aids progression is associated with the emergence of il-17producing cells early after simian...
TRANSCRIPT

The Journal of Immunology
AIDS Progression Is Associated with the Emergence ofIL-17–Producing Cells Early After Simian ImmunodeficiencyVirus Infection
Laure Campillo-Gimenez,* Marie-Christine Cumont,† Michele Fay,‡ Hassen Kared,*
Valerie Monceaux,† Ousmane Diop,x Michaela Muller-Trutwin,{ Bruno Hurtrel,† Yves Levy,*,‖
John Zaunders,# Michel Dy,** Maria C. Leite-de-Moraes,** Carole Elbim,*,††,1 and
Jerome Estaquier*,†,‖,1
IL-17 is a potent effector cytokine involved in inflammatory response and antimicrobial defense. We report that SIV infection of
rhesus macaques (RMs) results in the emergence of IL-17–expressing cells during the acute phase. This subpopulation appears at
day 14 postinfection concomitantly with an increase in TGF-b and IL-18 expression. This subset, which exhibits phenotypic
markers of NK T cells (NKT), rather than Th17 CD4 cells, persists during the chronic phase and is higher in noncontrollers SIV-
infected RMs compared with controllers SIV-infected RMs. In contrast, in the nonpathogenic model of SIVagm infection of
African green monkeys, no change in the level of IL-17–expressing cells is observed in lymphoid organs. Consistent with the
emergence of TGF-b and IL-18 during the acute phase in SIV-infected RMs, but not in SIV-infected African green monkeys, we
demonstrate that in vitro TGF-b and IL-18 induce the differentiation and expansion of IL-17+NKT+. Altogether, these results
demonstrate that IL-17–producing NKT are associated with the pathogenesis of SIV in RMs and suggest that TGF-b and IL-18
play a role in their development. The Journal of Immunology, 2010, 184: 984–992.
Immunopathology mediated by inappropriate or poorly con-trolled effector T cell responses has typically been viewed inthe context of the Th cells (Th1 and Th2) paradigm. More
recently, a CD4+ T cell subset characterized by the production ofIL-17 and crucially involved in certain autoimmune, allergic, andinflammatory diseases was identified (1–4). Invariant NK T cells(NKT), which have a role in antitumor immune responses andantiviral immunity (5), have also been recently reported to pro-duce IL-17 in the context of inflammatory diseases (6, 7).TGF-b and inflammatory cytokines together induce the de-
velopment of Th17 cells from CD4+ T cells in mice and humans (8–10). In the immune system, TGF-b affects multiple cell lineages byeither promoting or opposing their differentiation, survival, and
proliferation (11). At present, TGF-b ismainly viewed as an immunesuppressive cytokinebecauseTGF-b is a critical factor for regulatoryT cells, and its loss is associated with a fatal lymphoproliferativedisease (12). TGF-b regulates the components of adaptive immunity,such as T cells, as well as innate immunity, such as NK cells (13). Incontrast, an inflammatory cytokine environment inhibits the gener-ation of regulatory T cells and instead leads to the differentiation ofTh17 cells. Inflammatory cytokines are strongly induced in cells ofthe innate immune system following engagement of specific pattern-recognition receptors such as TLRs and C-type lectin receptors.Thus, in addition to IL-6, TNF-a and IL-1 have been proposed tohave an additional role in the amplification of Th17 responses (14).The elevation of TGF-b (15–17) plus an inflammatory envi-
ronment may suggest the possible induction of Th17 population inthis context. However, recent reports indicated a lower frequencyof Th17 CD4+ T cells at mucosal and systemic sites during HIVinfection and SIV infection (18–21). Given the role of IL-17 incontrolling commensal bacteria, it has been proposed that de-pletion of Th17 may participate in the disruption of the mucosalbarrier. However, the absence of Th17 CD4+ during the acutephase is not associated with an increase in LPS translocation (22),which suggests the possibility of additional IL-17+–expressingcells that control intestinal flora translocation compensating forthe defect in Th17 CD4+ T cells. However, whether the loss ofIL-17–expressing cells is also true in intact secondary lymphoidtissues and whether other cells, such as NKT expressing IL-17,emerge during SIV infection is presently unknown.
Materials and MethodsAnimals and virus infection
Twenty-four rhesus macaques (RMs; Macaca mulatta) were inoculated i.v.with the pathogenic SIVmac251 strain (10 50% animal infectious doses).All the animals were challenged with the same batch of virus, titrated invivo in RMs, and stored in liquid nitrogen. Six African green monkeys
*Institut National de la Sante et de la Recherche Medicale (U 955), Faculte deMedecine de Creteil, ‖Assistance Publique Hopitaux de Paris, Hopital Henri Mondor,Creteil; †Unite de Physiopathologie des Infections Lentivirales, {Unite de Regulationdes Infections Retrovirales, Institut Pasteur; ‡Universite Paris 7 Denis Diderot, Fac-ulte de Medecine, Site Bichat; **Unite Mixte de Recherche 8147, Faculte de Mede-cine Rene Descartes, Paris V, Hopital NeckerParis, Cedex 15; ††Centre de Recherchedes Cordeliers, Universite Pierre et Marie Curie–Paris 6, UMR S 872, Paris, France;xInstitut Pasteur, Dakar, Senegal; and #Centre for Immunology, St. Vincent’s Hospi-tal, Darlinghurst, New South Wales, Australia
1C.E. and J.E. contributed equally to this work.
Received for publication July 17, 2009. Accepted for publication November 12,2009.
L.C.-G. was supported by a grant from Ministere de la Recherche et de l’Enseigne-ment of Paris XI University. Funding from the Agence Nationale de Recherches surle SIDA and Fondation pour la Recherche Medicale to J.E. supported this work.
Address correspondence and reprint requests to Dr. Jerome Estaquier, INSERM (U955), Faculte de Medecine de Creteil, Hopital Henri Mondor, 8 Rue du GeneralSarrail, 94010 Creteil, France. E-mail address: [email protected]
Abbreviations used in this paper: AGM, African green monkey; Ax, axillary; FSC,forward light scatter; LN, lymph node; Mes, mesenteric; NKT, NK T cell; RM,rhesus macaque; SIV+ RM, rhesus macaque infected with the pathogenic SIVmac251strain; SSC, side scatter.
Copyright� 2010 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0902316
(AGMs) of sabaeus species were experimentally infected with 300 50%tissue culture-infective dose of SIVagm.sab92018 strain. As previouslydescribed (23), this virus derives from a naturally infected AGM and hasnever been cultured in vitro. Animals were demonstrated as being sero-negative for simian T leukemia virus type-1), SRV-1 (type D retrovirus),herpes B viruses, and SIVmac. Animals were housed and cared for incompliance with existing French regulations (Institut Pasteur, Paris,France). The organs were collected and frozen in isopentane cooled inliquid nitrogen. Organs were cut into 4-mm sections on a cryostat, and thesections were stored at 280˚C until use (16).
Viral quantification
RNA was extracted from plasma of SIV-infected monkeys using the TRIReagent BD Kit (Molecular Research Center, Cincinnati, OH). Real-timequantitative RT-PCR was used to determine viral loads as previously de-scribed (24). Productively infected cells were assessed in lymph nodes(LNs) by in situ hybridization using a [35S]-labeled RNA probe derivedfrom the SIVmac nef gene, and the frequency of SIV infected cells wasmeasured by limiting dilution PCR as described (24).
Immunohistochemical analysis
Tissues were prepared for immunohistochemistry as described (16). Briefly,cryostat sections (4 to 5 mm) were fixed in acetone for 10 min and stored at220˚C until use. For staining, tissue sections were fixed in 2% para-formaldehyde, permeabilized with Triton 0.05%, then incubated withmAbs against IL-17 (eBioscience, San Diego, CA), and TGF-b (9016,R&D Systems, Minneapolis, MN), for 2 h, followed by biotinylated goat
anti-mouse Ab and streptavidin peroxidase complex (Vectastain ABC kit,Vector Laboratories, Burlingame, CA). Analyses were performed on fourdifferent slides, in a blind manner, by two investigators using a Nikon-FXAmicroscope (Nikon, Melville, NY) and the Optilab Pro 2.5 software(Graftek Imaging, Austin, TX). The numbers of cells counted were dividedby the surface of the entire tissue section. The results were expressed ascell numbers/2 mm2 sections. Semiquantitative analyses were also de-termined by measuring the level of staining (numbers of pixels of the entiretissue section) divided by the total surface. The results were expressed asa number of positive cells.
Cytokine assays
Blood from monkeys was collected in sterile EDTA-treated vacuum tubesand immediately centrifuged at 400 g for 15 min at 4˚C to avoid cytokinesynthesis in vitro. IL-6, TNF-a, and IL-1b were detected simultaneously inplasma by using the human inflammatory cytokine cytometric bead arraykit (BD Pharmingen, San Diego, CA), which has been validated for cy-tokine measurement in RMs and AGMs. The cytometric bead arrayworking range was 20–5000 pg/ml for each cytokine. IL-18 in plasma wasmeasured by using an ELISA kit (MBL Biomedical, Clinisciences), andstimulation of cells from healthy monkeys demonstrated no difference inIL-18 expression between RM and AGM. We also used ELISA to mea-sure type I IFN (PBL interferon source, Clinisciences) validated for bothspecies.
PBMCs from monkeys were isolated from peripheral blood by densitygradient centrifugation LymphoPrep (PAA Laboratories, Pasching, Austria)and cultured in RPMI 1640 supplemented with 10% FCS, 1% glutamine,1% pyruvate, and 1% antibiotics. Up to 500,000 PBMCs were cultured
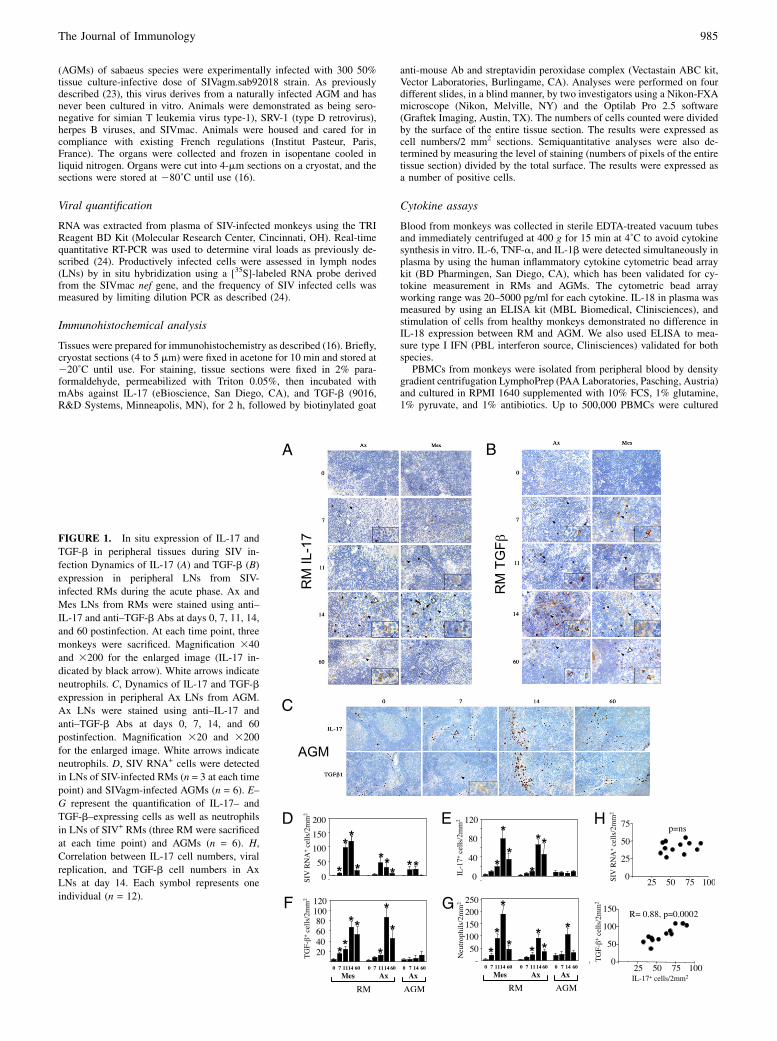
FIGURE 1. In situ expression of IL-17 and
TGF-b in peripheral tissues during SIV in-
fection Dynamics of IL-17 (A) and TGF-b (B)
expression in peripheral LNs from SIV-
infected RMs during the acute phase. Ax and
Mes LNs from RMs were stained using anti–
IL-17 and anti–TGF-b Abs at days 0, 7, 11, 14,
and 60 postinfection. At each time point, three
monkeys were sacrificed. Magnification 340
and 3200 for the enlarged image (IL-17 in-
dicated by black arrow). White arrows indicate
neutrophils. C, Dynamics of IL-17 and TGF-b
expression in peripheral Ax LNs from AGM.
Ax LNs were stained using anti–IL-17 and
anti–TGF-b Abs at days 0, 7, 14, and 60
postinfection. Magnification 320 and 3200
for the enlarged image. White arrows indicate
neutrophils. D, SIV RNA+ cells were detected
in LNs of SIV-infected RMs (n = 3 at each time
point) and SIVagm-infected AGMs (n = 6). E–
G represent the quantification of IL-17– and
TGF-b–expressing cells as well as neutrophils
in LNs of SIV+ RMs (three RM were sacrificed
at each time point) and AGMs (n = 6). H,
Correlation between IL-17 cell numbers, viral
replication, and TGF-b cell numbers in Ax
LNs at day 14. Each symbol represents one
individual (n = 12).
The Journal of Immunology 985
overnight. Cells were incubated in the presence of brefeldin A (5 mg/mlfor the last 12 h) before being stained for FITC–anti-CD4 and PerCP–anti-CD3 (BD Biosciences, San Jose, CA), then washed twice in PBS, andfurther incubated with PE–anti-IFN-g or PE–anti-TNF-a and APC–anti-IL-17A. Cells were also stained with PE-CD8 (RPA-T8), PE-CD56(MY31), PE-HLA-DR (G46-6), FITC-CD161 (DX12), PE-CD25 (M-A251), PE-CD27 (M-T271), and PE-CD95 (DX2) from BD Biosciences;PE-CXCR6 (56811) and PE-CCR6 (53103) from R&D Systems; PE-CD127 (R34.34) and FITC–anti-Va24 (clone C15) from Beckman Coulter(Fullerton, CA); and PE–anti-human invariant NKT (clone 6B11, BDPharmingen), PerCP–anti-CD4, and APC–anti-IL-17A. We also used anti-human FITC–anti-CD45RA (clone 2H4, Beckman Coulter, Fullerton, CA)and PE–anti-CD62L (clone SK11, BD Biosciences). Samples were ana-lyzed on an FACScalibur cytometer with CellQuest software (CellQuest,Tampa, FL).
In vitro differentiation of NKT
Human PBMCs were isolated from peripheral blood by density gradientcentrifugation on LymphoPrep (PAA Laboratories). Up to 500,000 PBMCswere stimulated with Con A (1 mg/ml) and then incubated with either TGF-b alone (5 ng/ml), IL-18 alone (50 ng/ml), or TGF-b plus IL-18 for 5 d at37˚C with 5% CO2 in RPMI 1640 with 5% of FCS. At day 6, cells wereactivated with PMA (100 ng/ml) plus ionomycin (300 ng/ml) for 5 h in thepresence of brefeldin A (5 mg/ml). This experiment was conducted usinghuman cells and human cytokines to preclude the possibility that thesecytokines might not have cross-species reactivity.
Statistical analysis
Data are reported as means 6 SEM. Comparisons were based on ANOVAand Tukey’s post hoc test, using Prism 3.0 software (GraphPad, San Diego,CA). Student t test and Mann-Whitney U test (Prism software, GraphPad)were also used to determine whether differences were significant if p ,0.05. Correlations were identified by means of the Spearman rank corre-lation coefficient (r). Differences were considered significant if p , 0.05.
ResultsEmergence of IL-17–expressing cells during primary SIVinfection
Byimmunochemistryanalyses,weclearlyobservedIL-17–expressingcells in axillary (Ax) andmesenteric (Mes) LNs of RMs infectedwiththe pathogenic SIVmac251 strain (SIV+ RM) (Fig. 1A). The quanti-fication of IL-17+–producing cells from RMs, killed at different timepoints postinfection (n = 3 RMs, at each time point), revealed a sig-nificant increase at day 14 (Fig. 1A, 1E) concomitantly with the peakof viral replication in these organs (Fig. 1D). These cells weremostlylocated within the paracortical T cell zone. TGF-b, assessed in thesame tissue sections, progressively increased and peaked at day 14(Fig. 1B, 1F). Thereafter, at day 60, IL-17– as well as TGF-b–expressing cells decreased both in Ax and Mes LNs but remainedhigher thanpreinfection.By analyzing agroupof 12monkeys (24, 25)at day 14, we found a positive correlation between the numbers ofIL-17–expressing cells and thenumbers ofTGF-b–expressingcells inAx LNs of SIV+ RM, whereas no correlation was observed with theextent of viral replication inLNs (Fig. 1H). Although SIVinfection ofRMs leads to progressive CD4+ T cell depletion and AIDS, SIVagminfection ofAGMis nonpathogenic despite levels of plasmaviral loadsimilar to those observed in SIVmac-infectedRMs (23, 26, 27). In thisstudy, in stark contrast toRMs,wedid not detect an increase in IL-17–and TGF-b–positive cells in peripheral LNs from six SIV-infectedAGMs (Fig. 1C, 1E, 1F). A role for IL-17 in the recruitment ofneutrophils in inflamed tissues hasbeenproposed (2). In this study,wefound, despite the absence of IL-17, the presence of neutrophils inLNs of SIV-infected AGMs at day 14, similar to that observed in AxLN of SIV-infected RMs (Fig. 1G), suggesting the possible role ofadditional cytokines (i.e., IL-8) in the recruitment of neutrophils innonpathogenic monkeys.These results demonstrate the differential expansion of IL-17–
expressing cells early postinfection in the pathogenic primatemodel of SIV infection in RMs.
Persistence of IL-17–expressing cells during the chronic phaseof SIV infection
We recently demonstrated during the chronic phase of SIV+ RMthat TGF-b expression is significantly increased in noncontrollersversus controllers (16). We thus quantified the expression of IL-17–expressing cells in Ax and Mes LNs of 10 SIV+ RMs (5 werenoncontrollers as previously described (16). Whereas no IL-17–expressing cells were found in a healthy monkey (#93750), weobserved increased levels of IL-17–expressing cells in Ax LNsfrom SIV+ monkeys; these levels were higher in noncontrollers (asshown in monkeys #94746 and #272) than in controllers (as shown
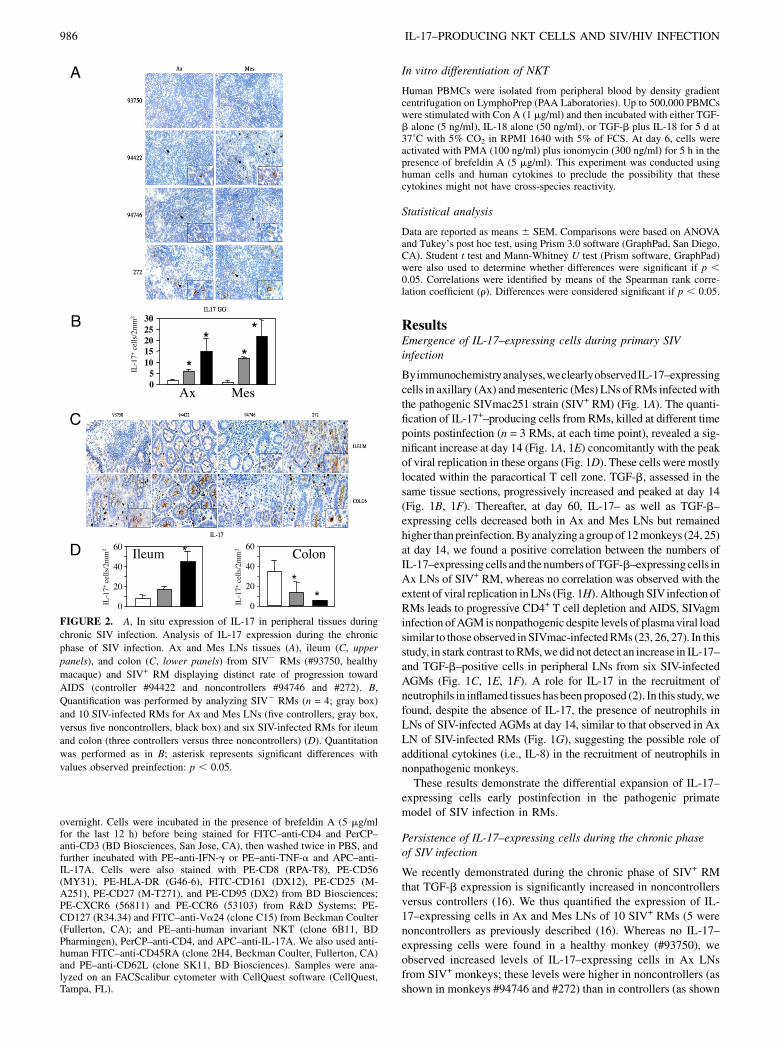
FIGURE 2. A, In situ expression of IL-17 in peripheral tissues during
chronic SIV infection. Analysis of IL-17 expression during the chronic
phase of SIV infection. Ax and Mes LNs tissues (A), ileum (C, upper
panels), and colon (C, lower panels) from SIV2 RMs (#93750, healthy
macaque) and SIV+ RM displaying distinct rate of progression toward
AIDS (controller #94422 and noncontrollers #94746 and #272). B,
Quantification was performed by analyzing SIV2 RMs (n = 4; gray box)
and 10 SIV-infected RMs for Ax and Mes LNs (five controllers, gray box,
versus five noncontrollers, black box) and six SIV-infected RMs for ileum
and colon (three controllers versus three noncontrollers) (D). Quantitation
was performed as in B; asterisk represents significant differences with
values observed preinfection: p , 0.05.
986 IL-17–PRODUCING NKT CELLS AND SIV/HIV INFECTION
in monkey #94422) (Fig. 2A, 2B). In Mes LNs, the levels of IL-17–expressing cells in SIV+ RMs were also higher compared withhealthy monkeys (Fig. 2A, 2B), a result that is consistent with theexpression of TGF-b (16). We also studied IL-17 expression inGALTs discriminating small and large intestine (ileum and colon,respectively). We found that SIV infection induces increasednumbers of IL-17+ cells in the ileum that is higher in non-controllers (monkeys #94746 and #272) than in controllers(monkey #94422) (Fig. 2C, 2D) and higher than that observed inhealthy macaques (#93750). Moreover, whereas we detected largenumbers of IL-17+ cells in the colon of healthy RMs (#93750),which may reflect the presence of commensal bacteria (28), weobserved a progressive depletion of these cells in animals pro-gressing toward AIDS (Fig. 2C, 2D). In Indian RMs, which aremore rapid progressors than Chinese RMs, it has also been shownthat there are differential dynamics between large and small in-testines (19). Thus, in large intestine, the depletion is higher.Altogether,our tissueanalysesdemonstrategreaterexpressionof IL-
17 cells associated with the progression to AIDS, except in the colon.
The IL-17+ subpopulation exhibits phenotypic markers of NKT
In addition to classical Th17 cells, NKT have been recently reportedto produce IL-17 (6, 7, 29). Therefore, we next investigated the
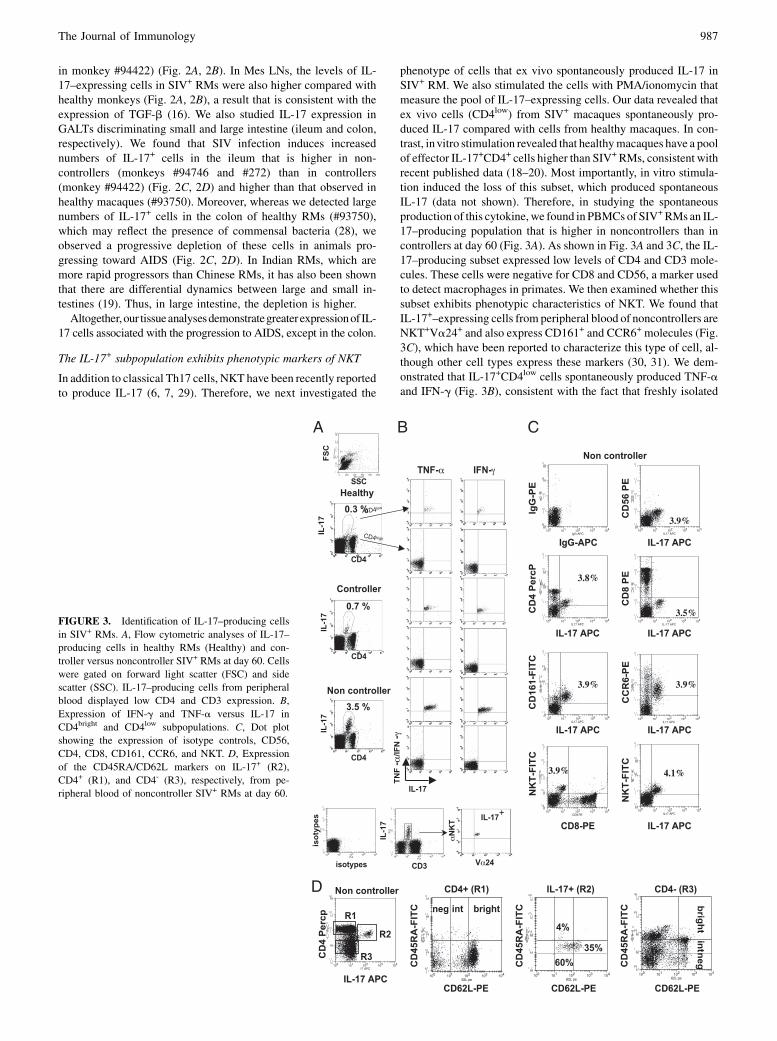
phenotype of cells that ex vivo spontaneously produced IL-17 inSIV+ RM. We also stimulated the cells with PMA/ionomycin thatmeasure the pool of IL-17–expressing cells. Our data revealed thatex vivo cells (CD4low) from SIV+ macaques spontaneously pro-duced IL-17 compared with cells from healthy macaques. In con-trast, invitro stimulation revealed that healthymacaques have a poolof effector IL-17+CD4+ cells higher than SIV+RMs, consistent withrecent published data (18–20). Most importantly, in vitro stimula-tion induced the loss of this subset, which produced spontaneousIL-17 (data not shown). Therefore, in studying the spontaneousproduction of this cytokine, we found in PBMCs of SIV+RMs an IL-17–producing population that is higher in noncontrollers than incontrollers at day 60 (Fig. 3A). As shown in Fig. 3A and 3C, the IL-17–producing subset expressed low levels of CD4 and CD3 mole-cules. These cells were negative for CD8 and CD56, a marker usedto detect macrophages in primates. We then examined whether thissubset exhibits phenotypic characteristics of NKT. We found thatIL-17+–expressing cells from peripheral blood of noncontrollers areNKT+Va24+ and also express CD161+ and CCR6+ molecules (Fig.3C), which have been reported to characterize this type of cell, al-though other cell types express these markers (30, 31). We dem-onstrated that IL-17+CD4low cells spontaneously produced TNF-aand IFN-g (Fig. 3B), consistent with the fact that freshly isolated
FIGURE 3. Identification of IL-17–producing cells
in SIV+ RMs. A, Flow cytometric analyses of IL-17–
producing cells in healthy RMs (Healthy) and con-
troller versus noncontroller SIV+ RMs at day 60. Cells
were gated on forward light scatter (FSC) and side
scatter (SSC). IL-17–producing cells from peripheral
blood displayed low CD4 and CD3 expression. B,
Expression of IFN-g and TNF-a versus IL-17 in
CD4bright and CD4low subpopulations. C, Dot plot
showing the expression of isotype controls, CD56,
CD4, CD8, CD161, CCR6, and NKT. D, Expression
of the CD45RA/CD62L markers on IL-17+ (R2),
CD4+ (R1), and CD4- (R3), respectively, from pe-
ripheral blood of noncontroller SIV+ RMs at day 60.
The Journal of Immunology 987
NKT from histologically tumor-bearing human tissues, were morepotent producers of TNF-a and IFN-g (32). We also analyzed theexpression of CD62L and CD45RAmarkers. Instead of being eitherCD62Lbright or CD62Lneg and either CD45RAbright or CD45RAneg
cells like CD4+ or CD42 T cells from peripheral blood of non-controllers (Fig. 3D), this population, as previously described (33),is mostly CD45RAint and CD62Lint (Fig. 3D). Gating on IL-17+
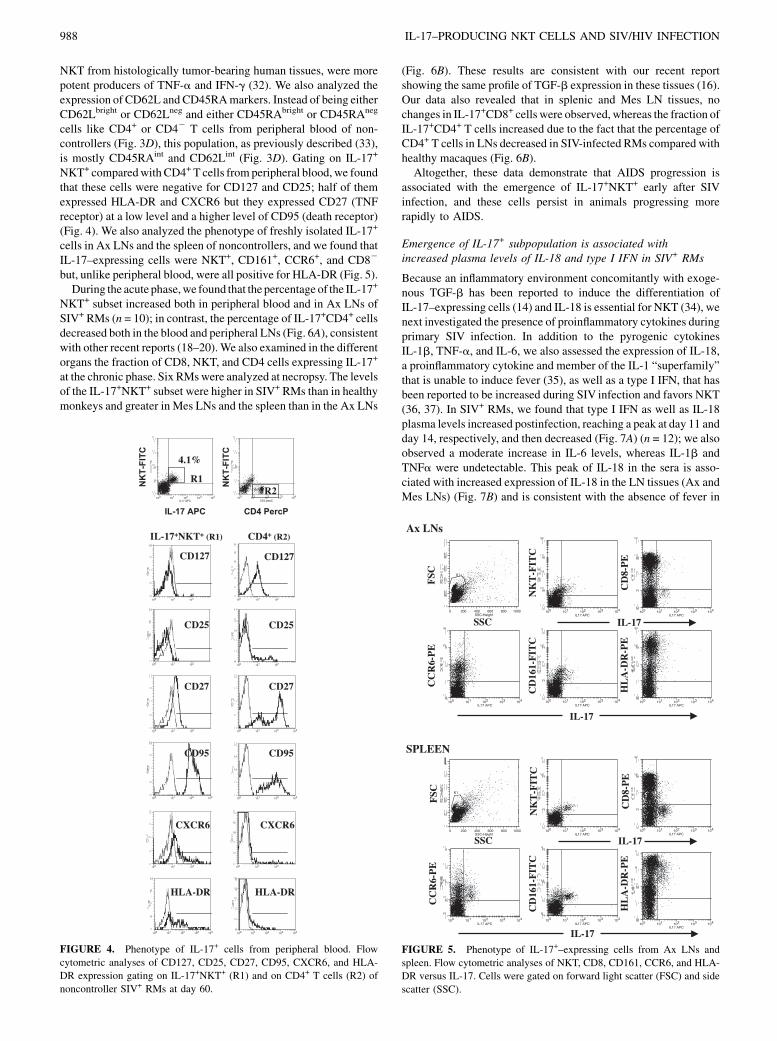
NKT+ comparedwith CD4+ T cells fromperipheral blood,we foundthat these cells were negative for CD127 and CD25; half of themexpressed HLA-DR and CXCR6 but they expressed CD27 (TNFreceptor) at a low level and a higher level of CD95 (death receptor)(Fig. 4). We also analyzed the phenotype of freshly isolated IL-17+
cells in Ax LNs and the spleen of noncontrollers, and we found thatIL-17–expressing cells were NKT+, CD161+, CCR6+, and CD82
but, unlike peripheral blood, were all positive for HLA-DR (Fig. 5).During the acute phase,we found that the percentage of the IL-17+
NKT+ subset increased both in peripheral blood and in Ax LNs ofSIV+ RMs (n = 10); in contrast, the percentage of IL-17+CD4+ cellsdecreased both in the blood and peripheral LNs (Fig. 6A), consistentwith other recent reports (18–20).We also examined in the differentorgans the fraction of CD8, NKT, and CD4 cells expressing IL-17+
at the chronic phase. Six RMswere analyzed at necropsy. The levelsof the IL-17+NKT+ subset were higher in SIV+ RMs than in healthymonkeys and greater in Mes LNs and the spleen than in the Ax LNs
(Fig. 6B). These results are consistent with our recent reportshowing the same profile of TGF-b expression in these tissues (16).Our data also revealed that in splenic and Mes LN tissues, nochanges in IL-17+CD8+ cells were observed, whereas the fraction ofIL-17+CD4+ T cells increased due to the fact that the percentage ofCD4+ T cells in LNs decreased in SIV-infected RMs compared withhealthy macaques (Fig. 6B).Altogether, these data demonstrate that AIDS progression is
associated with the emergence of IL-17+NKT+ early after SIVinfection, and these cells persist in animals progressing morerapidly to AIDS.
Emergence of IL-17+ subpopulation is associated withincreased plasma levels of IL-18 and type I IFN in SIV+ RMs
Because an inflammatory environment concomitantly with exoge-nous TGF-b has been reported to induce the differentiation ofIL-17–expressing cells (14) and IL-18 is essential for NKT (34), wenext investigated the presence of proinflammatory cytokines duringprimary SIV infection. In addition to the pyrogenic cytokinesIL-1b, TNF-a, and IL-6, we also assessed the expression of IL-18,a proinflammatory cytokine and member of the IL-1 “superfamily”that is unable to induce fever (35), as well as a type I IFN, that hasbeen reported to be increased during SIV infection and favors NKT(36, 37). In SIV+ RMs, we found that type I IFN as well as IL-18plasma levels increased postinfection, reaching a peak at day 11 andday 14, respectively, and then decreased (Fig. 7A) (n = 12); we alsoobserved a moderate increase in IL-6 levels, whereas IL-1b andTNFa were undetectable. This peak of IL-18 in the sera is asso-ciated with increased expression of IL-18 in the LN tissues (Ax andMes LNs) (Fig. 7B) and is consistent with the absence of fever in
FIGURE 4. Phenotype of IL-17+ cells from peripheral blood. Flow
cytometric analyses of CD127, CD25, CD27, CD95, CXCR6, and HLA-
DR expression gating on IL-17+NKT+ (R1) and on CD4+ T cells (R2) of
noncontroller SIV+ RMs at day 60.
FIGURE 5. Phenotype of IL-17+–expressing cells from Ax LNs and
spleen. Flow cytometric analyses of NKT, CD8, CD161, CCR6, and HLA-
DR versus IL-17. Cells were gated on forward light scatter (FSC) and side
scatter (SSC).
988 IL-17–PRODUCING NKT CELLS AND SIV/HIV INFECTION

SIV-infected RMs during the acute phase. Most interestingly, de-spite similar levels of viral replication, IL-18 and type I IFN wereonly slightly increased in SIV-infected AGMs.Thus, we found cytokine profiles that were distinct in pathogenic
compared with nonpathogenic SIV infection.
In vitro TGF-b and IL-18 induces the differentiation andexpansion of IL-17+NKT+
Because IL-18 is essential for NKT (34) and because it has beenproposed that IL-6 is dispensable for IL-17+NKT (29), we thendetermined the involvement of IL-18 in the expansion and in theinduction of IL-17+NKT in SIV+ RMs. Thus, we investigatedwhether IL-18 concomitantly with TGF-b might induce in vitrothe differentiation of IL-17+NKT. PBMCs from human healthydonors were used to test the effect of these cytokines to avoid anypossible defect in cross-reactivity in monkeys. Cells were firstactivated with Con A and then incubated in the absence or pres-ence of TGF-b and IL-18. At day 6, the cells were restimulatedwith PMA plus ionomycin (Fig. 8A, 8B). We found that TGF-balone increased the proportion of the cells that differentiated intoIL-17+NKT as well as IL-17+CD4+ cells. Given the role of stim-ulated APCs in the release of inflammatory cytokines, it is possiblethat the initial Con A stimulation also contributed to the in vitrodifferentiation of IL-17+ cells. IL-18 alone also increased thepercentage of IL-17+NKT, whereas it had no effect on the per-centage of IL-17+CD4+ cells. The combination of both TGF-b andIL-18 enhanced the percentage of both IL-17+CD4+ and IL-17+
NKT as compared with each cytokine alone. To determine whetherthis increase was reflected by the expansion and/or differentiationof these subsets, we evaluated the numbers of these cells in cul-ture. Each cytokine, TGF-b and IL-18 alone, reduced the numbersof IL-172NKT+, leading to a relative increase in the percentage ofIL-17–expressing NKT (Fig. 8C); in contrast, TGF-b together withIL-18 induced the expansion of these IL-17+NKT. Whereas TGF-balone induced the expansion of IL-17+CD4+, the addition of IL-18in culture with TGF-b enhanced their expansion. Thus, increasedIL-18 expression concomitantly with elevated TGF-b in SIV+
RMs may contribute to the induction of the IL-17+NKT subset.
DiscussionRecently, several groups have shown a decline of Th17CD4+ T cellsduring HIVand SIV infection and proposed that the absence of thissubset may participate in the disruption of mucosal barrier integrity(18–21). In this study, our results demonstrated the early expansion
of IL-17–expressing cells in SIV+ RMs that is correlated with TGF-b expression. Thus, an innate IL-17 production by NKT is rapid andprecedes the adaptive Th17 response. The emergence of this IL-17+
NKT+ population in SIV-infected RMs, therefore, could compen-sate for the defect in Th17 CD4+ T cells (19, 21), preventing mi-crobial translocation (no LPS was detected during the acute phase(20) and the occurrence of a wasting syndrome early postinfection.Moreover, in chronically SIV-infected monkeys (19) as well as inHIV-infected Africans (38), however, microbial translocation wasnot correlated with immune activation and viral replication. Thus,microbial translocation might be a symptom of the defect of IL-17populations and not a direct cause of HIV-1 disease. The observa-tion that SIV-infected AGMs that do not progress to disease have noexpansion of IL-17–expressing cells is consistent with the resultsfrom another group (20) that found to be unchanged numbers ofIL-17 cells in peripheral blood in the nonpathogenic model of Sootymangabey. The possible difference in the previous reports could bethe origin of the monkeys used, given that we used RMs of Chineseorigin instead of RMs of Indian origin, the latter being more sus-ceptible to SIV infection (26), and we used freshly isolated cellsinstead of frozen samples to perform these assays. Another point isour observation that ex vivo activation induced the loss of thissubset, suggesting the hypothesis that cells might be more prone todie (activation-induced cell death) because they express CD95 andCD27. Finally, to our knowledge, no other groups have assessed theexpression of IL-17 in the tissues, and therefore this is the firstreport of this observation. Therefore, although we cannot excludethe possibility that other cells in the tissues express IL-17, our datastrongly suggest that most of the freshly isolated cells that producedIL-17 were NKT.It is now clear that IL-17 is associated with a number of human
autoimmune disorders by inducing excessive inflammation. IL-17
FIGURE 6. Dynamics of NKT+, IL-17+NKT+, and IL-17+CD4+ T cells.
A, Cells were analyzed from peripheral blood and LNs at days 0, 14, and 60
from SIV+ RMs (n = 10). The percentages of these subsets of cells, re-
spectively, are expressed as mean 6 SEM. B, Graphs show the fractions of
IL-17–expressing cells gating on CD8, NKT, and CD4 cells in Ax and Mes
LNs and the spleen of healthy (SIV2, n = 4) and chronically SIV-infected
RMs (SIV+) (n = 6). IL-17–producing cells are expressed as mean6 SEM.
FIGURE 7. Dynamics of proinflammatory cytokines during primary
infection in SIV-infected monkeys. A, Levels of IL-18, IL-6, IL-1b, TNF-
a, and type I IFN were measured in the sera of SIV+ RMs (n = 12) and in
SIV+ AGMs (n = 6) at days 0, 7, 11, 14, 21, 30, and 60 postinfection. B,
Expression of IL-18 in Mes and Ax LNs. Analyses were performed at days
0, 7, and 14 postinfection.
The Journal of Immunology 989

exerts a potent effect against fibroblasts, epithelial cells, and en-dothelial cells and promotes the infiltration of inflammatory T cellsand neutrophils (39, 40). Thus, the early emergence of this novelpopulation expressing IL-17 could contribute to local tissue dam-age and favor viral dissemination. Despite this side effect, there isalso considerable evidence, in mice, that IL-17 is important in hostresponses to infection by Gram-negative bacteria, specificallyKlebsiella, Pseudomonas, Escherichia coli, Salmonella, andBordetella species (41, 42). Whereas we found higher levels ofIL-17–expressing cells in the ileum, our data demonstrated theabsence of IL-17–expressing cells in the colon of noncontrollersSIV+ RMs during the chronic phase. This could contribute to theabsence of bacterial control, consistent with the clinical observationthat animals progressing toward AIDS develop a wasting syndromecharacterized by a loss of weight and frequent diarrhea. These re-sults may be related to a distinct dynamic of IL-17+ cells in thesmall and large intestine of SIV-infected monkeys or a re-distribution of IL-17+ cells from the colon to the ileum.TGF-b and inflammatory cytokines together induce the de-
velopment of IL-17–expressingCD4+ T cells (8–10) aswell as NKT(6, 7). Our results demonstrate for the first time a role for IL-18 inthe genesis of IL-17+ cells consistent with its role in the emergenceof NKT (34). Caspase-1, a member of the cysteine protease familyinvolved in apoptosis, is involved in the maturation and secretion of
IL-18 (35). Therefore, the absence of IL-18 in SIV-infected AGMscould reflect the absence of apoptosis that we and others have de-scribed in this nonpathogenic primate model, compared withpathogenic primate models, during both the acute (26) and thechronic phase (24, 43–46). Thus, a vicious cycle may be generatedby the occurrence of apoptotic cells in the tissues of SIV+ RMs,favoring not only the expression of IL-18, but also, through theirphagocytosis by macrophages and dendritic cells, the production ofTGF-b and the absence of TNF-a and IL-1b (47), favoring theemergence of IL-17+NKT+ over IL-17+CD4+ T cells.However, how is the expansion of NKT early after SIV infection
generated? Viral danger signals through the type I IFN pathwaytriggerNKTactivation bymyeloid dendritic cells (48, 49). DNAandRNA viruses induce type I IFN through TLR7 and TLR9, and, inparticular, HIV has been shown to induce type I IFN (50). Recently,several groups (36, 37) have shown increased levels of type I IFNearly after SIV infection of RMs, whereas in nonpathogenic SI-Vagm infection, these levels are relatively low (23). Thus, the dy-namics of IL-17+NKT is consistent with the expression of type IIFN during the acute phase observed in SIV-infected RMs.In conclusion, we demonstrated the emergence of IL-17–pro-
ducing cells early after SIV infection. The levels of these cells,and their subsequent persistence in peripheral blood and in a largenumber of tissues, except colon, were associated with progression
FIGURE 8. Involvement of IL-18 in the expansion
of IL-17+NKT+. A, Human PBMCs were stimulated
with Con A (1 mg/ml) and then incubated with RPMI
1640 (Med), TGF-b (5 ng/ml), IL-18 (50 ng/ml), or
TGF-b plus IL-18 for 6 d at 37˚C. Cells were then
stimulated with PMA (100 ng/ml) and ionomycin (300
ng/ml) for 5 h at 37˚C in the presence of brefeldin A (5
mg/ml). Cells were stained with PE–anti-NKT, PerCP–
anti-CD4, and APC–anti-IL-17. Cells were gated on
live CD4+ T cells and NKT+ and dot plots show IL-17
expression. The cutoff reflects isotype controls. B,
Percentages of IL-17–expressing cells among NKT+
and CD4+ populations. Results are the mean 6 SD of
three independent xperiments. C, Cell numbers were
measured in parallel to A to determine the expansion of
NKT+IL-172, NKT+IL-17+, CD4+IL-172, and CD4+
IL-17+. One experiment is shown, out of three, giving
similar results.
990 IL-17–PRODUCING NKT CELLS AND SIV/HIV INFECTION

to AIDS. Moreover, our results show for the first time the ex-pansion of the IL-17+NKT+ subset in response to microbial in-fections. Our results strongly suggest the role of IL-18, in additionto TGF-b, in the differentiation of this novel subpopulation.
AcknowledgmentsWe thank J.M. Panaud (Institut Pasteur) for assistance with micro-
photographs.
DisclosuresThe authors have no financial conflicts of interest.
References1. Nakae, S., Y. Komiyama, A. Nambu, K. Sudo, M. Iwase, I. Homma,
K. Sekikawa, M. Asano, and Y. Iwakura. 2002. Antigen-specific T cell sensiti-zation is impaired in IL-17-deficient mice, causing suppression of allergic cel-lular and humoral responses. Immunity 17: 375–387.
2. Kolls, J. K., and A. Linden. 2004. Interleukin-17 family members and in-flammation. Immunity 21: 467–476.
3. Bettelli, E., T. Korn, and V. K. Kuchroo. 2007. Th17: the third member of theeffector T cell trilogy. Curr. Opin. Immunol. 19: 652–657.
4. Aujla, S. J., P. J. Dubin, and J. K. Kolls. 2007. Th17 cells and mucosal hostdefense. Semin. Immunol. 19: 377–382.
5. Bendelac, A., P. B. Savage, and L. Teyton. 2007. The biology of NKT cells.Annu. Rev. Immunol. 25: 297–336.
6. Michel, M. L., A. C. Keller, C. Paget, M. Fujio, F. Trottein, P. B. Savage,C. H. Wong, E. Schneider, M. Dy, and M. C. Leite-de-Moraes. 2007. Identifi-cation of an IL-17-producing NK1.1(neg) iNKT cell population involved inairway neutrophilia. J. Exp. Med. 204: 995–1001.
7. Pichavant, M., S. Goya, E. H. Meyer, R. A. Johnston, H. Y. Kim,P. Matangkasombut, M. Zhu, Y. Iwakura, P. B. Savage, R. H. DeKruyff, et al.2008. Ozone exposure in a mouse model induces airway hyperreactivity thatrequires the presence of natural killer T cells and IL-17. J. Exp. Med. 205: 385–393.
8. Bettelli, E., Y. Carrier, W. Gao, T. Korn, T. B. Strom, M. Oukka, H. L. Weiner,and V. K. Kuchroo. 2006. Reciprocal developmental pathways for the generationof pathogenic effector TH17 and regulatory T cells. Nature 441: 235–238.
9. Manel, N., D. Unutmaz, and D. R. Littman. 2008. The differentiation of human T(H)-17 cells requires transforming growth factor-b and induction of the nuclearreceptor RORgammat. Nat. Immunol. 9: 641–649.
10. Mangan, P. R., L. E. Harrington, D. B. O’Quinn, W. S. Helms, D. C. Bullard,C. O. Elson, R. D. Hatton, S. M. Wahl, T. R. Schoeb, and C. T. Weaver. 2006.Transforming growth factor-b induces development of the T(H)17 lineage.Nature 441: 231–234.
11. Wan, Y. Y., and R. A. Flavell. 2007. ‘Yin-Yang’ functions of transforming growthfactor-b and T regulatory cells in immune regulation. Immunol. Rev. 220: 199–213.
12. Kulkarni, A. B., C. G. Huh, D. Becker, A. Geiser, M. Lyght, K. C. Flanders,A. B. Roberts, M. B. Sporn, J. M. Ward, and S. Karlsson. 1993. Transforminggrowth factor b 1 null mutation in mice causes excessive inflammatory responseand early death. Proc. Natl. Acad. Sci. USA 90: 770–774.
13. Letterio, J. J., and A. B. Roberts. 1998. Regulation of immune responses byTGF-b. Annu. Rev. Immunol. 16: 137–161.
14. Veldhoen, M., R. J. Hocking, C. J. Atkins, R. M. Locksley, and B. Stockinger.2006. TGFbeta in the context of an inflammatory cytokine milieu supports denovo differentiation of IL-17-producing T cells. Immunity 24: 179–189.
15. Kekow, J., W. Wachsman, J. A. McCutchan, M. Cronin, D. A. Carson, andM. Lotz. 1990. Transforming growth factor b and noncytopathic mechanisms ofimmunodeficiency in human immunodeficiency virus infection. Proc. Natl.Acad. Sci. USA 87: 8321–8325.
16. Cumont, M. C., V. Monceaux, L. Viollet, S. Lay, R. Parker, B. Hurtrel, andJ. Estaquier. 2007. TGF-b in intestinal lymphoid organs contributes to the deathof armed effector CD8 T cells and is associated with the absence of viruscontainment in rhesus macaques infected with the simian immunodeficiencyvirus. Cell Death Differ. 14: 1747–1758.
17. Estes, J. D., S. Wietgrefe, T. Schacker, P. Southern, G. Beilman, C. Reilly,J. M. Milush, J. D. Lifson, D. L. Sodora, J. V. Carlis, and A. T. Haase. 2007.Simian immunodeficiency virus-induced lymphatic tissue fibrosis is mediated bytransforming growth factor b 1-positive regulatory T cells and begins in earlyinfection. J. Infect. Dis. 195: 551–561.
18. Raffatellu, M., R. L. Santos, D. E. Verhoeven, M. D. George, R. P. Wilson,S. E. Winter, I. Godinez, S. Sankaran, T. A. Paixao, M. A. Gordon, et al. 2008.Simian immunodeficiency virus-induced mucosal interleukin-17 deficiencypromotes Salmonella dissemination from the gut. Nat. Med. 14: 421–428.
19. Cecchinato, V., C. J. Trindade, A. Laurence, J. M. Heraud, J. M. Brenchley,M. G. Ferrari, L. Zaffiri, E. Tryniszewska, W. P. Tsai, M. Vaccari, et al. 2008.Altered balance between Th17 and Th1 cells at mucosal sites predicts AIDSprogression in simian immunodeficiency virus-infected macaques. MucosalImmunol 1: 279–288.
20. Brenchley, J. M., M. Paiardini, K. S. Knox, A. I. Asher, B. Cervasi, T. E. Asher,P. Scheinberg, D. A. Price, C. A. Hage, L. M. Kholi, et al. 2008. Differential
Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral in-fections. Blood 112: 2826–2835.
21. Favre, D., S. Lederer, B. Kanwar, Z. M. Ma, S. Proll, Z. Kasakow, J. Mold,L. Swainson, J. D. Barbour, C. R. Baskin, et al. 2009. Critical loss of the balancebetween Th17 and T regulatory cell populations in pathogenic SIV infection.PLoS Pathog. 5: e1000295.
22. Brenchley, J. M., D. A. Price, T. W. Schacker, T. E. Asher, G. Silvestri, S. Rao,Z. Kazzaz, E. Bornstein, O. Lambotte, D. Altmann, et al. 2006. Microbialtranslocation is a cause of systemic immune activation in chronic HIV infection.Nat. Med. 12: 1365–1371.
23. Diop, O. M., M. J. Ploquin, L. Mortara, A. Faye, B. Jacquelin, D. Kunkel,P. Lebon, C. Butor, A. Hosmalin, F. Barre-Sinoussi, and M. C. Muller-Trutwin.2008. Plasmacytoid dendritic cell dynamics and a interferon production duringSimian immunodeficiency virus infection with a nonpathogenic outcome. J. Vi-rol. 82: 5145–5152.
24. Monceaux, V., J. Estaquier, M. Fevrier, M. C. Cumont, Y. Riviere,A. M. Aubertin, J. C. Ameisen, and B. Hurtrel. 2003. Extensive apoptosis inlymphoid organs during primary SIV infection predicts rapid progression to-wards AIDS. AIDS 17: 1585–1596.
25. Monceaux, V., L. Viollet, F. Petit, R. Ho Tsong Fang, M. C. Cumont,J. Zaunders, B. Hurtrel, and J. Estaquier. 2005. CD8+ T cell dynamics duringprimary simian immunodeficiency virus infection in macaques: relationship ofeffector cell differentiation with the extent of viral replication. J. Immunol. 174:6898–6908.
26. Cumont, M. C., O. Diop, B. Vaslin, C. Elbim, L. Viollet, V. Monceaux, S. Lay,G. Silvestri, R. Le Grand, M. Muller-Trutwin, et al. 2008. Early divergence inlymphoid tissue apoptosis between pathogenic and nonpathogenic simian im-munodeficiency virus infections of nonhuman primates. J. Virol. 82: 1175–1184.
27. Hurtrel, B., F. Petit, D. Arnoult, M. Muller-Trutwin, G. Silvestri, andJ. Estaquier. 2005. Apoptosis in SIV infection. Cell Death Differ. 12(Suppl 1):979–990.
28. Ivanov, I. I., Rde. L. Frutos, N. Manel, K. Yoshinaga, D. B. Rifkin, R. B. Sartor,B. B. Finlay, and D. R. Littman. 2008. Specific microbiota direct the differen-tiation of IL-17-producing T-helper cells in the mucosa of the small intestine.Cell Host Microbe 4: 337–349.
29. Rachitskaya, A. V., A. M. Hansen, R. Horai, Z. Li, R. Villasmil, D. Luger,R. B. Nussenblatt, and R. R. Caspi. 2008. Cutting edge: NKT cells constitutivelyexpress IL-23 receptor and RORgammat and rapidly produce IL-17 upon re-ceptor ligation in an IL-6-independent fashion. J. Immunol. 180: 5167–5171.
30. Exley, M., S. Porcelli, M. Furman, J. Garcia, and S. Balk. 1998. CD161 (NKR-P1A) costimulation of CD1d-dependent activation of human T cells expressinginvariant V a 24 J a Q T cell receptor a chains. J. Exp. Med. 188: 867–876.
31. Thomas, S. Y., R. Hou, J. E. Boyson, T. K. Means, C. Hess, D. P. Olson,J. L. Strominger, M. B. Brenner, J. E. Gumperz, S. B. Wilson, and A. D. Luster.2003. CD1d-restricted NKT cells express a chemokine receptor profile indicativeof Th1-type inflammatory homing cells. J. Immunol. 171: 2571–2580.
32. Kenna, T., L. Golden-Mason, S. A. Porcelli, Y. Koezuka, J. E. Hegarty,C. O’Farrelly, D. G. Doherty, and L. G. Mason. 2003. NKT cells from normaland tumor-bearing human livers are phenotypically and functionally distinctfrom murine NKT cells. [Published erratum appears in 2003 J. Immunol. 171:5631.] J. Immunol. 171: 1775–1779.
33. van Der Vliet, H. J., N. Nishi, T. D. de Gruijl, B. M. von Blomberg, A. J. van denEertwegh, H. M. Pinedo, G. Giaccone, and R. J. Scheper. 2000. Human naturalkiller T cells acquire a memory-activated phenotype before birth. Blood 95:2440–2442.
34. Leite-De-Moraes, M. C., A. Hameg, A. Arnould, F. Machavoine, Y. Koezuka,E. Schneider, A. Herbelin, and M. Dy. 1999. A distinct IL-18-induced pathwayto fully activate NK T lymphocytes independently from TCR engagement.J. Immunol. 163: 5871–5876.
35. Lee, J. K., S. H. Kim, E. C. Lewis, T. Azam, L. L. Reznikov, and C. A. Dinarello.2004. Differences in signaling pathways by IL-1beta and IL-18. Proc. Natl.Acad. Sci. USA 101: 8815–8820.
36. Mandl, J. N., A. P. Barry, T. H. Vanderford, N. Kozyr, R. Chavan, S. Klucking,F. J. Barrat, R. L. Coffman, S. I. Staprans, and M. B. Feinberg. 2008. DivergentTLR7 and TLR9 signaling and type I interferon production distinguish patho-genic and nonpathogenic AIDS virus infections. Nat. Med. 14: 1077–1087.
37. Malleret, B., B. Maneglier, I. Karlsson, P. Lebon, M. Nascimbeni, L. Perie,P. Brochard, B. Delache, J. Calvo, T. Andrieu, et al. 2008. Primary infection withsimian immunodeficiency virus: plasmacytoid dendritic cell homing to lymphnodes, type I interferon, and immune suppression. Blood 112: 4598–4608.
38. Redd, A. D., D. Dabitao, J. H. Bream, B. Charvat, O. Laeyendecker,N. Kiwanuka, T. Lutalo, G. Kigozi, A. A. Tobian, J. Gamiel, et al. 2009. Mi-crobial translocation, the innate cytokine response, and HIV-1 disease pro-gression in Africa. Proc. Natl. Acad. Sci. USA 106: 6718–6723.
39. McGeachy, M. J., and D. J. Cua. 2008. Th17 cell differentiation: the long andwinding road. Immunity 28: 445–453.
40. Ouyang, W., J. K. Kolls, and Y. Zheng. 2008. The biological functions of Thelper 17 cell effector cytokines in inflammation. Immunity 28: 454–467.
41. O’Quinn, D. B., M. T. Palmer, Y. K. Lee, and C. T. Weaver. 2008. Emergence ofthe Th17 pathway and its role in host defense. Adv. Immunol. 99: 115–163.
42. Tesmer, L. A., S. K. Lundy, S. Sarkar, and D. A. Fox. 2008. Th17 cells in humandisease. Immunol. Rev. 223: 87–113.
43. Estaquier, J., T. Idziorek, F. de Bels, F. Barre-Sinoussi, B. Hurtrel,A. M. Aubertin, A. Venet, M. Mehtali, E. Muchmore, P. Michel, et al. 1994.Programmed cell death and AIDS: significance of T-cell apoptosis in pathogenicand nonpathogenic primate lentiviral infections. Proc. Natl. Acad. Sci. USA 91:9431–9435.
The Journal of Immunology 991

44. Finkel, T. H., G. Tudor-Williams, N. K. Banda, M. F. Cotton, T. Curiel,C. Monks, T. W. Baba, R. M. Ruprecht, and A. Kupfer. 1995. Apoptosis occurspredominantly in bystander cells and not in productively infected cells of HIV-and SIV-infected lymph nodes. Nat. Med. 1: 129–134.
45. Viollet, L., V. Monceaux, F. Petit, R. Ho Tsong Fang, M. C. Cumont, B. Hurtrel,and J. Estaquier. 2006. Death of CD4+ T cells from lymph nodes during primarySIVmac251 infection predicts the rate of AIDS progression. J. Immunol. 177:6685–6694.
46. Silvestri, G., D. L. Sodora, R. A. Koup, M. Paiardini, S. P. O’Neil,H. M. McClure, S. I. Staprans, and M. B. Feinberg. 2003. Nonpathogenic SIVinfection of sooty mangabeys is characterized by limited bystander immuno-pathology despite chronic high-level viremia. Immunity 18: 441–452.
47. Fadok, V. A., D. L. Bratton, A. Konowal, P. W. Freed, J. Y. Westcott, andP. M. Henson. 1998. Macrophages that have ingested apoptotic cells in vitro
inhibit proinflammatory cytokine production through autocrine/paracrine
mechanisms involving TGF-b, PGE2, and PAF. J. Clin. Invest. 101: 890–898.48. Paget, C., T. Mallevaey, A. O. Speak, D. Torres, J. Fontaine, K. C. Sheehan,
M. Capron, B. Ryffel, C. Faveeuw, M. Leite de Moraes, et al. 2007. Activation of
invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires
type I interferon and charged glycosphingolipids. Immunity 27: 597–609.49. Raftery, M. J., F. Winau, T. Giese, S. H. Kaufmann, U. E. Schaible, and
G. Schonrich. 2008. Viral danger signals control CD1d de novo synthesis and
NKT cell activation. Eur. J. Immunol. 38: 668–679.50. Beignon, A. S., K. McKenna, M. Skoberne, O. Manches, I. DaSilva,
D. G. Kavanagh, M. Larsson, R. J. Gorelick, J. D. Lifson, and N. Bhardwaj.
2005. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like
receptor-viral RNA interactions. J. Clin. Invest. 115: 3265–3275.
992 IL-17–PRODUCING NKT CELLS AND SIV/HIV INFECTION