cortical/amygdalar glutamatergic-circuit breakers alleviate tics in a transgenic tourette's...
TRANSCRIPT

1
Cortical/amygdalar glutamatergic-circuit breakers alleviate 1
tics in a transgenic Tourette's syndrome model 2
3
Eric J. Nordstroma,b, Katie C. Bittnera,1, Michael J. McGratha,2, Clinton R. Parks IIIa,3, 4
Frank H. Burton§,a,b 5
6
aDepartment of Pharmacology, University of Minnesota, 6-120 Jackson Hall, 321 7
Church Street SE, Minneapolis MN 55455-0217 USA 8
bMinneapolis Medical Research Foundation, Hennepin County Medical Center, 701 9
Park Ave, Shapiro S3.111, Minneapolis MN 55415-1623 USA 10
11
1Present Address: HHMI Janelia Farm Research Campus, 19700 Helix Dr, Ashburn, VA 12
20147, USA 13
2Present Address: Seager, Tufte & Wickem, 1221 Nicollet Avenue, Suite 800, 14
Minneapolis, MN 55403-2420 USA 15
3Present Address: SpaceNews, Inc. 1414 Prince Street, Suite 300, Alexandria, VA 22314, 16
USA 17
18
§Corresponding Author: Frank H. Burton, Ph.D., Department of Pharmacology, 19
University of Minnesota, 6-120 Jackson Hall, 321 Church Street S.E., Minneapolis MN 20
55455-0217 USA; E-mail: [email protected] 21

2
22
Running Title: Cortico/amygdalostriatal circuit breakers alleviate tics 23
24
Keywords: Tics, glutamate, transgenic, D1CT-7, mice, ritanserin, prazosin, moxonidine, 25
bromocriptine 26
27
Total number of pages: 57 28
29
Number of Figures: 5 30
31

3
Abbreviations 32
33
5-HT, 5-hydroxytryptamine; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; 34
AMY; amygdala; ANOVA (analysis of variance); cAMP, 3',5'-cyclic adenosine 35
monophosphate; CGN, "cortical/limbic glutamatergic neuron" hyperactivity model of tics and 36
compulsions; CSTC, cortico/amygdalo-striato-thalamo-cortical; CT, cholera toxin; CTX, 37
cortex; D1, dopamine receptor subtype 1; D1+, D1 receptor-containing; D1CT-7, Dopamine 38
receptor 1 gene (DRD1)-promoter/cholera toxin A1 subunit transgenic sub-strain 7 mouse; 39
D2, dopamine receptor subtype 2; DA, dopamine; DP, striatal direct pathway; DTM, 40
dermatillomania; GABA, gamma-amino butyric acid; Glu, glutamate; GS, stimulatory G 41
protein; I-1, imidazoline-1; IP, striatal indirect pathway; NMDA, N-methyl-D-aspartate; OC; 42
obsessive-compulsive; OCD; obsessive-compulsive disorder; PCP, phencyclidine; SEM, 43
standard error of the mean; SNc, substantia nigra pars compacta; STR, striatum; Tg, 44
transgenic; TS, Tourette's syndrome; TS+OCD, Tourette's syndrome and obsessive-45
compulsive disorder; TTM, trichotillomania. 46

4
ABSTRACT 47
48
The brain circuit that evokes the tic symptoms of Tourette's syndrome (TS) is unknown 49
but thought to involve hyperactivity of the cortico/amygdalo-striato-thalamo-cortical 50
(CSTC) circuit loop. We previously engineered a transgenic mouse model of TS by 51
expressing an artificial neuropotentiating transgene (encoding the cAMP-elevating, 52
intracellular A1 subunit of cholera toxin) within a cortical-amygdalar subset of dopamine 53
D1 receptor-expressing neurons whose potentiation excites pyramidal glutamatergic 54
somatosensory-motor-orbitofrontal cortical and limbic output circuits to the striatum that 55
are thought to be hyperactive in TS and comorbid obsessive-compulsive (OC) spectrum 56
disorders. These D1CT-7 ("Ticcy") transgenic mice's tics responded to clonidine, a 57
therapeutic TS drug, and their corticostriatal circuit was, like TS patients', sensitive to 58
inhibition by postsynaptic D2 receptor antagonists. To test the hypothesis that 59
cortical/amygdalar glutamatergic output circuit hyperactivity mediates tics, we've now 60
examined in these mice the tic-alleviating ability of drugs that counter different elements 61
of this circuit -- namely, anti-serotonoceptive and anti-noradrenoceptive corticostriatal 62
pyramidal output blockers, agmatinergic (imidazoline receptor-mediated) anti-63
GABAergic striatothalamic presynaptic inhibitors, and anti-dopaminergic nigrostriatal 64
presynaptic inhibitors. Drugs of each class (the serotonin 5-HT2a,c receptor antagonist 65
ritanserin, the NE alpha-1 receptor antagonist prazosin, the presynaptic 66
agmatine/imidazoline I1 receptor agonist moxonidine, and the presynaptic dopamine D2 67
receptor agonist bromocriptine) each fully alleviate tic symptoms in the Ticcy mice. This 68

5
supports a hyperglutamatergic "tic circuit" wherein cortical/amygdalar pyramidal 69
projection neurons' glutamatergic overexcitation of both striatal effector neurons and 70
nigral modulatory neurons hyperactivates these target subcircuits and unbalances their 71
integration to create tics, illuminating novel drug strategies for treating TS. 72
73
INTRODUCTION 74
75
Touretteʼs syndrome (TS) is characterized by voluntarily suppressible, urge-driven 76
motor and/or vocal tics and repeated complex movements, which are more prevalent 77
and severe in males than females, and often arising in childhood (Robertson, 2000). TS 78
is also frequently (>50%) co-morbid with obsessive-compulsive disorder (OCD), 79
including its related OC-spectrum hair- and skin- pulling and picking disorders 80
trichotillomania (TTM) and dermatillomania (DTM) or excoriation disorder (American 81
Psychiatric Association, 2013), which suggests all these neurological disorders share 82
overlapping brain circuitry. For example, analogous to motor urges in TS, OCD is 83
characterized by anxiogenic thought-urges, or “obsessions,” and urge-driven ritualistic 84
or repeated actions, or “compulsions,” arising either as compensatory behaviors to 85
reduce anxiety (American Psychiatric Association, 2013) or, more so in childhood-onset 86
TS+OCD and OCD, as primary compulsions without obsessions (Rapoport et al., 1992). 87
Both animal and clinical studies support a role for hyperactivity of cortical and 88
amygdalar forebrain glutamatergic output neurons in causing or mediating tic and OC 89
disorders (Campbell et al., 1999a; McGrath et al., 2000; Carlsson, 2000; Rosenberg et 90

6
al., 2000; Nordstrom and Burton, 2002; Singer et al., 2010; Milad and Rauch, 2012). TS 91
and OCD are associated with cortical neuron hyperexcitability and impaired 92
sensorimotor gating (Swedo et al., 1992; Breiter et al., 1996; Ziemann et al., 1997; 93
Edgley and Lemon, 1999; Gilbert et al., 2004), possibly from impaired neural inhibition 94
in some inherited forms, e.g., CNTNAP2 gene deletion (Verkerk et al., 2003), or from 95
diminished activity of cortical inhibitory (GABAergic) interneurons (Minzer et al., 2004). 96
Observed reductions in forebrain metabolic activity and blood flow in TS (Swerdlow and 97
Sutherland, 2005) may be consistent with diminished metabolic activity of cortical 98
inhibitory interneurons, where the interneuron "brake's" failure permits increased 99
excitatory glutamate output from the smaller population of corticostriatal pyramidal 100
projection neurons, as has been postulated to occur in other psychotic-spectrum 101
disorders and glutamate-modulating psychogenic drug symptoms (Homayoun and 102
Moghaddam, 2007). Consistent with that interpretation, elevated glutamatergic efflux to 103
the striatum is observed in TS and OCD (Campbell et al., 1999a; McGrath et al., 2000; 104
Carlsson, 2000; Rosenberg et al., 2000; Nordstrom and Burton, 2002; Singer et al., 105
2010). Possibly as a response to corticostriatal glutamatergic hyperexcitation, TS and 106
OCD patients also display striatal desensitization and volumetric damage (Peterson et 107
al., 1993; Peterson et al., 1998; Menzies et al., 2008), wherein glutamate may either 108
trigger cross-talk pharmacodynamic up-regulation (supersensitization) of neuroinhibitory 109
striatal dopamine D2 receptors (Wolf et al., 1996) or directly up-regulate nigrostriatal 110
dopamine release (Singer et al., 2010) and conversely desensitize striatal D2 receptors 111
(Denys et al., 2013), but in either case dopaminergically counter-inhibiting the 112

7
glutamatergically overexcited D2+ striatal neurons (also then explaining why TS 113
symptoms are sensitive to inhibition by D2 antagonists) (Campbell et al., 1999b). The 114
apparently distinct mechanisms of action of two early classes of TS drug -- inhibitory 115
postsynaptic D2 receptor antagonists like haldol or pimozide, and the stress-relieving 116
inhibitory norepinephrine (NE) alpha-2 receptor agonist clonidine -- also are consistent 117
with a hyperglutamatergic forebrain circuit model of TS and OCD. Both classes counter 118
corticostriatal glutamatergic circuit activity and its movement-stimulating effects: D2 119
antagonists by disinhibiting both cortical GABAergic inhibitory interneurons and striatal 120
movement-suppressing indirect pathway neurons (Campbell et al., 1999b; Nordstrom 121
and Burton, 2002; Minzer et al., 2004), and clonidine by possibly blocking noradrenergic 122
excitation of amygdalostriatal and/or corticostriatal glutamatergic pyramidal output 123
neurons (Lichter and Jackson, 1996; Nordstrom and Burton, 2002). 124
The first (and so far only) genetically-engineered model of TS that exhibits tics is 125
the D1CT-7 "Ticcy" transgenic mouse (Campbell et al., 1999a; Nordstrom and Burton, 126
2002) -- created as an early, pre-millennial foray into "brain circuit-testing" with an 127
artificial gene to induce neuropotentiation. Preceding by a decade the development of 128
optogenetic light-activated artificial channel transgenes designed to directly depolarize 129
and fire neurons, the artificial transgene in Ticcy mice in contrast chronically induces the 130
intracellular second-messenger, cAMP (3',5'-cyclic adenosine monophosphate), to 131
potentiate neurons' responsiveness to their own endogenous neurotransmitters 132
(Campbell et al., 1999a). This was achieved by D1 receptor gene promoter-targeted 133
expression of an artificial transgene encoding the exclusively intracellular A1 subunit of 134

8
cholera toxin (CT), which, by covalently activating the stimulatory G protein GS, 135
chronically stimulates adenylate cyclase activity and cAMP production (Burton et al., 136
1991; Zeiger et al., 1997). Furthermore, D1CT expression in the D1CT-7 ("Ticcy") line 137
of such transgenic mice is regionally restricted to a cortical/limbic subset of brain D1 138
receptor-containing (D1+) neurons, with no expression in striatum. These cortical/limbic 139
D1+ neurons, once potentiated by intracellular CT, induce in D1CT-7 mice voluntarily-140
suppressible, juvenile-onset tics (Nordstrom and Burton, 2002) and compulsions 141
(Campbell et al., 1998; Campbell et al., 1999a), with increased tic severity in males 142
(Nordstrom and Burton, 2002), stress sensitivity (McGrath et al., 1999a,b), and 143
alleviation by TS drugs of multiple classes (Nordstrom and Burton, 2002; Campbell et 144
al., 1999b) -- thus showing the greatest behavioral homology to TS+OCD of any animal 145
model reported so far (Burke and Lombroso, 2004). Based on their cortical circuit's 146
hyperglutamatergic status (normal mice show slower onset and calmer 147
pentylenetetrazole-kindled cortical seizures, and less pronounced glutamatergic drug-148
induced locomotion) (Campbell et al., 2000; McGrath et al., 2000) and associated tic-149
compulsion phenotype (Campbell et al., 1999a; Nordstrom and Burton, 2002), and on 150
the known cortical and limbic excitatory output triggered by these transgenically-151
potentiated D1+ neurons (which comprise cortical somatosensory/insular/piriform D1+ 152
glutamatergic pyramidal output neurons and amygdalar intercalated nucleus D1+ 153
GABAergic stellate interneurons, that respectively directly and indirectly trigger 154
glutamatergic excitation of the striatum from deep-layer somatosensory-motor-155
orbitofrontal cortical pyramidal and amygdalar pyramidal output neurons) (Campbell et 156

9
al., 1999a,c; Campbell et al., 2000), the Ticcy D1CT-7 transgenic mice comprised a 157
direct test of the hypothesis that corticostriatal and/or amygdalostriatal glutamatergic 158
circuit hyperactivity can cause tics and compulsions (Campbell et al., 1999a; Carlsson, 159
2000; Nordstrom and Burton, 2002). 160
Studies and reviews of the D1CT-7 mice (Sah and Sallee, 2002; Burke and 161
Lombroso, 2004; Swerdlow and Sutherland, 2005; Joel, 2006; Ting and Feng, 2008; 162
Wang et al., 2009; Wu et al., 2012) have so far helped inspire several clinical studies. 163
These include studies of glutamate's role in tics and compulsions (Chakrabarty et al., 164
2005; Singer et al., 2010), successful clinical trials of antiglutamatergic drugs for the 165
OC-spectrum disorders OCD (Lafleur et al., 2006; Grant et al., 2007) and TTM (Grant et 166
al., 2009); and successful clinical trials of the D1 antagonist, ecopipam, for TS (Gilbert 167
et al., 2014) and OC-spectrum gambling (impulse-control) disorder (Grant et al., 2014). 168
Additional clinical study recommendations could be provided by understanding what 169
pharmacological targets within the Ticcy mice's cortical/amygdalar glutamate-dependent 170
hyperactive circuitry can act as "circuit breakers" to suppress tics. In this study we have 171
confirmed that four such postulated circuit breakers -- serotonin 5-HT2a,c receptor 172
antagonists, NE alpha-1 receptor antagonists, presynaptic agmatine/imidazoline I1 173
receptor agonists, and presynaptic dopamine D2 receptor agonists -- are capable of 174
short-circuiting tics elicited by neurogenic hyperactivity of forebrain 175
cortico/amygdalostriatal and cortico/amygdalonigral glutamatergic outputs, and thus are 176
prospective sources of new clinical treatments to alleviate neurogenic tics in TS. 177
178

10
EXPERIMENTAL PROCEDURES 179
180
Animal subjects 181
182
Studies of drug effects on tic incidence and locomotion used 30 adult Balb/c-inbred (JAX 183
labs, Bar Harbor, ME, USA) female wild-type control (C) mice and 32 adult Balb/c-184
inbred female hemizygous D1CT-7 ("Ticcy") transgenic (Tg) sibling mice. Because Tg 185
females breed and nurse poorly due to Tg-induced anxiogenic fleeing from males and 186
over-grooming and biting of pups (Campbell et. al., 1999a; Nordstrom and Burton, 187
2002), Tg males must be used as breeders to maintain the Ticcy mouse colony, while 188
Tg females are used for drug studies. All animals were naive to behavioral or drug 189
assays prior to testing, and experiments were carried out with the investigators blinded 190
as to the animals' transgenic or control genotype status and drug injection status. All 191
mice were housed in groups of 2-5 in a temperature-controlled room on a 12-hour day-192
night cycle, allowed unrestricted access to food and water with the exception of testing 193
times, and drug-treated and videotaped at the same daytime range of hours to control 194
for the possibility of circadian fluctuation in drug response. Care was taken to ensure 195
that the animals used in this study received no unnecessary discomfort. All animals 196
were maintained and procedures were performed in accordance with the Animal 197
Welfare Act and the NIH Guide for the Care and Use of Laboratory Animals, under the 198
approval of the University of Minnesota Institutional Animal Care and Use Committee. 199

11
The University of Minnesota Research Animal Resources facility is fully accredited by 200
the American Association for the Accreditation of Laboratory Animal Care. 201
202
Drugs & Injections 203
204
Except where indicated, for each drug study mice in both genotype groups were 205
administered stock solutions of drug or saline vehicle, 24 hours apart, at the injection 206
volumes and at the acute doses, as well as assayed behaviorally at the post-injection 207
times, that were previously reported to induce a maximal behavioral effect; and the 208
behavioral observation of videotapes were performed blinded as to the animals' 209
transgenic or control genotype status and drug or saline injection status, while 210
observation counts were confirmed by a repeat observer. 211
Ritanserin (Research Biochemicals International, Natick, MA, USA) was prepared 212
as a stock solution (0.1 mg/ml ritanserin, 0.04% Tween-80 in saline) by suspending 10 213
mg drug in 2 ml of 2% Tween-80 followed by 50-fold dilution in saline vehicle (0.9% 214
NaCl). All administrations of ritanserin or vehicle were delivered intraperitoneally (i.p.) in 215
an injection volume of 10 ml/kg body weight. The 1 mg/kg ritanserin dosage was chosen 216
for this study based on its reported efficacy in alleviating abnormal behaviors triggered 217
via serotonin 5-HT2a receptors without any concomitant inhibition of spontaneous 218
locomotor activity (Ninan and Kulkarni, 1998), which we confirmed as described in 219
Results. Likewise, Tween-80/saline vehicles ranging in Tween-80 concentrations of up 220
to 32% reportedly have no motor-inhibiting effects (Castro et al., 1995), which we 221

12
confirmed by comparison of both tics and locomotor activity levels in saline-injected 222
versus 0.04% Tween-80/saline-injected transgenic mice (not shown). 223
Prazosin hydrochloride was obtained from Research Biochemicals International 224
(Natick, MA) and dissolved in 0.9% saline. The drug was administered in a volume of 225
10ml/kg body weight, at a previously reported effective dosage of 3mg/kg (i.p.) 226
(Wellman and Davies, 1992; Wellman et al., 1997; Cheng and Kuo, 2003). 227
Moxonidine hydrochloride was obtained from Research Biochemicals 228
International (Natick, MA) and dissolved in 0.9% saline. The drug was administered i.p. 229
in a volume of 10ml/kg body weight, at a previously reported effective dosage of 230
0.5mg/kg (Zhu et al., 1999). 231
Bromocriptine methanesulfonate was obtained from Research Biochemicals 232
International (Natick, MA) and dissolved in 0.9% saline. The drug was administered in a 233
volume of 10ml/kg body weight, at a previously reported effective dosage of 5 mg/kg 234
(i.p.) (Jackson et al., 1988). At this dosage, bromocriptineʼs D2 agonist action is 235
reported to be on presynaptic autoreceptors at two hours post-injection (Jackson et al., 236
1988). 237
238
Tic Behavior Quantification 239
240
The incidence of tic-like behavior was determined in videotapes of transgenic versus 241
control non-transgenic littermate female mice. Tics were defined as any very brief (0.05-242
0.1 sec, as determined by a duration of 1.5 to 3 frames in 30 fps videotape recordings) 243

13
isolated head and/or body jerk or shake, other than those associated with acoustic 244
startle or obvious shedding of litter visible on the coat. By this definition normal mice 245
exhibit tic-like twitches only infrequently, compared to 3- to 5-fold more frequent tic-like 246
twitches in D1CT-7 transgenic mice (Nordstrom and Burton, 2002). The effect on tic 247
incidence of vehicle versus drug treatment was determined as the mean number of 248
tics/15 min in transgenic or control mice observed over a 15 minute period beginning 30 249
minutes after either vehicle or drug injection and 15 minutes after introduction of the 250
mice into a new cage (i.e., after a 15 minute cage habituation period), with these 251
exceptions: 1) In the ritanserin study, 15 minute cage habituation was omitted to avoid 252
confounding a reported anxiolytic effect of ritanserin (Gao and Cutler, 1993) with any 253
potential anxiolytic effect of cage habituation, while tic counts were instead observed 254
and analyzed over a 30 minute period beginning 30 minutes after either vehicle or drug 255
injection -- however, because post-hoc analysis confirmed there was no significant 256
effect of 15 min cage habituation upon control or transgenic tic incidence in the 257
presence or absence of ritanserin treatment (not shown), the tics/30min data were 258
adjusted for figure display to the standard tics/15 min; 2) In the bromocriptine study, the 259
post-injection observations commenced not 30 minutes but two hours after injection of 260
bromocriptine, in keeping with the reported maximal presynaptic agonist action of the 261
drug in mice (Jackson et al., 1998), while pre-injection observational data was obtained 262
15 minutes prior to bromocriptine injection with no saline vehicle injection, to match 263
Jackson et al.'s prior reported drug design. No significant difference was observed in 264
Ticcy mouse tic counts between the bromocriptine study's "15 min no-drug pre-injection 265

14
vs. 2 hr post-injection drug" design and the remaining drug studies' "24 hr-separated 266
vehicle- vs. drug- injection" design. Videotapes and/or drug & vehicle samples were 267
coded to blind observers to the mice's genotype and drug-injection status, and logged 268
data were confirmed by at least one additional independent observer. 269
270
Locomotion Behavior Quantification 271
272
In all studies, to measure spontaneous locomotor activity levels during the observation 273
periods the same videotapes as described above were analyzed for the number of 274
cage-midline crossings (the observed number of locomotion-dependent cage midline 275
incursions, which reproduces an automated beam-break design, as described by 276
Nordstrom and Burton, 2002). Data are displayed as the number of midline crossings/15 277
min. At least two observers blinded to subject genotype and treatment independently 278
scored the numbers of midline crossings from the original videotapes, confirming 279
excellent interrater reliability (Intraclass Correlation Coefficient [ICC] > 0.8). Additionally, 280
for the bromocriptine study, because this dopaminergic motor output-inhibiting drug 281
(unlike ritanserin, prazosin and moxonidine) is reported to also diminish mouse motor 282
activity (Jackson et al., 1998), a more comprehensive behavioral analysis of this drug's 283
locomotor suppressing effects was also performed, as described in the next section. 284
285
Waveform display analysis of bromocriptine treated mice 286
287

15
Waveform display analysis was performed as previously described (Campbell et al., 288
1998). Briefly, the above-described videotapes of drug-naive D1CT-7 or control females 289
littermates were continuously observed for 15 minutes prior to bromocriptine injection 290
and 2 hours post-injection, in each case after 15 minutes of habituation to a new cage. 291
EthoMac (v1.10, © The University of Minnesota) software was used for behavioral state 292
entry, and for calculation and tabulation of behavioral state event timing, number, and 293
duration. Scored behaviors included: 1) climbing/leaping (animal standing on its hind 294
paws in the corner of the cage moving at least three limbs); 2) still (remaining in one 295
position with an occasional head movement); 3) rear; 4) gnaw (gnawing against the side 296
of the Plexiglas cage); 5) (horizontal) locomotion; 6) dig (into the sawdust bedding); 7) 297
groom; 8) hang (from the wire bar cage lid); 9) eat (bedding picked from the cage 298
bottom and put into the mouth); 10) sniff; 11) other (any activity that does not fit into the 299
previous categories). The total observer-scored number of locomotion events for each 300
mouse under each drug condition, as tabulated by the EthoMac logs, was confirmed by 301
at least one independent, genotype- and treatment- blinded observer from original 302
coded videotapes, confirmed to have ICC > 0.8 interrater reliability, then statistically 303
compared for the extent of bromocriptine and genotype effects and displayed as the 304
mean number of locomotion events/15 min. 305
306
Statistical Analyses 307
308
Overall statistical significance of a ritanserin, prazosin, moxonidine, or bromocriptine 309

16
drug (within-groups) effect, Ticcy transgenic (Tg) vs. control (C) wild-type genotype 310
(between-groups) effect, or a drug by genotype interaction, was determined in Statview 311
4.5 (Abacus Corp., Berkeley CA, USA) by initial repeated-measures analysis of variance 312
(repeated-measures ANOVA) on both tic and locomotion behavioral measures, followed 313
by individual between-group unpaired, two-tailed Studentʼs t-test comparisons of 314
genotype effect and within-group paired, two-tailed Studentʼs t-test comparisons of drug 315
effect, with significance assumed at P < 0.05, for parametrically-distributed locomotion 316
data; or individual between-group Mann-Whitney U-tests of genotype effect and within-317
group Wilcoxon Signed Rank tests of drug effect, with significance assumed at tied P < 318
0.05, for the non-parametrically-distributed tic incidence data. Because elevated tic 319
counts in the Ticcy genotype population routinely sort into a non-parametric biphasic 320
distribution caused by the presence within the Ticcy group of epigenetically-variable but 321
individually-consistent "super (6-fold) ticcers" and "elevated (3-fold) ticcers," the use of a 322
repeated-measures drug design, as performed by Nordstrom and Burton (2002) and 323
herein, where each subjects' behavior is tested both with and without drug injection, 324
permits drug effects to be tested reliably on such populations even though the mean 325
elevation and standard error of tic incidence may vary from one drug study to another 326
depending on each study population's random percentage of "super-ticcers." All data 327
were expressed as the mean plus standard error of the mean (S.E.M.) of the number of 328
tics per 15 minutes, cage midline crossings per 15 minutes, or locomotion events per 15 329
minutes, occurring during the videotaped windows of observation. 330
331

17
RESULTS 332
333
Deep-layer cortical pyramidal glutamatergic output neurons are known to express 334
excitatory serotonin 5-HT2a,c receptors (Sheldon and Aghajanian, 1991; Nestler, 1997; 335
Jakab and Goldman-Rakic, 1998; Marek and Aghajanian, 1998; Marek and Aghajanian, 336
1999; Aghajanian and Marek, 1999), suggesting a potential therapeutic role in TS for 5-337
HT2a,c antagonists like ritanserin or ketanserin (shown effective in a small trial of six TS 338
patients by Bonnier et al., 1999). Ritanserin also has anxiolytic activities both clinically 339
and in rodents (Ceulemans et al., 1985; Danjou et al., 1992; Gao and Cutler, 1993), 340
putatively due to its similar inhibition of amygdalar 5-HT2c receptors (Gibson et al., 341
1994) -- which may also trigger reduced excitatory amygdalar glutamatergic output to 342
the limbic cortex, orbitofrontal cortex, and motor striatal circuits. The ability of this pure 343
5-HT2a,c antagonist, ritanserin (Ceulemans et al., 1985), to suppress tics at a 344
concentration not inhibitory to mouse locomotor activity was tested in Fig. 1. While the 345
Ticcy D1CT-7 transgenic (Tg) mice show multiple times the number of TS-like twitches 346
compared to control non-transgenic control (C) mice, their tics are restored to control 347
levels by acute ritanserin (1 mg/kg, i.p.) treatment (Fig.1, Panel A, black bars). 348
Ritanserin treatment did not significantly decrease the control mice's normal, baseline 349
level of infrequent twitching (Fig. 1, Panel A, white bars). Nor did ritanserin treatment 350
significantly reduce in either Ticcy or control mice the level of general locomotor activity 351
(Fig. 2, Panel B), which is displayed as the number of cage midline crossings/15 min. 352
This is consistent with previous reports that this 1mg/kg ritanserin dose in rodents, 353

18
although psychoactive in reducing anxiety, does not inhibit spontaneous locomotion 354
(Ninan and Kulkarni, 1998). These data indicate that acute ritanserin treatment 355
selectively suppresses abnormal ticcing without inhibiting normal, baseline spontaneous 356
locomotor activities. 357
A drug thought to decrease corticostriatal glutamatergic output is the alpha-1 358
antagonist, prazosin (Fig. 2), whose alpha-1 NE receptors were shown to be co-359
expressed, with the 5-HT2a,c receptor targets of ritanserin, on deep-layer cortical 360
pyramidal glutamatergic output neurons (Marek and Aghajanian, 1999). Consequently, 361
the ability of prazosin to suppress corticostriatal glutamatergic tics, at a concentration 362
reportedly not inhibitory to mouse locomotor activity but capable of psychoactively 363
countering dopamine-dependent anorexia (Wellman and Davies, 1992; Wellman et al., 364
1997; Cheng and Kuo, 2003), was tested (Fig. 2). Like ritanserin, acute prazosin 365
treatment (3 mg/kg, i.p.) treatment of the Ticcy D1CT-7 transgenic (Tg) mice restored 366
their elevated tic counts to the level of control (C) littermates (Fig. 2, Panel A, black 367
bars). Prazosin treatment, also like ritanserin, didn't significantly decrease the control 368
mice's normal, baseline level of infrequent twitches (Fig. 2, Panel A, white bars), nor 369
significantly alter locomotion in either the Ticcy transgenic mice or control mice (Fig. 2, 370
Panel B). These data indicate that acute treatment with prazosin, whose alpha 1 NE 371
receptors are known to co-localize with ritanserin-targeted serotonin 5-HT2a,c receptors 372
on cortical glutamatergic output neurons, selectively suppresses, as does ritanserin, 373
abnormal ticcing without inhibiting normal, baseline spontaneous locomotor activities. 374
The agmatine/imidazoline-1 agonist moxonidine (Fairbanks and Wilcox, 1999; 375

19
Zhu et al., 1999; Taksande et al., 2010; Dixit et al., 2014), a less-sedating, less-376
hypotensive and less alpha-2 NE receptor-specific relative of the TS-drug clonidine, 377
may exert distinct central nervous system actions due to its imidazoline-1 (I-1) receptor 378
specificity. For example, whereas clonidine's presynaptic agonist action on alpha-2 NE 379
receptors is thought to decrease NE stimulation of anxiogenic amygdalar glutamatergic 380
output to the limbic cortex and striatum (Lichter and Jackson, 1996; Nordstrom and 381
Burton, 2002), moxonidine, by selectively acting as a presynaptic I-1 receptor agonist to 382
inhibit the striatal GABAergic direct- and indirect- pathway neurons targeted by 383
glutamate (Tanabe et al., 2006) and to reduce glutamate-triggered neurotoxicity 384
(Bakuridze et al., 2009), may suppress the more distal, striatal-output element within the 385
cortico/amygdalo-striato-thalamo-cortical (CSTC) tic-circuit. Consequently, we tested 386
the ability of acute i.p. moxonidine to block hyperglutamatergic-mediated tics at a 0.5 387
mg/kg dose sufficient for peripheral reduction of blood pressure (Zhu et al., 1999), as 388
well as for CNS reduction of drug withdrawal-induced anxiety and endogenous anxiety-389
dependent compulsive (marble-burying) behavior in rodents (Taksande et al., 2010; 390
Dixit et al., 2014) (Fig. 3). Like acute ritanserin and prazosin treatments, acute 391
moxonidine treatment of the Ticcy D1CT-7 transgenic (Tg) mice restored their elevated 392
tic counts to the level of control (C) littermates (Fig. 3, Panel A, black bars) without 393
decreasing the control mice's normal, baseline level of infrequent twitches (Fig. 3, Panel 394
A, white bars) or altering locomotion in either the transgenic or control mice (Fig. 3, 395
Panel B). These data suggest that acute treatment with moxonidine, whose imidazoline 396
I-1 receptors are known to localize presynaptically on and inhibit striatal GABAergic 397

20
neurons, can suppress cortico/amygdalostriatal glutamate-induced abnormal ticcing 398
without inhibiting normal, baseline spontaneous locomotor activities. 399
The final candidate "tic circuit breaker" drug we examined was the D2 receptor 400
agonist, bromocriptine (Fig. 4), which at low doses is selectively presynaptic in its action 401
on striatonigral dopaminergic axon terminals (Ceccherini-Nelli and Guazzelli, 1994) -- 402
thus reducing nigral dopaminergic efflux onto motion-activating, DA receptor-expressing 403
striatal neurons (Campbell et al., 1999b), which should counter the tic-inducing effect of 404
coincident hyperglutamatergic stimulation of the striatal D1+ direct pathway neurons. 405
Bromocriptine, however, is known to also reduce general locomotor activity as a 406
consequence of its retardation of DA input to the striatum (Jackson et al., 1988), as do 407
more classical postsynaptic D2 antagonist drugs (such as haloperidol) for tic and OC-408
spectrum disorders (Cohen et al., 1992). We consequently examined bromocriptine's 409
ability to diminish tics and, in this case, total locomotion events, modulated by 410
dopaminergic inputs to the same GABAergic striatal neurons that stimulate motion and 411
urges through the direct pathway and suppress them through the indirect pathway, and 412
that in the Ticcy mice are co-excited by cortical/amygdalar glutamatergic inputs (Fig. 4). 413
At 2 hours post-injection, when in rodents bromocriptine is documented to exert its 414
highest presynaptic D2 agonist effect to inhibit nigrostriatal axonal DA release (Jackson 415
et al., 1988), the elevated tics in the Ticcy D1CT-7 transgenic (Tg) mice were reduced to 416
the level of untreated control (C) littermates (Fig. 4, Panel A, black bars). But unlike 417
acute ritanserin, prazosin, or moxonidine, the acute bromocriptine treatment also 418
significantly decreased control mice's normal, baseline level of infrequent twitches (Fig. 419

21
4, Panel A, white bars) as well as their locomotion events (Fig. 4, Panel B, white bars), 420
indicating that bromocriptine likely exerts some of its tic-suppressing effects by more 421
generalized inhibition of baseline levels of locomotor initiation being mediated by the 422
striatal subcircuit targeted by the convergent cortical/amygdalar glutamate and nigral DA 423
inputs. This is consistent with the presumed role of presynaptic striatal dopamine D2 424
receptors to reduce various dopamine-dependent motor activities and increase 425
catalepsy in mice (Jackson et al., 1988). It is also consistent with DA's known role as a 426
modulatory inducer of locomotion, both normally and in DA-replacement therapies for 427
parkinsonism, and with mild parkinsonian effects being within the known clinical side-428
effect profile of presynaptic D2 agonist therapeutic drugs (including pergolide and 429
aripiprazole). The diminishment of locomotion events induced by bromocriptine in 430
control mice was not evident in Ticcy mice (Fig. 4, Panel B), although their incidence of 431
vertical motor events (climbing-leaping) was diminished (not shown), suggesting that 432
bromocriptine indeed exerts an indiscriminate motor-circuit inhibiting influence on both 433
Ticcy and control mice. The incidence of locomotion events in all untreated or 434
bromocriptine treated mice (Fig. 4, Panel B) was roughly twice their incidence of cage 435
midline crossings (not shown), consistent with prior reports that locomotor initiations in 436
both control and Ticcy mice represent initiations of extended locomotor sequences 437
(Campbell et al., 1999a; Nordstrom and Burton, 2002). 438
439
DISCUSSION 440
441

22
The D1CT-7 transgenic "Ticcy mouse" model of tics and compulsions was the first 442
performed brain "circuit test" of a complex psychiatric or psychomotor disease -- the first 443
symptomatic model to be created by transgenic neuropotentiation of a molecularly-specified 444
and regionally-restricted circuit element in the brain (Campbell et al., 1999a; Nordstrom and 445
Burton, 2002). The particular circuit element potentiated in the brains of Ticcy mice is D1+ 446
somatosensory cortical and limbic neurons that trigger deep-layer cortical and amygdalar 447
glutamatergic excitation of efferent striatothalamic GABAergic circuits and of efferent 448
nigrostriatal dopaminergic circuits, and that are thought to be hyperactive in human TS and 449
OCD. Hence the Ticcy mice permit examination of the biological role in tics of coincident 450
glutamatergic and dopaminergic action upon striatal neurons, as well as permit examination 451
of new classes of drugs that may therapeutically block the circuitry eliciting or mediating 452
Tourette's like tic symptoms. 453
What is the role in tics of coincident glutamate and dopamine action upon striatal 454
neurons, and what is its circuitry? In isolation, glutamate- and excitatory DA D1- receptor 455
coexpressing striatal direct-pathway GABAergic neurons would respond to either glutamate 456
or dopamine by triggering tics, while glutamate- and inhibitory DA D2- receptor 457
coexpressing striatal indirect-pathway GABAergic neurons would respond to glutamate by 458
suppressing tics or to DA by triggering tics. (Locomotor induction by psychoactive NMDA 459
glutamate receptor antagonists may be due to these drugs either primarily inhibiting the 460
indirect-pathway striatal neurons the suppress motion and urges, or primarily inhibiting 461
cortical GABAergic interneurons that suppress corticostriatal glutamate output to both 462
striatal NMDA and AMPA glutamate receptors (Homayoun and Moghaddam, 2007)). 463

23
But what happens if hyperglutamatergic output to both the striatum and substantia 464
nigra is initiated from a hyperactive cortex and/or amygdala (whether it be by transgenic 465
potentiation in the Ticcy mice of D1+ intermediate-cortical layer glutamatergic neurons and 466
amygdalar neurons that activate these deep-layer cortical and amygdalar glutamatergic 467
output neurons, or by cortical GABAergic interneurons' inhibition by psychoactive drugs like 468
PCP, or by various mutations or epigenetic changes of cortical/amygdalar glutamate output 469
in subtypes of neurogenic TS)? If such "cortical/limbic glutamatergic neuron hyperactivity" 470
occurs, the convergence of its hyperglutamatergic input to striatal circuits with its parallel 471
hyperglutamatergic-triggered nigral dopamine input to those same striatal circuits is 472
predicted to elicit chronic tics by chronically unbalancing those striatal circuits, in favor of tic 473
induction by the striatal direct pathway, as detailed below: 474
Our current model of the "tic circuit," with the sites and mechanisms of action of the 475
four tested "circuit breakers," is diagrammed in Fig. 5. This tic circuit is based on ours and 476
others' prior "cortical/limbic glutamatergic neuron (CGN) hyperactivity" circuit model of tics 477
and compulsions (Campbell et al., 1999a; Carlsson, 2000; Rosenberg et al., 2000; 478
Nordstrom and Burton, 2002), with a recent refinement adding corticonigral glutamate 479
excitation of nigrostriatal DA efflux (Singer et al., 2010). 480
In this hyperglutamatergic circuit model of neurogenic tics, balanced circuit 481
element outputs control motor activities and urges in normal individuals (Fig. 5, Panel 482
A). In contrast, excess cortical/amygdalar glutamate output to the striatum and 483
substantia nigra is proposed to initiate tics in the Ticcy mice and in some forms of 484
neurogenic TS (Fig. 5, Panel B), by exciting the GABAergic striatal "direct pathway" 485

24
neurons that co-express excitatory dopamine D1 receptors, and by simultaneously 486
exciting nigral neurons to release reinforcing DA onto those same striatal neurons' D1 487
receptors. Meanwhile, the simultaneous cortico/amygdalostriatal glutamatergic 488
excitation of tic-suppressing GABAergic striatal "indirect pathway" neurons, which co-489
express inhibitory dopamine D2 receptors, would be counteracted. Either of two 490
mechanisms (one pharmacodynamic, the other circuit-based) would block the ability of 491
the indirect pathway, once stimulated by forebrain glutamate output, to suppress tics: 1) 492
Forebrain glutamatergic hyperexcitation of these D2+ striatal neurons could trigger their 493
inhibitory D2 receptors to pharmacodynamically cross-supersensitize (Wolf et al.,1996), 494
countering the over-excitation by glutamate; or 2) The forebrain's parallel 495
cortico/amygdalonigral hyperglutamatergic excitation of the substantia nigra would 496
trigger excessive nigrostriatal DA release to those same striatal indirect pathway 497
neurons' inhibitory D2 receptors (Singer et al., 2010), again countering the neurons' 498
over-excitation by forebrain glutamate. Either glutamate-induced mechanism still would 499
cause tics to be chronically expressed, by unbalancing the tic-stimulating direct pathway 500
and tic-inhibiting indirect pathway in favor of chronic direct pathway activity (Fig. 5, 501
Panel A vs. B). 502
Given this neurogenic hyperglutamatergic "tic circuit" (Fig. 5, Panel B), where 503
glutamate and dopamine synergize with each other at the striatal direct pathway to elicit 504
tics, but antagonize each other at the indirect pathway to fail in suppressing tics, the 505
mechanisms by which the four tested "circuit-breaker" drugs would most likely then act 506
on the tic circuit to alleviate or suppress tics are also diagrammed (Fig. 5, Panel B vs. 507

25
Panels C-F): 508
First, ritanserin and prazosin both act as antagonists of different, but co-509
expressed, excitatory receptors located on the deep-layer cortical pyramidal output 510
neurons that glutamatergically excite the striatum, while prazosin may furthermore 511
inhibit amygdalar glutamate output to limbic cortex and directly to the striatum (Fig. 5, 512
Panels C and D). Blocking cortico/amygdalostriatal hyperglutamatergic output would 513
then "short-circuit" these neurons' chronic excitation of their target striatal and nigral 514
neurons, alleviating tics. Moxonidine would be proposed to act more distally, on the 515
striatal GABAergic neurons themselves, by presynaptically inhibiting their 516
hyperactivated striatothalamic output that excites tics (Fig. 5, Panel E). And 517
bromocriptine would act on input nigrostriatal dopaminergic neurons' D2 autoreceptors 518
to presynaptically inhibit their dopamine efflux both to the excitatory D1 receptors on the 519
glutamatergically co-excited striatal direct pathway GABAergic neurons that stimulate 520
tics, and to the inhibitory D2 receptors on the glutamatergically excited but 521
dopaminergically cross-inhibited indirect pathway GABAergic neurons that suppress tics 522
(Fig. 5, Panel F). The effectiveness of these drugs in either selectively alleviating tics 523
(i.e., ritanserin, prazosin, moxonidine) or, as in humans with TS, less selectively 524
suppressing both tics and locomotion (i.e., bromocriptine) undergirds the potential 525
validity of this forebrain hyperglutamatergic circuit model of neurogenic tics and 526
topographically parallel circuit-triggered OC- and psychotic- spectrum disorders. 527
The reported effects of drugs that should conversely "overload" rather than 528
"circuit-break" this tic-circuit also bolster its validity. For example, a PCP-like drug that 529

26
aggravated, rather than diminished, abnormal motor-urge symptoms in the Ticcy mice 530
(McGrath et al., 2000) has since been established to aggravate, rather than diminish, 531
corticostriatal glutamatergic output (Homayoun and Moghaddam, 2007). 532
533
Clinical Implications for Tourette's syndrome drug discovery 534
What is the therapeutic potential of these four "tic circuit-breakers" for the treatment of 535
human TS? The TS-like tic behavior of these mice, and thus perhaps of some 536
neurogenic forms of TS, is initiated by increased cortical/limbic glutamatergic output 537
from serotonoceptive plus noradrenoceptive pyramidal output neurons, which should be 538
amenable to inhibition by at least some 5-HT2 serotonin receptor as well as alpha-1 NE 539
receptor antagonists. Hence tics might respond not only to weak-to-strong-D2/strong-5-540
HT2 antagonists, such as the atypical neuroleptics risperidone (Bruun and Budman, 541
1996) and ziprasidone (Sallee et al., 2000), which showed effectiveness in TS pilot 542
studies, but may also respond to "pure" 5-HT2 antagonists like ritanserin, which is now 543
used in humans for other psychological disorders including anxiety (Ceulemans et al., 544
1985; Barone et al., 1986; Danjou et al., 1992), putatively reflecting the drug's 5-HT2c-545
antagonist-mediated suppression of amygdalar and limbic-cortex glutamatergic output 546
(Gibson et al., 1994). Furthermore, ketanserin, a ritanserin-related, non-anxiolytic, 547
hypotensive antagonist of both 5-HT2a and alpha-1 NE receptors (Hosie, et al., 1987), 548
has proven partially effective in a small clinical trial of childhood-onset TS (Bonnier, et 549
al., 1999), which suggests both ritanserin, as a 5-HT2a,c antagonist, and prazosin, as 550
an alpha-1 NE antagonist, should be clinically studied individually as separate 551

27
prospective human TS treatments. To our knowledge neither ritanserin nor prazosin 552
have yet been tested clinically for their efficacy in alleviating TS. 553
Could ritanserin be superior to ketanserin for TS, given these drugs' distinct 554
receptor specificities and affinities? The receptor binding profiles of ritanserin vs. 555
ketanserin suggests that ritanserin could prove therapeutically more effective. For 556
example, the glutamatergic pyramidal cortical output neurons our data suggest may be 557
hyperactive in TS and its comorbid OC-spectrum disorders carry both 5-HT2a and 5-558
HT2c receptors, as well as alpha-1 NE receptors (Sheldon and Aghajanian, 1991; 559
Marek and Aghajanian, 1998; Marek and Aghajanian, 1999). Ketanserin, as a 5-HT2a 560
antagonist but only weak alpha-1 NE antagonist (Brogden and Sorkin, 1990), would 561
thus be predicted to more weakly block cortical glutamate output from these neurons 562
than ritanserin, which efficiently inhibits both 5-HT2a and 5-HT2c receptors. Moreover, 563
ritanserin, as a 5-HT2c antagonist, unlike ketanserin, should also reduce tic severity by 564
reducing amygdalar glutamate output and consequent anxiety (Gibson et al., 1994). In 565
TS, tic severity correlates with the level of anxiety, and other anxiolytic drugs can lessen 566
tics (Goetz, 1992). Finally, ritanserin counters drug-dependent tic- and 567
compulsion/craving- like symptoms, associated with its ability to block 5-HT2a,c 568
receptor stimulation of the glutamatergic cortical output neurons whose hyperactivity 569
underlies tics in the Ticcy mice and, we propose, in some forms of neurogenic TS 570
(Sheldon and Aghajanian, 1991; Willins and Meltzer, 1997; Marek and Aghajanian, 571
1998; Marek and Aghajanian, 1999; Ciccocioppo et al., 1999; Campbell et al., 1999a; 572
Nordstrom and Burton, 2002). 573

28
The alpha-1 NE antagonist, prazosin, should also prove worthy to test for human 574
TS. As an central alpha-1 adrenergic (but more NE selective) receptor antagonist, 575
prazosin reduces the excitatory influence of NE on its limbic and cortical targets and, 576
like ritanserin (which antagonizes the same neurons but through 5-HT2a,c receptors), 577
thus reduces subsequent cortical/amygdalar excitatory glutamatergic output to the 578
striatum and substantia nigra. Prazosin clinically has served as an antihypertensive 579
drug, but more recently was found to alleviate alcohol craving (Simpson et al., 2009), 580
which is thought to involve striatal circuits shared with impulse control disorder, an OC-581
spectrum disorder (Grant et al., 2014). Given that TS is an urge-driven twitch disorder 582
often comorbid with OCD (Frankel et al., 1988), and given that target receptors for both 583
ritanserin and prazosin are co-expressed in the tic-circuit glutamatergic neurons, 584
prazosin too is a good candidate to vet for the ability to alleviate tics in human TS. 585
The proposed shared neuroanatomical basis of tics in the Ticcy mice and in (at 586
least some forms of) human TS suggests that both ritanserin and prazosin will be 587
capable of alleviating tics, by suppression of cortical (and also amygdalar, for ritanserin) 588
glutamate output. This prediction was supported by our present study, where both drugs 589
were able to completely normalize tics in the Ticcy D1CT-7 transgenic mice. But what of 590
drugs, like moxonidine and bromocriptine, that work downstream of the primary cortical 591
and amygdalar sites that glutamatergically elicited TS-like tic behavior in these mice? 592
The Ticcy mice's tics are not initiated, but are subsequently mediated, by 593
increased striatothalamic GABAergic output from glutamate-excited striatal neurons of 594
the direct pathway -- neurons which should be amenable to inhibition by presynaptic 595

29
imidazoline I-1 receptor agonists like moxonidine. In humans, moxonidine has so far 596
been approved only as a centrally acting antihypertensive treatment, but is contra-597
indicated for use in parkinsonian patients -- suggesting it may reduce motor initiation 598
events, which would include tics. In mice, moxonidine is not only antihypertensive but 599
inhibits anxiogenic and also anxiety-dependent compulsive behaviors (Taksande et al., 600
2010; Dixit et al., 2014), suggesting moxonidine is a good clinical candidate to treat TS. 601
Finally, because striatal dopamine D2 receptors are not only postsynaptic but 602
also presynaptic, existing as inhibitory autoreceptors on dopamine-releasing 603
nigrostriatal terminals, at low doses bromocriptine acts selectively as a presynaptic D2 604
dopamine autoreceptor agonist (Jackson et al., 1988), similarly to pergolide, a 605
presynaptic dopamine D1/D2 receptor agonist recently shown to reduce human TS 606
symptoms (Gilbert et al., 2000). Only at higher doses does bromocriptine act as a 607
postsynaptic D2 receptor agonist, mimicking dopamineʼs effects on the indirect striatal 608
pathway (Jackson et al., 1988). Low-dose bromocriptine has previously been shown to 609
reduce symptoms of OCD (Ceccherini-Nelli and Guazzelli, 1994), hence it could be 610
tested for TS, although it might have no better clinical effect than current presynaptic DA 611
receptor agonists like pergolide and the novel atypical neuroleptic, aripiprazole (which 612
has a mild postsynaptic D2 antagonist and presynaptic D2 agonist action). 613
Nevertheless, the Ticcy mice's responsiveness to bromocriptine as well as postsynaptic 614
D2 antagonist TS drugs like pimozide (Campbell et al., 1999b) or the presynaptic alpha-615
2 NE receptor agonist clonidine (Nordstrom and Burton, 2002) supports the predictive 616
validity of this tic-circuit for candidate TS drug selection. 617

30
618
Clinical Implications for OC-spectrum disorders 619
How effective would these drugs be on compulsions in humans? Although our findings 620
predict effectiveness of these drugs in tic alleviation in TS and TS+OCD, one limitation of 621
this study is that it has no implications for these drugs' treatment of OCD. Of these four 622
acute drug administration studies, we only observed one, bromocriptine, to depress a 623
compulsive behavior in Ticcy mice (data not shown). But bromocriptine was also the one 624
drug that also suppressed normal mouse locomotion, and the compulsive behavior it 625
suppressed, climbing-leaping, is a (vertical) locomotion-dependent behavior, meaning it 626
could have been attenuated due to bromocriptine's broader attenuation of locomotion 627
(Jackson et al., 1998). Broader locomotor suppression may likewise be the mechanism by 628
which anti-dopaminergic drugs alleviate human compulsions and tics, which might be 629
triggered by cortical/limbic glutamate hyperexcitation but still require co-stimulation of 630
striatal dopamine receptors -- explaining why drugs that attenuate the nigrostriatal release 631
of dopamine confer therapeutic benefits but also parkinsonian adverse effects (Ceccherini-632
Nelli and Guazzelli, 1994; Gilbert et al., 2000). Interestingly, no bromocriptine-elicited 633
decrease was seen in horizontal locomotion in the Ticcy mice. One possible explanation is 634
that the drug depressed the mice's vertical climbing/leaping motor behaviors to a more 635
horizontal locomotion behavior. An alternative possibility is that there may be some 636
selectivity of action of dopamine receptor blockade upon different subsets of the 637
topographically parallel striatal motor circuits that, when excessively stimulated by 638
cortical/limbic glutamate, trigger distinct motor symptoms. 639

31
Another possible reason why we didn't observe pronounced suppressive effects on 640
Ticcy mouse compulsions at drug doses that don't suppress locomotion is that, unlike 641
tics, human compulsions usually don't respond to single acute drug administrations, but 642
only to long-term repeated drug administration -- a design we rejected due to the 643
deleterious effects on the anxiogenic Ticcy mice of repeated physical injections or 644
surgical pump implantation procedures. We conjecture that compulsions might respond 645
slower than tics because tics may originate solely from a narrow psychogenic (e.g., 646
hyperglutamatergic somatosensory cortical) subcircuit, while compulsions may originate 647
integratively from a broad convergence of parallel psychogenic (e.g., 648
hyperglutamatergic orbitofrontal cortical) plus anxiogenic (e.g., hyperglutamatergic 649
amygdalar) subcircuits. Hence drugs other than direct cortical/amygdalar anti-650
glutamatergics must act longer, or exert broader (e.g., combined antipsychotic-651
anxiolytic) effects to additionally counter compulsions. 652
Nevertheless, some data is available on the therapeutic effect of these drugs 653
exclusively on compulsion-like behaviors in normal mice: Acute moxonidine reportedly 654
reduces endogenous anxiety-dependent compulsive (marble-burying) behavior in mice 655
(Dixit et al., 2014) -- an effect we could not study in Ticcy mice due to their ignoring all 656
marble-burying behavior in lieu of more immediate locomotion-dependent compulsions 657
(not shown). However, the ability of moxonidine to suppress both compulsive marble-658
burying in normal mice, and, in this study, tics in Ticcy mice, offers convergent evidence 659
to support a therapeutic trial of moxonidine for not just TS but OC-spectrum disorders. 660
Acute ritanserin has been reported to variously inhibit (Bruins Slot et al., 2008), activate 661

32
(Njung'e and Handley, 1991), or have no effect (Ichimaru et al., 1995; Gaikwad et al., 662
2010) on normal mice's marble-burying, although it has anxiolytic, and thus potentially 663
anti-compulsive, effects in both rodents and humans (Ceulemans et al., 1985; Danjou et 664
al., 1992; Gao and Cutler, 1993). We speculate that it may be fruitful to examine the 665
action of all four of our tested drugs on not only human TS but comorbid TS+OCD, OC-666
spectrum and impulse control disorders, psychomotor side effects of therapeutic drugs 667
and drugs of abuse, and psychotic-spectrum disorders -- all of which we believe may 668
involve hyperactivation of glutamatergic circuit output from topographically-parallel 669
CSTC circuit loops. 670
671
CONCLUSION 672
673
Human tic disorders may be induced, as in the Ticcy transgenic mouse model, by 674
abnormally high levels of cortical/amygdalar glutamate deposition and consequent 675
coincident dopamine deposition onto target striatal direct and indirect pathway circuits. 676
Interestingly, our model merges prior glutamate, dopamine, serotonin, norepinephrine, 677
and agmatine/imidazoline models of tic and related psychotic-spectrum, drug-abuse, 678
and OC/impulse control-spectrum disorders. In this "five neurotransmitter" hypothesis 679
(Fig. 5), symptoms like tics, obsessions, compulsions, impulses, cravings, and 680
hallucinations could be triggered initially by excessive forebrain glutamatergic excitation 681
of the striatum and of the substantia nigra -- the latter triggering consequent 682
dopaminergic unbalancing of the glutamate-excited striatal neurons' motion/urge-683

33
activating (direct) vs. motion/urge-suppressing (indirect) striatothalamic outputs, 684
chronically favoring activated motion/urge symptoms. These symptoms should then 685
accordingly be counteracted not only by drugs that directly block forebrain glutamatergic 686
neurons' output, but by antagonists of these neurons' co-expressed excitatory forebrain 687
serotonin (ritanserin) and norepinephrine (prazosin) receptors; and by presynaptic 688
agmatinergic or dopaminergic drugs that, respectively, would block the downstream 689
glutamate-triggered target striatothalamic neurons' GABA output (moxonidine), or the 690
downstream glutamate-triggered target nigrostriatal neurons' co-modulatory dopamine 691
output (bromocriptine). Hence our observation that the Ticcy transgenic mice's tics are 692
fully alleviated, albeit with differing specificity, by acute treatment with all four drugs 693
confirms the drugs may be "short-circuiting" these mice's initial hyperactive 694
cortico/amygdalostriatal and cortico/amygdalonigral glutamate output; their target striatal 695
neurons' consequent glutamate-triggered hyperactive striatothalamic GABAergic output; 696
and their target nigral neurons' consequent glutamate-triggered hyperactive nigrostriatal 697
DA output. Our findings suggest that the "cortical/limbic glutamatergic neuron (CGN) 698
hyperactivity" model of neurogenic tics is a valid tic-circuit model for designing future 699
interventional therapies for human TS, and suggest new drugs that should be useful to 700
test in clinical trials. 701
702
CONFLICT OF INTEREST 703
704
The authors declare no actual or potential conflict of interest. 705
706

34
ACKNOWLEDGEMENTS 707
708
This work was supported by NIH training grant T32DA07097 to MJM; and by NIH 709
research grant R03MH53553, the Jeff Sutton Memorial Young Investigator Award from 710
the National Alliance for Research on Schizophrenia and Depression and the Rochester 711
Area Alliance for the Mentally Ill, and grants from the Tourette Syndrome Association 712
and the University of Minnesota Foundation to FHB. 713
714
REFERENCES 715
716
Aghajanian GK, Marek GJ (1999) Serotonin, via 5-HT2A receptors, increases EPSCs in 717
layer V pyramidal cells of prefrontal cortex by an asynchronous mode of glutamate 718
release. Brain Res 825:161-171. 719
720
American Psychiatric Association (2013) Diagnostic and Statistical Manual of Mental 721
Disorders, 5th Edn. Washington, DC: APA. 722
723
Bakuridze K, Savli E, Gongadze N, Baş DB, Gepdiremen A (2009) Protection in 724
glutamate-induced neurotoxicity by imidazoline receptor agonist moxonidine. Int J 725
Neurosci 119(10):1705-1717. 726
727
Barone JA, Bierman RH, Cornish JW, Hsuan A, Drake ND, Colaizzi JL (1986) Safety 728

35
evaluation of ritanserin--an investigational serotonin antagonist. Drug Intell Clin Pharm 729
20(10):770-775. 730
731
Bonnier C, Nassogne MC, Evrard P (1999) Ketanserin treatment of Tourette's syndrome 732
in children. Am J Psychiatry 156:1122-1123. 733
734
Breiter HC, Rauch SL, Kwong KK, Baker JR, Weisskoff RM, Kennedy DN, Kendrick AD, 735
Davis TL, Jiang A, Cohen MS, Stern CE, Belliveau JW, Baer L, O'Sullivan RL, Savage 736
CR, Jenike MA, Rosen BR (1996) Functional magnetic resonance imaging of symptom 737
provocation in obsessive-compulsive disorder. Arch Gen Psychiatry 53(7):595-606. 738
739
Brogden RN, Sorkin EM (1990) Ketanserin. A review of its pharmacodynamic and 740
pharmacokinetic properties, and therapeutic potential in hypertension and peripheral 741
vascular disease. Drugs 40(6):903-949. 742
743
Bruins Slot LA, Bardin L, Auclair AL, Depoortere R, Newman-Tancredi A (2008) Effects 744
of antipsychotics and reference monoaminergic ligands on marble burying behavior in 745
mice. Behav Pharmacol 19(2):145-152. 746
747
Bruun RD, Budman CL (1996) Risperidone as a treatment for Tourette's syndrome. J 748
Clin Psychiatry 57(1):29-31. 749
750

36
Burke K, Lombroso PJ (2004) Animal models of Tourette syndrome. In: Animal Models 751
of Movement Disorders (LeDoux M, ed), pp 441-448. Burlington, MA: Elsevier Academic 752
Press. 753
754
Burton FH, Hasel KW, Bloom FE, Sutcliffe JG (1991) Pituitary hyperplasia and 755
gigantism in mice caused by a cholera toxin transgene. Nature 350:74-77. 756
757
Campbell KM, Rohland RM, McGrath MJ, Satoskar SD, Burton FH (1998) Detecting 758
subtle differences in behavior using waveform display analysis. Physiol Behav 64:83-91. 759
760
Campbell KM, de Lecea L, Severynse DM, Caron MG, McGrath MJ, Sparber SB, Sun 761
LY, Burton FH (1999a) OCD-Like behaviors caused by a neuropotentiating transgene 762
targeted to cortical and limbic D1+ neurons. J Neurosci 19(12):5044-5053. 763
764
Campbell KM, McGrath MJ, Burton FH (1999b) Differential response of cortical-limbic 765
neuropotentiated compulsive mice to D1 and D2 antagonists. Eur J Pharmacol 371:103-766
111. 767
768
Campbell KM, McGrath MJ and Burton FH (1999c) Behavioral effects of cocaine on a 769
transgenic mouse model of cortical-limbic compulsion. Brain Res 833:216-224. 770
771
Campbell MJ, Veldman MB, McGrath MJ, Burton FH (2000) TS+OCD-like 772

37
neuropotentiated mice are supersensitive to seizure induction. NeuroReport 11:2335-773
2338. 774
775
Carlsson ML (2000) On the role of cortical glutamate in obsessive-compulsive disorder 776
and attention-deficit hyperactivity disorder, two phenomenologically antithetical 777
conditions. Acta Psychiatr Scand 102:401-413. 778
779
Castro CA, Hogan JB, Benson KA, Shehata CW, Landauer MR (1995) Behavioral 780
effects of vehicles: DMSO, ethanol, Tween-20, Tween-80, and emulphor-620. 781
Pharmacol Biochem Behav 50(4):521-526. 782
783
Ceccherini-Nelli A, Guazzelli M (1994) Treatment of refractory OCD with the dopamine 784
agonist bromocriptine. J Clin Psychiatry 55:415-416. 785
786
Ceulemans DL, Hoppenbrouwers ML, Gelders YG, Reyntjens AJ (1985) The influence 787
of ritanserin, a serotonin antagonist, in anxiety disorders: a double-blind placebo-788
controlled study versus lorazepam. Pharmacopsychiatry 18:303-305. 789
790
Chakrabarty K, Bhattacharyya S, Christopher R, Khanna S (2005) Glutamatergic 791
Dysfunction in OCD. Neuropsychopharmacol 30:1735-1740. 792
793
Cheng JT, Kuo DY (2003) Both alpha1-adrenergic and D(1)-dopaminergic 794

38
neurotransmissions are involved in phenylpropanolamine-mediated feeding suppression 795
in mice. Neurosci Lett 347(2):136-138. 796
797
Ciccocioppo R, Angeletti S, Colombo G, Gessa G, Massi M (1999) Autoradiographic 798
analysis of 5-HT2A binding sites in the brain of Sardinian alcohol-preferring and 799
nonpreferring rats. Eur J Pharmacol 373(1):13-19. 800
801
Cohen DJ, Riddle MA, Leckman JF (1992) Pharmacotherapy of Tourette's syndrome 802
and associated disorders. Psychiatr Clin North Am 15:109-129. 803
804
Danjou P, Warot D, Hergueta T, Lacomblez L, Bouhours P, Puech AJ (1992) 805
Comparative study of the psychomotor and antistress effects of ritanserin, alprazolam 806
and diazepam in healthy subjects: some trait anxiety-independent responses. Int Clin 807
Psychopharmacol 7(2):73-79. 808
809
Denys D, de Vries F, Cath D, Figee M, Vulink N, Veltman DJ, van der Doef TF, 810
Boellaard R, Westenberg H, van Balkom A, Lammertsma AA, van Berckel BN (2013) 811
Dopaminergic activity in Tourette syndrome and obsessive-compulsive disorder. Eur 812
Neuropsychopharmacol 23(11):1423-1431. 813
814
Dixit MP, Thakre PP, Pannase AS, Aglawe MM, Taksande BG, Kotagale NR (2014) 815
Imidazoline binding sites mediates anticompulsive-like effect of agmatine in marble-816

39
burying behavior in mice. Eur J Pharmacol 732:26-31. 817
818
Edgley SA, Lemon RN (1999) Experiments using transcranial magnetic brain stimulation 819
in man could reveal important new mechanisms in motor control. J Physiol (Lond) 820
521:565. 821
822
Fairbanks CA, Wilcox GL (1999) Moxonidine, a selective alpha2-adrenergic and 823
imidazoline receptor agonist, produces spinal antinociception in mice. J Pharmacol Exp 824
Ther 290:403-412. 825
826
Frankel M, Cummings JL, Robertson MM, Trimble MR, Hill MA, Benson DF (1986) 827
Obsessions and compulsions in Gilles de la Tourette's syndrome. Neurology 36:378-828
382. 829
830
Gaikwad U, Parle M, Kumar A, Gaikwad D (2010) Effect of ritanserin and leuprolide 831
alone and combined on marble-burying behavior of mice. Acta Pol Pharm 67(5):523-832
527. 833
834
Gao B, Cutler MG (1993) Effects of acute and subchronic administration of ritanserin on 835
the social behaviour of mice. Neuropharmacology 32(3):265-272. 836
837
Gibson EL, Barnfield AM, Curzon G (1994) Evidence that mCPP-induced anxiety in the 838

40
plus-maze is mediated by postsynaptic 5-HT2C receptors but not by sympathomimetic 839
effects. Neuropharmacology 33(3-4):457-465. 840
841
Gilbert DL, Sethuraman G, Sine L, Peters S, Sallee FR (2000) Tourette's syndrome 842
improvement with pergolide in a randomized, double-blind, crossover trial. Neurology 843
54:1310-1315. 844
845
Gilbert DL, Bansal AS, Sethuraman G, Sallee FR, Zhang J, Lipps T, Wassermann EM 846
(2004) Association of cortical disinhibition with tic, ADHD and OCD severity in Tourette 847
Syndrome. Mov Disord 19:416-425. 848
849
Gilbert DL, Budman CL, Singer HS, Kurlan R, Chipkin RE (2014) A D1 receptor 850
antagonist, ecopipam, for treatment of tics in Tourette syndrome. Clin Neuropharmacol 851
37(1):26-30. 852
853
Goetz CG (1992) Clonidine and clonazepam in Tourette syndrome. Adv Neurol 58:245-854
251. 855
856
Grant JE, Odlaug BL, Kim SW (2009) N-acetylcysteine, a glutamate modulator, in the 857
treatment of trichotillomania: a double-blind, placebo-controlled study. Arch Gen 858
Psychiatry 66(7):756-763. 859
860

41
Grant JE, Odlaug BL, Black DW, Fong T, Davtian M, Chipkin R, Kim SW (2014) A 861
single-blind study of 'as-needed' ecopipam for gambling disorder. Ann Clin Psychiatry 862
26(3):179-186. 863
864
Grant P, Lougee L, Hirschtritt M, Swedo SE (2007) An open-label trial of riluzole, a 865
glutamate antagonist, in children with treatment-resistant obsessive-compulsive 866
disorder. J Child Adolesc Psychopharmacol 17(6):761-767. 867
868
Homayoun H, Moghaddam B (2007) NMDA receptor hypofunction produces opposite 869
effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 870
27(43):11496-11500. 871
872
Hosie J, Stott DJ, Robertson JI, Ball SG (1987) Does acute serotonergic type-2 873
antagonism reduce blood pressure? Comparative effects of single doses of ritanserin 874
and ketanserin in essential hypertension. J Cardiovasc Pharmacol 10 Suppl 3:S86-S88. 875
876
Ichimaru Y, Egawa T, Sawa A (1995) 5-HT1A-receptor subtype mediates the effect of 877
fluvoxamine, a selective serotonin reuptake inhibitor, on marble-burying behavior in 878
mice. Jpn J Pharmacol 68(1):65-70. 879
880
Jackson DM, Jenkins OF, Ross SB (1988) The motor effects of bromocriptine--a review. 881
Psychopharmacology (Berl) 95:433-446. 882

42
883
Jakab RL, Goldman-Rakic PS (1998) 5-Hydroxytryptamine2A serotonin receptors in the 884
primate cerebral cortex: possible site of action of hallucinogenic and antipsychotic drugs 885
in pyramidal cell apical dendrites. Proc Natl Acad Sci U S A 95(2):735-740. 886
887
Joel D (2006) Current animal models of obsessive compulsive disorder: A critical 888
review. Prog Neuropsychopharmacol Biol Psychiatry 30(3):374-388. 889
890
Lafleur DL, Pittenger C, Kelmendi B, Gardner T, Wasylink S, Malison RT, Sanacora G, 891
Krystal JH, Coric V (2006) N-acetylcysteine augmentation in serotonin reuptake inhibitor 892
refractory obsessive-compulsive disorder. Psychopharmacology (Berl) 184(2):254-256. 893
894
Lichter DG, Jackson LA (1996) Predictors of clonidine response in Tourette syndrome: 895
implications and inferences. J Child Neurol 11:93-97. 896
897
Marek GJ, Aghajanian GK (1998) The electrophysiology of prefrontal serotonin systems: 898
therapeutic implications for mood and psychosis. Biol Psychiatry 44(11):1118-1127. 899
900
Marek GJ, Aghajanian GK (1999) 5-HT2A receptor or alpha1-adrenoceptor activation 901
induces excitatory postsynaptic currents in layer V pyramidal cells of the medial 902
prefrontal cortex. Eur J Pharmacol 367(2-3):197-206. 903
904

43
McGrath MJ, Campbell KM, Veldman MB, Burton FH (1999a) Anxiety in a transgenic 905
mouse model of cortical-limbic neuropotentiated compulsive behavior. Behav 906
Pharmacol 10:435-443. 907
908
McGrath MJ, Campbell KM, Burton FH (1999b) The role of cognitive and affective 909
processing in a transgenic mouse model of cortical-limbic neuropotentiated compulsive 910
behavior. Behav Neurosci 113:1249-1256. 911
912
McGrath MJ, Campbell KM, Parks CR III and Burton FH (2000) Glutamatergic drugs 913
exacerbate symptomatic behavior in a transgenic model of comorbid Tourette's 914
Syndrome and obsessive-compulsive disorder. Brain Res 877:23-30. 915
916
Menzies L, Chamberlain SR, Laird AR, Thelen SM, Sahakian BJ, Bullmore ET (2008) 917
Integrating evidence from neuroimaging and neuropsychological studies of obsessive-918
compulsive disorder: the orbitofronto-striatal model revisited. Neurosci Biobehav Rev 919
32(3):525-549. Review. 920
921
Milad MR, Rauch SL (2012) Obsessive-compulsive disorder: beyond segregated 922
cortico-striatal pathways. Trends Cogn Sci 16(1):43-51. Review. 923
924
Minzer K, Lee O, Hong JJ, Singer HS (2004) Increased prefrontal D2 protein in Tourette 925
syndrome: a postmortem analysis of frontal cortex and striatum. J Neurol Sci 219:55-61. 926

44
927
Nestler EJ (1997) Schizophrenia. An emerging pathophysiology [news; comment]. 928
Nature 385(6617):578-579. 929
930
Ninan I, Kulkarni SK (1998) 5-HT2A receptor antagonists block MK-801-induced 931
stereotypy and hyperlocomotion. Eur J Pharmacol 358(2):111-116. 932
933
Njung'e K, Handley SL (1991) Effects of 5-HT uptake inhibitors, agonists and 934
antagonists on the burying of harmless objects by mice; a putative test for anxiolytic 935
agents. Br J Pharmacol 104(1):105-112. 936
937
Nordstrom EJ, Burton FH (2002) A transgenic model of comorbid Tourette's syndrome 938
and obsessive-compulsive disorder circuitry. Mol Psychiatry 7:617-625. 939
940
Peterson B, Riddle MA, Cohen DJ, Katz LD, Smith JC, Hardin MT, Leckman JF (1993) 941
Reduced basal ganglia volumes in Tourette's syndrome using three-dimensional 942
reconstruction techniques from magnetic resonance images. Neurology 43:941-949. 943
944
Peterson BS, Skudlarski P, Anderson AW, Zhang H, Gatenby JC, Lacadie CM, 945
Leckman JF, Gore JC (1998) A functional magnetic resonance imaging study of tic 946
suppression in Tourette syndrome. Arch Gen Psychiatry 55:326-333. 947
948

45
Rapoport JL, Swedo SE, Leonard HL (1992) Childhood obsessive compulsive disorder. 949
J Clin Psychiatry 53:11-16. 950
951
Robertson MM (2000) Tourette syndrome, associated conditions and the complexities of 952
treatment. Brain 123:425-462. 953
954
Rosenberg DR, MacMaster FP, Keshavan MS, Fitzgerald KD, Stewart CM, Moore GJ 955
(2000) Decrease in caudate glutamatergic concentrations in pediatric obsessive-956
compulsive disorder patients taking paroxetine. J Am Acad Child Adolesc Psychiatry 957
39(9):1096-1103. 958
959
Sah R, Sallee FR (2002) Dopaminergic neurotransmission in Touretteʼs syndrome: a 960
current update. In: Dopamine Receptors and Transporters: Function, Imaging, and 961
Clinical Implication, 2nd Edition (Sidhu A, Laruelle M, Vernier P, eds), pp 369-389. New 962
York, NY: Marcel Dekker Inc. 963
964
Sallee FR, Kurlan R, Goetz CG, Singer H, Scahill L, Law G, Dittman VM, Chappell PB 965
(2000) Ziprasidone treatment of children and adolescents with Tourette's syndrome: a 966
pilot study. J Am Acad Child Adolesc Psychiatry 39(3):292-299. 967
968
Sheldon PW, Aghajanian GK (1991) Excitatory responses to serotonin (5-HT) in 969
neurons of the rat piriform cortex: evidence for mediation by 5-HT1C receptors in 970

46
pyramidal cells and 5-HT2 receptors in interneurons. Synapse 9:208-218. 971
972
Simpson TL, Saxon AJ, Meredith CW, Malte CA, McBride B, Ferguson LC, Gross CA, 973
Hart KL, Raskind M (2009) A pilot trial of the alpha-1 adrenergic antagonist, prazosin, 974
for alcohol dependence. Alcohol Clin Exp Res 33(2):255–263. 975
976
Singer HS, Morris C, Grados M (2010) Glutamatergic modulatory therapy for Tourette 977
syndrome. Med Hypotheses 74(5):862-867. 978
979
Swedo SE, Pietrini P, Leonard HL, Schapiro MB, Rettew DC, Goldberger EL, Rapoport 980
SI, Rapoport JL, Grady CL (1992) Cerebral glucose metabolism in childhood-onset 981
obsessive-compulsive disorder. Revisualization during pharmacotherapy. Arch Gen 982
Psychiatry 49:690-694. 983
984
Swerdlow NR, Sutherland AN (2005) Using animal models to develop therapeutics for 985
Tourette Syndrome. Pharmacol Ther 108(3):281-293. Review. 986
987
Taksande BG, Kotagale NR, Patel MR, Shelkar GP, Ugale RR, Chopde CT (2010) 988
Agmatine, an endogenous imidazoline receptor ligand modulates ethanol anxiolysis and 989
withdrawal anxiety in rats. Eur J Pharmacol 637(1-3):89-101. 990
991
Tanabe M, Kino Y, Honda M, Ono H (2006) Presynaptic I1-imidazoline receptors reduce 992

47
GABAergic synaptic transmission in striatal medium spiny neurons. J Neurosci 993
26(6):1795-1802. 994
995
Ting JT, Feng G (2008) Glutamatergic synaptic dysfunction and obsessive-compulsive 996
disorder. Curr Chem Genomics 2:62-75. 997
998
Verkerk AJ, Mathews CA, Joosse M, Eussen BH, Heutink P, Oostra BA (2003) Tourette 999
Syndrome Association International Consortium for Genetics. CNTNAP2 is disrupted in 1000
a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. 1001
Genomics 82:1-9. 1002
1003
Wang L, Simpson HB, Dulawa SC (2009) Assessing the validity of current mouse 1004
genetic models of obsessive–compulsive disorder. Behav Pharmacol 20(2):119-133. 1005
1006
Wellman PJ, Davies BT (1992) Reversal of cirazoline- and phenylpropanolamine-1007
induced anorexia by the alpha1-receptor antagonist prazosin. Pharmacol Biochem 1008
Behav. 42(1):97-100. 1009
1010
Wellman PJ, McMahon LR, Green T, Tole A (1997) Effects of the alpha 1a-1011
adrenoceptor antagonist RS-17053 on phenylpropanolamine-induced anorexia in rats. 1012
Pharmacol Biochem Behav 57(1-2):281-284. 1013
1014

48
Willins DL, Meltzer HY (1997) Direct injection of 5-HT2A receptor agonists into the 1015
medial prefrontal cortex produces a head-twitch response in rats. J Pharmacol Exp Ther 1016
282(2):699-706. 1017
1018
Wolf SS, Jones DW, Knable MB, Gorey JG, Lee KS, Hyde TM, Coppola R, Weinberger 1019
DR (1996) Tourette syndrome: prediction of phenotypic variation in monozygotic twins 1020
by caudate nucleus D2 receptor binding. Science 273(5279):1225-1227. 1021
1022
Wu K, Hanna GL, Rosenberg DR, Arnold PD (2012) The role of glutamate signaling in 1023
the pathogenesis and treatment of obsessive-compulsive disorder. Pharmacol Biochem 1024
Behav 100(4):726-735. Review. 1025
1026
Zeiger MA, Saji M, Gusev Y, Westra WH, Takiyama Y, Dooley WC, Kohn LD, Levine 1027
MA (1997) Thyroid-specific expression of cholera toxin A1 subunit causes thyroid 1028
hyperplasia and hyperthyroidism in transgenic mice. Endocrinology 138:3133-3140. 1029
1030
Zhu QM, Lesnick JD, Jasper JR, MacLennan SJ, Dillon MP, Eglen RM, Blue DR Jr 1031
(1999) Cardiovascular effects of rilmenidine, moxonidine and clonidine in conscious 1032
wild-type and D79N alpha2A adrenoceptor transgenic mice. Br J Pharmacol 1033
126(6):1522-1530. 1034
1035
Ziemann U, Paulus W, Rothenberger A (1997) Decreased motor inhibition in Tourette's 1036

49
disorder: evidence from transcranial magnetic stimulation. Am J Psychiatry 154:1277-1037
1284. 1038
1039
FIGURE LEGENDS 1040
1041
Fig 1. Ritanserin alleviates tics in a transgenic model of Tourette's syndrome. 1042
Panel A. Ritanserin (1 mg/kg, i.p.) normalizes tics in D1CT-7 "Ticcy" transgenic mice. 1043
Data are shown as a bar graph of the mean number (+ S.E.M.) of head or body twitches 1044
occurring over 15 min of videotaped observation. Overall significance of genotype effect 1045
[F(1,17) = 8.771; P = 0.0087, n = 8 Tg, 11 C], drug effect [F(1,17) = 14.113; P = 0.0016, 1046
n = 8 Tg, 11 C], and genotype x drug interaction [F(1,17) = 8.487; P = 0.0097, n = 8 Tg, 1047
11 C] was established by repeated measures ANOVA, followed by individual between-1048
group Mann-Whitney U-test of genotype effect and within-group Wilcoxon Signed Rank 1049
test of drug effect, with significance established at tied P < 0.05, which revealed both 1050
elevated tics in transgenic mice and reduction of their tics by ritanserin treatment. 1051
Panel B. Tic reduction by ritanserin is not associated with reduced locomotion. Data are 1052
shown as a bar graph of the mean number (+S.E.M.) of cage midline crossings, an 1053
assay of locomotion, occurring over 15 min of videotaped observation. Non-significance 1054
of all effects and interactions was established by repeated measures ANOVA, which 1055
revealed that 1 mg/kg i.p. ritanserin did not alter locomotor activity, indicating that the 1056
tic-suppressing effect of ritanserin in Ticcy mice occurs in the absence of general 1057
locomotor inhibition or sedation. 1058

50
Statistics: Initial repeated measures ANOVA (n = 8 transgenic, 11 control non-1059
transgenic mice) was performed to establish overall significance on tics or locomotion of 1060
genotype effect, drug effect, or genotype x drug interaction, after which individual 1061
comparisons of the non-parametrically distributed tic data (see Methods) or the 1062
parametrically distributed locomotion data were performed by between-group non-1063
parametric Mann-Whitney U-tests or parametric unpaired 2-tailed Student's t-tests of 1064
genotype effects, and within-group non-parametric Wilcoxon Signed Rank tests or 1065
parametric paired 2-tailed Student's t-tests of drug effects, with significance of effect on 1066
non-parametrically distributed tic counts assumed at tied P < 0.05 and on 1067
parametrically-distributed locomotor event counts assumed at P < 0.05. 1068
Abbreviations: Tg (D1CT- 7 "Ticcy" transgenic female mice); C (non-transgenic control 1069
female mice); Veh (saline vehicle i.p. injection); Rit (1 mg/kg i.p. ritanserin injection); **P 1070
< 0.01 for between-group (Tg vs C, Veh) comparison (unpaired Mann-Whitney U-test), 1071
+P < 0.05 for within-group, between-treatment (Tg, Veh vs Rit) comparison (paired 1072
Wilcoxon Signed Rank test), n = 8 Tg, 11 C mice. 1073
1074
Fig 2. Prazosin alleviates tics in a transgenic model of Tourette's syndrome. 1075
Panel A. Prazosin (3 mg/kg, i.p.) normalizes tics in D1CT-7 "Ticcy" transgenic mice. 1076
Data are shown as a bar graph of the mean number (+ S.E.M.) of head or body twitches 1077
occurring over 15 min of videotaped observation. Overall significance of genotype effect 1078
[F(1,11) = 10.259; P = 0.0084, n = 7 Tg, 6 C], drug effect [F(1,11) = 21.495; P = 0.0007, 1079
n = 7 Tg, 6 C], and genotype x drug interaction [F(1,11) = 18.424; P = 0.0013, n = 7 Tg, 1080

51
6 C] was established by repeated measures ANOVA, followed by individual between-1081
group Mann-Whitney U-test of genotype effect and within-group Wilcoxon Signed Rank 1082
test of drug effect, with significance established at tied P < 0.05, which revealed both 1083
elevated tics in transgenic mice and reduction of their tics by prazosin treatment. 1084
Panel B. Tic reduction by prazosin is not associated with reduced locomotion. Data are 1085
shown as a bar graph of the mean number (+ S.E.M.) of cage midline crossings, an 1086
assay of locomotion, occurring over 15 min of videotaped observation. Non-significance 1087
of all effects and interactions was established by repeated measures ANOVA, which 1088
revealed that 3 mg/kg i.p. prazosin did not alter locomotor activity, indicating that the tic-1089
suppressing effect of prazosin in Ticcy mice occurs in the absence of general locomotor 1090
inhibition or sedation. 1091
Statistics: Initial repeated measures ANOVA (n = 7 transgenic, 6 control non-transgenic 1092
mice) was performed to establish overall significance on tics or locomotion of genotype 1093
effect, drug effect, or genotype x drug interaction, after which individual comparisons of 1094
the non-parametrically distributed tic data (see Methods) or the parametrically 1095
distributed locomotion data were performed by between-group non-parametric Mann-1096
Whitney U-tests or parametric unpaired 2-tailed Student's t-tests of genotype effects, 1097
and within-group non-parametric Wilcoxon Signed Rank tests or parametric paired 2-1098
tailed Student's t-tests of drug effects, with significance of effect on non-parametrically 1099
distributed tic counts assumed at tied P < 0.05 and on parametrically-distributed 1100
locomotor event counts assumed at P < 0.05. 1101
Abbreviations: Tg (D1CT- 7 "Ticcy" transgenic female mice); C (non-transgenic control 1102

52
female mice); Veh (saline vehicle i.p. injection); Praz (3 mg/kg prazosin i.p. injection); 1103
**P < 0.01 for between-group (Tg vs C, Veh) comparison (unpaired Mann-Whitney U-1104
test), +P < 0.05 for within-group, between-treatment (Tg, Veh vs Praz) comparison 1105
(paired Wilcoxon Signed Rank test), n = 7 Tg, 6 C mice. 1106
1107
Fig 3. Moxonidine alleviates tics in a transgenic model of Tourette's syndrome. 1108
Panel A. Moxonidine (0.5 mg/kg, i.p.) normalizes tics in D1CT-7 "Ticcy" transgenic mice. 1109
Data are shown as a bar graph of the mean number (+ S.E.M.) of head or body twitches 1110
occurring over 15 min of videotaped observation. Overall significance of genotype effect 1111
[F(1,12) = 8.753; P = 0.012, n = 9 Tg, 5 C], drug effect [F(1,12) = 39.656; P < 0.0001, n 1112
= 9 Tg, 5 C], and genotype x drug interaction [F(1,12) = 18.344; P = 0.0011, n = 9 Tg, 5 1113
C] was established by repeated measures ANOVA, followed by individual between-1114
group Mann-Whitney U-test of genotype effect and within-group Wilcoxon Signed Rank 1115
test of drug effect, with significance established at tied P < 0.05, which revealed both 1116
elevated tics in transgenic mice and reduction of their tics by moxonidine treatment. 1117
Panel B. Tic reduction by moxonidine is not associated with reduced locomotion. Data 1118
are shown as a bar graph of the mean number (+ S.E.M.) of cage midline crossings, an 1119
assay of locomotion, occurring over 15 min of videotaped observation. Non-significance 1120
of all effects and interactions was established by repeated measures ANOVA, which 1121
revealed that 0.5 mg/kg i.p. moxonidine did not alter locomotor activity, indicating that 1122
the tic-suppressing effect of moxonidine in Ticcy mice occurs in the absence of general 1123
locomotor inhibition or sedation. 1124

53
Statistics: Initial repeated measures ANOVA (n = 9 transgenic, 5 control non-transgenic 1125
mice) was performed to establish overall significance on tics or locomotion of genotype 1126
effect, drug effect, or genotype x drug interaction, after which individual comparisons of 1127
the non-parametrically distributed tic data (see Methods) or the parametrically 1128
distributed locomotion data were performed by between-group non-parametric Mann-1129
Whitney U-tests or parametric unpaired 2-tailed Student's t-tests of genotype effects, 1130
and within-group non-parametric Wilcoxon Signed Rank tests or parametric paired 2-1131
tailed Student's t-tests of drug effects, with significance of effect on non-parametrically 1132
distributed tic counts assumed at tied P < 0.05 and on parametrically-distributed 1133
locomotor event counts assumed at P < 0.05. 1134
Abbreviations: Tg (D1CT- 7 "Ticcy" transgenic female mice); C (non-transgenic control 1135
female mice); Veh (saline vehicle i.p. injection); Mox (0.5 mg/kg moxonidine i.p. 1136
injection); **P < 0.01 for between-group (Tg vs C, Veh) comparison (unpaired Mann-1137
Whitney U-test), ++P < 0.01 for within-group, between-treatment (Tg, Veh vs Mox) 1138
comparison (paired Wilcoxon Signed Rank test), n = 9 Tg, 5 C mice. 1139
1140
Fig 4. Bromocriptine alleviates tics in a transgenic model of Tourette's 1141
syndrome. 1142
Panel A. Bromocriptine (5 mg/kg, i.p.) normalizes tics in D1CT-7 "Ticcy" transgenic 1143
mice. Data are shown as a bar graph of the mean number (+ S.E.M.) of head or body 1144
twitches occurring over 15 min of videotaped observation beginning 15 minutes before 1145
(-Bromo) vs. two hours after (+Bromo) drug injection. Repeated measures ANOVA 1146

54
showed both a significant overall effect on ticcing incidence of bromocriptine treatment 1147
[F(1,14) = 42.215; P < 0.0001, n = 8 Tg, 8 C], and a significant genotype x 1148
bromocriptine interaction [F(1,14) = 5.385; P = 0.0359, n = 8 Tg, 8 C ], justifying 1149
individual comparisons of the non-parametrically distributed tic count data by between-1150
group Mann-Whitney U-test of genotype effect and within-group Wilcoxon Signed Rank 1151
test of drug effect with significance established at tied P < 0.05, which revealed elevated 1152
tics in transgenic mice and reduction of their tics by bromocriptine treatment, as well as 1153
reduction of control mice's baseline twitch count by bromocriptine treatment. 1154
Panel B. Bromocriptine is associated with reduced locomotion in control mice. Data are 1155
shown as a bar graph of the mean number (+ S.E.M.) of locomotion events occurring 1156
over 15 min of videotaped observation beginning 15 minutes before (-Bromo) vs. two 1157
hours after (+Bromo) drug injection. Repeated measures ANOVA showed a significant 1158
overall effect on locomotion events of drug treatment [F(1,14) = 12.427; P = 0.0034; n = 1159
8 Tg, 8 C], justifying individual comparison of bromocriptine's effects on the 1160
parametrically distributed locomotion event count data by within-group, paired 2-tailed 1161
Studentʼs t-test, which revealed that 5 mg/kg i.p. bromocriptine suppressed locomotion 1162
events in control mice, indicating that the tic-suppressing effect of bromocriptine in Ticcy 1163
and control mice occurs in conjunction with a general locomotor inhibiting or sedating 1164
effect evident in control mice. 1165
Statistics: Initial repeated measures ANOVA (n = 8 transgenic, 8 control non-transgenic 1166
mice) was performed to establish overall significance on tics or locomotion of genotype 1167
effect, drug effect, or genotype x drug interaction, after which individual comparisons of 1168

55
the non-parametrically distributed tic data (see Methods) or the parametrically 1169
distributed locomotion data were performed by between-group non-parametric Mann-1170
Whitney U-tests or parametric unpaired 2-tailed Student's t-tests of genotype effects, 1171
and within-group non-parametric Wilcoxon Signed Rank tests or parametric paired 2-1172
tailed Student's t-tests of drug effects, with significance of effect on non-parametrically 1173
distributed tic counts assumed at tied P < 0.05 and on parametrically-distributed 1174
locomotion event counts assumed at P < 0.05. 1175
Abbreviations: Tg (D1CT- 7 "Ticcy" transgenic female mice); C (non-transgenic control 1176
female mice); -Bromo (15 mins pre-injection); +Bromo (5 mg/kg i.p. bromocriptine, 2 hrs 1177
post-injection); *P < 0.05 for between-group (Tg vs C, -Bromo) comparison of genotype 1178
effect on non-parametrically distributed tic counts (unpaired Mann-Whitney U-test), +P < 1179
0.05 for within-group, between-treatment (-Bromo vs +Bromo) comparisons of drug 1180
effect on non-parametrically distributed Tg and C tic counts (paired Wilcoxon Signed 1181
Rank tests), ++P < 0.01 for within-group, between-treatment (C, -Bromo vs +Bromo) 1182
comparison of drug effect on parametrically-distributed locomotion event counts (paired 1183
2-tailed Students' t-test), n = 8 Tg, 8 C mice. 1184
1185
Figure 5. Predicted hyperglutamatergic tic circuit and circuit-breaker drugs' 1186
actions. 1187
Panel A. Normal circuit controlling motion and urges. 1188
Panel B. Abnormal cortical/amygdalar hyperglutamatergic circuit triggers tics. 1189
Panel C. Ritanserin breaks tic circuit as a cortical/amygdalar 5-HT2a,c antagonist. 1190

56
Panel D. Prazosin breaks tic circuit as a cortical/amygdalar alpha-1 NE antagonist. 1191
Panel E. Moxonidine breaks tic circuit as a striatothalamic I-1 presynaptic agonist. 1192
Panel F. Bromocriptine counters tic circuit as a nigrostriatal D2 DA presynaptic agonist. 1193
Symbols: Triangles, excitatory glutamatergic pyramidal cortical/amygdalar output 1194
neurons; Filled triangles, hyperactivated glutamatergic output neurons (due to D1CT-7 1195
transgene-potentiated excitatory afferents in Ticcy mice or genetic/epigenetic alterations 1196
in neurogenic TS; Circles, target striatal GABAergic neurons; Squares, modulatory 1197
substantia nigra dopaminergic neurons; , move-urge-exciting or striatal neuron-1198
exciting neurotransmission; ---|, move-urge-inhibiting or striatal-neuron inhibiting 1199
neurotransmission; Thicker arrows, increased neurotransmission; Thicker "move-urge" 1200
box, move-urge excitation (e.g., tics, obsessions, compulsions, impulses, cravings, or 1201
hallucinations, depending on topographic parallelism of the circuit). 1202
Abbreviations: GLU, glutamate (excitatory); DA, dopamine (modulatory); CTX, cortex; 1203
AMY, amygdala; STR, striatum; SNc, substantia nigra pars compacta; D1, dopamine D1 1204
excitatory postsynaptic receptors; D2, dopamine D2 inhibitory postsynaptic (left) or 1205
presynaptic (middle) receptors; DP, striatal direct pathway (D1 receptor excited, 1206
motor/urge-activating); IP, striatal indirect pathway (D2 receptor inhibited, motion/urge-1207
suppressing); 5-HT2a,c, serotonin 5-HT2a,c excitatory postsynaptic receptors; alpha-1, 1208
norepinephrine alpha-1adrenergic excitatory postsynaptic receptors; I-1, imidazoline-1 1209
(agmatine) inhibitory presynaptic receptors; RIT, ritanserin; PRAZ, prazosin; MOX, 1210
moxonidine; BROMO, bromocriptine. 1211
1212

57
1213
1214
1215
1216
1217
1218
1219
1220
1221
1222
1223
1224
1225
Fig 1. Ritanserin alleviates tics in transgenic model of Tourette's syndrome. 1226
1227
1228
0
5
10
15
Veh Rit0
20
40
60
80
Veh Rit
Nu
mb
er
of T
ics
Num
ber
of M
idlin
e C
rossin
gs
A B
+
**
Tg
C
Tg
C
+
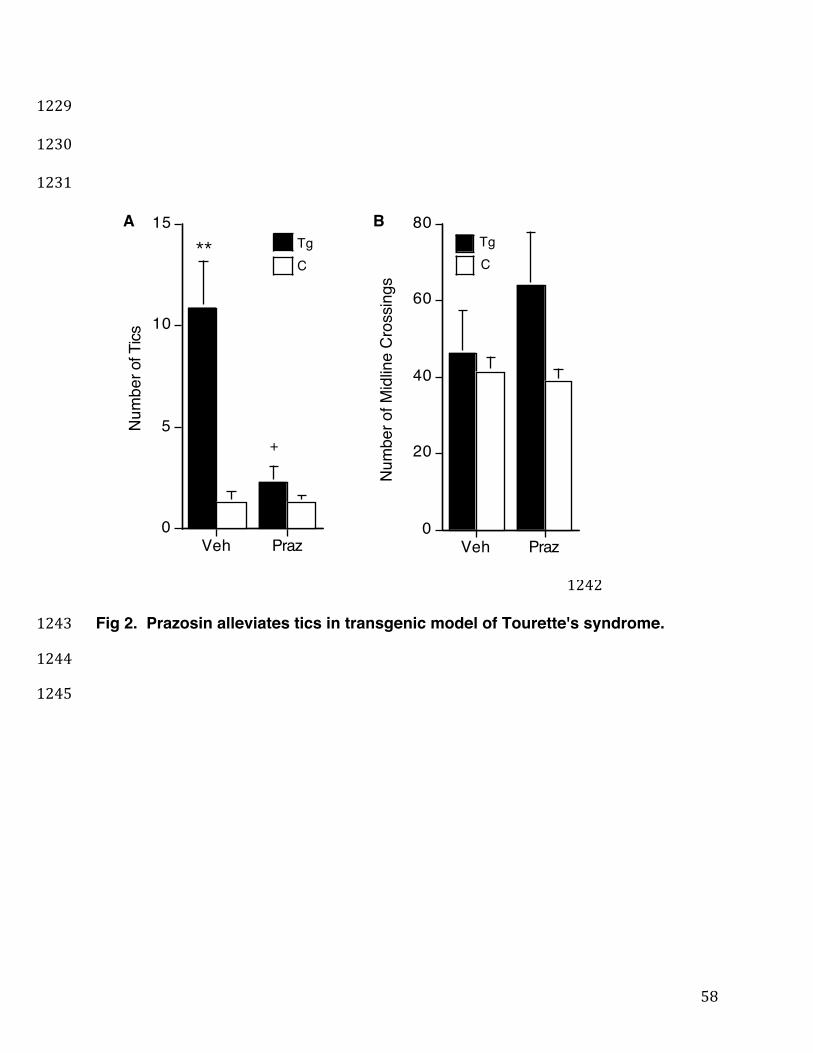
58
1229
1230
1231 1232
1233
1234
1235
1236
1237
1238
1239
1240
1241
1242
Fig 2. Prazosin alleviates tics in transgenic model of Tourette's syndrome. 1243
1244
1245
0
5
10
15
Veh Praz0
20
40
60
80
Veh Praz
Nu
mb
er
of T
ics
Num
ber
of M
idlin
e C
rossin
gs
A B
+
**Tg
C
Tg
C
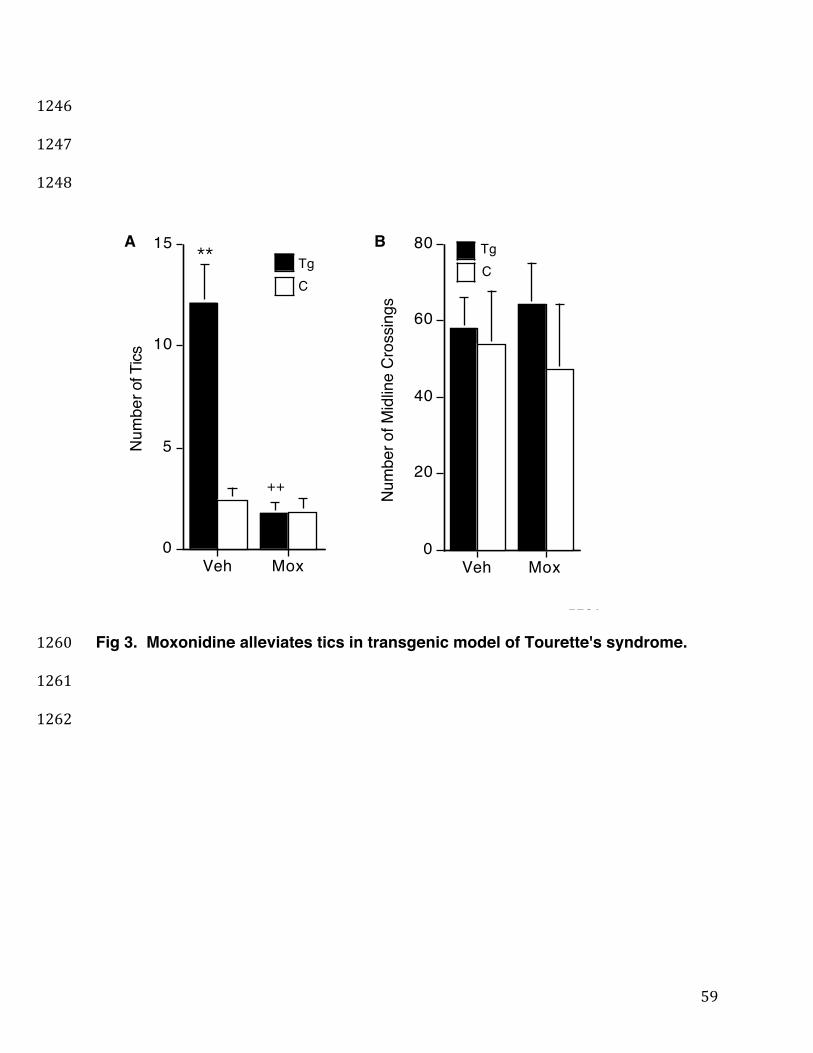
59
1246
1247
1248
1249
1250
1251
1252
1253
1254
1255
1256
1257
1258
1259
Fig 3. Moxonidine alleviates tics in transgenic model of Tourette's syndrome. 1260
1261
1262
0
5
10
15
Veh Mox0
20
40
60
80
Veh Mox
Nu
mb
er
of T
ics
Num
ber
of M
idlin
e C
rossin
gs
A B
++
**Tg
C
Tg
C

60
1263
1264
1265
1266
1267
1268
1269
1270
1271
1272
1273
1274
1275
1276
Fig 4. Bromocriptine alleviates tics in transgenic model of Tourette's syndrome. 1277
1278
1279
0
2.5
5
7.5
10
-Bromo +Bromo0
25
50
75
100
125
-Bromo +Bromo
Nu
mb
er
of T
ics
Num
ber
of Locom
otion E
vents
A B
Tg
C
Tg
C*
+
+
++

61
1280
1281
1282
1283
Fig 5. Predicted hyperglutamatergic tic circuit and circuit-breaker drugs' actions. 1284
1285
1286
MOVE-URGE
D2 D1
CTX/AMY
SNc
GLU
DA
5-HT2a,c
alpha-1
I-1 I-1
D2 STR
DP STR
IP
MOVE-URGE
SNc
GLU
D2 D1 DA
CTX/AMY 5-HT2a,c
alpha-1
I-1 I-1
D2 STR
DP STR
IP
MOVE-URGE
D2 D1
CTX/AMY
SNc
GLU
DA
5-HT2a,c RIT
alpha-1
I-1 I-1
D2 STR
DP STR
IP
MOVE-URGE
D2 D1
CTX/AMY
SNc
GLU
DA
5-HT2a,c PRAZ alpha-1
I-1 I-1
D2 STR
DP STR
IP
SNc
GLU
D2 D1 DA
CTX/AMY 5-HT2a,c
alpha-1
I-1 I-1 MOX
MOVE-URGE
D2
MOX
STR
DP STR
IP
SNc
GLU
D2 D1 DA
CTX/AMY 5-HT2a,c
alpha-1
I-1 I-1
D2 BROMO
STR
DP STR
IP
MOVE-URGE
A B C!
!
!
!
!
!
!
!
!
D E F!