dietary n-6 and n-3 polyunsaturated fatty acids and colorectal carcinogenesis: results from cultured...
TRANSCRIPT

Dietary n-6 and n-3 polyunsaturated fatty acids and colorectalcarcinogenesis: results from cultured colon cells, animal models and
human studies�
Yvonne E.M. Dommels a,b,*, Gerrit M. Alink b, Peter J. van Bladeren c,Ben van Ommen c
a WUR/TNO Centre for Food Toxicology, The Netherlandsb Toxicology Group, Division of Toxicology, Wageningen University, Tuinlaan 5, PO Box 8000, 6700 EA Wageningen, The Netherlands
c Toxicology Division, TNO Nutrition and Food Research, Department of Explanatory Toxicology, Utrechtseweg 48, PO Box 360, 3700 AJ Zeist, The
Netherlands
Received 19 October 2001; received in revised form 18 December 2001; accepted 11 January 2002
Abstract
During the past few decades, many studies have been conducted to evaluate the effects of n-6 and n-3 polyunsaturated fatty acids
(PUFAs) on colorectal carcinogenesis. This report provides a brief overview of the recent studies that have been performed in
cultured colon cells, animal models as well as of the population-based and short-term biomarker studies with humans. No
differential effect between n-6 and n-3 PUFAs has been observed in vitro. Results from animal models indicate that n-6 PUFAs have
a tumor enhancing effect, predominantly during the post-initiation phase. n-3 PUFAs may protect against colorectal carcinogenesis
during both the initiation and post-initiation phase. Population-based human studies show little or no associations between n-6 or n-
3 PUFA intake and colorectal cancer. Short-term biomarker studies in humans suggest though that fish oil (FO) supplementation
with high amounts of n-3 PUFAs may protect against colorectal carcinogenesis and that n-6 PUFA supplementation may increase
the risk. # 2002 Elsevier Science B.V. All rights reserved.
Keywords: Colorectal carcinogenesis; Dietary n-6 and n-3 fatty acids; Human colorectal carcinoma cell lines; Animal models; Biomarkers;
Epidemiology
1. Introduction
Colorectal cancer is one of the most common causes
of cancer deaths in the industrialized Western countries.
It is the most prevalent cancer form for men after lung-
and prostate cancer. For females, colorectal cancer is the
second form after breast cancer (Woutersen et al. , 1999).
In the Netherlands the colon cancer incidence is 8600
new cases per year (Visser et al., 2001). In 1998, 4400
people died of colorectal cancer, which is around 12% of
Abbreviations: AA, arachidonic acid (20:4n-6); ACF, aberrant crypt foci; AgNOR, silver-stained nucleolar organizer region protein; AIN,
American Institute of Nutrition; ALA, a-linolenic acid (18:3n-3); AOM, azoxymethane; APC, adenomatous polyposis coli; BHT, butylated hydroxy
toluene; CO, corn oil; COX, cyclooxygenase; COX-1, cyclooxygenase 1; COX-2, cyclooxygenase 2; DAG, diacylglycerol; DBA, Dolichos biflorus
agglutinin; DHA, docosahexaenoic acid (22:6n-3); EPA, eicosapentaenoic acid (20:5n-3); FAP, familial adenomatous polyposis; FO, fish oil; FPTase,
farnesyl protein transferase; GJIC, gap junctional intercellular communication; GLA, g-linolenic acid (18:3n-6); HFCO, high-fat corn oil; HFFO,
high-fat fish oil; HFML, high-fat mixed lipids; HNPCC, Hereditary Non-Polyposis Colorectal Cancer; IM, indomethacin; LA, linoleic acid (18:2n-
6); LFCO, low-fat corn oil; MDA, malondialdehyde; MO, menhaden oil; O, olive oil; OA, oleic acid (18:1n-9); ODC, ornithine decarboxylase; P,
perilla oil (18:3n-3); PGE2, prostaglandin E2; PGE3, prostaglandin E3; PI-PLC, phosphatidylinositol-specific phospholipase C; PKC, protein kinase
C; PLA2, phospholipase A2; PUFAs, polyunsaturated fatty acids; S, safflower oil (18:2n-6); TPK, tyrosine protein kinase.�
PII of original article: S 1 3 8 2 - 6 6 8 9 ( 0 2 ) 0 0 0 0 6 - 6
* Corresponding author. Tel.: �/31-317-484357; fax: �/31-317-484931
E-mail address: [email protected] (Y.E.M. Dommels).
Environmental Toxicology and Pharmacology 12 (2002) 233�/244
www.elsevier.com/locate/etap
1382-6689/02/$ - see front matter # 2002 Elsevier Science B.V. All rights reserved.
PII: S 1 3 8 2 - 6 6 8 9 ( 0 2 ) 0 0 0 9 5 - 9

the total cancer deaths (Health Council of the Nether-
lands, 2001). Many factors can be responsible for the
development of colon cancer. Genetic predisposition is
considered as an important risk factor. Two groups ofgenetic predisposition are Hereditary Non-Polyposis
Colorectal Cancer (HNPCC) and Familial Adenoma-
tous Polyposis (FAP). HNPCC is a genetic disease,
which is characterized by an early age of onset and
mutational inactivation of mismatch repair genes.
Patients with FAP may develop hundreds of polyps,
also at early age, due to an inactivation mutation of the
Apc gene (Kinzler and Vogelstein, 1998; Roncucci et al.,2000).
Besides genetic predisposition, also diet is an impor-
tant risk factor for colon cancer. Already in the early
1980s Doll and Peto (1981) estimated that 90% of the
risk of colorectal cancer could be attributed to environ-
mental factors, mostly dietary factors. Accumulating
evidence suggests an association between a high fat
intake and increased risk of colorectal cancer (Doll andPeto, 1981; Woutersen et al., 1999). Moreover, epide-
miological and experimental studies do provide much
evidence that not only the amount of fat consumed, but
also the type of fat is very important.
This mini-review will provide a brief overview of the
current knowledge on the differential effects between n-6
and n-3 polyunsaturated fatty acids (PUFAs) on color-
ectal carcinogenesis as determined in cultured coloncells, animal models and human studies.
2. Nomenclature, dietary sources and metabolism of n-6
and n-3 PUFAs
N-6 and n-3 fatty acids are PUFAs with two or more
double bonds in the carbon atom chain. N-6 and n-3
fatty acids are named after the position of the firstdouble bond from the methyl end of the molecule. For
example, linoleic acid (LA, 18:2n-6) has 18 C-atoms and
two double bonds, with the first double bond at the 6th
carbon atom counted from the methyl end. Linoleic acid
is the parent compound of the n-6 family, whereas a-
linolenic acid (ALA, 18:3n-3) is the parent compound of
the n-3 fatty acid family. These two PUFA families are
considered as essential and must be derived from the diet(Gurr, 1996). Linoleic acid is mostly found in vegetable
seeds and oils such as safflower, soybeans, corn and
sunflower oil. Perilla oil from the Asian beefsteak plant
(Perilla frutescens), linseed oil, rapeseed, walnuts and
blackcurrant oil are rich in a-linolenic acid. a-Linolenic
acid is also present in dark green leafy plants (Bartsch et
al., 1999).
Linoleic acid and a-linolenic acid can be metabolizedto more polyunsaturated fatty acids by a combination of
desaturation and elongation enzymes (Gurr, 1996).
Desaturation enzymes introduce a new double bond in
the carbon chain and elongase enzymes introduce two
new C-atoms. Arachidonic acid (AA, 20:4n-6) is the
major long-chain n-6 PUFA. Eicosapentaenoic acid
(EPA, 20:5n-3) and docosahexaenoic acid (DHA,
22:6n-3) are the major PUFAs of the n-3 family. Marine
fish such as salmon, tuna, herring, mackerel and
anchovy are rich sources of EPA and DHA (Rose and
Connolly, 1999). The marine food chain is based on n-3
fatty acids, which are present in plankton and algae on
which fish feed (Bartsch et al., 1999). Dietary LA is
considered to be the main source of tissue AA, although
lean meats and meat fat are direct dietary AA sources
(Rose and Connolly, 1999). Production of AA and EPA
from LA and ALA, respectively, is thought to proceed
preferentially by D6-desaturation followed by a two-
carbon atom chain elongation and D5-desaturation
(Grammatikos et al., 1994) (see Fig. 1). Metabolism of
n-6 and n-3 fatty acids follows a series of competitive
elongation and desaturation steps, which is limited by
the activity of D6-desaturase, with the n-3 fatty acids
having greater affinities for the enzyme. In consequence,
increasing dietary intake of n-3 fatty acids reduces the
desaturation of LA and so, the production of AA (Rose
and Connolly, 1999).
AA and EPA also show competition for cyclooxy-
genase (COX) enzyme activities. Cyclooxygenase 1 and -
2 (COX-1, COX-2, respectively) introduce two atoms of
oxygen into AA to form the hydroperoxy endoperoxide,
PGG2, which is then reduced by the endoperoxidase
moiety of the enzyme to the hydroxy endoperoxide
PGH2. PGH2 can be a substrate for several enzymes
including PGE synthase, which forms prostaglandin E2
(PGE2) (Fischer, 1997) (see Fig. 2). EPA is a precursor
of the prostaglandins of the 3-series, with three double
bonds. The n-3 fatty acid is converted by the same COX
enzyme to the 3-series endoperoxide PGH3, by way of
PGG3, which can be further metabolized to PGE3 (Rose
and Connolly, 1999; see Fig. 2). EPA supplementation
can thus lead to competitive inhibition of arachidonic
acid metabolism as well as the production of metabolites
such as PGE3. These metabolites are biologically less
active than the corresponding arachidonic acid metabo-
lites like PGE2 (Fischer, 1997). AA and EPA can besides
the cyclooxygenase pathway, also be metabolized by the
Fig. 1. Metabolism of n-6 and n-3 PUFAs.
Erratum234
lipoxygenase pathway and the cytochrome P450 mono-
oxygenase pathway (Krause and Dubois, 2000) to
prostaglandins, thromboxanes and leukotrienes, collec-
tively referred to as eicosanoids. In general, the potency
of eicosanoids derived from the n-3 PUFAs is less than
of those derived from the n-6 PUFAs (Gurr, 1996).
3. In vitro effects of n-6 and n-3 PUFAs on human
colorectal carcinoma cell lines
Several in vitro studies have been carried out to
investigate the effects of essential fatty acids on various
cells in culture (Grammatikos et al., 1994; Jiang et al.,
1998).
In the present review we will focus on the effects of
individual n-6 and n-3 PUFAs on human colorectal
carcinoma cell lines. Table 1 gives a summary of some of
the recent studies.
Biomarkers of carcinogenesis, which are mostly
determined in vitro are cell proliferation and apoptosis.
From the responses of the different cell lines, it can be
concluded that there is no obvious differential effect
between n-6 and n-3 PUFAs on colon cancer cell lines.
In most studies, LA (18:2n-6) and ALA (18:3n-3)
showed no effect on cell proliferation (Awad et al.,
1995; Chen and Istfan, 2000; Collett et al., 2001; Tsai et
al., 1998). Other PUFAs with more double bonds, such
as AA (20:4n-6), EPA (20:5n-3) and DHA (22:6n-3)
caused an overall decrease in cell proliferation (Men-
geaud et al., 1992; Tsai et al., 1998; Clarke et al., 1999;
Chen and Istfan, 2000; Collett et al., 2001; Kim et al.,
2000; Palozza et al., 2000) or increase in apoptosis
(Clarke et al., 1999; Chen and Istfan, 2000). These fatty
acids appear to act directly, because, indomethacin
(IM), an inhibitor of prostaglandin synthesis, did not
Fig. 2. Formation of prostaglandin E from arachidonic acid (AA;
20:4n-6) and eicosapentaenoic acid (EPA; 20:5n-3).
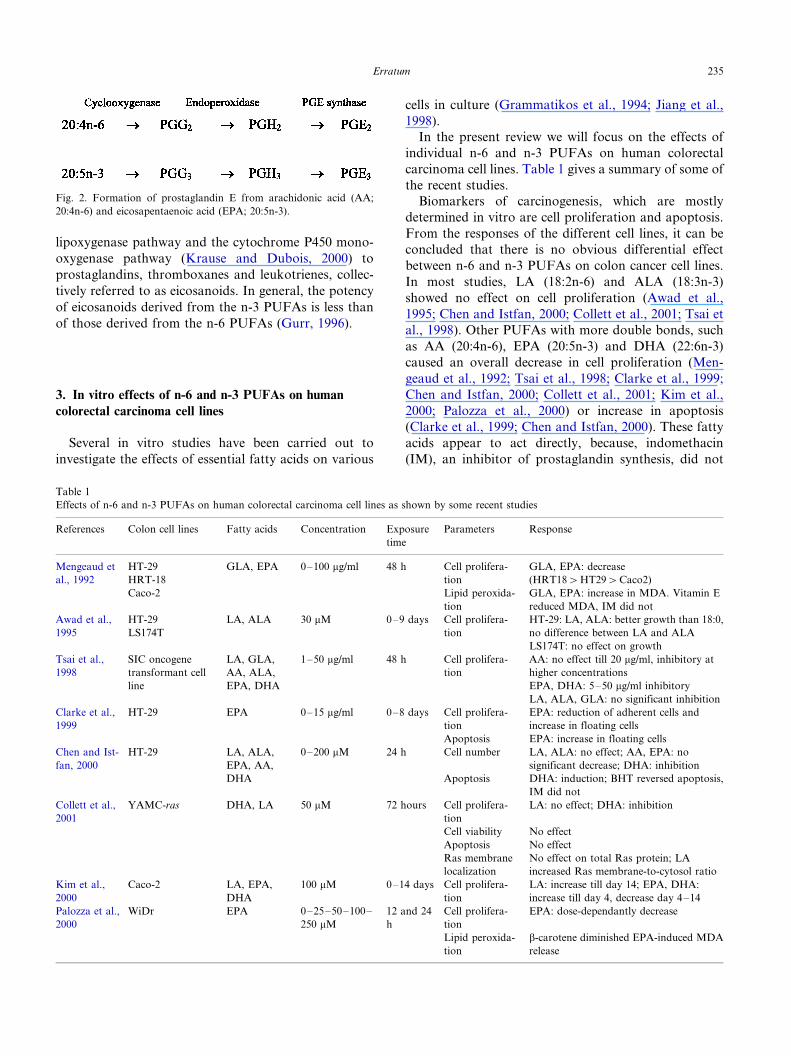
Table 1
Effects of n-6 and n-3 PUFAs on human colorectal carcinoma cell lines as shown by some recent studies
References Colon cell lines Fatty acids Concentration Exposure
time
Parameters Response
Mengeaud et
al., 1992
HT-29
HRT-18
GLA, EPA 0�/100 mg/ml 48 h Cell prolifera-
tion
GLA, EPA: decrease
(HRT18�HT29�Caco2)
Caco-2 Lipid peroxida-
tion
GLA, EPA: increase in MDA. Vitamin E
reduced MDA, IM did not
Awad et al.,
1995
HT-29
LS174T
LA, ALA 30 mM 0�/9 days Cell prolifera-
tion
HT-29: LA, ALA: better growth than 18:0,
no difference between LA and ALA
LS174T: no effect on growth
Tsai et al.,
1998
SIC oncogene
transformant cell
line
LA, GLA,
AA, ALA,
EPA, DHA
1�/50 mg/ml 48 h Cell prolifera-
tion
AA: no effect till 20 mg/ml, inhibitory at
higher concentrations
EPA, DHA: 5�/50 mg/ml inhibitory
LA, ALA, GLA: no significant inhibition
Clarke et al.,
1999
HT-29 EPA 0�/15 mg/ml 0�/8 days Cell prolifera-
tion
EPA: reduction of adherent cells and
increase in floating cells
Apoptosis EPA: increase in floating cells
Chen and Ist-
fan, 2000
HT-29 LA, ALA,
EPA, AA,
DHA
0�/200 mM 24 h Cell number
Apoptosis
LA, ALA: no effect; AA, EPA: no
significant decrease; DHA: inhibition
DHA: induction; BHT reversed apoptosis,
IM did not
Collett et al.,
2001
YAMC-ras DHA, LA 50 mM 72 hours Cell prolifera-
tion
LA: no effect; DHA: inhibition
Cell viability No effect
Apoptosis No effect
Ras membrane
localization
No effect on total Ras protein; LA
increased Ras membrane-to-cytosol ratio
Kim et al.,
2000
Caco-2 LA, EPA,
DHA
100 mM 0�/14 days Cell prolifera-
tion
LA: increase till day 14; EPA, DHA:
increase till day 4, decrease day 4�/14
Palozza et al.,
2000
WiDr EPA 0�/25�/50�/100�/
250 mM
12 and 24
h
Cell prolifera-
tion
EPA: dose-dependantly decrease
Lipid peroxida-
tion
b-carotene diminished EPA-induced MDA
release
Erratum 235

modify the effects. The decrease in cell proliferation was,
however, highly related to lipid peroxidation, as anti-
oxidants such as vitamin E (Mengeaud et al., 1992),
BHT (Chen and Istfan, 2000) and b-carotene (Palozza etal., 2000) diminished the effects.
To better compare the differential effects between n-6
and n-3 PUFAs, the effects of fatty acids with compar-
able chain lengths such as LA versus ALA and AA
versus EPA, should be determined together in one colon
cell line. Only Awad et al. (1995), Tsai et al. (1998) and
Chen et al. (2000) performed these kinds of experiments.
From this limited number of studies it can, however,also be concluded that there is no differential effect
between n-6 and n-3 PUFAs in colon cancer cell lines.
We investigated the effects of n-6 and n-3 PUFAs on
gap junctional intercellular communication (GJIC),
another biomarker of carcinogenesis, in human colon
adenocarcinoma Caco-2 cells. Also no differential
effects between LA versus ALA and AA versus EPA
were observed. All fatty acids inhibited GJIC after long-term incubation (Dommels et al., 2002).
4. Effects of n-6 and n-3 PUFAs in animal models of
colorectal carcinogenesis
Many in vivo studies have been performed to evaluate
the effects of n-6 and n-3 PUFAs on colorectal
carcinogenesis. In 1992, Reddy reviewed the knowledgeat that time on the relationship between different types
of dietary fat and colon carcinogenesis in laboratory
animal models with emphasis on n-3 fatty acids. He
concluded that lack of a colon tumor enhancing effect of
dietary fish oil (FO) was observed during both the
initiation and postinitiation phases. This was mediated
by an effect of FO on ornithine decarboxylase activity
(ODC), colonic secondary bile acids and/or prostaglan-din synthesis. Klurfeld and Bull wrote a review article in
1997 about fatty acids and colon cancer in experimental
animal models. In the present mini-review we will give
the state of the art knowledge on n-6 and n-3 PUFAs on
colorectal carcinogenesis in animal models till 2002.
In Table 2, several recent experimental animal studies
on the effects of n-6 and n-3 PUFAs on colorectal
carcinogenesis are summarized. From these studies itbecame indeed clear that the colon tumor enhancing
effect of corn oil (CO) is most prevalent during the
postinitiation phase and that the inhibiting effect of FO
is observed during both the initiation and postinitiation
phase (Reddy et al., 1991). Not only FO with a high
amount of EPA and DHA but also perilla oil (Onogi et
al., 1996) with high levels of ALA, and individual n-3
fatty acids such as DHA (Takahashi et al., 1997; Oshimaet al., 1995) can inhibit colorectal carcinogenesis.
Reddy and Sugie (1988) performed a study to
investigate the modulating effects of varying levels of
n-3 and n-6 fatty acids during the promotional phase of
colon carcinogenesis in order to determine the optimum
dietary levels of these fatty acids that elicit maximum
inhibition of colon tumors. Inhibition of colon tumorincidence by decreasing the level of dietary CO in the
high fat diets or increasing the ratio of n-3 to n-6 fatty
acids in the diet was, however, not dose-dependant. A
23.5% high- fat diet with only 5.9% Menhaden FO and
17.6% CO had no enhancing effect on the incidence of
total colon tumors and colon adenocarcinomas as
compared with a low-fat 5% CO diet, whereas the
incidence of total colon tumors and adenocarcinomaswas increased in animals fed a high-fat diet containing
23.5% CO. These results indicate that high fat intake is a
necessary but not a sufficient condition for colon tumor
promotion, and that the relative proportions of n-3 and
n-6 fatty acids in the diet are determinants of the high
fat effect (Reddy and Sugie, 1988).
Besides chemically-induced colon tumors, effects of n-
3 and n-6 PUFAs have been investigated on thedevelopment of aberrant crypt foci (ACF) and in
transgenic mice. Oshima et al. (1995) have investigated
the effects of DHA on mouse intestinal polyposis using
Apc gene knockout mice. Dietary DHA decreased
tumor number in female, but not in male mice. This
was the first study that demonstrated that DHA also
inhibits intestinal polyposis induced by an Apc muta-
tion. According to the authors, this may open apossibility for chemopreventive intervention of FAP
by dietary supplementation with DHA. In addition,
Petrik et al. (2000) were the first to report that dietary
EPA also has anti-tumorigenic properties in the
ApcMin/� mice.
ACF have been used as intermediate biomarkers of
colon cancer development in animal studies (Roncucci
et al., 2000). Aberrant crypts are crypts, which appear tobe larger, thicker and darker than normal crypts and
cluster in aggregates, foci. However, it is not definitely
proven that ACFs are true precursors of colon tumors
(Klurfeld and Bull, 1997), because, no correlation
between the number of ACF and the incidence of
carcinomas in rats was found or it was found only in
the left colon (Roncucci et al., 2000). On the other hand,
crypt multiplicity in ACF, i.e. the number of crypts perfocus, seems to be a predictor of tumor incidence. The
explanation of this apparent discrepancy is that most
ACF regress and that only larger foci progress towards
cancer (Roncucci et al., 2000). The prognostic value of
ACF is, however, still a matter of debate. Furihata et al.
(2001) did find a positive correlation between the
incidence of ACF and the incidence of tumors in F344
rats.There are relatively few studies on effects of n-6 and
n-3 PUFAs on ACF formation. Those that have been
performed show a decrease in total ACF incidence and
multiplicity by perilla oil (Onogi et al., 1996), DHA
Erratum236
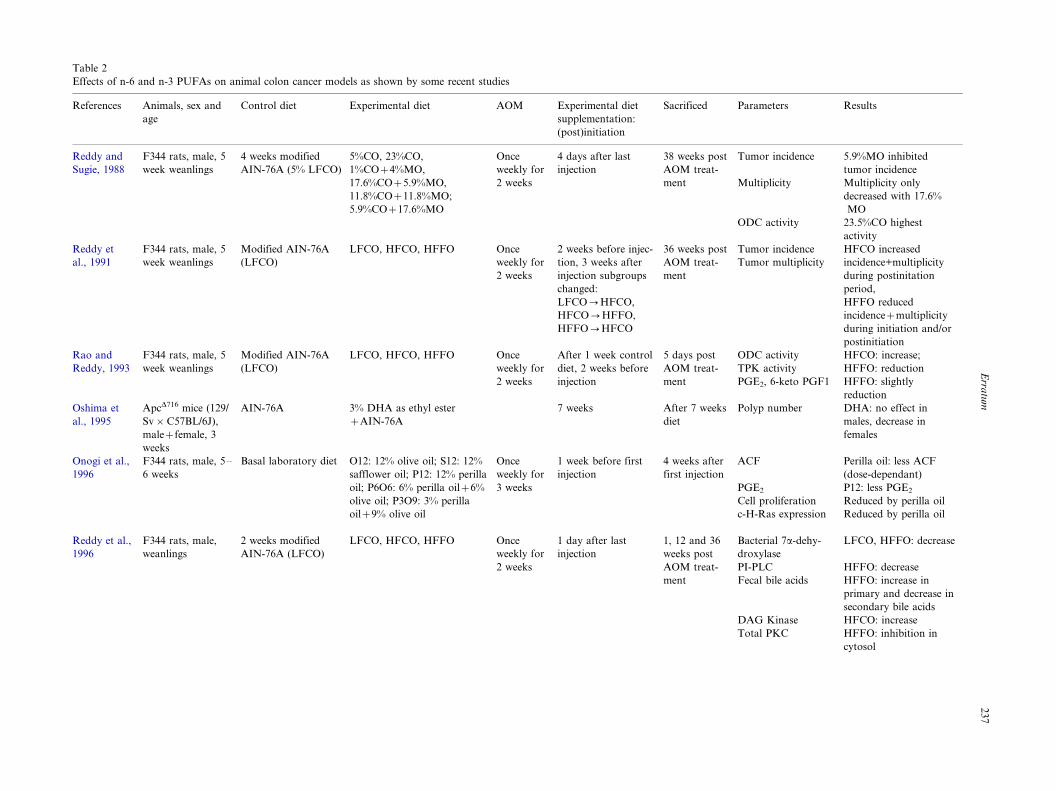
Table 2
Effects of n-6 and n-3 PUFAs on animal colon cancer models as shown by some recent studies
References Animals, sex and
age
Control diet Experimental diet AOM Experimental diet
supplementation:
(post)initiation
Sacrificed Parameters Results
Reddy and
Sugie, 1988
F344 rats, male, 5
week weanlings
4 weeks modified
AIN-76A (5% LFCO)
5%CO, 23%CO,
1%CO�4%MO,
17.6%CO�5.9%MO,
11.8%CO�11.8%MO;
5.9%CO�17.6%MO
Once
weekly for
2 weeks
4 days after last
injection
38 weeks post
AOM treat-
ment
Tumor incidence
Multiplicity
ODC activity
5.9%MO inhibited
tumor incidence
Multiplicity only
decreased with 17.6%
MO
23.5%CO highest
activity
Reddy et
al., 1991
F344 rats, male, 5
week weanlings
Modified AIN-76A
(LFCO)
LFCO, HFCO, HFFO Once
weekly for
2 weeks
2 weeks before injec-
tion, 3 weeks after
injection subgroups
changed:
LFCO0HFCO,
HFCO0HFFO,
HFFO0HFCO
36 weeks post
AOM treat-
ment
Tumor incidence
Tumor multiplicity
HFCO increased
incidence+multiplicity
during postinitation
period,
HFFO reduced
incidence�multiplicity
during initiation and/or
postinitiation
Rao and
Reddy, 1993
F344 rats, male, 5
week weanlings
Modified AIN-76A
(LFCO)
LFCO, HFCO, HFFO Once
weekly for
2 weeks
After 1 week control
diet, 2 weeks before
injection
5 days post
AOM treat-
ment
ODC activity
TPK activity
PGE2, 6-keto PGF1
HFCO: increase;
HFFO: reduction
HFFO: slightly
reduction
Oshima et
al., 1995
ApcD716 mice (129/
Sv�C57BL/6J),
male�female, 3
weeks
AIN-76A 3% DHA as ethyl ester
�AIN-76A
7 weeks After 7 weeks
diet
Polyp number DHA: no effect in
males, decrease in
females
Onogi et al.,
1996
F344 rats, male, 5�/
6 weeks
Basal laboratory diet O12: 12% olive oil; S12: 12%
safflower oil; P12: 12% perilla
oil; P6O6: 6% perilla oil�6%
olive oil; P3O9: 3% perilla
oil�9% olive oil
Once
weekly for
3 weeks
1 week before first
injection
4 weeks after
first injection
ACF
PGE2
Cell proliferation
c-H-Ras expression
Perilla oil: less ACF
(dose-dependant)
P12: less PGE2
Reduced by perilla oil
Reduced by perilla oil
Reddy et al.,
1996
F344 rats, male,
weanlings
2 weeks modified
AIN-76A (LFCO)
LFCO, HFCO, HFFO Once
weekly for
2 weeks
1 day after last
injection
1, 12 and 36
weeks post
AOM treat-
ment
Bacterial 7a-dehy-
droxylase
PI-PLC
Fecal bile acids
LFCO, HFFO: decrease
HFFO: decrease
HFFO: increase in
primary and decrease in
secondary bile acids
DAG Kinase HFCO: increase
Total PKC HFFO: inhibition in
cytosol
Erra
tum
23
7
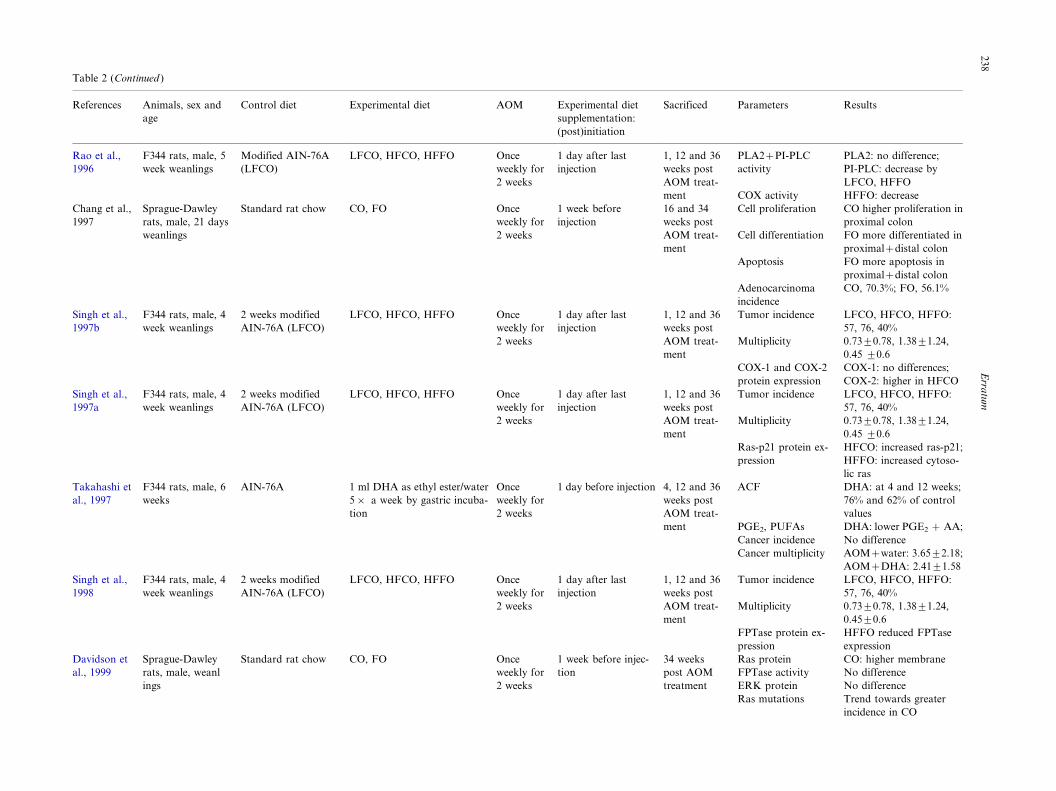
Table 2 (Continued )
References Animals, sex and
age
Control diet Experimental diet AOM Experimental diet
supplementation:
(post)initiation
Sacrificed Parameters Results
Rao et al.,
1996
Chang et al.,
1997
F344 rats, male, 5
week weanlings
Sprague-Dawley
rats, male, 21 days
weanlings
Modified AIN-76A
(LFCO)
Standard rat chow
LFCO, HFCO, HFFO
CO, FO
Once
weekly for
2 weeks
Once
weekly for
2 weeks
1 day after last
injection
1 week before
injection
1, 12 and 36
weeks post
AOM treat-
ment
16 and 34
weeks post
AOM treat-
ment
PLA2�PI-PLC
activity
COX activity
Cell proliferation
Cell differentiation
Apoptosis
PLA2: no difference;
PI-PLC: decrease by
LFCO, HFFO
HFFO: decrease
CO higher proliferation in
proximal colon
FO more differentiated in
proximal�distal colon
FO more apoptosis in
proximal�distal colon
Adenocarcinoma
incidence
CO, 70.3%; FO, 56.1%
Singh et al.,
1997b
F344 rats, male, 4
week weanlings
2 weeks modified
AIN-76A (LFCO)
LFCO, HFCO, HFFO Once
weekly for
2 weeks
1 day after last
injection
1, 12 and 36
weeks post
AOM treat-
ment
Tumor incidence
Multiplicity
COX-1 and COX-2
protein expression
LFCO, HFCO, HFFO:
57, 76, 40%
0.7390.78, 1.3891.24,
0.45 90.6
COX-1: no differences;
COX-2: higher in HFCO
Singh et al.,
1997a
F344 rats, male, 4
week weanlings
2 weeks modified
AIN-76A (LFCO)
LFCO, HFCO, HFFO Once
weekly for
2 weeks
1 day after last
injection
1, 12 and 36
weeks post
AOM treat-
ment
Tumor incidence
Multiplicity
Ras-p21 protein ex-
pression
LFCO, HFCO, HFFO:
57, 76, 40%
0.7390.78, 1.3891.24,
0.45 90.6
HFCO: increased ras-p21;
HFFO: increased cytoso-
lic ras
Takahashi et
al., 1997
F344 rats, male, 6
weeks
AIN-76A 1 ml DHA as ethyl ester/water
5� a week by gastric incuba-
tion
Once
weekly for
2 weeks
1 day before injection 4, 12 and 36
weeks post
AOM treat-
ment
ACF
PGE2, PUFAs
Cancer incidence
Cancer multiplicity
DHA: at 4 and 12 weeks;
76% and 62% of control
values
DHA: lower PGE2 � AA;
No difference
AOM�water: 3.6592.18;
AOM�DHA: 2.4191.58
Singh et al.,
1998
F344 rats, male, 4
week weanlings
2 weeks modified
AIN-76A (LFCO)
LFCO, HFCO, HFFO Once
weekly for
2 weeks
1 day after last
injection
1, 12 and 36
weeks post
AOM treat-
ment
Tumor incidence
Multiplicity
LFCO, HFCO, HFFO:
57, 76, 40%
0.7390.78, 1.3891.24,
0.4590.6
FPTase protein ex-
pression
HFFO reduced FPTase
expression
Davidson et
al., 1999
Sprague-Dawley
rats, male, weanl
ings
Standard rat chow CO, FO Once
weekly for
2 weeks
1 week before injec-
tion
34 weeks
post AOM
treatment
Ras protein
FPTase activity
ERK protein
Ras mutations
CO: higher membrane
No difference
No difference
Trend towards greater
incidence in CO
Erra
tum
23
8
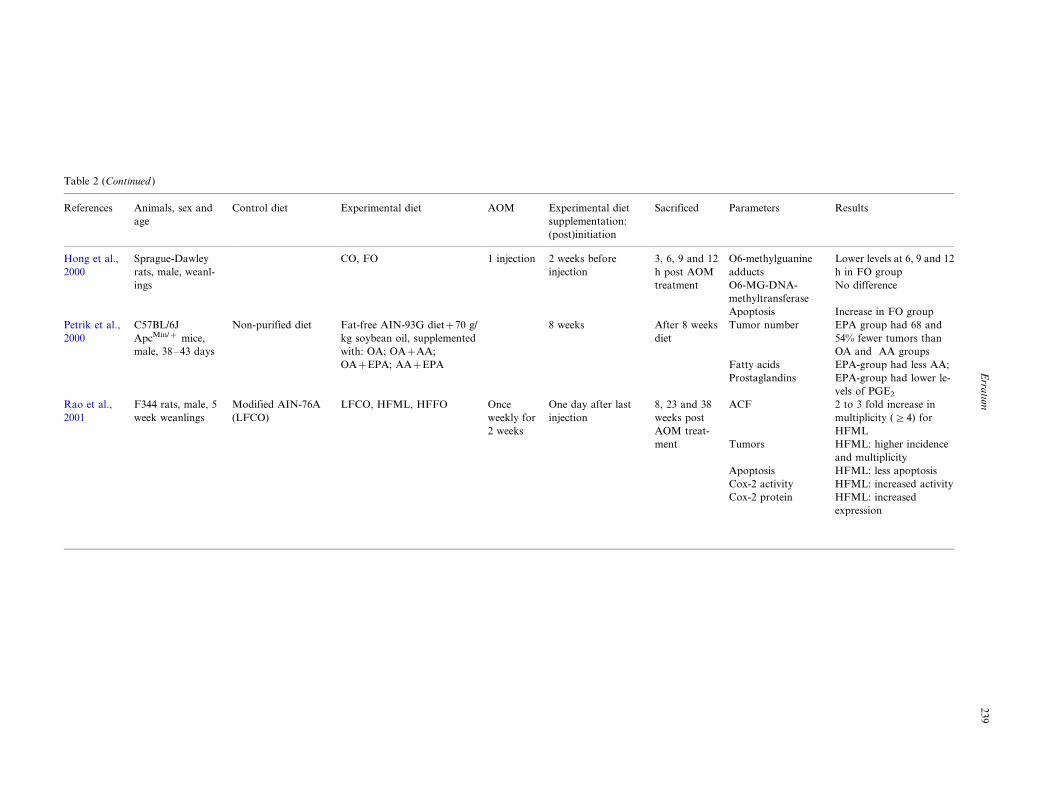
Table 2 (Continued )
References Animals, sex and
age
Control diet Experimental diet AOM Experimental diet
supplementation:
(post)initiation
Sacrificed Parameters Results
Hong et al.,
2000
Sprague-Dawley
rats, male, weanl-
ings
CO, FO 1 injection 2 weeks before
injection
3, 6, 9 and 12
h post AOM
treatment
O6-methylguanine
adducts
O6-MG-DNA-
methyltransferase
Apoptosis
Lower levels at 6, 9 and 12
h in FO group
No difference
Increase in FO group
Petrik et al.,
2000
C57BL/6J
ApcMin/� mice,
male, 38�/43 days
Non-purified diet Fat-free AIN-93G diet�70 g/
kg soybean oil, supplemented
with: OA; OA�AA;
OA�EPA; AA�EPA
8 weeks After 8 weeks
diet
Tumor number
Fatty acids
Prostaglandins
EPA group had 68 and
54% fewer tumors than
OA and AA groups
EPA-group had less AA;
EPA-group had lower le-
vels of PGE2
Rao et al.,
2001
F344 rats, male, 5
week weanlings
Modified AIN-76A
(LFCO)
LFCO, HFML, HFFO Once
weekly for
2 weeks
One day after last
injection
8, 23 and 38
weeks post
AOM treat-
ment
ACF
Tumors
Apoptosis
Cox-2 activity
Cox-2 protein
2 to 3 fold increase in
multiplicity (]4) for
HFML
HFML: higher incidence
and multiplicity
HFML: less apoptosis
HFML: increased activity
HFML: increased
expression
Erra
tum
23
9

(Takahashi et al., 1997) and HFFO (Rao et al., 2001).
These studies only focussed on the protecting effect of n-
3 PUFAs during the postinitiation phase. This experi-
mental design, however, superimposes effects on initia-tion such as carcinogen metabolism. It has been
demonstrated that FO reduced the azoxymethane
(AOM)-induced k-Ras mutations and decreased mem-
brane Ras expression (Davidson et al., 1999) when given
for and after initiation. These results indicate that FO
may protect against colon carcinogenesis by either
decreasing DNA adduct formation and/or enhancing
DNA repair. Hong et al. (2000) determined the ability ofFO and CO to simultaneously modulate O6-methylgua-
nine DNA adduct formation (DNA damage), removal
by O6-methylguanine-DNA-methyltransferase (repair)
and deletion (apoptosis). No main effect of diet on O6-
methylguanine-DNA-methyltransferase was found.
However, FO enhanced apoptosis combined with a
reduction in adduct formation. This may account, in
part, for the observed protective effect of n-3 PUFAsagainst experimentally induced colon cancer during the
initiation phase (Reddy et al., 1991). The protective
effect may also be explained by modulation of bio-
transformation enzymes related to carcinogen activa-
tion, thereby altering the amounts and activities of
oxidative (Phase 1) and conjugative (Phase 2) xenobiotic
metabolizing enzymes (Hong et al., 2000; Reddy, 1992).
Additional experiments are needed to address thishypothesis.
Various mechanisms have been postulated to explain
the enhancing effect of a high fat CO diet and the
protecting effect of a high fat FO diet during the
promotion phase of carcinogenesis. These mechanisms
include as stated above modulation of colonic mucosal
ODC activity, colonic secondary bile acids and/or PGE2
synthesis. Secondary bile acids can increase ODCactivity and cell proliferation and act as tumor promo-
ters (Rao and Reddy, 1993). It has been shown that
arachidonic acid metabolites are involved in increased
secondary bile acid production and the induction of
tissue ODC activity (Reddy et al., 1996; Rao and Reddy,
1993). It is possible that diets rich in n-3 fatty acids
result in decreased levels of arachidonic acid and its
metabolites and thereby inhibit tissue ODC activity andcell proliferation.
n-3 PUFAs have been reported to inhibit the produc-
tion of the type-2 series of eicosanoids, including PGE2,
from arachidonic acid (Onogi et al., 1996; Rao and
Reddy, 1993; Takahashi et al., 1997). Endogenous PGE2
has been shown to promote rat colon tumors and COX-
inhibitors that prevent prostaglandin production such as
IM can block the development of colon carcinomas(Inaba et al., 1999). Therefore, the mechanism respon-
sible for the inhibitory effects of n-3 PUFAs on color-
ectal tumors may partly be related to inhibition of PGE2
synthesis from AA and reduction of the AA levels itself
(Takahashi et al., 1997). Singh et al. (1997b) observed
that n-3 PUFAs inhibit AOM-induced expression of
COX-2, whereas n-6 PUFAs enhance levels of AOM-
induced COX-2 expression. Overexpression of COX-2has been reported in 90% of colon tumors and pre-
malignant colorectal adenomas. Therefore, the assay of
COX-2 expression may be used to monitor the process
of colon carcinogenesis (Singh et al., 1997b). Also Rao
et al. (2001) found that n-3 PUFAs in a high-fat fish oil
(HFFO) diet inhibited the levels of COX-2 and AA
metabolites (eicosanoids). They suggest that modulation
of AA metabolism through COX activity play a role inapoptosis, since the HFFO diet also enhanced apopto-
sis.
Chang et al. (1997) investigated whether the protec-
tive effect of dietary FO is mediated through changes in
proliferation, differentiation or apoptosis, all intermedi-
ate biomarkers for colon tumor development during the
promotion phase of tumorigenesis. Dolichos biflorus
agglutinin (DBA) binding (marker of differentiation)was higher for FO versus CO fed animals in both
proximal and distal colon. There was also a greater
number of apoptotic cells/crypt column in both prox-
imal and distal colon after feeding with FO compared
with CO. However, changes in cell proliferation did not
predict the beneficial effect of FO versus CO. Though,
Onogi et al. (1996) found that perilla oil significantly
reduced silver-stained nucleolar organizer region protein(AgNORs) count/nucleus, suggesting that perilla oil
decreased the number of cells in S-phase and thus
decreased cell proliferation.
Another inconsistent relationship exists between FO
and farnesyl protein transferase (FPTase) expression.
FPTase is an enzyme which catalyses the biological
activation of Ras proteins. The first step in this process
is the transfer of a 15-carbon isoprene, farnesyl, to thecysteine residue of the C-terminal tetrapeptide
sequence*/CAAX, of Ras precursors, which is cata-
lysed by FPTase. Farnesylation of Ras precursors is a
critical step during post-translational modification of
Ras oncoproteins, thereby enabling their anchorage to
the plasma membrane. Singh et al. (1998) demonstrated
that consumption of high amounts of FO reduced the
levels of FPTase comparing to high CO levels, thusinhibiting post-translational processing of Ras precur-
sors resulting in decreased Ras functioning. However,
Davidson et al. (1999) reported that perturbation in the
farnesylation of Ras is not a decisive factor regulating
membrane localization during malignant transforma-
tion in the colon. They found no differences in farnesyl
protein transferase activity and prenylation state of Ras
between tumors and uninvolved mucosa.Overall results from the animal models suggest that n-
3 fatty acids may protect against colorectal carcinogen-
esis and that n-6 fatty acids may enhance the risk of
colorectal carcinogenesis. The effects of these fatty acid
Erratum240

families on the complex and multistage process of
carcinogenesis can, however, not easily be explained
by one mechanism.
5. Effects of n-6 and n-3 PUFAs on colon cancer in
humans
Epidemiological studies have provided much of the
information available about diet and cancer risk in
humans. Migration studies have shown that the low
mortality rates from colon cancer in Japan increase
when Japanese migrate to the US and adapt to aWestern diet, which contains for example higher levels
of n-6 PUFAs, such as linoleic acid. Like Japanese, also
Eskimos have a relatively low incidence of colon cancer
(Woutersen et al., 1999). Both populations consume
large quantities of fish (1000�/3000 mg per day), which is
rich in n-3 fatty acids, such as EPA and DHA, whereas
the average consumption of n-3 PUFAs in western
countries is only 100 mg per day (Hong et al., 2000). Adetailed correlation analysis of international data ex-
plored the relationship between estimates of per capita
disappearance of PUFAs (total, fish n-3 and n-6) and
the incidence of cancers of the breast, colon, prostate,
lung and cervix (Hursting et al., 1990). In this study, an
estimate of n-6 PUFA intake was obtained by subtract-
ing fish n-3 PUFAs from total PUFA intake. Fish n-3
PUFAs were used, because, the database used for n-3fatty acids in foods other than fish was substantially
incomplete. Fish n-3 PUFAs showed a nonsignificant
negative association with the cancer sites studied. The
estimate of n-6 PUFAs gave virtually the same results as
total PUFAs, namely a positive correlation with the
incidence of breast and prostate cancers but not for
colon cancer. Zock and Katan (1998) reviewed the
epidemiological literature on linoleic acid (18:2n-6)intake and cancer risk. From this meta-analysis of
case-control and prospective cohort studies, it seemed
unlikely that a high intake of linoleic acid substantially
raises the risk of colorectal cancer in humans but a small
increase in risk could not be excluded. Terry et al. (2001)
examined the relation between total fat, fat types,
specific fatty acids and the risk of colorectal cancer in
a population-based prospective cohort of Swedishwomen. They observed no associations for any of the
specific PUFAs, including linoleic acid, a-linolenic acid,
EPA, DHA or the sum of n-6 fatty acids or n-3 fatty
acids and colorectal cancer.
An estimate of the dietary intake of individual fatty
acids by for example food composition tables or food
questionnaires can, however, be biased in several ways.
Assessment of fatty acid composition in lipid subfrac-tions in relation to widely varying fatty acid intake
patterns will provide more precise indicators of recent
dietary intake (Bartsch et al., 1999). Bakker et al. (1997)
used adipose tissue fatty acid composition as a biomar-
ker for long-term dietary exposure to fatty acids. In this
study, no associations between n-3 and n-6 fatty acids in
adipose tissue and incidences of cancer of the breast,prostate and colon were found. Thus, despite the
findings in animal models, data from population-based
studies show little or no associations between PUFA
intake and colorectal cancer risk.
There have been a couple of short-term human
intervention studies on FO supplementation and bio-
markers of colon cancer risk. Bartram et al. (1993)
investigated the effect of FO on cell proliferation, ODCactivity and PGE2 release in 12 healthy volunteers. In
addition to a controlled basal diet, the test subjects
received either 550 mg FO (4,4 g n-3 fatty acids per day)
or CO supplements (4�/5 capsules per day) for two 4-
week periods in a double-blind crossover trial. A
washout period of 4 weeks was allowed between the
two study periods to maintain basal conditions for each
supplementation. All three biomarkers were signifi-cantly lower during the FO than the CO period,
suggesting an inhibiting effect of FO supplementation
on colon cancer development. In another short-term
intervention study, Bartram et al. (1996) investigated the
effect of FO consumption in healthy subjects on fecal
parameters. No significant differences were noted for
fecal activities of b-glucuronidase, b-glucosidase and
sulfatase, nor did FO or CO consumption significantlyaffect fecal bile acid excretion. However, daily excretion
of the putative colon carcinogen 4-cholesten-3-one was
significantly lower in the FO than in the CO period. The
authors suggest that this may be another mechanism by
which FO may inhibit colon cancer development.
The effect of n-3 fatty acids on patients who have had
a bowel neoplasm and thus at high risk of developing a
second neoplasm was studied by Huang et al. (1996).Patients with stage 1 or stage 2 colon carcinoma or
adenomatous polyps were randomized to consume
either 9 g per day n-3 fatty acid or placebo capsules.
Patients in the n-3 group experienced significant reduc-
tions in BrdU labeling index (cell proliferation) after 3
months of supplementation. The effect was only found
in patients with hyperproliferative baseline levels. There-
fore, characteristics of mucosal proliferation at baselinemay be a crucial factor for the effect of n-3 fatty acid
supplementation. Anti et al. (1994) attempted to identify
an optimum dose for FO supplementation and evalu-
ated the persistence of its effects during long-term
administration. Sixty patients with sporadic adenomas
received 2.5, 5.1 or 7.7 g FO per day or placebo for 30
days. Significantly reduced proliferation was also only
observed in patients with abnormal baseline patterns.The effects persisted during long-term (6 months) FO
treatment (2.5 g per day). The data stated above suggest
that if the n-3/n-6 fatty acid equilibrium indeed affects
colonic tumor development, its mode of action seems to
Erratum 241

be based on inhibition of cell proliferation, most likely
through the cyclooxygenase/prostaglandin pathways.
Other mechanisms may also be involved, but have not
been investigated yet in the human population.Moreover, there is a lack of consistency between the
various studies. Gee et al. (1999) determined the effect of
FO on the fatty acid composition of colonic mucosa and
on rectal crypt cell proliferation in patients (28 males
and 21 females) undergoing surgery for colonic carci-
noma. Consumption of FO capsules (1.4 g EPA and 1.0
g DHA) for an average of 12.3�//�/0.5 days lead to
incorporation of EPA into colonic mucosa but had nodetectable effect on epithelial cytokinetics. During a
long-term intervention trial of DHA-concentrated fish
oil capsules (2.2 g DHA and 0.6 g EPA per day), three
patients with FAP and two patients with more than 30
colorectal polyps received these FO capsules for 1 or 2
years. A marked increase or decrease in the number of
polyps was not observed, but the three patients with
FAP developed either endometrial or lung cancer after12 months or colon cancer after 24 months, respectively,
(Akedo et al., 1998). Both studies showed no protective
effect of FO consumption. In these studies, however,
beneficial effects of n-3 fatty acids may have been missed
due to either a short supplementation period or a limited
number of subjects.
Nair et al. (1997) performed a dietary study on n-6
PUFA intake and biomarkers of cancer risk. This studyrevealed that high intake of n-6 PUFAs increased
malonaldehyde-derived adducts in both male and female
subjects. Etheno adducts in white blood cells were not
elevated in males but about 40 times higher in females.
This might be a possible link between increased intake of
dietary n-6 PUFAs, DNA damage and elevated risk of
for example colon cancer.
Taken together, the data of the population-basedstudies suggest that n-6 and n-3 PUFA intake do not
influence colorectal cancer. If associations exist, they are
likely to be weak. Short-term intervention studies reveal
though that fish oil supplementation may protect
against colorectal carcinogenesis in humans and that
n-6 PUFA intake may increase the risk.
6. General discussion
In this review we provided a brief overview of
experimental and epidemiological studies that have
been performed in the past decade on the influence of
n-6 and n-3 PUFAs on colorectal carcinogenesis. No
differential effects between n-6 and n-3 PUFAs on in
vitro colonic cell proliferation and apoptosis, both
biomarkers of carcinogenesis, have been observed.Results from animal models indicate that n-6 PUFAs
have a tumor enhancing effect, predominantly during
the postinitiation phase. n-3 PUFAs may protect against
colorectal carcinogenesis during both the initiation and
postinitiation phase. Population-based studies show
little or no associations between dietary n-6 or n-3
PUFA intake and colorectal cancer. If associations existin the human population, they are likely to be weak.
Short-term biomarker studies in humans suggest though
that fish oil supplementation may protect against color-
ectal carcinogenesis and that n-6 PUFA intake may
increase the risk.
Several explanations could be given for the discre-
pancy in results between in vitro, animal and human
studies. Colon cell lines are used to investigate themechanistic effects of individual n-6 and n-3 PUFAs on
colorectal carcinogenesis in vitro. However, lipid per-
oxidation is a complication that must be considered in
PUFA-supplemented cell cultures with low levels of
antioxidants or enzymatic detoxification enzymes
(Grammatikos et al., 1994; Schonberg et al., 1997)
compared with the in vivo situation. This may be an
explanation why AA (20:4n-6) and EPA (20:5n-3) donot show a differential effect and are both cytotoxic for
human colon carcinoma cell lines. Furthermore, con-
clusions from in vitro experiments are obtained from
colorectal carcinoma cell lines and not from normal
cells, which develop to cancer cells, contrary to most
animal studies. Carcinoma cell lines may not be sensitive
anymore to fatty acid treatment, or react in a different
way due to mutations which turn cell signaling pathwayson or off. Therefore, it would be better to also
investigate the effects of n-6 and n-3 PUFAs on primary
cultures of normal colon cells or better differentiated
colon tumor cells. A good example may be the human
colonic adenocarcinoma cell line, HCA-7. These cells
retained some of the morphological and functional
polarity exhibited by normal colonic epithelium (Kirk-
land, 1985).Another reason for the discrepancy between the in
vitro and in vivo results may be the lack of interaction in
vitro with other cell systems and integrative functions
involved in the process of carcinogenesis. In general, cell
lines can be useful to study mechanistic pathways and
parts of cell biological and physiological processes, but
care should be taken in interpretation of the results.
As generally known, animal models have severaladvantages over in vitro models in studying colorectal
carcinogenesis. They can for example be used to test
hypotheses about mechanisms in a physiological con-
text. Another advantage, also comparing to human
studies, is the possibility to quantify the development
of tumors in a short time. Inherent to these animal
experiments is the use of high amounts of isolated
dietary constituents. In most animal experiments de-scribed in this review, rats were fed experimental diets,
supplemented with high amounts (9�/23.5 wt.%) of CO
or fish oil as the only fat source at the expense of starch.
Zock et al. (1998) revealed that in the population-based
Erratum242

studies they reviewed, the intake difference for linoleic
acid was between 5�/25 g per day, which is comparable
to a range intake of 4�/10% of daily energy. This is much
lower than the n-6 or n-3 PUFA intake in the animalexperiments, which is about 18�/48% of daily energy.
Therefore, a narrow range of n-6 or n-3 PUFA intake in
the population may be a reason why associations
between PUFA intake and colorectal cancer were not
found in population-based studies compared with the
animal data. It can thus not be excluded that cancer risk
within populations would be affected by larger differ-
ences in PUFA intake, which is the case in the humanshort-term biomarker studies on for example cell pro-
liferation. These studies revealed that extra supplemen-
tation of fish oil (2.5�/9 g n-3 PUFAs per day) in
addition to a normal diet may protect against colorectal
carcinogenesis in humans. Intervention studies on real
disease endpoints such as polyps or tumors have,
however, not been conducted yet for PUFAs intake
and colorectal cancer.The efficacy of dietary n-3 PUFAs can depend on the
levels of AA that are found in mixed diets and may
therefore, account for some of the variability of results
observed among studies using human subjects (Whelan,
1996). The efficacy of dietary n-3 PUFAs is also
dependant on the baseline patterns of for example cell
proliferation, as described in the short-term biomarker
studies. In two studies described, significantly reducedproliferation after fish oil supplementation was only
observed in patients at high risk of developing colon
cancer due to abnormal baseline patterns of cell
proliferation. This situation is comparable to the in
vivo studies in which the animals are also at higher risk
of developing colon cancer due to chemically induced
AOM mutations or germline Apc mutations. Therefore,
this may be another reason why there were no associa-tions found between n-6 or n-3 PUFA intake and
colorectal carcinogenesis in the population-based stu-
dies of the healthy population.
In conclusion, despite the lack of a differential effect
in vitro, animal studies and moreover, data from short-
term human biomarker studies indicate a promising
beneficial effect of fish oil supplementation, with high
amounts of n-3 PUFAs, on colorectal carcinogenesis.However, more human dietary intervention studies are
needed to evaluate the real implication of fish oil
supplementation on colon tumor development in hu-
mans at high and low risk of developing colon cancer.
References
Akedo, I., Ishikawa, H., Nakamura, T., Kimura, K., Takeyama, I.,
Suzuki, T., Kameyama, M., Sato, S., Matsuzawa, Y., Kakizoe, T.,
Otani, T., 1998. Three cases with familial adenomatous polyposis
diagnosed as having malignant lesions in the course of a long-term
trial using docosahexaenoic acid (DHA)-concentrated fish oil
capsules. Jpn. J. Clin. Oncol. 28, 762�/765.
Anti, M., Armelao, F., Marra, G., Percesepe, A., Bartoli, G.M.,
Palozza, P., Parrella, P., Canetta, C., Gentiloni, N., De Vitis, I.,
1994. Effects of different doses of fish oil on rectal cell proliferation
in patients with sporadic colonic adenomas. Gastroenterology 107,
1709�/1718.
Awad, A.B., Kamei, A., Horvath, P.J., Fink, C.S., 1995. Prostaglandin
synthesis in human cancer cells: influence of fatty acids and
butyrate. Prostaglandins Leukotrienes Essent Fatty Acids 53,
87�/93.
Bakker, N., Van’t Veer, P., Zock, P.L., 1997. Adipose fatty acids and
cancers of the breast, prostate and colon: an ecological study. Int.
J. Cancer 72, 587�/591.
Bartram, H.P., Gostner, A., Kelber, E., Dusel, G., Weimer, A.,
Scheppach, W., Kasper, H., 1996. Effects of fish oil on fecal
bacterial enzymes and steroid excretion in healthy volunteers:
implications for colon cancer prevention. Nutr. Cancer 25, 71�/78.
Bartram, H.P., Gostner, A., Scheppach, W., Reddy, B.S., Rao, C.V.,
Dusel, G., Richter, F., Richter, A., Kasper, H., 1993. Effects of fish
oil on rectal cell proliferation, mucosal fatty acids, and prosta-
glandin E2 release in healthy subjects. Gastroenterology 105,
1317�/1322.
Bartsch, H., Nair, J., Owen, R.W., 1999. Dietary polyunsaturated fatty
acids and cancers of the breast and colorectum: emerging evidence
for their role as risk modifiers. Carcinogenesis 20, 2209�/2218.
Chang, W.C., Chapkin, R.S., Lupton, J.R., 1997. Predictive value of
proliferation, differentiation and apoptosis as intermediate markers
for colon tumorigenesis. Carcinogenesis 18, 721�/730.
Chen, Z.Y., Istfan, N.W., 2000. Docosahexaenoic acid is a potent
inducer of apoptosis in HT-29 colon cancer cells. Prostaglandins
Leukotrienes Essent Fatty Acids 63, 301�/308.
Clarke, R.G., Lund, E.K., Latham, P., Pinder, A.C., Johnson, I.T.,
1999. Effect of eicosapentaenoic acid on the proliferation and
incidence of apoptosis in the colorectal cell line HT29. Lipids 34,
1287�/1295 (published erratum appears in Lipids 2000
Jan;35(1):113).
Collett, E.D., Davidson, L.A., Fan, Y.Y., Lupton, J.R., Chapkin,
R.S., 2001. n-6 and n-3 polyunsaturated fatty acids differentially
modulate oncogenic Ras activation in colonocytes. Am. J. Physiol.
Cell. Physiol. 280, C1066�/C1075.
Davidson, L.A., Lupton, J.R., Jiang, Y.H., Chapkin, R.S., 1999.
Carcinogen and dietary lipid regulate ras expression and localiza-
tion in rat colon without affecting farnesylation kinetics. Carcino-
genesis 20, 785�/791.
Doll, R., Peto, R., 1981. The causes of cancer: quantitative estimates of
avoidable risks of cancer in the United States today. J. Natl.
Cancer Inst. 66, 1191�/1308.
Dommels, Y.E.M., Alink, G.M., Linssen, J.P.H., van Ommen, B.
2002. Effects of n-6 and n-3 polyunsaturated fatty acids on gap
junctional intercellular communication (GJIC) during spontaneous
differentiation of the human colon adenocarcinoma cell line Caco-
2, Nutrition and Cancer (accepted for the publication).
Fischer, S.M., 1997. Prostaglandins and cancer. Front. Biosci. 2,
d482�/d500.
Furihata, T., Kawamata, H., Ohsugi, R., Sato, S., Kubota, K.,
Fujimori, T., 2001. Colitis enhances the colorectal carcinogenesis
in rats: correlation between the incidence of aberrant crypt foci and
the incidence of tumors. Nippon Shokakibyo Gakkai Zasshi 98,
525�/532.
Gee, J.M., Watson, M., Matthew, J.A., Rhodes, M., Speakman, C.J.,
Stebbings, W.S., Johnson, I.T., 1999. Consumption of fish oil leads
to prompt incorporation of eicosapentaenoic acid into colonic
mucosa of patients prior to surgery for colorectal cancer, but has
no detectable effect on epithelial cytokinetics. J. Nutr. 129, 1862�/
1865.
Erratum 243

Grammatikos, S.I., Subbaiah, P.V., Victor, T.A., Miller, W.M., 1994.
Diverse effects of essential (n-6 and n-3) fatty acids on cultured
cells. Cytotechnology 15, 31�/50.
Gurr, M., 1996. Fats. In: Garrow, J.S., James, W.P.T. (Eds.), Human
Nutrition and Dietetics. Churchill Livingstone, London, pp. 77�/
102.
Health Council of the Netherlands, 2001. Population screening for
colorectal cancer. The Hague: Health Council of the Netherlands
Publication number 2001/01.
Hong, M.Y., Lupton, J.R., Morris, J.S., Wang, N., Carroll, R.J.,
Davidson, L.A., Elder, R.H., Chapkin, R.S., 2000. Dietary fish oil
reduces O6-methylguanine DNA adduct levels in rat colon in part
by increasing apoptosis during tumor initiation [In Process
Citation]. Cancer Epidemiol. Biomarkers. Prev. 9, 819�/826.
Huang, Y.C., Jessup, J.M., Forse, R.A., Flickner, S., Pleskow, D.,
Anastopoulos, H.T., Ritter, V., Blackburn, G.L., 1996. n-3 Fatty
acids decrease colonic epithelial cell proliferation in high- risk
bowel mucosa. Lipids 31 (Suppl), S313�/S317.
Hursting, S.D., Thornquist, M., Henderson, M.M., 1990. Types of
dietary fat and the incidence of cancer at five sites. Prev. Med. 19,
242�/253.
Inaba, A., Uchiyama, T., Oka, M., 1999. Role of prostaglandin E2 in
rat colon carcinoma. Hepatogastroenterology 46, 2347�/2351.
Jiang, W.G., Bryce, R.P., Horrobin, D.F., 1998. Essential fatty acids:
molecular and cellular basis of their anti-cancer action and clinical
implications. Crit. Rev. Oncol. Hematol. 27, 179�/209.
Kim, E., Kim, W., Hang, Y., Ha, Y., Bach, L., Park, J., 2000.
Inhibition of Caco-2 cell proliferation by (n-3) fatty acids: possible
mediation by increased secretion of insulin-like growth factor
binding protein-6. Nutr. Res. 20, 1409�/1421.
Kinzler, K.W., Vogelstein, B., 1998. Colorectal Tumors. In: Vogel-
stein, B., Kinzler, K.W. (Eds.), The Genetic Basis of Human
Cancer. McGraw Hill, Baltimore.
Kirkland, S.C., 1985. Dome formation by a human colonic adeno-
carcinoma cell line (HCA-7). Cancer Res. 45, 3790�/3795.
Klurfeld, D.M., Bull, A.W., 1997. Fatty acids and colon cancer in
experimental models. Am. J. Clin. Nutr. 66, 1530S�/1538S.
Krause, W., Dubois, R.N., 2000. Eicosanoids and the large intestine.
Prostaglandins Lipid Mediat. 61, 145�/161.
Mengeaud, V., Nano, J.L., Fournel, S., Rampal, P., 1992. Effects of
eicosapentaenoic acid, gamma-linolenic acid and prostaglandin E1
on three human colon carcinoma cell lines. Prostaglandins
Leukotrienes Essent Fatty Acids 47, 313�/319.
Nair, J., Vaca, C.E., Velic, I., Mutanen, M., Valsta, L.M., Bartsch, H.,
1997. High dietary v-6 polyunsaturated fatty acids drastically
increase the formation of etheno-DNA base adducts in white blood
cells of female subjects. Cancer Epidemiol Biomarkers Prev. 6(8),
597�/601.
Onogi, N., Okuno, M., Komaki, C., Moriwaki, H., Kawamori, T.,
Tanaka, T., Mori, H., Muto, Y., 1996. Suppressing effect of perilla
oil on azoxymethane-induced foci of colonic aberrant crypts in
rats. Carcinogenesis 17, 1291�/1296.
Oshima, M., Takahashi, M., Oshima, H., Tsutsumi, M., Yazawa, K.,
Sugimura, T., Nishimura, S., Wakabayashi, K., Taketo, M.M.,
1995. Effects of docosahexaenoic acid (DHA) on intestinal polyp
development in Apc delta 716 knockout mice. Carcinogenesis 16,
2605�/2607.
Palozza, P., Calviello, G., Maggiano, N., Lanza, P., Raneletti, F.O.,
Bartoli, G.M., 2000. Beta-carotene antagonizes the effects of
eicosapentaenoic acid on cell growth and lipid peroxidation in
WiDr adenocarcinoma cells. Free Radic. Biol. Med. 28, 228�/234.
Petrik, M.B., McEntee, M.F., Chiu, C.H., Whelan, J., 2000. Antagon-
ism of arachidonic acid is linked to the antitumorigenic effect of
dietary eicosapentaenoic acid in ApcMin/� mice. J. Nutr. 130 (5),
1153�/1158.
Rao, C.V., Reddy, B.S., 1993. Modulating effect of amount and types
of dietary fat on ornithine decarboxylase, tyrosine protein kinase
and prostaglandins production during colon carcinogenesis in male
F344 rats. Carcinogenesis 14, 1327�/1333.
Rao, C.V., Simi, B., Wynn, T.T., Garr, K., Reddy, B.S., 1996.
Modulating effect of amount and types of dietary fat on colonic
mucosal phospholipase A2, phosphatidylinositol-specific phospho-
lipase C activities, and cyclooxygenase metabolite formation during
different stages of colon tumor promotion in male F344 rats.
Cancer Res. 56, 532�/537.
Rao, C.V., Hirose, Y., Indranie, C., Reddy, B.S., 2001. Modulation of
experimental colon tumorigenesis by types and amounts of dietary
fatty acids. Cancer Res. 61, 1927�/1933.
Reddy, B.S., 1992. Dietary fat and colon cancer: animal model studies.
Lipids 27, 807�/813.
Reddy, B.S., Sugie, S., 1988. Effect of different levels of omega-3 and
omega-6 fatty acids on azoxymethane-induced colon carcinogenesis
in F344 rats. Cancer Res. 48, 6642�/6647.
Reddy, B.S., Burill, C., Rigotty, J., 1991. Effect of diets high in omega-
3 and omega-6 fatty acids on initiation and postinitiation stages of
colon carcinogenesis. Cancer Res. 51, 487�/491.
Reddy, B.S., Simi, B., Patel, N., Aliaga, C., Rao, C.V., 1996. Effect of
amount and types of dietary fat on intestinal bacterial 7 a-
dehydroxylase and phosphatidylinositol-specific phospholipase C
and colonic mucosal diacylglycerol kinase and PKC activities
during stages of colon tumor promotion. Cancer Res. 56, 2314�/
2320.
Roncucci, L., Pedroni, M., Vaccina, F., Benatti, P., Marzona, L., De
Pol, A., 2000. Aberrant crypt foci in colorectal carcinogenesis. Cell
and crypt dynamics. Cell. Prolif. 33, 1�/18.
Rose, D.P., Connolly, J.M., 1999. Omega-3 fatty acids as cancer
chemopreventive agents. Pharmacol. Ther. 83, 217�/244.
Schonberg, S.A., Rudra, P.K., Noding, R., Skorpen, F., Bjerve, K.S.,
Krokan, H.E., 1997. Evidence that changes in Se-glutathione
peroxidase levels affect the sensitivity of human tumour cell lines
to n-3 fatty acids. Carcinogenesis 18 (10), 1897�/1904.
Singh, J., Hamid, R., Reddy, B.S., 1997a. Dietary fat and colon
cancer: modulating effect of types and amount of dietary fat on ras-
p21 function during promotion and progression stages of colon
cancer. Cancer Res. 57, 253�/258.
Singh, J., Hamid, R., Reddy, B.S., 1997b. Dietary fat and colon
cancer: modulation of cyclooxygenase-2 by types and amount of
dietary fat during the postinitiation stage of colon carcinogenesis.
Cancer Res. 57, 3465�/3470.
Singh, J., Hamid, R., Reddy, B.S., 1998. Dietary fish oil inhibits the
expression of farnesyl protein transferase and colon tumor devel-
opment in rodents. Carcinogenesis 19, 985�/989.
Takahashi, M., Fukutake, M., Isoi, T., Fukuda, K., Sato, H., Yazawa,
K., Sugimura, T., Wakabayashi, K., 1997. Suppression of azox-
ymethane-induced rat colon carcinoma development by a fish oil
component, docosahexaenoic acid (DHA). Carcinogenesis 18,
1337�/1342.
Terry, P., Bergkvist, L., Holmberg, L., Wolk, A., 2001. No association
between fat and fatty acids intake and risk of colorectal cancer.
Cancer Epidemiol. Biomarkers Prev. 10, 913�/914.
Tsai, W.S., Nagawa, H., Kaizaki, S., Tsuruo, T., Muto, T., 1998.
Inhibitory effects of n-3 polyunsaturated fatty acids on sigmoid
colon cancer transformants. J. Gastroenterol 33, 206�/212.
Visser, O., Coebergh, J., Schouten, L., Van Dijck, J., 2001. Incidence
of cancer in The Netherlands. In: (Eds.), Verening van Integrale
Kankercentra, Utrecht, 86.
Whelan, J., 1996. Antagonistic effects of dietary arachidonic acid and
n-3 polyunsaturated fatty acids. J. Nutr. 126 (Suppl. 4), 1086S�/
1091S.
Woutersen, R.A., Appel, M.J., Van Garderen-Hoetmer, A., Wijnands,
M.V., 1999. Dietary fat and carcinogenesis. Mutat. Res. 443, 111�/
127.
Zock, P.L., Katan, M.B., 1998. Linoleic acid intake and cancer risk: a
review and meta-analysis. Am. J. Clin. Nutr. 68, 142�/153.
Erratum244