biophysics of selectin–ligand interactions in inflammation and cancer
TRANSCRIPT

Biophysics of selectin–ligand interactions in inflammation and cancer
This article has been downloaded from IOPscience. Please scroll down to see the full text article.
2011 Phys. Biol. 8 015013
(http://iopscience.iop.org/1478-3975/8/1/015013)
Download details:
IP Address: 128.220.16.101
The article was downloaded on 07/02/2011 at 20:44
Please note that terms and conditions apply.
View the table of contents for this issue, or go to the journal homepage for more
Home Search Collections Journals About Contact us My IOPscience

IOP PUBLISHING PHYSICAL BIOLOGY
Phys. Biol. 8 (2011) 015013 (14pp) doi:10.1088/1478-3975/8/1/015013
Biophysics of selectin–ligand interactionsin inflammation and cancerLuthur Siu-Lun Cheung1,4, Phrabha S Raman1,4, Eric M Balzer1,2,3,Denis Wirtz1,2,3 and Konstantinos Konstantopoulos1,2,3,5
1 Department of Chemical and Biomolecular Engineering, The Johns Hopkins University, Baltimore,MD 21218, USA2 NCI PS-OC Johns Hopkins Engineering in Oncology Center, The Johns Hopkins University,Baltimore, MD 21218, USA3 Institute for NanoBioTechnology, The Johns Hopkins University, Baltimore, MD 21218, USA
E-mail: [email protected]
Received 21 September 2010Accepted for publication 15 November 2010Published 7 February 2011Online at stacks.iop.org/PhysBio/8/015013
AbstractSelectins (L-, E- and P-selectin) are calcium-dependent transmembrane glycoproteins that areexpressed on the surface of circulating leukocytes, activated platelets, and inflamed endothelialcells. Selectins bind predominantly to sialofucosylated glycoproteins and glycolipids(E-selectin only) present on the surface of apposing cells, and mediate transient adhesiveinteractions pertinent to inflammation and cancer metastasis. The rapid turnover ofselectin–ligand bonds, due to their fast on- and off-rates along with their remarkably hightensile strengths, enables them to mediate cell tethering and rolling in shear flow. This paperpresents the current body of knowledge regarding the role of selectins in inflammation andcancer metastasis, and discusses experimental methodologies and mathematical models usedto resolve the biophysics of selectin-mediated cell adhesion. Understanding the biochemistryand biomechanics of selectin–ligand interactions pertinent to inflammatory disorders andcancer metastasis may provide insights for developing promising therapies and/or diagnostictools to combat these disorders.
Introduction
Cell adhesion in shear flow is instrumental in diverse biologicalprocesses including inflammation and cancer metastasis.Leukocyte recruitment to sites of inflammation or infectionis mediated by highly specific receptor–ligand interactionsthat allow leukocytes to first tether and roll on activatedendothelium under hydrodynamic shear and then firmly adhereprior to their extravasation into the tissue space. Duringthe metastatic process, tumor cells invade the surroundingtissues to reach and penetrate the vascular endothelium. Oncethey enter the circulatory system, these rogue cancerous cells,referred to as circulating tumor cells (CTCs), are subjectedto shear forces and immunological stresses, which may affecttheir ability to metastasize. Only tumor cells uniquely fitto overcome or even exploit the effects of hemodynamic
4 Both authors contributed equally to this work.5 Author to whom any correspondence should be addressed.
forces and immunosurveillance will adhere to the vascularendothelium of distant organs, extravasate and successfullycolonize these sites. Thus, only a tiny fraction of CTCsis capable of establishing secondary colonies (Fidler et al2002); most CTCs die or remain dormant. Understanding themolecular and biophysical underpinnings of CTC adhesion tohost cells in shear flow may provide guidelines for developingpromising anti-metastatic therapies when initiated early in thecourse of disease progression.
Accumulating evidence suggests that the adhesiveinteractions of CTCs with host cells, such as platelets,leukocytes and endothelial cells, modulate their extravasationfrom the vasculature, and thus the development of secondarymetastatic foci. For instance, CTCs may escape immunesurveillance and promote their extravasation from thecirculatory system by co-opting platelets. Direct evidence forthe involvement of platelets in the facilitation of hematogenousdissemination of tumor cells stems from studies showinginhibition of metastasis by either pharmacological (Gasic et al
1478-3975/11/015013+14$33.00 1 © 2011 IOP Publishing Ltd Printed in the UK

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
1968, Karpatkin et al 1988) or genetic depletion (Camereret al 2004) of platelets, and the restoration of metastaticpotential by platelet infusion in a mouse model (Karpatkinet al 1988). It is believed among others that platelets, byforming heterotypic adhesive clusters with CTCs (Borsig et al2001, 2002), mask and protect CTCs from immune-mediatedmechanisms of clearance (Nieswandt et al 1999, Palumboet al 2005). Alternatively, platelets may potentiate tumor celladhesion to the vessel wall via a platelet bridging mechanism,in which platelets adherent to an endothelial-bound tumorcell capture free-flowing CTCs that subsequently attach tothe vessel wall downstream or next to the already adherentcell (Burdick and Konstantopoulos 2004). Platelets may alsosecrete an array of bioactive compounds, such as vascularendothelial growth factor (VEGF) at points of attachment toendothelium, thereby promoting vascular hyperpermeabilityand extravasation (Nash et al 2002). Once tumor cellshave exited the circulation, factors released from activatedplatelets are capable of inducing angiogenesis and stimulatinggrowth at the metastatic site (Pinedo et al 1998). CTCscan also hijack polymorphonuclear leukocytes (PMNs) forarrest in the endothelium of distant organs. PMN-facilitatedCTC arrest under hydrodynamic shear involves initial PMNtethering on the endothelium and subsequent capture of free-flowing tumor cells by tethered PMNs. CTCs may alsomasquerade as immune cells and directly bind to vascularendothelium in a manner analogous to leukocyte recruitment,which involves tethering, rolling and firm adhesion (Burdicket al 2003, Burdick and Konstantopoulos 2004). Selectinsmediate the initial tethering and rolling events during leukocyteaccumulation to sites of inflammation. Similarly, selectinsfacilitate cancer metastasis (Borsig et al 2001, 2002, Mannoriet al 1997) and tumor cell arrest in the microvasculatureby mediating the specific interactions between selectin-expressing host cells and ligands on tumor cells (Burdick andKonstantopoulos 2004, Burdick et al 2003, Jadhav et al 2001,Jadhav and Konstantopoulos 2002, McCarty et al 2000, 2002).Indeed, a variety of tumor cells, such as colon and pancreaticcarcinoma cells, express sialofucosylated molecules that arerecognized by selectins (Mannori et al 1995, Park et al 2003,Satomura et al 1991).
The adhesion of leukocytes and CTCs to endothelialcells involves highly regulated molecular events such asselectin–ligand interactions that rely on the local circulatoryhemodynamics and the micromechanical and kineticproperties of participating adhesive molecular constituents.Fluid shear generated by blood flow, on one hand, inducescollisions among free-flowing cells as well as between free-flowing cells and the vessel wall, thereby increasing theencounter rate between membrane-bound receptors and theircognate ligands. On the other hand, fluid shear reduces thecell–cell contact duration, and exerts tensile forces tendingto disrupt the receptor–ligand bonds that are responsible forcell adhesion. Particle size and compliance also modulatethe extent of cell adhesion in shear flow (Jadhav et al 2005,Pawar et al 2008). The ability to predict cell–cell bindingwill be vital to the optimization of design parameters, such assite density, affinity and tensile strength of targeted receptor–ligand interactions, for developing effective targeted drug
delivery strategies to combat inflammatory disorders andcancer metastasis.
In this paper we discuss the critical roles of selectinsand newly discovered functional selectin ligands in cancermetastasis. We proceed with a description of experimentaltechniques and mathematical models used to resolve thebiophysics of selectin-mediated cell adhesion pertinent toinflammation. These biophysical/mathematical approachescan be readily applied to resolve the selectin–ligand bindinginteractions in the area of cancer metastasis. Understandingthe biochemical and biophysical underpinnings of selectin–ligand interactions pertinent to inflammation and cancermetastasis may provide insights for combating these disorders.
Selectins and selectin ligands on host cells andtumor cells
Selectins are calcium-dependent transmembrane glycopro-teins that recognize specific glycoconjugates on apposing cellsurfaces (Kansas 1996, Varki 1997), and play pivotal roles inthe cell–cell interactions pertinent to inflammation and cancermetastasis. All three known members of the selectin family(L-, E-, and P-selectin) share a similar cassette structure: anN-terminal, calcium-dependent lectin domain, an epidermal-growth-factor (EGF)-like domain, a variable number of con-sensus repeat units (2, 6, and 9 for L-, E-, and P-selectin, respec-tively), a transmembrane domain (TM) and an intracellularcytoplasmic tail (cyto). Though they share common elements,their tissue distribution and binding kinetics are quite different,reflecting their divergent roles in various pathophysiologicalprocesses.
L-selectin (CD62L) is constitutively expressed on thesurface of almost all types of leukocytes but gets rapidlyshed upon cell activation with cytokines, chemokines, orformyl peptides. Expression of P-selectin (CD62P) onendothelium and platelet surfaces is inducible. E-selectin(CD62E) expression is induced on vascular endothelial cellsand requires de novo mRNA and protein synthesis. Maximallevel of E-selectin expression typically occurs 4–6 h afteractivation with inflammatory stimuli, such as interleukin-1,tumor necrosis factor-alpha or endotoxin in vitro.P-selectin is stored preformed in the Weibel–Palade bodiesof endothelial cells and alpha-granules of platelets, and israpidly mobilized to the plasma membrane upon activationwith agonists like histamine or thrombin. However, P-selectinexpression on vascular endothelium may also be regulated atthe transcriptional level after stimulation with cytokines suchas interleukin-4 or oncostatin M (Yao et al 1996).
Selectins, like all C-type lectins, bind to carbohydrateligands in a calcium-dependent manner. Detailed studiesinvolving site-directed mutagenesis, domain swapping, andantibody inhibition have revealed that carbohydrate ligandsbind to the lectin domain on a shallow region that overlapsa single calcium coordination site opposite where the EGFdomain is located (Kansas 1996, Varki 1997). Evidencealso suggests that the EGF domain and the short consensusrepeat domains not only contribute to ligand specificity butalso confer unique kinetic and mechanical properties on
2

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
Figure 1. Schematic diagram of selectin–ligand interactions between tumor cells and host vascular cells. CD44 on free-flowing tumor cellsbinds to P-selectin on activated endothelial cells or platelets, whereas CD44, CEA, and PCLP mediate E-selectin-dependent tumor celladhesion to endothelial cells. CD44, CEA, and PCLP also bind to L-selectin expressed by leukocytes. Adopted from Konstantopoulos andThomas (2009).
each selectin when binding to its ligand (Patel et al 1995b).All selectins recognize the tetrasaccharide sialyl Lewis x(sLex; NeuAc α2,3 Gal β1,4 [Fuc α1,3] GlcNAc-R) andits isomer sialyl Lewis a (sLea; NeuAc α2,3 Gal β1,3 [Fucα1,4] GlcNAc-R) (Kansas 1996, Varki 1997). sLex is aterminal component of glycans attached to glycoproteinsand glycolipids on most circulating leukocytes and someendothelial cells. In contrast, sLea is expressed on some tumorcells but not on normal leukocytes. The affinity of selectinsfor isolated monovalent sLex and sLea oligosaccharides isvery low (Nicholson et al 1998, Poppe et al 1997). Thus,neither expression of the sLex nor the sLea groups correlatewith the properties of endogenous selectin ligands on cellulartargets per se. Instead, sLex or related structures arepart of more extensive binding determinants. Selectinscan also recognize other classes of molecules, namely non-sialylated or non-fucosylated molecules such as heparansulfate glycosaminoglycans, sulfated glycolipids such assulfatides, and sulfoglucuronosyl glycosphingolipids (Varki1997).
High affinity ligands for P- and L-selectin have beenidentified (figure 1). P-selectin binds to the amino terminusof P-selectin glycoprotein ligand-1 (PSGL-1; CD162),which is expressed on nearly all blood leukocytes, humanhematopoietic progenitor cells (McEver and Cummings1997a), and to a much lesser extent on blood platelets (Frenetteet al 2000). The extracellular domain of PSGL-1 (∼60 nmlong) has the hallmarks of a mucin, since it is rich in serines,
threonines and prolines, and includes 16 decameric repeats(McEver and Cummings 1997b). High affinity binding ofPSGL-1 to P- and L-selectin requires three clustered tyrosinesulfate residues, adjacent peptide components, and fucose andsialic acid residues on an optimally positioned short core-2O-glycan within the anionic amino-terminal region of PSGL-1 (Leppanen et al 2000). Crystallographic studies reveal apatch of positive electrostatic potential on P-selectin designedto engage the tyrosine sulfate residues and increase the bindingaffinity (Somers et al 2000). Although PSGL-1 is recognizedby all three selectins (Hanley et al 2004, Patel et al 1995a),it serves as functional ligand only for P- and L-selectin, sinceselective removal or blockade of PSGL-1 on intact humanleukocytes nearly abrogates their binding to P- and L-selectin(Patel et al 1995b, Walcheck et al 1996). In contrast, PSGL-1 appears dispensable for E-selectin-dependent binding ofhuman PMNs (Nimrichter et al 2008).
L-selectin also binds to a series of mucin-typeglycoproteins such as CD34, podocalyxin-like protein (PCLP)and MadCAM-1 that contain O-linked glycans in which thesLex determinant is present and further substituted with asulfate ester on the 6-hydroxyl group of the GlcNAc (NeuAca2,3 Gal ß1,4 [Fuc a1,3] SO36-GlcNAc; 6-sulfo-sLex) and/orGal (NeuAc a2,3 SO36-Gal ß1,4 [Fuc a1,3] GlcNAc; 6′-sulfo-sLex) residues (Rosen 2004). The sulfate residue on GlcNAc isrequired for high-affinity L-selectin-dependent binding, whilethe contribution of Gal-6-sulfation is controversial (Rosen2004).
3

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
Although E-selectin binds to PSGL-1 (Hanley et al2004), accumulating evidence supports the concept thatsialylated fucosylated glycolipids on human PMNs representthe physiologically relevant E-selectin ligands (Alon et al1995a, Burdick et al 2001, Nimrichter et al 2008). Also,L-selectin itself has been reported as a high-affinity ligand forE-selectin. Direct evidence for this interaction was providedin affinity isolation experiments with E-selectin-Ig, which alsodocumented that the binding was dependent on sialic acid onL-selectin (Zollner et al 1997). Although the inhibitory effectsof anti-L-selectin mAbs on PMN binding to E-selectin-coatedsurfaces were initially ascribed to the blockade of the so-calledsecondary tethering mediated by L-selectin on free-flowingcell and L-selectin ligands on already adherent leukocytes(Alon et al 1996), it has been reported that antibody blockadeinterferes with primary interactions between L- and E-selectin(Zollner et al 1997).
A variety of tumor cells, including colon and pancreaticcarcinoma cells, express sialofucosylated molecules (Mannoriet al 1995, Park et al 2003, Satomura et al 1991,Kannagi 1997) that are recognized by selectins. However,the characterization of selectin ligands on tumor cellsbeyond general classifications (i.e. sialofucosylated mucin-like glycoproteins) has recently begun to emerge. Asdescribed by Varki (1997), distinctions must be drawn betweenmolecules that can bind to selectins under static conditionsin vitro and those (i.e. the functional ligands) that actuallyinteract with selectins in vivo. To this end, a functional selectinligand should fulfill certain criteria: it should be expressedin the right place and at the right time, the ligand shouldbind with some selectivity and relatively high affinity, andselective removal or absence of the ligand should prevent celladhesive interactions. CD44 variant isoforms, CD44v, havebeen identified as P-selectin ligands that fit the aforementionedcriteria (Napier et al 2007) (figure 1). CD44 proteins are typeI transmembrane molecules encoded by a single gene thatcomprises at least 20 exons. Exons 1–5, 16–18 and 20 arespliced together to form the smallest CD44 transcript, knownas standard isoform (CD44s). However, CD44s displays arather low affinity for P-selectin (Hanley et al 2006). Itis believed that the structural biology of CD44 is such thatsplicing of variant exons into CD44 by metastatic tumor cellsextends its molecular length and inserts additional sites forglycosylation, thereby transforming CD44v into a mucin-likesialofucosylated glycoprotein capable of functioning as anefficient P-selectin ligand. CD44v also serves as an ancillaryL-selectin ligand by stabilizing L-selectin-dependent tumorcell rolling (Napier et al 2007). In contrast, CD44 appearsdispensable for E-selectin-dependent binding (Napier et al2007). Of note, CD44v overexpressing in certain types ofcancer confers resistance to apoptosis, metastatic potential invivo and leads to prognosis (Harada et al 2001, Wielenga et al1993).
Carcinoembryonic antigen (CEA) was recently shownto possess L- and E-, but not P-, selectin ligand activity oncolon carcinoma cells (Thomas et al 2008a) (figure 1). Mostimportantly, CEA and CD44v cooperate to mediate coloncarcinoma cell adhesion to L- and E-selectin in shear flow
(Thomas et al 2008a). Interestingly, CEA is expressed ina number of tumors of epithelial origin including colorectalcarcinoma, lung adenocarcinoma and mucinous ovariancarcinoma, and has been reported to promote the metastaticpotential of colon carcinoma cells (Hashino et al 1994,Minami et al 2001). PCLP was also found to support L-and E-, but not P-selectin-dependent tethering and rollingof metastatic colon carcinoma cells in shear flow (Thomaset al 2009a) (figure 1). The selectin-binding determinants onPCLP expressed by tumor cells are non-sulfated (MECA-79-negative) sialofucosylated structures displayed on O-linkedglycans (Thomas et al 2009a, 2009b), distinct from theMECA-79-reactive O-glycans expressed by high endothelialvenules, which possess L-selectin ligand activity. Of note,PCLP is expressed by a number of metastatic tumor cells suchas colon and breast carcinoma (Somasiri et al 2004, Thomaset al 2009a). PCLP overexpression has been reported as anindependent predictor of breast cancer progression (Somasiriet al 2004).
The role of selectins in cancer metastasis
Mounting evidence suggests that selectins facilitate thehematogenous dissemination of tumor cells and their arrestin the microvasculature by mediating specific interactionsbetween selectin-expressing host cells and ligands on tumorcells. The most direct evidence for the involvement of P-selectin in the metastatic process is the marked inhibition ofmetastasis in P-selectin-deficient mice compared to wild-type(wt) controls in a colon carcinoma cell model (Borsig et al2001, Kim et al 1998). Microscopic observations of tumorcells arrested in the lungs of wt mice reveal the presence ofa dense coat of platelets surrounding the colon carcinomacells that is diminished in P-selectin-deficient mice (Borsiget al 2001). Moreover, the initial seeding and subsequentlodging of metastatic cells in target organs was mitigatedin P-selectin-knockout mice compared to wt controls (Borsiget al 2001, Kim et al 1998). Although these observationssuggest that platelet P-selectin plays a critical role not only incolon carcinoma-platelet adhesion but also in the facilitationof metastasis, they cannot rule out an additional role forendothelial P-selectin that could potentially tether tumor cellsand mediate their extravasation from the vasculature. In vivostudies also disclose the role of L-selectin in cancer metastasis(Borsig et al 2002). It is believed that tumor cells can formmulticellular complexes with platelets and leukocytes (via anL-selectin-dependent mechanism) (Jadhav et al 2001, Jadhavand Konstantopoulos 2002), which can then arrest in themicrovasculature of distant organs, and eventually extravasateand establish metastatic colonies. Selectins can thus actsynergistically to facilitate tumor cell–host cell interactionsand cancer metastasis. To date, the cooperative effects of P-and L-selectin on cancer metastasis have been demonstratedin vivo (Borsig et al 2002). Interestingly, leukocyte L-selectincan also enhance metastasis by interacting with endothelial L-selectin ligands induced adjacent to established intravascularcolon carcinoma cell emboli (Laubli et al 2006). EndothelialE-selectin has been shown to support metastatic spread
4

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
in vivo (Mannori et al 1997). Since selectins recognize sLex-and/or sLea-decorated glycoproteins such as CD44v, CEAand PCLP, and over-expression of these moieties on tumorcells correlates with poor prognosis and tumor progression(Kannagi 1997, Konstantopoulos and Thomas 2009, Thomaset al 2009b), it appears that selectin-mediated adhesion tothese sialofucosylated target molecules on tumor cells is animportant determinant for metastatic spread.
The ability of selectin–ligand bonds to initiate tetheringand rolling in shear flow is dictated by their micromechanicaland kinetic properties, and their responses to external force.Below, we review quantitative models used to resolve thebiophysics of receptor–ligand interactions.
Mathematical models of two-dimensional (2D)receptor–ligand binding affinity
Several techniques, such as surface plasmon resonance andradio-immunoassays, that have been developed to studyreceptor–ligand binding kinetics require at least one moleculepresent in solution, which limits their application to themeasurement of three-dimensional (3D) binding constants.Yet, cell adhesive interactions are mediated by the bindingof receptors to ligands, which are anchored on apposingcell membrane surfaces. Consequently, the motion of bothmolecules is restricted to 2D (Piper et al 1998). Not only thebinding mechanism is different but also their respective on-rates have different units (M−1 s−1 in 3D and μm2 s−1 in 2D).Even though 3D kinetic rates have been extensively reportedin the literature, they are inadequate to describe the 2D kineticsof cell–cell adhesive interactions.
Sophisticated biophysical assays have been developedto quantify the 2D receptor–ligand binding kinetics. In thewidely used micropipette aspiration assay (Evans et al 1991),a red blood cell, serving as a picoforce transducer, coatedwith a low density of the adhesion receptor of interest isheld at a fixed position by a micropipette. Also held bya second micropipette, a cell (PMN) or a ligand-bearingbead is translated to contact the RBC by precision-piezodisplacements for prescribed durations of time, and thenretracted from the contact position at defined pulling velocities.The deformation of the RBC induced by the receptor–ligandbond is recorded using a video-enhanced microscope. Thebond rupture force is readily calculated by multiplying the totalextension of RBC with the stiffness of membrane transducerat the moment of bond failure (Evans et al 1991, 1995).In addition to measuring force-dependent 2D off-rates, themicropipette aspiration assay can also be used to quantify the2D affinity constant, AcKa
0, of receptor–ligand bonds (Huanget al 2010, Chesla et al 1998), as described below.
In this assay, the small contact zone between the target cell(or ligand-bearing bead) and the receptor-coated RBC alongwith the low site density of adhesion molecules dictate thatreceptor–ligand binding occurs as a random event, even thoughall experimental conditions can be kept identical includingcontact area (Ac), contact duration (t), surface density ofreceptors (mr ) and ligands (ml). Hence, the probabilistic
model, rather than the deterministic model, is more suitablefor this scenario (Chesla et al 1998, McQuarrie et al 1964).
For a 1-step reversible kinetic system involving vr
receptors (R) binding to vl ligands (L) to produce vb bonds(B), its reaction equation is given by
0
0
f
r
k
blrk
v Bv Lv R + , (1)
where k0f and k0
r represent the unstressed on- and off-rates, respectively. The rates of change of the probabilitycomponents (dpn/dt) are defined as (Chesla et al 1998)
dpn
dt= (n + 1)vb
k0r
Avb−1c
pn+1
−[(
Acmr − vr
vb
n
)vr(Acml − vl
vb
n
)vl k0f
Avr +vl−1c
+ nvbk0r
Avb−1c
]pn
+
[Acmr − vr
vb
(n − 1)
]vr[Acml − vl
vb
(n − 1)
]vl k0f
Avr +vl−1c
pn−1
(2a)
where the first term on the right-hand side of equation (2a)represents the probability of (n + 1) bonds losing one bondduring time t; the second and third terms represent theprobability of n bonds adding one or losing one during timet, respectively; and the last term represents the probability of(n − 1) adding one bond during time t.
Assuming that the formation of a small number ofreceptor–ligand bonds will have no appreciable effect on theavailability of unbound receptors and ligands inside the contactarea, equation (2a) can be simplified by neglecting the n and(n − 1) terms in [Acmj − (vj/vb)n] and [Acmj − (vj/vb)(n− 1)] (subscript j = r or l) as given by equation (2b) (Cheslaet al 1998):
dpn
dt= (n + 1)vb
k0r
Avb−1c
pn+1
−[(Acmr)
vr (Acml)vl
k0f
Avr +vl−1c
+ nvbk0r
Avb−1c
]pn
+ (Acmr)vr (Acml)
vlk0f
Avr +vl−1c
pn−1. (2b)
The solution of equation (2b) for the special case, vr = vl =vb = 1, is of the form of the Poisson distribution (Chesla et al1998, Long et al 1999):
Pn(t) = 〈n〉nn!
exp(−〈n〉) (3)
where 〈n〉 is the average number of bonds at time t given by
〈n〉 = Acmvr
r mvl
l K0a
[1 − exp
(−k0r t
)]. (4)
Equations (3) and (4) will deviate significantly from the exactsolution when the numbers of available receptors and ligandsare similar (i.e. mr ≈ ml). The comparison between theapproximated solution and exact solution against the ratioof receptor to ligand site densities has been discussed inthe literature (Chesla et al 1998). The analysis reveals that
5

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
0.0
0.2
0.4
0.6
0.8
1.0
0 1 2 3 4 5 6 7 8
Contact time [s]
Ad
hes
ion
Pro
bab
ility
, Pa
t 50
P a,ss
m l ×m r =300
m l ×m r =150
m l ×m r =50
Figure 2. Estimation of the 2D binding kinetics by plotting theadhesion probability as a function of contact duration time fordifferent receptor–ligand site densities (mr × ml). The 2D bindingaffinity AcK
0a is estimated from the steady-state value of the
adhesion probability (Pa,ss) using equation (6). The unstresseddissociation rate k0
r is determined from the half-time (t50) usingequation (7).
the Poisson distribution given by equation (3) is in excellentagreement with the exact solution if the ratio Acmmin/〈n〉 is>10.
In most chemical reactions, the concentration of theproduct is proportional to the reaction time until the systemreaches equilibrium. Similar reaction characteristics havebeen observed for receptor–ligand-mediated cell adhesion toa surface (Chesla et al 1998). An example plot showingthe dependence of adhesion probability (Pa) on the contactduration (t) for different site densities of membrane-boundreceptors and immobilized ligands is presented in figure 2.Taking into consideration that the adhesion event is definedas having n � 1 bonds, the dependence of receptor–ligandbinding probability on the contact duration time is given by
Pa(t) = 1 − P0(t)
= 1 − exp{−AcmrmlK
0a
[1 − exp
(−k0r t
)]}. (5)
The 2D binding affinity k0a and the unstressed off-rate k0
r can beobtained by a simple graphic representation method, as shownin figure 2. By setting t → ∞ in equation (5), the steady-state solution Pa,ss, which represents the maximum adhesionprobability for a given set of conditions, is given by
Pa,ss = 1 − exp(−AcmrmlK
0a
). (6)
The 2D affinity constant AcK0a can be estimated directly from
the experiment data for a given set of receptor and ligandsite densities. The unstressed off-rate k0
r can be expressed by(Chesla et al 1998)
k0r ≈ 0.5
t50, (7)
where t50 represents the duration time for Pa(t) to achieve 50%of steady-state value Pa,ss as shown in the figure 2. Althoughwe cannot directly estimate k0
f , an alternative expression ofthe on-rate, Ack
0f , can be obtained by multiplying 2D binding
affinity AcK0a with unstressed off-rate k0
r .
Effects of molecular length and orientation on 2Dbinding kinetics
Cell adhesion is regulated not only by the intrinsic kineticand mechanical properties of receptor–ligand pairs but alsoother geometric factors such as the length and orientation ofadhesion molecules. The critical role of the molecular lengthin receptor–ligand binding kinetics was first demonstratedby rolling PSGL-1-expressing PMNs on different P-selectinconstructs whose lengths were engineered by controllingthe number of CR domains (Patel et al 1995b). P-selectinconstructs containing only two or three CR domains failed tosupport PMN binding in shear flow even though they exhibiteda similar binding efficiency as that of native P-selectin understatic conditions (Patel et al 1995b). The impact of themolecular length on the 2D selectin–ligand binding kineticswas also investigated by the micropipette aspiration assayusing two types of soluble P-selectin: P-selectin consistingof Lec-EGF domains plus 9 CR but no transmembrane andcytoplasmic domains, and P-selectin consisting of only theLec-EGF domains with an added C-terminal epitope. The2D affinity AcK
0a of the long P-selectin molecule is ∼twofold
larger than that of the short molecule; however, the molecularlength did not have an appreciable effect on the unstressedoff-rates k0
r (Huang et al 2004). Cumulatively, these findingssuggest that the absence of PMN binding to immobilized P-selectin constructs bearing two or three CR domains (Patel et al1995b) can be attributed to the poor accessibility of the shortmolecule to the ligand-coated surface. Once P-selectin–ligandbonds form, their dissociation is not altered by the length ofthe molecules.
The effect of molecular orientation on the 2D bindingkinetics was also investigated using the micropipette aspirationassay. Soluble P- or E-selectin was either adsorbed randomlyor uniformly coupled to non-blocking anti-P- or E-selectinantibodies, respectively, on the RBC surface. The 2D bindingaffinity of uniformly oriented selectins is markedly higher thanthat of randomly absorbed selectins, whereas no differencewas noted in their respective unstressed off-rates (Huang et al2004). Taken together, the molecular length and orientationof selectins modulate the 2D selectin–ligand binding affinitywithout altering the unstressed off-rate.
Receptor–ligand dissociation: slip bond dissociationkinetics
The kinetic and micromechanical properties of receptor–ligand bond dissociation have been studied by the use ofsingle-molecule force spectroscopy. There are two typesof ultrasensitive probes: the biomembrane force probe(BFP) where force is sensed by the displacement of a glassmicrosphere attached to a pressurized membrane capsule (i.e.RBC) (Evans et al 2001, 2004), and the molecular forceprobe (MFP) where force is sensed by the deflection of a thincantilever (Evans 2001, Hanley et al 2003, 2004, Marshallet al 2003). Both techniques can quantify the tensile strengthof receptor–ligand pairs at the single molecule level, and allowdetection of a wide force spectrum ranging from 5 pN to
6

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
>1 nN, which covers the typical biological forces from themolecular to the cellular level (Dobrowsky et al 2008, Evanset al 2001, 2004, Evans and Calderwood 2007, Hanley et al2003, 2004, Marshall et al 2003, Sarangapani et al 2004,Helenius et al 2008, Muller et al 2009). In this section,we review the kinetic models of slip bond dissociation usedto extract the biophysical properties of receptor–ligand pairsfrom BFP and MFP experiments. Slip bonds are defined asthose whose bond lifetimes decrease with the application ofan external force, because force reduces the energy barrierbetween bound and free states. Of note, all receptor–ligandpairs exhibit the slip bond dissociation kinetics in the highforce regime.
When a receptor is in close proximity with a ligand, thebond formation can be described by a reversible bimolecularreaction with two discrete steps: receptor–ligand encounterand reaction. This conceptual design (Bell 1978) is expressedby
Bondrd
rdRLLR
++
−−
+ (8)
where R is a free receptor molecule, L is a free ligand molecule,d+ and d− are the rates of formation and dissociation of theencounter complex RL, r+ and r− are the forward and reverserates for the bond formation. In most cases, the concentrationof RL complex is significantly lower than that of free receptor Rand free ligand L. Consequently, equation (8) can be simplifiedby assuming each bond is formed directly from the pair of freemolecules (Bell 1978). The overall binding process is thusrepresented by
Bondf
r
k
kR L+ (9)
where kf and kr are the on- and off-rates of the bondresulting from a free receptor and a free ligand pair. In 1978,Bell proposed the force dependence of receptor–ligand bonddissociation, which is given by
kr(f ) = k0r exp
[xβf
kBT
](10)
where k0r is the unstressed off-rate, xβ is the reactive
compliance, f is the rupture force acting on the bond, kBTis the Boltzmann constant multiplied by the temperature (orthermal energy). The off-rate, kr , which is related to bondlifetime tb by tb = 1/kr , exponentially increases with theapplied force. In the literature, kr is also given in the followingform (Merkel et al 1999, Evans and Calderwood 2007):
kr(f ) = k0r exp
[f
fβ
]. (11)
The new parameter f β = kBT/xβ has units of force, and canbe considered as the characteristic force scale at which thedissociation rate changes 2.7-fold. Equations (10) and (11)are commonly used to model slip bond dissociation underforce.
In practice, it is essentially impossible to apply a forceinstantaneously. Therefore, there is always a finite loading raterf . Evans and colleagues (Evans and Ritchie 1997) proposeda model to estimate the dissociation parameters when a bond
is ruptured by a finite loading rate. The probability of a singlebond rupture in the time interval (t, t + dt) as a function offorce is given by (Evans and Ritchie 1997)
p(t, f ) = kr(f ) exp
{−
∫ t
0kr [f (t ′)] dt ′
}(12)
where the exponential term represents the probability of bondsurvival up to time t, whereas the kr (f ) term represents theprobability of bond rupture in the next short time interval dt.The peak value of this probability distribution can be obtainedby taking the derivative of equation (12) with respect to f . Bysetting ϑp/ϑf = 0 and using the linear ramp of force, f (t) =rf ·t, the off-rate at the critical rupture force 〈fb〉 is given by
kr (〈fb〉) = rf
∂
∂f[ln kr(f )]f =〈fb〉 . (13)
This equation suggests that the bond off-rate increases withthe loading rate rf . Substituting the Bell model equation intoequation (13) results in equation (14):
〈fb〉 = kBT
xβ
ln
(xβ
k0r kBT
)+
kBT
xβ
ln(rf ). (14)
By plotting the rupture force 〈fb〉 against the logarithm ofloading rate ln(rf ), the Bell model parameters, k0
r and xβ , canbe estimated from the slope and intercept of experimental datain the linear region by using the following equations (Merkelet al 1999, Tees et al 2001, Hanley et al 2004):
slope :kBT
xβ
; intercept :kBT
xβ
ln
(xβ
k0r kBT
). (15)
Alternatively, k0r and xβ can also be estimated by a nonlinear
least-squares fit of equation (16) to the experimental data overthe entire range of loading rates (Evans and Ritchie 1997,Hanley et al 2003, Tees et al 2001):
〈fb〉 = kbT
xβ
exp
(k0r kbT
xβrf
) ∫ ∞
1
exp(−k0
r kbT
xβrft)
tdt . (16)
The accuracy of the Bell model parameters can be validatedby Monte Carlo simulations of receptor–ligand bond ruptureunder constant loading rates (Hanley et al 2003, 2004). Inbrief, given values for k0
r and xβ in each simulation, the ruptureforce (Frup = rf ×n�t) at a prescribed loading rate can becalculated for which the probability of bond rupture, Prup, isgreater than Pran, a random number between 0 and 1:
Prup = 1 − exp
[−k0
r exp
(xβrf n�t
kbT
)�t
]. (17)
where n = 1, 2, 3, . . . , �t is the interval and n�t is the timestep.
Receptor–ligand dissociation: catch bonddissociation kinetics
The Bell model was proposed to describe the dissociationkinetics of the so-called slip bonds (Bell 1978). Ten years
7

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
later, Dembo proposed an alternative model, in which theoff-rate increases exponentially with the square of appliedforce (Dembo et al 1988). As a theoretical possibility,Dembo suggested that the bond lifetime may also increasewith the applied force or remain unchanged; these bondswere classified as catch and ideal bonds, respectively. Catchbinding is an intriguing and counter-intuitive phenomenonin which receptor–ligand bonds resist breakage and becomestronger under the influence of a low externally applied force.Application of higher forces is expected to reduce the energybarrier between the bound and free states, and as such receptor–ligand bonds follow the slip dissociation kinetics. To date,at least four distinct receptor–ligand pairs show evidence ofcatch bond behavior in the low force regime and transitionto slip bond dissociation at higher forces: selectin–ligand(Marshall et al 2003, Sarangapani et al 2004, Yago et al2004), glycoprotein Ib-von Willebrand factor (Yago et al2008), the bacterial adhesion protein FimH-mannose (Thomaset al 2002), and integrin–ligand bonds (Kong et al 2009).This seemingly paradoxical phenomenon at the nanoscale leveltranslates into complex behavior witnessed at the cellular level,such as shear-threshold phenomenon in which the extent ofcell binding to selectins, for instance, first increases and thendecreases while monotonically increasing the wall shear stress(Finger et al 1996, Lawrence et al 1997).
The energy landscape that describes the dissociation ofa slip bond has a bound state, which is separated from thefree state by a potential energy barrier corresponding to thetransition state. According to the Dembo model, the boundstate and the transition state of a receptor–ligand bond areHookean springs with specific elastic constants (k) and restinglengths (λ). If the elastic constant is the same for both springsbut the difference between the resting lengths of the transitionstate and bound state springs, δλ, is positive, the off-rate ofthe receptor–ligand pair can be expressed as an exponentialfunction of the applied force f (Dembo et al 1988), as shownin equation (18). If the resting lengths are the same for bothsprings but the difference between the elastic constants of thetransition state and bound state springs, δκ , is negative, the off-rate increases exponentially as the square of the force (Demboet al 1988):
kr(f ) = k0r exp
{(δλ) f − δk
2k2 f2
kBT
}, (18)
where kr is the off-rate in the presence of an applied forcef , k0
r is the unstressed off-rate and k is the elastic constantof the bound state. To account for the distinctions betweenslip and catch bonds, the possibilities of δλ < 0 and δκ > 0were considered. In the latter scenarios, the off-rate decreasesexponentially with the applied force or the square of force. Itwas thought that the application of external force on the catchbond decreases its failure rate in the low force regime, butthis scenario switches to the traditional slip binding at higherforces where the applied force increases the failure rate.
Various physical and mathematical models have beenproposed to describe the experimentally observed catchand slip bond behavior exhibited by selectin–ligand bonds.Conceptually, the receptor–ligand bond can be thought to be
shaped like a harpoon or a hook. This configuration canlock ‘tightly’ when pulled apart by the two ends. Initiallythe unbinding time (or bond lifetime) increases with theapplied force, indicative of catch bond behavior. Progressivelyincreasing forces are capable of bending the elastic hook,thereby promoting dissociation via the slip pathway (Thomaset al 2008b). Alternatively, the receptor can undergo aligand-induced switch to an active conformation (Thomas et al2008b). The transition state in the catch pathway would reflectreversion to the inactive conformation followed by immediatereceptor–ligand unbinding. The slip pathway would involveunbinding through the active conformation, similar to theforceful bending of the elastic hook.
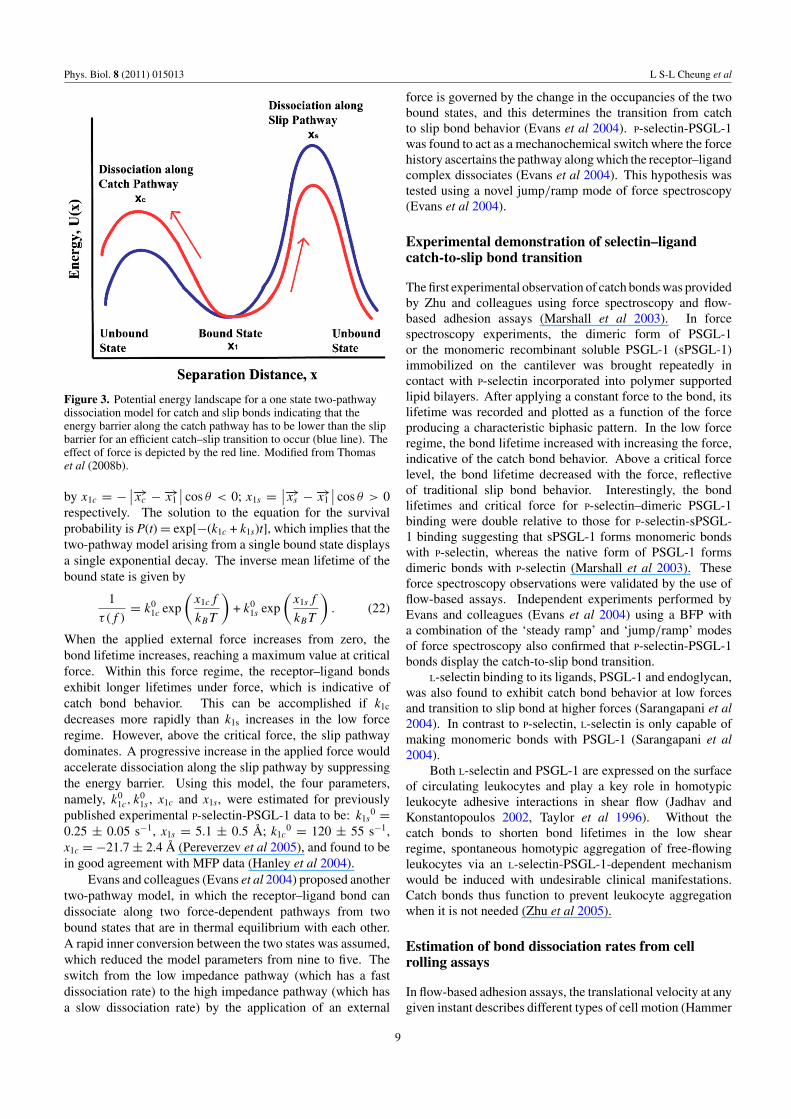
A simple four-parameter model (Pereverzev et al 2005)was proposed to describe the unbinding of the selectin–ligandcomplex from a single bound state either along the catch bondpathway over a low-energy barrier or the slip bond pathwayover a high-energy barrier. In the absence of an externalforce, the transition of the receptor–ligand complex from thebound to the free state is related to the thermal probabilityof reaching the top of the barrier. However, in the presenceof an applied force, f , the height of the energy barrier �Echanges depending on the distance between the bound state1 and the transition state 2 projected on to the direction ofthe externally applied force. When the applied tensile forcepulls the bond from the bound state to the transition state byperforming positive work on the ligand, the energy barrier islowered (figure 3). The bond breakage in this case is througha single pathway where the projected distance is positive(x12 > 0) and the receptor–ligand complex forms a slip bond.But force can also pull the ligand away from the transition statein such a way that the projected distance becomes negative(x12 < 0). Under this circumstance, the force performsnegative work on the ligand, thereby increasing the depth ofthe energy barrier �E and decreasing the off-rate (figure 3). Inother words, the applied tensile force increases the lifetime ofthe receptor–ligand bound complex thereby making it a catchbond (Dembo et al 1988, Pereverzev et al 2005).
The bound complex can dissociate from the bound statevia two alternative paths that can be represented as two finiteenergy barriers on either side, for a catch–slip transition tooccur. The probability that a ligand is still bound to its receptorat a later time t > 0, P(t), decreases with time according toequation (19) (Pereverzev et al 2005):
dP
dt= − (k1c + k1s) P (t), (19)
where k1c and k1s are the dissociation rate constants forunbinding through the catch and slip pathways with thecoordinates xc and xs , respectively. Along the two pathways,the exponential dependence of the dissociation rates on theapplied force is given by the following relations:
k1c = k01c exp
(x1cf
kBT
); (20)
k1s = k01s exp
(x1sf
kBT
), (21)
where k01c and k0
1s are the unstressed dissociation rates alongthe catch and slip pathways, respectively. x1c and x1s are given
8
Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
Figure 3. Potential energy landscape for a one state two-pathwaydissociation model for catch and slip bonds indicating that theenergy barrier along the catch pathway has to be lower than the slipbarrier for an efficient catch–slip transition to occur (blue line). Theeffect of force is depicted by the red line. Modified from Thomaset al (2008b).
by x1c = − ∣∣−→xc − −→x1
∣∣ cos θ < 0; x1s = ∣∣−→xs − −→x1
∣∣ cos θ > 0respectively. The solution to the equation for the survivalprobability is P(t) = exp[−(k1c + k1s)t], which implies that thetwo-pathway model arising from a single bound state displaysa single exponential decay. The inverse mean lifetime of thebound state is given by
1
τ(f )= k0
1c exp
(x1cf
kBT
)+ k0
1s exp
(x1sf
kBT
). (22)
When the applied external force increases from zero, thebond lifetime increases, reaching a maximum value at criticalforce. Within this force regime, the receptor–ligand bondsexhibit longer lifetimes under force, which is indicative ofcatch bond behavior. This can be accomplished if k1c
decreases more rapidly than k1s increases in the low forceregime. However, above the critical force, the slip pathwaydominates. A progressive increase in the applied force wouldaccelerate dissociation along the slip pathway by suppressingthe energy barrier. Using this model, the four parameters,namely, k0
1c, k01s , x1c and x1s, were estimated for previously
published experimental P-selectin-PSGL-1 data to be: k1s0 =
0.25 ± 0.05 s−1, x1s = 5.1 ± 0.5 A; k1c0 = 120 ± 55 s−1,
x1c = −21.7 ± 2.4 A (Pereverzev et al 2005), and found to bein good agreement with MFP data (Hanley et al 2004).
Evans and colleagues (Evans et al 2004) proposed anothertwo-pathway model, in which the receptor–ligand bond candissociate along two force-dependent pathways from twobound states that are in thermal equilibrium with each other.A rapid inner conversion between the two states was assumed,which reduced the model parameters from nine to five. Theswitch from the low impedance pathway (which has a fastdissociation rate) to the high impedance pathway (which hasa slow dissociation rate) by the application of an external
force is governed by the change in the occupancies of the twobound states, and this determines the transition from catchto slip bond behavior (Evans et al 2004). P-selectin-PSGL-1was found to act as a mechanochemical switch where the forcehistory ascertains the pathway along which the receptor–ligandcomplex dissociates (Evans et al 2004). This hypothesis wastested using a novel jump/ramp mode of force spectroscopy(Evans et al 2004).
Experimental demonstration of selectin–ligandcatch-to-slip bond transition
The first experimental observation of catch bonds was providedby Zhu and colleagues using force spectroscopy and flow-based adhesion assays (Marshall et al 2003). In forcespectroscopy experiments, the dimeric form of PSGL-1or the monomeric recombinant soluble PSGL-1 (sPSGL-1)immobilized on the cantilever was brought repeatedly incontact with P-selectin incorporated into polymer supportedlipid bilayers. After applying a constant force to the bond, itslifetime was recorded and plotted as a function of the forceproducing a characteristic biphasic pattern. In the low forceregime, the bond lifetime increased with increasing the force,indicative of the catch bond behavior. Above a critical forcelevel, the bond lifetime decreased with the force, reflectiveof traditional slip bond behavior. Interestingly, the bondlifetimes and critical force for P-selectin–dimeric PSGL-1binding were double relative to those for P-selectin-sPSGL-1 binding suggesting that sPSGL-1 forms monomeric bondswith P-selectin, whereas the native form of PSGL-1 formsdimeric bonds with P-selectin (Marshall et al 2003). Theseforce spectroscopy observations were validated by the use offlow-based assays. Independent experiments performed byEvans and colleagues (Evans et al 2004) using a BFP witha combination of the ‘steady ramp’ and ‘jump/ramp’ modesof force spectroscopy also confirmed that P-selectin-PSGL-1bonds display the catch-to-slip bond transition.
L-selectin binding to its ligands, PSGL-1 and endoglycan,was also found to exhibit catch bond behavior at low forcesand transition to slip bond at higher forces (Sarangapani et al2004). In contrast to P-selectin, L-selectin is only capable ofmaking monomeric bonds with PSGL-1 (Sarangapani et al2004).
Both L-selectin and PSGL-1 are expressed on the surfaceof circulating leukocytes and play a key role in homotypicleukocyte adhesive interactions in shear flow (Jadhav andKonstantopoulos 2002, Taylor et al 1996). Without thecatch bonds to shorten bond lifetimes in the low shearregime, spontaneous homotypic aggregation of free-flowingleukocytes via an L-selectin-PSGL-1-dependent mechanismwould be induced with undesirable clinical manifestations.Catch bonds thus function to prevent leukocyte aggregationwhen it is not needed (Zhu et al 2005).
Estimation of bond dissociation rates from cellrolling assays
In flow-based adhesion assays, the translational velocity at anygiven instant describes different types of cell motion (Hammer
9

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
Fx
Ty
R
θFb l
S
x
z
Cell
d
Fo
Figure 4. Force balance on a tethered cell in shear flow. A cell ofradius R experiences a hydrodynamic force Fx and torque Ty
induced by the shear rate S. On the other hand, a tether which haslength d and oriented at an angle θ with the substrate induces a forceFb on the cell. The length of lever arm l is the distance betweenbinding points.
and Apte 1992, Chang et al 2000, Krasik and Hammer 2004).Specifically, a cell is in free motion when its translationalvelocity is close to the prevailing hydrodynamic velocity.When the cell mean velocity is significantly slower thanthe hydrodynamic velocity due to the formation/dissociationof receptor–ligand bonds, the cell is classified as in rollingmotion. If a cell is stationary for at least several seconds, itis considered as firmly adherent. Although cell rolling is acomplex process controlled by many different physical andchemical parameters, such as cell deformation, microvillusextension, receptor and ligand site densities, and receptor–ligand bond formation and dissociation, several simplificationscan still possibly be made. In the low shear regime, the celldeformation can be neglected such that the cell can be modeledas a hard sphere in a Couette flow. The hydrodynamic forceand torque exerted on the cell can be determined by Fx = 1.7 ×(6πμR2S) and Ty = 0.944×(4πμR3S), respectively (Goldmanet al 1967), where μ is the viscosity of the suspended buffer,R is the radius of cell and S is the shear rate. If ligandsare immobilized on the substrate at very low site densities,the binding interaction can be assumed to involve a singlereceptor–ligand bond complex. Thus, the bond lifetime canbe obtained by simply recording the cell tethering time.
The bond rupture force Fb can be estimated by balancingforces and torques on the cell at static equilibrium, as shownin figure 4. The sum of forces in x- or z-directions must equalzero:
Fx = Fb cos θ, (23)
Fo = Fb sin θ, (24)
where Fo is the contact force between cell and flow chamber inthe y-direction. The balance of torque in the y-direction aboutthe contact point of Fo is given by
Ty + FxR = l (Fb sin θ) . (25)
The length of the lever arm is equal to the distance betweenthe binding point and the contact point of Fo (figure 4) which
can be measured by the distance that bound cells move duringflow reversal assays (Chen et al 1997, Alon et al 1997). Thetether length d (figure 4) is the total length of an extendedmicrovillus and the receptor–ligand complex (Alon et al 1997,Chen et al 1997, Shao et al 1998). The oriented angle θ of thetether is expressed by (Shao et al 1998)
θ = tan−1
(R
l
)+ cos−1
(d2 + l2
2d√
R2 + l2
). (26)
To ensure that cell tethering is mediated by a single receptor–ligand pair, flow-based adhesion assays must be performedusing a substrate (i.e. chamber wall) coated with very lowselectin site densities, typically, lower than those supportingstable cell rolling. The cell tethering time, tb, represents thebond lifetime. Under a constant force, the dissociation oftethered cells follows the first order kinetics (Alon et al 1995b).By plotting the natural logarithm of the number of cells thatremained tethered as a function of lifetime, the dissociationrate kr can be extracted by the negative slope of the fittingcurve as shown in figure 5(a) (Alon et al 1995b, Alon et al1997). By performing the flow chamber assays under differentwall shear stresses, the off-rate kr can be obtained as a functionof applied force, which is calculated from equations (23)–(26). The independence of off-rate on the selectin site densityon the substrate is indicative of a quantum unit mediatingselectin-dependent cell tethering (Alon et al 1995b). Thekinetic parameters of selectin–ligand bond dissociation can beobtained by fitting the appropriate kinetic model (e.g. two-pathway model (Evans et al 2004)) to the off-rate data as afunction of force (figure 5(b)).
Effects of fluid shear on cell rolling
In the absence of viscous fluid moving around a cell, itstranslational velocity Ucell would be synchronized with theangular velocity when the modeled cell is in the limitof touching the surface. Hence, R /Ucell would be equalto unity. However, the numerical solutions of R /Ucell
obtained by Goldman et al (1967) showed that the valueof R /Ucell approaches a finite limit of ∼0.5676 whenz → R (e.g. the modeled cell ‘touches’ the surface) (figure 4).Consequently, the cell translational velocity Ucell is alwayslarger than the surface tangential velocity R , leading toa cell slipping motion relative to the chamber wall. Thisslipping velocity has been shown to enhance the receptor–ligand encounter rate (Chang and Hammer 1999). In theabsence of a slipping motion between the cell surface andthe chamber wall, each cell receptor could only interact witha limited number of immobilized counter-receptors locatedwithin its reactive zone. In contrast, when a cell surfacemoves with a finite slipping velocity, each cell receptor canpotentially react with any counter-receptor passing its reactivezone. Although fluid shear exerts forces tending to disrupt thereceptor–ligand bonds responsible for cell tethering/adhesion,it also induces collisions between free-flowing cells and thevessel wall, thereby increasing the encounter rate betweenmembrane-bound receptors and their ligands (Caputo et al2007, Chang and Hammer 1999). This mechanism potentiates
10

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
Nat
ura
l lo
g o
f b
ou
nd
cel
ls
Bond lifetime tb [s]
F2
Shear force: F1 < F2 < F3
F1F3
Nat
ura
l lo
g o
f b
ou
nd
cel
ls
Bond lifetime tb [s]
F2
Shear force: F1 < F2 < F3
F1F3 Bin
din
g o
ff-r
ate,
kr
Rupture force, Fb
Bin
din
g o
ff-r
ate,
kr
Rupture force, Fb
(a)(b)
Figure 5. (a), Example of distribution of bond lifetimes obtained from cell tethering assays at different shear forces. The receptor–ligandbond off-rate kr can be estimated from the slope of the logarithm of the number of bound cells versus the bond lifetime (Alon et al 1995b).This example plot illustrates that bonds exhibit catch bond behavior at low forces (F1) and transition to slip bonds at higher forces (F3).(b), Dependence of the off-rate on the rupture force. The catch–slip bond kinetic parameters can be obtained by fitting the appropriate model(e.g. two-pathway model) to the data points (Evans et al 2004).
leukocyte attachment to inflamed endothelial cells duringthe inflammatory response, but can also enhance tumor celladhesion to vascular endothelium, thereby increasing the riskof tumor cell extravasation to secondary tissues.
Selectin-PSGL-1-dependent interactions require a shearthreshold to mediate optimal leukocyte tethering and rolling(Lawrence et al 1997, Yago et al 2004). In other words,leukocyte tethering rate first increases and then decreases whilemonotonically increasing wall shear stress. This so-calledshear threshold phenomenon seems counter-intuitive becauseincreasing levels of shear stress increase the dissociation forceon the bonds, thereby increasing the probability of the cellto detach from the substrate. Both in vitro and in vivoassays reveal that this phenomenon may be characteristic ofall three selectins binding to their respective glycoproteinligands (Finger et al 1996, Lawrence et al 1997). Shear-induced cell deformation was initially proposed to explain thisphenomenon by increasing the contact area between the celland the substrate; as such, the probability of bond formationis increased (Lawrence et al 1997). However, numericalstudies show that cell deformation plays a modest role inthe shear threshold phenomenon (Pawar et al 2008). Inlight of observations showing that cell-free flow assays usingpurified selectins and their respective ligands successfullyrecapitulate the shear threshold phenomenon (Marshall et al2003, Yago et al 2004), it is now accepted that the originof this phenomenon is primarily molecular, intimately linkedwith the kinetic and micromechanical properties of receptor–ligand bonds.
Based on the analysis of various biophysical parametersthat control selectin-mediated adhesion under flow, Zhu andcolleagues identified two major mechanisms that contribute tothe shear threshold phenomenon (Zhu et al 2008): transport-dependent acceleration of bond formation and force-dependentdeceleration of bond dissociation. The first mechanismencompasses three distinct modes of transport (Zhu et al 2008):(i) the relative sliding between the cell and the surface; (ii)the Brownian motion, which alters the gap distance betweenthe cell and the surface and increases their collisions; and(iii) the molecular diffusivity of the receptors and ligands,
which orients their binding sites for molecular docking.The second mechanism refers to the catch bond kineticsin which the bond lifetime is prolonged by the tetherforce. Interestingly, simulation studies using the adhesivedynamics model for cell rolling reveal that the shear-thresholdphenomenon observed in L-selectin-dependent cell rolling ispredominantly attributed to the catch–slip bond kinetics andto a lesser extent to the shear-controlled on rate (Caputo et al2007). Dimensional analysis studies also disclose that thetether force, but not the wall shear rate or wall shear stress,is the critical parameter controlling flow-enhanced cell rolling(Zhu et al 2008).
Concluding remarks
The mechanisms used for trafficking of leukocytes may beappropriated for the dissemination of metastatic tumor cells viathe bloodstream and lymphatics. In particular, the paradigmof the coordinated action of a ‘rapid’ selectin-dependentbinding followed by a ‘slow’ integrin-mediated adhesion hasbeen extended to account for maximal binding of tumorcells to activated endothelium (Burdick and Konstantopoulos2004, Burdick et al 2003), platelets (McCarty et al 2000,2002), and PMNs (Jadhav et al 2001, Jadhav et al 2007,Jadhav and Konstantopoulos 2002) under physiological shearconditions. Significant advances have recently been madein the identification and characterization of functional selectinligands expressed by metastatic tumor cells (Napier et al 2007,Thomas et al 2008a), which appear to be distinct from theselectin ligands found on the leukocyte surface. Although thebiophysics of selectin-mediated leukocyte binding has beenextensively studied, little is known about the biomechanics ofselectin–ligand interactions in the context of cancer metastasis(Hanley et al 2003). Since these interactions precede and arethus necessary for tumor cell extravasation, and facilitate CTCsurvival and metastatic outgrowth, a quantitative analysis ofselectin–ligand binding will further our understanding of themetastatic process and aid the development of new screeningmethods and therapeutic strategies that exploit these bindinginteractions. Elucidating the molecular nature of tumor cell
11

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
adhesion in the dynamic setting of the vasculature requiresa multidisciplinary approach that integrates fundamentals ofhydrodynamics, modeling of receptor–ligand binding kineticsand cell mechanics, with concepts and techniques frombiochemistry and molecular and cell biology.
Acknowledgments
This work was supported by NIH/NCI R01 CA101135 andU54 CA143868.
References
Alon R, Chen S, Puri K D, Finger E B and Springer T A 1997 Thekinetics of L-selectin tethers and the mechanics ofselectin-mediated rolling J. Cell Biol. 138 1169–80
Alon R, Feizi T, Yuen C T, Fuhlbrigge R C and Springer T A 1995aGlycolipid ligands for selectins support leukocyte tethering androlling under physiologic flow conditions J. Immunol.154 5356–66
Alon R, Fuhlbrigge R C, Finger E B and Springer T A 1996Interactions through L-selectin between leukocytes andadherent leukocytes nucleate rolling adhesions on selectins andVCAM-1 in shear flow J. Cell Biol. 135 849–65
Alon R, Hammer D A and Springer T A 1995b Lifetime of theP-selectin-carbohydrate bond and its response to tensile force inhydrodynamic flow Nature 374 539–42
Bell G I 1978 Models for the specific adhesion of cells to cellsScience 200 618–27
Borsig L, Wong R, Feramisco J, Nadeau D R, Varki N M andVarki A 2001 Heparin and cancer revisited: mechanisticconnections involving platelets, P-selectin, carcinoma mucins,and tumor metastasis Proc. Natl Acad. Sci. USA 98 3352–7
Borsig L, Wong R, Hynes R O, Varki N M and Varki A 2002Synergistic effects of L- and P-selectin in facilitating tumormetastasis can involve non-mucin ligands and implicateleukocytes as enhancers of metastasis Proc. Natl Acad. Sci.USA 99 2193–8
Burdick M M, Bochner B S, Collins B E, Schnaar R Land Konstantopoulos K 2001 Glycolipids supportE-selectin-specific strong cell tethering under flow Biochem.Biophys. Res. Commun. 284 42–9
Burdick M M and Konstantopoulos K 2004 Platelet-inducedenhancement of LS174T colon carcinoma and THP-1monocytoid cell adhesion to vascular endothelium under flowAm. J. Physiol. Cell Physiol. 287 C539–47
Burdick M M, Mccaffery J M, Kim Y S, Bochner B Sand Konstantopoulos K 2003 Colon carcinoma cell glycolipids,integrins, and other glycoproteins mediate adhesion toHUVECs under flow Am. J. Physiol. Cell Physiol.284 C977–87
Camerer E, Qazi A A, Duong D N, Cornelissen I, Advincula Rand Coughlin S R 2004 Platelets, protease-activated receptors,and fibrinogen in hematogenous metastasis Blood 104 397–401
Caputo K E, Lee D, King M R and Hammer D A 2007 Adhesivedynamics simulations of the shear threshold effect forleukocytes Biophys. J. 92 787–97
Chang K C and Hammer D A 1999 The forward rate of binding ofsurface-tethered reactants: effect of relative motion betweentwo surfaces Biophys. J. 76 1280–92
Chang K C, Tees D F and Hammer D A 2000 The state diagram forcell adhesion under flow: leukocyte rolling and firm adhesionProc. Natl Acad. Sci. USA 97 11262–7
Chen S, Alon R, Fuhlbrigge R C and Springer T A 1997 Rolling andtransient tethering of leukocytes on antibodies revealspecializations of selectins Proc. Natl Acad. Sci. USA94 3172–7
Chesla S E, Selvaraj P and Zhu C 1998 Measuring two-dimensionalreceptor–ligand binding kinetics by micropipette Biophys. J.75 1553–72
Dembo M, Torney D C, Saxman K and Hammer D 1988 Thereaction-limited kinetics of membrane-to-surface adhesion anddetachment Proc. R. Soc. Lond. B. Biol. 234 55–83
Dobrowsky T M, Panorchan P, Konstantopoulos K and Wirtz D2008 Live-cell single-molecule force spectroscopy MethodsCell Biol. 89 411–32 chapter 15
Evans E 2001 Probing the relation between force—lifetime—andchemistry in single molecular bonds Annu. Rev. Biophys.Biomol. Struct. 30 105–28
Evans E, Berk D and Leung A 1991 Detachment ofagglutinin-bonded red blood cells: I. Forces to rupturemolecular-point attachments Biophys. J. 59 838–48
Evans E, Leung A, Hammer D and Simon S 2001 Chemicallydistinct transition states govern rapid dissociation of singleL-selectin bonds under force Proc. Natl Acad. Sci. USA98 3784–9
Evans E, Leung A, Heinrich V and Zhu C 2004 Mechanicalswitching and coupling between two dissociation pathways in aP-selectin adhesion bond Proc. Natl Acad. Sci. USA101 11281–6
Evans E and Ritchie K 1997 Dynamic strength of molecularadhesion bonds Biophys. J. 72 1541–55
Evans E, Ritchie K and Merkel R 1995 Sensitive force technique toprobe molecular adhesion and structural linkages at biologicalinterfaces Biophys. J. 68 2580–7
Evans E A and Calderwood D A 2007 Forces and bond dynamics incell adhesion Science 316 1148–53
Fidler I J, Yano S, Zhang R D, Fujimaki T and Bucana C D 2002The seed and soil hypothesis: vascularisation and brainmetastases Lancet Oncol. 3 53–7
Finger E B, Puri K D, Alon R, Lawrence M B, Von Andrian U Hand Springer T A 1996 Adhesion through L-selectin requires athreshold hydrodynamic shear Nature 379 266–9
Frenette P S, Denis C V, Weiss L, Jurk K, Subbarao S, Kehrel B,Hartwig J H, Vestweber D and Wagner D D 2000 P-Selectinglycoprotein ligand 1 (PSGL-1) is expressed on platelets andcan mediate platelet–endothelial interactions in vivo J. Exp.Med. 191 1413–22
Gasic G J, Gasic T B and Stewart C C 1968 Antimetastatic effectsassociated with platelet reduction Proc. Natl Acad. Sci. USA61 46–52
Goldman A J, Cox R G and Brenner H 1967 Slow viscous motion ofa sphere parallel to a plane wall: 2. Couette flow Chem. Eng.Sci. 22 653–60
Hammer D A and Apte S M 1992 Simulation of cell rolling andadhesion on surfaces in shear flow: general results and analysisof selectin-mediated neutrophil adhesion Biophys. J.63 35–57
Hanley W, McCarty O, Jadhav S, Tseng Y, Wirtz Dand Konstantopoulos K 2003 Single molecule characterizationof P-selectin/ligand binding J. Biol. Chem. 278 10556–61
Hanley W D, Napier S L, Burdick M M, Schnaar R L, Sackstein Rand Konstantopoulos K 2006 Variant isoforms of CD44 are P-and L-selectin ligands on colon carcinoma cells FASEB J.20 337–9
Hanley W D, Wirtz D and Konstantopoulos K 2004 Distinct kineticand mechanical properties govern selectin–leukocyteinteractions J. Cell Sci. 117 2503–11
Harada N, Mizoi T, Kinouchi M, Hoshi K, Ishii S, Shiiba K, SasakiI and Matsuno S 2001 Introduction of antisense CD44S CDNAdown-regulates expression of overall CD44 isoforms andinhibits tumor growth and metastasis in highly metastatic coloncarcinoma cells Int. J. Cancer 91 67–75
Hashino J, Fukuda Y, Oikawa S, Nakazato H and Nakanishi T 1994Metastatic potential of human colorectal carcinoma SW1222cells transfected with cDNA encoding carcinoembryonicantigen Clin. Exp. Metastasis 12 324–8
12

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
Helenius J, Heisenberg C P, Gaub H E and Muller D J 2008Single-cell force spectroscopy J. Cell Sci. 121 1785–91
Huang J, Chen J, Chesla S E, Yago T, Mehta P, McEver R P, Zhu Cand Long M 2004 Quantifying the effects of molecularorientation and length on two-dimensional receptor–ligandbinding kinetics J. Biol. Chem. 279 44915–23
Huang J, Zarnitsyna V I, Liu B, Edwards L J, Jiang N, Evavold B Dand Zhu C 2010 The kinetics of two-dimensional TCR andpMHC interactions determine T-cell responsiveness Nature464 932–6
Jadhav S, Bochner B S and Konstantopoulos K 2001 Hydrodynamicshear regulates the kinetics and receptor specificity ofpolymorphonuclear leukocyte–colon carcinoma cell adhesiveinteractions J. Immunol. 167 5986–93
Jadhav S, Eggleton C D and Konstantopoulos K 2005 A 3Dcomputational model predicts that cell deformation affectsselectin-mediated leukocyte rolling Biophys. J.88 96–104
Jadhav S, Eggleton C D and Konstantopoulos K 2007 Mathematicalmodeling of cell adhesion in shear flow: application to targeteddrug delivery in inflammation and cancer metastasis Curr.Pharmaceutical Des. 13 1511–1526
Jadhav S and Konstantopoulos K 2002 Fluid shear- andtime-dependent modulation of molecular interactions betweenpolymorphonuclear leukocytes and colon carcinomas Am. J.Physiol. Cell Physiol. 283 C1133–43
Kannagi R 1997 Carbohydrate-mediated cell adhesion involved inhematogenous metastasis of cancer Glycoconj. J. 14 577–84
Kansas G S 1996 Selectins and their ligands: current concepts andcontroversies Blood 88 3259–87
Karpatkin S, Pearlstein E, Ambrogio C and Coller B S 1988 Role ofadhesive proteins in platelet tumor interaction in vitro andmetastasis formation in vivo J. Clin. Invest. 81 1012–9
Kim Y J, Borsig L, Varki N M and Varki A 1998 P-selectindeficiency attenuates tumor growth and metastasis Proc. NatlAcad. Sci. USA 95 9325–30
Kong F, Garcia A J, Mould A P, Humphries M J and Zhu C 2009Demonstration of catch bonds between an integrin and itsligand J Cell Biol. 185 1275–84
Konstantopoulos K and Thomas S N 2009 Cancer cells in transit:the vascular interactions of tumor cells Annu. Rev. Biomed.Eng. 11 177–202
Krasik E F and Hammer D A 2004 A semianalytic model ofleukocyte rolling Biophys. J. 87 2919–30
Laubli H, Stevenson J L, Varki A, Varki N M and Borsig L 2006L-selectin facilitation of metastasis involves temporal inductionof Fut7-dependent ligands at sites of tumor cell arrest CancerRes. 66 1536–42
Lawrence M B, Kansas G S, Kunkel E J and Ley K 1997 Thresholdlevels of fluid shear promote leukocyte adhesion throughselectins (CD62L, P, E) J. Cell Biol. 136 717–27
Leppanen A, White S P, Helin J, McEver R P and Cummings R D2000 Binding of glycosulfopeptides to P-selectin requiresstereospecific contributions of individual tyrosine sulfate andsugar residues J. Biol. Chem. 275 39569–78
Long M A, Goldsmith H L, Tees D F J and Zhu C 1999 Probabilisticmodeling of shear-induced formation and breakage of doubletscross-linked by receptor–ligand bonds Biophys. J. 76 1112–28
Mannori G, Crottet P, Cecconi O, Hanasaki K, Aruffo A, Nelson RM, Varki A and Bevilacqua M P 1995 Differential colon cancercell adhesion to E-, P-, and L-selectin: role of mucin-typeglycoproteins Cancer Res. 55 4425–31
Mannori G, Santoro D, Carter L, Corless C, Nelson R Mand Bevilacqua M P 1997 Inhibition of colon carcinoma celllung colony formation by a soluble form of E-selectin Am. J.Pathol. 151 233–43
Marshall B T, Long M, Piper J W, Yago T, McEver R P and Zhu C2003 Direct observation of catch bonds involving cell-adhesionmolecules Nature 423 190–3
McCarty O J, Mousa S A, Bray P F and Konstantopoulos K 2000Immobilized platelets support human colon carcinoma celltethering, rolling, and firm adhesion under dynamic flowconditions Blood 96 1789–97
McCarty O J T, Jadhav S, Burdick M M, Bell W Rand Konstantopoulos K 2002 Fluid shear regulates the kineticsand molecular mechanisms of activation-dependent plateletbinding to colon carcinoma cells Biophys. J. 83 836–48
McEver R P and Cummings R D 1997a Perspectives series: celladhesion in vascular biology. Role of PSGL-1 binding toselectins in leukocyte recruitment J. Clin. Invest. 100 485–91
McEver R P and Cummings R D 1997b Role of PSGL-1 binding toselectins in leukocyte recruitment J. Clin. Invest.100 S97–103
McQuarrie D A, Russell M E and Jachimowicz C J 1964 Kinetics ofsmall systems: II J. Chem. Phys. 40 2914–21
Merkel R, Nassoy P, Leung A, Ritchie K and Evans E 1999 Energylandscapes of receptor–ligand bonds explored with dynamicforce spectroscopy Nature 397 50–3
Minami S, Furui J and Kanematsu T 2001 Role ofcarcinoembryonic antigen in the progression of colon cancercells that express carbohydrate antigen Cancer Res. 61 2732–5
Muller D J, Helenius J, Alsteens D and Dufrene Y F 2009 Forceprobing surfaces of living cells to molecular resolution Nat.Chem. Biol. 5 383–90
Napier S L, Healy Z R, Schnaar R L and Konstantopoulos K 2007Selectin ligand expression regulates the initial vascularinteractions of colon carcinoma cells: the roles of CD44v andalternative sialofucosylated selectin ligands J. Biol. Chem.282 3433–41
Nash G F, Turner L F, Scully M F and Kakkar A K 2002 Plateletsand cancer Lancet Oncol. 3 425–30
Nicholson M W, Barclay A N, Singer M S, Rosen S D andVan Der Merwe P A 1998 Affinity and kinetic analysis ofL-selectin (CD62L) binding to glycosylation-dependentcell-adhesion molecule-1 J. Biol. Chem. 273 763–70
Nieswandt B, Hafner M, Echtenacher B and Mannel D N 1999 Lysisof tumor cells by natural killer cells in mice is impeded byplatelets Cancer Res. 59 1295–300
Nimrichter L et al 2008 E-selectin receptors on human leukocytesBlood 112 3744–52
Palumbo J S, Talmage K E, Massari J V, La Jeunesse C M,Flick M J, Kombrinck K W, Jirouskova M and Degen J L 2005Platelets and fibrin(ogen) increase metastatic potential byimpeding natural killer cell-mediated elimination of tumorcells Blood 105 178–85
Park H U, Kim J W, Kim G E, Bae H I, Crawley S C, Yang S C,Gum J R Jr, Batra S K, Rousseau K, Swallow D M, SleisengerM H and Kim Y S 2003 Aberrant expression of MUC3 andMUC4 membrane-associated mucins and sialyl Le(x) antigenin pancreatic intraepithelial neoplasia Pancreas 26 e48-54
Patel K D, Moore K L, Nollert M U and McEver R P 1995aNeutrophils use both shared and distinct mechanisms to adhereto selectins under static and flow conditions J. Clin. Invest.96 1887–96
Patel K D, Nollert M U and McEver R P 1995b P-selectin mustextend a sufficient length from the plasma membrane tomediate rolling of neutrophils J. Cell Biol. 131 1893–902
Pawar P, Jadhav S, Eggleton C D and Konstantopoulos K 2008Roles of cell and microvillus deformation and receptor–ligandbinding kinetics in cell rolling Am. J. Physiol. Heart Circ.Physiol. 295 H1439–50
Pereverzev Y V, Prezhdo O V, Forero M, Sokurenko E Vand Thomas W E 2005 The two-pathway model for thecatch–slip transition in biological adhesion Biophys. J.89 1446–54
Pinedo H M, Verheul H M, D’amato R J and Folkman J 1998Involvement of platelets in tumour angiogenesis? Lancet352 1775–7
13

Phys. Biol. 8 (2011) 015013 L S-L Cheung et al
Piper J W, Swerlick R A and Zhu C 1998 Determining forcedependence of two-dimensional receptor–ligand bindingaffinity by centrifugation Biophys. J. 74 492–513
Poppe L, Brown G S, Philo J S, Nikrad P V and Shah B H 1997Conformation of sLe(x) tetrasaccharide, free in solution andbound to E-, P-, and L-selectin J. Am. Chem. Soc.119 1727–36
Rosen S D 2004 Ligands for L-selectin: homing, inflammation, andbeyond Annu. Rev. Immunol. 22 129–56
Sarangapani K K, Yago T, Klopocki A G, Lawrence M B,Fieger C B, Rosen S D, McEver R P and Zhu C 2004 Low forcedecelerates L-selectin dissociation from P-selectin glycoproteinligand-1 and endoglycan J. Biol. Chem. 279 2291–8
Satomura Y, Sawabu N, Takemori Y, Ohta H, Watanabe H, Okai T,Watanabe K, Matsuno H and Konishi F 1991 Expression ofvarious sialylated carbohydrate antigens in malignant andnonmalignant pancreatic tissues Pancreas 6 448–58
Shao J Y, Ting-Beall H P and Hochmuth R M 1998 Static anddynamic lengths of neutrophil microvilli Proc. Natl Acad. Sci.USA 95 6797–802
Somasiri A et al 2004 Overexpression of the anti-adhesinpodocalyxin is an independent predictor of breast cancerprogression Cancer Res. 64 5068–73
Somers W S, Tang J, Shaw G D and Camphausen R T 2000 Insightsinto the molecular basis of leukocyte tethering and rollingrevealed by structures of P- and E-selectin bound to SLe(X) andPSGL-1 Cell 103 467–79
Taylor A D, Neelamegham S, Hellums J D, Smith C W andSimon S I 1996 Molecular dynamics of the transition fromL-selectin- to beta 2-integrin-dependent neutrophil adhesionunder defined hydrodynamic shear Biophys. J.71 3488–500
Tees D F, Waugh R E and Hammer D A 2001 A microcantileverdevice to assess the effect of force on the lifetime ofselectin–carbohydrate bonds Biophys. J. 80 668–82
Thomas S N, Schnaar R L and Konstantopoulos K 2009aPodocalyxin-like protein is an E-/L-selectin ligand on coloncarcinoma cells: comparative biochemical properties ofselectin ligands in host and tumor cells Am. J. Physiol. CellPhysiol. 296 C505–13
Thomas S N, Tong Z, Stebe K J and Konstantopoulos K 2009bIdentification, characterization and utilization of tumor cell
selectin ligands in the design of colon cancer diagnosticsBiorheology 46 207–25
Thomas S N, Zhu F, Schnaar R L, Alves C S and KonstantopoulosK 2008a Carcinoembryonic antigen and CD44 variant isoformscooperate to mediate colon carcinoma cell adhesion to E- andL-selectin in shear flow J. Biol. Chem. 283 15647–55
Thomas W E, Trintchina E, Forero M, Vogel V and Sokurenko E V2002 Bacterial adhesion to target cells enhanced by shear forceCell 109 913–23
Thomas W E, Vogel V and Sokurenko E 2008b Biophysics of catchbonds Annu. Rev. Biophys. 37 399–416
Varki A 1997 Selectin ligands: will the real ones please stand up?J. Clin. Invest. 100 S31–5
Walcheck B, Moore K L, McEver R P and Kishimoto T K 1996Neutrophil–neutrophil interactions under hydrodynamic shearstress involve L-selectin and PSGL-1. A mechanism thatamplifies initial leukocyte accumulation on P-selectin in vitroJ. Clin. Invest. 98 1081–7
Wielenga V J, Heider K H, Offerhaus G J, Adolf G R, Van Den BergF M, Ponta H, Herrlich P and Pals S T 1993 Expression ofCD44 variant proteins in human colorectal cancer is related totumor progression Cancer Res. 53 4754–6
Yago T et al 2008 Platelet glycoprotein Ibalpha forms catch bondswith human WT vWF but not with type 2B von Willebranddisease vWF J. Clin. Invest. 118 3195–207
Yago T, Wu J, Wey C D, Klopocki A G, Zhu C and McEver R P2004 Catch bonds govern adhesion through L-selectin atthreshold shear J. Cell Biol. 166 913–23
Yao L, Pan J, Setiadi H, Patel K D and McEver R P 1996 Interleukin4 or oncostatin M induces a prolonged increase in P-selectinmRNA and protein in human endothelial cells J. Exp. Med.184 81–92
Zhu C, Lou J and McEver R P 2005 Catch bonds: physical models,structural bases, biological function and rheological relevanceBiorheology 42 443–62
Zhu C, Yago T, Lou J, Zarnitsyna V I and McEver R P 2008Mechanisms for flow-enhanced cell adhesion Ann. Biomed.Eng. 36 604–21
Zollner O, Lenter M C, Blanks J E, Borges E, Steegmaier M,Zerwes H G and Vestweber D 1997 L-selectin from human, butnot from mouse neutrophils binds directly to E-selectin J. CellBiol. 136 707–16
14