clinical-ultrastructural study of thioridazine retinopathy
TRANSCRIPT

Clinical-Ultrastructural Study of Thioridazine Retinopathy FREDERICK S. MILLER III, MD, ANN H. BUNT-MILAM, PhD, ROBERT E. KALINA, MD
Abstract: Advanced thioridazine retinopathy was observed clinically and histopathologically in a 61-year-old man with progressive, severe loss of vision who had received thioridazine in high dosages 18 years previously. The ocular fundi showed multiple confluent, nummular areas of hypopigmentation and choroidal atrophy posterior to the equator. Atrophy and disorganization of the photoreceptor outer segments appeared to be the initial degenerative event, followed by loss of the retinal pigment epithelium and choriocapillaris. [Key words: thioridazine (Mellaril®), toxic retinopathy.] Ophthalmology 89:1478-1488, 1982
Thioridazine is a phenothiazine commonly used in the treatment of psychoses. Shortly after its introduction into clinical use in 1959, thioridazine was found to cause a pigmentary retinopathy associated with visual loss.I,2 While the currently recommended maximum dose is 800 mg/day, dosages exceeding 1600 mg/day were common before the dose-related retinal toxicity was recognized widely. 3-5 This report describes a patient with clinically advanced thioridazine retinopathy whose eyes were preserved immediately postmortem for light and electron microscopic study.
CASE REPORT
A 61-year-old white man was hospitalized for schizophrenia from 1960-1961 and from 1962-1963. During his first admission he received prochlorperazine (Compazine®), trihexyphenidyl (Artane®), promazine (Sparine®), reserpine, and imipramine (Tofranil®). During his second admission, he was treated with the same medications at various times and additionally with trifluoperazine (Stelazine®), chlorpromazine (Thorazine®), and thioridazine (Mellaril®). Over 11 months, he received 163.6 g of thioridazine, with a
From the Department of Ophthalmology, RJ-10, University of Washington School of Medicine, Seattle, Washington.
Presented as a poster at the Annual Meeting of the Association for Research in Vision and Opthalmology, Sarasota, Florida, May 3, 1982.
Reprint requests to Robert E. Kalina, MD, Department of Ophthalmology, RJ-10, University of Washington, Seattle, WA 98195.
1478
maximum dosage of 1200 mg/day for one six-week period, after which the drug was discontinued. The patient was then given an average dose of 350 mg/day of chlorpromazine for 57 days and discharged. An eye examination was performed four weeks after the thioridazine was discontinued in response to the patient's complaints of "blindness" but the results are not available. His family did not recall any prior history of eye disease nor did they observe any obvious visual dysfunction over the next two years. He subsequently became a transient for 14 years and was lost to follow-up.
The patient's eyes were examined elsewhere in 1979, at which time he said he had noticed reduced vision in the right eye for 12 years, and in the left eye for 8 years. The visual acuity was no light perception, right eye, and 20/50, left eye. The right pupil was amaurotic; the left reacted to light. The intraocular pressure was 15 mm Hg in both eyes. The right disc was markedly pale; the left disc was not described. Atrophic areas of the choroid and retina, with some associated pigmentation, were noted by indirect ophthalmoscopy as being "marked and most unusual."
We first saw the patient in December 1980 after he had been struck by an automobile at night. A partial avulsion of the left upper lid was repaired, and a subdural hematoma was evacuated.
A family history obtained from a sister revealed no significant eye disease (Fig 1). The patient had no children.
At an initial bedside examination, the vision in the right eye was no light perception, and the left eye had poor fixation and following movements. The patient's mental status did not allow more precise testing. The refractive error determined by streak retinoscopy was right eye:plano; left eye: -0.75 + 1.00 x 180. The right pupil was amaurotic; the left pupil showed hippus and questionable reaction to light. The anterior segment of each eye was normal and the media were clear. Indirect ophthalmoscopy of the right fundus
0161-6420/8211200/1478/$1.35 © American Academy of Ophthalmology

MILLER, et al • THIORIDAZINE RETINOPATHY
Fig 1. The pedigree does not suggest an inherited disease. No family members are known to have visualloss or symptoms similar to the propositus.
* EXAMINED - NORMAL
~ PATIENT WITH RETINOPATHY
showed disc pallor, slightly attenuated arterials and a striking mosaic pattern of chorioretinal atrophy. Posterior to the equator there were multiple, 1-4 disc diameter, nummular hypopigmented lesions. In these areas, large choroidal vessels were prominent due to apparent absence of the choriocapiIIaris, choroidal pigment, and retinal pigment epithelium (RPE). There was mild pigment clumping at the margins of some lesions. The lesions in the macula were almost confluent. The fundus appeared normal anterior to the equator. The left fundus showed mild disc pallor, normal vessels, and chorioretinal changes similar to the right eye except that the macula was less affected.
Serum was obtained between tube feedings of Isocal® (a complete dietary solution that includes arginine); the ornithine/lysine ratio was normal (147/340, in micromoles/L).
The patient died of pneumonia in January 1981. The eyes were injected through the pars plana with 0.1 ml of Yanoffs fixative immediately after death, enucleated promptly, placed in the same fixative and processed for light and electron microscopy. Autopsy diagnoses included pneumonia, blunt impact injuries to the head, and moderate coronary atherosclerosis.
PATHOLOGY
Gross examination. Right eye: The globe was opened horizontally. Multiple scalloped, semi-circular and circular pale yellow lesions extended from the posterior pole to the equator, with involvement of the macula (Fig 2). The disc appeared pale and cupped, and the retinal vessels were attenuated. The remainder of the globe was unremarkable.
Left eye: The gross findings were similar to those of the right eye except the optic nerve head appeared normal and macular pigmentation was better preserved (Fig 3).
Microscopic examination. Right eye: Anterior to the equator, the retina, retinal pigment epithelium (RPE), cho-
roid, and sclera appeared normal, except for the loss of retinal ganglion cells also found in the remaining retina (Fig 4). Posterior to the equator, the predominant feature was multiple areas of retinal thinning. In the center of each lesion, the RPE and choriocapillaris were absent and the photoreceptors were either absent or lacking outer segments. When present, the photoreceptor layer frequently assumed a rosette-like pattern with inner segments and Muller cell processes turned 90 to 180°, resting against granular deposits in Bruch's membrane (Figs 5, 9) that were prominent but comparable to those found in an age-matched normal eye (Fig 10). At the margin of the lesions, the RPE was present but thin and amelanotic. Occasional RPE cells appeared to be undergoing coagulative necrosis next to normal-appearing choriocapillaris (Fig 11). Mitotic figures rarely were found in the RPE. Beyond the margins of the lesions, the RPE contained normal amounts of melanin and lipofuscin for the patient's age. 6 However, the rod and cone outer segments were short and disorganized against this normal-appearing RPE, and macrophages were present in the interphotoreceptor matrix (Fig 12). The optic nerve head contained no axons and was gliotic (Fig 6). The choroid was of normal thickness and pigmentation, and the large choroidal vessels appeared normal.
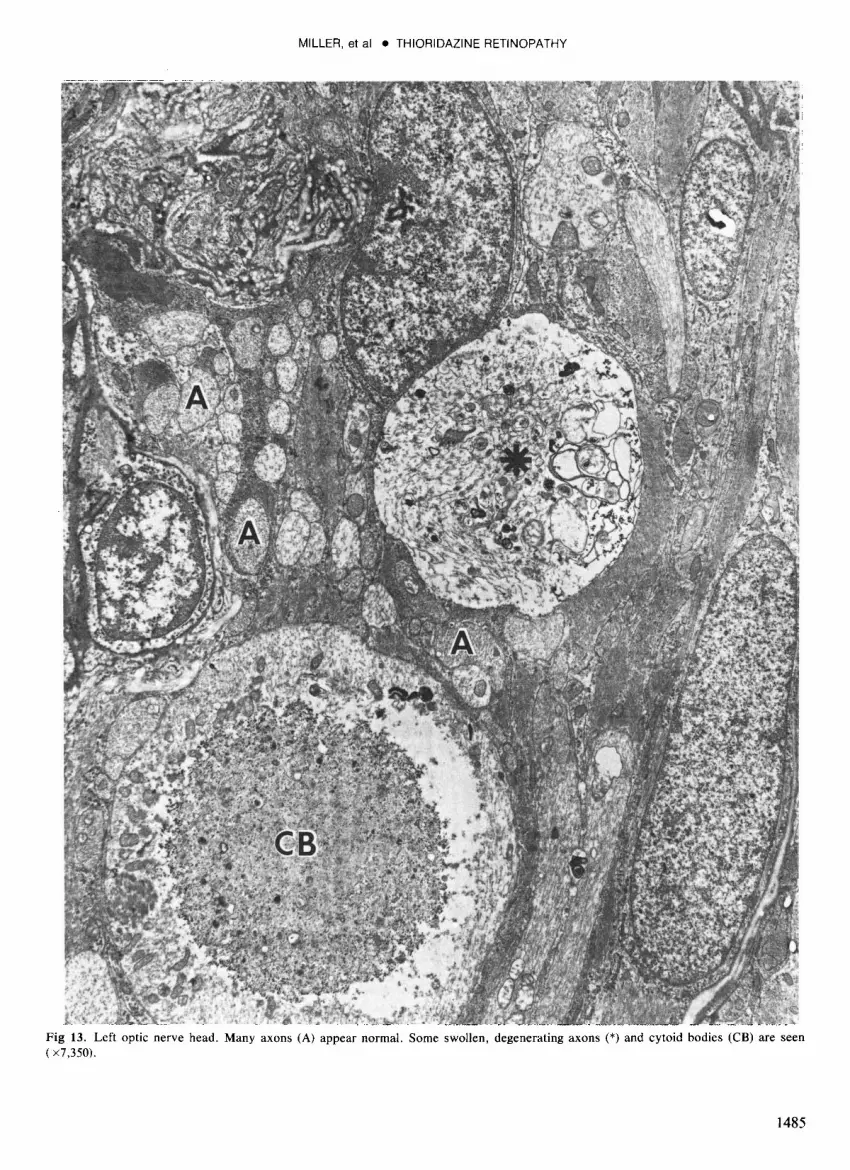
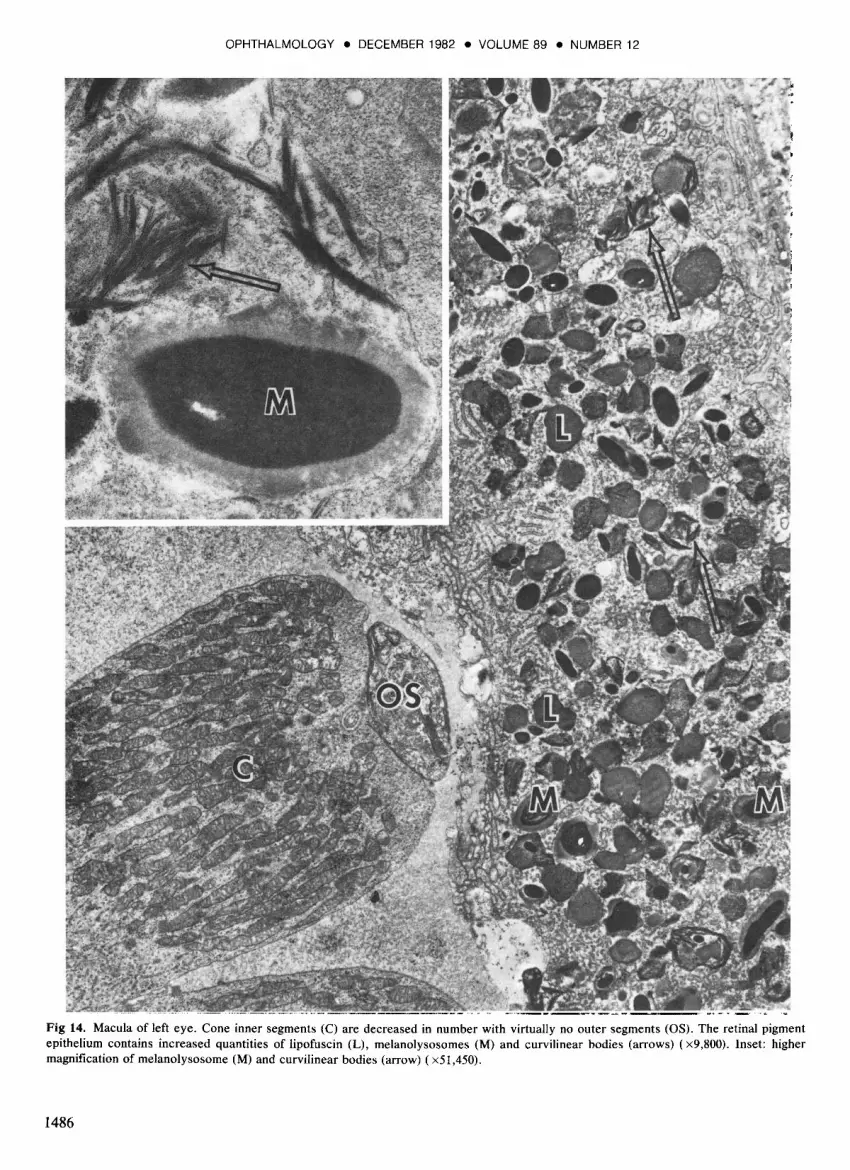
Left eye: The left eye showed similar changes in the photoreceptors, RPE, and choriocapiIIaris, including the rosette-like formations in the centers of the circular lesions. The nerve fiber layer and the optic nerve head (Fig 7) were more normal than in the right eye but degenerating axons and cytoid bodies occasionally were seen (Fig 13). In the macula, the cones were reduced in number and had virtually no outer segments. The RPE was continuous, variably pigmented, and contained increased numbers of lipofuscin and melanolysosome granules, as well as unusual curvilinear bodies free in the cytoplasm (Fig 14). Patchy loss of the choriocapiIIaris was noted (Fig 8).
1479

1480
Figs 2 (right eye) and 3 (left eye). Top left and top right, many circular hypo pigmented lesions occupy the entire fundus of each eye except for the area anterior to the equator. A lesion appears to involve the fovea of the right eye but not the left. Fig 4. Second row left, the retina of each eye is normal anterior to the equator except for the loss of ganglion cells. The choroid is of normal thickness and pigmentation throughout and the large choroidal vessels appear normal (paraffin section, periodic acid-Schiff stain x64). Fig S. Second row right, in the center of each circular hypopigmented lesion, the retinal pigment epithelium and choriocapillaris are absent and the photoreceptors are either totally absent or lack outer segments. Remaining photoreceptors frequently assume a rosettelike pattern, with inner segments and MOller cells turned 90 to 180°, resting against Bruch's membrane. Lipid laden macrophages are found in the enclosed space (epoxy section, Richardson's stain, x 160). Fig 6. Third row left, the right optic nerve head contains no axons and is gliotic. Epoxy section, Richardson's stain. x25. Fig 7. Third row right, the left optic nerve head shows preservation of some axons presumed functional during life (epoxy section, Richardson's stain x25). Fig 8. Bottom, left macula shows decreased numbers of cone inner segments and occasional hypertrophied retinal pigment epithelial cells. Note pale cystic spaces corresponding to loss of approximately half of the choriocapillaris (x 160).

MILLER, et al • THIORIDAZINE RETINOPATHY
Fig 9, Transmission electron micrograph of edge of rosette-like structure as shown in Figure 5. Note inner segments of cone (C) and rod (R) photoreceptors, a cone outer segment (OS), Moller cell nuclei (M) and cytoplasmic processes (*). B, Bruch's membrane ; choroid (Ch) ( x4,900).
1481

OPHTHALMOLOGY • DECEMBER 1982 • VOLUME 89 • NUMBER 12
Fig 10. Electron micrograph of photoreceptor-retinal pigment epithelium complex from a normal 61-year-old eye that had been removed surgically and processed identically to serve as an age-matched control eye. OS, photoreceptor outer segments ; RPE, retinal pigment epithelium containing melanin and lipofuscin granules; B, Bruch' s membrane with granular deposits ( x 5,240) .
1482

MILLER, et al • THIORIDAZINE RETINOPATHY
Fig 11. Transmission electron micrograph at the margin of a retinal lesion. The retinal pigment epithelium (RPE) is thin and amelanotic . An RPE cell appears necrotic ('*) next to normal appearing choriocapillaris (Ce). B, Bruch's membrane ; R, rod inner segment which lacks an outer segment ( x9,800).
1483

OPHTHALMOLOGY. DECEMBER 1982 • VOLUME 89 • NUMBER 12
Fig 12. Transmission electron micrograph of region between the circular lesions. The retinal pigment epithelium and choriocapillaris appear normal. The rod (R) and cone (C) outer segments (OS) are short and disorganized . Macrophages (*) are noted occasionally in the interphotoreceptor matrix ( x7 ,350).
1484
MILLER, et al • THIORIDAZINE RETINOPATHY
Fig 13. Left optic nerve head. Many axons (A) appear normal. Some swollen, degenerating axons (*) and cytoid bodies (CB) are seen ( x7,350).
1485
OPHTHALMOLOGY • DECEMBER 1982 • VOLUME 89 • NUMBER 12
Fig 14. Macula of left eye. Cone inner segments (C) are decreased in number with virtually no outer segments (OS). The retinal pigment epithelium contains increased quantities of lipofuscin (L), melanolysosomes (M) and curvilinear bodies (arrows) (x9,800). Inset: higher magnification of melanolysosome (M) and curvilinear bodies (arrow) (xSl,4S0).
1486

MILLER, et al • THIORIDAZINE RETINOPATHY
DISCUSSION
Patients with acute thioridazine retinopathy typically report blurred vision, dyschromatopsia, or nyctalopia three to eight weeks after receiving the drug in excess of 800 mg/day, 2,7,8 rarely in lower dosages. 9,10 The fundus examination may be normal at the onset of such symptoms, but within weeks to a few months a diffuse appearance of fine, deep retinal pigment posterior to the equator may be seen. The pigmentation later usually becomes coarsely granular and may form large plaques. Visual acuity may be normal or variably reduced. Color vision often is abnormal, the visual field is constricted or shows irregular paracentral or ring scotomas, and dark adaptation is impaired. The ERG and EOG may be abnormal. 8,11,12
If the drug is discontinued promptly, mildly affected patients may have good recovery of visual function and the fundus may revert to a normal or nearly normal appearance. 7.11 •13 On the other hand, despite the discontinuation of thioridazine, progressive retinopathy with deterioration of visual acuity, visual fields, dark adaptation, and electrical function is known to occur in patients who had received high dosages. 5,14 Clumps of pigment may coalesce and subsequently clear over many years, leaving multiple, depigmented lacunae from the posterior pole to the midperiphery. The progressive loss of the RPE and choriocapillaris that corresponds to this distinctive appearance of the fundus has been demonstrated by fluorescein angiography. 14
Our patient showed the clinical appearance typical of late, advanced thioridazine retinopathy. The ultrastructural examination of the multiple atrophic areas revealed a sequence of degenerative changes from the margin to the center of a given lesion. Near the margin, shortening and loss of photoreceptor outer segments overlying morphologically intact RPE appeared to be the initial degenerative event, although the functional integrity of this RPE is unknown. More centrally, this was followed by loss of RPE cells and the choriocapillaris. This sequence is compatible with the development and enlargement of nummular areas of chorioretinal atrophy observed clinically in other cases of progressive thioridazine retinopathy. 14
To our knowledge, no histopathologic or ultrastructural study of early thioridazine retinopathy in humans is available. Animal studies have shown that many phenothiazines, including thioridazine, are concentrated in uveal tissue and RPE by adsorption to melanin granules where they may remain for months. 15,16 A recent report confirmed that thioridazine uptake occurs in the human uvea as well; the concentration of thioridazine was over 21h times greater in the choroid than in the clinically normal retina of a patient without visual symptoms who was receiving 600 mg/day Y Retinal toxicity has been attributed to uveal binding and the specific configuration of the piperidyl side chain possessed by thioridazine and
a related drug, NP-207. 2 The latter drug was found to have such adverse effects on the retina during clinical trials in the 1950s that it never was marketed. Both NP-207 and thioridazine have been shown to alter retinal enzyme kinetics, inhibiting the oxidation of retinol. 18,19
Several feline studies on NP-207 have demonstrated acute alterations that include vacuolated photoreceptor outer segments and increased lamellar inclusions in the RPE arising from rod and cone degeneration,20,21 The dark pigment seen clinically in NP-207 retinopathy in cats was thought to be an accumulation of visual pigments from the degenerating photoreceptors. 18 This pigment forms in clumps and eventually is followed by a mosaic pattern of atrophy, similar to thioridazine retinopathy in human eyes.
Our patient was exposed to other drugs in addition to the large daily doses of thioridazine. Prochlorperzaine, promazine, and trifluoperazine are phenothiazines that are not known to be associated with a pigmentary retinopathy. 22 A possible exception is a case of retinopathy attributed to trifluoperazine, in which a mean dose of 15 mg/day was taken for 153 days.23 Our patient received only 10 mg/day for nine days. .
Our patient also received chlorpromizine two years before and immediately following the thioridazine. Although pigmentation of the anterior segment is seen commonly in association with high chlorpromazine dosage, pigmentary retinopathy is unusual but has been reported with dosages of 1200-2400 mg/day for 12 - 24 months. 24,25 This abnormal fundus pigmentation was said to disappear largely within a year. By comparison, the dose of chloropromazine that our patient received immediately prior to his visual symptoms was quite small. The various other medications (trihexyphenidyl, reserpine, imipramine) this patient received are not known to cause retinopathy. We believe that thioridazine was the primary cause of this patient's retinopathy, based on the typical history and the characteristic appearance of the fundus. However, we cannot rule out the possibility of an additional cause of blindness in this patient's left eye, in which there was ongoing optic atrophy, and in the right eye where there was total loss of retinal ganglion cells.
Although not suspected in our patient, several other conditions can cause a mosaic or nummular pattern of chorioretinal degeneration. Gyrate atrophy, an autosomal-recessive dystrophy, results in a scalloped pattern of atrophy in the peripheral fundus that progresses posteriorly and is associated with hyperornithinemia and presenile cataracts, neither of which was seen in our case. Myopic degeneration may cause lacunar choroidal defects limited to the posterior fundus but our patient was essentially emmetropic. Central areolar or geographic atrophy of the retinal pigment epithelium ("choroidal sclerosis") usually reduces central acuity during the third to fourth decades because of early macular involvement and may have an autosomal dominant inheritance. The history, the
1487

OPHTHALMOLOGY. DECEMBER 1982 • VOLUME 89 • NUMBER 12
wide distribution, and appearance of the fundus lesions and the pedigree of our patient would be atypical for that entity. Finally, the differential diagnosis includes choroideremia. In the early stages of this autosomal-recessive dystrophy, patchy loss of RPE and choriocapillaris may underlie the mosaic pattern seen clinically. In fact, even older patients whose choroideremia is only slowly progressive may have this appearance and good visual acuity may persist well into middle life. Although some of these features are seen in the present case, the absent family history argues strongly against a diagnosis of choroideremia.
We believe our case of advanced thioridazine retinopathy confirms the damaging effects of thioridazine on the RPE/photoreceptor complex and the eventual loss of the choriocapillaris that may be progressive even after the drug is discontinued. It also emphasizes the importance of a careful drug history when evaluating patients with an acute or chronic retinopathy.
ACKNOWLEDGMENT
The authors are grateful to D. F. Milam, MD, and M. J. Reeh, MD, for histopathologic consultation, R. Patterson, C. Meligro, I. Klock, and D. Ichikawa for technical assistance, J. Foltz and B. Clifton for photographic help, and J. Seng for secretarial assistance. T. Jackson, MD, Montgomery, Alabama, kindly provided portions of the case history.
REFERENCES
1. Kinross-Wright VJ. Scientific exhibit. Academy of Psychosomatic Medicine. Cleveland, OH, October, 1959.
2. Weekley RD, Potts AM, Reboton J, May RH. Pigmentary retinopathy in patients receiving high doses of a new phenothiazine. Arch Ophthalmol 1960; 64:65-76.
3. deMargerie J. Ocular changes produced by a phenothiazine drug: thioridazine. Trans Can Ophthalmol Soc 1962; 25:160-75.
4. Hagopian V, Stratton DB, Busiek RD. Five cases of pigmentary retinopathy associated with thioridazine administration. Am J Psychiatry 1966; 123:97-100.
5. Davidorf FH. Thioridazine pigmentary retinopathy. Arch Ophthalmol 1973; 90:2.51-5.
1488
6. Feeney L. Lipofuscin and melanin of human retinal pigment epithelium. Fluorescence, enzyme cytochemical, and ultrastructural studies. Invest Ophthalmol Vis Sci 1978; 17:583-600.
7. Scott AW. Retinal pigmentation in a patient receiving thioridazine. Arch Ophthalmol 1963; 70:775-8.
8. Connell MM, Poley BJ, McFarlane JR. Chorioretinopathy associated with thioridazine therapy. Arch Ophthalmol 1964; 71:816-21.
9. Heshe J, Engelstoft FH, Kirk L. Retinal injury developing under thioridazine treatment. Nord Psykiatr Tidskr 1961; 15:442- 7.
10. Applebaum A. An ophthalmoscopic study of patients under treatment with thioridazine. Arch Ophthalmol 1963; 69:578-80.
11. Leinfelder PJ, Burian HM. Mellaril intoxication of retina with full restitution of function. (Abstract) Invest Ophthalmol 1964; 3:466.
12. Potts AM. Drug-induced macular disease. Trans Am Acad Ophthalmol Otolaryngol 1966; 70: 1 054-7.
13. Campbell JM, Gralnick A. Pigmentary retinopathy associated with thioridazine administration. Behav Neuropsychiat 1971; 3(9-10):14,24.
14. Meredith TA, Aaberg TM, Willerson WD. Progressive chorioretinopathy after receiving thioridazine. Arch Ophthalmol 1978; 96:1172-6.
15. Potts AM. Uveal pigment and phenothiazine compounds. Trans Am Ophthalmol Soc 1962; 60:517-52.
16. Potts AM. The reaction of uveal pigment in vitro with polycyclic compounds. Invest Ophthalmol 1964; 3:405-16.
17. Kimbrough BO, Campbell RJ. Thioridazine levels in the human eye. Arch Ophthalmol 1981; 99:2188-9.
18. Meier-Ruge W, Cerletti A. Experimental pathology of chloroquine and phenothiazine retinopathy. Concilium Ophthalmologicum, 20th, 1966, Germany. 1967; 2:1129-36.
19. Muirhead JF. Drug effects on retinol oxidation: retinal alcohol: NAD+ oxidoreductase. Invest Ophthalmol 1967; 6:635-41.
20. Cerletti A, Meier-Ruge W. Toxicological studies on phenothiazine induced retinopathy. In: Proceedings of the European Society for the Study of Drug Toxicity. Amsterdam: Excerpta Medica Foundation, 1968; 9:170-88.
21. Gregory MH, Rutty DA, Wood RD. Differences in the retinotoxic action of chloroquine and phenothiazine derivatives. J Pathol 1970; 102:139-50.
22. Grant WM. Toxicology of the Eye, 2nd ed. Springfield: Charles C Thomas, 1974; 812-4.
23. Reboton J Jr, Weekley RD, Bylenga NO, May RH. Pigmentary retinopathy and iridocycloplegia in psychiatric patients. J Neuropsychiatry 1962; 3:311-6.
24. Zelickson AS, Zeller HC. A new and unusual reaction to chlorpromazine: JAMA 1964; 188:394-6.
25. Siddall JR. The ocular toxic findings with prolonged and high dosage chlorpromazine intake. Arch Ophthalmol 1965; 74:460-4.