bacteriophage lambda dna maturation
TRANSCRIPT

.I. Mol. Riol. (1981) 145, 375404
Bacteriophage Lambda DNA Maturation
The Functional Relationships Among the Products of Genes Nul, A and FZ
HELIO~ MURIALDO~, WENDY L. FIFE, ANDREW BECKER
Department of Medical Genetics l’niversity of Toronto, Toronto, Ontario, (‘anada
M5S IA8
AND
MICHAEL FEISS AND JOHN YOCHEM$
Department of Microbiology, College of Medicine T’niversity of Iowa, Iowa City, Iowa 52242, U.S.A.
(Received 12 May 1980)
The FI gene of bacteriophage h functions in head assembly, but its exact role is not well understood. FZ mutants are leaky, producing between @l and @5 viable particles per infected cell. In order to investigate the function of the FI product (gpF1) in &o, mutants of h were isolated that are able to grow in the absence of gpF1. These mutants, called fin (for FI independence) map in the region of gene Nul and the beginning of gene A.
Proteins made in cells infected with the fin mutants were labelled with [35S]methionine and analysed by polyacrylamide gel electrophoresis. In addition, the levels of activity of the A product were measured in the in vitro DNA packaging assay. As a result of these experiments, the fin mutants can be classified in two groups. Upon infection, fin mutants of one group selectively produce three to fivefold more gpA than do wild-type phage. fin mutants of the second group do not overproduce any A late gene product detectable by the autoradiographic technique.
gpA overproducers can also be isolated by selecting for hAam Warn phages that can plate on a weak suI1 cell strain. The mutation responsible for this pseudoreversion is called Aop and maps in the Nul-A region. Aop is also a fin mutation, since its presence in hFI- enables it to plate on non-permissive hosts.
Therefore, it seems that one condition sufficient for normal growth of FI- phage ix the overproduction of gpA. The nature of the fin mutations that do not result in gpA overproduction is discussed.
1, Introduction
The bacteriophage h genome is a linear DNA duplex of about 49,000 base-pairs (Daniels et al., 1980) with its two 5’ ends extending out as single strands of 12
t Author to whom correspondence should be addressed. $ Present address: Department of Genetics, University of Washington. Seattle. Wash. 98195, U.S.A.
375 (~2%2836/81/02037.%30 $02.00/O ?Y 1981 Academic Press Inc. (London) Ltd.

376 H. MURIALDO ET AL.
nucleotides. These terminal extensions are complementary and can anneal to convert the linear molecule into a ring (Wang & Davidson, 1966: Wu & Taylor, 1971; Hershey & Burgi, 1965). Soon after injection into the bacterium, h DNA indeed circularizes (Bode & Kaiser, 1965; Ogawa Oz Tomizawa, 1967) and the two terminal phosphodiester bond interruptions are closed by ligase action (Gellert, 1967). The duplex DNA derived from the cohesive ends in this way is called cos, for cohesive-end site (Emmons, 1974).
Replication of these circular molecules gives rise to daughter duplex circles during the early stages of the latent period (Tomizawa & Ogawa, 1968). Later, the rolling-circle mode of replication takes over and concatemers of h chromosomes are generated (Carter & Smith, 1970; Gilbert & Dressler, 1968; Enquist & Skalka, 1973: Skalka et al., 1972; McClure et al., 1973; Wake et al., 1972). These concatemers are the substrates from which monomeric chromosomes are cut and packaged into preformed proheads (Emmons, 1974: Hohn & Hohn, 1974; Kaiser et al., 1975). The cutting event is known as the ter (for terminase) reaction (Mousset & Thomas, 1969). In this reaction, the single-stranded ends of the h chromosome are regenerated by the introduction of two specific single-strand breaks, placed 12 nucleotides apart on opposite strands at cos.
Heads Tails -- NUI F& J ii-u red cl S I ---a_ I --- I I
-----A_____
I ----ma_
---me_
FIG. 1. Genetic map of bacteriophage h. The region containing the head genes is expanded. The length of the genes is proportional to the molecular weight of the polypeptides coded by them, as estimated by mobility in sodium dodecyl sulfate/polyacrylamide gel electrophoresis (Echols & Murialdo, 1978). The products of genes Nul and W have not yet been identified, although approximate estimates of their sizes exist (Echols & Murialdo, 1978; Weisberg et al., 1979).
In vivo, the ter reaction is coupled to DNA packaging into proheads. Thus, when prohead morphogenesis is blocked by mutations in genes E, R, (7, or N113 (Fig. 1) or the host gene groE, cos cleavage does not take place (Dove, 1966; MacKinlay & Kaiser, 1969; Wake et al., 1972: McClure et al., 1973; Georgopoulos et al., 1973). Mutations in four other h genes, Nd, A, D and PI (Fig. 1 ), similarly lead to an accumulation of uncut concatemers, even though competent proheads are present (Dove, 1966 ; MacKinlay & Kaiser, 1969; Skalka et al., 1972 : Boklage et al., 1973 ; Becker et al., 19776).
Of these four latter gene products, gpD is a major structural component of the mature phage particle (Murialdo & Siminovitch, 1971 : Casjens & Hendrix, 1974) and is added to an expanded capsid shell at about the time of the DNA encapsulation (Kaiser et al., 1975; Sternberg & Weisberg, 1977b). It has been shown that gpD is not a component of the nuclease that cuts h concatemers at cos during

LAMBDA DNA MATURATION 377
packaging. Its role in cos cutting seems to be indirect through its effect on head stability (Sternberg & Weisberg, 1977b).
The role of the other three gene products involved in cos cutting has been examined both in vivo and in vitro. The A gene product acts transiently and is required for DNA packaging into proheads. gpA provides a specific function in the initiation of DNA packaging, determining which DNA can enter the prohead (Sternberg & Weisberg, 1977a). During DNA packaging and maturation in vitro. gpA interacts with concatemers of h DNA to form a DNA-gpA complex. This complex then binds proheads to form a DNA-gpA-prohead intermediate which can be converted to infectious phage particles upon the addition of gpD, gpW, gpFI1, and tails (Becker et al., 1977aJ). It has also been shown in vitro that gpA is an essential component of the cos cutting enzyme (terminase) (Wang & Kaiser, 1973 : Becker & Gold, 1978). For its packaging and nucleolytic activities, gpA requires the function of gpNu1. The exact role of gpNu1 in these reactions is still uncertain: it may be a subunit of the terminase or it may modify gpA to render it active (Becker & Gold, 1978: Becker et al., 1977b).
Although gpF1 is essential to achieve a normal burst size in vivo, amber mutants in FI are much more leaky than amber mutants in other head morphogenetic genes. Furthermore, gpF1 action is not normally required for DNA maturation and packaging in vitro (Becker et al., 19773), which occurs efficiently in the presence of h FIam-infected cell extracts. The FI-independent packaging of A DNA in vitro occurs even when h deletion mutants reaching into the FI gene are used to prepare the packaging extracts (cited in Hohn & Katsura, 1977) or when double FI amber mutants are used for the same purpose (Benchimol, personal communication), indicating that the leakiness in vitro is not due to low levels of gpF1 present in XFZam,-infected cell extracts as a result of readthrough past the FI amber site in /*i/lo.
One peculiarity of in vitro packaging is that cos cutting occurs at the earliest step that can be measured, namely the binding of gpA to DNA concatemers, even before proheads are added (Becker & Gold, 1978). Recently, Benchimol et al. (1978) have shown that single-stranded DNA &haves as an inhibitor of cos cutting in this early step, and that the inhibition of tLe ter reaction by single-stranded DNA is reversed by the addition of gpF1 to the reaction mixture. They have also shown that the inhibitory effect of single-stranded DNA on terminase-catalysed cos cutting can be overcome by the addition of an excess amount of terminase. Based on these observations, and on the known ability of h to carry out limited growth in vivo in the absence of the FI gene product (Dove, 1966; Weigle, 1966: Boklage et al., 1973), we reasoned that if it were possible to construct phage mutants that overproduce terminase upon infection, the requirement of gpF1 might be bypassed. Alternatively, if it were possible to isolate second-site revertants of FI amber mutants, it might be expected that these suppressors would map in the genes specifying terminase (Nzrl, A) and that some might be gpA overproducers. In this way, a relationship between gpF1 and the component(s) of the terminase enzyme could be revealed as operating in vivo. The present paper describes the results of such a search.

378 H. MURIALDO ET AL.
2. Materials and Methods
(a) Media and buffers
L broth is 10 g Bacto Tryptone (Difco), 5 g yeast extract and 5 g NaCl/‘l of distilled water neutralized with NaOH. TB is 10 g Tryptone, 5 g NaCl, 1 mg of thiamine hydrochloride/l of distilled water (Campbell, 1961). RM medium is 18.7 mM-NH,CI, 1 mM-MgSO,, 20 mM-KCl, @l mM-Call,, 3 PM-FeC13, 494 mM-Na,HPO,, 22 mM-KH,PO,, 22 mllr-glycerol and 04% maltose. RM-Mg is RM medium containing 10 mM-MgSO, instead of 1 IIlM. Lambda diluent (h-dil) is 10 mM-MgSO,, 10 mM-TrisHCl (pH 7.4). The solid medium used for bacterial colony formation and phage plaque assays has been described (Campbell, 1961; Murialdo & Siminovitch, 1971).
Buffer A is 20m~-Tris-HCl (pH 8.0), 1 mM-EDTA, 3 mm-M&l,, 5 mM-2-mer- ceptoethenol.BufferBislO”/O (w/v)sucrosein005 M-Tris.HCl(pH 7.4).SMAsolutionis60 mM- spermidine, 18 mM-Mgcl,, 15 mM-ATP, 30 mM-2-mercaptoethanol in 5 mM-TrigHCl (pH 7.4).
(b) Bacterial strains
The Escherichia coli K12 derivatives used are listed in Table 1. Strains 594 or R594 are SmR derivatives of W3350 in which the degree of leakiness of amber mutations is at least one order of magnitude less than in W3350 (Gorini, 1971). QR23 carries several mutations: relevant to this work is the weakening of its au11 suppressor phenotype due to a streptomycin resistance mutation (Couturier et al., 1964; Gorini et al., 1966; Otsuji $ Aono, 1968). Whereas the efficiency of suppression of an amber codon is between 10 and 30% for the suII in C600 (or TCSOO) (Garen et al., 1965; Kaplan et aZ., 1965; Sol1 & Berg, 1969), it is about 1 to 2% in QR23 in the absence of streptomycin (Gorini et al., 1966). The first 7 strains listed in Table 1 will be referred to colloquially as follows : sup0 (W3350), tight sup0 (594 and R594), au11 (CSOO and TC600), au111 (QD5003 and MF327), weak au11 (QR23), and weak suII1 (MF848).
(c) Phagr strains
AFIawd,, , AFIam471cI,,, FIam730cI,,, ~I~amso4cI,l, hWam403cId and hAamg,,Wam,,,cI,, were a gift from J. S. Parkinson (Parkinson, 1968), and hAam,, and hAawl were obtained from A. Campbell (Campbell, 1961). They were stored in the prophege state in TC600, and lysates were prepared from these lysogens by thermal induction (Sussman & Jacob, 1962), or by U.V. light induction (Lwoff et al., 1950). A derivative, AAam9,4Wam403cI,,, was obtained from a cross between /\Aam9,4Wum4,,3cI,, and heI,,,. The lysate wa.s plated on QD5003 at 38°C and clear plaques were replicated onto lawns of QD.5003 (master plate) and 594. The plates were incubated at 38°C overnight. One isolate that did not grow on 594 was cloned twice, checked by marker rescue and used to lysogenize TC600. hAts,,14Eam,,5~Z,,, was constructed by means of a cross between hAts,,,cI,,,Sam, obtained from C. R. Fuerst (McClure & Gold, 1973) and AEam,,,cZ,5, (from Parkinson, 1968). hcl,, (Kaiser, 1957) and a hvir isolate (obtained from C. R. Fuerst), both from our collection, were used to check for lysogens. himm2’cI and himm2’ were obtained from D. Kaiser (Liedke-Kulke & Kaiser, 1967). h~&~cI,,~ is described by Signer & Weil (1968). 4801am, from Sato et al. (1968) was a gift from N. Sternberg. A h-480 hybrid derivative, no. 410 = r\hy~801am3red3cIs,,, contains head genes of $80 and the tail specificity of X. It was made in a cross between 4801am, and /\red,cI,,,. Phages hred,imm”cI XWam 403redds57 and hAamJ2red,cI,,, the aforemeniioned phages. XAam12, b,3,redam270cI
were constructed by crosses between s5, was obtained as a recombinant from
a cross between &mm” Aam,21b,,, and hcIs5, redam,,, (Feiss & Bublitz, 1975). himm” Aam ,2,b538 was from a cross between hilam,,, (Campbell, 1961) and himm2’b,38 (Feiss & Bublitz, 1975). /\imm434TcI,o, is a clear derivative of /\imm4j4T (Pirrotta & Pteshne, 1969).

LAMBDA DNA MATURATION
(d) Plaque-forming method
379
A 3-ml culture of cells in L broth was grown to stationary phase at 37°C’ in a shaker water bath. The cells were then diluted to about 5 x lO*/ml (about 5-fold) with h-dil. A volume of @2 ml was added to tubes containing phage (0.1 ml) and adsorption allowed to occur at 37°C for 15 min. Melted top agar (3 ml) kept at 46°C was then added to the complexes and the mixture poured into Petri dishes containing about 60 ml of solid bottom agar. After the top agar had solidified, the plates were incubated overnight at appropriate temperatures.
(e) Preparation of phage lysates
Whenever it was possible, phage stocks were grown by thermal induction of lysogens (Lieb, 1966). For this, 3 ml cultures of cells growing in L broth at 32°C were induced when they reached a cell concentration of about 2 x 10s to 3 x 10s (by direct visual estimation). For induction, the culture tube was incubated with shaking at 45°C for 15 min and then transferred to a 37°C shaker water bath until lysis. After vortexing with a few drops of CHCl,, the lysates were cleared free of cell debris by centrifugation for 15 min at 8000 revs/ min in the SE12 rotor of the Sorvall centrifuge.
For lytic growth of phage, a lysis plaque was picked with a Pasteur pipette and transferred into a tube containing 3 ml of L broth supplemented with MgSO, to a final concentration of @02 M. Cells (20 ~1) in stationary phase (about 5 x 10’ cells) were added if the plaque was big ; if, however, indicator cells were carried from the plate, as in the case of small plaques, then no extra cells were added. After a few hours of incubation at 32°C or 38”C, depending on the phage genetic make-up, lysie was observed. The lysate was chloroformed and cleared of cell debris as above.
(f) Construction of lysogens
If the host was permissive for the phage, then the phage was simply plated at the appropriate temperature and lysogens were isolated from the turbid centre of an “average” looking plaque. Lysogens were cloned at least 3 times, checking several isolates each time for immunity by cross-streaking through hcZ,’ and hvir to make sure that the lysogens were stable (Lederberg & Lederberg, 1953).
If the host was not permissive for growth of the phage, cells were diluted in &dil to a concentration of approx. log/ml and infected with phage at a multiplicity of infection of 5. Adsorption was at 30°C for 20 min. The complexes were then diluted l/lo4 and @l ml was spread on a Petri dish containing about 30 ml of solid medium. After incubation for 20 h at 31”C, individual colonies were checked for immunity by the cross-streaking technique. Lysogens were cloned and re-checked as described above.
.Permissive lysogens were characterized after induction of the prophage. The markers in the prophage were checked in a marker rescue test by spotting the lysate on a series of sup0
lysogens carrying known prophage mutations. Non-permissive lysogens were characterized by spotting a series of phages carrying known mutations on a lawn of the lysogen.
(g) Marker rescue spot test and phage crosses
Lysates of known X amber mutants (about 10s phage/ml) were prepared, and 30 ~1 of such a lysate was used in spot-testing. Spotting was done on a top agar overlay containing about 2 x 10’ total cells, consisting of a 1 : 1 mixture of 594 cells and the lysogenic cells to be tested. After the spot had dried, the plates were incubated at 42°C if the prophage was cZs5’. If the prophage was cl+ or cZ,~, the lawn was irradiated with ultraviolet light at an incident dose of .500 ergs/mm’. After induction, the plates were incubated at 38°C. A clear zone of lysis indicates that there is recombination with the production of non-defective phages. Absence of lysis, or presence of only a few plaques, presumably revertants, indicates that both prophage and superinfecting phage are mutated in the same allele. Positive and negative

TABL
E 1
List
of
bac
teria
l st
rain
s
Stra
in
Prop
ertie
s R
efer
ence
1 w
3350
2
594
and
R59
4
3 C
600
and
TC60
0 4
QD
5003
5
MF3
27
6 Q
R23
7
MF8
48
8 w
3805
9
AB
1157
sup0
, ga
l. O
ur
oolle
ctio
n C
ampb
ell
(196
1)
sup0
, S
m’.
Our
co
llect
ion
Cam
pbel
l (1
961)
; W
eigl
e (1
966)
w
pEf
(~11
). O
ur
colle
ctio
n Ap
pley
ard
( 195
4)
supF
+ (~
~111
). O
ur
colle
ctio
n Ya
nofs
ky
& Ito
(1
966)
su
pF+,
H
frH,
laca
m,
trpam
, th
y.
Obt
aine
d fro
m
S.
Adhy
a su
pE+,
S
m’.
From
I.
Her
kow
itz
Man
ly
et a
l. (1
969)
su
pC,
HfrH
. Fr
om
D.
H.
Wal
ker
sup0
, ga
l E
,, Ad
ler
& Te
mpl
eton
(1
963)
nu
pE44
Ba
chm
ann
(197
2)
Thes
e st
rain
s w
ere
wed
fo
r pl
atin
g,
lyso
gen
cons
truct
ion
and
isol
atio
n of
ps
eudo
reve
rtant
s
10 M
F611
11 p
3478
12 N
S428
sup0
, re
cA,.
This
st
rain
w
as
Feis
s et
al.
(197
7)
used
to
perfo
rm
cros
ses
in t
he a
bsen
ce
of
gene
raliz
ed
reco
mbi
natio
n th
y,
polA
,. Th
is
stra
in
was
us
ed
Del
ucia
&
Cai
rns
(196
9)
to
chec
k fo
r th
e re
d3
mar
ker
in
reco
mbi
nant
ph
ages
YU
PO,
recA
(h
Aam
,, b,
red3
cZ,,,
Sam
,) St
ernb
erg
et al
. (1
977)
us
ed a
s a
sour
ce
of p
rohe
ads
for
in
vitro
D
NA
pack
agin
g as
says

13 M
S504
su
p0.
A(ga
lk-h
(‘).
A de
letio
n de
rirat
,ive
of a
h ly
soge
n 14
NT3
805
(hdg
al,c
Z*5,
) 15
W38
0.5
(Adg
al,,
imm
2’)
I6
w33
50
(Adg
als
* 5 im
my
17 ~
~380
5(hd
gaZ,
,,red
,cZ,
,,)
1X U
7380
5(hd
gaZ~
reda
m,,~
cl,,7
nin5
) 19
W38
05(h
dgal
,ooe
Z,,,)
sup0
, A(
hAam
;;v
;: w
;~w
;;;;ft
sup0
, A(
hAam
, , -
att)
sup0
, A(
Aam
,, -a
tt).
Isol
ated
by
Adl
er
& Te
mpl
eton
(1
963)
.
Shap
iro
& Ad
hya
(196
9)
Feis
s Br
Vam
pbel
l (1
974)
Th
is
wor
k C
ampb
ell
(196
1) :
Mur
iald
o R
r Sirn
inov
itch
(197
2)
Cam
pbel
l (1
961)
: M
uris
ldo
& Si
min
ovitc
h (1
972)
Fe
iss
& C
ampb
ell
(197
4)
Ray
&
Pear
son
(197
4)
20 S
erie
s 59
4 (h
amel
,,)
and
594
(ham
) 21
R
594
(hFZ
am,,,
imm
2’)
22 R
594
(XFZ
am,&
mm
2’)
The
prop
hage
in
ea
ch
stra
in
c+rry
a
Mur
isld
o &
Beck
er
(197
&z)
mut
atio
n in
a l
ate
gene
. O
ur
colle
ctio
n Th
e pr
opha
ge
in
stra
ins
nos
21 a
nd
22
This
w
ork
wer
e co
nstru
cted
by
rec
ombi
natio
n us
ing
stan
dard
pr
oced
ures
w
ith
phag
es
liste
d un
der
phag
e st
rain
s Th
ese
stra
ins
wer
e us
ed f
or
mar
ker
resc
ue
test
s to
ch
eck
for
the
pres
ence
of
am
ber
mut
atio
ns
23
159T
-(XEt
s,,e
Z,,,S
am,)
24 Y
lO-l(
hlJt
s,,,e
Z,,,)
25
QD
5003
(h
Aam
,3cZ
,,,)
26 5
94(h
eZ,,)
28
TC6O
O(h
Aam
,, im
m2L
rIt.~
5Sla
m7)
29 (
‘600
(A
Earn
,,)
Obt
aine
d fro
m
C.
R.
Fuer
st
McC
lure
&
Gol
d (1
973)
O
btai
ned
from
C
. R
. Fu
erst
O
ur
colle
ctio
n.
.Jar
a &
Mur
iald
o (1
975)
M
ade
with
a
phag
e se
nt
by
J.
S.
Park
inso
n (1
968)
Pa
rkin
son
Con
stru
cted
by
co
nven
tiona
l m
etho
ds
star
ting
from
X
Aam
,,cZ,
,57S
7,
from
ou
r co
llect
ion,
an
d hi
mm
”cZt
s,
isol
ated
by
In
okuc
hi
(see
Roe
hrda
nz
& D
ove.
19
77),
a gi
ft fro
m
.J. S
haw
A
gift
from
A
. C
ampb
ell
Cam
pbel
l (1
961)

382 H. MURIALDO ET AL
controls were always included in the test. The same test was also used to check the markers in a mutant phage lysate against a series of lysogens with known prophage mutations.
For phage crosses the method of Parkinson (1968) was used.
(h) Measurement of phuge growth
Several hFI mutants and a AFII (used as a control) were examined for phage production under non-permissive conditions. R594 cells were grown in TB+@2% (w/v) maltose at 37°C to about 1 x 10s cells/ml. The cells were spun down at low speed and resuspended in 62 vol. 001 M-MgSO,. Cell number was determined in a Petroff-Hausser counter and the cells were infected at various multiplicities. Adsorption was for 15 min at room temperature, followed by the addition of anti-l\ serum (K = 1.7 min-’ ). The infected cells were diluted in TB and incubated with shaking at 37°C. 20 min after the beginning of the incubation, the complexes were plated on MF327. After an additional 50 min of incubation at 37”C, CHC13 was added to the cultures to ensure lysis and the phage were plated on MF327 after suitable dilution.
(i) Phage complementation
Four h&al prophages were examined for their ability to complement amber mutations in the 1 gene of $80 and the A gene of bacteriophage A. Depending on the immunity region of the hdgal prophage being studied, 1 of 2 protocols was followed.
If the prophage carried a heat-inducible repressor (hcfs,,), cells were grown at 30°C in TB + 0.2% maltose to - 1 x 10s cells/ml. The cells were then resuspended in 92 vol. 601 M-
MgSO, and cell number was determined by counting in a Petroff-Hausser counter. The cells were infected at a multiplicity of infection of 5, and adsorbed with phage for 15 min at room temperature. After adsorption, anti-h serum (K = 1.7 min-‘) was added to inactivate free phage and the cells were diluted l/10’ in TB. Cultures were incubated 10 min at 42°C to induce the prophage and then transferred to 37°C for 60 min. CHCl, was added to complete lysis and the yield of infecting phage determined by titering on C600.
If the dgal prophage carried a wild-type immunity region (imm434), cells were grown in TB plus 92% maltose at 37°C to - 1 x 10s cells/ml, resuspended in an equal volume of 601 M-
MgSO,, irradiated, resuspended in TB plus 92% maltose, and grown for an additional 25 min at 37°C to allow induction of the prophage. Infection with the phage then followed according to the protocol for the hcI ss7 dgaZ lysogens with a final incubation for 70 min at 37°C. To determine the yield of amber phage grown in the absence of the hdgal prophages, infections in non-lysogenic analogues of the host bacteria were also done.
(j) Marker rescue of the jin+ allele iz liquid culture
The lysogens (or non-lysogens, as controls) were grown in TB plus 62% maltose at either 30 or 37°C (depending on the immunity region of the prophage) to 1 x 10s cells/ml, resuspended in 92 vol. 901 M-MgSO,, and infected with the appropriate phage at a multiplicity of infection of 2. Cell number was determined by counting in a Petroff-Hausser bacteria counter. After adsorption for 15 min at room temperature, cells were diluted l/100 into 0.01 M-MgSO,, irradiated, diluted l/100 into TB, and shaken at 37°C for 70 min. This was followed by CHCl 3 addition to complete lysis. The lysates were plated on either C600 or AB1157. Small, scruffy plaques were picked into grid wells and printed on lawns of C600 and 594. The superinfecting phage carrying the fin mutation also carried 2 amber mutations in the FI gene. The fin mutation enables these phages to grow on sup0 or sull hosts. If a phage has picked up the fin+ allele it will make a small unhealthy plaque on C600 and no plaques on 594.
(k) Construction of phage strains
Construction of a phage with 2 amber mutations in FI. Whereas the right-most FI mutant (UIam730cI,,) was used by both groups, the left-most FI mutant used by the Iowa group was hl”Iam,,,cI,,, while the Toronto group used AFIam7sscI,1. Further tests have

LAMBDA DNA MATURATIOS 3X3
failed to show any recombination between the FIam471 and FIamTS5 markers. Therefore. t,hese 2 mutations are either in the same site, or they are extremely close. For the construction of the double amber FZ mutant, the 2 groups followed different procedures.
Iowa group procedure: hFam,,lcI,l was crossed with kFam730cI,, in C6OO. The cross was stimulated by ultraviolet irradiation (400 ergs/mm*). The frequency of crossing over between the FI markers was @5% as measured by plating the burst on R594 to score am+ recombinants. The lysate was plated on C600 and small plaques were picked and cloned on IvlF327 where they produce middle-size plaques. The plaques were picked into grid wells and rraplicated onto lawns of R594(himmz1 Flam 471) and R594(himm2’Flam,,,). The lawns were treated with ultraviolet light (400 ergs/mm’) to stimulate recombination and incubated at 37°C’. One well contained a recombinant phage that did not give ham+ recombinant)s on chither lawn. The FI double amber genotype of the recombinant was confirmed by crosses in liquid culture between the derivative and its parents, with crosses between the parents scsrving as the positive control. This isolate was designated hFZam,71am,30cI,,.
Toronto group procedure: For the construction of AFlam47,am730cl,,. two intermediate lthage recombinants were constructed first. These were hFlam,85 i’t~,,,~cI~~, and AEts,, FIam73,,cl,l. To construct the first intermediate, hFlam7,5cI,, was crossed with Xf~tS,0HCls5, in TC6OO as host and the lysate plated on TC6oO at 32°C’. Plaques were then rchplicated on 2 plates containing about 10’ seeded TC600 cells and on a 3rd plate seeded with an equal number of 594 cells. One of the plates with a TC600 lawn was incubated overnight at 32°C’ to serve as the master plate; the other was incubated at 42°C’ to check for t#emperature sensitivity. The 594 replica was incubated overnight at 32°C’ to check for isolates carrying the amber mutation. Of 286 plaques tested, one failed t,o grow on TO600 at 42°C and on 594 at 32°C. The recombinant, hFZam7,, I~ts,,,cl,,,. was cloned twice and used to make a TC6OO lysogen. This lysogen was then induced and the phage was checked for thus FI marker by marker rescue. The presence of the ts marker was checked by plating the lysat)e at 42°C’ on TV600 and QD5OO3. The efficiency of plating at 42°C‘ is < IO- 5 in comparison tjo the plating efficiency at 32°C’.
An analogous procedure was followed for the construction of hEtso,FIam7,,cf,, starting with the cross XEts,,cI,,, Sam, x XFIam,,o cl,, Of 1497 plaques tested, one carried both am and ts mutations. The isolate was purified and checked in a way analogous to th(B XFlanl,,,l!ts,,,cl,,, recombinant.
Next, the 2 intermediate phages, hFIam,,,7its,,,cI,,, and AEtuO,FIam,,,cI,,. werfl crossed using QD5003 as host and the lysate plated on QD5OO3 at 42°C. Under these conditions, both parents are unable to plate, and only recombinants between the Ets and 1’l.s markers (and ts+ revertants) are able to plate. Forty plaques were picked and used to make small tysate stocks. The lysates were checked by marker rescue, and 2 isolates that by this test scored as double amber mutants in gene FZ were retained. The isolates were clonrd twict=. and stored as prophages in QD5003.
(I ) Isolation of mutands able to grow in the absence of the gene FI product
Lysates were prepared by thermal induction of the two QD5OO3 (AFfam7,,am,,ocft/) tysogms. The 2 lysates were plated on QD5003 at 32°C and, from the centres of 3 plaques of I Iysatr, and 2 of the 2nd, lysogens were isolated and purified through 3 clonings. From these 5 lysogens, lysates were prepared by thermal induction and plated on 594 and W3350 at about 107 phage/platr. Only a few small plaques were found/plate. A total of 14 of them were purified and were stored as prophages in QD5003. Lysates were prepared from these lysogens to characterize the mutants as described in Results. The mutation that presumably allows them to plate on 594 or W3350 is called fin (for FZ independent). The isolation of fin mutants from hF lam 47,am730cZtZ was carried out in a similar manner, by plating about, IO’ phagcb/plate on a lawn of R594 and incubating at 37°C’.
(m) Isolation of a gpil o~wprodwxr
In several instances, phagr mutants regain the ability to grow under non-permissive

384 H. MURIALDO ET .4L.
conditions, not due to the reversion of the original mutation, but rather to the acquisition of a mutation in a different, 2nd gene (Floor, 1970: Sternberg, 1976). Such a phage will be referred to as a pseudorevertant and the suppressing mutation will be referred to as a 2nd. site mutation. One example has been the isolation of pseudorevertants of Dam mutants on a cell carrying a weak suII1 suppressor. The 2nd.site mutation lies in the -4 gene (Sternberg, 1976).
The isolation of gpA overproducers involves the selection of pseudorevertants of hAam mutants able to form plaques in a strain carrying a weak suI1 suppressor. The rationale for this approach is based on the premise that deficiency in gpA production in /\Aam-infected cells carrying a weak suI1 suppressor could be overcome by one of several mechanisms : (i) by enhancing the frequency of transcription of the gene which is mutated (promotor mutation), (ii) by increasing the frequency of translation (ribosome binding site mutation, (iii) by increasing the functional stability of the mRNA to a level such that the intracellular gpA concentration becomes adequate, (iv) by increasing the activity of gpA, or (v) by increasing the stability of gpA. This approach was followed, except that a phage carrying mutations in 2 different, but contiguous genes was used in order to avoid the possible selection of pseudorevertants with a Dam 2nd.site mutation (Sternberg, 1976).
hAamgl,W’am403cZ,,7 plates on QR23 with a frequency of 3 x 10m7 (efficiency of plating on TC600 is taken as 1). Four big and four medium sized plaques were picked, purified, and small lysates of each were prepared. Each of the 8 isolates still carried at least 1 amber mutation since none plated on 594. On further testing, the 4 large plaque formers proved to be A+ revertants but were still Warn 403. (On checking 2 other Warn mutants isolated by Parkinson (1968), it was shown that all of them plate on QR23 with the same efficiency as on TCSOO.) The 4 middle-sized plaque formers were still Aam9,4Wam403. They did not carry any defective mutations to the left of gene R as checked by marker rescue against a series of hdgal lysogens with left-arm deletions of various lengths. One of these 4 isolates called hilop,ilam,,,Wam,,,cls,, was selected for further study as described in Results.
(n) Preparation of phage stocks for radioactive label&g
For the preparation of phage stocks for radioactive labelling of h-induced proteins, 100 or sometimes 500 ml cultures of the lysogens in L broth were grown with vigorous shaking at 32°C until a cell density of about 3 x lO*/ml was reached. The prophage was induced by a 15 min pulse at 45°C and the cultures were further incubated with vigorous shaking at 37°C until lysis. If a lysogen was not available, the phage was grown lytically in a way similar to that described for the preparation of small phage stocks (see section (e), above). The starting cell concentration was about 2 x lO’/ml and the multiplicity of infection was 0.1. After lysis, a few ml of CHC13 and a few crystals of DNase were added and shaking was continued for 10 min at 37°C. The lysates were cleared of debris by centrifugation for 20 min at 7500 revs/min in the GSA rotor of the Sorvall centrifuge. Phage were then pelleted by centrifugation, either for 3 h at 21,000 revs/min in a Spinco 21 rotor, or for 4 h at 19,000 revs/min in a Spinco 19 rotor. The phage were resuspended by overlaying with 1.5 ml of X-dil and allowing diffusion overnight at 4°C. The suspension was collected. The remaining pellet was disrupted with 1 ml of h-dil and left on ice for 1 h. The 2 suspensions were then pooled and centrifuged at low speed to eliminate remaining cell debris. Finally, these phage suspensions were dialysed extensively against several changes of h-dil.
(0) Preparation of labelled extracts of infected cells
Cells were grown at 37°C in RM medium to a density of about 3 x lO*/ml. They were harvested by low-speed centrifugation and resuspended at a concentration of 109/ml in RM- Mg medium. The cells were then irradiated with ultraviolet light. The suspension (6 ml) was placed in a 109mm diameter Petri dish and irradiated for about 45 s at 90 ergs/s per mm’. Next, 0.5 ml of cells were infected at O”C, at a multiplicity of infection of 5 phage/cell. The mixtures were incubated for 20 min at O”C, diluted with 2 ml of warmed RM-Mg and incubated with shaking at 37°C. About 30 &i of 35S-labelled E. coli hydrolysate, prepared as

LAMBDA DNA MATURATION 3x.5
described by Crawford & Gesteland (1973), were added 25 min after the beginning of the incubation at 37°C. Incorporation was arrested 10 min later by the addition of 14 ml of RM medium in the form of crushed ice. The following operations were all carried out at 0°C‘.
After the addition of a drop of log-phase E. coEi cells to act as carrier during centrifugation 1 the cells were pelleted at 9000 revs/min for 15 min in the Sorvall SS34 rotor. The pellets were resuspended in 63 ml of sample buffer (Laemmli, 1970), transferred to Eppendorf tubes. and incubated in a boiling water bath for 3 min. The samples were loaded into the acrylamide gel wells immediately or stored at -20°C until the next day when. after thawing, they were loaded without further treatment.
For the pulse-and-chase type of experiment, cultures (2 ml) of infected cells were labelled with 75 &i of [35S]Met (Amersham Corp., SJ204) 24 min after the addition of warmed R?rl- Mg. This was followed by the addition of a 105-fold excess of cold Met 2 min later. Samples (400 ~1) were withdrawn 10, 150, 300 and 600 s thereafter into Eppendorf tubes containing 50 ~1 of lu-NaN,, the tubes were closed and submerged in liquid N,. The tubes were put on ice until the samples thawed, at which time the cells were collected by centrifugation in the Eppendorf centrifuge for 3 min in the cold and resuspended in 60 ~1 of sample buffer.
(p) Gel electrophoresis and radkmrtography
The method of Laemmli (1970) was used for electrophoresis with a stacking gel of 596 and a separating gel of 75% or 12.5%. The initial voltage was 40 V for 2 h and then 100 \ overnight. After completion of electrophoresis, the gels were washed with 2 changes of 500 ml of water for 20 min each (Murialdo, 1979), they were dried under vacuum and exposed to Kodak SB-5 X-ray film for 1 to 20 days. Scanning was performed in a Gilford spectrophotometer provided with a linear transport. Care was taken to ensure that the optical density of the bands was within the linear response of the film.
(q) =1ssay of g+ activity
The method of Becker & Gold (1975) was used (see also Murialdo & Becker. 197%) Preparation of prohead-donor extract: 600 ml of L broth were innoculated with strain
NS42X and the cells were grown at 32°C in a shaker to a cell concentration of about 7 x lO’/ml. For thermal induction of the prophage, the cells were incubated with shaking at 70°C until the temperature in the cell suspensions reached 43°C’. Subsequently, the flasks were shaken at 44°C for 15 min followed by cooling in an ice/water mixture until the temperature dropped to 38°C. The cells were incubated at 37°C for another 45 min: they were then chilled and spun down at 13,000 revs/min for 10 min. The pellets were resuspended in 0% ml of buffer A. Before sonication the volume was adjusted to about 2.5 ml with buffer A. Sonication of the concentrated suspension was done by 2 pulses of 10 s each in an ice/salt bath. The sonicates were cleared by centrifugation at 406Og for IO min. Extracts of lysogens to be tested for gpA activity were prepared in an identical way.
Preparation of LFT extracts: these extracts were prepared by the lysozyme-freeze-thaw method first described by Kaiser & Masuda (1973). The lysogen NS428 was grown, induced. and the cells collected by centrifugation as described above for the preparation of the sonicates. The pellets were subsequently resuspended in 0% ml of buffer B. The final volume of this mixture was about 1.5 ml. To 1 ml of this suspension, 0.05 ml of a lysozyme solution (2 mg/ml in 025 M-TrisHC1, pH 7.4) was added, and this was followed by freezing in liquid N, and shaking in a water bath at 37°C just to the point of complete thawing. The viscous extract was incubated on ice for 45 min, then 100~1 of SMA solution was added.
The mixture was then centrifuged at 35,000 revs/min in the 50Ti rotor of the Spinco ultracentrifuge for 25 min. The supernatant was used as the LFT extract, providing gpD, gpW, gpFLI and tail function to the assay (Becker et al., 19776).
Assay of gpA : one volume of prohead-donor extract was mixed with one volume of a gpA- donor extract (or a gp.4-donor extract diluted in buffer A) : 20 ~1 of this mixture was then added to a mixture of 30 ~1 of buffer A. 4 ~1 of SMA solution, and 4 ~1 of a solution

3% H. MURIALDO ET AL.
(185 pg/ml) of mature himm434Tc1700 DNA, a gift from M. Sumner-Smith. This mixture was incubated at 22°C for I5 min. For the 2nd stage of the reaction, 150~1 of LFT extract was added, followed by 60 min of incubation at 22°C. The phages assembled in this system were plated on 594 after appropriate dilutions. Phage yields are expressed as phage concentration in the final reaction mixture.
3. Results
(a) Rehaviow of FI mutants of lambda
A number of experiments were performed to examine the growth of FZ amber mutants after infection of sup0 cells. The results are given in Table 2. The number of infectious centers and the yield are surprisingly high. The values for FZ mutants are two orders of magnitude greater than the values obtained for other lambda head amber mutants. A typical result, included for comparison, is that obtained in an infection by AFZZam,,,. The results in Table 2 also show that the leakiness is not due to a particular FZam mutation: AFZam,,, and AFZam,,, give similar results. The leakiness is not affected by change in multiplicity, a result which could be expected for mutants defective in a late gene. Most importantly. AFZam 47,am730 is just as leaky as the single PI amber mutants, indicating that leakiness results from lambda’s partial independence of FZ function.
Table 2
Growth of hFIam strains in non-permissicr conditions
Phage Multiplicity Infective centers/ of infection cell Burst size
XFZZamso4cZ,, AFZam4,,eZ,, hFZam730cZ,,
WZam 47,am730cZ,,
10 < 04wi? 0.0% 10 0.13 0.13 10 0%
5 0.32 0.1 0%
10 023 0.11
(b) Isolation of mutants able to grow in the absence of gene FI product
AFIam 785am730cZ,, plates with an efficiency of 10Y6 on 594 and 3 x 10Y5 on W3350 (efficiency of plating on QD5003 = I), producing rather small plaques. Similarly AFZam,,,am,3,, cl,, plates at a frequency of 10m6 on R594. Since the frequency of revertants among most single amber mutants of h is usually of the order of 10e6 or less (Campbell, 1961; Jara & Murialdo, 1975), one would expect that the frequency of revertants in a double amber mutant would be of the order of lo- I2 (if the mutations revert independently). Therefore, the finding of phages able to plate on sup0 strains at a frequency of 10e6 or higher and the small size of the plaques formed, suggested that these plaques were not due to FI + revertants, but rather to phages carrying a second-site mutation that was called fin. A total of 19 such isolates was selected for study.

+++
+++
+++
+++
+t+
I ++
++++
+ I +
13

388 H. MURIALDO 397’ AL
In order to characterize the mutants, they were plated at different temperatures on sup0 strains and on other strains carrying different suppressor genes. The results of this analysis are presented in Table 3.
In general, the plaques are medium sized on QD5003 at 38”(!, small on TC600 and very small on 594 and W3350. At 31”C, the plaque size decreases on all the strains.
As can be seen, most fin mutants display some degree of temperature sensitivity : they are sensitive to high temperatures (ts), or to low temperatures (cs), or both (cts). This effect can be observed only in the absence of gpF1, that is, upon infection of sup0 cells. A partial cs character can also be observed on plating the FZ- fin isolates on SUII cells. fin mutant phages which are cold-sensitive will be designated fines; if heat-sensitive, fints. Mutant fin, ,2 will be designated fin&, ,2.
(c) Genetic stru~cture of jin mutants
We presumed that the fin mutations would lie in the head-gene region but outside the FZ gene. We further presumed that the fin mutations might lie in the Nul-A region, because gpF1 seems to act at the same stage in head assembly as gpA (and gpNu1) (Benchimol et al., 1978). Genetic crosses were performed to confirm the expected structure of hfin FZam,7, am73,,cZ,l isolates.
fintse F-zbm74,730 aft red3 imm2’cI I II I I I
--------_- ------------,
I
Warn403
L------- ____- -----
aft red3 a357
FIG. 2. Phage croons to determine whether the fin, mutation lies on the right or left arm.
The first cross was performed to show that fin mutations mapped in the morphogenetic region of the lambda chromosome. The cross, diagrammed in Figure 2, was between a red3imm2’cZ derivative of hfintssFIam471am730cZtl and hWam403red3cZ,,, and was carried out in the recA, sup0 strain, MF611. The burst was plated on C600 at 33°C and turbid plaques (cl,,,) were picked and tested for ability to grow on R594. Approximately 1% of the turbid progeny of the cross were able to grow on R594 and hence were Afints,FZam,,,am,,ored,cZ,,7 recombinants. Because only site-specific recombination took place in the cross (controls not presented), the fin marker must be in the chromosomal left arm together with the FZ mutations.
Additional crosses were performed using hfincs, FZam,,,am730cZ,I in order to (i) separate the FZ markers from the fines, marker,-and (ii) to reconstruct the starting phage.
First, a cross was performed to separate and recover the FZam,,,am,,, mutations from the fines, mutation present in the 6ncsFamFam derivative, as diagrammed in Figure 3(a). W3805 (hgal,,imm2’) was u.v.-induced and infected with hfincs, FZam 471am,30cZ,, and, after growth, the lysate was plated on C600.

LAMBDA DSA MATUKATIOS 389
fincsl F~m41.730 att cIt1 I I, I I
I--- ____________...------ --------------
(a) 1 I
g% imm 21
fines Franhpo att 4, I II I I
----------< ,------ - _______ -___-___
(b) : s ------_---_-- #’
I I 1 504 del att bia
FJam41,730 CIt, II I
r _________________ ____ -__ --- --___--__----- --
(cl :’ - ---, 1 fin Cs, crt,
Frc: 3. Separation of the fin mutation from the 2 amber mutants in PI and reconstitution of hfinc.~, FIam4,,am,30cI,,.
The infecting phage makes large plaques on C600 and hfin+FIam4,1am730 recombinants form very small plaques. Small plaques on C600 were picked, and some of these were readily shown to be unable to grow on R594 and to contain both FI amber mutations.
To isolate the fines, mutation, a cross was performed between hfmcs, FIam,7,am73,, cZ,, and the deletion prophage in strain MS504. The cross is diagrammed in Figure 3(b). Recombinants forming large plaques on R594 were picked and presumed to be hfincs,cI,, , based on the fact that the parent makes very small plaques on sup0 hosts. These recombinants were indistinguishable from hfinfcl,l in their plating ability using various host cells and various incubation temperatures.
The presumed structure of hfincs,FIam,,,am,30cI,, was then confirmed by crossing the strains isolated in the previous two crosses, hFIama71am730cI,l and hfincs, cI,l, as diagrammed in Figure 3(c). Recombinants indistinguishable from Xfincs, FIam,7,am,,ocI,, were found as small plaque formers on R594. These recombinants were cold-sensitive on R594 at 31”C, like the original isolate. The results of these crosses show that AFIam,, 1 am1 3o can mutate to FI independence by mutation at a site in the Nul-D interval present in the hgal,,imm2’.
(d) Mapping of the $n mutation
Two fin mutations, fin,, a cold-sensitive isolate which produces normal amounts of gpA. and fin,,,, also a cold-sensitive isolate that overproduces gpA. were mapped

390 H. MURIALDO ET AL.
TABLE 4
Complementation of hgal strains with 480 amber mutant in 1 and h mutants in A
Infected bacteria lam3
Yield (phage/cell) Infecting phaget
Aam Aam,,,
w3350 2.3 x lo-* 7.1 x 1o-4 W3805 1.4x 10-3 W3805 (~~al,redam2,0cZ,,,nin,) 33 < 8 x 1o-2 W3350 (XdgaZ,,,imm434) 25 9 x 1o-2 W3805 (hdgal,,,red3cZ,5,) 0.51 1.7 x 1o-3 W3805 W~4oocZ,,,) 2.8 x lo-’ 1.3 x 10-3
t The complete genotype of the superinfecting phages is: lam, = ~hy@Olam3red3cZ,,7 ; Aam,, = hAam3,red3cZ,,,; A am 12, = AAam,z, redam2&3ds5~.
by marker rescue studies utilizing hgal prophages broken in the Nul to A region. Four gal strains were used: Xga14, hga&,,, hgal,i, and hgal,oo. These were first characterized by complementation experiments with amber mutants that had been previously mapped (Campbell 1961; Murialdo & Siminovitch 1972; Sato et al., 1968 ; Weisberg et al., 1979). Table 4 gives the results of complementation tests between the Agal prophages and hA amber mutant phages, as well as the JL#SO hybrid that carries an amber mutation in 480 gene 1. This latter mutant was used since no amber mutants in h gene Nul are available and it has been shown that gene 1 function of 480 can substitute for the gene Nul function of X (Weisberg et al., 1979). None of the Xgal strains used complemented hA - Two hgal prophages, hgal, and hga16i 5, complemented the 1 -mutant well. Agal,,, complemented the 1 - mutant to a limited, but significant extent, and hgalloo did not complement the 1 - mutant. The weak complementation by hgals15 w&9 also observed under conditions of reduced recombination, ruling out recombination as the source of phage production. Therefore, it can be concluded that none of the Xgal strains contains a functional A gene, and that /\gal4 and Xgals15 contain an intact Nul gene. Xgal,,, gave weak complementation for Nul function. The weak complementation by this prophage could be due to the production of a truncated or hybrid polypeptide that possesses some activity. Finally, the deletion-substitution of hgalIoo apparently breaks within the Nul gene.
To map the fin mutants, marker rescue of the tin+ allele was carried out using the four hgal strains. These crosses were identical to that described in Figure 3(b). The results are presented in Figure 4 and they locate the fm mutations to adjacent segments that include the end of the Nul gene and the beginning of the A gene. The fincs,o, mutation maps to the left of the fines, mutation. If one assumes that the hgal,,, prophage is broken within the Nul gene, then fincsio6 probably maps in the Nul gene. The assumption is subject to serious reservations, however, for hgal,l5 could have picked up a Nul mutation during its propagation as a prophage. Additional work is required to resolve this point. We conclude that the tin

LAMBDA DNA MATURATIOS
Map of NUI - A region Rescue of fin+ alleles
I(NuJ) A k I o/0 fin phages
391
am3 041 am121 amI9 am32 I I I I I fin, fin 106
A&d4 l-72( ,s) + I-52& )+
WaJ,,, o-44 (A) + o.31 Ed +
WaJloo - x- 0.0+%) - o-07&-)-
FIG. 4. Mapping of the fin mutations. The upper lines indicate the size of genes Nul and A. The molecular weight of gpA has been estimated
at 79,ooO (Murialdo & Siminovitch, 1972). The size of gpNu1 is not known, but it could have a maximum &f, of41,(@9 (Weisberg et al., 1979). Whether there is an interval between Nul and A is unknown. The second line shows the correct order of amber markers on the DNA, but the distances between them in the map are arbitrary. The subsequent 4 lines represent the X genetic material present in 4 Xdgal transducing phages in this region of the h genetic map. X- stands for non-lysogen. The endpoints of the Adgal strains are not known precisely, but their location between known amber markers is indicated. The bottom line shows the possible region of the chromosome where the fin mutations lie. In the right part of the Figure the frequencies of marker rescue of the fin+ allele from the corresponding hdgal phages and from the non-lysogen are listed. The genotype of the bacterial strains is as in Table 4.
mutations map in a segment that includes the end of Nul and the beginning of A, and that fines,,, maps to the left of tlncs,.
(e) Levels of gpFI and gpA in cells infected with A$fin mutants
To confirm that the fm isolates are indeed pseudorevertants and, therefore, still carry at least one of the Flam mutations, we did experiments to directly examine if gpF1 was or was not being produced in sup0 cells. This was done by labelling with [35S]Met the phage-specific proteins made after infection of cells preirradiated with a heavy dose of ultraviolet light. The proteins in the extracts prepared from such cells were separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and the pattern visualized by autoradiography. The results of this analysis are shown in Figure 5. Electrophoretograms from only a few representative experimental cases are shown. Figure 5(a) shows the pattern obtained after infection of cells with wild-type A. Figure 5(b) to 5(e) shows that gpF1 is indeed absent in cells infected with AFIam,,,, AFIarnTS5 and hFIam730 as well as in cells infected with the double mutant hFIam785am730. It can be seen in channels (f), (g) and (h) that all the pseudorevertants were still FI- since no detectable gpFI was made upon infection. This result strongly suggests that the isolates are indeed pseudorevertants that are able to plate in the absence of the FI-gene product due to a mutation at a second site.

-4P
-4P
-9P
(a) (by (c) (d)(e) (f) (9) (h) (i) (i) (k)
FIG. 5. Levels of gpA in cells infected by wild-type and mutant phage. Experimental details are given in Materials and Methods. 159 cells were irradiated with ultraviolet light and subsequently infected with the phages listed above the autoradiograph. The infected cells were pulselabelled for 10 min with the addition of a hydrolysate containing 35S-labelled amino acids at 25 min after infection. Extracts were prepared and electrophoresed in a 125% acrylamide/sodium dodecyl sulfate gel. The dried gel was exposed to X-ray film for 4 days prior to development. The phages used in (a) to (h) carried, in addition to the mutations specified on top of the autoradiograph, a PI,, mutation. The genotype of the other phages were: (i) /\Aop,cZ,,,Sam7: (j) hAnm,,i2’elt.~,Sam,: (k) AcI,,,Sum,.

LAMBDA DNA MATURATIOS 393
Figure 5(f) also shows that, while the fin,,, derivative of hFl~rn~s,arn~~~cZ,, produced increased amounts of gpA, an essential component of the terminase enzyme, another derivative, fin, 10 (Fig. 5 (h)), produced normal amounts of gpA. It thus appears that there are at least two ways of bypassing the requirement for gpF1 in h growth. One way is by mutation that increases the level of the gene A product. and another by a mutation in which there seems to be no alteration in the level of any h late gene product identifiable by this technique of protein labelling. electrophoresis and autoradiography. For convenience, the fin mutants that overproduce gpA will be called fin type A, and those that do not overproduce gpA, fin type B, or more simply, finA and finB, respectively.
In order to compare the amount of gpA present in cells infected by the tin mutants with the amount present in cells infected by wild-type /\, both gpA mass and gpA activity were measured. To measure mass, the amount of radioactivity in the gpA band was estimated by scanning autoradiograms similar to those shown in Figure 5 and integrating the area under the curve. In order to standardize the measurement internally, the area of the gpA peak was divided by the area under the gplJ peak and under the sum of gpH and gpH* peaks in t)he same channel of the gel (that is, the amount of gpJ and gpH+gpH* present in the same extract). Table 5 summarizes the data obtained. gpJ and gpH +gpH* were used for standardization because they migrate very close to gpA in the polyacrylamide gel and. more importantly, because they migrate to an area of the gel free of host and other h-induced protein bands. The intensity of all these bands is close enough so that one gel exposure can accommodate their intensities within the range of linear response of the film. Although the conversion of gpH to gpH* varies from phage to phage, the effect seems to be independent of the presence of fin mutations. Besides, the total amount of gene product, that is, gpH+gpH*, as well as of gpJ does not
TABLE Fi
Levels of gpA in phage-infected cells
Inmsme relative to wild type gpJ ss ~PH +gpH*
7$ 7p@ lp@* gpA gpA/gpT .w%pH +BPH* standard as standard

fin106 fin+
(a) (b) (cl (dl (a) (b) (4 (d)
wJ
FIG. 6. Stability of gpA. Complete experimental details are given in Materials and Methods. 159 cells were irradiated with
ultraviolet light and infected with hfin,c,Flam,,,am,,,el,57 Sam, or Ad,,. [3sS]Met was added to the infected cells 25 min after infection. Two minutes later, a large excess of non-radioactive Met was added to the cultures and samples were withdrawn into tubes containing azide and these were immediately submerged in liquid N2 at (a) 10 s; (b) 150 s; (c) 300 s: (d) 600 s after the addition of cold Met. Extracts were prepared and electrophoresed in a 75% acrylamide/sodium dodecyl sulfate gel. The gel was dried and exposed for 2 days to X-ray film prior to development. In the same experiment, but not shown here, the cells were also infected with hfin,,,FIam,s,am,,,cZs,,Sam, and with hAop,cls,,Sam,. The patterns obtained with these phages were identical to the hCI,l and the ~fin,csFZam,,,am,,ceIs~,Sam,, respectively.

LAMBDA DNA MATURATION 395
seem to be altered by the fin mutation. Although the gpA to standard ratios vary slightly when gpJ or gpH+gpH* are used, it is clear that the amount of gpA in
hfinll0FIaw85fm30 cI,, extracts is essentially the same as that produced in cells infected by wild-type X, whereas Xfincs,06FIam,s,am,J,cl,, infection resulted in the production of several-fold more gpA. The presence of the fines,,, mutation results in a fourfold increase in gpA levels. In a similar way, the levels of gpA were checked for the following 14 fin mutants: cs,, cs2, tss, ts,,, cslol, cslo2, csio3, cslo4. csio6, csio9, cslio, cts,,,, cts,,, and csl13. Of these c81c6, cslol and 104 showed increased amounts of gpA. fin,,, may induce the synthesis of gpA in levels intermediate between that found in the wild-type and in &icsic6 and hfincslo, cases.
The increased levels of gpA observed in the autoradiograms in hfincsio6-infected cells is not due to a decrease in the rate of degradation of the pgA polypeptide. Experiments using a similar protocol were done except that the pulse of [35S]Met was for only two minutes, and the pulse was followed by a chase using a IO’-fold excess of non-radioactive Met. The results show that the amount of gpA remained unchanged for at least ten minutes in both the wild-type and finlo cases, whereas cleavage of other proteins such as gpB took place (Fig. 6).
To see if this increase in gpA mass is also associated with an increase in gpA activity, measurements of gpA activity were carried out using the assay described by Becker & Gold (1975). In these experiments, the amounts of A - prohead-donor extract and of h DNA were kept constant, while the concentration of gpA in the assay was varied by adding a constant volume from a set of successive dilutions of t,he gpA-donor extract. In the second stage of the assay, the concentration of the A - extract providing the remaining essential gene products, and tails, was always kept constant. In this way, the only variable is the amount of gpA provided in the gpA-donor extract. Dose-response curves on gpA generated in this manner are non- linear and have the form of a power function over a wide range of gpA concentrations (Hohn, 1975; Becker et al., 1977a).
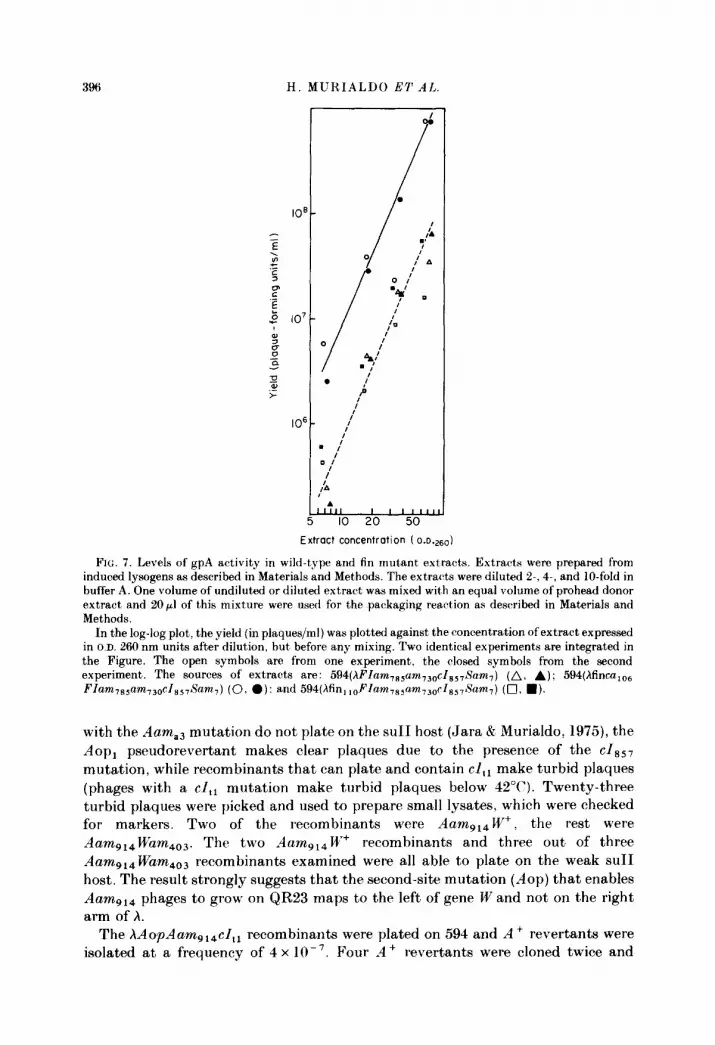
Figure 7 shows the gpA activities in extracts of induced lysogens containing a fin+ prophage, and prophages carrying either the f&s,,, mutation or the fin 110 mutation, in addition to two amber mutations in FI. The levels of gpA activity in the fin lloFI- extract are very similar, if not identical, to the levels in the FI- extracts. The gpA activity level in the fincs,,,FI-, however, is about, tenfold higher. These results are in complete agreement with what can be predicted from the results shown in Figures 5 and 6, which indicate that fincs,,,FI- extracts, but not fin i loFI- extracts, contain elevated levels of the A-specific polypeptide.
(f) Behaviour of Aop mutants of lambda
As described in Materials and Methods, gpA overproducer strains can be isolated by selecting for a mutation that allows hAam,,,Wam,03cI,,, to grow on a weak suI1 strain. Having obtained such an Aop variant, we first had to transfer this mutation into a wild-type background. This was done as follows: /\AoplAamg14Wam,03~I,,, was crossed with AAama3cItl using QD5003 as a host (Fig. 8(a)). The lysate was plated on TC600 at 38°C. Under these conditions, phages
396 H. MURIALDO ET AL.
Extract concentration ( o.D.260)
FIG. 7. Levels of gpA activity in wild-type and fin mutant extracts. Extracts were prepared from induced lysogens as described in Materials and Methods. The extracts were diluted 2-, 4-, and IO-fold in buffer A. One volume of undiluted or diluted extract was mixed with an equal volume of prohead donor extract and 20~1 of this mixture were used for the packaging reaction as described in Materials and Methods.
In the log-log plot, the yield (in plaques/ml) was plotted against the concentration of extract expressed in O.D. 260 nm units after dilution, but before any mixing. Two identical experiments are integrated in the Figure. The open symbols are from one experiment, the closed symbols from the second experiment. The sources of extracts are: 594(hFlam 7ssam,3Dc~s,,~~am~) (A, A); 534(hfinca,,, FIam,s+m,,o cl ss7Sam,) (0, a): and 594(hfin,,DFlam,s,am,~D~Zs~,~~am,) (Cl, n l.
with the Aama3 mutation do not plate on the suI1 host (Jara & Murialdo, 1975), the Aop, pseudorevertant makes clear plaques due to the presence of the cl,,, mutation, while recombinants that can plate and contain cI,r make turbid plaques (phages with a cI,~ mutation make turbid plaques below 42°C). Twenty-three turbid plaques were picked and used to prepare small lysates, which were checked for markers. Two of the recombinants were Aamg14W+, the rest were Aamg14Wam403. The two Aam,,,W+ recombinants and three out of three
Aam Warn403 recombinants examined were all able to plate on the weak suI1 host. The result strongly suggests that the second-site mutation (Aop) that enables Aamg14 phages to grow on QR23 maps to the left of gene W and not on the right arm of A.
The XAoPAamg14 cI,, recombinants were plated on 594 and A+ revertants were isolated at a frequency of 4 x lo- ‘. Four -4 + revertants were cloned twice and

LAMBDA DNA MATURATIOS 3%
AOP Aamg,&‘am403 I I I
.----- - ---. . . . . . . . . . . . . . . . . . ..~
cIs57 I
(0) \a ‘;b -----_---- 2 ----------, - . _ _ _ - - - _ - -
AOP, CI857 Sam,
Aw Em315 C&57
I I I
-------3 ,----- ________ -_
Cd) : L __-_- ____._-.____---__ 2 -_--______.-__--_
FIam 705.730 Gl
AOPI F~ams5.730 CI857
I II I r____ _______________-____ - _-____---_- --
(0) : __---.., I
Earn43
FIG. 8. Isolation of the Aop, mutation from hAo~,ilam,,,W,~~rl,,, and t,ransfer of the mutation to a FZ- hackground.
further characterized. hAop,cZ,, could not be distinguished from Xcl,, by simple plating at different temperatures on strains with or without suppressors. Thus, as in the case of fines,, the Aop mutation by itself has no easily discernible phenotype.
(g) The Aop, mutation is a $n type A mutation
I f Aop, can behave as a fin mutation, then a FI- phage carrying the Aop, mutation should plate on sup0 cells. To study this possibility, hAoplcZss7Sam,
was first built by crossing hAop,cl,, with hAtso,4cZ85,Sam, (Fig. X(b)). The lysat,e of this cross was plated on QD5003 at 42°C. Seven out of ten clear (cl,,,) plaques were Sam,. Four of these putative hAoplcI,,,Sam, were checked for gpA

398 H. MURIALDO ET AL.
overproduction by electrophoresis-autoradiography of radioactively labelled infected-cell extracts. They all proved to be gpA overproducers (data not shown).
In order to transfer the Aop, mutation, the following procedure was used. hAop,~I~~~Sarn~ was crossed with hAts,,,Eam,,scI,,7 and the progeny of the cross was plated at 42°C on TC600, conditions under which neither parent can plate (Fig. 8(c)). Among 51 recombinants, six were unable to grow on 594. Four of these recombinants (nos 3, 4, 5 and 6) were cloned twice and checked further. They were all Earn,,, by the marker rescue spot test. However, when the polypeptides synthesized by these phages were examined by electrophoresis-autoradiography of radioactively labelled infected-cell extracts, only isolates 4 and 6 were gpA overproducers (data not shown). Therefore, isolates 3 and 5 are AEam8,5c1857, whereas isolates 4 and 6 are hAop, Earns1 +Igs7. Isolates 5 and 6 were crossed with hFIam785am730cI,, and the lysates were plated on 594 at 38°C (Fig. 8(d)). About two-thirds of the plaques of the cross involving no. 6 were very small, presumably hAopiFIam 7ssam,3,,cI,, plus hAop,FIam,85am730cI,,7, and one-third were of large size, presumably /\cI,,, and hcl,,. The progeny of the cross with isolate 5 gave only big plaques. These results are in agreement with the results on the measurement of gpA present in extracts of cells infected with isolates 5 and 6, which showed that isolate 5 was not a gpA overproducer.
To make sure that the small plaques on 594 were indeed ~Aop,FIam7s5am7sc recombinants, it was essential to show that the two amber mutations in FI were still present. Therefore, two of the small plaques were cloned and crossed with hEamd3 and the lysates plated on QD5003 at 38°C (Fig. 8(e)) (hEamd3 does not plate on suII1 strains). When the plaques were replicated on 594 (and QD5003 as master), about 2% of the progeny were not able to grow in the sup0 strain. These isolates were shown to carry the two FI amber mutations by marker rescue spot tests. These results show that the small plaques on 594 obtained in the cross between /\AoplEams15cIs57 and hFIam,,,am,,,cI,, carried the two FI amber mutations and that they were able to plate on sup0 due to the presence of the Aop, mutation.
Further tests showed that hAop,FIam7ssam730cI,,7 is cs when plated on sup0 cells, as is the case with another gpA overproducer selected as an FI-independent revertant, Mincs,,-,6FIam78,am,3,cI,, (Table 3).
(h) Mapping of the Aop, mutation
The mapping with Aop, has not been as extensive as in the case of the tin mutants. The selection procedure used for plating the progeny of the cross diagrammed in Figure 8(c) assured that only recombinants between the Sam7 marker of one parent and the At+,,, marker of the other parent would plate. In six of 51 such recombinants examined, the recombination event took place to the left of the EarnsIs marker. The fact that in the four recombinants carrying EamB15 chosen at random, two did not carry the Aop, mutation can be explained in either of two ways. Either Aop, maps to the right of the Ats014 and one crossover took place; or alternatively, Aop, maps to the left of Atsol and, in addition to a crossover in the interval between ALsol and Earns1 5, a second crossover between
LAMBDA DNA MATURATIOS 399
Aop, and Atsol took place. In the first case, Aop, would map in gene A, or to the right of gene A. In the second case, Aop, could map to the left of gene A, as would a promoter or ribosome-binding site mutation.
Starting from strain TC600(hAop,cZsS, Sam,), a hdgal transducing phage (hdgaZ800cZ8s,Sam7) has been isolated by standard procedures (Campbell, 1957). The left endpoint of the deletion in this phage is between the markers Aam,, and Aam,, (see Fig. 4 for location of mutations). W3350(hAop,dga1900cZ,,7s,) was crossed with hFZam7,,am 730cZ,l, giving 62% recombinants able to plate on 594. The recombination frequency with the control Aop, + strain W3350 (~dgaE615cZss7) with deletion endpoint between markers Aam,, and AamJz gave < 0*0025% recombinants able to plate on 594 (see Fig. 4 for location of mutations). These results clearly show that the Aop, mutation maps to the left of Aam,,.
(i) Level of gpA synthesis in Aop,-infected cells
As for the case of hfin mutants, the amount of radiolabelled A polypeptide present in extracts of hAop,-infected cells was measured, and A protein activity was determined in the DNA packaging assay.
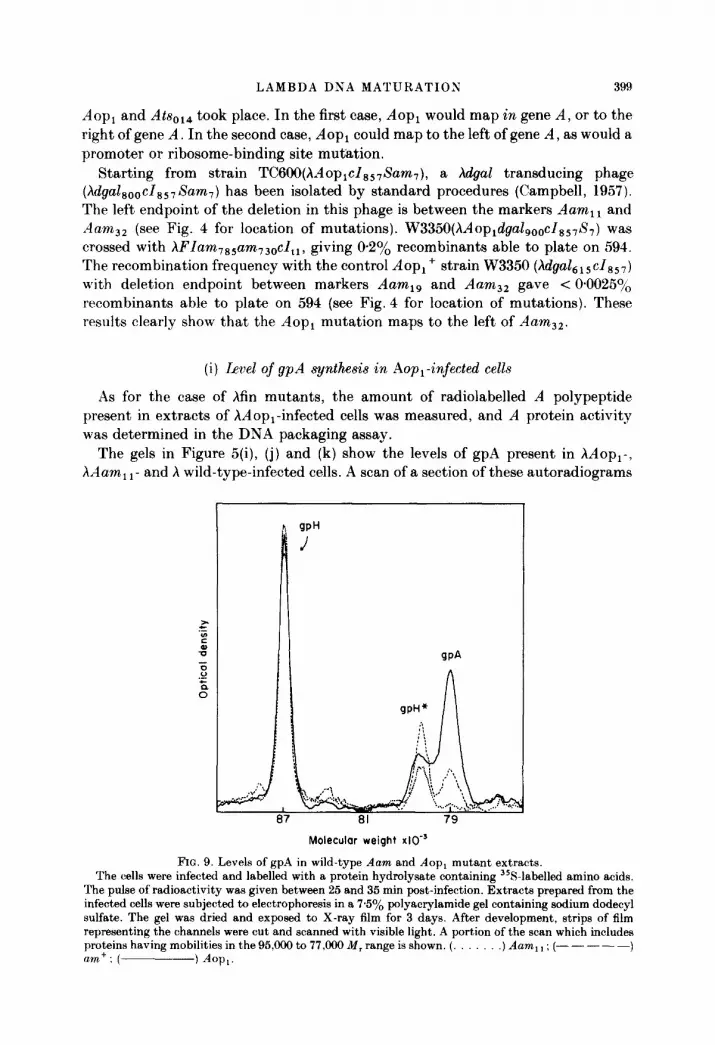
The gels in Figure 5(i), (j) and (k) show the levels of gpA present in hAop,-, hAam, 1- and X wild-type-infected cells. A scan of a section of these autoradiograms
I ! JPH
/
L 87
Molecular weight x10-
FIG. 9. Levels of gpA in wild-type Aam and Aop, mutant extracts. The cells were infected and labelled with a protein hydrolysate containing 358-labelled amino acids.
The pulse of radioactivity was given between 25 and 35 min post-infection. Extracts prepared from the infected cells were subjected to electrophoresis in a 75% polyacrylamide gel containing sodium dodecyl sulfate. The gel was dried and exposed to X-ray film for 3 days. After development, strips of film representing the channels were cut and scanned with visible light. A portion of the scan which includes proteins having mobilities in the 95,ooO to 77,909 M, range is shown. (. .) Aam, 1 ; (- - - - -) am+: ( ) Am.

400 H. MURIALDO El’ AI,
as presented in Figure 9 shows that the amount found in hdopi-infected cells is about fourfold the amount found in wild-type-infected cells. This was determined by integrating the areas under the curves for the gpA band and using as background the curves obtained for the hAam i ,-infected cell extract (Table 5).
In pulse and chase experiments, the stability of the gpA synthesized in hAop,- infected cells was found to be the same as in the wild-type and fin,,, cases (data not shown).
1n U&O packaging experiments showed that extracts of cells infected with /\Aopl were more active in terminase, ranging in activity from 6 to 20 times that of wild- type extracts (results not shown). All in all, these data for /\Aop, extracts are similar to those obtained for the hfin,,h extracts in so far as gpA overproduction is concerned.
4. Discussion
Of the ten known gene products that participate in phage /\ head assembly, the role of gpF1 is one of the least understood (Murialdo & Becker, 1978b). In order to pursue the action of this protein in viva, we relied chiefly on the broad implication of two observations. The first observation was that FI mutants of /\ were leaky, suggesting that gpFI served some role in the pathway which was ancillary. The second observation, made in a cell-free system (Benchimol et al., 1978), indicated that there exists an interaction, however indirect, between gpF1 and the terminase enzyme. Thus, it appeared feasible to isolate variants of /\ which were FI-independent (fin) and that the second site mutation conferring this independence would involve the terminase genes (S~rl and/or .+I). More precisely, the in vitro observations suggested that one category of such pseudorerertants would have increased intracellular levels of terminase as the basis for the fin phenotype.
(a) jn mutants that overproduce gpA
We have isolated single-step fin mutants that overproduce gpA, an essential component of terminase. In another approach, a pseudorevertant of an 4 -W- phage that was able to plate on a weak suppressor turned out to be a gpA overproducer (Aop,). When the Aop, mutation was crossed into an FI- phage, it allowed the phage to plate in sup0 cells. These results show that one condition sufficient for the growth of phage in the absence of gpF1 is overproduction of gpA.
It has been shown that gpA activity in vitro is not a linear function of gpA concentration (Becker &, Gold, 1975; Hohn, 1975: Becker et al., 1977a). This can also be observed in Figure 8, where a doubling in the gpA concentration results in a 4Sfold increase in phage yield. One might expect, therefore, that the 3.7-fold increase in the concentration of gpA mass as measured in the autoradiograms of the fin io6-infected cell extracts (Table 5) would result in a 26fold increase in packaging activity. Clearly, this was not the case. The increase in activity as measured by packaging is tenfold (Fig. 8). The discrepancy suggests that other factor(s) essential for the activity of gpA are limiting. One prime candidate as limiting factor is

LAMBDA DNA MATUKATIOX 4)l
gpNuI, which has recently been identified by polyacrylamide gel electrophoresis (M. Sumner-Smith, personal communication).
There are two broad categories of mechanism by which the activity of an enzyme can be selectively increased by mutation: (1) through a change in the structural gene of the enzyme resulting in an enhancement of its catalytic action, or (2) by one of several mechanisms that result in a high intracellular concentration of the enzyme: for example, (i) by enhancing the frequency of transcription of the structural gene of the enzyme (promotor mutation), (ii) by increasing the frequency of its translation (ribosome binding site mutation), (iii) by increasing the functional stability of the mRPlu’A and (iv) by increasing the stability of the protein itself. For those single-step mutations where the increase in terminase activity is associated with an increase in gpA mass, it appears sufficient to consider only the second category mechanisms. We have presented evidence that the structural stabilities of gpA synthesized in cells infected with wild-type A, or with hfin and AAop mutants were the same. (:onsequently, it appears unlikely that terminase accumulation due to enhanced protein stability is the reason for the increase in protein mass. We have also investigated the possibility that the fin type A mutations generate a new promoter. To do this, h lysogens were infected with /\FI - and with a finA derivative of it, and the synthesis of h-induced proteins was examined. So /\-specific polypeptides were observed in either case. (Infection by a heteroimmune phage served as the positive control.) Thus, it appears unlikely that a new promoter for the A gene had been created. We consider as remote the possibility that a new promoter had been created, but that this promoter is subject to h repressor control.
We favour the idea that the increase in gpA mass is the result of an increase in the rate of initiation of translation such as would result from an alteration in the rihosomal binding site for the A cistron. This model would adequately explain the selective overproduction of gpA from a polycistronic mRSA (Guha & Chowdhury. 1973 ; Ray & Pearson, 1974). gpA overproduction as the result of an increase in the functional stability of the mRNA cannot be excluded at present.
(b) jin mdants that do not owrprod~rce gpA
The nature of the fin mutants that do not overproduce gpA (finB) is unknown. This t,ype of fin mutation could map in the genes coding for terminase. Limited genetic mapping placed the finB, mutation to the right of the finA,,, mutation. It is possible, therefore, that fin, maps in the A structural gene. It is possible that many. if not all, of the fin type B mutations map in the genes that specify terminase. that is, in A (or ~Vul) and that the mutations allow terminase to operate more efficiently in GVO in the absence of gpF1.
The majority of the fin mutants and the Aop, mutant are cold-sensitive (cs). Among the cs fin mutants, we find both gpA overproducers (finA) and non overproducers (finB). In addition, some finB isolates are ts. Our recovery of many cs fin mutations is perhaps related to the unusual susceptibility of gp-A to cold- sensitive alterations that was discovered by (“ox Rr Strack (1971). They studied 17 independent cs mutants of lambda and found the mutations to map in only four genes A. Q, *J and S, with almost half the isolates being Acs mutants. Considering

402 H. MURIALDO ET AL.
possible explanations for the cold-sensitive phenotypes points the way for further experiments. Among the explanations for this cold sensitivity are: (1) improper assembly, (2) inactivation due to a cold-induced conformational change, and (3) temperature dependence of the level of activity.
(1) Improper assembly. The active form of gpA is likely to be a multimer (Becker et al., 1977a) which may also include gpNu1 subunits. Thus, functional terminase may be a hetero-oligomer. The proper association of subunits to form the multimer, or the proper conformation by the multimer, may include a temperature-dependent step. fin mutations could alter the structure of a subnit in such a way as to increase the temperature required for this temperature-dependent step. We draw an analogy here to the ribosome assembly mutants of Guthrie et al. (1969). Should the gpA overproducing mutants have an altered ribosome binding sequence, then improper assembly seems less likely, because such a change would not be expected to alter the primary structure of the A polypeptide. A ribosome binding site change for gpA translation, however, could change gpNu1, if the A binding site were part of the Nul structural information. Such an overlap has been found in the tryptophan operon of E. coli: the trpB polypeptide coding sequence also contains the ribosome binding sequence for the next gene product, the trpA polypeptide (Platt & Yanofsky, 1975). In this case, the defect in assembly would be made correct when gpF1 is present.
(2) Inactivation due to a cold-induced conformational change. The effect of fin mutations might be on the activity of terminase at low temperature rather than on the assembly of terminase as in the preceding case. For example, one might postulate that at low temperature the specificity of terminase from a fin mutant for cos DNA decreases relative to its ability to bind to inhibitory ligands such as single-stranded nucleic acid. Thus, at low temperature, terminase functions are lost in the absence of gpF1, which acts to reverse this non-specific binding. This hypothesis can be distinguished from the previous one by a study of terminase isolated after growth of cells at 37°C. In the first case the active fin terminase should be indistinguishable from the wild-type enzyme with respect to temperature and inhibition by single-stranded DNA. In the second case, the fin terminase should be less active or more inhibitable than the wild-type enzyme.
(3) Temperature dependence of the level of activity. Finally, the functional level of gpA that is attained in infected cells may be sharply temperature-dependent. Such dependence could be due to a decreased rate of synthesis or stability of the mRNA or terminase at low temperature. The level obtained would be sufficient only in the presence of gpF1 to prevent, for example, inhibition by single-stranded nucleic acid. This possibility can be tested by comparing the terminase activities of fin and wild-type phages grown at 37 and 31°C. All of these models imply an interaction between gpNu1 and/or gpA on the one hand, and gpF1 on the other. To establish directly whether such an interaction indeed takes place, further experiments in vitro are required.
We thank Deborah L. Siegele for excellent assistance with the mapping and complement&ions studies. This work was supported by research grants from the National Institute of Health (GM24361) and the Medical Research Council of Canada (MT-3325) (to

LAMBDA DNA MBTURATIOS 103
H. M. and A. B.), and from the National Institute of Health (A112851) and National Science Foundation (37310) (to M. F.).
REFERENCES
Adler, J. & Templeton, B. (1963). J. Mol. Biol. 7, 71(t720. Appleyard, R. K. (1954). Genetics, 39, 44&452. Bachmann, B. J. (1972). Bacterial Rev. 36, 525-557. Becker, A. & Gold, M. (1975). Proc. Nat. Acad. Sci., U.S.A. 72, 581-585. Becker, A. & Gold, M. (1978). Proc. Nat. Acud. Sci., U.S.A. 75, 41994203. Becker, A.: Marko, M. & Gold, M. (1977a). Virology, 78, 291-305. Becker, A., Murialdo, H. & Gold, M. (19773). Virology, 78, 277-290. Benchimol, S., Becker, A., Murialdo, H. & Gold, M. (1978). Virology, 91, 205-221. Bode, V. C. & Kaiser, A. D. (1965). J. Mol. Biol. 14, 399-417. Boklage, C. E., Wong, E. C. & Bode, V. C. (1973). Genetics, 75, 221-230. Campbell, A. (1957). Virology, 4, 366384. (‘ampbell, A. (1961). Virology, 14, 22-32. Carter, B. J. h Smith, M. G. (1970). J. Mol. Biol. 50, 713-718. (‘asjens, 8. R. & Hendrix, R. W. (1974). J. Mol. Biol. 88, 535-545. Couturier, M., Desmet, L. Rr Thomas, R. (1964). Biochem. Biophys. Res. Commun. 16. 244
248. (‘ox, J. H. & Strack, H. B. (1971). Genetics, 67, 5-17. (‘rawford. L. V. & Gesteland, R. F. (1973). J. Mol. Biol. 94, 627-634. Daniels, D. L., de Wet, J. R. & Blattner, F. R. (1980). J. Viral. 33, 390-400. DeLuctia, P. & Cairns, J. (1969). Nature (London), 224, 116&1166. Dove, W. F. (1966). J. Mol. Biol. 19, 187-201. Echols, H. & Murialdo, H. (1978). Microbial. Rev. 42, 577-591. Emmons, S. W. (1974). J. Mol. Biol. 83, 511-525. Enquist, L. W. & Skalka, A. (1973). J. Mol. Biol. 75, 185-212. Feiss, M. & Bublitz, A. (1975). J. Mol. Biol. 94, 583-594. Feiss, M. & Campbell, A. (1974). J. Mol. Biol. 83, 527-540. Feiss, M., Fisher, R. A., Crayton, M. A. & Egner, C. (1977). Virology, 77, 281-293. Floor. E. (1970). J. Mol. Biol. 47, 293-306. (Karen, A., Garen, S. & Wilhelm, R. C. (1965). J. Mol. Biol. 14, 167-178. Gellert. M. (1967). Proc. Nat. Acud. Sci., U.S.A. 57, 14&155. (:eorgopoulos, C. R.. Hendrix, R. W., Casjens, S. R. & Kaiser, A. D. (1973). J. Mol. Biol. 76,
45-60. Gilbert, W. 8: Dressler, D. (1968). Cold Spring Harbor Symp. Quunt. Biol. 33, 473484. (iorini, L. (1971). Suture New Biol. 234, 261-264. (>orini, L., Jacoby. G. A. & Breckenridge, L. (1966). Cold Spring Harbor Symp. &ant. Biol.
31, 577-664. Guha, A. 8 Chowdhury, D. M. (1973). Nature New Biol. 241, 196-198. (iuthrie, C., Nashimoto, H. & Nomura, M. (1969). Proc. i~at.Acud. Sci., ZT.S.il, 6X384-391. Hershey, A. D. & Burgi, E. (1965). Proc. Nut. Acud. Sci., F.S.3. 53, 325-328. Hohn, B. (1975). J. Mol. Biol. 98, 93-106. Hohn, B. & Hohn, T. (1974). Proc. Nut. Acod. Sci., ZT.S.A. 71, 2372-2376. Hohn. T. & Katsura, I. (1977). Curr. Top. Microbial. Zmmunol. 78, 69-110. ,Jara, L. & Murialdo, H. (1975). Virology, 64, 264-268. Kaiser, A. D. (1957). Virology, 3, 4241. Kaiser, A. D. & Masuda, T. (1973). Proc. Xut. Acud. Sci., I:.S.il. 70, 260-264. Kaiser, A. D., Syvanen, M. & Masuda, T. (1975). J. Mol. Biol. 91, 175-186. Kaplan. S., Stretton, A. 0. W. & Brenner, S. (1965). J. Mol. Biol. 14, 528-533. Laemmli, U. K. (1970). Nature (London), 277, 68&685. Lederberg, E. M. & Lederberg, J. (1953). Genetics, 38, 5144. Lieb, M. (1966). J. Mol. Biol. 16, 149-163.

404 H. MURIALDO ET AL.
Liedke-Kulke, M. & Kaiser, A. D. (1967). Virology, 32, 465481. Lwoff, A., Siminovitch, L. & Kjeldgaard, N. (1950). .4 nm. Inst. Pasteur. 79, 815-859. MacKinlay, A. G. & Kaiser, A. D. (1969). J. Mol. Biol. 39, 679-683. McClure, S. C. C. & Gold, M. (1973). Virology, 54, 19-27. McClure, S. (1. C., MacHattie, L. & Gold, M. (1973). Virology, 54, l-18. Manly, K. F., Signer, E. R. 8: Radding, C. M. (1969). Virology, 37, 177-188. Mousset, S. & Thomas, R. (1969). Nature (London), 221, 242-244. Murialdo, H. (1979). Virology, 96, 341-367. Murialdo, H. 8: Becker, A. (1978u). J. Mol. Biol. 125, 57-74. Murialdo, H. 8: Becker, A. (19783). Microbial. Rev. 42, 529-576. Murialdo, H. & Siminovitch, L. (1971). In The Bacteriophage Lambda (Hershey, A. D., ed.),
pp. 711-723, Cold Spring Harbor Press, Cold Spring Harbor, N. Y. Murialdo, H. & Siminovitch, L. (1972). Virology, 48, 78,&823. Ogawa, H. & Tomizawa, J. -1. (1967). J. Mol. Biol. 34, 26.5276. Otsuji, N. 8 Aono, H. (1968). J. Bucteriol. 96, 43-50. Parkinson, J. S. (1968). Genetics, 59, 311-325. Pirrotta, V. & Ptashne, M. (1969). iliature (London), 222, 541-544. Platt, T. & Yanofsky, C. (1975). Proc. i2’at. Acad. Sci., U.S.A. 72, 2399-2403. Ray, P. N. & Pearson, M. L. (1974). J. Mol. BioZ. 85, 163-175. Roehrdanz, R. L. 8r Dove, W. F. (1977). Virology, 79, 4&49. Sato, L., Niskimune, Y., Sato, M., Numich, R., Matsushiro, A.. Inokuchi, H. & Ozeki, H.
(1968). Virology, 34, 637449. Shapiro, J. A. & Adhya, S. (1969). Genetics, 62, 249271. Signer, E. R. & Weil, H. (1968). J. Mol. BioZ. 34, 261-271. Skalka, A., Poonian, M. & Bartl, P. (1972). J. Mol. Biol. 64, 541-550. Soil, L. & Berg. P. (1969). Proc. ,I’&. ilcad. Sci.. l’.S.A. 63, 392-399. Sternberg, N. (1976). Virology, 71, 568-582. Sternberg, N. & Weisberg, R. (1977a). J. Mol. BioZ. 117, 717-731. Sternberg, S. & Weisberg, R. (1977b). J. Mol. Biol. 117, 733-759. Sternberg, S., Tiemeirer, D. & Enquist, L. (1977). Gene, 1, 255-280. Sussman, R. & Jacob, F. (1962). C. R. H. Acad. 8ci. 254, 1517-1519. Tomizawa, J. -I. & Ogawa, S. (1968). Cold Spring Harbor Symp. Quant. Biol. 33, 533151. Wake, R. G., Kaiser, A. D. & Inman, R. B. (1972). J. Mol. BioZ. 64, 519-540. Wang, J. C. & Davidson, N. (1966). J. Mol. BioZ. 15, 111-123. Wang, J. (1. & Kaiser, A. D. (1973). h’ature Kew Biol. 241, 16-17. Weigle, J. (1966). Proc. Nat. Acud. hi., IT.S.,4. 55, 146221466. Weisberg, R. A., Steinberg, N. & Gallay, E. (1979). Virology, 95, 99106. Wu, R. & Taylor, E. (1971). J. Mol. BioZ. 57, 491-511. Yanofsky, C. & Ito, ,J. (1966). J. Mol. BioZ. 21, 313-334.