csra post-transcriptionally represses pgaabcd , responsible for synthesis of a biofilm...
TRANSCRIPT

Molecular Microbiology (2005)
56
(6), 1648–1663 doi:10.1111/j.1365-2958.2005.04648.x
© 2005 Blackwell Publishing Ltd
Blackwell Science, LtdOxford, UKMMIMolecular Microbiology0950-382XBlackwell Publishing Ltd, 2005
? 2005
56
616481663
Original Article
Repression of biofilm formation by CsrAX. Wang
et al.
Accepted 11 March, 2005. *For correspondence. [email protected]; Tel. (
+
1) 404 727 3734; Fax(
+
1) 404 727 3659.
CsrA post-transcriptionally represses
pgaABCD
, responsible for synthesis of a biofilm polysaccharide adhesin of
Escherichia coli
Xin Wang,
1
Ashok K. Dubey,
2
Kazushi Suzuki,
1
Carol S. Baker,
2
Paul Babitzke
2
and Tony Romeo
1
*
1
Department of Microbiology and Immunology, Emory University School of Medicine, 3105 Rollins Research Center, 1510 Clifton Road N.E., Atlanta, GA 30322, USA.
2
Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA.
Summary
The RNA-binding protein CsrA represses biofilm for-mation, while the non-coding RNAs CsrB and CsrCactivate this process by sequestering CsrA. We nowprovide evidence that the
pgaABCD
transcript,required for the synthesis of the polysaccharideadhesin PGA (poly-
bbbb
-1,6-
N
-acetyl-
D
-glucosamine) of
Escherichia coli
, is the key target of biofilm regula-tion by CsrA.
csrA
disruption causes an approxi-mately threefold increase in PGA production and anapproximately sevenfold increase in expression of a
pgaA
¢¢¢¢
–
¢¢¢¢
lacZ
translational fusion. A
DDDD
csrB
DDDD
csrC
mutant exhibits a modest decrease in
pgaA
¢¢¢¢
–
¢¢¢¢
lacZ
expression, while the response regulator UvrY, a tran-scriptional activator of
csrB
and
csrC
, stimulates thisexpression. Biofilm formation is not regulated by
csrA
,
csrB
or
uvrY
in a
DDDD
pgaC
mutant, which cannotsynthesize PGA. Gel mobility shift and toeprint anal-yses demonstrate that CsrA binds cooperatively to
pgaA
mRNA and competes with 30S ribosome sub-unit for binding. CsrA destabilizes the
pgaA
tran-script
in vivo
. RNA footprinting and boundaryanalyses identify six apparent CsrA binding sites inthe
pgaA
mRNA leader, the most extensive arrange-ment of such sites in any mRNA examined to date.Substitution mutations in CsrA binding sites overlap-ping the Shine–Dalgarno sequence and initiationcodon partially relieve repression by CsrA. Thesestudies define the crucial mechanisms, though not
the only means, by which the Csr system influencesbiofilm formation.
Introduction
An important microbial survival strategy is to form surface-associated multicellular communities known as biofilms(Costerton
et al
., 1995). This behaviour enables bacteriato establish persistent relationships with their surround-ings, and in various settings, provides protection againststresses, predation, the immune system and antimicrobialtherapies (Costerton
et al
., 1999; Donlan and Costerton,2002; Hogan and Kolter, 2002; Mah
et al
., 2003; Hall-Stoodley
et al
., 2004). Biofilm development typicallyinvolves attachment to a surface, replication, cell–celladhesion to form microcolonies, maturation of a structuredbiofilm and eventual return of cells to the planktonic state.It is a genetically programmed process, in part involvingbacterial surface factors and the regulatory systems thatgovern their expression in response to nutritional andenvironmental cues (e.g. O’Toole and Kolter, 1998; Vidal
et al
., 1998; Jackson
et al
., 2002a,b; Stoodley
et al
., 2002;Branda
et al
., 2004; Sauer
et al
., 2004).Attachment of bacteria to abiotic surfaces and to each
other is paramount for biofilm to form. In
Escherichia coli
,this is facilitated by motility and proteinaceous adhesins(Pratt and Kolter, 1998; Vidal
et al
., 1998; Danese
et al
.,2000). In addition, a cell-bound polysaccharide of
E. coli
,poly-
b
-1,6-GlcNAc or PGA, promotes attachment to solidsurfaces, cell–cell adherence and stabilization of biofilmstructure (Wang
et al
., 2004; Itoh
et al
., 2005). Structurallyrelated polysaccharides have been implicated as biofilmadhesins in diverse species (e.g. Gotz, 2002; Kaplan
et al
., 2003; Itoh
et al
., 2005). Such studies recommendthese polysaccharides, their biosynthetic pathways andregulatory elements as potential targets for biofilm controlstrategies.
Carbon metabolism regulators play important, largelyunresolved roles in biofilm formation (O’Toole
et al
., 2000;Jackson
et al
., 2002a,b; Stanley
et al
., 2003). In
E. coli
and related species, the Csr (carbon storage regulatory)system exerts profound effects on biofilm development(Jackson
et al
., 2002a,b). Its key component, CsrA, is asmall RNA-binding protein that recognizes sequences
Repression of biofilm formation by CsrA
1649
© 2005 Blackwell Publishing Ltd,
Molecular Microbiology
,
56
, 1648–1663
located within the leaders of target mRNAs and alters theirtranslation and stability (Liu
et al
., 1995; 1997; Wei
et al
.,2001; Baker
et al
., 2002; Dubey
et al
., 2003). CsrArepresses biofilm formation and a variety of stationary-phase processes (reviewed by Romeo, 1998), includinggluconeogenesis (Sabnis
et al
., 1995), glycogen synthe-sis and glycogen catabolism (Romeo
et al
., 1993; Yang
et al
., 1996). Glycogen synthesis and turnover in the earlystationary phase of growth influence biofilm, perhaps byaffecting metabolic flux into precursors of adhesins orother factors (Jackson
et al
., 2002a). Conversely, CsrAactivates biofilm dispersal (Jackson
et al
., 2002a), glyco-lysis (Sabnis
et al
., 1995), motility (Wei
et al
., 2001) andacetate metabolism (Wei
et al
., 2000). Two untranslatedRNAs, CsrB and CsrC, antagonize CsrA activity bysequestering this protein (Fig. 1A; Liu
et al.
1997; Weil-bacher
et al
., 2003). Previously, CsrA was found to regu-late biofilm formation independently of several surfacefactors, leaving its effects on biofilm to be determined(Jackson
et al
., 2002a).Bacteria use two-component signal transduction sys-
tems (TCS) extensively for sensing and responding to
environmental conditions (Hoch, 2000; West and Stock,2001). Several of these systems exhibit complex influ-ences on
E. coli
biofilm development (Dorel
et al
.,1999; Prigent-Combaret
et al
., 2001; Otto and Silhavy,2002; Suzuki
et al
., 2002; Ferrieres and Clarke, 2003;Gerstel
et al
., 2003; Weilbacher
et al
., 2003). The BarA-UvrY TCS activates biofilm formation and is requiredfor
csrB
and
csrC
transcription (Fig. 1A; Suzuki
et al.
2002; Weilbacher
et al
., 2003). Thus, this TCS influ-ences biofilm formation by relieving CsrA repression.Whether this is its only role in biofilm formation wasnot known.
Results of the present study revealed that biofilm for-mation by a mutant that is unable to synthesize PGA nolonger responds to components of the Csr system,including CsrA, CsrB/C or UvrY. This suggested to usthat CsrA directly or indirectly represses production ofPGA or that PGA and CsrA-repressed factor(s) arerequired in combination for biofilm to form. The results ofthe present study support the first hypothesis and estab-lish the signalling circuitry and molecular mechanisms ofthis regulation.
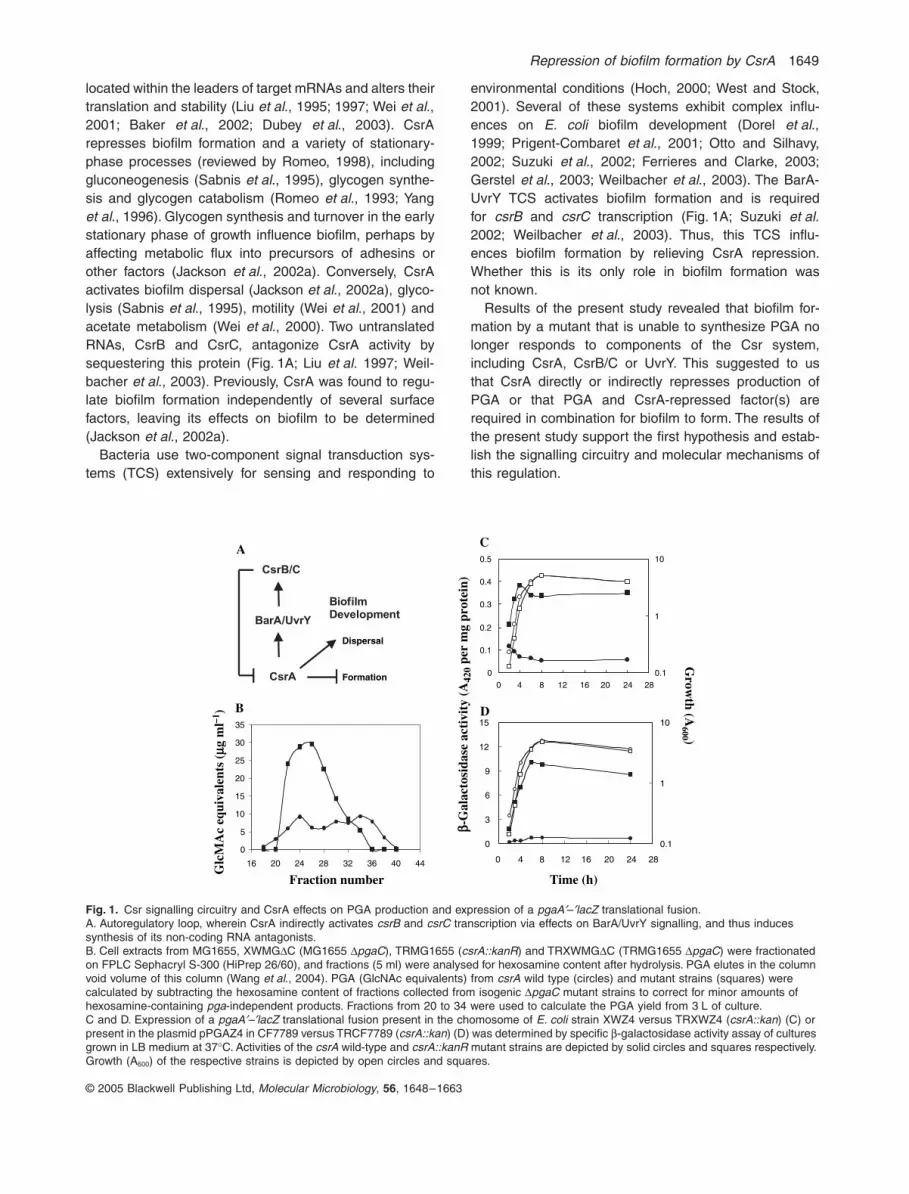
Fig. 1.
Csr signalling circuitry and CsrA effects on PGA production and expression of a
pgaA
¢
–
¢
lacZ
translational fusion.A. Autoregulatory loop, wherein CsrA indirectly activates
csrB
and
csrC
transcription via effects on BarA/UvrY signalling, and thus induces synthesis of its non-coding RNA antagonists.B. Cell extracts from MG1655, XWMG
D
C (MG1655
D
pgaC
), TRMG1655 (
csrA::kanR
) and TRXWMG
D
C (TRMG1655
D
pgaC
) were fractionated on FPLC Sephacryl S-300 (HiPrep 26/60), and fractions (5 ml) were analysed for hexosamine content after hydrolysis. PGA elutes in the column void volume of this column (Wang
et al
., 2004). PGA (GlcNAc equivalents) from
csrA
wild type (circles) and mutant strains (squares) were calculated by subtracting the hexosamine content of fractions collected from isogenic
D
pgaC
mutant strains to correct for minor amounts of hexosamine-containing
pga
-independent products. Fractions from 20 to 34 were used to calculate the PGA yield from 3 L of culture.C and D. Expression of a
pgaA
¢
–
¢
lacZ
translational fusion present in the chomosome of
E. coli
strain XWZ4 versus TRXWZ4 (
csrA::kan
) (C) or present in the plasmid pPGAZ4 in CF7789 versus TRCF7789 (
csrA::kan
) (D) was determined by specific
b
-galactosidase activity assay of cultures grown in LB medium at 37
∞
C. Activities of the
csrA
wild-type and
csrA::kanR
mutant strains are depicted by solid circles and squares respectively. Growth (A
600
) of the respective strains is depicted by open circles and squares.
0
5
10
15
20
25
30
35
16 20 24 28 32 36 40 44
Fraction number
B
0
0.1
0.2
0.3
0.4
0.5
0 4 8 12 16 20 24 280.1
1
10
0
3
6
9
12
15
0 4 8 12 16 20 24 28
0.1
1
10
b-G
alac
tosi
dase
act
ivit
y (A
420
per
mg
prot
ein)
Grow
th (A600 )
Time (h)
C
D
A
Dispersal
Formation
Glc
MA
c eq
uiva
lent
s (m
g m
l–1)

1650
X. Wang
et al.
© 2005 Blackwell Publishing Ltd,
Molecular Microbiology, 56, 1648–1663
Results
Effect of csrA on PGA and pgaA¢–¢lacZ expression
To determine whether CsrA influences biofilm formationby regulating PGA production, we analysed polysaccha-rides from wild-type and csrA mutant cultures (Fig. 1B;Experimental procedures). Based on this approach, a cul-ture of the wild-type strain produced 140 mg l-1 PGA, whilethe isogenic csrA mutant produced 430 mg l-1 PGA; a dif-ference of 3.1-fold.
To determine whether the increase in PGA in the csrAmutant results from an increase in pga gene expression,b-galatosidase-specific activity from a pgaA¢–¢lacZ trans-lational fusion containing the upstream non-coding regionthrough the initiation codon of pgaA was monitored in csrAwild-type and mutant strains. Disruption of csrA resultedin an approximately sevenfold increase in expression ofthis genomic fusion (Fig. 1C). A similar, though slightlygreater effect of csrA disruption was observed when thepgaA¢–¢lacZ fusion was carried on the plasmid pPGAZ4(Fig. 1D). Together, these experiments establish that CsrArepresses pga gene expression and the production ofPGA.
Effects of Csr components and other potential regulators on biofilm formation and pgaA¢–¢lacZ expression
The Csr signalling circuitry constitutes an autoregulatoryloop, wherein CsrA indirectly activates transcription ofcsrB and csrC, which specify the non-coding RNA antag-onists of CsrA (Fig. 1A; Gudapaty et al. 2001; Weilbacheret al., 2003). These effects of CsrA are mediated by theresponse regulator UvrY (Suzuki et al., 2002; Weilbacheret al., 2003). Disruption of csrA activates biofilm forma-tion, while disruption of pgaC, uvrY or csrB and csrC isinhibitory (Fig. 2A, lanes 1–6). The DcsrB DcsrC doublemutant was used in these experiments, rather than thesingle mutants, because CsrB levels exhibit a compensa-tory increase in response to csrC disruption and viceversa (Weilbacher et al., 2003). Importantly, csrA disrup-tion no longer represses (Fig. 2A, lanes 5 and 6) and uvrYor csrB overexpression no longer stimulates biofilm forma-tion in a pgaC mutant (Fig. 2A, compare lanes 7 and 8with 9 and 10; lanes 11 and 12 with 13 and 14).
The roles of csrB, csrC and uvrY in biofilm formationwere further defined by examining their effects on pgaA¢–¢lacZ expression. Disruption of csrB and csrC or uvrYmodestly (~30–40%), but significantly, reduced pgaA¢–¢lacZ expression (Fig. 2B, lanes 1–3). These modesteffects are consistent with previous results showing thatCsrA is present in the cell in excess of the binding capacityof its RNA antagonists (Gudapaty et al., 2001). Ectopicexpression of uvrY or csrB from a multicopy plasmidsignificantly stimulated pgaA¢–¢lacZ expression (Fig. 2B,
lanes 5–8). Together, the results of Figs 1 and 2 indicatethat Csr components influence biofilm formation by regu-lating pga gene expression.
Escherichia coli biofilm formation is subject to cataboliterepression by cyclic AMP (cAMP) and catabolite activatorprotein (CAP) (Jackson et al., 2002b). However, Dcrp andDcya mutations did not affect expression of the pga¢–¢lacZfusion (data not shown). We also examined effects ofycdT, a gene oriented divergently with respect topgaABCD (Fig. 3C). This gene is predicted to encode asignalling protein of the GGDEF domain family, membersof which are being increasingly recognized as regulatorsof biofilm formation and extracellular polysaccharide syn-thesis (reviewed in D’Argenio and Miller, 2004). A homol-
Fig. 2. Effects of Csr components on biofilm formation and pgaA¢–¢lacZ expression at 26∞C.A. Biofilm formation in polystyrene microtitre plates. Numbered bars represent strains as follows: 1, MG1655; 2, XWMGDC (DpgaC); 3, TWRGMG1655 (csrB::cam csrC::tet); 4, UYMG1655 (uvrY::cam); 5, TRMG1655 (csrA::kan); 6, TRXWMGDC (csrA::kan DpgaC); 7, MG1655[pBR322]; 8, MG1655[pUY14] (uvrY ++); 9, XWMGDC[pBR322] (DpgaC); 10, XWMGDC[pUY14] (DpgaC uvrY++); 11, MG1655[pCR2.1-TOPO]; 12, MG1655[pCB44] (csrB++); 13, XWMGDC[pCR2.1-TOPO]; 14, XWMGDC [pCB44]. Biofilms were grown in LB medium at 26∞C for 24 h.B. Expression of the pgaA¢–¢lacZ translational fusion. Strain (geno-type) identities are as follows: 1, XWZ4 (pgaA¢–¢lacZ); 2, XWBCZ4 (pgaA¢–¢lacZ csrB::cam csrC::tet); 3, XWUYZ4 (pgaA¢–¢lacZ uvrY::cam); 4, TRXWZ4 (pgaA¢–¢lacZ csrA::kan); 5, XWZ4[pBR322] (pgaA¢–¢lacZ); 6, XWZ4[pUY14] (pgaA¢–¢lacZ uvrY ++); 7, XWZ4[pDLE11] (pgaA¢–¢lacZ); 8, XWZ4[pCB44] (csrB++).The asterisks denote significant differences relative to the corre-sponding parent in each strain set (P < 0.001; Tukey multigroup analysis).
bb-G
alac
tosi
dase
act
ivit
y(A
420
per
mg
prot
ein)
Cry
stal
vio
let
stai
ning
(A
630)
1 2 3 4 5 6 7 8 9 10 11 12 13 14
1 2 3 4 5 6 7 8
A
B
*
*
*
*
*
*
**
* **
*
21.81.61.41.2
0.80.60.4
1.4
1
1.2
0.8
0.6
0.4
0.2
0
0.20
1

Repression of biofilm formation by CsrA 1651
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
ogous gene, hmsT, of Yersinia pestis is located in thesame genetic context as ycdT, and is needed at lowtemperature (26∞C) for biofilm formation and the produc-tion of a congo red- and calcofluor white-staining material(Kirillina et al., 2004). This material is dependent on thehmsHFRS (pgaABCD) locus and is most likely PGA (Itohet al., 2005). Nevertheless, targeted replacement of theycdT open reading frame in the E. coli chromosome didnot affect biofilm formation or pgaA¢–¢lacZ expression ateither 26∞C or 37∞C (data not shown).
Primer extension mapping of pgaA mRNA
To identify the transcriptional initiation site of the apparent
pgaABCD operon, primer extension analysis was per-formed using primers PEXT1, PEXT3 or PEXT5 (Table S1in Supplementary material). The csrA wild-type strainexhibited a single weak signal with the first two primers,corresponding to an A residue 234 nucleotides upstreamfrom the initiation codon of pgaA (Fig. 3A, lane 2; data notshown). Disruption of csrA generated no new signals, butincreased the level of this transcript (Fig. 3A, lane 1). Noproduct was detected using PEXT5, designed to anneal~220 nucleotides upstream of PEXT3 (data not shown).The -10 and -35 regions of pgaA exhibited 3 and 4 bp ofidentity with respect to the consensus sequences forthese elements, a spacing of 18 bp between the elements,and transcription initiated 7 bp downstream of the -10
Fig. 3. Analyses of pga mRNA.A. Primer extension products were generated with radiolabelled primer PEXT3 and resolved on a 6% polyacrylamide sequencing gel containing 6 M urea and detected by autoradiography. Products from csrA::kanR mutant and csrA wild-type transcripts are shown in lanes 1 and 2 respectively. The dideoxy-sequencing ladder (lanes G, A, T and C) was generated with PEX3 using pPGA372 as a template.B. Partial sequence of pgaA showing the promoter sequence, transcription initiation site, Shine–Dalgarno sequence, initiation codon and primers used in these experiments.C. pgaABCD locus indicating the regions of mRNA that were analysed by rt-qRT-PCR.D. Results of rt-qRT-PCR expressed as DCT (difference in cycle threshold) and fold difference in transcript levels in csrA wild type (MG1655) versus mutant (TRMG1655), as determined in Experimental procedures.E. Decay of pgaA mRNA in MG1655 and TRMG1655, as determined using rt-qRT-PCR.
G A T C 1 2
AAAACTTTTTCCGTAACCCTAAA
TTTTGAAAAAGGCATTGGGATTT
*
Region DCT Fold difference
pgaL-A 3.0 0.0 8.3 ±±±±±±±±
0.3pgaA-B 3.4 0.2 11.0 1.2pgaB-C 4.0 0.1 16.0 1.1pgaC-D 3.6 0.2 12.6 1.3
1
10
100
0 1 2 3 4 5 6 7
Time (min)
phoH pgaD pgaC pgaB pgaA ycdT
C
D
EB
ARegion pgaC-D pgaB-C pgaA-B pgaL-A
+1
-10 -35CGGAATTTATCTGATTTAATTATTTTAATCCTAATTTATTTTG
AAAAAGGCATTGGGATTTATGCCGTATTCCTGAAGATCCTCAT
CATTGGAATGGATTTTCGGGCGAGAAAAGGATTTTATATGGACACTCTGCTCATCATTTCTTCTTCTCATCATCAACAATTCACGT
PEXT3CTCTCTTCCGCGTTTAATAACGGATTATGAGGTGCAAAAATATCTTTCTTTTCAGTTACCTGTAATTAGATACAGAGAGAGATTTT
S-D initiation codonGGCAATACATGGAGTAATACAGGATGTATTCAAGTAGCAGAAA
PEXT1AAGGTGCCCGAAAACCAAATGGGCTTTGAAACTTCTTACTGCCGCATTTTTA
% p
gaA
mR
NA
rem
aini
ng

1652 X. Wang et al.
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
element (Fig. 3B). These findings are relatively typical ofE. coli promoters (Harley and Reynolds, 1987), and sug-gest that a single promoter drives pgaA transcription.
CsrA affects pgaABCD mRNA steady-state levels and chemical stability
To test whether CsrA co-ordinately regulates genes of theapparent pgaABCD operon, the junction regions betweenthe pga untranslated leader and pgaA (pgaL-A), pgaA andpgaB (pgaA-B), pgaB and pgaC (pgaB-C), pgaC andpgaD (pgaC-D) were analysed using quantitative real-timereverse transcription polymerase chain reaction (rt-qRT-PCR; Fig. 3C). As shown in Fig. 3D, all four regions wereamplified, suggesting that these four genes comprise anoperon. Furthermore, the steady-state levels of these seg-ments were higher in the csrA mutant than its wild-typeparent.
Because genes that are regulated by CsrA generallyshow effects on transcript stability, the chemical decayrate of pgaL-A was measured, following the addition ofrifampicin to csrA wild-type and mutant strains, usingrt-qRT-PCR (Experimental procedures). As shown inFig. 3E, decay of this transcript was more rapid in thewild-type strain (~0.4 min) compared with the isogeniccsrA mutant (~1.1 min), while the decay of the controlicd transcript (~2 min) was not affected by the disrup-tion of csrA (data not shown).
CsrA binds specifically to pgaA mRNA
The results described above suggest that CsrA interactswith the pga transcript. To characterize the interaction ofCsrA with pgaA mRNA, quantitative gel mobility shiftassays were performed with a transcript containing the234 nt untranslated leader and the first 26 nucleotides ofthe pgaA coding sequence (+1 to +260) and CsrA protein,purified as described previously (Baker et al., 2002). CsrAbinding to this transcript was detected as a distinct bandin native gels between 5 and 20 nM CsrA (Fig. 4, top). At40 nM CsrA we observed two distinct shifted complexes.Complete shifting was not observed until the concentrationof CsrA reached 80 nM; however, at this concentrationessentially all of the RNA was present in a third complex.A non-linear least-squares analysis of these data yieldedan estimated Kd value of 22 nM with a cooperativity coef-ficient of 2.7. This positive cooperativity suggested thatthe binding of CsrA to one binding site stimulated CsrAinteraction at additional sites. As the concentration of CsrAwas increased further, additional shifted species of highermolecular weights were observed (Fig. 4, top). This gelshift pattern suggested that multiple CsrA molecules werebound to each pgaA transcript at these higher concentra-tions, consistent with positive cooperativity.
The specificity of the CsrA–pgaA RNA interaction wasinvestigated by performing competition experiments withspecific (pgaA and CsrB) and non-specific (Bacillus sub-tilis trp leader) unlabelled RNA competitors (Fig. 4, bot-tom). CsrA was previously shown to bind specifically toCsrB RNA with a Kd value of approximately 0.5 nM (Weil-bacher et al., 2003). Both pgaA and CsrB RNAs wereeffective competitors, whereas the B. subtilis trp leaderRNA did not compete with the CsrA–pgaA RNA interac-tion. These results establish that CsrA binds specificallyto pgaA RNA.
CsrA competes with ribosome binding to the pga transcript
A toeprint analysis was performed to identify the posi-tion(s) of bound CsrA in the pgaA transcript (Figs 5 and6). As the concentration of CsrA was increased, fourCsrA-dependent toeprint bands were observed at posi-tions A250, U238, A215 and A207. Identification of sev-eral CsrA toeprints is consistent with the gel shift analysis,suggesting that multiple CsrA binding sites are present inthe pgaA transcript.
Similar toeprint experiments were performed to identifythe position of bound 30S ribosomal subunits. Weobserved a cluster of three consecutive tRNAfMet-
Fig. 4. Gel mobility shift analysis of CsrA–pgaA transcript interaction. 5’-end-labelled pgaA transcript (0.5 nM) was incubated with CsrA at the concentration shown beneath each lane. Gel shift assays were performed in the absence (top) or presence (bottom) of various com-petitor RNAs. The concentrations of specific (pgaA and CsrB) and non-specific (trpL from B. subtilis) competitor RNAs are shown beneath each lane. Positions of free (F) and bound (B) RNAs are shown.

Repression of biofilm formation by CsrA 1653
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
dependent toeprint bands that were centred 15 nt down-stream from the A of the AUG initiation codon (G251,A250 and A249) (Figs 5 and 6). Interestingly, the 3¢ mostCsrA toeprint band was also at A250. The positions of theCsrA and 30S ribosomal toeprints suggested that boundCsrA would inhibit ribosome binding. When CsrA wasbound to the pgaA transcript before the addition of 30Sribosomes and tRNAfMet, all four of the CsrA toeprint bandswere observed, whereas the ribosome toeprint was con-siderably reduced (Fig. 5). These results demonstrate thatbound CsrA inhibits ribosome binding to the pgaA tran-
script. When taken together with the in vivo expressionresults (Fig. 1), the toeprint results provide strong evi-dence that CsrA regulates translation of pgaA by blockingribosome binding.
Identification of CsrA binding sites in the pga transcript
The gel shift and toeprint results indicated that multiplemolecules of CsrA can bind to the pgaA transcript (Figs 4and 5). CsrA–pgaA RNA footprint experiments were car-ried out to identify the CsrA binding sites. Three single-strand-specific RNases were used as probes for thesestudies. As the concentration of CsrA was increased from0 to 4 mM, protection of several nucleotides from RNaseA (C- and U-specific), RNase T1 (G-specific) and RNaseT2 (A preference), cleavage was observed. The protectedresidues were clustered in three RNA segments (146–
Fig. 5. CsrA and 30S ribosomal subunit toeprints of pgaA RNA. The presence, as well as the order of addition, of CsrA and/or 30S ribo-somal subunits is shown at the top of each lane. Arrowheads and arrows indicate bands corresponding to CsrA and 30S ribosomal subunit toeprints respectively. The regions of the gel corresponding to the pgaA Shine–Dalgarno (S–D) sequence and start codon (Met) are shown. Sequencing lanes to reveal G, U, A and C residues are marked.
Fig. 6. Summary of the in vitro binding results and a comparison of the CsrA binding sites in pgaA. Arrowheads and small arrows mark the positions of the CsrA and 30S ribosomal subunit toeprints respec-tively. Nucleotides in which bound CsrA decreases (–) or increases (+) cleavage by sequence-specific ribonucleases are indicated. Large arrows mark the positions of the 3¢ boundaries. I to VI, six CsrA binding sites. Positions of the pgaA Shine–Dalgarno (S–D) sequence and start codon (Met) are shown. Numbering is from the start of transcrip-tion. A sequence comparison of the six CsrA binding sites in the pga leader is shown. M = A or C; N = A, C, G or U; K = G or U; W = A or U.

1654 X. Wang et al.
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
158, 199–206 and 217–247), again consistent with thepresence of multiple CsrA binding sites in the pgaA tran-script leader (Figs 6 and 7). Despite the presence of threeGGA motifs upstream of position 110, we did not observeany CsrA-dependent protection from cleavage in thisregion (data not shown).
A 3¢ boundary analysis was performed to more pre-cisely define the CsrA binding sites. The cut-offs for the3¢ boundaries were relatively sharp, revealing four bound-aries at positions 68, 85–87, 147 and 202–206 (Figs 6 and8). Additional boundary experiments were carried out inwhich the native gel was electrophoresed for a longer timeto resolve potential complexes at the top of the gel. In thiscase a 3¢ boundary was detected between 233 and 238(Fig. 6 and data not shown). Thus, the 3¢ boundary studiesidentified five likely CsrA binding sites in the pgaA leader.Attempts to identify the 5¢ boundaries of the CsrA bindingsites were unsuccessful.
Taken together, the in vitro experiments led to the iden-tification of six apparent CsrA binding sites in the
pgaABCD transcript. The presence of two CsrA bindingsites overlapping the Shine–Dalgarno sequence and startcodon, combined with the finding that bound CsrA inhibits30S ribosomal subunit binding, suggests that CsrArepresses biofilm formation by preventing translation ofpgaA, thereby promoting accelerated degradation of thepgaABCD transcript.
Mutations in CsrA binding sites overlapping the pgaA translation initiation region reduce CsrA-dependent repression
We suspected that the CsrA binding sites overlapping theShine–Dalgarno sequence and initiation codon of pgaAshould be especially important for repression, as CsrAbound at these sites should interfere with ribosome bind-ing. To examine the in vivo function of these two sites,substitution mutations were introduced at each individualbinding site and at both sites in the pgaA¢–¢lacZ transla-tional fusion in pPGAZ4, which is repressed by CsrA
Fig. 7. CsrA–pgaA RNA footprint. 5¢-end-labelled pgaA RNA was treated with RNase A, RNase T1 or RNase A. Reactions were carried out in the absence or presence of CsrA at the concentrations indicated at the top of the lanes (mM). Control lanes (C) in the absence of nuclease treatment, as well as partial alkaline hydrolysis (OH) and RNase T1 digestion (T1) ladders are shown. The RNase T1 ladder was generated under denaturing conditions so that every G residue in the transcript could be visualized. Nucleotides that were protected from RNase cleavage (–), as well as those enhanced for cleavage (+), by bound CsrA, are shown at the right of each panel. Numbering is from the start of transcription.

Repression of biofilm formation by CsrA 1655
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
(Fig. 1C). Previous studies have suggested a consensussequence for CsrA binding, ACA-GGAUG, with ACA andGGA motifs representing the most highly conservednucleotides (Baker et al., 2002; Dubey et al., 2003). TheGGA motifs of the two distal CsrA binding sequences
overlap the Shine–Dalgarno sequence and initiationcodon of pgaA mRNA. Nucleotide substitutions intro-duced at these sites eliminated the ACA motif of theCsrA consensus at the Shine–Dalgarno sequence andthe GGA motif near the initiation site (Table 1). Thesemodest changes were designed to cause minimal effectson translation, and remnants of the CsrA bindingsequences were retained at both sites. The expression ofpgaA¢–¢lacZ in parental and mutant plasmids was mea-sured in csrA wild-type and mutant strains (Table 1). Bothsubstitution mutations caused modest CsrA-independenteffects on expression, which most likely reflect changesin translation efficiency. We corrected for these effects bycalculating the ratio of expression in the csrA mutant ver-sus wild-type strain for each construct. As shown inTable 1, mutants in either of the individual binding sitesexhibited 70–80% of full CsrA repression, whereas thecombined mutant exhibited ~40% of full repression. Thus,60% of the repression was relieved in the construct con-taining four wild-type and two mutant CsrA binding sites.Although not dramatic, these results were reproducible,and establish a role for the latter two sites in CsrA-medi-ated repression in vivo.
Discussion
Biofilm development is associated with sweeping changesin gene expression (e.g. Whiteley et al., 2001; Beloinet al., 2004; Sauer et al., 2004), yet a variety of studiessupport the notion that a limited number of global regula-tory systems are central in governing this process. Basedon: (i) the quantitative effect of CsrA on biofilm formation(Fig. 2), arguably the strongest of any known regulator, (ii)
Fig. 8. 3¢ boundary analysis of CsrA–pgaA RNA interaction. Limited alkaline hydrolysis ladders of pgaA RNA were incubated with CsrA. CsrA–RNA complexes were separated from unbound RNA on a native gel and subsequently fractionated through 6% denaturing gels (shown). Lanes corresponding to distinct CsrA–pgaA RNA complexes purified from the native gel (B1, B2, B3, B4) and unbound RNA (U) are shown. Lanes corresponding to limited alkaline hydrolysis (OH) and RNase T1 digestion (T1) ladders are indicated. Arrows, positions of the boundaries. Roman numerals correspond to the CsrA binding sites depicted in Fig. 6. Numbering is from the start of transcription.
Table 1. Effects of site-directed mutations of CsrA binding sites onpga¢–¢lacZ expression.a
Plasmid (genotype)
Genotype of strainb
csrA+ csrA::kanRRepressionfactorc
pPGAZ4 (wild type) 0.76 ± 0.00 9.4 ± 0.1 12.4pSD23b (S–D site)d 1.2 ± 0.0 12 ± 0 10.0pINI21b (initiation site)d 0.81 ± 0.01 6.9 ± 0.0 8.5pSI5 (double mutant) 1.5 ± 0.0 8.0 ± 0.0 5.3
a. Cultures were grown in LB medium for 24 h at 37∞C with shakingand b-galactosidase-specific activity (A420/mg protein·h ± standarddeviation) was determined as described in Experimental procedures.b. The wild-type strain (csrA+) was CF7789.c. Repression factor represents the ratio of specific b-galactosidaseactivity from the csrA mutant versus the csrA wild-type strain.d. Nucleotide changes of CsrA binding sequences at the Shine–Dalgarno sequence (bold) or initiation codon (bold) of pgaA areindicated in lower case letters below. The highly conserved nucle-otides (GGA) of the CsrA binding site at each location are underlined.S–D site: CAATACATGGAGÆCAAattATGGAGInitiation site: ACAGGATGTATTÆACAaaATGTATT

1656 X. Wang et al.
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
its consistent role under diverse growth conditions and (iii)its involvement in a variety of strains and species (Jacksonet al., 2002a,b; Agladze et al., 2003; Wang et al., 2004),CsrA is a key regulator of E. coli biofilm development.Thus far, CsrA is the only regulator known to induce bio-film dispersal (Jackson et al., 2002a), although the mech-anism(s) responsible for this activity remains to bedetermined.
Our past studies have revealed that the Csr system ofE. coli co-ordinates metabolism and behaviour on a glo-bal scale (e.g. Romeo et al., 1993; Sabnis et al., 1995;Yang et al., 1996; Wei et al., 2001; Jackson et al., 2002a;Dubey et al., 2003). We estimate that >400 genes aremodulated directly or indirectly by CsrA (our unpublishedgenomic array studies). Here, CsrA is shown to regulatebiofilm formation by post-transcriptionally repressinggenes required for the production of a recently describedpolysaccharide adhesin, PGA (Figs 1–3; Wang et al.,2004). Several genes and operons that are regulated byCsrA are transcriptionally activated by cAMP-CAP(Romeo and Preiss, 1989; Yang et al., 1996: Wei et al.,2001; Dubey et al., 2003), and biofilm formation itself isactivated by cAMP-CAP (Jackson et al., 2002b); however,cAMP-CAP does not activate pgaA gene expression(data not shown).
While CsrA represses pgaABCD expression, and PGAsynthesis is necessary for CsrA and other Csr signallingcomponents to affect biofilm formation (Figs 1–3), this isnot the only role of CsrA in this process. Glycogen syn-thesis and turnover are needed for optimum biofilmformation (Jackson et al., 2002a), both of which arerepressed by CsrA (Yang et al., 1996). Thus, CsrA mayalso repress PGA biosynthesis indirectly, by inhibitingglgCAP expression, necessary for stationary-phase car-bon flux into glycogen and subsequent conversion of gly-cogen into glucose 1-phosphate, which can be used togenerate the precursor of PGA (Wang et al., 2004).
The signalling circuitry of the Csr system includes theBarA/UvrY TCS as well as the regulatory non-codingRNAs CsrB and CsrC (Suzuki et al., 2002; Weilbacheret al., 2003; Fig. 1A). Our present findings suggest thatthe full effect of the BarA-UvrY TCS on biofilm formationis mediated via CsrA (Fig. 2). They also suggest possibleregulatory circuitry and molecular mechanisms for a broadrange of host–microbe and microbial community interac-tions that involve Csr (Rsm) or BarA-UvrY (GacS-GacA)components (e.g. Mukherjee et al., 1996; Wong et al.,1998; Cui et al., 1999; Altier et al., 2000; Parkins et al.,2001; Haas and Keel, 2003; Liaw et al., 2003; Molofskyand Swanson, 2003; Whistler and Ruby, 2003; Zuberet al., 2003; Barnard et al., 2004; Goodman et al., 2004;Heurlier et al., 2004; Newton and Fray, 2004).
Previously, we have shown that CsrA binds to multiplesites in the glgCAP and cstA leader transcripts, and inhib-
its translation by blocking ribosome binding (Baker et al.,2002; Dubey et al., 2003). Footprinting and 3¢ boundaryanalyses identified six apparent CsrA binding sites in thepgaABCD leader transcript (Figs 6–8). This is the mostextensive arrangement of CsrA binding sites yet observedwithin an mRNA leader, and provides for relatively strongrepression in vivo (Fig. 1, Table 1). This is also the firstexample of CsrA binding to a site that overlaps the initia-tion codon (Figs 6–8). The CsrA–pga leader RNA gel shiftpattern indicated that multiple CsrA molecules are boundto each pga transcript at high CsrA concentrations(Fig. 4). Although the stoichiometry of these complexeswas not examined, we presume that the first shifted spe-cies contained one pga leader transcript and one CsrAdimer and that the additional shifted species containedmultiple CsrA dimers bound to RNA. However, we cannoteliminate the formal possibility that one or more of theslower-migrating species in the gel shift analysis mighthave resulted from CsrA–CsrA interactions. The findingthat CsrA interacts cooperatively with pga leader mRNAis consistent with the interaction of several CsrA dimersper pga transcript. Interestingly, CsrA binding to cstA,CsrB and CsrC RNAs was shown to be cooperative(Dubey et al., 2003; Weilbacher et al., 2003). In contrast,CsrA–glgCAP leader mRNA interaction was not coopera-tive (Baker et al., 2002).
Results from CsrA and 30S ribosomal toeprint experi-ments indicate that bound CsrA inhibits ribosome bindingto pgaA (Fig. 5). Thus, it appears that CsrA inhibits trans-lation of pgaA by a mechanism that is related to thoseused for translational inhibition of glgC and cstA. As afurther test of this model, we mutated the distal bindingsites (5 and 6), which overlap the Shine–Dalgarnosequence and initiation codon of pgaA, and examined theeffects of these mutations on expression of a pgaA¢–¢lacZtranslational fusion (Table 1). The finding that these twosites are required for full repression by CsrA in vivo, com-bined with the toeprint, footprint and boundary results,provides strong evidence in support of this model. Studieson the turnover and steady-state levels of the pgaABCDtranscript are also consistent with previous studies on theglgC message (Liu et al., 1995), and suggest that CsrAbinding, perhaps secondary to translation inhibition, leadsto mRNA destablization.
We previously published a consensus sequence of theknown or presumed CsrA targets in the CsrB, CsrC, glgleader and cstA leader transcripts (YANGGANR) (Dubeyet al., 2003). The CsrA binding sites that were identifiedin the pga leader fit this consensus reasonably well(Fig. 6); however, the binding sites in pga exhibited moreextended conservation further upstream: AUAMANGGAKW. GGA is the most highly conserved sequence inthe known E. coli CsrA binding sites. Site IV providesinformation that may prove to be useful in defining the

Repression of biofilm formation by CsrA 1657
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
rules for CsrA binding, as it establishes that the predom-inant GGA motif is not invariably present. Interestingly, theGGA motif is also conserved in the known or presumedbinding sites of the CsrA homologue of Pseudomonas sp.(Blumer et al., 1999; Valverde et al., 2004).
In addition to primary sequence conservation, it appearsthat RNA secondary structure plays an important role inCsrA–RNA interaction. The majority of the GGA motifs inthe putative CsrB and CsrC binding sites are in the loopsof predicted short hairpins (Liu et al., 1997; Weilbacheret al., 2003). In addition, one of the two published CsrAbinding sites in the glgCAP leader contains GGA in theloop of a hairpin; however, this pattern is not evident forany of the four binding targets in the cstA transcript (Dubeyet al., 2003). Interestingly, computer modelling usingMFOLD (Mathews et al., 1999; Zuker, 2003) predicts thatthe GGA motifs of binding sites I, V and VI in the pgaleader are located in the loops of short hairpins. Further-more, several of the GA residues in binding site IV arepredicted to be in the loop of a hairpin. Finally, the resultsof SELEX experiments establish that the presence of theGGA motif in the loop of a short hairpin is required forhigh-affinity CsrA–RNA interaction at solitary binding sites(A.K. Dubey, T. Romeo and P. Babitzke, unpubl.).
As predicted for any post-transcriptional regulation, theeffects of CsrA on pga expression are superimposed uponthose of other control mechanisms. The threefold effect ofCsrA on pga transcript half-life does not appear to fullyaccount for the several-fold increase in pgaABCD tran-script levels in a csrA mutant (Fig. 3). As inhibition oftranslation can lead to Rho-mediated transcription termi-nation, polarity in the csrA wild-type strain may contributeto some of the differences in transcript levels. Neverthe-less, it is also possible that CsrA may regulate a transcrip-tional regulator of pgaABCD. The pgaA¢–¢lacZ genomicfusion described in this study exhibits increased expres-sion at low growth temperatures (e.g. 26∞C versus 37∞C)and under elevated cation concentrations in both csrAwild-type and mutant strains (Goller et al., 2004). Themechanisms for these effects are being investigated togain further insight into the complex environmental cuesand signalling pathways that govern biofilm development.
Experimental procedures
Bacterial strains, phage, plasmids and growth conditions
The bacterial strains, phage and plasmids used in this studyare listed in Table 2. Unless otherwise indicated, bacteriawere routinely grown at 37∞C in Luria–Bertani (LB) medium(Miller, 1972). Biofilms were grown at 26∞C in LB or coloni-zation factor antigen (CFA) medium (1% casamino acids,0.15% yeast extract, 0.005% MgSO4, and 0.0005% MnCl2,pH 7.4; Jackson et al., 2002a). Media were supplementedwith antibiotics, as needed, at the following final concentra-
tions: ampicillin, 100 mg ml-1; chloramphenicol, 25 mg ml-1;kanamycin, 100 mg ml-1; and tetracycline, 10 mg ml-1.
Plasmid construction
Plasmid pCB44, which expresses csrB under the control ofthe lac promoter, was constructed using the vector pCR2.1-TOPO (Invitrogen, Carlsbad, CA). First, the csrB gene wasamplified by PCR from E. coli MG1655 genomic DNA usingthe primers lacZcsrBF2 and csrBR. The resulting PCR prod-uct contained 18 base pairs of lacZ sequence, the full-lengthtranscribed region of csrB, and 42 nt downstream of thepredicted 3¢-end of csrB transcription. Next, the lacZ pro-moter region was amplified from pUC19 using primers lacZFand csrBlacZR2. The latter primer is complementary insequence to primer lacZcsrBF2. The resulting PCR productcontains from -174 to +1 of lacZ and from +1 to +18 nt ofcsrB. The two PCR products were gel-purified, and used asoverlapping templates for a PCR reaction containing primerslacZF and csrBR. The resulting 0.58 kb PCR product was gel-purified and ligated into pCR2.1-TOPO by TA cloning to gen-erate plasmid pCB04. This clone contains duplicated lac pro-moter sequences, one coming from the insert and the otherfrom pCR2.1-TOPO vector. To delete the lac promoter frompCR2.1-TOPO, pCB04 was digested with ScaI/HindIII, whichgenerated a 1.6 kb fragment containing the lac promoter fromthe vector and a 2.7 kb fragment containing the PCR-ampli-fied region. The 1.6 kb fragment was digested with SapI toremove the lac promoter. The SapI and 2.7 kb fragmentswere treated with T4 DNA polymerase to create blunt-endsand ligated to yield the final product, pCB44. The insert wasconfirmed to be free of PCR-generated mutations by DNAsequencing.
To generate a plasmid to be used as a ‘vector control’ forpCB44, the pCR2.1-TOPO plasmid was digested with EcoRI,which excised a single 14 bp fragment of lacZ from pCR2.1-TOPO. The larger fragment was gel-purified, made blunt-ended by treatment with DNA polymerase Klenow fragment,and self-ligated to generate plasmid pDLE11, which containsan out-of-frame lacZ gene. This plasmid was transformed intoE. coli TOP10 (Invitrogen) and the resulting strain was con-firmed to be lacZ minus on an X-gal-containing LB agar.
Plasmid pAD4 contains the 234 nt pga leader and the first26 nt of the pgaA coding region (+1 to +260) cloned into theEcoRI/BamHI sites of the pTZ18U polylinker (Strategene).pAD4 was generated by amplifying the pga-specific chromo-somal region using primers PGA5¢ and PGA3¢ (Table S1 inSupplementary material), digestion of the PCR product withEcoRI and BamHI, and ligating into the same sites ofpTZ18U.
PGA determination
Strains were grown for PGA isolation in LB medium for 24 hat 37∞C with shaking at 250 r.p.m. A lysate from 3 l of eachculture was prepared and fractionated by gel filtration fast-protein liquid chromatography (FPLC) on Sephacryl S-300(HiPrep 26/60; Amersham Pharmacia Biotech) as previouslydescribed (Wang et al., 2004). Fractions (5 ml) were col-lected, hydrolysed and assayed in duplicate for hexosamine

1658 X. Wang et al.
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
content using 3-methyl-2-benzothiazolone hydrazone hydro-chloride (MBTH) with N-acetyl-D-glucosamine as a standard(Smith and Gilkerson, 1979). Extracts from four isogenicstrains were compared, MG1655, its csrA::kan mutant(TRMG1655), and their pgaC deletion strains, XWMGDC andTRXWMGDC. The pgaC gene encodes a predicted glycosyl-transferase (GT-2 family) and is required for PGA synthesis(Wang et al., 2004). The minor amounts of hexosamine-con-taining components that were detected from DpgaC mutantextracts were considered to be co-purifying contaminants ofPGA, and were subtracted from those of the isogenic pgawild-type strains to generate difference profiles. The com-plete experiment was conducted independently twice, withessentially the same results.
Construction of a pgaA¢–¢lacZ translational fusion and integration into the genome
A 690 bp fragment containing the upstream regulatory regionand initiation codon of pgaA was amplified by PCR using theprimer pair PGAZFW/PGAZRV and pPGA372 as a template(sequences provided in Table S1 in Supplementary material).
The PCR product was gel-purified, treated with T4 DNA poly-merase and polynucleotide kinase and cloned into the SmaIsite of pMB1034 (Silhavy et al., 1984). The resulting plasmid,pPGAZ4, was confirmed to contain the correct pgaA¢–¢lacZfusion, free of PCR-generated mutations, by DNA sequenc-ing (DNA Sequencing Facility, University of Arizona). ThepgaA¢–¢lacZ fusion in pPGAZ4 was moved into the E. coliCF7789 chromosome using lInCh1 and confirmed by PCRanalysis, as described previously (Boyd et al., 2000).
Construction of ycdT in-frame deletion mutant
The chromosomal ycdT gene was replaced with the chloram-phenicol acetyltransferase (cat) gene using targeted genesubstitutions (Datsenko and Wanner, 2000). The cat gene,flanked by FLP recognition target (FRT), was PCR-amplifiedfrom pKD3 using primers TH1P1 and TH2P2 (Table S1 inSupplementary material). The PCR products, containing thecat gene flanked by ycdT sequences at both ends, werepurified and then introduced by electroporation into arabi-nose-treated MG1655 containing pKD46. Transformantswere selected for chloramphenicol resistance (CamR), and
Table 2. Strains, plasmids and bacteriophages used in this study.
Strain, plasmid or phage Description or genotype Source or reference
E. coli K-12 strainsDH5a supE44 DlacU169 (f80lacZDM15) hsdR17 relA1 recA1 endA1 gyrA96 thi-1 Ausubel et al. (1989)MG1655 F– l- Michael CashelTRMG1655 MG1655 csrA::kan Romeo et al. (1993)CF7789 MG1655 DlacI-Z (MluI) Michael CashelXWMGDC MG1655 DpgaC Wang et al. (2004)TRXWMGDC TRMG1655 DpgaC Wang et al. (2004)XWMGDT MG1655 DycdT::cam This studyTWRGMG1655 MG1655 DcsrB::cam DcsrC::tet Weilbacher et al. (2003)UYMG1655 MG1655 uvrY::cam Suzuki et al. (2002)XWZ4 CF7789 pgaA¢–¢lacZ This studyTRXWZ4 XWZ4 csrA::kan This studyXWBCZ4 XWZ4 DcsrB::cam DcsrC::tet This studyXWUYZ4 XWZ4 uvrY::cam This studyXWYTZ4 XWZ4 DycdT::cam This studyXWCYAZ4 XWZ4 Dcya::kan This studyXWCRPZ4 XWZ4 Dcrp::cam This studyML2 met gal hsdKR supE supF Dcya::kan Jackson et al. (2002b)SA2777 F-rpsl relA Dcrp::cam Jackson et al. (2002b)
PlasmidspMLB1034 ¢lacZ translational fusion vector Silhavy et al. (1984)pPGAZ4 pMLB1034 f (pgaA¢–¢lacZ) This studypINI21 Mutant of pPGAZ4 This studypSD23 Mutant of pPGAZ4 This studypSI5 Mutant of pPGAZ4 This studypPGA372 pgaABCD in pUC19 Wang et al. (2004)pKD46 For arabinose induction of l Red system Datsenko and Wanner (2000)pKD3 Contains the cat gene Datsenko and Wanner (2000)pUY14 uvrY in pBR322 Suzuki et al. (2002)pBR322 Cloning vector Sambrook et al. (1989)pTZ18U Cloning vector StratagenepAD4 +1 to +260 of pgaA in pTZ18U This studypCR2.1-TOPO Cloning vector InvitrogenpCB44 csrB gene in pCR2.1-TOPO This studypDLE11 pCR2.1-TOPO with disrupted lacZ gene This study
BacteriophageP1vir Strictly lytic P1 Carol GrosslInCh1 For genomic insertions Boyd et al. (2000)

Repression of biofilm formation by CsrA 1659
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
their insertion sites were confirmed by PCR. The CamR
marker was subsequently moved into TRMG1655 and strainscontaining pgaA¢–¢lacZ fusion by P1vir transduction.
Isolation of total RNA
Total cellular RNA was prepared using the MasterPurTM RNAPurification Kit (Epicentre Technologies, Madison, WI)according to the manufacturer’s instructions. DNA wasremoved from the RNA preparations that were to be analysedby RT-PCR, using two DNase I digestion steps, as recom-mended (Epicentre Technologies). RNA was quantified by itsabsorbance at 260 and 280 nm, and rRNA integrity wasassessed on formaldehyde agarose gels. RNA samples werestored at -80∞C in 70% ethanol.
Primer extension analysis
Cells were grown in LB at 37∞C to the exponential phase ofthe growth (A600 ª 0.4), harvested and total RNA was pre-pared. Three primers that anneal at position +44 to +65, -78to -100 and -324 to -343 (PEXT1, PEXT3, PEXT5, respec-tively; Table S1 in Supplementary material) relative to thetranslation initiation codon of pgaA were 5¢-end-labelled with[g-32P]-ATP (3000 Ci mmole-1, NEN Life Science Products)using T4 polynucleotide kinase (Promega) as described inthe manufacturer’s manual. Unincorporated [g-32P]-ATP wasremoved using MicroSpinTM G-25 Column (Amersham Bio-sciences). Approximately 6 pmol of labelled primer wasadded to 37 mg of total RNA. Subsequent cDNA synthesiswas performed using the ThermoScriptTM RT-PCR system(Invitrogen) according to the manufacturer’s instructions. Thesame labelled primer and template pPGA372A was used togenerate a corresponding DNA sequencing ladder using theSequiTherm EXCELTM II DNA Sequencing Kit (Epicentre).The primer extension products were analysed alongside theDNA sequencing ladder on a 6% polyacrylamide sequencinggel containing 6 M urea. The sequencing gel was dried andsubjected to autography using a phosphorimager (Storm®Gel and Blot Imaging system, Amersham Bioscience).
rt-qRT-PCR of pgaA-D transcript levels
To measure the steady-state level of pgaA-D transcripts, csrAwild-type (MG1655) and mutant (TRMG1655) strains weregrown at 37∞C in LB medium to exponential phase(OD600 ª 0.7). Cells from 1 ml of culture were harvested andfrozen in ethanol-dry ice. Total RNA was isolated, and thesteady levels of pgaL-A, pgaA-B, pgaB-C and pgaC-D weredetermined by rt-qRT-PCR analyses using the primer pairsPGART1/PAGRT2, PGABFW/INTERRV, PGACFW/PGABRVand PGACDFW/PGACDRV respectively (Table S1 in Supple-mentary material). rt-qRT-PCR was performed using theiScript one-step RT-PCR Kit with SYBR Green, according tothe manufacturer’s guidelines (Bio-Rad, Hercules, CA).Reactions for each sample were performed in triplicate using100 ng RNA template. A reaction lacking reverse tran-scriptase was included for each sample, which served as acontrol for DNA contamination. Reactions were conductedusing the iCycler iQ real-time system (Bio-Rad) under the
following conditions: 65∞C – 5 min, 53∞C – 60 min, 95∞C –5 min (95∞C – 10 s, 57∞C – 30 s) for 45 cycles. The differencein cycle threshold (DCT) between samples from the csrA wild-type and mutant strains was calculated. The PCR productidentities were confirmed by electrophoresis on 1% agarosegels with ethidium bromide staining and product uniformitywas determined using melting curves (iCycler InstructionManual, Bio-Rad). Previous studies have shown that theexpression level of icd mRNA is not affected by CsrA (Weiet al., 2000). Therefore, this transcript was amplified as acontrol for each RNA sample using primers icdRT1 andicdRT2. The resulting DCT values for icd were subtracted asbackground corrections from the pga DCT values (Livak andSchmittgen, 2001). These experiments were conducted inde-pendently twice, with similar results, and the mean values ofthe two experiments were determined.
Analysis of pgaL-A mRNA stability using rt-qRT-PCR
To analyse the stability of pgaL-A or icd mRNA (control), csrAwild-type (MG1655) and mutant TRMG1655 strains weregrown at 37∞C in LB medium to exponential phase(OD600 ª 0.5), and treated with rifampicin at a final concen-tration of 200 mg ml-1 to inhibit transcription initiation. Sam-ples (1 ml) of TRMG1655 culture were collected at 0, 2, 4, 6,8 and 12 min following rifampicin addition. Samples ofMG1655 culture were collected at 0, 0.5, 1, 1.5 and 2 min foranalysing pgaL-A mRNA or 0, 2, 4, 6, 8 and 12 min for icdmRNA following rifampicin addition. The cells were harvestedand frozen in ethanol-dry ice, allowing no more than 40 s toelapse between sampling and freezing. Total RNA was iso-lated, and the amount of pgaL-A or icd mRNA in each samplewas determined using rt-qRT-PCR as described above andquantified based on a standard curve (below). The percent-age of remaining mRNA through the time-course was calcu-lated. To generate a standard curve, pgaL-A or icd DNAtemplate was PCR-amplified from chromosomal DNA ofMG1655 using the same prime pairs in rt-qRT-PCR and gel-purified. Standard reactions were set up containing 10-foldserial dilutions of pgaL-A or icd DNA template. The calculatedCt values were plotted versus the log of the initial amount ofpgaL-A or icd molecules (2–2 ¥ 108) to generate the standardcurve.
Gel mobility shift assay
Gel mobility shift assays followed previously published pro-cedures (Baker et al., 2002; Dubey et al., 2003; Weilbacheret al., 2003). Plasmid pAD4 contains the 234 nt pgaA leaderand the first 26 nt of the coding region (+1 to +260) relativeto the start of pgaA transcription cloned into the pTZ18Upolylinker (Strategene). This plasmid was used as a templateto generate transcripts used for in vitro studies. RNA wassynthesized in vitro with the Ambion MEGAscript kit andplasmid pAD4, which had been linearized with BamHI as thetemplate. Gel-purified RNA was dephosphorylated with calfintestinal alkaline phosphatase and subsequently 5¢-end-labelled using [g-32P]-ATP and polynucleotide kinase.Labelled RNA was gel-purified, suspended in TE (10 mMTris-HCl, pH 8.0, 1 mM EDTA) and renatured by heating to

1660 X. Wang et al.
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
85∞C and slowly cooling to room temperature. Binding reac-tion mixtures (10 ml) contained 10 mM Tris-HCl, pH 7.5,100 mM KCl, 10 mM MgCl2, 32.5 ng of yeast RNA, 10%glycerol, 20 mM dithiothreitol, 4 U of RNase inhibitor(Ambion), 0.5 nM 5¢-end-labelled pgaA RNA, various con-centrations of purified CsrA, and 0.1 mg of xylene cyanol permillilitre. Competition assay mixtures also contained unla-belled RNA competitor (see Results for details). Reactionmixtures were incubated at 37∞C for 30 min to allow CsrA–pgaA RNA complex formation. Samples were then fraction-ated on native 12% polyacrylamide gels. Radioactive bandswere visualized with a phosphorimager (Molecular Dynam-ics). Free and bound RNA species were quantified withImageQuant software (Molecular Dynamics), and the appar-ent equilibrium binding constant (Kd) for CsrA–pgaA RNAcomplex formation was calculated.
Toeprint assay
Toeprint assays were carried out by modifying published pro-cedures (Hartz et al., 1988; Baker et al., 2002; Dubey et al.,2003). Unlabelled pAD4-derived pga transcripts used in thisanalysis were generated as described for the gel mobility shiftassay. Gel-purified pga RNA (250 nM) in TE was renaturedand hybridized to a 5¢-end-labelled DNA oligonucleotide(250 nM) complementary to the 3¢-end of the transcript byheating to 85∞C and slowly cooling to room temperature.Toeprint assays were performed with 1, 2 and 4 mM CsrAand/or 250 nM 30S ribosomal subunits and 10 mM tRNAfMet.Toeprint reactions (20 ml) contained 2 ml of the hybridizationmixture, 375 mM each dNTP and 10 mM dithiothreitol in toe-print buffer (10 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 60 mMNH4OAc, 6 mM 2-mercaptoethanol). Reaction mixtures con-taining CsrA were incubated for 30 min at 37∞C to allowCsrA–RNA complex formation. 30S ribosomal subunit toe-print reactions were carried out by incubating RNA, 30Sribosomal subunits and tRNAfMet in toeprint buffer asdescribed previously (Hartz et al., 1988). Following the addi-tion of 0.6 U of avian myeloblastosis virus reverse tran-scriptase (Roche) the reaction mixture was further incubatedfor 15 min at 37∞C. Reactions were terminated by adding12 ml of stop solution (85% formamide, 70 mM EDTA, 0.1¥Tris-Borate-EDTA, 0.025% xylene cyanol, 0.025% bromophe-nol blue). Samples were fractionated through 6% sequencinggels. Sequencing reactions were carried out using pAD4 asthe template and the same end-labelled oligonucleotide as aprimer.
RNA footprinting
5¢-end-labelled pga transcripts were generated as describedfor the gel shift assay. Titrations of RNase T1 (Roche), RNaseT2 (Sigma) and RNase A (Ambion) were performed to opti-mize the amount of enzyme to prevent multiple cleavages inany one transcript. Binding reaction mixtures (10 ml) contain-ing 2 nM pga RNA, various concentrations of CsrA wereotherwise identical to those described for the gel mobility shiftassay. RNase T1 (0.2 U), RNase T2 (0.015 U) or RNase A(1 ¥ 10-6 mg m-1l) was added in separate reactions, and thereaction mixtures were further incubated for 15 min at 37∞C.
Reactions were terminated by adding 10 ml of stop solution(95% formamide, 20 mM EDTA, 0.025% sodium dodecyl sul-phate, 0.025% xylene cyanol, 0.025% bromophenol blue).Partial alkaline hydrolysis and RNase T1 digestion ladderswere prepared as described previously (Bevilacqua and Bev-ilacqua, 1998). Samples were fractionated through 6% dena-turing polyacrylamide gels.
Boundary analysis
3¢ boundary experiments were carried out by modifying apreviously published procedure (Dubey et al., 2003). Todetermine the 3¢ boundary required for CsrA binding, 5¢-end-labelled RNA was generated as described above. To gener-ate 5¢-labelled alkaline hydrolysis ladders, 50 ml of RNA sam-ples (10 pmol) were incubated for 4 min at 95∞C in alkalinehydrolysis buffer (100 mM NaHCO3-Na2CO3, pH 9.0, 2 mMEDTA) and then recovered by ethanol precipitation. Hydroly-sed RNAs (70 nM) were mixed with 1 mM CsrA (20 ml reac-tion volume) and incubated for 30 min at 37∞C to allow CsrA–pga RNA complex formation. Samples were fractionatedthrough 8% native polyacrylamide gels. Bound and unboundtranscripts were visualized by autoradiography, excised fromthe gel and subsequently eluted from the gel. RNAs wereethanol precipitated and fractionated through 6% denaturingpolyacrylamide gels. Partial alkaline hydrolysis and RNaseT1 digestion ladders of the same transcript were used asmolecular size standards.
Site-directed mutagenesis of CsrA binding sites in pga leader region
The pPGAZ4 plasmid containing the pgaA¢–¢lacZ transla-tional fusion was used as a template for site-directedmutagenesis of two potential CsrA binding sequences in thepga leader region using QuickChange® II Site-DirectedMutagenesis Kit (Stratagene). The nucleotides at position+217 to +219 relative to transcription start (+1) within theCsrA binding site that overlaps the pgaA Shine–Dalgarnosequence were changed from TAC to ATT using primers SD-FW and SD-RV, generating plasmid pSD23b. Two Gs at posi-tion +233 and +234 within the CsrA binding site that overlapspgaA translation initiation codon were replaced with two Aresidues using primers INI-FW and INI-RV, resulting in plas-mid pINI21. To introduce substitutions at both CsrA bindingsites, pSD23b was used as a template, and the two Gs atpositions +233 and +234 were replaced with two A residuesusing primers INI-FW and INI-RV, creating plasmid pSI5. Thesequences of the mutagenic primers are listed in Table S1 inSupplementary material. The pgaA¢–¢lacZ translationalfusions of all the plasmid constructs were confirmed to befree of PCR-generated mutations by DNA sequencing anal-yses conducted at SeqWright DNA Technology Service(Houston, TX).
b-Galactosidase activity and total protein assays
b-Galactosidase was determined as described previously(Romeo et al., 1990), except that the activity (A420 per mgprotein) was based on a 1 h incubation time, due to low

Repression of biofilm formation by CsrA 1661
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
expression of pgaA¢–¢lacZ. Total cellular protein was mea-sured by the bicinchoninic acid method using bovine serumalbumin as the standard (Smith et al., 1985).
Quantitative biofilm assay
Biofilms were assayed using crystal violet staining, asdescribed previously (Jackson et al., 2002a). Overnight cul-tures were diluted 1:100 into fresh medium and grown in 96-well microtitre plates for these assays. At least six replicaswere conducted for each sample, and each experiment wasperformed at least twice. The results were calculated asaverages and standard errors of two or more experiments.Tukey multigroup analysis (StatView; SAS Institute, Cary, NC)was used for statistical analysis of data.
Genetics and molecular biology
Standard procedures were used for transformation, transduc-tion, plasmid isolation, restriction digestion and ligation(Miller, 1972; Sambrook et al., 1989).
Acknowledgements
We wish to thank Lindsay Stevenson for help with the con-struction of the pga¢–¢lacZ fusion and Bhavana Achary forconstruction of pAD4. These studies were funded in part bythe National Institutes of Health (GM066794; GM59969).Kane Biotech may develop applications related to the findingsherein. T. Romeo serves as Chief Scientific Advisor for, ownsequity in, and may receive royalties from this company. Theterms of this arrangement have been reviewed and approvedby Emory University in accordance with its conflict of interestpolicies.
Supplementary material
The following supplementary material is available for thisarticle online:Table S1. Oligonucleotides used in this study.
References
Agladze, K., Jackson, D., and Romeo, T. (2003) Periodicityof cell attachment patterns during Escherichia coli biofilmdevelopment. J Bacteriol 185: 5632–5638.
Altier, C., Suyemoto, M., Ruiz, A.I., Burnham, K.D., and Mau-rer, R. (2000) Characterization of two novel regulatorygenes affecting Salmonella invasion gene expression. MolMicrobiol 35: 635–646.
Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D.,Seidman, J.G., Smith, J.A., and Struhl, K. (eds) (1989)Current Protocols in Molecular Biology. New York: JohnWiley & Sons.
Baker, C.S., Morozov, I., Suzuki, K., Romeo, T., andBabitzke, P. (2002) CsrA regulates glycogen biosynthesisby preventing translation of glgC in Escherichia coli. MolMicrobiol 44: 1599–1610.
Barnard, F.M., Loughlin, M.F., Fainberg, H.P., Messenger,M.P., Ussery, D.W., Williams, P., and Jenks, P.J. (2004)Global regulation of virulence and the stress response byCsrA in the highly adapted human gastric pathogen Heli-cobacter pylori. Mol Microbiol 51: 15–32.
Beloin, C., Valle, J., Latour-Lambert, P., Faure, P., Kzremin-ski, M., Balestrino, D., et al. (2004) Global impact of maturebiofilm lifestyle on Escherichia coli K-12 gene expression.Mol Microbiol 51: 659–674.
Bevilacqua, J.M., and Bevilacqua, P.C. (1998) Thermody-namic analysis of an RNA combinatorial library containedin a short hairpin. Biochemistry 37: 15877–15884.
Blumer, C., Heeb, S., Pessi, G., and Haas, D. (1999) GlobalGacA-steered control of cyanide and exoprotease produc-tion in Pseudomonas fluorescens involves specific ribo-some binding sites. Proc Natl Acad Sci USA 96: 14073–14078.
Boyd, D., Weiss, D.S., Chen, J.C., and Beckwith, J. (2000)Towards single-copy gene expression systems makinggene cloning physiologically relevant: lambda InCh, a sim-ple Escherichia coli plasmid-chromosome shuttle system.J Bacteriol 182: 842–847.
Branda, S.S., Gonzalez-Pastor, J.E., Dervyn, E., Ehrlich,S.D., Losick, R., and Kolter, R. (2004) Genes involved information of structured multicellular communities by Bacil-lus subtilis. J Bacteriol 186: 3970–3979.
Costerton, J.W., Lewandowski, Z., Caldwell, D.E., Korber,D.R., and Lappin-Scott, H.M. (1995) Microbial biofilms.Annu Rev Microbiol 49: 711–745.
Costerton, J.W., Stewart, P.S., and Greenberg, E.P. (1999)Bacterial biofilms: a common cause of persistent infec-tions. Science 284: 1318–1322.
Cui, Y., Mukherjee, A., Dumenyo, C.K., Liu, Y., and Chat-terjee, A.K. (1999) rsmC of the soft-rotting bacteriumErwinia carotovora subsp. carotovora negatively controlsextracellular enzyme and harpin(Ecc) production and vir-ulence by modulating levels of regulatory RNA (rsmB)and RNA-binding protein (RsmA). J Bacteriol 181: 6042–6052.
D’Argenio, D.A., and Miller, S.I. (2004) Cyclic di-GMP as abacterial second messenger. Microbiology 150: 2497–2502.
Danese, P.N., Pratt, L.A., Dove, S.L., and Kolter, R. (2000)The outer membrane protein, antigen 43, mediates cell-to-cell interactions within Escherichia coli biofilms. Mol Micro-biol 37: 424–432.
Datsenko, K.A., and Wanner, B.L. (2000) One-step inactiva-tion of chromosomal genes in Escherichia coli K-12 usingPCR products. Proc Natl Acad Sci USA 97: 6640–6645.
Donlan, R.M., and Costerton, J.W. (2002) Biofilms: survivalmechanisms of clinically relevant microorganisms. ClinMicrobiol Rev 15: 167–193.
Dorel, C., Vidal, O., Prigent-Combaret, C., Vallet, I., andLejeune, P. (1999) Involvement of the Cpx signal transduc-tion pathway of E. coli in biofilm formation. FEMS MicrobiolLett 178: 169–175.
Dubey, A.K., Baker, C.S., Suzuki, K., Jones, A.D., Pandit, P.,Romeo, T., and Babitzke, P. (2003) CsrA regulates trans-lation of the Escherichia coli carbon starvation gene, cstA,by blocking ribosome access to the cstA transcript. J Bac-teriol 185: 4450–4460.

1662 X. Wang et al.
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
Ferrieres, L., and Clarke, D.J. (2003) The RcsC sensorkinase is required for normal biofilm formation in Escheri-chia coli K-12 and controls the expression of a regulon inresponse to growth on a solid surface. Mol Microbiol 50:1665–1682.
Gerstel, U., Park, C., and Romling, U. (2003) Complex reg-ulation of csgD promoter activity by global regulatory pro-teins. Mol Microbiol 49: 639–654.
Goller, C., Wang, X., Desplas, R., and Romeo, T. (2004)Role of the sodium pump regulator NhaR in Escherichiacoli biofilm formation. American Society for Microbiology104th General Meeting. New Orleans, LA. 23–27 May2004.
Goodman, A.L., Kulasekara, B., Rietsch, A., Boyd, D., Smith,R.S., and Lory, S. (2004) A signaling network reciprocallyregulates genes associated with acute infection andchronic persistence in Pseudomonas aeruginosa. Dev Cell7: 745–754.
Gotz, F. (2002) Staphylococcus and biofilms. Mol Microbiol43: 1367–1378.
Gudapaty, S., Suzuki, K., Wang, X., Babitzke, P., andRomeo, T. (2001) Regulatory interactions of Csr compo-nents: the RNA binding protein CsrA activates csrBtranscription in Escherichia coli. J Bacteriol 183: 6017–6027.
Haas, D., and Keel, C. (2003) Regulation of antibiotic pro-duction in root-colonizing Pseudomonas spp. and rele-vance for biological control of plant disease. Annu RevPhytopathol 41: 117–153.
Hall-Stoodley, L., Costerton, J.W., and Stoodley, P. (2004)Biofilms: from the natural environment to infectious dis-eases. Nat Rev Microbiol 2: 95–108.
Harley, C.B., and Reynolds, R.P. (1987) Analysis of E. colipromoter sequences. Nucleic Acids Res 15: 2343–2361.
Hartz, D., McPheeters, D.S., Traut, R., and Gold, L. (1988)Extension inhibition analysis of translation initiation com-plexes. Methods Enzymol 164: 419–425.
Heurlier, K., Williams, F., Heeb, S., Dormond, C., Pessi, G.,Singer, D., et al. (2004) Positive control of swarming, rham-nolipid synthesis, and lipase production by the posttran-scriptional RsmA/RsmZ system in Pseudomonasaeruginosa PAO1. J Bacteriol 186: 2936–2945.
Hoch, J.A. (2000) Two-component and phosphorelay signaltransduction. Curr Opin Microbiol 3: 165–170.
Hogan, D., and Kolter, R. (2002) Why are bacteria refractoryto antimicrobials? Curr Opin Microbiol 5: 472–477.
Itoh, Y., Wang, X., Hinnebusch, B.J., Preston, J.F., III, andRomeo, T. (2005) Depolymerization of b-1,6-N-acetyl-D-glucosamine disrupts the integrity of diverse bacterial bio-films. J Bacteriol 187: 382–387.
Jackson, D.W., Suzuki, K., Oakford, L., Simecka, J.W., Hart,M.E., and Romeo, T. (2002a) Biofilm formation and dis-persal under the influence of the global regulator CsrA ofEscherichia coli. J Bacteriol 184: 290–301.
Jackson, D.W., Simecka, J.W., and Romeo, T. (2002b)Catabolite repression of Escherichia coli biofilm formation.J Bacteriol 184: 3406–3410.
Kaplan, J.B., Ragunath, C., Ramasubbu, N., and Fine, D.H.(2003) Detachment of Actinobacillus actinomycetemcomi-tans biofilm cells by an endogenous beta-hexosaminidaseactivity. J Bacteriol 185: 4693–4698.
Kirillina, O., Fetherston, J.D., Bobrov, A.G., Abney, J., andPerry, R.D. (2004) HmsP, a putative phosphodiesterase,and HmsT, a putative diguanylate cyclase, control Hms-dependent biofilm formation in Yersinia pestis. Mol Micro-biol 54: 75–88.
Liaw, S.J., Lai, H.C., Ho, S.W., Luh, K.T., and Wang, W.B.(2003) Role of RsmA in the regulation of swarming motilityand virulence factor expression in Proteus mirabilis. J MedMicrobiol 52: 19–28.
Liu, M.Y., Yang, H., and Romeo, T. (1995) The product of thepleiotropic Escherichia coli gene csrA modulates glycogenbiosynthesis via effects on mRNA stability. J Bacteriol 177:2663–2672.
Liu, M.Y., Gui, G., Wei, B., Preston, J.F., 3rd, Oakford, L.,Yuksel, U., et al. (1997) The RNA molecule CsrB binds tothe global regulatory protein CsrA and antagonizes itsactivity in Escherichia coli. J Biol Chem 272: 17502–17510.
Livak, K.J., and Schmittgen, T.D. (2001) Analysis of relativegene expression data using real-time quantitative PCR andthe 2-DDCT method. Methods 25: 402–408.
Mah, T.F., Pitts, B., Pellock, B., Walker, G.C., Stewart, P.S.,and O’Toole, G.A. (2003) A genetic basis for Pseudomonasaeruginosa biofilm antibiotic resistance. Nature 426: 306–310.
Mathews, D.H., Sabina, J., Zuker, M., and Turner, D.H.(1999) Expanded sequence dependence of thermody-namic parameters improves prediction of RNA secondarystructure. J Mol Biol 288: 911–940.
Miller, J. (1972) Experiments in Molecular Genetics. ColdSpring Harbor, NY: Cold Spring Harbor Laboratory Press.
Molofsky, A.B., and Swanson, M.S. (2003) Legionellapneumophila CsrA is a pivotal repressor of transmissiontraits and activator of replication. Mol Microbiol 50: 445–461.
Mukherjee, A., Cui, Y., Liu, Y., Dumenyo, C.K., and Chat-terjee, A.K. (1996) Global regulation in Erwinia speciesby Erwinia carotovora rsmA, a homologue of Escherichiacoli csrA: repression of secondary metabolites, pathoge-nicity and hypersensitive reaction. Microbiology 142:427–434.
Newton, J.A., and Fray, R.G. (2004) Integration of environ-mental and host-derived signals with quorum sensing dur-ing plant–microbe interactions. Cell Microbiol 6: 213–224.
O’Toole, G.A., and Kolter, R. (1998) Initiation of biofilm for-mation in Pseudomonas fluorescens WCS365 proceedsvia multiple, convergent signalling pathways: a geneticanalysis. Mol Microbiol 28: 449–461.
O’Toole, G.A., Gibbs, K.A., Hager, P.W., Phibbs, P.V., Jr, andKolter, R. (2000) The global carbon metabolism regulatorCrc is a component of a signal transduction pathwayrequired for biofilm development by Pseudomonas aerug-inosa. J Bacteriol 182: 425–431.
Otto, K., and Silhavy, T.J. (2002) Surface sensing and adhe-sion of Escherichia coli controlled by the Cpx-signalingpathway. Proc Natl Acad Sci USA 99: 2287–2292.
Parkins, M.D., Ceri, H., and Storey, D.G. (2001) Pseudomo-nas aeruginosa GacA, a factor in multihost virulence, isalso essential for biofilm formation. Mol Microbiol 40:1215–1226.
Pratt, L.A., and Kolter, R. (1998) Genetic analysis of Escher-

Repression of biofilm formation by CsrA 1663
© 2005 Blackwell Publishing Ltd, Molecular Microbiology, 56, 1648–1663
ichia coli biofilm formation: roles of flagella, motility, chemo-taxis and type I pili. Mol Microbiol 30: 285–293.
Prigent-Combaret, C., Brombacher, E., Vidal, O., Ambert, A.,Lejeune, P., Landini, P., and Dorel, C. (2001) Complexregulatory network controls initial adhesion and biofilm for-mation in Escherichia coli via regulation of the csgD gene.J Bacteriol 183: 7213–7223.
Romeo, T. (1998) Global regulation by the small RNA-bindingprotein CsrA and the non-coding RNA molecule CsrB. MolMicrobiol 29: 1321–1330.
Romeo, T., and Preiss, J. (1989) Genetic regulation of gly-cogen biosynthesis in Escherichia coli: in vitro effects ofcyclic AMP and guanosine 5¢-diphosphate 3¢-diphosphateand analysis of in vivo transcripts. J Bacteriol 171: 2773–2782.
Romeo, T., Black, J., and Preiss, J. (1990) Genetic regulationof glycogen biosynthesis in Escherichia coli: in vitro effectsof catabolite repression and stringent response systems inglg gene expression. Curr Micribiol 21: 131–137.
Romeo, T., Gong, M., Liu, M.Y., and Brun-Zinkernagel, A.M.(1993) Identification and molecular characterization ofcsrA, a pleiotropic gene from Escherichia coli that affectsglycogen biosynthesis, gluconeogenesis, cell size, andsurface properties. J Bacteriol 175: 4744–4755.
Sabnis, N.A., Yang, H., and Romeo, T. (1995) Pleiotropicregulation of central carbohydrate metabolism in Escheri-chia coli via the gene csrA. J Biol Chem 270: 29096–29104.
Sambrook, J., Fritsch, E.F., and Maniatis, T. (eds) (1989)Molecular Cloning: A Laboratory Manual. Cold Spring Har-bor, NY: Cold Spring Harbor Laboratory.
Sauer, K., Cullen, M.C., Rickard, A.H., Zeef, L.A., Davies,D.G., and Gilbert, P. (2004) Characterization of nutrient-induced dispersion in Pseudomonas aeruginosa PAO1 bio-film. J Bacteriol 186: 7312–7326.
Silhavy, T.J., Berman, M.L., and Enquist, L.W. (1984) Exper-iments with Gene Fusions. Cold Spring Harbor, NY: ColdSpring Harbor Laboratory Press.
Smith, R.L., and Gilkerson, E. (1979) Quantitation of gly-cosaminoglycan hexosamine using 3-methyl-2-benzothia-zolone hydrazone hydrochloride. Anal Biochem 98: 478–480.
Smith, P.K., Krohn, R.I., Hermanson, G.T., Mallia, A.K.,Gartner, F.H., Provenzano, M.D., et al. (1985) Measure-ment of protein using bicinchoninic acid. Anal Biochem150: 76–85.
Stanley, N.R., Britton, R.A., Grossman, A.D., and Lazazzera,B.A. (2003) Identification of catabolite repression as aphysiological regulator of biofilm formation by Bacillus sub-tilis by use of DNA microarrays. J Bacteriol 185: 1951–1957.
Stoodley, P., Sauer, K., Davies, D.G., and Costerton, J.W.(2002) Biofilms as complex differentiated communities.Annu Rev Microbiol 56: 187–209.
Suzuki, K., Wang, X., Weilbacher, T., Pernestig, A.K., Mele-fors, O., Georgellis, D., et al. (2002) Regulatory circuitry of
the CsrA/CsrB and BarA/UvrY systems of Escherichia coli.J Bacteriol 184: 5130–5140.
Valverde, C., Lindell, M., Wagner, E.G., and Haas, D. (2004)A repeated GGA motif is critical for the activity and stabilityof the riboregulator RsmY of Pseudomonas fluorescens. JBiol Chem 279: 25066–25074.
Vidal, O., Longin, R., Prigent-Combaret, C., Dorel, C., Hoo-reman, M., and Lejeune, P. (1998) Isolation of an Escher-ichia coli K-12 mutant strain able to form biofilms on inertsurfaces: involvement of a new ompR allele that increasescurli expression. J Bacteriol 180: 2442–2449.
Wang, X., Preston, J.F., III, and Romeo, T. (2004) ThepgaABCD locus of Escherichia coli promotes the synthesisof a polysaccharide adhesin required for biofilm formation.J Bacteriol 186: 2724–2734.
Wei, B., Shin, S., LaPorte, D., Wolfe, A.J., and Romeo, T.(2000) Global regulatory mutations in csrA and rpoS causesevere central carbon stress in Escherichia coli in the pres-ence of acetate. J Bacteriol 182: 1632–1640.
Wei, B.L., Brun-Zinkernagel, A.M., Simecka, J.W., Pruss,B.M., Babitzke, P., and Romeo, T. (2001) Positive regula-tion of motility and flhDC expression by the RNA-bindingprotein CsrA of Escherichia coli. Mol Microbiol 40: 245–256.
Weilbacher, T., Suzuki, K., Dubey, A.K., Wang, X., Gudapaty,S., Morozov, I., et al. (2003) A novel sRNA component ofthe carbon storage regulatory system of Escherichia coli.Mol Microbiol 48: 657–670.
West, A.H., and Stock, A.M. (2001) Histidine kinases andresponse regulator proteins in two-component signalingsystems. Trends Biochem Sci 26: 369–376.
Whistler, C.A., and Ruby, E.G. (2003) GacA regulates sym-biotic colonization traits of Vibrio fischeri and facilitates abeneficial association with an animal host. J Bacteriol 185:7202–7212.
Whiteley, M., Bangera, M.G., Bumgarner, R.E., Parsek, M.R.,Teitzel, G.M., Lory, S., and. Greenberg, E.P. (2001) Geneexpression in Pseudomonas aeruginosa biofilms. Nature413: 860–864.
Wong, S.M., Carroll. P.A., Rahme, L.G., Ausubel, F.M., andCalderwood, S.B. (1998) Modulation of expression of theToxR regulon in Vibrio cholerae by a member of the two-component family of response regulators. Infect Immun 66:5854–5861.
Yang, H., Liu, M.Y., and Romeo, T. (1996) Coordinate geneticregulation of glycogen catabolism and biosynthesis inEscherichia coli via the CsrA gene product. J Bacteriol178: 1012–1017.
Zuber, S., Carruthers, F., Keel, C., Mattart, A., Blumer, C.,Pessi, G., et al. (2003) GacS sensor domains pertinent tothe regulation of exoproduct formation and to the biocontrolpotential of Pseudomonas fluorescens CHA0. Mol PlantMicrobe Interact 16: 634–644.
Zuker, M. (2003) Mfold web server for nucleic acid foldingand hybridization prediction. Nucleic Acids Res 31: 3406–3415.