controlling the density of nucleic acid oligomers on fiber optic sensors for enhancement of...
TRANSCRIPT

Controlling the density of nucleic acid oligomers on ®ber opticsensors for enhancement of selectivity and sensitivity
James H. Wattersona, Paul A.E. Piunnoa,b, Christopher C. Wustb, Ulrich J. Krulla,*
aDepartment of Chemistry, Chemical Sensors Group, University of Toronto at Mississauga,
3359 Mississauga Road North, Mississauga, Ont., Canada L5L 1C6bFONA Technologies Inc., 855 Matheson Blvd. East, Unit #14, Mississauga, Ont., Canada L4W 4L6
Abstract
The immobilization of oligonucleotides to solid surfaces is relevant to the development of biosensor and microarray technologies. The
density of oligonucleotide immobilization determines the charge density at the surface by means of ionizable phosphate groups, and may
result in an interfacial dielectric constant, pH and ionic strength that are unlike those of bulk solution. The density of immobilization may
affect the extent of interactions between neighbouring oligomers, as well as interactions between the immobilized oligomers and the
substrate surface. Experiments were done to examine the effects of immobilization density and solution conditions on the sensitivity,
selectivity and dynamic range of hybridization assays done using a ®ber optic nucleic acid biosensor based on total internal re¯ection
¯uorescence (TIRF). Such immobilized nucleic acid ®lms ®rst required activation by thermal denaturation cycling to reach full activity. The
effects of non-selective adsorption of oligonucleotides were dependent on ionic strength, and could not be removed independently of
hybridization. Increased immobilization density resulted in signi®cantly higher sensitivity but reduced dynamic range in all hybridization
assays done. Sensitivity and selectivity were a function of temperature, however, the selectivity of hybridization assays done using these
sensors could not be predicted by consideration of thermal denaturation temperatures alone. # 2001 Elsevier Science B.V. All rights
reserved.
Keywords: Biosensor; Oligonucleotide; Immobilization; Nucleic acid; Fluorescence; Optical ®ber
1. Introduction
The immobilization of biomolecules to solid surfaces is
widely used in the preparation of analytical sensors. Appli-
cations include immunosensor techniques [1±3], which tend
to rely on protein binding as the means of molecular
`̀ recognition'', as well as those which make use of nucleic
acid hybridization [4±9] as the basis for selective recogni-
tion. The use of immobilized nucleic acids to provide for
selective binding interactions is attractive since the selec-
tivity of nucleic acid binding interactions can be quite high
and the advent of polymerase chain reaction and solid phase
nucleic acid synthesis has allowed for relatively simple
nucleic acid preparation and immobilization.
The utility of immobilized selective molecular recogni-
tion elements is dependent upon the retention of selective
binding capacity after the immobilization process is
complete. The binding capacity is dependent upon the
structure of the immobilized molecules in their local
environments, which can be signi®cantly different from
those experienced in bulk solution. The density of immo-
bilization of single-stranded DNA (ssDNA) onto the sur-
face of a solid substrate affects the charge density at the
surface, and the extent to which the immobilized oligomers
interact with the surface of the solid substrate and
with neighbouring nucleic acid oligomers. This has
consequences to issues of selectivity and the extent of
hybridization, as well as the orientation of the immobilized
ssDNA, and therefore affects the kinetics of hybridization
[10]. Clearly, the control of selectivity of binding and the
dynamic range that could be achieved by control of the
concentration of oligonucleotide sequences at an interface
can be complex.
The elucidation of the orientation and packing structure
of nucleic acids immobilized on gold and polystyrene
surfaces has been attempted [11±13]. It was suggested
that the alignment of immobilized oligonucleotides with
respect to the substrate surface may be controlled by
selection of oligonucleotide immobilization density, as
well as through control of the chemical environment at
Sensors and Actuators B 74 (2001) 27±36
* Corresponding author. Tel.: �1-905-828-5437; fax: �1-905-828-5425.
E-mail address: [email protected] (U.J. Krull).
0925-4005/01/$ ± see front matter # 2001 Elsevier Science B.V. All rights reserved.
PII: S 0 9 2 5 - 4 0 0 5 ( 0 0 ) 0 0 7 0 8 - 5

the surface. Adsorptive interactions of oligonucleotides
immobilized by sulfur±gold interactions on a gold surface
were reduced by blocking unreacted surface sites with
mercaptohexanol [10]. The reduction in oligonucleotide
adsorption to gold resulted in extension of the immobilized
oligonucleotides away from the substrate surface [11]. The
extent of hybridization was found to be affected by the
packing density of immobilized oligonucleotides, with
hybridization being inhibited at higher packing densities
where steric hindrance and electrostatic repulsion were
thought to reduce the stability of hybrids that could
form [11].
In previous work [14,15], we described an examination of
the thermodynamics of interfacial nucleic acid hybridiza-
tion. The thermal denaturation temperature, Tm (temperature
at which 50% of all duplexes formed are denatured), of the
hybrids formed at a fused silica surface was used to inves-
tigate the thermodynamics of the hybridization process. It
was observed that short immobilized nucleic acid strands
(20mers) that were immobilized on hexaethylene glycol
linkers that were covalently attached to fused silica sub-
strates underwent thermal denaturation transitions that were
signi®cantly broader than those observed for experiments
done in bulk solution. The immobilized nucleic acids exhib-
ited a two±three-fold reduction in the enthalpy change that
accompanied the thermal denaturation transition compared
with experiments done in bulk solution. It was also observed
that immobilized nucleic acids could provide larger devia-
tions in the Tm between fully complementary sequences and
those containing single base-pair mismatches (SBPMs) than
could similar hybridization reactions in bulk solution, with
the effects becoming exacerbated at higher immobilization
densities. These results corroborated the notion that inter-
facial hybridization occurs in a signi®cantly different envir-
onment than that of bulk solution, and that surface-strand
interactions and nearest neighbour interactions may become
signi®cant.
In the present work, we report the effects of oligonucleo-
tide immobilization density on the selectivity and sensitivity
of transduction of hybridization using a fused silica ®ber
optic biosensor based on total internal re¯ection ¯uores-
cence (TIRF). From an analytical standpoint, it is not
possible to simply extrapolate experimental conditions for
a hybridization assay for such biosensors (i.e. temperature,
ionic strength and immobilization density) from data
obtained from experiments done in bulk solution. Conven-
tionally, solution temperature and ionic strength are con-
trolled in order to maximize differences in Tm between fully
complementary and partially complementary sequences,
thereby reducing the effects of non-selective hybridization.
The experiments described herein, were done in an effort to
better characterize the effects of immobilization density on
the sensitivity and selectivity of hybridization assays done in
a variety of conditions using the ®ber optic biosensor.
Particular attention was paid to issues of reproducibility
and non-selective adsorption.
2. Experimental
2.1. Chemicals
Solvents were obtained from BDH (Toronto, Ont.) as
reagent grade and were further puri®ed or dried, when
necessary, by standard distillation methods. Reagent grade
salts were purchased from BDH (Toronto, Ont.). DNA
synthesis reagents were from Dalton Chemical Laboratories
Inc. (Toronto, Ont.). Anhydrous acetonitrile (Dalton) was
dried by distillation from P2O5 prior to receipt, and was
further distilled from calcium hydride under a dry argon
atmosphere prior to use. Tetrahydrofuran (BDH) was ®rst
dried over CaH2, ®ltered and ®nally distilled immediately
prior to use from sodium metal (Aldrich)/benzophenone
(Aldrich). Sterile water for use on its own and with hybri-
dization buffer was produced with the water ®rst double-
distilled in glass, then subsequently treated with diethyl
pyrocarbonate (Aldrich) and sterilized by autoclave. Mole-
cular biology grade polyacrylamide gel electrophoresis
reagents and apparatus were obtained from Bio-Rad (Her-
cules, CA, USA). Silica gel (Toronto Research Chemicals,
Toronto, Ont.) that was used for puri®cation had a particle
size of 30±70 mm.
2.2. Preparation of optical fiber segments
Fused silica optical ®bers of 400 mm core diameter (3M
Powercore Series Optical Fiber, FT-400-URT or FP-400-
UHT) were acquired from Thor Labs Inc., Newton, NJ,
USA. The polymeric outer cladding was removed mechani-
cally by means of a ®ber-stripping tool also obtained through
Thor Labs, Inc. Removal of the outer cladding exposed the
inner cladding layer that coated the core of fused silica.
Individual sensor elements were then made by cutting
optical ®ber pieces of 48 mm length with a ®ber-scoring
device. Each ®ber was cleanly scored by rotating a diamond
pencil about the optical ®ber. Removal of the top portion of
the scored ®ber from the remainder of the optical ®ber
secured in the pin-chuck then yielded cylindrical optical
®ber segments with clean, ¯at termini as evidenced by visual
inspection of the termini at 40� magni®cation.
The fused silica ®ber segments and controlled pore glass
(CPG) (CPG Inc., Lincoln Park, NJ, USA) that were used as
solid substrates for automated DNA synthesis were cleaned
prior to modi®cation of the surface according to the two-
stage method of Kern and Puotinen [16]. The ®rst stage
consisted of immersing the solid substrates in a 1:1:5 (v/v)
solution of 30% ammonium hydroxide/30% hydrogen per-
oxide/water and gently agitating at 808C for 5 min. In the
second stage, the substrates were then recovered, thoroughly
washed with sterile water and then gently agitated in a
solution of 1:1:5 (v/v/v) conc. HCl/30% hydrogen perox-
ide/water for 5 min at 808C. The substrates were then
recovered and washed with successive 100 ml portions of
water, methanol, dichloromethane and diethyl ether. The
28 J.H. Watterson et al. / Sensors and Actuators B 74 (2001) 27±36

substrates were then dried under vacuum and stored in vacuo
and over P2O5 until required.
2.3. Surface modification of solid substrates Ð functiona-
lization of substrates with 3-glycidoxypropyltrimethoxy-
silane (GOPS)
The cleaned solid substrates were suspended in an anhy-
drous solution of xylene/3-glycidoxypropyltrimethoxysi-
lane/diisopropylethylamine (100:30:1 v/v/v). The reaction
took place at 808C with stirring over 24 h under an argon
atmosphere. The substrates were then collected and succes-
sively washed with two 50 ml portions of each of methanol,
dichloromethane, diethyl ether, and then were dried and
stored under vacuum and over P2O5 at room temperature
until required.
2.4. Surface modification of solid substrates Ð linkage of
dimethoxytrityl (DMT)±hexaethylene glycol (HEG) onto
GOPS functionalized substrates
DMT±HEG was synthesized as outlined previously [19].
DMT±HEG (700 mg DMT±HEG/100 mg CPG) that had
been dried under vacuum and over P2O5 (>72 h) was dis-
solved in 20 ml of anhydrous pyridine. An excess of NaH
(10 eq.) that had been thoroughly washed with dry hexane
was then introduced to the mixture. The subsequent reaction
was permitted to proceed with stirring for 1 h at room
temperature under an argon atmosphere. The reaction mix-
ture was ®ltered through a sintered glass frit under a positive
pressure of argon into a vessel containing the GOPS func-
tionalized substrates. GOPS functionalized substrates were
separated into three batches, containing both optical ®bers
and CPG. The three batches then underwent the DMT±HEG
coupling reaction, which was permitted to proceed under a
positive pressure of argon at room temperature with gentle
agitation on an oscillating platform stirrer for durations of 4
and 12 h, respectively. Following the coupling reaction, the
substrates were quickly recovered and washed with succes-
sive 150 ml portions of methanol, water, methanol, and
diethyl ether to quench the coupling reaction and remove
any reactants that were non-speci®cally adsorbed. The
DMT-protected HEG-functionalized substrates were dried
under vacuum and over P2O5 and were maintained under
these conditions until further required.
2.5. Surface modification of solid substrates Ð capping of
unreacted silanol and hydroxyl functionalities with
chlorotrimethylsilane (TMS-Cl)
Unreacted silanol and hydroxyl functionalities on the
surface of the solid substrates where undesired oligonucleo-
tide synthesis could occur were capped prior to oligonucleo-
tide synthesis using TMS-Cl according to the method of Pon
[17]. The dried substrates were suspended in a solution
of 1:10 (v/v) TMS-Cl/pyridine for 16 h under an argon
atmosphere at room temperature. The substrates were sub-
sequently recovered and washed with three successive 20 ml
portions of pyridine, methanol and diethyl ether and were
then stored under vacuum and over P2O5 at room tempera-
ture until required.
2.6. Solid phase phosphoramidite synthesis of
oligonucleotides
All solid phase oligonucleotide synthesis was done using
a PE-ABI 392 DNA synthesizer (Perkin-Elmer Applied
Biosystems, Foster City, CA, USA). The pre-programmed
synthesis cycles employed for oligonucleotide assembly
were modi®ed to adjust the reagent delivery times in order
to ensure that the synthesis columns used were completely
®lled. The column used for oligonucleotide synthesis onto
optical ®bers segments was a custom manufactured Te¯on1
synthesis column (6 mm i:d:� 50 mm) capable of holding
eight ®bers. The ®bers were secured by means of insertion
into cylindrical bores (400 mm i:d:� 2 mm deep) machined
into one of the end caps. All end-caps were secured onto the
column bodies with aluminum crimp seals. The columns
used for oligonucleotide synthesis onto DMT±HEG±GOPS
functionalized CPG were custom manufactured Te¯on1
columns (8 mm i:d:� 10 mm). Te¯on1 end ®lters
(0.22 mm pore size, PE-ABI) were used to retain the glass
beads within the column. Synthesis of oligonucleotides for
use as complementary material for immobilized DNA was
carried out on nucleoside functionalized LCAA±CPG sub-
strates pre-packed in polyethylene columns as supplied by
the manufacturer, and has been described previously [14].
Polythymidylic acid icosanucleotides (dT20) were
assembled onto all of the optical ®ber and CPG substrates
functionalized with DMT±HEG linker molecules. Determi-
nation of the density of surface coverage of CPG substrates
with covalently immobilized oligonucleotide±HEG conju-
gates was done by anion-exchange HPLC following meth-
ods developed in our research group that have been reported
elsewhere [18].
Icosanucleotides labeled at the 50-terminus with a ¯uor-
escein moiety were used as complementary material to
hybridize with immobilized dT20 sequences. The 50-¯uor-
escein labeled oligonucleotides were prepared by use of a
¯uorescein phosphoramidite synthon (Dalton) and standard
protocols for oligonucleotide preparation [19]. Additionally,
unlabeled complementary icosanucleotides were prepared
by standard protocols for use in studies of hybridization in
bulk solution.
2.7. Instrumentation for studies of immobilized nucleic
acid films
Fluorescence-based studies of nucleic acid hybridization
at the surface of optical ®bers were carried out via an
automated spectro¯uorimeter instrument developed in our
research group that has been described previously[14]. Laser
J.H. Watterson et al. / Sensors and Actuators B 74 (2001) 27±36 29

radiation (488 nm) from a Coherent Innova 70 CW argon ion
laser (Coherent Laser Products, Palo Alto, CA, USA) was
coupled into a sensing ®ber by ®rst guiding the beam of
source radiation such that it was incident upon the surface of
a dichroic mirror (505 nm cut-off, Omega Optical, Battle-
boro, VT, USA) oriented at 458 to the incident beam. The
sensing ®ber was secured via a Te¯on1 holder with a
waterproof compression-type seal within a stainless steel
hybridization cell with small volume (1 mm i:d:� 50 mm)
that permitted a solution volume of 137 ml to be exposed to
the sensing ®ber. Fluorescence emission from the sensing
®ber with wavelength >505 nm was then directed back
through the dichroic mirror into a Bentham M300 mono-
chromator (f=# � 4:2, 2.5 nm bandwidth, distributed by
Optikon Corporation Limited, Waterloo, Ont.). Fluores-
cence emission exiting the emission monochromator was
detected by a side-on photomultiplier tube (106 A/W respon-
sivity, Model 77348, Oriel Corp.) operated at a potential of
500VDC (PMT Power Supply Model 5502, Products for
Research, Danvers, MA, USA).
The temperature of the solution within the ¯ow cell was
determined by use of a glass-encapsulated bead thermistor
(Fenwal Electronics Inc., distributed by Electrosonic Inc.,
Toronto, Ont.) embedded within the stainless steel block at a
distance not more than 1 mm from the internal wall of the
solution compartment surrounding the sensing ®ber. The
temperature of the ¯ow cell was regulated by use of a Peltier
temperature control accessory (Model 89090A, Hewlett-
Packard Corp., Mississauga, Ont.). Analyte sampling and
delivery was done using an automated sampler and pump
system as previously described [14].
2.8. Acquisition of thermal denaturation profiles from
nucleic acid films immobilized onto fused silica optical
fibers
All thermal denaturation pro®les for hybridization occur-
ring at the surface of the optical ®ber sensors were acquired
by monitoring the intensity of ¯uorescence emission at
542 nm over the temperature range of ca. 20±808C using
a temperature ramp rate of 0.38C minÿ1 to ensure that
equilibrium conditions were satis®ed, as described pre-
viously [15]. All sensors were cleaned by sonication in
ethanol in a 40 W bath sonicator for 90 min to remove
adsorbed impurities from the sensor surface prior to ana-
lysis. Thermal denaturation pro®les were obtained for
optical sensors that were exposed to ¯uorescein labelled
cDNA in various dilutions of a stock phosphate buffered
saline (PBS) hybridization buffer (1.0 M NaCl, 50 mM
PO4ÿn, pH 7.0). Removal of complementary oligonucleo-
tide associated with the sensor surface from previous ana-
lyses was done prior to each subsequent experiment by
¯ushing 15 ml of 808C water through the ¯ow-cell
(3 ml minÿ1, 5 min) and by ¯ushing 1 ml of 90% forma-
mide in TE buffer (10 mM Tris±HCl, 5 mM EDTA, pH 8.3)
through the ¯ow cell.
2.9. Hybridization assays of nucleic acid films immobilized
onto fused silica optical fibers
All sensors were cleaned by sonication in ethanol in a
40 W bath sonicator for 90 min to remove adsorbed impu-
rities from the sensor surface prior to analysis. Hybridization
assays were done for optical sensors that were exposed to
solution phase oligonucleotides labelled on the 50-terminus
with ¯uorescein. In all cases, oligonucleotides were dis-
solved in various dilutions of a stock PBS hybridization
buffer (1.0 M NaCl, 50 mM PO4ÿn, pH 7.0), with oligonu-
cleotide concentrations in the range of 0.001±0.1 mM. Dilu-
tions of the stock buffer by factors of 1.0, 0.5 and 0.1 were
used for ionic strength studies. Each sample solution had a
total volume of 1 ml, and was washed past the sensor at a
¯ow rate of 3 ml minÿ1. Following the addition of each
sample, a 1 ml solution of PBS buffer identical to that used
to prepare the oligonucleotide solution was washed past the
sensor to remove non-selectively adsorbed material. Assays
were done using solution temperatures of 28, 36, and 448C.
All hybridization assays were done in triplicate. Removal of
complementary oligonucleotide that had associated with a
sensor surface from previous analyses were done prior to
each subsequent experiment by ¯ushing 15 ml of 808C water
through the ¯ow-cell (3 ml minÿ1, 5 min) and by ¯ushing
1 ml of 90% formamide in TE buffer (10 mM Tris±HCl,
5 mM EDTA, pH 8.3) through the ¯ow cell.
3. Results and discussion
3.1. Control and characterization of oligonucleotide
immobilization density
The immobilization of dT20 onto the surface of fused
silica optical ®ber substrates was achieved by means of a
modi®cation to the method of Maskos and Southern [20].
The fused silica optical ®ber substrates were ®rst functio-
nalized with glycidoxypropyltrimethoxysilane (GOPS).
Linker molecules were then covalently attached to the
GOPS layer. The linker species used was HEG, protected
on one terminus with DMT in order to ensure single-site
reactivity and to minimize the risk of formation of closed-
ring structures with unreacted sites on the epoxysilane ®lm.
The modi®ed optical ®ber substrates were then subjected to
standard b-cyanoethyl-phosphoramidite oligonucleotide
synthesis protocols to prepare by stepwise synthesis the
dT20 oligonucleotides on the surface of the substrates.
The packing density of immobilized oligonucleotides was
altered by means of controlling the duration of the reaction
of DMT±HEG conjugates with the GOPS-functionalized
substrates. Two different immobilized densities were
obtained by permitting the DMT±HEG coupling reaction
to proceed for 4 and 8 h, respectively. In order to character-
ize the density of immobilization, oligonucleotide synthesis
was carried out as described above on the ®bers and
30 J.H. Watterson et al. / Sensors and Actuators B 74 (2001) 27±36

concurrently on GOPS-functionalized controlled-pore glass
(CPG), which has a well-de®ned surface area. The oligo-
nucleotide±HEG conjugates were then cleaved from the
surface of the CPG by means of exposure to concentrated
ammonium hydroxide for approximately 3 h, lyophilized
and re-dissolved in water. The quantity of immobilized
ssDNA as well as the quality of the products prepared by
automated synthesis were subsequently determined by
anion-exchange HPLC. Quantitation of the cleaved HEG±
dT20 conjugates was achieved by co-injection with a known
quantity of dT20. The results of the HPLC analysis are shown
in Table 1. The peaks corresponding to HEG±dT20 were
determined to have retention times of ca. 33 min. Distribu-
tions of peaks in the region of 33 min were due to the
formation of a HEG-linker ®lm in which some of the HEG
molecules had coupled onto one another to provide a dis-
tribution of linker lengths, as was described earlier [15,18].
These data show that these two densities provide two
different physical environments for the immobilized oligo-
nucleotides. The low-density sample consisted of immobi-
lized dT20±HEG conjugates separated by approximately
263 AÊ between adjacent strands, assuming uniform oligo-
nucleotide distribution. Since the length of the dT20±HEG
conjugate is ca. 100 AÊ in length, the low-density sample then
represents the system wherein there may be the onset of
some interaction between neighbouring strands. The high-
density sample consisted of immobilized dT20±HEG con-
jugates separated by approximately 100 AÊ between adjacent
strands. This close packing is much more likely to facilitate
interactions between neighbouring strands than at the lower
packing density.
3.2. Activation of nucleic acid films for hybridization
Regardless of the density of immobilization of ssDNA,
initial hybridization assays done using freshly prepared
®bers resulted in no signi®cant ¯uorescence increase fol-
lowing introduction of ¯uorescently labelled complemen-
tary DNA (cDNA). In attempts to achieve activation of
hybridization, ®bers were washed sequentially with water,
a solution of 90% formamide in TE buffer, and again with
sterile water, all at 808C. Sensors showed no response to
complementary material even after 10 such hybridization
assay cycles were attempted. The results indicate that heat-
ing of the immobilized nucleic acid ®lm alone is not
suf®cient to provide activation for response to cDNA. It
was observed that the process of selective hybridization
could be activated by subjecting freshly prepared ®bers to a
series of thermal denaturation experiments. The sensor
surfaces were exposed to a phosphate buffered solution
containing cDNA, and subsequently underwent a tempera-
ture ramp of 0.38C minÿ1, over a range from 20 to 808C.
Fig. 1 illustrates the response of a sensor coated with dT20 at
low packing density to a solution containing 10ÿ7 M dA20-
50-¯uorescein, before and after the sensor was subjected to
two thermal denaturation cycles using a solution containing
10ÿ7 M dA20-50-¯uorescein as the complementary material.
It may be that the presence of cDNA throughout the thermal
denaturation experiments provided a driving force for the
removal of some impurities adsorbed during the synthesis of
the nucleic acid ®lm. Evidence for this was that the back-
ground ¯uorescence was observed to decrease as a result of
the activating thermal denaturation experiments, and spec-
tral data suggested that trityl cation might have been the
species that was desorbed [21].
The response of a biosensor to successive hybridization
assays with 10ÿ7 M solutions of cDNA did not remain
constant after the initial activation cycles, but rather
decreased to a steady value after approximately three cycles,
as may be observed in Fig. 1. This suggests that the binding
activity of the immobilized oligonucleotides is dynamic
even after the process of activation. It was originally pos-
tulated that this reduction in signal was due to occlusion of
the surface by impurities present in the buffer solutions, but
this may only partially account for the loss of sensitivity
Table 1
Density of immobilization of dT20±HEG conjugate onto GOPS-functionalized substrates as determined by anion-exchange high performance liquid
chromatography [18]
Sample Reaction duration
(DMT±HEG-substrate) (h)
Total surface area
of CPG used (AÊ 2)
Molecules dT20±HEG
immobilized
Mean separation
distance (AÊ )
Low density 4 3.04 � 1019 5.59 � 1014 263
High density 8 4.12 � 1019 5.25 � 1015 100
Fig. 1. Response of sensor with low dT20 immobilization density to
10ÿ7 M solutions of dA20-50-fluorescein in 1� PBS buffer, before and
after activation of the film. Activation of the sensor was achieved by
exposure of the sensor to two thermal denaturation cycles.
J.H. Watterson et al. / Sensors and Actuators B 74 (2001) 27±36 31
since full signal (relative to maximum sensitivity shown)
was not achieved even after sonication of the sensor and
reintroduction of identical analyte solutions. It may be that
loss of the trimethylsilane capping species was occurring,
since the susceptibility of this functionality to cleavage from
the substrate at elevated temperatures was established by
wettability experiments (data not shown). In this case, the
variation in signal may be the result of the changing nature
of the interactions between the immobilized oligonucleo-
tides and the substrate surface.
3.3. Non-selective adsorption of oligonucleotides
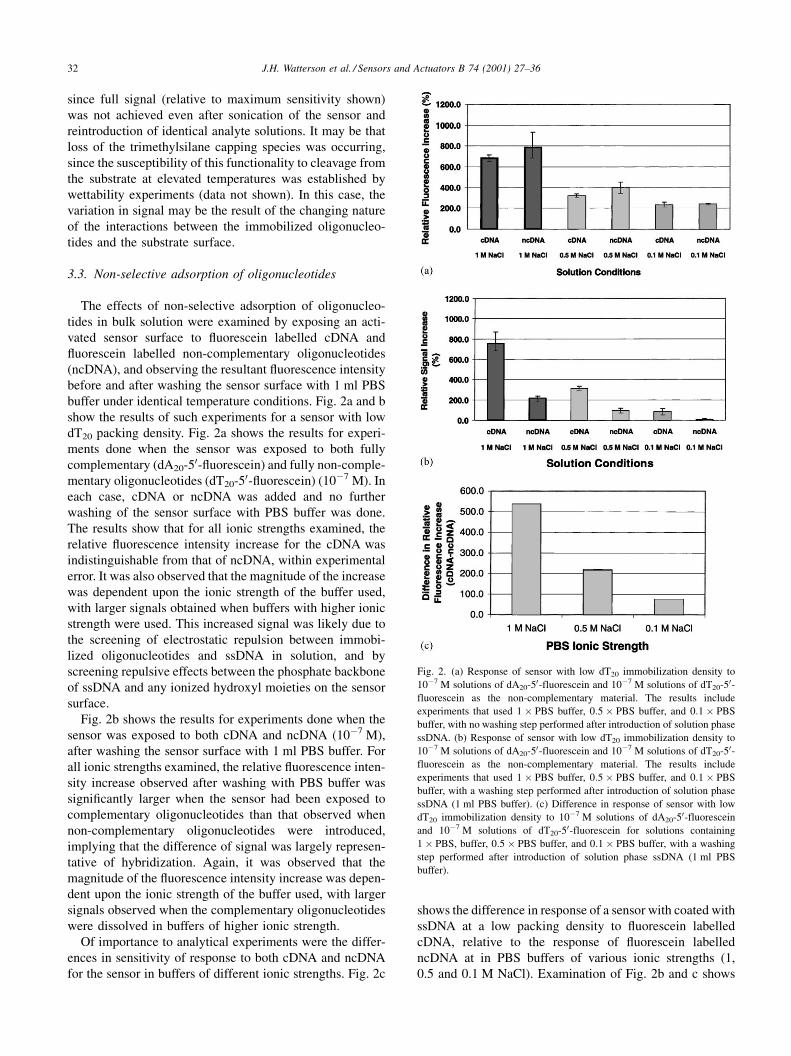
The effects of non-selective adsorption of oligonucleo-
tides in bulk solution were examined by exposing an acti-
vated sensor surface to ¯uorescein labelled cDNA and
¯uorescein labelled non-complementary oligonucleotides
(ncDNA), and observing the resultant ¯uorescence intensity
before and after washing the sensor surface with 1 ml PBS
buffer under identical temperature conditions. Fig. 2a and b
show the results of such experiments for a sensor with low
dT20 packing density. Fig. 2a shows the results for experi-
ments done when the sensor was exposed to both fully
complementary (dA20-50-¯uorescein) and fully non-comple-
mentary oligonucleotides (dT20-50-¯uorescein) (10ÿ7 M). In
each case, cDNA or ncDNA was added and no further
washing of the sensor surface with PBS buffer was done.
The results show that for all ionic strengths examined, the
relative ¯uorescence intensity increase for the cDNA was
indistinguishable from that of ncDNA, within experimental
error. It was also observed that the magnitude of the increase
was dependent upon the ionic strength of the buffer used,
with larger signals obtained when buffers with higher ionic
strength were used. This increased signal was likely due to
the screening of electrostatic repulsion between immobi-
lized oligonucleotides and ssDNA in solution, and by
screening repulsive effects between the phosphate backbone
of ssDNA and any ionized hydroxyl moieties on the sensor
surface.
Fig. 2b shows the results for experiments done when the
sensor was exposed to both cDNA and ncDNA (10ÿ7 M),
after washing the sensor surface with 1 ml PBS buffer. For
all ionic strengths examined, the relative ¯uorescence inten-
sity increase observed after washing with PBS buffer was
signi®cantly larger when the sensor had been exposed to
complementary oligonucleotides than that observed when
non-complementary oligonucleotides were introduced,
implying that the difference of signal was largely represen-
tative of hybridization. Again, it was observed that the
magnitude of the ¯uorescence intensity increase was depen-
dent upon the ionic strength of the buffer used, with larger
signals observed when the complementary oligonucleotides
were dissolved in buffers of higher ionic strength.
Of importance to analytical experiments were the differ-
ences in sensitivity of response to both cDNA and ncDNA
for the sensor in buffers of different ionic strengths. Fig. 2c
shows the difference in response of a sensor with coated with
ssDNA at a low packing density to ¯uorescein labelled
cDNA, relative to the response of ¯uorescein labelled
ncDNA at in PBS buffers of various ionic strengths (1,
0.5 and 0.1 M NaCl). Examination of Fig. 2b and c shows
Fig. 2. (a) Response of sensor with low dT20 immobilization density to
10ÿ7 M solutions of dA20-50-fluorescein and 10ÿ7 M solutions of dT20-50-fluorescein as the non-complementary material. The results include
experiments that used 1� PBS buffer, 0:5� PBS buffer, and 0:1� PBS
buffer, with no washing step performed after introduction of solution phase
ssDNA. (b) Response of sensor with low dT20 immobilization density to
10ÿ7 M solutions of dA20-50-fluorescein and 10ÿ7 M solutions of dT20-50-fluorescein as the non-complementary material. The results include
experiments that used 1� PBS buffer, 0:5� PBS buffer, and 0:1� PBS
buffer, with a washing step performed after introduction of solution phase
ssDNA (1 ml PBS buffer). (c) Difference in response of sensor with low
dT20 immobilization density to 10ÿ7 M solutions of dA20-50-fluorescein
and 10ÿ7 M solutions of dT20-50-fluorescein for solutions containing
1� PBS, buffer, 0:5� PBS buffer, and 0:1� PBS buffer, with a washing
step performed after introduction of solution phase ssDNA (1 ml PBS
buffer).
32 J.H. Watterson et al. / Sensors and Actuators B 74 (2001) 27±36

that the largest response to both cDNA and ncDNA occurred
when the PBS buffer containing 1 M NaCl was used.
Although the non-selective response of the sensor to non-
complementary ssDNA was essentially removed when the
low ionic strength buffer was used, the difference in
response to cDNA relative to that of ncDNA was largest
when the high ionic strength buffer was used. For the
experiments using the PBS buffer containing 1 M NaCl,
the difference in response to complementary and non-com-
plementary oligonucleotides was 10-fold larger than that
observed in experiments done using PBS buffer with low
ionic strength (0.1 M NaCl). Thus, greater sensitivity is
obtained when using buffers of higher ionic strength, despite
the increased contribution from non-selective adsorption of
oligonucleotides in solution. Consequently, PBS buffer con-
taining 1 M NaCl was used for the remaining experiments to
examine other experimental parameters (sensitivity and
selectivity).
3.4. Comparison of sensitivity of biosensors with different
oligonucleotide packing densities
Experiments were done to examine the effect of oligo-
nucleotide packing density on the response calibration
curves of the ®ber optic sensors with respect to the con-
centration of complementary oligonucleotide concentration.
Fig. 3 shows calibration curves generated using activated
sensors with low and high oligonucleotide packing density,
obtained using solution temperatures of 35 and 448C, in PBS
buffer containing 1 M NaCl to maximize sensitivity. After
the addition of complementary oligonucleotides and incu-
bation for 15 min, the sensors were rinsed with 1 ml PBS
buffer at the stated temperature, in order to remove non-
selectively adsorbed oligonucleotides. It is apparent from
the results shown in Fig. 3 that at each temperature exam-
ined, the sensor with high oligonucleotide immobilization
density exhibited a signi®cantly greater response to the
presence of complementary oligonucleotides than did the
sensor with low immobilization density. Interestingly, it was
also observed that the dynamic range had an upper limit of
about 1� 10ÿ8 M cDNA for the sensor with high oligonu-
cleotide packing density, whereas deviations from linearity
did not begin until cDNA concentrations reached about
3� 10ÿ8 M for the sensor with low packing density. This
result can be explained in terms of the variation in thermal
stability of hybrids formed at surfaces with different oligo-
nucleotide packing densities. In a previous paper [15], we
reported the thermal denaturation temperatures of dA20-50-¯uorescein hybridized to immobilized dT20 with different
packing densities. We found that the Tm values for hybrids
formed at interfaces with high oligonucleotide packing
density were consistently lower (�68C) than those observed
using sensors with low and medium packing densities. It was
postulated that at higher packing densities, interactions
between neighbouring oligonucleotides become signi®cant,
and these interactions reduce thermodynamic stability of
hybrids formed in the interfacial environment. Conse-
quently, at comparable temperatures, the hybrids formed
at the surface with high oligonucleotide packing density are
less stable than those formed in an environment with lower
packing density where nearest-neighbour interactions are
not as likely to occur. The larger absolute number of
immobilized oligonucleotides may account for greater sen-
sitivity for sensors coated with higher oligomer packing
densities. However, the onset of deviations from linearity
occurs at a lower cDNA concentration for surfaces with high
immobilization density in comparison to those with low
packing density, suggesting that the optimal oligonucleotide
immobilization density will require a compromise between
sensitivity and dynamic range. High oligonucleotide pack-
ing density is better suited for low cDNA concentrations,
while higher cDNA concentrations may be more easily
quanti®ed with a sensor coated with a low oligonucleotide
packing density.
3.5. Biosensor selectivity
A comparison of the selectivity of hybridization was done
by exposing an optical ®ber coated with a high density of
dT20 to solutions containing ¯uorescein labelled ssDNA
with varying degrees of complementarity. In each experi-
ment, the ®ber was exposed sequentially to cDNA solutions
containing dA20-50-¯uorescein, d(A19G)-50-¯uorescein,
d(A9GA10)-50-¯uorescein, d(A9G2A9)-50-¯uorescein and a
mixture of equimolar amounts of all of these oligonucleo-
tides. All samples were prepared in PBS buffer containing
1 M NaCl to maximize sensitivity. The sensor was incubated
in the cDNA sample for 10 min, rinsed with 1 ml PBS buffer
and the resulting ¯uorescence was then measured. The
sensor was subsequently cleaned by washing sequentially
with 15 ml sterile water, 1 ml 90% formamide in TE buffer
and again with 15 ml sterile water, all at 808C. Two different
cDNA concentrations were chosen, and they represented
two different regions of the calibration curves in Fig. 3. The
2:5� 10ÿ9 M cDNA concentration was suf®ciently low that
Fig. 3. Response of sensors with low and high dT20 immobilization
densities to solutions of various concentrations of dA20-50-fluorescein in
1� PBS buffer at 28 and 408C. Wash steps were performed after each
addition of cDNA.
J.H. Watterson et al. / Sensors and Actuators B 74 (2001) 27±36 33
it was expected that incremental additions of cDNA and
therefore the mixed cDNA (all four types of cDNA in
equimolar quantities) sample would produce a linear cali-
bration response with respect to cDNA concentration. At the
higher cDNA concentration, it was expected that the mixed
analyte sample would not provide a signal that would fall on
the linear portion of the calibration curves. Hybridization
experiments were done using individual component con-
centrations of 2:5� 10ÿ9 and 2:5� 10ÿ8 M. The mixed
component (all four types of cDNA in equimolar quantities)
hybridization experiments were done using total oligonu-
cleotide concentrations of 1:0� 10ÿ8 and 1:0� 10ÿ7 M.
Experiments using cDNA samples of 2:5� 10ÿ9 M con-
centration regime were done at 28 and 448C. The lower
temperature was chosen such that all types of cDNA were
expected to be totally hybridized with immobilized dT20.
The higher temperature was chosen since the Tm of immo-
bilized d(A19G)-50-¯uorescein:dT20 was found to be 448C,
and this species was expected to have a Tm closest to that of
the fully complementary sequence (Tm � 48�C). Thus,
experiments done at the higher temperature were expected
to show enhanced selectivity relative to those done at the
lower temperature.
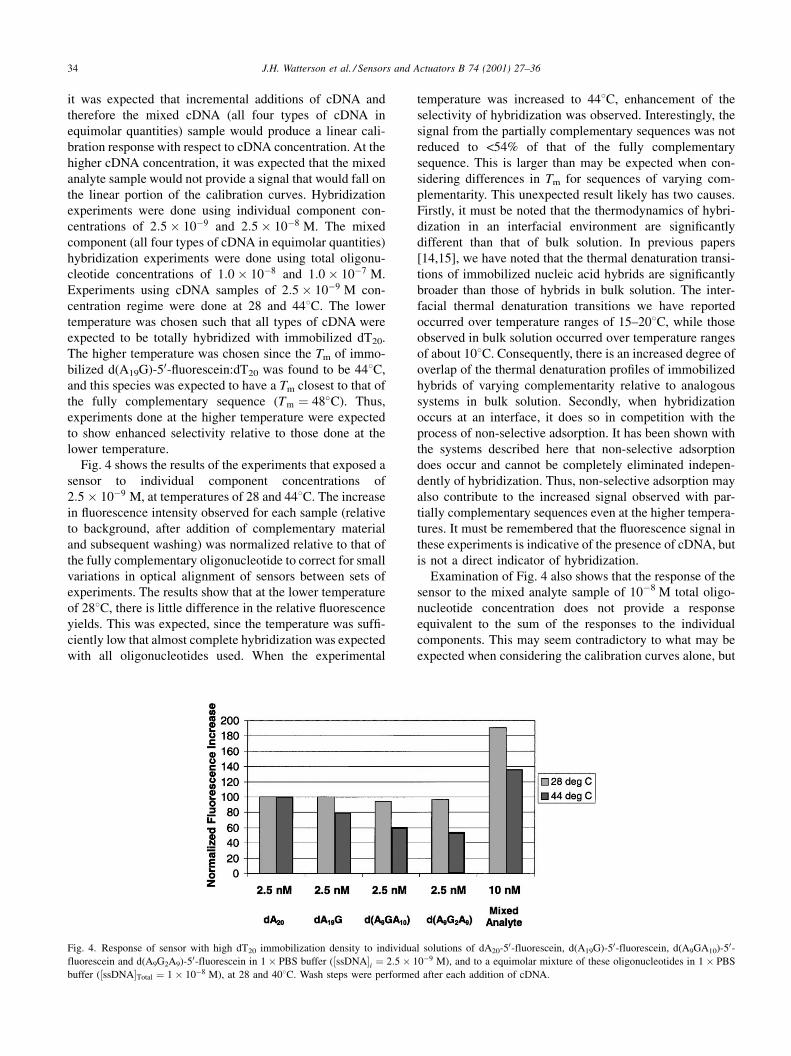
Fig. 4 shows the results of the experiments that exposed a
sensor to individual component concentrations of
2:5� 10ÿ9 M, at temperatures of 28 and 448C. The increase
in ¯uorescence intensity observed for each sample (relative
to background, after addition of complementary material
and subsequent washing) was normalized relative to that of
the fully complementary oligonucleotide to correct for small
variations in optical alignment of sensors between sets of
experiments. The results show that at the lower temperature
of 288C, there is little difference in the relative ¯uorescence
yields. This was expected, since the temperature was suf®-
ciently low that almost complete hybridization was expected
with all oligonucleotides used. When the experimental
temperature was increased to 448C, enhancement of the
selectivity of hybridization was observed. Interestingly, the
signal from the partially complementary sequences was not
reduced to <54% of that of the fully complementary
sequence. This is larger than may be expected when con-
sidering differences in Tm for sequences of varying com-
plementarity. This unexpected result likely has two causes.
Firstly, it must be noted that the thermodynamics of hybri-
dization in an interfacial environment are signi®cantly
different than that of bulk solution. In previous papers
[14,15], we have noted that the thermal denaturation transi-
tions of immobilized nucleic acid hybrids are signi®cantly
broader than those of hybrids in bulk solution. The inter-
facial thermal denaturation transitions we have reported
occurred over temperature ranges of 15±208C, while those
observed in bulk solution occurred over temperature ranges
of about 108C. Consequently, there is an increased degree of
overlap of the thermal denaturation pro®les of immobilized
hybrids of varying complementarity relative to analogous
systems in bulk solution. Secondly, when hybridization
occurs at an interface, it does so in competition with the
process of non-selective adsorption. It has been shown with
the systems described here that non-selective adsorption
does occur and cannot be completely eliminated indepen-
dently of hybridization. Thus, non-selective adsorption may
also contribute to the increased signal observed with par-
tially complementary sequences even at the higher tempera-
tures. It must be remembered that the ¯uorescence signal in
these experiments is indicative of the presence of cDNA, but
is not a direct indicator of hybridization.
Examination of Fig. 4 also shows that the response of the
sensor to the mixed analyte sample of 10ÿ8 M total oligo-
nucleotide concentration does not provide a response
equivalent to the sum of the responses to the individual
components. This may seem contradictory to what may be
expected when considering the calibration curves alone, but
Fig. 4. Response of sensor with high dT20 immobilization density to individual solutions of dA20-50-fluorescein, d(A19G)-50-fluorescein, d(A9GA10)-50-fluorescein and d(A9G2A9)-50-fluorescein in 1� PBS buffer (�ssDNA�i � 2:5� 10ÿ9 M), and to a equimolar mixture of these oligonucleotides in 1� PBS
buffer (�ssDNA�Total � 1� 10ÿ8 M), at 28 and 408C. Wash steps were performed after each addition of cDNA.
34 J.H. Watterson et al. / Sensors and Actuators B 74 (2001) 27±36

may be understood when the thermal denaturation pro®les
are considered. In a previous paper [15], we reported that
®bers with high oligonucleotide immobilization density
(where nearest neighbour interactions between immobilized
oligonucleotides become signi®cant) have reduced Tm
values relative to analogous systems with lower oligonu-
cleotide immobilization densities, where the probability of
nearest neighbour interactions is lower. At higher packing
densities, the formation of hybrids at the surface increases
the charge density at the interface and therefore affects the
nearest neighbour interactions, as observed in the calibration
curves presented in Fig. 3. Non-selective adsorption also
affects the surface charge distribution, and cannot be entirely
eliminated independently of hybridization. These effects
may combine to cause further reductions in the stability
of the partially complementary hybrids at the temperatures
used herein, resulting in smaller contributions to the net
signal than would be expected on the basis of the calibration
curves of Fig. 3.
Experiments were done using individual component
concentrations of 2:5� 10ÿ8 M, using the temperature of
448C to enhance selectivity. These concentrations corre-
spond to a region on the calibration curves where deviations
from linearity were observed. Consequently, there was
little or no signi®cant change in ¯uorescence yield as
the complementarity of the oligonucleotides in solution
was adjusted. It was observed that the mixed analyte sample
showed only a very small increase in ¯uorescence yield
relative to the individual component samples, also as
expected. These results are consistent with the correspond-
ing concentration response range of the calibration curves of
Fig. 3, and serve as further con®rmation of the limited
dynamic range of the sensors with high oligonucleotide
packing density.
3.6. Conclusions and analytical consequences
The results that are presented here suggest that the control
of the density of nucleic acid ®lms may impart some control
of sensitivity and selectivity for a given hybridization assay
when optimization is done concurrently by adjustment of
other experimental conditions such as temperature and
solution ionic strength. Careful attention must be given to
activation of the ®lm for hybridization. Once activated, it
was found that higher immobilization densities provided
increased sensitivity and selectivity. Enhancements in sen-
sitivity and selectivity afforded by increasing the oligonu-
cleotide immobilization density may be realized only in
lower analyte concentration regimes, since higher oligonu-
cleotide immobilization density resulted in a smaller
dynamic range for such sensors. Consequently, it is of
fundamental importance to characterize the calibration
and thermal denaturation behavior of any nucleic acid sensor
before use with a given sample. It has been shown that
selectivity cannot be predicted from thermal denaturation
experiments done in bulk solution, nor can it be predicted
on the basis of Tm considerations alone, even if the Tm
values that are associated with speci®c sensors are mea-
sured. Non-selective adsorption occurs concurrently with
hybridization, and the dynamic response of the sensors due
to incomplete activation, surface occlusion and due to the
changing surface environment must be accounted for in each
experiment.
Acknowledgements
We are grateful to the Natural Sciences and Engineering
Research Council of Canada, and to FONA Technologies,
Inc., for ®nancial support of this research.
References
[1] R. Blonder, E. Katz, Y. Cohen, N. Itzhak, A. Riklin, I. Willner, Anal.
Chem. 68 (1996) 3151.
[2] R. Granzow, R. Reed, Biotechnology 10 (1992) 390.
[3] B. KoÈnig, M. GraÈtzel, Anal. Chim. Acta 309 (1995) 19.
[4] K.M. Millan, A. Saraullo, S.M. Mikkelsen, Anal. Chem. 66 (1994)
2943.
[5] J. Wang, S. Bollo, J.L. Lopez Paz, E. Sahlin, B. Mukherjee, Anal.
Chem. 71 (1999) 1910.
[6] H. Su, K.M.R. Kallury, M. Thompson, A. Roach, Anal. Chem. 66
(1994) 769.
[7] F. Caruso, E. Rodda, D.N. Furlong, K. Niikura, Y. Okahata, Anal.
Chem. 69 (1997) 2043.
[8] A.P. Abel, M.G. Weller, G.L. Duveneck, M. Ehrat, H.M. Widmer,
Anal. Chem. 68 (1996) 2905.
[9] P.A.E. Piunno, U.J. Krull, R.H.E. Hudson, M.J. Damha, H. Cohen,
Anal. Chem. 67 (1995) 2635.
[10] H. Su, P. Williams, M. Thompson, Anal. Chem. 67 (1995) 1010.
[11] T.M. Herne, M.J. Tarlov, J. Am. Chem. Soc. 119 (1997) 8916.
[12] R. Levicky, T.M. Herne, M.J. Tarlov, S.K. Satija, J. Am. Chem. Soc.
120 (1998) 9787.
[13] M.-T. Charreyre, O. Tcherkassaya, et al., Langmuir 13 (1997) 3103.
[14] P.A.E. Piunno, J.H. Watterson, C.C. Wust, U.J. Krull, Anal. Chim.
Acta 400 (1999) 73.
[15] J.H. Watterson, P.A.E. Piunno, C.C. Wust, U.J. Krull, Langmuir 16
(2000) 4984.
[16] W. Kern, D.A. Puotinen, RCA Rev. 6 (1970) 187.
[17] R.T. Pon, in: S. Agrawal (Ed.), Methods in Molecular Biology:
Protocols for Oligonucleotides and Analogs, Vol. 20, Humana Press,
Totowa, 1993, p. 465.
[18] B. Sojka, P.A.E. Piunno, C.C. Wust, U.J. Krull, Anal. Chim. Acta 395
(1999) 273.
[19] A.H. Uddin, P.A.E. Piunno, R.H.E. Hudson, M.J. Damha, U.J. Krull,
Nucleic Acids Res. 25 (1997) 4139.
[20] U. Maskos, E.M. Southern, Nucleic Acids Res. 20 (1992) 1679.
[21] P.A.E. Piunno, Ph.D. Thesis, 1999.
Biographies
Ulrich J. Krull received his PhD from the University of Toronto in 1983.
He is appointed as Professor of Analytical Chemistry at the University of
Toronto, where he holds the position of Astra Zeneca Chair in
Biotechnology. His current research interests are in the development of
nucleic acid biosensors based on optical fiber technology and also bilayer
lipid membrane electrochemistry.
J.H. Watterson et al. / Sensors and Actuators B 74 (2001) 27±36 35

Paul A.E. Piunno received his PhD in Analytical Chemistry from the
University of Toronto in 1999. He currently holds the position of Director of
Research at FONA Technologies, Inc., in Mississauga, Canada. His research
interests are in the development of fiber optic nucleic acid biosensors.
Christopher C. Wust received his BSc in Chemistry from the University of
Toronto in 1995. He is currently serving as a Product Development
Scientist at FONA Technologies, Inc., in Mississauga, Canada. His
research interests are in the development of fiber optic nucleic acid
biosensors.
James H. Watterson received his MSc in Analytical Chemistry from the
University of Toronto in 1999, where he is currently working toward the
completion of his PhD. His research interests are focussed on investigation
of thermodynamic and kinetic aspects of nucleic acid hybridization at
interfaces for the development of fiber optic nucleic acid biosensors.
36 J.H. Watterson et al. / Sensors and Actuators B 74 (2001) 27±36