 15 F 1 9 88,954 474 JX860418M923 TNPRC 16 F 1 11 1,909,700 1002 JX860419M935 TNPRC 16 M 1 13 279,283 506 JX860422M947 TNPRC 15 M 1 8 19,073 670 JX860425M926 TNPRC 16 M 2 9 126,374 842 JX860420M934 TNPRC 15 M 2 7 127,900 467 JX860421M946 TNPRC 13 F 2 9 30,823 1124 JX860424M950 TNPRC 15 M 2 8 49,692 1043 JX860427M952 TNPRC 13 F 2 6 54,147 886 JX860429M940 TNPRC 15 M 3 8 1,870 850 JX860423M949 TNPRC 14 M 3 6 16,267 852 JX860426M951 TNPRC 20 M 3 14 28,747 588 JX860428G932 TNPRC 30 F 4 21 128,846 1218 JX860416FTq YNPRC 13 M 5 7 113,000 890 JX860414D215 TNPRC 24 F 6 21 20,393 480 JX860413CFU212 NIRC NA NA 7 NA NA NA JX860407CFU226 NIRC NA NA 7 NA NA NA JX860408, JX860409CFU233 NIRC NA NA 7 NA NA NA JX860410, JX860411, JX860412a All animals were sampled during chronic infection; all infections were natural except for the New Iberia Primate Center (NIRC) samples, deemed “accidental”; the NIRC viralnucleic acids were isolated from serum from chronically infected sooty mangabeys. Tulane National Primate Research Center (TNPRC) and Yerkes National Primate ResearchCenter (YNPRC) isolates were derived from tissue culture supernatant from short-term-infected sooty mangabey PBMC cultures. ID, identity; M, male; F, female; NA, notavailable.b In years between the first positive test and sampling for DNA sequencing.
TABLE 2 Origins of newly sequenced genomes from previouslypublished SIVsmm isolates
Isolate nameCountry oforigin Reference(s)
GenBankaccession no.
SIVsmCI2 Côte D’Ivoire 69 JX860430SIVsmLIB1 Liberia 59 JX860431SIVsmSL92A Sierra Leone 11, 12 JX860432SIVsmSL92B Sierra Leone 11, 12 JX860433
Fischer et al.
13218 jvi.asm.org Journal of Virology
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from

had been sampled between 1980 and 1986, most probably during chronicinfection. Using standard procedures (38, 68, 69), viral RNA purifiedfrom tissue culture supernatants or serum was reverse transcribed, andsingle-genome amplification (SGA) was performed by limiting dilution ofcDNA followed by nested PCR. Nucleotide sequences of single-half-ge-nome amplicons (HGAs) were determined by direct sequencing. Onesequence per isolate was used for the study. For the 17 isolates of lineages1 to 6, nucleotide sequences of HGAs overlapped in integrase by 49 bp(D215, FTq, and G932) or 65 bp extending from U5 in the 5= long terminalrepeat (LTR) to U3 in the 3= LTR, thus including the complete viral pro-teome excepting the carboxyl-terminal amino acid and stop codon of Nef.When multiple HGA-derived sequences were available from a single ani-mal, we favored inclusion of sequences with identity in the overlap be-tween corresponding 5= and 3= HGA sequences and with intact open read-ing frames. For lineage 7, only single HGAs have been obtained to date forCFU212 (3= HGA) and CFU226 (5= HGA). Therefore, additional attemptswere made to obtain SGA-derived subgenomic fragments in gag (1.4 kb),pol (2.5 kb), and gp41-nef (1.1 kb). Individual SGA-derived amplicons areidentified in Table S1 in the supplemental material.
Sequence selection, alignment, and phylogenetic analysis. To assem-ble a data set of HIV-1 sequences to compare with the SIVsmm sequence set,a subset of sequences was selected from the Los Alamos National Laboratory(LANL) HIV database’s web reference alignments (2007 edition [http://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html]). Starting with se-quences with complete reading frames for Gag, Pol, Env, and Nef, sequenceswere down-sampled to the point where simple neighbor-joining trees of the
HIV-1 sequences were superficially similar in clade size and distribution totrees inferred from the SIVsmm data set. A full-genome codon-based align-ment of HIV-1, SIVsmm, and SIVmac sequences was constructed using cu-rated sequence alignments from the LANL HIV database (http://hiv.lanl.gov)as a starting point; individual genes were extracted as needed from the finalalignment. Phylogenetic trees were constructed from both nucleotide andamino acid data sets using Garli (95) with GTR and WAG models, respec-tively; for both amino acids and DNA, site-to-site rate variation was modeledwith a gamma distribution with invariant positions (GTR � � � I; WAG �� � I). Base frequencies were estimated, and amino acid frequencies werefixed; random starting trees were used, with 16 replicates for DNA trees and 4replicates for amino acid analyses. Programs from the Newick Utilities pack-age (33) were used to process phylogenetic trees for presentation (e.g., root-ing, scaling, and branch coloring).
Selection analysis. Codon-aligned nucleotide sequence sets werechecked for recombination using genetic algorithm recombination detec-tion (GARD) (49). Alignments with putative recombinants were dividedinto phylogenetically coherent partitions that were submitted individuallyto MEME (48, 62), FEL, and IFEL (47) analyses on the Data Monkey webserver (http://www.datamonkey.org).
Mosaic sequence generation. Mosaic sequences were generated fromsets of SIV isolates derived from naturally infected sooty mangabeys (orviruses minimally removed from such isolates). Mosaics were generatedusing a slight modification of our previously reported method (19): in-stead of generating a multiple-sequence mosaic cocktail in a single step,we first generated a single mosaic sequence and then sequentially added
SIVsmCI2
SIVsm92A
SMM.SL.1992.SL92B.AF334679
SIVsmLIB1
STM.US.x.STM.M83293
D215 D7 D8
CFU212 D10 A2
CFU226 F5 C7
CFU233 F1 F11 F6
FTq F9 E5
G078 A12 A10
M940 B2 E11
M949 D3 B2
M951 H10 D5
M926 A9 D6
M934 F3 A12
M946 A12 C12
M950 E4 D4
M952 C11 C2
G932 A12 E1
MNE.US.1982.MNE 8.M32741
MNE.US.x.MNE027.U79412
MAC.US.-.MM142 IVMXX.Y00277
MAC.US.-.251 32H PJ5.D01065
MAC.US.x.251 BK28.M19499
MAC.US.x.251 1A11.M76764
MAC.US.-.MAC239-87801.AY587015
MAC.US.x.239.M33262
M919 F2 A2
SMM.US.x.PGM53.AF077017
M922 E12 B1
M935 G10 C6
SMM.US.-.F236 H4.X14307
SMM.US.-.SME543.U72748
SMM.US.x.SIVsmH635F L3.DQ201172
CG7G
CG7V
M947 C11 A8
M923 B4 B11
SMM.US.-.PBJ 143.M80193
SMM.US.x.H9.M80194
SMM.US.-.PBJA.M31325
SMM.US.-.PBJ14 15.L03295
SMM.US.-.PBJ 6P12.L09211
0 0.1 0.2 0.3 0.4substitutions/site
SIVmac239/251 lineage
SIVmacE660
(a) SIVsmm with representative SIVmac lineages
H.CF.1990.056.AF005496
J.SE.1993.SE9280 7887.AF082394
J.SE.1994.SE9173 7022.AF082395
G.SE.1993.SE6165 G6165.AF061642
04 cpx.CY.1994.94CY032 3.AF049337
A2.CY.1994.94CY017 41.AF286237
01 AE.TH.1990.CM240.U54771
02 AG.NG. .IBNG.L39106
A.SN.1996.DDJ360.AY521630
A.SN.2001.DDI579.AY521629
A1.RW.1992.92RW008.AB253421
A1.UG.1999.99UGA07072.AF484478
C.IN.1995.95IN21068.AF067155
C.ZA.2004.04ZASK164B1.DQ056405
C.BR.2004.04BR013.AY727522
C.ET.1986.ETH2220.U46016
C.ET.2002.02ET 288.AY713417
K.CM.1996.96CM MP535.AJ249239
F1.BE.1993.VI850.AF077336
F1.ES. .P1146.DQ979023
F2.CM.1997.CM53657.AF377956
F2.CM.2002.02CM 0016BBY.AY371158
D.CM.2001.01CM 0009BBY.AY371155
D.YE.2002.02YE516.AY795907
D.CD.1983.ELI.K03454
D.ZA.1990.R1.EF633445
D.KE.2001.NKU3006.AF457090
D.YE.2001.01YE386.AY795903
03 AB.RU.1997.KAL153 2.AF193276
B.BO.1999.BOL0122.AY037270
B.GE.2003.03GEMZ010.DQ207942
B.CN.2005.05CNHB hp3.DQ990880
B.TH.2000.00TH C3198.AY945710
B.US.1999.PRB959 03.AY331296
B.CA.1997.CANB3FULL.AY779553
B.AU.1987.MBC925.AF042101
B.FR.1983.HXB2 LAI IIIB BRU.K03455
0.05 0.150 0.1 0.2substitutions/site
(b) HIV-1 M group
FIG 1 Whole-genome maximum likelihood phylogenetic trees inferred from nucleotide data for HIV-1 and SIVsmm data sets. Trees were inferred using Garli(95); both trees are drawn to the same scale (number of expected substitutions per site). The SIVsmm tree (a) is rooted on SMM.SL.1992.SL92B.AF334679 �SIVsm92A; the HIV-1 tree (b) is rooted at the midpoint. Branches with �95% bootstrap support are thickened. Tree files were processed with Newick Utilities(version 1.6 [33]) and sumtrees (version 3.3.1; DendroPy, version 3.11.0 [81]). GenBank accession numbers are included in sequence names, with the exceptionof the two SIVmacE660 isolates CG7G and CG7V (JX648291 and JX648292, respectively [M. Lopker and G. M. Shaw, unpublished data]) and sequences newlyreported in this work (Tables 1 and 2).
Evolutionary Comparisons of SIVsmm and HIV-1 Diversity
December 2012 Volume 86 Number 24 jvi.asm.org 13219
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from
Fischer et al.
13220 jvi.asm.org Journal of Virology
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from

three more sequences, at each step using the sequence(s) from the previ-ous step as a “fixed” sequence(s), i.e., preexisting cocktail members. Theintent of this alteration was to allow incremental testing of increasingmosaic cocktail size. The first generated mosaic alone would be compara-ble to a single consensus sequence; adding cocktail sequences in orderwould increase coverage in a stepwise manner. Compared to that of si-multaneously generated mosaics, the incremental cocktails’ 9-mer cover-age was reduced very slightly (data not shown). Incremental cocktailscontaining 4 sequences were generated for each of gag, pol, env, and nef;coverage data are presented here for 2-sequence cocktails only.
Nucleotide sequence accession numbers. All sequences have beensubmitted to GenBank (accession numbers JX860407 to JX860433[Tables1 and 2]).
RESULTSExtended sampling of SIVsmm diversity. Previous studies ofSIVsmm diversity and evolution (2, 93) suggested that the levels ofwithin-group sequence diversity were roughly comparable be-tween SIVsmm and HIV-1 M group viruses, implying thatSIVsmm vaccine efficacy models could approximate real-worldconditions for HIV-1. We performed full-length sequencing of 20new SIVsmm strains from sooty mangabeys (Table 1) and 4 exist-ing isolates (Table 2) and combined these with previously avail-able sequence data. This data set was assembled with the intent ofexploring the evolutionary pressures on SIVsmm and of designingSIVsmm mosaic vaccine inserts (19) to enable efficacy testing ofmosaic vaccines in the rhesus macaque model (mosaic proteinsare artificial proteins produced by a computational design strategythat optimizes coverage of potential epitope variants in a diversepopulation of viruses). To exclude effects of evolution in nonnat-ural hosts (i.e., evolution during pathogenic passage in ma-caques), and so that standard laboratory stocks could serve asheterologous challenges, we excluded macaque-adapted challengestrains (such as SIVmac239, SIVmac251, and SIVmacE660) andtheir derivatives from the mosaic design input data sets.
For comparisons of HIV-1 and SIVsmm evolution, and of po-tential epitope coverage of various potential vaccines, we selecteda set of HIV-1 M group sequences comparable in size and diversityto the SIVsmm set, including a cross-section of clades (seeMaterials and Methods). Like HIV-1, SIVsmm has a well-definedclade structure with phylogenetically distinct lineages (Fig. 1),though this may be due in part to founder effects (since U.S. pri-mate center sooty mangabey populations represent a small,nonrandom sampling of wild African populations). Of note, asingle SIVsmm sequence with a very long branch length,SMM.SL.1992.SL92B.AF334679, isolated from a sooty mangabey,is typical of SIVsmm in Env and Nef but highly divergent in Poland Gag (Fig. 2a); it is therefore thought to have originated via
recombination between an SIVsmm strain and an SIV of un-known lineage (P. Sharp, personal communication).
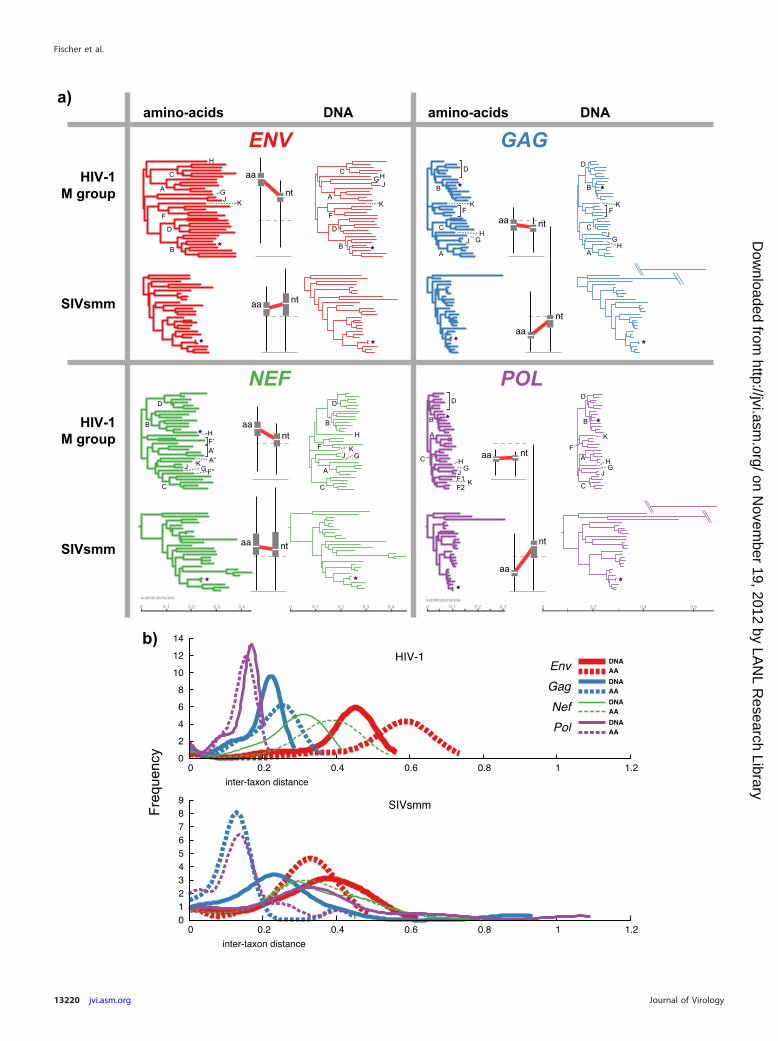
Distinct evolutionary pressures in SIV and HIV lineages. Weinferred maximum likelihood phylogenetic trees for the Gag, Pol,Env, and Nef genes for both the SIVsmm and HIV lineages, usingboth DNA and amino acid data (Fig. 2a). Overall differences be-tween the two viral groups were evident in this simple visualiza-tion, suggestive of distinct selective pressures acting on the differ-ent coding regions over evolutionary time. The inferredevolutionary rates differed between the two viruses. Moreover, theratios of amino acid branch lengths to DNA branch lengths variedfrom gene to gene and between the two viral groups (Fig. 2). Thereare distinct patterns of relative branch length between the fourdifferent genes in the two lineages: for HIV-1, amino acid branchlengths exceeded DNA branch lengths for Gag, Nef, and Env,while Pol amino acid branch lengths were slightly shorter thanDNA branch lengths. This contrasted with SIVsmm: Pol, Gag, andEnv all had amino acid branch lengths shorter than DNA branchlengths (substantially so for Pol and Gag), and Nef had amino acidbranch lengths only slightly longer than DNA branch lengths. Atthe amino acid level, as well, SIVsmm did not match HIV-1 ste-reotypes: Env protein diversity in SIVsmm is much lower than inHIV-1, and while HIV-1 Gag is more diverse than HIV-1 Pol,SIVsmm Pol is more diverse than SIVsmm Gag (Fig. 2b). Thesedistinctions in phylogenetic patterns prompted us to investigateand compare the selective regimes that gave rise to the high diver-sity in these two retroviral lineages.
Codon-based selection analysis. (i) SNAP. To explore the po-tential overall differences in pressure on different genes, we ana-lyzed the spatial occurrences of synonymous and nonsynonymousmutations using SNAP (45, 46), which plots the cumulative oc-currence of each type of substitution from start to end of a gene. Inthe absence of regional differences in selective pressure, the slopeis linear (as is generally the case for synonymous substitutions[Fig. 3b]). Regions of positive pressure (diversifying selection)yield a steeply rising curve in the nonsynonymous plot (Fig. 3a),showing mutation accumulation, while regions under strong neg-ative (stabilizing) selection are level. SIVsmm Pol, Env, and Gaggenes have accumulated many more synonymous substitutionsthan their HIV counterparts (Fig. 3b), as would be expected in amuch older epidemic, in which silent mutations may be nearlysaturated. Nonsynonymous substitutions, on the other hand, ap-pear to have accumulated much more rapidly, relatively speaking,in HIV-1 Env than in SIVsmm Env (Fig. 3a): compared to HIV-1Pol, SIVsmm Pol appears to be subject to slightly reduced stabi-lizing selection, while SIVsmm Gag has both fewer nonsynony-
FIG 2 Comparisons of four genes in HIV-1 and SIVsmm based on phylogenetic trees inferred from amino acid and nucleotide data. (a) Maximum likelihoodtrees (95). Each panel contains 4 trees for a single gene (two viruses, HIV-1 and SIVsmm, and two data types, amino acid and nucleotide sequences); all trees inall panels are drawn to the same scale (number of expected substitutions per site). HIV-1 trees are midpoint rooted, SIVsmm trees are rooted onSMM.SL.1992.SL92B.AF334679 � SIVsm92A. The distributions of intertaxon distances within trees are presented using box-and-whisker plots, with interquar-tile ranges (25th to 75th percentile) as heavy bars, medians as white lines, and extreme values as thin vertical lines; a heavy red line is drawn between the medianof each AA tree branch-length distribution to the median of the DNA tree branch-length distribution. The median value for all HIV/HIV and SIV/SIV pairwiseintertaxon distances (0.2646 expected substitution per site) is shown with a dotted line; a thin gray solid line marks zero. The long branches in the SIV Gag andPol trees (which are broken into two additive pieces) represent sequence SMM.SL.1992.SL92B.AF334679, which is possibly recombinant with an unknown virallineage in these genes (Sharp, personal communication). HIV clades with more than 2 taxa are identified by a letter at the base of the clade or with a bracket if notrecovered as monophyletic; clades represented by 1 or 2 taxa are labeled at the branch tips. Small stars mark the HIV-1 reference sequence HXB2/K03455 and thetwo SIVsmm E660 sequences that are most closely related to the prospective SIV challenge strain. (b) Branch length distributions for each tree (kernel densityestimates based on all pairwise intertaxon distances). Intertaxon pairwise distances are represented by solid lines for DNA trees, and dotted lines for amino acid(AA) trees.
Evolutionary Comparisons of SIVsmm and HIV-1 Diversity
December 2012 Volume 86 Number 24 jvi.asm.org 13221
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from
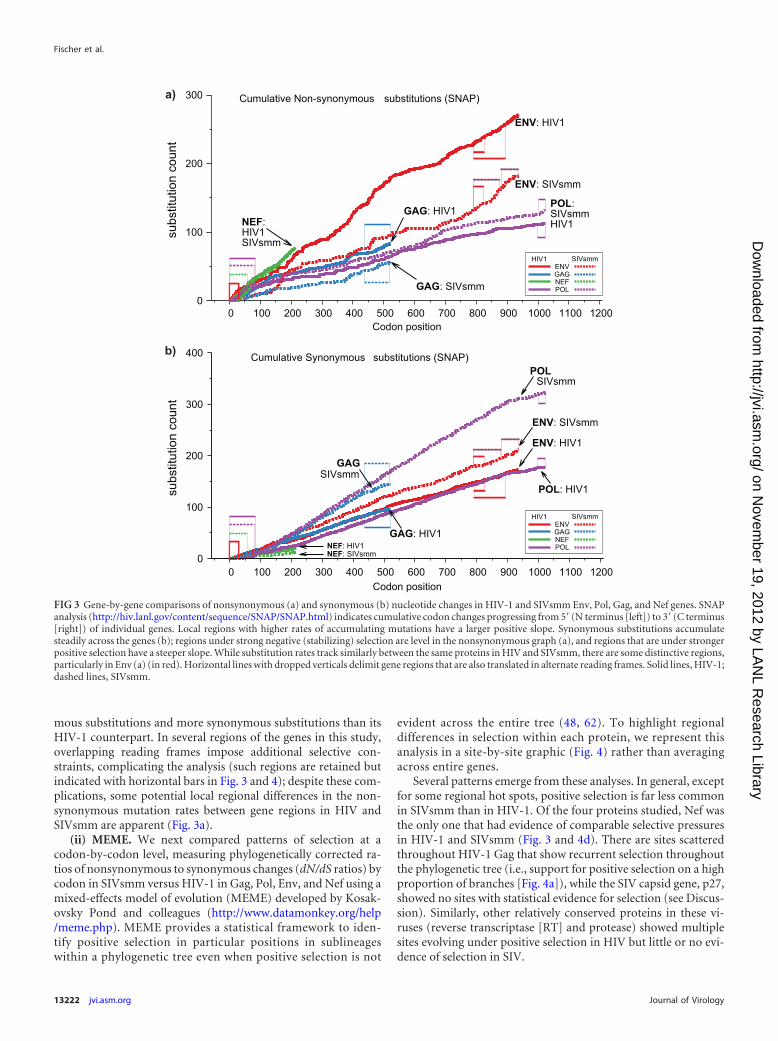
mous substitutions and more synonymous substitutions than itsHIV-1 counterpart. In several regions of the genes in this study,overlapping reading frames impose additional selective con-straints, complicating the analysis (such regions are retained butindicated with horizontal bars in Fig. 3 and 4); despite these com-plications, some potential local regional differences in the non-synonymous mutation rates between gene regions in HIV andSIVsmm are apparent (Fig. 3a).
(ii) MEME. We next compared patterns of selection at acodon-by-codon level, measuring phylogenetically corrected ra-tios of nonsynonymous to synonymous changes (dN/dS ratios) bycodon in SIVsmm versus HIV-1 in Gag, Pol, Env, and Nef using amixed-effects model of evolution (MEME) developed by Kosak-ovsky Pond and colleagues (http://www.datamonkey.org/help/meme.php). MEME provides a statistical framework to iden-tify positive selection in particular positions in sublineageswithin a phylogenetic tree even when positive selection is not
evident across the entire tree (48, 62). To highlight regionaldifferences in selection within each protein, we represent thisanalysis in a site-by-site graphic (Fig. 4) rather than averagingacross entire genes.
Several patterns emerge from these analyses. In general, exceptfor some regional hot spots, positive selection is far less commonin SIVsmm than in HIV-1. Of the four proteins studied, Nef wasthe only one that had evidence of comparable selective pressuresin HIV-1 and SIVsmm (Fig. 3 and 4d). There are sites scatteredthroughout HIV-1 Gag that show recurrent selection throughoutthe phylogenetic tree (i.e., support for positive selection on a highproportion of branches [Fig. 4a]), while the SIV capsid gene, p27,showed no sites with statistical evidence for selection (see Discus-sion). Similarly, other relatively conserved proteins in these vi-ruses (reverse transcriptase [RT] and protease) showed multiplesites evolving under positive selection in HIV but little or no evi-dence of selection in SIV.
FIG 3 Gene-by-gene comparisons of nonsynonymous (a) and synonymous (b) nucleotide changes in HIV-1 and SIVsmm Env, Pol, Gag, and Nef genes. SNAPanalysis (http://hiv.lanl.gov/content/sequence/SNAP/SNAP.html) indicates cumulative codon changes progressing from 5= (N terminus [left]) to 3= (C terminus[right]) of individual genes. Local regions with higher rates of accumulating mutations have a larger positive slope. Synonymous substitutions accumulatesteadily across the genes (b); regions under strong negative (stabilizing) selection are level in the nonsynonymous graph (a), and regions that are under strongerpositive selection have a steeper slope. While substitution rates track similarly between the same proteins in HIV and SIVsmm, there are some distinctive regions,particularly in Env (a) (in red). Horizontal lines with dropped verticals delimit gene regions that are also translated in alternate reading frames. Solid lines, HIV-1;dashed lines, SIVsmm.
Fischer et al.
13222 jvi.asm.org Journal of Virology
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from
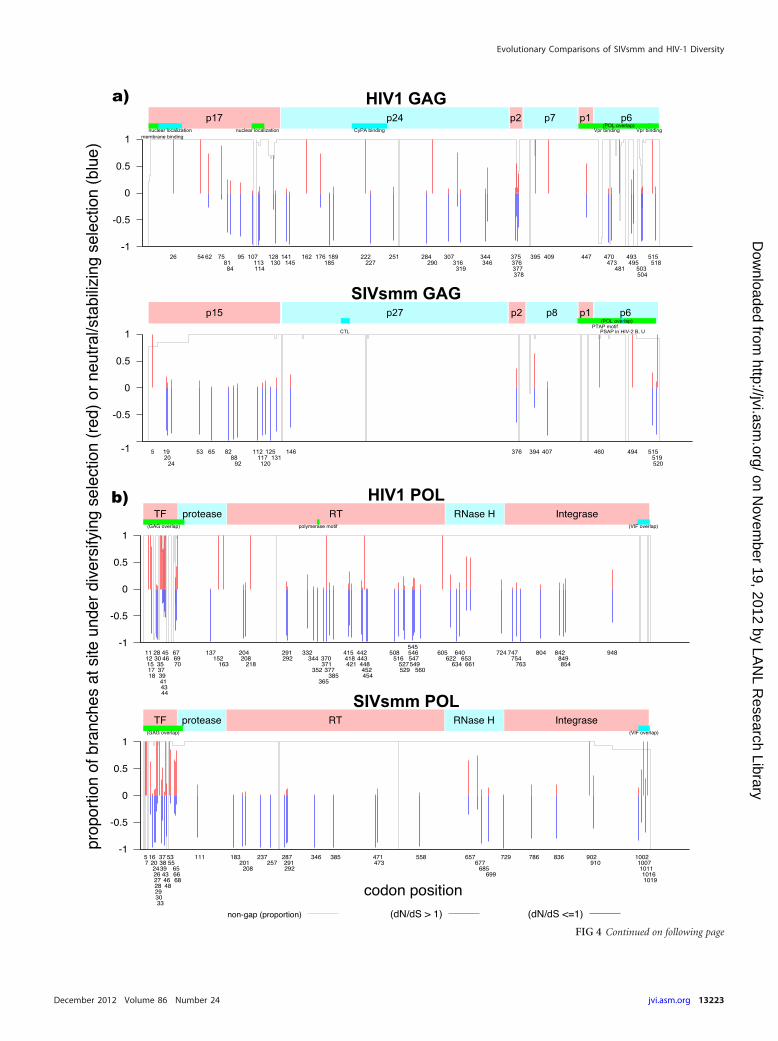
FIG 4 Continued on following page
Evolutionary Comparisons of SIVsmm and HIV-1 Diversity
December 2012 Volume 86 Number 24 jvi.asm.org 13223
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from
Fischer et al.
13224 jvi.asm.org Journal of Virology
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from

In contrast, the upstream transframe (TF) region of Pol poly-protein, which precedes protease and overlaps with Gag in a dif-ferent reading frame, has many sites with strong selective signals inboth SIV and HIV (Fig. 4b); it is also subject to length variation.This small stretch of protein is autocatalytically cleaved by pro-tease and is not very constrained in terms of mutational fitnesscosts (51), so many changes might be tolerated; however, the se-lective signal from MEME may be an artifact caused by overlap-ping reading frames (see Discussion below).
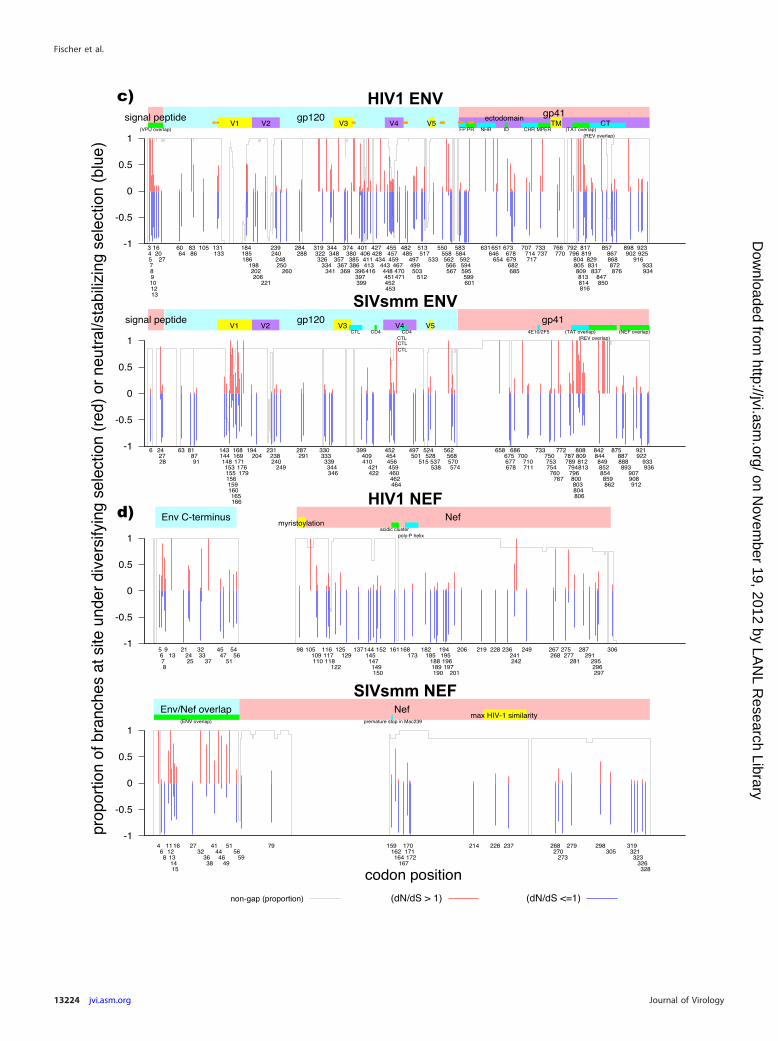
Overall, HIV-1 Env appears to be under greater selective pres-sure than SIVsmm Env, with apparent exceptions of V1 and tworegions in the SIVsmm cytoplasmic tail (Fig. 4c). The region span-ning V1 is hypervariable and in HIV-1 is more commonly mu-tated via insertions and deletions (indels) than via base substitu-tions (88). Frequent indels in V1 introduce extensive lengthvariation and overlays of nonhomologous regions. Therefore, theapparently regional increase in variation in SIVsmm relative toHIV-1 is likely to be misleading, since the dN/dS ratio as a measureof evolutionary selective pressure is based on aligned codons andhence does not take into account indels and length variation.While SIVsmm V1 also shows length variation, it is to a muchlesser degree and the region is much more readily aligned. Conse-quently, selective pressure on the HIV-1 V1 region is likely to beunderestimated relative to SIVsmm by this measure. In contrast,the focused regions of positive selection in the cytoplasmic do-main of SIVsmm gp41 are in regions that are readily aligned; likethe Pol TF region, the cytoplasmic tail of Env may tolerate change,though it is subject to structural constraints (79).
It is likely that some regionally focused MEME-detected in-creases in positive selection are spurious: when reading framesoverlap, stabilizing selection in one reading frame will suppressapparently synonymous substitutions in the other frame(s) andthereby give rise to a false signal of positive selection. Most regionsof strongly elevated SIVsmm selection detected by MEME in theseanalyses, including the Pol TF region and the Env cytoplasmic tail,do in fact map to areas with active genes in multiple overlappingreading frames (Fig. 4), supporting this hypothesis.
Coverage of potential epitopes by candidate SIVsmm andHIV-1 immunogens. Coverage of potential T cell epitopes basedon sequence substrings (i.e., k-mers) provides a metric of se-quence diversity that is likely to be both immunologically andepidemiologically relevant: the overlap of amino acid 9-mers be-tween vaccine candidates and target viral populations provides arough indication of the potential for broad CTL-mediated protec-tion (19, 53). Thus, 9-mer coverage of the different genes of theSIVsmm and HIV-1 provides a metric for the possible breadth anddepth of potential vaccine-induced epitope responses. As we haveseen before (19), we find that in comparisons of within-clade,cross-clade, and overall lineage coverage by different putative vac-cine immunogens (Fig. 5 and 6; see also Fig. S1 in the supplemen-tal material), single-natural-sequence immunogens are very lim-ited in terms of population coverage of potential epitope diversity
in natural strains; polyvalent mosaic designs can provide dramaticincreases in coverage of natural circulating strains. Theoreticalk-mer-based estimates have consistently translated to cross-reac-tive potential of immune responses in macaque models, as well asin mice (5, 42, 71, 72).
As there is a particular interest in Gag and Env as potentialimmunogens (44), we compared potential epitope coverage forthese two proteins, simulating homologous and heterologouschallenge experiments in SIV and within-clade and cross-cladevaccination/exposure in HIV-1. For evaluation of coverage inchallenge experiments, we used SIVmac sequences obtained fol-lowing low-dose infection of macaques (40). In Fig. 5, we present9-mer coverage distributions based on target virus populations,showing the potential coverage of a given population by a givenvaccine. For HIV-1, we compared coverage of groups of B-cladeand C-clade sequences and of a diverse HIV M group sequence setby, in turn, a single B-clade sequence, a single C-clade sequence, ora 2-sequence mosaic HIV cocktail (Fig. 5a and c). For SIV, wecalculated coverage of swarm sequences of SIVmac239/251 and ofSIVmacE660, as well as our diverse SIVsmm population sequenceset (as a comparison to the HIV-1 M group [Fig. 5]), evaluatingSIVmac239, SIVmacE660, and a 2-sequence mosaic SIV cocktailas candidate vaccine inserts.
In HIV-1, for both Env and Gag, cross-clade coverage is poor,within-clade coverage is moderate, and mosaic coverage is supe-rior to within-clade coverage (Fig. 5a and c). For both SIVmac239and SIVmacE660 swarm sequences, coverages of cross-clade(“heterologous challenge”) and within-clade (“homologous chal-lenge”) are both considerably higher (and more tightly peaked)than the corresponding HIV-1 coverages: within-clade coverage isvery high, cross-clade coverages are moderate, and mosaic cover-age values for SIVmac239 and SIVmacE660 are between cross-clade and within-clade values. Compared to mosaic single-cladecoverage values for HIV-1, SIVsmm mosaic coverage values areequivalent for Gag and much higher for Env (Fig. 5b and d).SIVsmm mosaic coverage of the diverse SIVsmm data set is sub-stantially greater than the comparable mosaic coverage of theHIV-1 M group.
The greatest potential for high-coverage optimized vaccine im-munogens appears to be at moderate levels of variability, e.g., forSIVsmm and HIV-1 Gag, SIVsmm Pol, and the more conservedSIVsmm Env (see Fig. S1c, d, g, k, and l in the supplemental ma-terial). The highly conserved HIV-1 Pol gene, which is reasonablywell covered by HXB2 (see Fig. S1i), is more broadly covered bythe 2-sequence mosaics (see Fig. S1k), but the advantage is moredramatic for Gag and SIVsmm Pol. The much more variableHIV-1 Env and both Nef proteins are covered at only a moderatelevel by the mosaics (see Fig. S1h, o, and p), though there is still asubstantial improvement compared to the single-sequence im-munogens.
Intriguingly, the potential epitope diversity that has evolved inHIV-1 in less than 100 years is more extensive than the diversity
FIG 4 Evidence of episodic diversifying selection in HIV-1, SIVsmm, and SIVmac Gag, Pol, Env, and Nef genes. Codon alignments for each gene were analyzedusing the mixed-effects model of evolution (MEME) (12, 48) via the DataMonkey website (http://www.datamonkey.org) (13). Thin red and blue bars indicatecodon positions with statistically significant evidence of positive selection: the relative lengths of the red and blue portions of each bar denote the relativeproportions of branches with positive selection (red) or neutral/stabilizing selection (blue). A thin gray line indicates the proportion of sequences with basespresent (in contrast to gaps inserted to maintain the alignment) at each codon position in the alignments used for analysis. Regions where alternate reading framesare present and other regions of interest are denoted by colored bars in the upper plot. Env residues involved in CD4 binding (94) are marked with orange circles.
Evolutionary Comparisons of SIVsmm and HIV-1 Diversity
December 2012 Volume 86 Number 24 jvi.asm.org 13225
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from
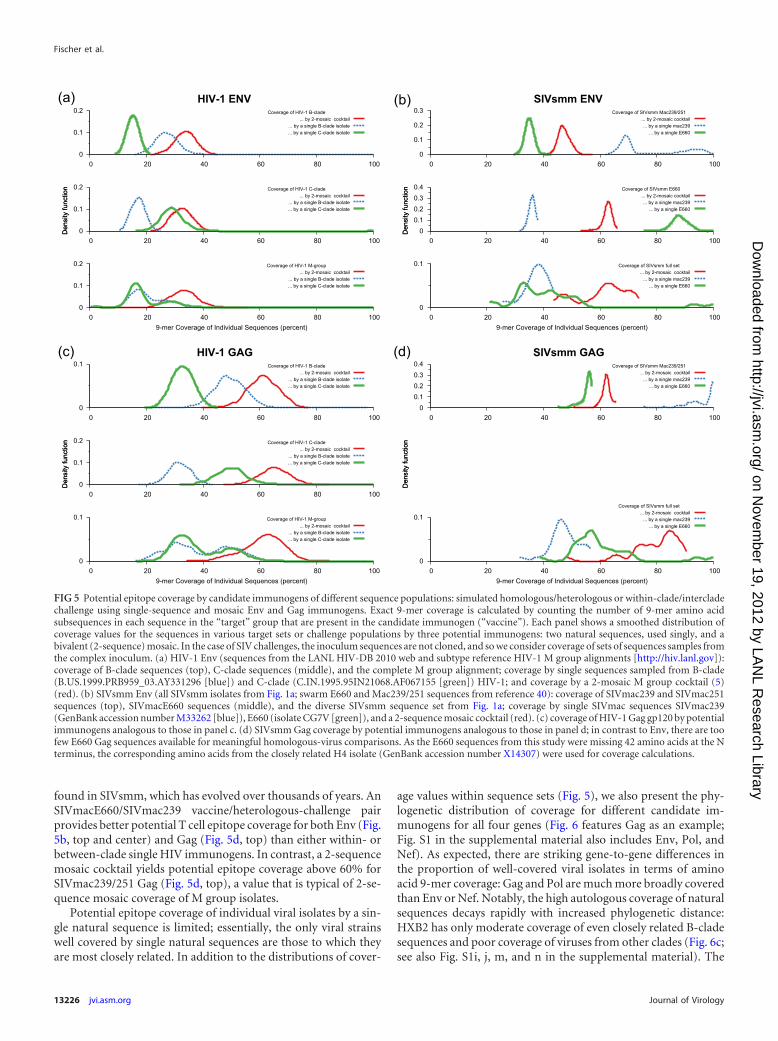
found in SIVsmm, which has evolved over thousands of years. AnSIVmacE660/SIVmac239 vaccine/heterologous-challenge pairprovides better potential T cell epitope coverage for both Env (Fig.5b, top and center) and Gag (Fig. 5d, top) than either within- orbetween-clade single HIV immunogens. In contrast, a 2-sequencemosaic cocktail yields potential epitope coverage above 60% forSIVmac239/251 Gag (Fig. 5d, top), a value that is typical of 2-se-quence mosaic coverage of M group isolates.
Potential epitope coverage of individual viral isolates by a sin-gle natural sequence is limited; essentially, the only viral strainswell covered by single natural sequences are those to which theyare most closely related. In addition to the distributions of cover-
age values within sequence sets (Fig. 5), we also present the phy-logenetic distribution of coverage for different candidate im-munogens for all four genes (Fig. 6 features Gag as an example;Fig. S1 in the supplemental material also includes Env, Pol, andNef). As expected, there are striking gene-to-gene differences inthe proportion of well-covered viral isolates in terms of aminoacid 9-mer coverage: Gag and Pol are much more broadly coveredthan Env or Nef. Notably, the high autologous coverage of naturalsequences decays rapidly with increased phylogenetic distance:HXB2 has only moderate coverage of even closely related B-cladesequences and poor coverage of viruses from other clades (Fig. 6c;see also Fig. S1i, j, m, and n in the supplemental material). The
HIV-1 ENV
0
0.1
0.2
0 20 40 60 80 100
Den
sity
func
tion
Coverage of HIV-1 B-clade... by 2-mosaic cocktail
... by a single B-clade isolate
... by a single C-clade isolate
0
0.1
0.2
0 20 40 60 80 100
Den
sity
func
tion Coverage of HIV-1 C-clade
... by 2-mosaic cocktail... by a single B-clade isolate... by a single C-clade isolate
0
0.1
0.2
0 20 40 60 80 1009-mer Coverage of Individual Sequences (percent)
Coverage of HIV-1 M-group... by 2-mosaic cocktail
... by a single B-clade isolate
... by a single C-clade isolate
(a) SIVsmm ENV
0
0.1
0.2
0.3
0 20 40 60 80 100
Den
sity
func
tion
Coverage of SIVsmm Mac239/251... by 2-mosaic cocktail... by a single mac239
... by a single E660
0 0.1 0.2 0.3 0.4
0 20 40 60 80 100
Den
sity
func
tion Coverage of SIVsmm E660
... by 2-mosaic cocktail... by a single mac239
... by a single E660
0
0.1
0 20 40 60 80 1009-mer Coverage of Individual Sequences (percent)
Coverage of SIVsmm full set... by 2-mosaic cocktail
... by a single mac239... by a single E660
(b)
HIV-1 GAG
0
0.1
0 20 40 60 80 100
Den
sity
func
tion
Coverage of HIV-1 B-clade... by 2-mosaic cocktail
... by a single B-clade isolate
... by a single C-clade isolate
0
0.1
0.2
0 20 40 60 80 100
Den
sity
func
tion Coverage of HIV-1 C-clade
... by 2-mosaic cocktail... by a single B-clade isolate... by a single C-clade isolate
0
0.1
0 20 40 60 80 1009-mer Coverage of Individual Sequences (percent)
Coverage of HIV-1 M-group... by 2-mosaic cocktail
... by a single B-clade isolate
... by a single C-clade isolate
(c) SIVsmm GAG
0 0.1 0.2 0.3 0.4
0 20 40 60 80 100
Den
sity
func
tion
Coverage of SIVsmm Mac239/251... by 2-mosaic cocktail
... by a single mac239... by a single E660
Den
sity
func
tion
0
0.1
0 20 40 60 80 1009-mer Coverage of Individual Sequences (percent)
Coverage of SIVsmm full set... by 2-mosaic cocktail
... by a single mac239... by a single E660
(d)
FIG 5 Potential epitope coverage by candidate immunogens of different sequence populations: simulated homologous/heterologous or within-clade/intercladechallenge using single-sequence and mosaic Env and Gag immunogens. Exact 9-mer coverage is calculated by counting the number of 9-mer amino acidsubsequences in each sequence in the “target” group that are present in the candidate immunogen (“vaccine”). Each panel shows a smoothed distribution ofcoverage values for the sequences in various target sets or challenge populations by three potential immunogens: two natural sequences, used singly, and abivalent (2-sequence) mosaic. In the case of SIV challenges, the inoculum sequences are not cloned, and so we consider coverage of sets of sequences samples fromthe complex inoculum. (a) HIV-1 Env (sequences from the LANL HIV-DB 2010 web and subtype reference HIV-1 M group alignments [http://hiv.lanl.gov]):coverage of B-clade sequences (top), C-clade sequences (middle), and the complete M group alignment; coverage by single sequences sampled from B-clade(B.US.1999.PRB959_03.AY331296 [blue]) and C-clade (C.IN.1995.95IN21068.AF067155 [green]) HIV-1; and coverage by a 2-mosaic M group cocktail (5)(red). (b) SIVsmm Env (all SIVsmm isolates from Fig. 1a; swarm E660 and Mac239/251 sequences from reference 40): coverage of SIVmac239 and SIVmac251sequences (top), SIVmacE660 sequences (middle), and the diverse SIVsmm sequence set from Fig. 1a; coverage by single SIVmac sequences SIVmac239(GenBank accession number M33262 [blue]), E660 (isolate CG7V [green]), and a 2-sequence mosaic cocktail (red). (c) coverage of HIV-1 Gag gp120 by potentialimmunogens analogous to those in panel c. (d) SIVsmm Gag coverage by potential immunogens analogous to those in panel d; in contrast to Env, there are toofew E660 Gag sequences available for meaningful homologous-virus comparisons. As the E660 sequences from this study were missing 42 amino acids at the Nterminus, the corresponding amino acids from the closely related H4 isolate (GenBank accession number X14307) were used for coverage calculations.
Fischer et al.
13226 jvi.asm.org Journal of Virology
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from
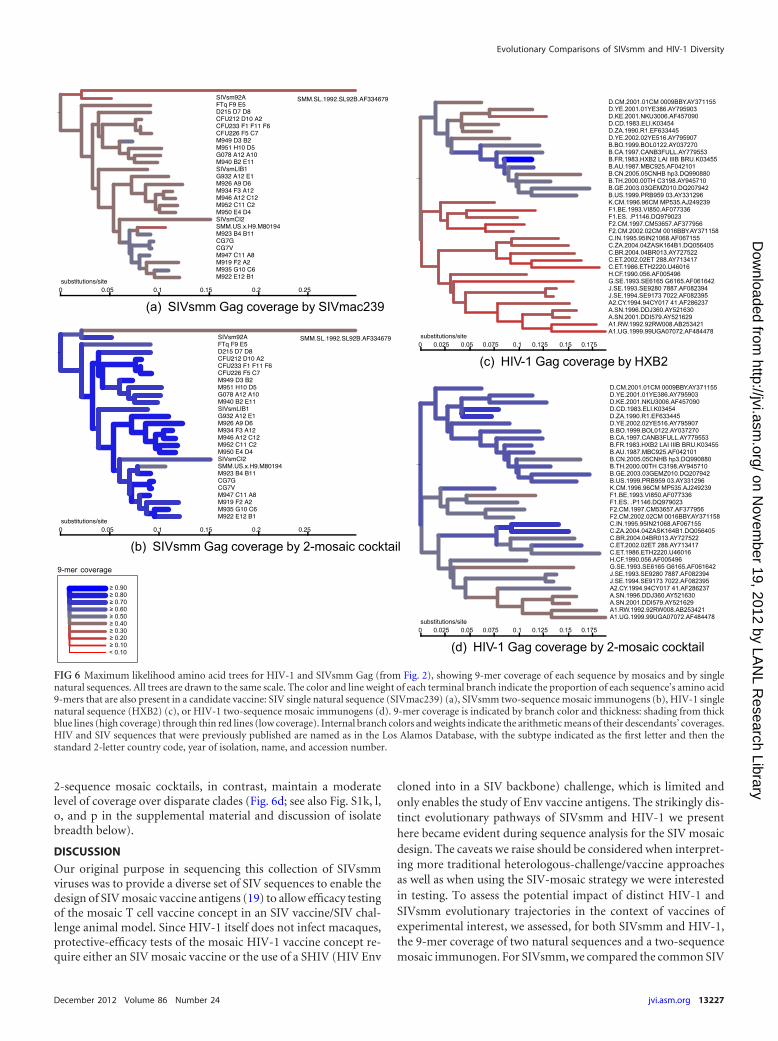
2-sequence mosaic cocktails, in contrast, maintain a moderatelevel of coverage over disparate clades (Fig. 6d; see also Fig. S1k, l,o, and p in the supplemental material and discussion of isolatebreadth below).
DISCUSSION
Our original purpose in sequencing this collection of SIVsmmviruses was to provide a diverse set of SIV sequences to enable thedesign of SIV mosaic vaccine antigens (19) to allow efficacy testingof the mosaic T cell vaccine concept in an SIV vaccine/SIV chal-lenge animal model. Since HIV-1 itself does not infect macaques,protective-efficacy tests of the mosaic HIV-1 vaccine concept re-quire either an SIV mosaic vaccine or the use of a SHIV (HIV Env
cloned into in a SIV backbone) challenge, which is limited andonly enables the study of Env vaccine antigens. The strikingly dis-tinct evolutionary pathways of SIVsmm and HIV-1 we presenthere became evident during sequence analysis for the SIV mosaicdesign. The caveats we raise should be considered when interpret-ing more traditional heterologous-challenge/vaccine approachesas well as when using the SIV-mosaic strategy we were interestedin testing. To assess the potential impact of distinct HIV-1 andSIVsmm evolutionary trajectories in the context of vaccines ofexperimental interest, we assessed, for both SIVsmm and HIV-1,the 9-mer coverage of two natural sequences and a two-sequencemosaic immunogen. For SIVsmm, we compared the common SIV
FIG 6 Maximum likelihood amino acid trees for HIV-1 and SIVsmm Gag (from Fig. 2), showing 9-mer coverage of each sequence by mosaics and by singlenatural sequences. All trees are drawn to the same scale. The color and line weight of each terminal branch indicate the proportion of each sequence’s amino acid9-mers that are also present in a candidate vaccine: SIV single natural sequence (SIVmac239) (a), SIVsmm two-sequence mosaic immunogens (b), HIV-1 singlenatural sequence (HXB2) (c), or HIV-1 two-sequence mosaic immunogens (d). 9-mer coverage is indicated by branch color and thickness: shading from thickblue lines (high coverage) through thin red lines (low coverage). Internal branch colors and weights indicate the arithmetic means of their descendants’ coverages.HIV and SIV sequences that were previously published are named as in the Los Alamos Database, with the subtype indicated as the first letter and then thestandard 2-letter country code, year of isolation, name, and accession number.
Evolutionary Comparisons of SIVsmm and HIV-1 Diversity
December 2012 Volume 86 Number 24 jvi.asm.org 13227
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from

vaccine/challenge strains SIVmacE660 and SIVmac239 and ournewly designed SIV mosaic vaccine set against our collection ofdiverse natural SIVsmm isolates (the most comprehensive whole-genome sequence set currently available). For HIV-1, we com-pared a single natural B-clade and single natural C-clade se-quences and a set of HIV-1 mosaic vaccine immunogens that arenow approaching human phase I safety and immunogenicity trialsagainst a set of HIV-1 M group isolates.
Genetic (DNA-based) distances were roughly comparable be-tween the two viruses, excepting SIVsmm Pol, which has largerdistance values (Fig. 2). At the protein level, however, the SIVsmmlineage is generally less diverse, and apparently under less positiveselective pressure (or under greater fitness constraints), than theHIV-1 M group. Furthermore, inferred positions subject to posi-tive selection were distributed differently among and within dif-ferent proteins in SIVsmm and HIV. The differences in positiveselection between HIV-1 and SIVsmm Gag were particularly strik-ing: there are sites scattered throughout HIV-1 Gag that showrecurrent selection throughout the phylogenetic tree (i.e., supportfor positive selection on a high proportion of branches [Fig. 4a]).In contrast, SIVsmm Gag was under substantially weaker positiveselection (or stronger negative selection, i.e., greater fitness con-straints) than Pol: the SIVsmm p27 capsid (equivalent to HIV-1p24) showed very little variation at the amino acid level and noevidence of positive selection at all (Fig. 4a). Capsid (p24/p27) isone of the more conserved proteins in both HIV and SIV, andcertain HIV p24 epitopes are targets for HLA-restricted T cellresponses associated with viral control and long-term survival (67,80). Despite the overall conservation of HIV-1 p24, these epitopesstill vary under CTL-mediated immune pressure, and it has beenhypothesized that immune escape from these epitopes comes at ahigh fitness cost for the virus, contributing to the beneficial effectassociated with particular HLAs (60). The lack of inferred selec-tion in SIVsmm p27 suggests either that the SIVsmm capsid pro-tein is generally less tolerant of mutation than the HIV-1 capsid orthat mangabeys lack the potent Gag-directed immune responsesseen in humans (67, 80) and in macaques (55, 86). The latterexplanation appears probable: although sooty mangabeys makeCTL responses to SIV infection that are strong enough to driveviral escape (34, 36), and although Gag-directed CTL responses inparticular do indeed exist in sooty mangabeys (59, 84), both themagnitude and breadth of CTL responses are reduced in SIVsmm-infected sooty mangabeys compared with those in HIV-infectedhumans (15). Furthermore, SIVsmm-infected sooty mangabeyshave lower neutralizing antibody titers than HIV-infected hu-mans (35, 52) or SIVmac-infected rhesus macaques. In SIVagm-infected African green monkeys (56), another “natural host,” anti-p27 antibody responses are also much weaker than in rhesusmacaques; this may be a general feature of nonpathogenic primatelentiviral infections (28, 35). Finally, CD8� T cell depletion exper-iments with SIVsmm-infected sooty mangabeys (6) showedsmaller effects on viral load than cognate experiments with ma-caques (32, 57, 75) and African green monkeys (22). These com-bined data suggest that immunological pressure on SIVsmm p27is much lower than on HIV-1 p24. Therefore, the observed varia-tion in SIVsmm more likely results from gradual accrual of muta-tions over time (i.e., genetic drift) than from rapid selection forimmune evasion. In contrast, rapid immune escape is a majoraspect of positive selection in HIV-1: most early mutations areconcentrated within T cell epitopes (17, 18, 25, 27), viruses within
individuals continually evolve to escape from neutralizing anti-bodies during chronic infection (3, 61, 91), and HLA imprinting isevident in viruses circulating in different human populations(37, 67).
The HIV-1 M group has likely been evolving in the humanpopulation for on the order of 100 years (43, 89). The muchgreater age of the “natural host” epidemics in African monkeys,and in sooty mangabeys in particular (90), is consistent with thegreater extent of silent mutations we observed in SIVsmm, and itwould furthermore allow the evolution of higher degrees of viraltolerance (virus/host coevolution for reduced pathogenicity).Sooty mangabeys, like African green monkeys, rarely progress toAIDS despite high-intensity SIVsmm infection; one mechanismby which tolerance is achieved in mangabeys is reduced immuneactivation (8, 15, 16, 74). Rates of progression to AIDS are associ-ated with chronic immune activation (24), and the failure todownregulate interferon-stimulated genes after acute infectionappears to be restricted to the pathological infections that occur inmacaques and humans (8). In any case, the combined immuno-logical and natural selection data argue strongly that the non-pathogenicity of SIVsmm in mangabeys is not due to a more ef-fective immune response, and SIVsmm diversity appears to haveevolved under lower immune pressure than HIV-1 diversity.
Diversity is of course a central problem in HIV vaccine design.A vaccine that protects against (or mitigates) HIV-1 infectionmust induce cross-reactive immunity against pandemic variants.We have proposed mosaic vaccines as a strategy for inducing suf-ficient numbers of T cell immune responses that cross-react withmany different HIV-1 variants (19).
To quantify T cell cross-reactivity, we previously distinguishedbetween breadth and depth of T cell responses (5), breadth refer-ring to the summed quantities (counts) of different epitope locirecognized and depth referring to the recognition of multiple vari-ants at individual epitope loci. In previous experimental work (70,71, 72), we assessed the cross-reactivity of vaccine-induced re-sponses by comprehensively testing peptides derived from diverseHIV-1 isolates. We here introduce the theoretical counterpart, theconcept of “isolate breadth,” meaning the proportion of viral iso-lates from a given population for which a vaccine candidate haspotential epitope coverage above a given threshold. A vaccine im-munogen (or immunogen set) with high isolate breadth willmatch large portions of many sequences in a target population,and hence the responses it induces are more likely to cross-reactwith epitopes of an infecting virus. As expected based on earlierfindings, a stark contrast in isolate breadth is evident between the2-mosaic immunogen cocktails and the single “standard strain”sequences (Fig. 6; see also Fig. S1 in the supplemental material).The contrast is consistent within both viruses and all four genestested, not only in the highly variable Env and Nef genes but also inthe relatively more conserved Pol and Gag. Since multiple vac-cine-induced epitope responses per gene appear to be correlatedwith protection against stringent heterologous challenge in ma-caques (55, 86), and the potential epitope overlap of any singlenatural sequence with any likely infective strain is low (72), opti-mized high-coverage immunogens such as mosaics are muchmore likely than single-sequence natural immunogens to haveprotective efficacy against HIV-1 exposure “in the wild.”
The importance of sufficient diversity in SIV heterologous-challenge studies must be emphasized: although SIVmac239 andSIVmac251 have been described by some authors as heterologous
Fischer et al.
13228 jvi.asm.org Journal of Virology
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from

challenge pairs, an SIVmac251 challenge of SIVmac239-vacci-nated animals (for examples, see references 14 and 31), or viceversa, cannot be considered heterologous. SIVmac239 andSIVmac251 originated in the same individual macaque (Mm251-79) and are far more closely related than circulating HIV-1 se-quences even within the same clade (Fig. 1); SIVmac239 is a cloneisolated after additional macaque passages of SIVmac251 (11, 30,63). For a heterologous challenge to resemble the distances ob-served in the HIV circulating population, more distant virusesmust be used as vaccine/challenge pairs (for instance, SIVmac239/SIVmac251 vaccination followed by SIVmacE660 challenge [7, 50,66, 92]). SIVmacE660 is phylogenetically distinct from theSIVmac251/239 lineage (Fig. 1), and the genetic distance betweenthe SIVmacE660 and SIVmac239/251 lineages is roughly compa-rable to the distances between HIV-1 strains circulating in thehuman population (93); the criteria of phylogenetic distinctnessand genetic distance should always be evaluated for proposed vac-cine/challenge pairs.
Using natural variation from SIV-infected sooty mangabeys(1) enables comparisons between polyvalent design strategies thatattempt to more comprehensively address diversity and other,more traditional, polyvalent or monovalent natural-strain im-munogens. Based on this premise, we gathered and sequencednovel SIVsmm strains, combined the results with existing data,and, using the full data from this augmented set, designed anSIVsmm polyvalent mosaic vaccine, which will be tested for effi-cacy in macaques (G.J.N., study in progress). Mosaic vaccineshave been shown to induce greater numbers of epitope responsesand enhanced cross-reactivity of responses (5, 71, 72) comparedto those of monovalent immunogens; the mosaic design presentedhere will enable us to test whether either or both of these improve-ments in immunological response may result in effective T cellresponses to pathogenic heterologous viral challenge.
Considerable thought has been applied to evaluating the dif-ferences between the macaque and human immune responses toimmunodeficiency viruses in the context of vaccine development(reviewed in reference 76). In contrast, we are here attempting tohighlight the potential importance of the viral component in as-sessing the applicability of the SIV/macaque model. The differ-ences between the immune responses of both macaque andhuman nonnatural hosts are of course significant, but while ma-caques’ immune responses to SIVsmm may indeed be more hu-man-like than those of mangabeys, the diversity against whichthey are challenged appears to be derived from a much differentset of selective pressures, and such differences may affect heterol-ogous challenge. For instance, as we discuss above, SIVsmm cap-sid (p27), Pol RT, and Env gp120 appear to have evolved underlow immunological pressure in sooty mangabeys, and they showreduced signatures of selection. The corresponding HIV-1 pro-teins, in contrast, have been subjected to intense immune pres-sure, and HIV-1 population diversity has adapted, to some degree,to the immunological diversity of its human host populations (67,73). Vaccine-induced CTL responses in macaques (e.g., to theSIVmac CM9 Gag epitope [92]) can be correlated with reducedviral load; furthermore, the overall number of Gag-directed CTLresponses is correlated with viral control (55, 86), and CTL re-sponses can control viral replication in the absence of neutralizingantibodies (87). However, the relative benefits of a Gag-directedresponse against SIV in macaques (e.g., versus Nef- or Env-di-rected responses) might not be paralleled in a human population:
whether particular benefits of particular responses to particularproteins observed in SIV/macaque heterologous-challenge mod-els will translate to HIV in humans remains an open question. Thisis because the sequence diversity in SIVsmm-derived heterolo-gous macaque challenges is not adapted to the macaque immunesystem, so even if the immune responses were qualitatively similarbetween macaques and humans, the viral diversity is not. Identicalconsiderations apply to neutralizing antibody responses, sinceSIVmac Env protein diversity has largely evolved in the absence ofstrong antibody responses.
In short, the origin of interstrain diversity in macaque-adaptedSIV strains is different from the origin of interclade and within-clade HIV diversity. HIV-1 diversifies under immune pressureand selection for rapid transmission; as we discuss above, SIVsmmappears to have diversified under reduced immune pressure, aswell as long-term selection for low pathogenicity (78). Through-out the spread of the HIV-1 pandemic, the initial low diversity ofsmall viral founder populations has expanded dramatically (bothwithin and outside Africa) via continuous passage in hosts withhighly active immune systems. Laboratory SIVmac variants orig-inated from sooty mangabey-adapted lineages, with a history ofreduced immune pressure, and they have not been continuouslypassaged in a host with strong immune responses. Therefore, evenif sequence diversity between two SIVsmm or SIVmac isolatesarithmetically approximates the diversity between two HIV-1 iso-lates, it may not be functionally equivalent in terms of inducedimmune responses, and the degree of heterology against which avaccine might be protective (i.e., the breadth of protection) maynot be easily predicted from a macaque model.
Nonhuman primate models are nevertheless an invaluable toolfor studying immune responses to HIV, and careful use of thevarious SIV/macaque models will advance the imperative goal of aprotective HIV vaccine. However, the differences in immunology,pathogenesis, and diversity (29, 85), as well as the distinctive evo-lutionary pressures in the SIVsmm and HIV-1 M groups discussedhere, should be considered when extrapolating from SIV/ma-caque experimental results to HIV/human vaccine applications.
ACKNOWLEDGMENTS
Many of the full-length SIVsmm sequences listed in Table 1 and in TableS1 in the supplemental material were generated by the late MatthiasKraus; other viral isolates (Table 2) were kindly provided by MartinePeeters and Preston Marx. We thank Michael Worobey and Paul Sharp foruseful comments on the manuscript.
This research was supported by the National Institutes of Health viathe following grants: AI-067854 (CHAVI) from the Division of AIDS,NIAID (W.F., B.T.K., G.M.S., B.H.H., M.L.S., and Y.L.), R01 AI-065325(C.A., I.P., and R.G.), and R37 AI-50529, R21 AI-087383, and P01 AI-088564 (G.M.S., B.H.H., M.L.S., and Y.L.), as well as by the Los AlamosNational Laboratory Directed Research and Development program (W.F.and B.T.K.), the Bill and Melinda Gates Foundation (G.M.S., B.H.H.,M.L.S., and Y.L.), and the intramural research program of the VaccineResearch Center, NIAID, NIH, through NIH/DOE interagency agree-ment NIH Y1-A1-8309 (W.F. and B.T.K.).
REFERENCES1. Apetrei C, et al. 2007. Virus subtype-specific features of natural simian
immunodeficiency virus SIVsmm infection in sooty mangabeys. J. Virol.81:7913–7923.
2. Apetrei C, et al. 2005. Molecular epidemiology of simian immunodefi-ciency virus SIVsm in U.S. primate centers unravels the origin of SIVmacand SIVstm. J. Virol. 79:8991–9005.
Evolutionary Comparisons of SIVsmm and HIV-1 Diversity
December 2012 Volume 86 Number 24 jvi.asm.org 13229
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from

3. Bar KJ, et al. 2012. Early low-titer neutralizing antibodies impede HIV-1replication and select for virus escape. PLoS Pathog. 8:e1002721. doi:10.1371/journal.ppat.1002721.
4. Barouch DH, et al. 2012. Vaccine protection against acquisition of neu-tralization-resistant SIV challenges in rhesus monkeys. Nature 482:89 –93.
5. Barouch DH, et al. 2010. Mosaic HIV-1 vaccines expand the breadth anddepth of cellular immune responses in rhesus monkeys. Nat. Med. 16:319 –323.
6. Barry AP, et al. 2007. Depletion of CD8� cells in sooty mangabey mon-keys naturally infected with simian immunodeficiency virus reveals lim-ited role for immune control of virus replication in a natural host species.J. Immunol. 178:8002– 8012.
7. Berry N, et al. 2011. Early potent protection against heterologousSIVsmE660 challenge following live attenuated SIV vaccination in Mau-ritian cynomolgus macaques. PLoS One 6:e23092. doi:10.1371/journal.pone.0023092.
8. Bosinger SE, Jacquelin B, Benecke A, Silvestri G, Müller-Trutwin M.2012. Systems biology of natural simian immunodeficiency virus infec-tions. Curr. Opin. HIV AIDS 7:71–78.
9. Brenchley JM, Paiardini M. 2011. Immunodeficiency lentiviral infectionsin natural and non-natural hosts. Blood 118:847– 854.
10. Brenchley JM, et al. 2004. CD4� T cell depletion during all stages of HIVdisease occurs predominantly in the gastrointestinal tract. J. Exp. Med.200:749 –759.
11. Daniel MD, et al. 1985. Isolation of T-cell tropic HTLV-III-like retrovirusfrom macaques. Science 228:1201–1204.
12. Delport W, Poon A, Frost SD, Kosakovsky Pond SL. 2011. Mixed EffectsModel of Evolution (MEME). http://www.datamonkey.org/help/meme.php.
13. Delport W, Poon AFY, Frost SDW, Kosakovsky Pond SL. 2010. Data-monkey 2010: a suite of phylogenetic analysis tools for evolutionary biol-ogy. Bioinformatics 26:2455–2457.
14. Dubie RA, et al. 2009. Co-immunization with IL-15 enhances cellularimmune responses induced by a vif-deleted simian immunodeficiencyvirus proviral DNA vaccine and confers partial protection against vaginalchallenge with SIVmac251. Virology 386:109 –121.
15. Dunham R, et al. 2006. The AIDS resistance of naturally SIV-infectedsooty mangabeys is independent of cellular immunity to the virus. Blood108:209 –217.
16. Estes JD, et al. 2008. Early resolution of acute immune activation andinduction of PD-1 in SIV-infected sooty mangabeys distinguishes non-pathogenic from pathogenic infection in rhesus macaques. J. Immunol.180:6798 – 6807.
17. Ferrari G, et al. 2011. Relationship between functional profile of HIV-1specific CD8 T cells and epitope variability with the selection of escapemutants in acute HIV-1 infection. PLoS Pathog. 7:e1001273. doi:10.1371/journal.ppat.1001273.
18. Fischer W, et al. 2010. Transmission of single HIV-1 genomes and dy-namics of early immune escape revealed by ultra-deep sequencing. PLoSOne 5:e12303. doi:10.1371/journal.pone.0012303.
19. Fischer W, et al. 2007. Polyvalent vaccines for optimal coverage of po-tential T-cell epitopes in global HIV-1 variants. Nat. Med. 13:100 –106.
20. Fukazawa Y, et al. 2012. Lymph node T cell responses predict the efficacyof live attenuated SIV vaccines. Nat. Med. [Epub ahead of print.] doi:10.1038/nm.2934.
21. Fultz PN, et al. 1990. Humoral response to SIV/SMM infection in ma-caque and mangabey monkeys. J. Acquir. Immune Defic. Syndr. 3:319 –329.
22. Gaufin T, et al. 2010. Experimental depletion of CD8� cells in acutelySIVagm-infected African green monkeys results in increased viral replica-tion. Retrovirology 7:42.
23. Gautam R, et al. 2007. In vitro characterization of primary SIVsmmisolates belonging to different lineages. In vitro growth on rhesus macaquecells is not predictive for in vivo replication in rhesus macaques. Virology362:257–270.
24. Giorgi JV, et al. 2002. Predictive value of immunologic and virologicmarkers after long or short duration of HIV-1 infection. J. Acquir. Im-mune Defic. Syndr. 29:346 –355.
25. Goonetilleke N, et al. 2009. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infec-tion. J. Exp. Med. 206:1253–1272.
26. Hansen SG, et al. 2011. Profound early control of highly pathogenic SIVby an effector memory T-cell vaccine. Nature 473:523–527.
27. Henn MR, et al. 2012. Whole genome deep sequencing of HIV-1 revealsthe impact of early minor variants upon immune recognition during acuteinfection. PLoS Pathog. 8:e1002529. doi:10.1371/journal.ppat.1002529.
28. Hirsch VM. 2004. What can natural infection of African monkeys withsimian immunodeficiency virus tell us about the pathogenesis of AIDS?AIDS Rev. 6:40 –53.
29. Hirsch VM, Johnson PR. 1994. Pathogenic diversity of simian immuno-deficiency viruses. Virus Res. 32:183–203.
30. Hunt RD, et al. 1983. Transmission of naturally occurring lymphoma inmacaque monkeys. Proc. Natl. Acad. Sci. U. S. A. 80:5085–5089.
31. Hutnick NA, et al. 2012. An optimized SIV DNA vaccine can serve as aboost for Ad5 and provide partial protection from a high-dose SIVmac251challenge. Vaccine 30:3202–3208.
32. Jin X, et al. 1999. Dramatic rise in plasma viremia after CD8(�) T celldepletion in simian immunodeficiency virus-infected macaques. J. Exp.Med. 189:991–998.
33. Junier T, Zdobnov EM. 2010. The Newick Utilities: high-throughputphylogenetic tree processing in the UNIX shell. Bioinformatics 26:1669 –1670.
34. Kaur A, et al. 2001. Emergence of cytotoxic T lymphocyte escape muta-tions in nonpathogenic simian immunodeficiency virus infection. Eur. J.Immunol. 31:3207–3217.
35. Kaur A, et al. 1998. Diverse host responses and outcomes followingsimian immunodeficiency virus SIVmac239 infection in sooty mangabeysand rhesus macaques. J. Virol. 72:9597–9611.
36. Kaur A, et al. 2000. Identification of multiple simian immunodeficiencyvirus (SIV)-specific CTL epitopes in sooty mangabeys with natural andexperimentally acquired SIV infection. J. Immunol. 164:934 –943.
37. Kawashima Y, et al. 2009. Adaptation of HIV-1 to human leukocyteantigen class I. Nature 458:641– 645.
38. Keele BF, et al. 2008. Identification and characterization of transmittedand early founder virus envelopes in primary HIV-1 infection. Proc. Natl.Acad. Sci. U. S. A. 105:7552–7557.
39. Keele BF, et al. 2009. Increased mortality and AIDS-like immunopathol-ogy in wild chimpanzees infected with SIVcpz. Nature 460:515–519.
40. Keele BF, et al. 2009. Low-dose rectal inoculation of rhesus macaques bySIVsmE660 or SIVmac251 recapitulates human mucosal infection byHIV-1. J. Exp. Med. 206:1117–1134.
41. Klatt NR, Silvestri G, Hirsch V. 2012. Nonpathogenic simian immuno-deficiency virus infections. Cold Spring Harb. Perspect. Med. 2:a007153.doi:10.1101/cshperspect.a007153.
42. Kong W-P, et al. 2009. Expanded breadth of the T-cell response to mosaichuman immunodeficiency virus type 1 envelope DNA vaccination. J. Vi-rol. 83:2201–2215.
43. Korber B, et al. 2000. Timing the ancestor of the HIV-1 pandemic strains.Science 288:1789 –1796.
44. Korber BT, Letvin NL, Haynes BF. 2009. T-cell vaccine strategies forhuman immunodeficiency virus, the virus with a thousand faces. J. Virol.83:8300 – 8314.
45. Korber BTM. 2000. HIV signature and sequence variation analysis, p55–72. In Rodrigo AG, Learn GH (ed), Computational analysis of HIVmolecular sequences. Kluwer Academic Publishers, Dordrecht, Nether-lands.
46. Korber BTM. 2000. SNAP: Synonymous Non-synonymous Analysis Pro-gram. http://www.hiv.lanl.gov/content/sequence/SNAP/SNAP.html.
47. Kosakovsky Pond SL, et al. 2006. Adaptation to different human popu-lations by HIV-1 revealed by codon-based analyses. PLoS Comput. Biol.2:e62. doi:10.1371/journal.pcbi.0020062.
48. Kosakovsky Pond SL, et al. 2011. A random effects branch-site model fordetecting episodic diversifying selection. Mol. Biol. Evol. 28:3033–3043.
49. Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SDW.2006. Automated phylogenetic detection of recombination using a geneticalgorithm. Mol. Biol. Evol. 23:1891–1901.
50. Lai L, et al. 2012. SIVmac239 MVA vaccine with and without a DNAprime, similar prevention of infection by a repeated dose SIVsmE660 chal-lenge despite different immune responses. Vaccine 30:1737–1745.
51. Leiherer A, Ludwig C, Wagner R. 2009. Uncoupling human immuno-deficiency virus type 1 Gag and Pol reading frames: role of the transframeprotein p6* in viral replication. J. Virol. 83:7210 –7220.
52. Li B, et al. 2010. Nonpathogenic simian immunodeficiency virus infec-tion of sooty mangabeys is not associated with high levels of autologousneutralizing antibodies. J. Virol. 84:6248 – 6253.
Fischer et al.
13230 jvi.asm.org Journal of Virology
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from

53. Li F, et al. 2006. Peptide selection for human immunodeficiency virustype 1 CTL-based vaccine evaluation. Vaccine 24:6893– 6904.
54. Ling B, et al. 2004. Classic AIDS in a sooty mangabey after an 18-yearnatural infection. J. Virol. 78:8902– 8908.
55. Liu J, et al. 2009. Immune control of an SIV challenge by a T-cell-basedvaccine in rhesus monkeys. Nature 457:87–91.
56. Lozano Reina J-M, et al. 2009. Gag p27-specific B- and T-cell responsesin simian immunodeficiency virus SIVagm-infected African green mon-keys. J. Virol. 83:2770 –2777.
57. Matano T, et al. 1998. Administration of an anti-cd8 monoclonal anti-body interferes with the clearance of chimeric simian/human immunode-ficiency virus during primary infections of rhesus macaques. J. Virol. 72:164 –169.
58. Mattapallil JJ, Smit-McBride Z, McChesney M, Dandekar S. 1998.Intestinal intraepithelial lymphocytes are primed for gamma interferonand MIP-1� expression and display antiviral cytotoxic activity despitesevere CD4(�) T-cell depletion in primary simian immunodeficiency vi-rus infection. J. Virol. 72:6421– 6429.
59. Meythaler M, et al. 2011. Early induction of polyfunctional simian im-munodeficiency virus (SIV)-specific T lymphocytes and rapid disappear-ance of SIV from lymph nodes of sooty mangabeys during primary infec-tion. J. Immunol. 186:5151–5161.
60. Miura T, et al. 2009. HLA-B57/B*5801 human immunodeficiency virustype 1 elite controllers select for rare Gag variants associated with reducedviral replication capacity and strong cytotoxic T-lymphocyte recognition.J. Virol. 83:2743–2755.
61. Moore PL, et al. 2009. Limited neutralizing antibody specificities driveneutralization escape in early HIV-1 subtype C infection. PLoS Pathog.5:e1000598. doi:10.1371/journal.ppat.1000598.
62. Murrell B, et al. 2012. Detecting individual sites subject to episodic diversi-fying selection. PLoS Genet. 8:e1002764. doi:10.1371/journal.pgen.1002764.
63. Naidu YM, et al. 1988. Characterization of infectious molecular clones ofsimian immunodeficiency virus (SIVmac) and human immunodeficiencyvirus type 2: persistent infection of rhesus monkeys with molecularlycloned SIVmac. J. Virol. 62:4691– 4696.
64. Pandrea I, Apetrei C. 2010. Where the wild things are: pathogenesis ofSIV infection in African nonhuman primate hosts. Curr. HIV/AIDS Rep.7:28 –36.
65. Pandrea I, Silvestri G, Apetrei C. 2009. AIDS in African nonhumanprimate hosts of SIVs: a new paradigm of SIV infection. Curr. HIV Res.7:57–72.
66. Reynolds MR, et al. 2008. Macaques vaccinated with live-attenuated SIVcontrol replication of heterologous virus. J. Exp. Med. 205:2537–2550.
67. Rousseau CM, et al. 2008. HLA class I-driven evolution of human im-munodeficiency virus type 1 subtype C proteome: immune escape andviral load. J. Virol. 82:6434 – 6446.
68. Salazar-Gonzalez JF, et al. 2008. Deciphering human immunodeficiencyvirus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 82:3952–3970.
69. Salazar-Gonzalez JF, et al. 2009. Genetic identity, biological phenotype,and evolutionary pathways of transmitted/founder viruses in acute andearly HIV-1 infection. J. Exp. Med. 206:1273–1289.
70. Santra S, et al. 2008. A centralized gene-based HIV-1 vaccine elicits broadcross-clade cellular immune responses in rhesus monkeys. Proc. Natl.Acad. Sci. U. S. A. 105:10489 –10494.
71. Santra S, et al. 2010. Mosaic vaccines elicit CD8� T lymphocyte re-sponses that confer enhanced immune coverage of diverse HIV strains inmonkeys. Nat. Med. 16:324 –328.
72. Santra S, et al. 2012. Breadth of cellular and humoral immune responseselicited in rhesus monkeys by multi-valent mosaic and consensus im-munogens. Virology 428:121–127.
73. Schellens IMM, et al. 2011. Loss of HIV-1-derived cytotoxic T lympho-cyte epitopes restricted by protective HLA-B alleles during the HIV-1 ep-idemic. AIDS 25:1691–1700.
74. Schindler M, et al. 2006. Nef-mediated suppression of T cell activationwas lost in a lentiviral lineage that gave rise to HIV-1. Cell 125:1055–1067.
75. Schmitz JE, et al. 1999. Control of viremia in simian immunodeficiencyvirus infection by CD8� lymphocytes. Science 283:857– 860.
76. Shedlock DJ, Silvestri G, Weiner DB. 2009. Monkeying around with HIVvaccines: using rhesus macaques to define ‘gatekeepers’ for clinical trials.Nat. Rev. Immunol. 9:717–728.
77. Silvestri G, et al. 2005. Divergent host responses during primary simianimmunodeficiency virus SIVsm infection of natural sooty mangabey andnonnatural rhesus macaque hosts. J. Virol. 79:4043– 4054.
78. Silvestri G, Paiardini M, Pandrea I, Lederman MM, Sodora DL. 2007.Understanding the benign nature of SIV infection in natural hosts. J. Clin.Invest. 117:3148 –3154.
79. Steckbeck JD, Craigo JK, Barnes CO, Montelaro RC. 2011. Highlyconserved structural properties of the C-terminal tail of HIV-1 gp41 pro-tein despite substantial sequence variation among diverse clades: implica-tions for functions in viral replication. J. Biol. Chem. 286:27156 –27166.
80. Streeck H, et al. 2007. Recognition of a defined region within p24 gag byCD8� T cells during primary human immunodeficiency virus type 1 in-fection in individuals expressing protective HLA class I alleles. J. Virol.81:7725–7731.
81. Sukumaran J, Holder MT. 2010. DendroPy: a Python library for phylo-genetic computing. Bioinformatics 26:1569 –1571.
82. VandeWoude S, Apetrei C. 2006. Going wild: lessons from naturallyoccurring T-lymphotropic lentiviruses. Clin. Microbiol. Rev. 19:728 –762.
83. Veazey RS, et al. 1998. Gastrointestinal tract as a major site of CD4� Tcell depletion and viral replication in SIV infection. Science 280:427– 431.
84. Wang Z, Metcalf B, Ribeiro RM, McClure H, Kaur A. 2006. Th-1-type cytotoxic CD8� T-lymphocyte responses to simian immunode-ficiency virus (SIV) are a consistent feature of natural SIV infection insooty mangabeys. J. Virol. 80:2771–2783.
85. Watkins DI, Burton DR, Kallas EG, Moore JP, Koff WC. 2008. Non-human primate models and the failure of the Merck HIV-1 vaccine inhumans. Nat. Med. 14:617– 621.
86. Wilson NA, et al. 2009. Vaccine-induced cellular responses control sim-ian immunodeficiency virus replication after heterologous challenge. J.Virol. 83:6508 – 6521.
87. Wilson NA, et al. 2006. Vaccine-induced cellular immune responsesreduce plasma viral concentrations after repeated low-dose challenge withpathogenic simian immunodeficiency virus SIVmac239. J. Virol. 80:5875–5885.
88. Wood N, et al. 2009. HIV evolution in early infection: selection pressures,patterns of insertion and deletion, and the impact of APOBEC. PLoS Pat-hog. 5:e1000414. doi:10.1371/journal.ppat.1000414.
89. Worobey M, et al. 2008. Direct evidence of extensive diversity of HIV-1 inKinshasa by 1960. Nature 455:661– 664.
90. Worobey M, et al. 2010. Island biogeography reveals the deep history ofSIV. Science 329:1487.
91. Wu X, et al. 2012. Selection pressure on HIV-1 envelope by broadlyneutralizing antibodies to the conserved CD4-binding site. J. Virol. 86:5844 –5856.
92. Yamamoto T, et al. 2012. Virus inhibition activity of effector memoryCD8(�) T cells determines simian immunodeficiency virus load in vacci-nated monkeys after vaccine breakthrough infection. J. Virol. 86:5877–5884.
93. Yeh WW, et al. 2009. Partial protection of simian immunodeficiencyvirus (SIV)-infected rhesus monkeys against superinfection with a heter-ologous SIV isolate. J. Virol. 83:2686 –2696.
94. Zhou T, et al. 2010. Structural basis for broad and potent neutralizationof HIV-1 by antibody VRC01. Science 329:811– 817.
95. Zwickl DJ. 2006. Genetic algorithm approaches for the phylogenetic anal-ysis of large biological sequence datasets under the maximum likelihoodcriterion. Ph.D. thesis.The University of Texas at Austin, Austin, TX.
Evolutionary Comparisons of SIVsmm and HIV-1 Diversity
December 2012 Volume 86 Number 24 jvi.asm.org 13231
on Novem
ber 19, 2012 by LAN
L Research Library
http://jvi.asm.org/
Dow
nloaded from