advances in api development - informa markets
TRANSCRIPT

PLUS:
PEER-REVIEWED
Statistical Tools to Aid in the Assessment of Critical Process Parameters
Vo
lum
e 4
0 N
um
be
r 3
PH
AR
MA
CEU
TIC
AL T
EC
HN
OLO
GY
MA
RC
H 2
016 P
harmTech.com
MARCH 2016 Volume 40 Number 3Extractables and Leachables
Process Control Data
Single-Use Technologies
FACILITIES Determining the Right Facility Size
SUPPLY CHAIN Redefining Pharma Agility
DATA INTEGRITY How Important is Data Integrity?
Advances in API Development

Join us at INTERPHEX New York, Booth #2341

132.A1.0102.B © 2016 Eppendorf AG.
ZZZ�HSSHQGRUI�FRP��ş��������������
New Brunswick™ shakers —
Defining shaker innovation
A Proud LegacyEppendorf shakers built on a foundation of excellence
> Many have been in operation for over
���\HDUV�DQG�UHPDLQ�RSHUDWLQJ�WRGD\
>��(DFK�JHQHUDWLRQ�KDV�RXWSHUIRUPHG�
LWV�SUHGHFHVVRU��IURP�WKRVH�GHYHORSHG�
LQ�������ULJKW��WR�WKH�ODWHVW�1HZ�
%UXQVZLFN�,QQRYD®���5�,QFXEDWRU�
6KDNHU��DERYH�
7KH�OHJDF\�EHJDQ�LQ�������WKH�ILUVW�
1HZ�%UXQVZLFN�VKDNHU�ZDV�GHYHORSHG�
IRU�'U��6DOPDQ�:DNVPDQ��ZKR�ZDV�
DZDUGHG�WKH�1REHO�3UL]H�LQ�0HGLFLQH�IRU�
GLVFRYHULQJ�WKH�ILUVW�DQWLELRWLF�VXFFHVVIXOO\�
XVHG�DJDLQVW�WXEHUFXORVLV��1HZ�%UXQVZLFN�
VKDNHUV�IURP�(SSHQGRUI�FRQWLQXH�WKDW�
WUDGLWLRQ�RI�LQQRYDWLRQ�DQG�UHOLDELOLW\�

4 Pharmaceutical Technology MARCH 2016 PharmTech .com
EDITORIAL
Editorial Director Rita Peters [email protected]
Senior Editor Agnes Shanley [email protected]
Managing Editor Susan Haigney [email protected]
Science Editor Adeline Siew, PhD [email protected]
Manufacturing Editor Jennifer Markarian [email protected]
Science Editor Randi Hernandez [email protected]
Community Manager Caroline Hroncich [email protected]
Art Director Dan Ward
Contributing Editors Jill Wechsler [email protected]; Jim Miller
[email protected]; Hallie Forcinio [email protected]; Susan J. Schniepp
[email protected]; Eric Langer [email protected];
and Cynthia A. Challener, PhD [email protected]
Correspondent Sean Milmo (Europe, [email protected])
485 Route One South, Building F, Second Floor, Iselin, NJ 08830, USA
Tel. 732.596.0276, Fax 732.647.1235, PharmTech.com
EDITORIAL ADVISORY BOARDPharmaceutical Technology publishes contributed technical articles that undergo a
rigorous, double-blind peer-review process involving members of our distinguished
Editorial Advisory Board. Manuscripts should be sent directly to the managing editor. Below is a partial list
of the Pharmaceutical Technology brand editorial advisory members. The full board, which includes advisory
members from Pharmaceutical Technology Europe, can be found online at PharmTech.com.
James P. AgallocoPresident, Agalloco & Associates
Larry L. Augsburger, PhDProfessor Emeritus University of Maryland
David H. Bergstrom, PhDSenior Vice-President, Pharmaceutical Development & Corporate Quality Assurance Antares Pharma, Inc.
Phil BormanQbD Lead & Data Management & Analysis Manager GlaxoSmithKline
Rory BudihandojoLachman Consultants
Metin Çelik, PhDPresident, Pharmaceutical Technologies International (PTI)
Zak T. Chowhan, PhDConsultant, Pharmaceutical Development
Suggy S. Chrai, PhDPresident and CEO,Chrai Associates, Inc.
Roger Dabbah, PhDPrincipal Consultant, Tri-Intersect Solutions
Robert DreamManaging DirectorHDR Company
Tim FreemanManaging Director, FreemanTechnology
Sanjay Garg, PhDProfessor and Director, Centre for Pharmaceutical Innovation and Development, University of South Australia
R. Gary Hollenbeck, PhDChief Scientific Officer, UPM Pharmaceuticals
Ruey-ching (Richard) Hwang, PhDSenior Director, Pharmaceutical Sciences,Pfizer Global R&D
Mansoor A. Khan, PhDProfessor & Vice DeanIrma Lerma Rangel College of Pharmacy, Texas A&M Health Science Center
Russell E. MadsenPresident, The Williamsburg Group, LLC
Heidi M. Mansour, PhDAssistant Professor College of Pharmacy & The BIO5 Research Institute, University of Arizona–Tucson
Jim MillerPresident, PharmSource Information Services Bio/Pharmaceutical Outsourcing Report
Colin Minchom, PhDSenior Director Pharmaceutical Sciences, Shire Pharmaceuticals
R. Christian Moreton, PhDPartner, Finnbrit Consulting
Fernando J. Muzzio, PhDDirector, NSF Engineering Research Center on Structured Organic Particulate Systems, Dept. of Chemical and Biochemical Engineering, Rutgers University
Moheb M. Nasr, PhDVice-President, CMC Regulatory Strategy, Global Regulatory Affairs, GlaxoSmithKline
Garnet E. Peck, PhDProfessor Emeritus of Industrial Pharmacy, Purdue University
Wendy Saffell-ClemmerDirector, ResearchBaxter Healthcare
Gurvinder Singh Rekhi, PhDDepartment of Pharmaceutical and Biomedical Sciences,The University of Georgia College of Pharmacy
Susan J. SchnieppFellow Regulatory Compliance Associates
David R. SchonekerDirector of Global Regulatory Affairs, Colorcon
Aloka SrinivasanPrincipal Consultant, PAREXEL International
Read board members’ biographies online at PharmTech.com/ pharmtech-editorial- advisory-board.
Pharmaceutical Technology’s eNewsletter Team:
t�ePT, Editor Caroline Hroncich, [email protected]
t�Sourcing and Management, Editor Rita Peters, [email protected]
t�Equipment & Processing Report, Editor Jennifer Markarian, [email protected]
t�Send news and product releases to [email protected]
EMBO CAPS® - DESIGN
EMBOs PREVENT
“SEMI-LOCK” BEFORE and
SEPARATION AFTER FILLING
Non-animal based capsulesMade from hypromellose (HPMC)
Extremely suitable for hygroscopic/ ŵŽŝƐƚƵƌĞ�ƐĞŶƐŝƟǀĞ�ŵĂƚĞƌŝĂůƐ�ĂŶĚ�ĨŽƌŵƵůĂƟŽŶƐ�ůƐŽ�ƐƵŝƚĂďůĞ�ĨŽƌ�ĮůůŝŶŐ�ůŝƋƵŝĚƐ
'ƌĞĂƚ�ĂůƚĞƌŶĂƟǀĞ�ƚŽ�ĐŽŶǀĞŶƟŽŶĂů�ĐĂƉƐƵůĞƐ�ĨŽƌ�ǀĂƌŝŽƵƐ�ĐƵůƚƵƌĂů��ƌĞůŝŐŝŽƵƐ��and dietary requirementsKh�<ŽƐŚĞƌ���/&�E���,ĂůĂů��ĞƌƟĮĞĚ
.
.
.
FOUR EMBO® SIX PRE-LOCK
AIR VENT
SLOPED EDGE
MAIN LOCK
X
X
X
X
X
www.suheung.com
428 E. SATURN ST. BREA CA 92821
Tel:714.854.9887 | Fax:714.854.9896
STERILE PRODUCT MANUFACTURING&� Contract Research and
manufacturing of SVPs&� (ZLW[PJ�ÄSS�ÄUPZO
&� 9HKPVSHILSLK�HUK�cytoxic product
&� 5V]LS�KY\N�KLSP]LY`�systems
INDUSTRIAL TRAINING COURSE&� ���OV\YZ�VM�SLJ[\YL�HUK�SHIVYH[VY`
&� /HUKZ�VU�[YHPUPUN�VU�aseptic processing
&� :[LYPSP[`�[LZ[PUN��LU]PYVUTLU[HS�TVUP[VYPUN��8*��(ZLW[PJ�ÄSS�ÄUPZO
Contact: Frank Horton [email protected] or (901) 448-6096
https://www.uthsc.edu/plough-center/

1700 Garments t High filtration efficiency t Low particulate and shedding performance t Excellent water repelency
1600 Garments t Breathable
t Comfortable t High bacterial efficiency
Face Masks t Breathable t Reduces goggle fogging due to absorption efficiency��t Soft and comfortable
Veltek Associates, Inc. offers two garment product lines, which are both pre-folded in our system. Comfortably styled and fitted with elastic thumb loops to reduce shifting, as well as tunnelized elastic wrists and ankles.

6 Pharmaceutical Technology March 2016 PharmTech .com
www.pharmacore.com
OUR EXPERIENCE
WORKS FOR
YOU.
GMP & non-GMP Manufacturing
Process Validation
GMP Analytical
Full Regulatory Support
Process Chemistry R&D
Process Engineering Support
Custom Synthesis & Contract Research
For more information see our website or call 336-841-5250
4180 Mendenhall Oaks ParkwayHigh Point, NC 27265
SEE US ATINTERPHEX BOOTH #1468
SALESPublisher Mike Tracey [email protected]
Mid-West Sales Manager Irene Onesto [email protected] Coast Sales Manager Joel Kern [email protected]
European Sales Manager Chris Lawson [email protected] Senior Sales Executive Stephen Cleland [email protected]
Executive Assistant Barbara Sefchick [email protected]
AddRESS485 Route One South
Building F, Second Floor Iselin, NJ 08830, USA
Tel. 732.596.0276, Fax 732.647.1235 PharmTech.com
Sr. Production Manager Karen Lenzen
International Licensing Maureen Cannon [email protected], tel. 440.891.2742 or toll-free 800.225.4569 ext 2742, fax. 440.756.5255
Audience Development Manager Rochelle Ballou [email protected]
© 2016 UBM. All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any
means, electronic or mechanical including by photocopy, recording, or information storage and retrieval without per-
mission in writing from the publisher. Authorization to photocopy items for internal/educational or personal use, or the
internal/educational or personal use of specific clients is granted by UBM for libraries and other users registered with
the Copyright Clearance Center, 222 Rosewood Dr. Danvers, MA 01923, 978-750-8400 fax 978-646-8700 or visit http://
www.copyright.com online. For uses beyond those listed above, please direct your written request to Permission Dept.
fax 440-756-5255 or email: [email protected].
UBM Life Sciences provides certain customer contact data (such as customers name, addresses, phone numbers,
and e-mail addresses) to third parties who wish to promote relevant products, services, and other opportunities that
may be of interest to you. If you do not want UBM Life Sciences to make your contact information available to third
parties for marketing purposes, simply call toll-free 866.529.2922 between the hours of 7:30 a.m. and 5 p.m. CST and
a customer service representative will assist you in removing your name from UBM Life Sciences’ lists. Outside the US,
please phone 218.740.6477.
Pharmaceutical Technology does not verify any claims or other information appearing in any of the advertisements
contained in the publication, and cannot take responsibility for any losses or other damages incurred by readers in
reliance of such content.
Pharmaceutical Technology welcomes unsolicited articles, manuscripts, photographs, illustrations, and other materials
but cannot be held responsible for their safekeeping or return.
Single issues, back issues: Call toll-free 800.598.6008. Outside the US call 218.740.6480. Reprints of all articles in this
issue and past issues of this publication are available. Call 877-652-5295 ext. 121 or email [email protected].
Outside US, UK, direct dial: 281-419-5725. Ext. 121. Direct mail lists: Contact Tamara Phillips, Marketing Services,
tel. 440.891.2773, [email protected]. Display, Web, Classified, and Recruitment Advertising: Contact Tod
McCloskey, tel. 440.891.2739, [email protected]. Permissions: Contact Maureen Cannon, tel. 440.891.2742
or toll-free 800.225.4569 ext 2742, fax. 440.756.5255, [email protected].
To subscribe: Call toll-free 888.527.7008. Outside the U.S. call 218.740.6477.

PFIZER CENTRESOURCE
Bringing the best of
together
Let's provide some clarity around how this affects YOU
Our Commitment
We’re bringing together the best of our contract manufacturing capabilities to grow our
CMO beyond what it would achieve as standalone businesses. That way, we can provide a
ZLGHU�DUUD\�RI�VHUYLFHV�DQG�WHFKQRORJLHV�IRU�\RX��DOO�ZLWK�3ú]HUÙV�XQZDYHULQJ�FRPPLWPHQW�
to quality, compliance and supply reliability.
What’s Next
New name, expanded capabilities, same focus: YOU

PharmTech.com
On
th
e c
ov
er
Pharmaceutical Technology is the authoritative source of peer-reviewed research
and expert analyses for scientists, engineers, and managers engaged in process
development, manufacturing, formulation and drug delivery, API synthesis, analytical
technology and testing, packaging, IT, outsourcing, and regulatory compliance in the
pharmaceutical and biotechnology industries.
March 2016 Volume 40 Number 3
FEATURES
API SYNTHESIS & MANUFACTURING
32 Polymorph Screening for Identification of Relevant Crystalline Forms
Gauging the adequate level and
type of screening is the challenge.
EXTRACTABLES AND LEACHABLES
46 Building Consensus for E&L Testing Standards
Standardized testing protocols are crucial
for acceptance of single-use systems.
DATA INTEGRITY
50 How Important is Data Integrity to Regulatory Bodies?
Data integrity is a widespread,
global problem that must be addressed.
PROCESS CONTROL
54 Gaining Insight from Process Control Data
Integrated data and cloud-based solutions
can be used for process optimization.
CRITICALITY ASSESSMENT
36 Statistical Tools to Aid in the Assessment of Critical Process Parameters
This article introduces a statistical approach to help determine when a statistically
significant relationship between a process parameter and a CQA is large
enough to make a practical meaningful impact (i.e., practical significance).
PEER-REVIEWED RESEARCH
SUPPLY CHAIN
56 Redefining Pharma Agility
Supply-chain success is measured by how
effectively new medications reach patients,
and how swiftly manufacturers can react to
internal and external changes.
FACILITY DESIGN
60 Flexibility vs. Right-Sizing: Determining the Right Facility Size
Choosing the right facility size requires
tailoring the design for current needs.
COMING IN MARCHVisit PharmTech.com/ebooks to
check out the 2016 Solid Dosage Drug
Development and Manufacturing eBook,
featuring articles on excipients, non-
gelatin capsules, tableting, continuous
manufacturing, regulations, and more!
Continued on page 10
COVER STORY
26 Specialty Markets and Services Drive API GrowthAs specialty API outsourcing grows, manufacturers
and CDMOs are investing for the long haul.
Cover Design by Dan Ward; Images: Ravitaliy/LAGUNA DESIGN/Getty Images


PharmTech.com
PHARMACEUTICAL TECHNOLOGY (Print ISSN: 1543-2521,
Digital ISSN: 2150-7376) is published monthly, except two
issues in June, by UBM Life Sciences 131 W. First St., Duluth
MN 55802-2065. Subscription rates: US and possessions —
1 year (13 issues), $76; 2 years (26 issues), $133. Canada
and Mexico — 1 year, $99; 2 years, $151. All other countries
1 year, $145; 2 years, $263. International price includes
air-expedited service. Single-copies (prepaid only) — US,
$15; Canada and Mexico, $16; outside the US, $19. Back
issues (if available): US and possessions — $34; Canada
and Mexico, $39; all other countries — $41. Include an
additional $6.50 per order plus $2 per additional copy for
US postage and handling. If shipping outside the US, include
an additional $10 per order plus $3 per additional copy.
Periodicals postage paid at Duluth, MN 55806 and additional
mailing offices. POSTMASTER: Please send address changes
to Pharmaceutical Technology, PO Box 6188, Duluth,
MN 55806-6188. PUBLICATIONS MAIL AGREEMENT NO.
40612608, Return Undeliverable Canadian Addresses to:
IMEX Global Solutions, P. O. Box 25542, London, ON N6C
6B2, CANADA. Canadian G.S.T. number: R-124213133RT001.
Printed in the U.S.A.
Pharmaceutical Technology is selectively abstracted or indexed in:
» Biological Sciences Database (Cambridge Scientific Abstracts)
» Biotechnology and Bioengineering Database (Cambridge Scientific Abstracts)
» Business and Management Practices (RDSI)
» Chemical Abstracts (CAS)
» Current Packaging Abstracts
» DECHEMA
» Derwent Biotechnology Abstracts (Derwent Information, Ltd.)
» Excerpta Medica (Elsevier)
» International Pharmaceutical Abstracts (ASHP)
» Science Citation Index (Thomson)
Pharmaceutical Technology is proud to be a member of DCAT, IPEC, and PDA.
NEWS & ANALYSIS
FROM THE EDITOR
12 Keynote Series Addresses Crucial Industry Issues
Thought leaders tackle drug shortages
and manufacturing innovations.
TROUBLESHOOTING
62 Designing a Biomanufacturing Facility Incorporating Single-Use Technologies
Asking the right questions is crucial.
OUTSOURCING
68 Pharma Outsourcing Market Expands
The pharma outsourcing market
starts 2016 with expansions,
acquisitions, and new offerings.
REGULATION & COMPLIANCE
US REGULATORY WATCH
18 Generic-Drug Production and Oversight Challenge FDA
and Manufacturers
Policy makers debate strategies for
promoting access to less costly medicines.
EUROPEAN REGULATORY WATCH
22 Tackling Regulatory Challenges of EU’s Variations Framework
The EU is striving to reduce the costs and
administrative burden when complying
with variations regulations for keeping
authorization dossiers up to date.
ASK THE EXPERT
78 Phase-Appropriate GMP
Siegfried Schmitt, principal consultant,
PAREXEL, discusses the regulatory
requirements for cGMPs in the
different phases of drug development
and manufacture.
DEPARTMENTS/PRODUCTS
14 Product Spotlight
72 INTERPHEX Planning Guide
77 Showcase/Marketplace
78 Ad Index
Continued from page 8

Fairfield, New Jersey
�����������������������
mgamerica.com
innovation
From processing to packaging, we’ll show you the way
Products with unique needs require carefully-engineered solutions, not a limited menu of options. That’s why
MG America brings unrivaled expertise in processing and packaging machinery to help custom-craft a solution
that is perfect for your individual product needs.
Innovation? We’ve got that too, from our groundbreaking MultiNETT™ Weight Control System to our
state-of-the-art packaging lines, we’re advancing into exciting new areas to meet your growing needs.
Put your vision in our hands...and start seeing things in a whole new way.
CAPSULE FILLING
MATERIAL HANDLING
ASEPTIC EQUIPMENT
PRIMARY PACKAGING
SECONDARY PACKAGING
LINE INTEGRATION
CHECKWEIGHING
TABLET & CAPSULE INSPECTION
See us at Interphex Booth #2221

12 Pharmaceutical Technology MARCH 2016 PharmTech .com
FROM THE EDITOR
JO
RG
GR
EU
EL
/PH
OT
OD
ISC
/GE
TT
Y I
MA
GE
S
PharmTech.com/forum
At INTERPHEX 2016 at the Javits Center in New York, Pharma-ceutical Technology will host a
Keynote Series on leading bio/pharma industry issues. Sessions will be pre-sented on the Innovation Stage in the exhibit hall. Admission is free to any attendee with an exhibit hall pass. The following is a preview of the topics.
Overcoming Bottlenecks inBiopharmaceutical Development(Tuesday, April 26, 10:15–11:45 am)Industry experts discuss how technol-ogy advances are addressing challenges in biopharmaceutical development.
Innovations in Solid Dosage Development and Manufacturing(Tuesday, April 26, 1–3:30 pm)Drop-On-Demand Manufacturing System for
the Flexible Production of Solid Oral Dosage
Forms. Dropwise additive manufactur-ing of pharmaceutical products uses drop-on-demand printing for auto-mated and controlled deposition of solution-, melt-, or suspension-based formulations onto edible substrates.
Polymer Thin Films for Drug Delivery: Pro-
cess and PAT Development. Polymer thin film is a platform technology for con-tinuous manufacturing, in which in-
line monitoring of product quality can be accomplished using various sensing and process analytical technologies, making the technology amenable for real-time release.
Minitablets: Manufacturing, Character-
ization Methods, and Future Opportunities.
Manufacturing techniques used to pro-duce minitablet-based dosage forms are essentially the same as manufac-turing of larger tablet images; however, some special considerations to manu-facturing techniques and analytical tests may be required.
Contract Services Market: 2016 Update(Wednesday, April 27, 10:30–11:30 am)How will consolidation in the bio/pharmaceutical and contract services market, a changing financial market, and an active political and regulatory year shape the fortunes of the con-tract services market? In his annual presentation, Jim Miller, founder and president of PharmSource Information Services, will offer his perspectives on the contract services landscape for the next few years.
Strategies and Innovations to Reduce Drug Shortages and Improve Availability of Medicines(Wednesday, April 27, 1:30–3:15 pm)An Interdisciplinary Approach to Address
Drug Shortages. The effects of drug shortages on patients, caregivers, hos-pitals, and medical professionals are often not observed or understood by the bio/pharmaceutical manufacturing segment. This presentation will explore
alternative, innovative, and cost-effec-tive ways to provide needed therapies to patients.
BARDA Innovation Initiatives in Medi-
c a l Co u n te r m e a s u r e M a n u f a c t u r i n g.
This presentation will describe the Biomedical Advanced Research and Development Authority’s (BARDA) program initiatives in manufactur-ing technologies for medical coun-termeasure advanced development, including opportunities with the Centers for Innovation in Advanced Development & Manufacturing and in continuous manufacturing.
Continuous Manufacturing for Rapid Pro-
cess Development. The pharmaceutical industry has begun to adopt continu-ous manufacturing technology for the manufacture of traditional solid-dos-age forms. Benefits include lower cost and expedited development due to the ability to develop at scale. This presen-tation will outline some of the new ap-proaches and potential benefits.
Panel Discussion: Addressing Sterile Manufacturing Challenges (Wednesday, April 27, 3:30–4:30 pm)
Sterile injectables have been in ex-tremely short supply, and industry efforts have been focusing on root causes involv-ing infrastructure, quality, and efficiency. Experts involved in this work discuss recent initiatives, and offer insights into what must be done to prevent injectables shortages in the future.
For more information about the Keynote Series, visit: www.PharmTech.com/pt/Interphex2016. PT
Keynote Series Addresses Crucial Industry Issues
Thought leaders tackle drug
shortages and manufacturing innovations.
Rita Peters
Rita Peters is editorial
director of Pharmaceutical
Technology. Send your
thoughts and story ideas
Pharmtech.com/pt/forum


14 Pharmaceutical Technology MARCH 2016 PharmTech .com
PRODUCT SPOTLIGHT
High-Speed Dispersers Prevent Explosions
High Speed Dispersers from Ross feature
a National Electrical Manufacturers
Association (NEMA) 7&9 Operator Panel
and grounding systems. The dispersers
can be used in hazardous environments
where flammable vapors, liquids, solids,
and/or dusts are present. The saw-tooth
blade of the High Speed Disperser turns
at tip speeds up to approximately 5000 ft/
min and creates flow in low to medium
viscosity formulations under 50,000
centipoise. The dispersers’ grounding
systems are designed to ensure that the
vessel is properly grounded before the
agitator is allowed to operate.
The dispersers’ control panel features start, stop, and emergency
stop pushbuttons; a speed potentiometer; digital readout for agitator
speed; and a 16-ft. grounding clamp device. Also supplied on the
mixer are explosion-proof limit switches for locking out the motor if
the blade is in a raised position or a mix vessel is not in place. The
dispersers are available in sizes from 1 to 500 HP for batches from a
gallon to more than 5000 gallons.
Ross, Charles & Son
www.mixers.com
Multimode Microplate Reader Improves Sensitivity
The Spark 20M multimode
microplate reader has the
ability to read 6- to 1536-
well microplates and
includes a high-frequency
xenon flash lamp that can
be combined with detec-
tion modules. The unit also
includes a fluorescence
module designed to
improve sensitivity and increase the speed of high-throughput
screening. The reader includes the Te-Cool cooling module that allows
the measurement chamber temperature to be set below the ambient
room temperature.
The Spark 20M features automated cell imaging and confluence
measurement, which allows incubation and monitoring of cell culture
microplates within the measurement chamber. The user can define
the confluence at which the assay starts or ends or the substrate is
injected.
Tecan
www.tecan.com/spark20m
Confocal Microscope for Surface AnalysisThe Zeiss Smartproof 5
wide-field confocal micro-
scope is designed for
applications in quality
assurance and quality
control, production, and
R&D laboratories. The
microscope system pro-
vides 3D reconstructions
and roughness measure-
ments for work piece surfaces.
The microscope has an embedded optics, electronics, and camera
minimizing the number of cables used. Smartproof 5 can be installed
and run in different working environments without anti-vibration
equipment. The machine can also complete roughness analyses in
2D (profile) and 3D (area)—both based on ISO standards. The
Smartproof 5’s software provides easy-to-operate workflow routines,
and a graphical user interface guides users through recurring tasks,
ensuring user-independent data acquisition.
Zeiss
www.zeiss.com
Cleanroom Oven The Class 100 cleanroom oven
from Grieve is used for a variety of
heat processes such as sterilizing,
depyrogenation, curing, and drying
workloads. The cleanroom oven
includes four-inch thick insulated
walls, aluminized steel exterior, and
a type 304 2B-finish stainless steel
interior with continuously back-
welded seams. The unit includes
a four-inch insulated floor with
removable truck wheel guide tracks.
In addition, the unit has two thick
stainless steel high-temperature HEPA recirculating air filters, digital
indicating temperature controller, manual reset excess temperature
controller with separate contactors, recirculating blower airflow
safety switch, and a circular chart recording temperature controller.
The Grieve Corporation
www.grievecorp.com

Emergent Contract Manufacturing:
Enhancing Life in Every Single Dose
BDSManufacture
Single-usePlatform
AsepticFill/Finish
Vials &Syringes
Clinical& Commercial
emergentcontractmanufacturing.com
800-441-4225
Emergent BioSolutions is a fully integrated Contract Development
& Manufacturing Organization, supporting both bulk drug substances
and sterile injectable drug products at clinical and commercial scale.

16 Pharmaceutical Technology MARCH 2016 PharmTech .com
PRODUCT SPOTLIGHT
Mixing Systems for Rehydration and Mixing ApplicationsThe QuaDrum rigid outer con-
tainers (ROCs) from Meissner are
used for rehydration and mixing
applications. The retaining lid,
stainless steel dolly, and bottom
drain allow recirculation-based
mixing for process volumes
between 50 L and 200L. The
retaining lid for mixing applica-
tions provides access to fluid
paths located on the top of the
biocontainer and can positively
locate and support a 3-inch TC
port on top of the biocontainer.
This port can be used for powder addition or other operations that
require large bore access to the biocontainer. The stainless-steel
dolly, which is required to facilitate bottom drain applications, can
be specified with swivel wheels that lock for added security.
Meissner
www.meissner.com
High-Pressure Ion Chromatography System The Dionex Integrion High-Pressure Ion
Chromatography system from Thermo
Scientific includes high-pressure capability
and electrochemical detection. The system
includes viper fittings, which minimize
peak dispersion and band broadening, a
detachable tablet with local language sup-
port, and a consumables device monitor
that regulates installation errors by logging
and tracking both system and consum-
able performance. The system’s thermally regulated detector
compartment provides extended life to consumables.
Thermo Scientific
www.thermoscientific.com/chromatography
Tool Management SystemThe IH-TMS Tool
Management System
from I Holland moni-
tors tool rotations,
tooling inventory,
and maintenance.
The stand-alone
system allows tablet
manufacturers to
keep a record of
tablet quantities by number of tablets, work order, or batch infor-
mation. The system incorporates a guide to tooling specification
and troubleshooting. In addition, the system has the capability
to archive tablet and tool images and drawings and incorporates
an alarm to alert users of any problems including over compres-
sion, tooling replacements, and when maintenance is required.
I Holland
www.tablettingscience.com


18 Pharmaceutical Technology MARCH 2016 PharmTech .com
GLO
BE
: Z
OO
NA
R R
F/G
ET
TY
IM
AG
ES
It’s well known that generic drugs account for 88% of prescrip-
tion drug sales in the United States and have saved billions for
patients and healthcare systems since Congress enacted the
Hatch-Waxman Act more than 30 years ago. That growth, though,
has created difficulties for FDA in processing the hundreds of
resulting abbreviated new drug applications (ANDAs) and in
inspecting an expanding number of generic-drug manufacturers
and ingredient producers all over the world.
Concerns about ensuring the quality and safety of medical
products, moreover, have led to the closure of outdated and
noncompliant facilities, contributing to shortages and price spikes
for certain widely used generics, particularly sterile injectables.
These developments have raised questions about whether
government regulatory policies limit competition in certain drug
classes and support monopoly pricing.
Fees add resources
FDA’s ability to expeditiously approve new generics was compro-
mised by a budget squeeze over many years. The Prescription
Drug User Fee program (PDUFA) of 1992 bolstered funding for
new drug review by the Center for Drug Evaluation and Research
(CDER), but also shifted resources away from generics. ANDA
approvals slowed to a crawl, resulting in an enormous backlog of
pending applications.
Generic-drug makers finally agreed to pay user fees in 2012 to
strengthen FDA regulation of generic-drug development, review,
and inspection. In its first three years, the Generic Drug User Fee
program (GDUFA I) has generated nearly $1 billion to support
CDER’s Office of Generic Drugs (OGD) and certain operations of
the new Office of Pharmaceutical Quality (OPQ). FDA’s field force
also has increased inspections of overseas producers to help
level the playing field between US and foreign manufacturers.
As FDA and industry negotiate GDUFA renewal in 2017,
Congressional committees and the broader healthcare
community are examining FDA policies and programs governing
generics and the prescription drug market, as seen at a January
2016 hearing before the Senate Health, Education, Labor and
Pensions (HELP) Committee. Chairman Lamar Alexander (R.Tenn.)
cited concerns about “unnecessary regulatory burdens” that can
slow drug development and the importance of a pharmaceutical
marketplace that “remains competitive” (1).
The panel also is developing a Senate version of the “21st
Century Cures” legislation, which the House approved
in July 2015. Instead of combining multiple proposals into
a comprehensive bill, Alexander and ranking Democrat Patty
Murray (D-Wash) are considering numerous individual measures
on FDA policies, disease research, and expanded use of
electronic data technology to support broader research goals.
With deliberations running through April 2016, though, there’s
not much chance that Congress will adopt any final “Cures”
legislation this year, but will wait until 2017 when action is
required to reauthorize FDA user fee programs.
No more backlog
A main issue explored at the HELP hearing is whether too-slow
FDA approval of new generics limits drug access and com-
petition. Some legislators suggested that a pharma company
would be less likely to buy up a small drug firm with the intent
of boosting product prices if it knew that FDA could quickly
approve a new competing drug.
Despite complaints from generics makers about still-delayed
ANDA approvals, CDER director Janet Woodcock made a strong
case for agency progress in addressing the backlog problem,
speeding important new generics through the approval process
and expanding timely inspections of manufacturing facilities.
She explained that generic-drug makers submitted nearly 2500
applications in 2013 and 2014, making it difficult for OGD to
process those documents and to tackle long-pending submissions,
while also restructuring and expanding its program (2).
Even so, in the past three years CDER was able to “take
action” on approximately 85% of 4600 overdue ANDAs and
post-approval supplements, Woodcock stated. She promised
that all the backlog would be gone by 2017 and that OGD would
meet its goal for taking a “first action” within 10 months on
ANDAs submitted this year. No applications in the backlog
are first generics, she emphasized, and OGD’s “express lane”
policy moves these products to the front of the queue. She also
highlighted CDER efforts to promote advanced manufacturing in
the generic-drug industry, as continuous, computer-controlled
production systems would enable fast ramp-up of new
production.
Key to achieving these goals is an FDA-industry effort to
achieve more first-cycle approvals. A “right-the-first-time” policy
permits rejection of notably incomplete applications when they
first come in. CDER also is issuing more guidance on what data
it wants from sponsors and encouraging manufacturers to
conduct all necessary tests and processes before sending in
applications. A new pre-ANDA process that addresses approval
challenges for particular drugs prior to application submission
may be included in GDUFA II.
Supporting competition
In highlighting FDA efforts to quickly approve first generics,
Woodcock acknowledged that multiple drugs per innovator
may drive down costs and facilitate patient access to more
affordable therapies. Yet FDA does not approve a new drug or
generic in response to rising prices, she noted, and does not
have the expertise to calculate what qualifies as a “price hike:”
would that involve doubling a price from 10 cents to 20 cents, or
possibly raising a list price by more than 1000%?, she queried,
Generic-Drug Production and Oversight Challenge FDA and ManufacturersPolicy makers debate strategies for promoting access to less costly medicines.
Jill Wechsler is Pharmaceutical Technology’s
Washington editor, tel. 301.656.4634,


20 Pharmaceutical Technology MARCH 2016 PharmTech .com
adding that a new report from the US Department of Health and
Human Services (HHS) better addresses generic-drug pricing (3).
The agency does keep a close eye on sole-source products
and those with only one or two competitors, as part of efforts
to anticipate drug shortages and supply disruptions. Woodcock
presented data indicating that 99 innovator drugs have only one
generic competitor; 66 drugs have two generics; and 623 drugs
have 3 or more generics. Of particular interest is the segment of
125 innovator drugs with no approved generics (and no patent or
exclusivity protections) (4).
These drugs may have limited competition, Woodcock
explained, because they are orphans or specialized therapies
that serve small patient populations. Many topical products,
inhalants, and complex substances also lack well-understood
methods for testing and documenting bioequivalence. To support
the development of generic versions of such therapies, GDUFA
provided FDA with approximately $35 million for research on
new bioequivalence test methods and guidances to “open up
previously blocked pathways” for new generics.
Woodcock acknowledged that, in some cases, innovator firms
take steps to block and delay generic drug entry. Generic-drug
makers have complained loudly about problems in obtaining
supplies for bioequivalence testing of brand products that are
subject to Risk Evaluation and Mitigation Strategies (REMS).
Woodcock said that FDA has advised brand firms that REMS don’t
warrant withholding drugs for research purposes, and indicated
that Congressional action would help address this problem more
directly. The Generic Pharmaceutical Association also wants
the legislators to repeal a recent budget provision that boosts
Medicaid rebates on generic drugs (5).
One strategy Woodcock strongly opposed is to turn to drug
compounders to provide less costly alternative medicines
when generics fail to meet demand. She emphasized that there
are “very great risks” in such proposals, citing two examples
of compounded drugs that sickened dozens of people. Mass
production of these drugs without adherence to GMPs, she
warned, “could have put thousands of people in the hospital.”
References
1. US Senate Committee on Health, Education, Labor & Pensions, “Alexander: Despite Extra $1 Billion to Speed Generic Drug Ap-provals, FDA Process Still Too Slow,” Press Release, Jan. 28, 2016, www.help.senate.gov/chair/newsroom/press/alexander-despite-extra-1-billion-to-speed-generic-drug-approvals-fda-process-still-too-slow., accessed Feb. 2, 2016.
2. Implementation of the Generic Drug User Fee Amendments of 2012 (GDUFA), Testimony of Janet Woodcock, MD, Before the Committee on Health, Education, Labor and Pensions, Jan. 28, 2016, www.help.senate.gov/imo/media/doc/Woodcock5.pdf, ac-cessed Feb. 2, 2016.
3. Office of the Assistant Secretary for Planning and Evaluation, Understanding Recent Trends in Generic Drugs, Jan. 27, 2016, https://aspe.hhs.gov/pdf-report/understanding-recent-trends-generic-drug-prices, accessed Feb. 2, 2016.
4. FDA, Slides from FDA GDUFA presentation before the Senate Health, Education, Labor and Pensions Committee, January 2016, www.pharmtech.com/generic-drug-production-and-oversight-challenge-fda-and-manufacturers.
5. GPhA, “GPhA to Congress: Embrace Five Opportunities for More Generic Drug Savings,” Statement by Chip Davis, Presi-dent and CEO, GPhA, Feb. 1, 2016, www.gphaonline.org/gpha-media/press/gpha-to-congress-embrace-five-opportunities-for-more-generic-drug-savings, accessed Feb. 2, 2016. PT
SimpleBlend™
Blenders
GP Cone Mill
www.globepharma.com
Sift-N-Blend™ Technology
New
GP-Mill-5™
GP-HSG-5
Granulator
MaxiBlend® Lab
Blender
VersaPress™
Formulation to Pilot Scale Blenders both Stand Alone &
Interchangeable Vessel Models - Granulators- Mills
-Powder, Liquid & Semi-Solid Samplers - Valves - Cleaning
Validation Tools- Powder Segregation Testers -
GlobePharma

ES744954_PT0316_021_FP.pgs 03.01.2016 00:33 ADV blackyellowmagentacyan

22 Pharmaceutical Technology MARCH 2016 PharmTech .com
Sean Milmo
is a freelance writer based in
Essex, UK, [email protected].
GLO
BE
: Z
OO
NA
R R
F/G
ET
TY
IM
AG
ES
The European Union’s medicines licensing agencies have
committed themselves in a strategy document in December
2015 (1) to ease the regulatory burden on the pharmaceutical
industry by being more efficient in their control of pharma-
ceuticals throughout product lifecycles. This pledge has been
raising the hope of the generic medicines industry. One of the
generic medicines industry’s priorities is the reduction of the
costs and administrative difficulties that occur when com-
plying with variations regulations for keeping authorization
dossiers up to date.
Problems with variations rules were a major topic at an
annual regulatory and scientific regulatory affairs conference
in London in January 2016, which was organized by the
European Generic and Biosimilar medicines Association (EGA).
Approximately a third of the 200 participants were regulators
with most of the remainder from the industry.
As the EU’s pharmaceuticals regulations have been
tightened up in recent years, the submissions on variations
to regulatory agencies—comprising the London-based
European Medicines Agency at the center and a network
of national agencies in the EU’s 28 member states—have
increased substantially. As a result, variations have been
taking up a rising proportion of the pharmaceutical industry’s
regulatory costs.
Variations requirements
Pharmaceutical producers and importers are required to pro-
vide regulators with even more information about changes to
their medicines, such as modifications to manufacturing proc-
esses, improvements or extensions to formulations, and even
minor additions such as alterations to names and addresses.
The industry is seeing many of the variations requirements as
being the imposition of an unfair weight of responsibility on
their shoulders.
The current regulatory requirements are laid down by an
EU legislation (2) approved in 2008, which came into force in
August 2013. The aim was to provide clearer rules by dividing
the variations into four main categories. Two of these are type
IA, which have “minimal or no impact at all” on the quality,
safety, or efficacy of medicines and type II, which can have a
“significant impact.” An “extension” category covers changes
to an active substance or the strength, pharmaceutical form,
and route of administration of a medicine. Type IB variations
are changes that are neither minor, major, nor an extension.
Although the legislation’s main objective was to simplify
rules on the notification and approval of variations, a series
of guidelines have had to be issued to clarify their application.
At the same time, new EU pharmacovigilance legislation (3)
was introduced, which brought in additional requirements for
information of variations in the quality, safety, and efficacy
of medicines after their launch on the market. This new
legislation also had to be clarified by guidelines, the latest
of which was issued in the form of a questions and answers
document (4) in January 2016.
“Guidance covering matters that should be clearly described
in legislation has increased the regulatory work load of the
industry,” Susana Pereira, a principal regulatory affairs officer
at Teva Pharmaceutical Industries, told the conference (5). An
increasing proportion of variations are now related to details
about the production, origin, and composition of APIs. This
follows the migration of API manufacture out of Europe and
North America to China and India, which has considerably
complicated the supply chains for active substances, mainly
used in generic medicines. At the same time, rules on good
manufacturing practice (GMP) have been made stricter and
broader while GMP inspections of API facilities have become
more rigorous. A lot of additional variations information is
being generated by the more stringent application of GMP.
API changes and GMP rules
The average number of variations per marketing authoriza-
tion per year had increased by 45% in the five years leading
up to 2014 (5), the equivalent to one additional variation per
authorization. Variations in APIs are a major factor behind
this increase. Up to 60% of quality variations submitted by
marketing authorization holders (MAHs) are now related to API
changes, said Pereira (5). New interpretation of regulatory data
requirements had led to rises of up to 300% in the variations
related to the API supply chain, with a high proportion relating
to GMP matters, she added (5).
With the addition of stricter GMP rules and the new
requirements under the pharmacovigilance legislation,
“variations have become a mechanism to implement new
legal obligations,” Pereira claimed. The result was rises in
regulatory costs covering the maintenance of a product’s
lifecycle to a level that is higher than the authorization costs
for the marketing of the medicine. “This imbalance can lead
to the withdrawal of a medicine from the market, particularly
Tackling Regulatory Challenges of EU’s Variations FrameworkThe EU is striving to reduce the costs and administrative burden facing pharmaceutical
manufacturers when complying with variations regulations for keeping authorization dossiers up to date.

The Parenteral Drug Association presents...
2016 PDA Biosimilars ConferenceCo-Sponsored by
June 20-21, 2016 | Baltimore, MD
Hilton Baltimore
Exibition: June 20-21 | Courses: June 22
Since the approval of the first biosimilars in the EU in the early 2000s, biosimilars regulatory science has
evolved quickly and continues to advance.
In order to meet industry’s need for information on this increasingly important topic, PDA is introducing a
new event, the 2016 PDA Biosimilars Conference. This Conference will offer practical guidance and best
practices for the development of biosimilar products and demonstration of analytical similarity.
Through presentations and case studies from industry experts and regulators, you’ll hear about new
development strategies and updates on recent regulatory expectations, and you will have the opportunity
to raise questions and concerns.
For more information and to register today, please visit pda.org/2016Bio
#2016Bio

24 Pharmaceutical Technology MARCH 2016 PharmTech .com
if the cost of maintenance is higher than the product’s profit,”
she warned (5). Instead of taking a drug off the market, a
company may delay process improvements or hold back other
changes that would reduce the medicine’s cost.
Companies are also choosing carefully the regulatory
authorities to which they are submitting variations, particularly
because of dif ferences in the fees they charge. These
costs can range from zero to up to $1544 (€1400) for type
IA variations; up to $1654 (€1500) for type IB; and up to
$19,852 (€18,000) for type II, Pereira said. “The current system
doesn’t create incentives for [national licensing authorities] to
implement cost-effective mechanisms,” she added.
Other speakers pointed out how certain aspects of API
manufacture were increasing the requirements for variations
information, often as a result of the interpretation of
regulations made in guidelines. Marieke van Dalen, global
regulatory specialist at Aspen Oss B.V., a Dutch API producer,
said that a recent trend was a requirement for information on
starting and even pre-starting materials, even including the
names and addresses of the suppliers. This information was
passed to the MAH before being added to the authorization
dossier. Some authorities also wanted information on the
analytical methods used in the validation of the starting
materials and their intermediates.
Joseph Bondin, executive director, quality operations, at
the generic-drug producer Actavis, gave examples of how
much information had to be provided when an outsourced
API manufacture introduced intermediates from new
producers with new testing sites. In one example, details
of as many as 12 additional players in the API supply chain
had to be submitted because of changes in the suppliers of
intermediates. As a result of new regulatory interpretations of
the information requirements on API intermediates, he warned
that there could be a two- to three-fold increase in the total
number of variations, which in 2015 averaged just under three
variations per marketing authorization per year (6).
“This increase seems contradic tor y to several EU
policies,” he said. “[These policies include] have effective
and fit regulatory systems that foster supply-chain resilience
to prevent temporary supply disruptions.” In addition, it
would seem to refute the value of the systems adopted by
pharmaceutical companies themselves to validate the quality
of intermediates, even though these could be based on quality
guidelines proposed by the International Conference on
Harmonization (ICH).
Recommendations for improvements
In a regulatory efficiency report (7) published in September
2015, the EGA recommended a series of specific improve-
ments to the EU regulatory system on variations. These rec-
ommendations include reliance on the new database system
of Identification of Medical Products (IDMP) to make the API
supply chain more transparent. It proposed that API informa-
tion should be limited to details about the final API manufac-
turer. Information on other involved sites, such as intermediate
producers, would be managed through the drug manufac-
turers quality systems and regulators’ GMP inspections.
Much greater use, as a source of required variations
data, could be made of the central European inventory of
information on medicines and active substances, which is
being set up under the EU’s pharmacovigilance legislation.
Under article 57 of the law, companies have to provide up-to-
date post-marketing information on their products relating to
quality, safety, and efficacy (3).
Regulators at the conference reiterated their commitment
to making regulatory compliance easier and less costly for
the pharmaceutical industry, particularly for the generic-drug
sector. Noel Wathion, the EMA’s deputy executive director,
stressed that the article 57 database and the implementation
of IDMP would be used to improve the cost-effectiveness of
regulations for generic and biosimilar medicines within the
EU’s regulatory network. The regulators, however, were unable
to make pledges about specific measures, particularly on the
vexed issue of variations. This issue seems likely to remain a
matter of grievance among generic-drug companies for some
time.
References
1. European Medicines Agency (EMA) and Heads of Medicines
Agencies (HMA), EU Medicines Agencies Network Strategy to
2020: Working Together to Improve Health (London, Dec. 17,
2015).
2. EC Regulation No. 1234/2008 The examination of variations to
the terms of marketing authorisations for medicinal products for
human use and veterinary medicinal products (Brussels, Nov. 24,
2008).
3. EC Regulation No. 1235/2010, Amending as regards pharmaco-
vigilance of medicinal products for human use, Regulation No.
726/2004 laying down community procedures for the authoriza-
tion and supervision of medicinal products for human and vet-
erinary use (Brussels, Dec. 15, 2010).
4. EMA, Practical questions and answers to support the implemen-
tation of the variations guidelines in the centralised procedure,
EMA/427505/2013 (London, Jan. 18, 2016).
5. S.Pereira, “Challenges of the Current Variations System,” presen-
tation at the EGA Regulatory and Scientific Affairs Conference
(London, Jan. 28–29, 2016).
6. J.Bondin, “Management of the Active Substance Regulatory Dos-
sier,” presentation at the EGA Regulatory and Scientific Affairs
Conference (London, Jan. 28–29, 2016).
7. European Generic and Biosimilar medicines Association (EGA),
An Efficient Regulatory System for Patient Access to New Generic
Medicines” (Brussels, September 2015). PT
Companies are choosing
carefully the regulatory
authorities to which they
are submitting variations.

0LNDUW�LV�WKH�&RQWUDFW�'HYHORSPHQW�DQG�0DQXIDFWXULQJ�
2UJDQL]DWLRQ��&'02��WKDW�GHOLYHUV�WKH�VHUYLFHV�\RX�
QHHG��SOXV�WKH�VSHHG�DQG�UHVSRQVLYHQHVV�\RX�ZDQW�
:K\�SXW�XS�ZLWK�LQቹH[LEOH�SURFHGXUHV�DQG�
XQUHWXUQHG�FDOOV"�$W�0LNDUW��\RX�KDYH�RXU�
IXOO�DWWHQWLRQ��$QG�ZH·UH�TXLFN�WR�UHVSRQG�
ZLWK�SHUVRQDOL]HG�VROXWLRQV�WKDW�JHW�\RXU�
SURMHFWV�FRPSOHWHG�IDVWHU�DQG�WR�\RXU�
LQGLYLGXDO�UHTXLUHPHQWV�
2XU�SURIHVVLRQDO�VHUYLFHV�LQFOXGH��)RUPXODWLRQ�'HYHORSPHQW��
&OLQLFDO�7ULDO�6XSSOLHV��5HJXODWRU\�)LOLQJ�6XSSRUW��SOXV�0DQX�
IDFWXULQJ�DQG�3DFNDJLQJ�VROXWLRQV�
<RX·OO�ቸQG�0LNDUW�KDV�HYHU\WKLQJ�\RX·UH�ORRNLQJ�IRU�LQ�D�
SKDUPD�SDUWQHU��LQQRYDWLYH�WHFKQRORJ\�DQG�HTXLSPHQW��
KLJKO\�VNLOOHG�SHRSOH�DQG����\HDUV�RI�VROLG�&'02�
H[SHULHQFH��
7R�H[SHULHQFH�MXVW�KRZ�UHVSRQVLYH�ZH�FDQ�EH��FRQWDFW�
XV�DW��������0,.$57�RU�VHQG�XV�DQ�HPDLO�WR�
LQIR#PLNDUW�FRP
��7KH�&XUH�)RU��&'02�
�$WWHQWLRQ�'HI�LFLW�'LVRUGHU�
0LNDUW��,QF���_�������&KDWWDKRRFKHH�$YHQXH��_��$WODQWD�*$������
ZZZ�PLNDUW�FRP��_���������0,.$57
5HDG\���5HVSRQVLYH�� 5LJKW�IRU�<RX�

26 Pharmaceutical Technology MARCH 2016 PharmTech .com
For decades, APIs, the foundation of the pharmaceutical industry, have been a staid, solid, if somewhat
invisible market. Some drug product manufacturers make APIs inhouse, while others outsource their manu-facturing to contract development and manufacturing partners.
Recently, however, as drug approvals have approached levels last seen in the early 2000s, APIs have become a dy-namic focal point for mergers and ac-quisitions, investments in innovation, lab and plant expansions, outsourcing growth, and the development of new chemistry platforms.
The past few months have seen sig-nificant merger and acquisition activ-ity. In July 2015, The Siegfried Group bought BASF’s API facilities in France,
Germany, and Switzerland for $300 million (1). In October 2015, Merck KGaA bought Sigma-Aldrich Corp., a supplier of APIs and fine and labo-ratory chemicals, for $17 billion (2). The acquisition solidifies its biophar-maceuticals business and emphasizes chemical manufacturing in its product portfolio.
At around the same time, Johnson Matthey bought Pharmorphix, a spe-cialist in solid dosage form chemistry, from Sigma-Aldrich (3). The company brings expertise in understanding polymorphism and cocrystallization, and helps to bridge the gap between toxicity testing through Phase I.
Drug product innovation challenges are driving new developments in APIs.
“Our customers must find new ways to
bring new therapies to market,” says Nick Johnson, strategic marketing di-rector at Johnson Matthey. The com-pany, once synonymous with chemical catalysis, has rebranded its fine chemi-cals business to focus on custom API development, manufacturing, and life cycle management; chiral chemistry and catalysts; and controlled substance manufacturing. “From a contract man-ufacturing perspective, [our customers’ innovation challenge] affects how we structure our business and services. Our goal must be speed to market, and we must structure on areas of R&D and around technologies,” Johnson says.
Currently, market estimates peg the global API market at $121 billion, and expect it to reach nearly $199 billion by 2022 (4). Generic pharma firms such as Teva and Mylan produce many of their own APIs inhouse, but also sell to other companies, a practice that is becoming more common.
More than 10% of the business is made up of specialty APIs outsourced by pharma innovator companies for clinical research (5). Contract com-panies that focus on API development and manufacturing include Cambrex, Hovione, Siegfried, Johnson-Matthey, and Lonza, are seeing business growth.
Roughly 99% of API volumes (when measured in kilograms) are based on small molecules and 1% from biologics, says Matthew Moorcroft, global mar-keting director for Cambrex, a con-tract development and manufacturing organization (CDMO) that specializes in small-molecule APIs.
However, prices for small-molecule, and even some generic drugs, have been increasing, he notes. Market value share, historically reported be-tween 80–85% small molecules and 25–20% biologics, might tip tempo-rarily even further in favor of small-molecule APIs, Moorcroft adds.
Quality has displaced cost as the top requirement, even in commodity businesses. As a result, the API busi-ness is “no longer a race to the bottom,” according to Peter Werth, CEO of the generic API supplier, Chemworth, in an interview with CPhI for CPhI’s
Specialty Markets and Services Drive API GrowthAgnes Shanley
RA
VIT
ALIY
/LA
GU
NA
DE
SIG
N/G
ET
TY
IM
AG
ES
; D
AN
WA
RD
Cover Story: API Development
As specialty API outsourcing grows, manufacturers and contract development and manufacturing organizations are investing for the long haul.

Non-sterile ointments, creams & liquids that come through our facility get the same high level of scrutiny and quality oversight that sterile products do.
Interphex NYC, 1830Bio San Francisco, 5753
Specialized capability to support multiple dosage forms
…Transfer and manufacture of complex formulations
…Available capacity for large scale manufacturing
…Potent products (up to SafeBridge 5)
…Milling and totally enclosed vessels
…Oxygen sensitive compounds
…Light sensitive products
…Explosion proof room
…Controlled substances
…Hormones
+5#,���$/-��9������ ��������� �������9���"."%"��9���.%*"[email protected]
Like us on Facebook

28 Pharmaceutical Technology MARCH 2016 PharmTech .com
Cover Story: API Development
2016 US Pharmaceutical Industry Outlook, which was released at the Informex show in February, 2016 (6).
“Four years ago, the main consideration that finished formulation companies would take when sourcing an API was cost. These past four years have seen a rapid rise in quality concerns, which are now at the forefront of everyone’s minds,” he said.
API outsourcing on the rise“Worldwide, large innovator companies are increasing outsourcing require-ments,” says Moorcroft. “Coupled with the increasing use of generics, and ad-ditional opportunities for increased penetration in developing markets, the industry will continue to experi-ence strong growth,” he adds.
Currently, API manufacturing is the largest business for pharmaceutical CDMOs, the operation most frequently outsourced by small-molecule manu-facturers, and the fifth most frequently outsourced operation for biopharma-ceutical manufacturers, according to
Kate Hammeke, research director with Industry Standard Research (ISR), who shared recent market research findings with Pharmaceutical Technology.
On the commodity API side, an aging population and government spending caps are driving global demand for ge-neric drugs and the APIs used to make them. Recently, Japan has been an area of high generics demand, according to a report by Nikkei Asian Review, as the Japanese government moves to boost use of generics from 50% to 80% of all prescription drugs by 2020 (7).
In Japan, a number of commodity APIs are in short supply, Nikkei Asian Review reported in February, and the pharmaceutical manufacturer, Eisai, plans to start selling APIs for allergy, high-cholesterol and other therapies to generic drug manufacturers in Japan. The company plans to double API pro-duction capacity at its plant in Andhra Pradesh, southern India (7), which had been previously used as a captive source. Eisai will use revenues to ex-pand its R&D pipeline, the report says.
Specialty API manufacturing moving back to the US and EuropeIn specialty areas, API outsourcing demand that had moved offshore is now coming back to the United States and Europe. “Ten to 15 years ago, its perceived economic benefits made offshoring very attractive. Now, com-panies are focusing more on quality, and regulations are becoming more stringent,” says Johnson.
While tax incentives have played some role in stimulating API capacity expansion in the US, insiders say that the onshoring trend has been fueled by concerns about ongoing compliance and data integrity issues at some offshore API manufacturing facilities (Sidebar). The Generic Drug User Fee Act (GDUFA), which charges a fee for manufacturers who want to sell their APIs in the US, is also said to be having an impact.
“Quality and regulatory concerns pertaining to China and India make the US market look increasingly at-tractive … We are in a renaissance for European and US manufacturing,”
Data integrity (see the article on p. 50 of this issue of Pharmaceutical Tech-
nology) has become a more serious compliance problem at pharmaceutical
manufacturing plants throughout the world. Over the past three years, as the
FDA has increased inspections of offshore facilities, the agency has penalized
a number of API manufacturers in India and China for cGMP violations, many
of them involving data integrity. In some of the FDA inspection reports, quality
control staffers said that they were ordered by superiors to back date lab data,
or to delete information and perform tests until samples passed. Some of the
companies whose plants were penalized were also put on an import ban list,
preventing them from shipping products to the United States.
As more of these plant issues became public, rumors spread in India in Febru-
ary 2016 that the US was banning all imports of API from India and China (1). In
fact, the ban only affects supplies that are covered by US government contract.
The panic might be understandable, given the growing number of regulatory
citations directed at offshore API facilities. Because India and China supply
80% of the APIs used in US pharmaceutical production, mostly commodity-
type products, the US government has been questioning FDA’s ability to moni-
tor quality in Chinese and Indian plants, according to a February 2016 report in
the Regulatory Affairs Professionals Society (RAPS) journal, Regulatory Focus.
API manufacturing company executives believe that data integrity issues and
perceptions that offshore suppliers are not sufficiently trained in current good
manufacuring practices (cGMPs) are driving a move back to outsourcing more
APIs manufactured in the U.S. and Europe.
According to the RAPS report (2), 41 manufacturing sites in China are now on
import alert, five are on alert in Hong Kong, and 42 sites in India (some of which
have only been cited for problems with specific products, not all of their manufac-
turing output). In December 2015, RAPS reported, US representatives wrote to the
US Government Accountability Office asking that it look into whether FDA could
handle the load of inspections now required in India and China. RAPS also pointed
out logistical issues that FDA has faced in getting visas that would allow more of its
new inspectors to work in China. The agency closed two Chinese offices in 2014 to
consolidate activities in Beijing, according to RAPS. In India, FDA plans to increase
the number of inspectors from nine to 19, RAPS reports. Below are just a few of
the offshore API manufacturing facilities involved and the penalties they received:
t� Indian-based Biocon Ltd. was placed on an import ban list
t� A Dr. Reddy’s facility in India received a warning letter for
documentation problems
t� Zhejiang Hisung’s plant in China was cited for data manipulation
t� Ipca Labs’ three API plants in India were cited for data integrity problems.
References1. D. Shenoy, “Misleading Reporting: Indian Pharma Company APIs are NOT
Banned in the U.S., “ CapitalMind.in, http://capitalmind.in/2016/02/mislead-ing-reporting-indian-pharma-company-apis-are-not-banned-in-the-us/
2. Z. Brennan, “US FDA Inspections in China: An Analysis of Form 483s from 2015,” Regulatory Affairs Focus, Regulatory Affairs Professionals (RAPS) Society, www.raps.org, http://www.raps.org/Regulatory-Focus/News/2016/02/10/24296/US-FDA-Inspections-in-China-An-Analysis-of-Form-483s-from-2015/
– Agnes Shanley
Concerns Mount Over Data Integrity and Compliance Issues Abroad

Pharmaceutical Technology MARCH 2016 29
said David Hoffman, president of US operations for the API manufacturer, Hovione, in an interview in the CPhI report previously cited (6).
US and European expansionsHovione plans to expand its facility in East Windsor, NJ, by the end of 2017 (8).The company will more than double API manufacturing capacity at the site, and will also add a new com-mercial spray dryer, as well as highly potent API capacity.
Meanwhile, Cambrex is expanding its Charles City, IA, facility. “In 2013, we added an additional 40,000 gallons of reactor capacity,” says Moorcroft,
“and during the first quarter of 2016, we will see the completion of a new manufacturing facility, which will ini-tially add a total of 70 cubic meters of glass-lined and Hastelloy reactors, sig-nificantly increasing the site’s current good manufacturing practices (cGMP)
manufacturing capabilities,” he says. Cambrex also plans to invest in its API site in Sweden to support new late-stage clinical projects, and in its Italian plant, to improve generic API develop-ment capacities, Moorcroft says.
As pharma’s drug products evolve, so are the drug substances that make them possible. Specialty actives are becoming more complex and struc-turally larger, reflecting the increased sophistication of today’s therapies, says Johnson.
Oncology drug growth drives investment in containment systemsOn the small-molecule side, Johnson notes, demand for oncology drugs, the fastest growing segment of the API market, is driving increased invest-ment in high containment capacity. Because fewer of today’s complex new API therapeutics can be separated by crystallization, Johnson Matthey is
emphasizing work in advanced separa-tions based on chromatography, John-son says.
R&D hubsJohnson Matthey is focusing some of its API research work and invest-ments in Cambridge, UK, a research hub for many Big Pharma companies, including AstraZeneca, GlaxoSmith-Kline, and Takeda. The company has expanded its facilities there to bring additional process R&D services and kilo-scale manufacture to the grow-ing site, and to integrate assets that it gained when it bought Pharmorphix.
The project has increased Johnson Matthey’s capacity to perform chem-istry and biocatalysis process R&D, route scouting, process development, optimization and scale-up of heteroge-neous, homogeneous and biocatalytic processes, as well as the non-GMP kilo-scale manufacture of APIs and
Award-Winning On-Dose
Authentication
& Brand Protection
Solutions
HE
ALT
H &
S
AFETY PRODUCT O
F T
HE
YE
AR
AWARDS2013
�����rt��������������sy����Appl��
��rt� ��������������� ������������
���st��������� �������t
7UX7DJ�R�HUV�DQ�HɝFLHQW��FRVW�H�HFWLYH�VROXWLRQ�
�������� ����������rf���������������rs�����
������������������������fr������������������t.
Contact us now: www.trutags.com/technologies

30 Pharmaceutical Technology MARCH 2016 PharmTech .com
intermediates. “We will now be able to scale up processes to the 100-L level,” says Johnson.
Developers have ambitious product-introduction goalsGiven robust drug product approval rates and pipelines, individual API suppliers are setting ambitious goals. Cambrex reportedly targets 10 new product introductions a year (9). For Neuland Labs, a specialized API sup-plier in India, the figure is 10 to 15, says Davaluri Suharsh Rao, director and president of contract research.
Neuland focuses on developing cus-tom manufacturing solutions for inno-vator companies in the US, Europe, and Japan. The company, which currently produces more than 75 APIs, has spent more than $2 million to expand its API R&D lab in Hyderabad, adding process engineering and parallel processing ca-pabilities, as well as such tools as reaction calorimeters, software and data mining capabilities. The expansion should be completed by the end of 2016, says Rao.
Neuland is investing in lab improve-ments and employee training to allow it to build skill sets around pharmaceuti-cal quality by design (QbD) and cGMPs.
“We’ve realized that QbD results in final product quality and reliability, so we are investing in the infrastructure required,” Rao says. “Companies getting ready to file investigational new drug (IND) and new drug (NDA) applications with FDA will need QbD data for these filings,” he adds.
QbD has also become important for Johnson-Matthey, says Johnson, particularly the use of design-of-ex-periments techniques. The company has also used modeling and rapid screening to evaluate the effectiveness of APIs for solid-dosage forms, he says.
Neuland’s strategy has been to avoid crowded areas where there is a lot of competition. “We prefer to focus on areas such as complex formulations where there is insufficient expertise, (e.g., metered dose inhalers for respira-tory therapies),” says Rao. “These ma-terials are hard to synthesize and have very complex particle size distribution requirements,” he says.
The company has also invested in micronization capacity, and in niches such as deuterated molecules for repur-posing approved APIs for new indica-tions. Deuteration is one of many drug repurposing strategies that are being explored today. The idea of repurpos-ing mature APIs in novel formulations may not be new, but Moorcroft says that it has been gaining more atten-tion, as companies develop drugs for rare diseases and unmet medical needs, or new dosage forms that would offer enhanced benefit in a new dosage form or application.
Repurposing challenges API de-velopers, both on the specialty and the commodity side. Deuteration, for instance, which replaces hydrogen atoms with deuterium, requires the use of special process and storage equip-ment, because deuterium is flammable. On the commodity side, repurposing challenges supplier agility, Moorcroft says. “It requires that generic API manufacturers respond more quickly to customer demands for material, and that they supply targeted volumes that address a smaller patient subset. Cam-brex has successfully supported many drug manufacturers working in the repurposing space,” he says.
Recently, Neuland has moved into peptides, an area where Rao sees great potential, because close to 1000 com-mercial drugs today are peptide-based.
“We started this business in 2010 in a very muted way, making building blocks,” he says, noting that the com-pany recently partnered with a US cli-ent, using solution-phase technology to synthesize a fairly long-chain peptide at 30 kilograms/year.
Another new area of interest is car-bohydrate chemistry, which Neuland is exploring through a partnership with a local university. Rao says the company started working on synthetic Vitamin D analogs about a year ago, challeng-ing compounds that are highly potent and require highly specialized synthe-sis and analysis.
Clearly, the market is becoming more competitive, and more consolida-tion can be expected as regulatory re-
quirements become more stringent and demands for complex APIs increase. For suppliers that have invested in the knowledge and infrastructure required, however, a stronger drug product pipe-line promises to yield more opportuni-ties in the future.
References 1. J. Miller, “CMO Consolidation Pace May
Slow Down, “PharmTech.com, January 6, 2016, accessed February 5, 2016, www.pharmtech.com/cmo-consolidation-pace-may-slow-down
2. C. Hroncich, “Merck KGA, Darm-stadt Germany, Announces Completion of Sigma-Aldrich Acquisition,” Pharm-Tech.com, November 18, 2015, accessed February 5, 2016, www.pharmtech.com/merck-kgaa-darmstadt-germany-announces-completion-sigma-aldrich-acquisition
3. Johnson Matthey Acquires Pharmorphix, Press Release, www.matthey.com/media_and_news/news/2015/pharmorphix
4. Active Pharmaceutical Ingredients Global Market Outlook –Trends, Forecast, and Opportunity, 2014-2022, Reportlinker Preview, Press Release, Accessed February 4, 2016, www.prnewswire.com/news-re-leases/active-pharmaceutical-ingredients-global-market-outlook---trends-forecast-and-opportunity-assessment-2014-2022--
-reportlinker-review-300159995.html 5. Cambrex Sell Off A Golden Oppor-
tunity, Seeking Alpha , p. 4, analyst report,accessed February 5, 2016, Site registration required for access. www.seekingalpha.com/article/3874116-cam-brex-sell-golden-opportunity?page=4
6. CPhI Worldwide Pharma Insights 2016, FiercePharma.com, www.fiercephar-mamanufacturing.com/press-releases/cphi-worldwide-launches-pharma-in-sights-2016-usa-report-informex-2016
7. “Eisai to Supply Generic Drug Makers With Bulk Ingredients,” Nikkei Asian Review, Feb. 9, 2016, accessed Feb. 19, 2016, www.asia.nikkei.com/Business/Companies/Eisai-to-supply-generic-drug-makers-with-bulk-ingredients
8. Hovione Announces the Expansion of its New Jersey Facility, Press Re-lease, accessed February 20, 2016, www.prnewswire.com/news-releases/hovione-announces-the-expansion-of-its-new-jersey-facility-529940241.html
9. Cambrex Sell Off A Golden Oppor-tunity, Seeking Alpha , p. 2, analyst report,accessed February 5, 2016, Site registration required for access, www.seekingalpha.com/article/3874116-cam-brex-sell-golden-opportunity?page=2 PT
Cover Story: API Development

cGMP MANUFACTURE OF COMPLEX API
DISCOVERY SERVICES | CHEMICAL DEVELOPMENT | API MANUFACTURING | DRUG PRODUCT MANUFACTURING
< Early Phase Clinical Batches (cGMP)
< Late Phase Clinical Batches through to Registration/Validation Batches (cGMP)
< Potent & Cytotoxic Compounds
< Controlled Substances
< Steroids
< Hormones
< Sterile API
Corporate Headquarters:
26 Corporate Circle, Albany, NY 12203 USA
Contact: [email protected]
Website: www.amriglobal.com
NORTH AMERICA | EUROPE | ASIA

32 Pharmaceutical Technology MARCH 2016 PharmTech .com
Solid-dosage drugs remain the pre-ferred dosage form due to ease of administration and typically
lower manufacturing costs compared to parenteral and other dosage forms. Many of the properties of a chemical relate directly to its physical form, and solid compounds can adopt numerous different crystalline forms (e.g., poly-morphs, solvates, and hydrates) and non-crystalline (e.g., mesophases or amorphous) forms. Extensive screening must be completed to identify the dif-ferent potential stable and meta-stable forms that an API may adopt during manufacturing of the drug substance and drug product, packaging, storage, and within the body. Potentially stable salts and cocrystals of the API may also be evaluated. In addition to the skills and knowledge required to conduct and evaluate the large quantities of struc-ture data generated during polymorph screening projects, the ability to design appropriate screening studies has a direct impact on their success.
Why polymorph screeningToxicology, efficacy, and stability are important criteria when selecting an appropriate candidate for the develop-ment of a solid-dosage drug. “These properties can vary depending on the physical structure of the API. It is necessary to understand the physical properties of each potential solid form and the relationship between these different forms in order to identify the candidates with the greatest like-lihood of success,” says Brett Cowans, director of materials science for SSCI (Solid State Chemical Information), a division of AMRI.
The chances of selecting the best candidate improve considerably the earlier these factors are known. “The challenge for companies is to balance the cost of development and risk of se-lecting the wrong solid form. Ideally, this balance will include understand-ing the true physical properties of the selected candidate compound, includ-ing polymorphism, and how these properties will affect the API manufac-turing and drug product efficacy and stability for each form, as well as the M
ALE
RA
PA
SO
/E+
/GE
TT
Y I
MA
GE
S
Gauging the adequate level and type of screening is the challenge.
intellectual property position for the formulated drug,” Cowans observes.
Many forms to identifyFrom a technical point of view, the main challenge of polymorph screen-ing is to identify all crystalline forms relevant and potentially crucial to de-velopment. Solid forms may, for exam-ple, be observed in the presence or ab-sence of certain impurities in the final isolation step of the API, upon storage of its intended dosage form, or in vivo when administered orally, according to Patricia Andres, director of particle en-gineering for SSCI. “The screening of such forms typically requires a rational design of experiments often not ame-nable to high-throughput screening, and the unambiguous identification of new phases necessitates the prepara-tion of pure phases in sufficient quanti-ties for characterization,” she notes.
Andres cautions, however, that while thorough and targeted screening will considerably reduce the chances that a new critical form is discovered later in development, it is not possible to know without a doubt that all relevant forms have been found. She cites ritonavir as a classic example of a crystalline form not discovered during development. The product was developed using a form designated as Form I, which was later found to be metastable with re-spect to a new form exhibiting consid-erably lower solubility and rendering the initially developed drug product bio-unavailable.
Achieving the right balance“The ability to gauge the level and type of screening that is adequate based on considerations of development phase, dosage form, physicochemical and biological properties of the com-pound, and regulatory requirements is paramount,” Andres asserts. Risks associated with the discovery of a new crystalline form at a later phase of de-velopment must be weighed against the possibility of the compound failing in development. This risk assessment re-quires both experience of solid-state is-sues that can affect the performance of
Cynthia A. Challener is a contributing
editor to Pharmaceutical Technology.
API Synthesis & Manufacturing
Polymorph Screening for Identification of Relevant Crystalline Forms Cynthia A. Challener

“I need a CDMO that’s
ACCOUNTABLE.”
With Rottendorf Pharma as your CDMO, you can be sure we will take owner-
ship of your projects from start to finish, fully understand your time-to-market
pressures, know your supply chain inside and out, and be able to grow with you.
Need someone you can count on for your solid oral doses? Rottendorf owns it.
��������� ������������!������������� ���������������������� ������ ���
� � � �� � � � � � � � � � � �� � � � � � � � � � � � � � � � �� � � � � � � �� � � � � � � � � � � � �� � � � � � � � � � � � �

34 Pharmaceutical Technology MARCH 2016 PharmTech .com
API Synthesis & Manufacturing
the drug substance and drug product and intimate knowledge of the drug substance and drug-product processes.
Another practical challenge in poly-morph screening, according to Marco Gil, a general manager with Hovione, is to identify processes for isolation of different forms that can be scaled in a robust manner to yield the selected polymorphs (or cocrystals).
Fit-for-purpose approachPolymorph screening is a complicated process that involves many individ-ual experiments and significant data analysis. Significant resources have been invested in efforts to accelerate the process. The dream of displacing rational experimental design in phar-maceutical form screening (e.g., salts, cocrystals, and polymorphs) with
high-throughput screening was pur-sued for about a decade by mid-sized and large pharmaceutical companies, as well as material science contract re-search organizations (CRO), according to Jon Selbo, director of preformula-tion with SSCI. “Significant internal resources were dedicated to develop-ing home-built screening instruments and/or purchasing equipment and software to implement this new strat-egy,” he says.
The justification for high-through-put screening was that significant im-provement in identification of other-wise unknown forms could be more easily obtained by conducting large ar-rays of experiments in parallel. In most cases, however, this strategy failed for both identification of new forms and desired forms for development. “In
general, crystallization quality was poor, there was a lack of experimen-tal diversity (regardless of significant increases in solvents used in crystalli-zation attempts), the analytical results were inadequate, and analyzing the large quantities of poor quality data generated in the experiments often proved difficult,” Selbo observes. In addition, polymorphs identified using high-throughput screening must be considered as general lead compounds and must be thoroughly assessed, in-cluding evaluation of their pharmaco-kinetics, solubility, and other proper-ties, according to Gil.
The driver for large-scale screening in early development has, for the most part, also disappeared with the new molecular entities failing more often today due to efficacy and toxicity issues
FDA warns firms on data integrity and cannabidiol claims
Data Integrity Violations Earn Warning Letter for Ipca Laboratories
Ipca Laboratories, a Mumbai, India-based drug manufacturer, has received a
warning letter from FDA for good manufacturing practice violations identi-
fied during inspections in 2014 at three facilities including numrous data
integrity violations.
The Ratlam API facility was cited for failure to have sufficient computer sys-
tem controls to prevent unauthorized access or changes to data. FDA investiga-
tors noted a lack of basic laboratory controls to prevent changes to electronically
stored data; and use of incomplete records to evaluate the quality of drugs and
to determine whether the drugs conformed with established specifications and
standards. The Jan. 29, 2016 letter notes that Ipca “routinely re-tested samples
without justification and deleted analytical data. We observed systemic data
manipulation across your facility, including actions taken by multiple analysts,
on multiple pieces of testing equipment, and for multiple drugs.”
FDA noted that laboratory analysts had administrator access to chromatog-
raphy systems and used the access to manipulate raw data and test results. The
agency noted that the firm’s quality unit was aware of the lack of controls in the
computerized systems, but “senior management failed to take sufficient cor-
rective action and prevent the recurrence of these problems.” FDA noted that
the company failed to ensure that laboratory records at the Pithampur drug
product manufacturing facility included complete data derived from all tests
to assure compliance with established specifications and standards. Inspectors
documented multiple instances of analytical test results without original data,
injections overwritten and deleted without justification, and chromatograms
related to original test results overwritten by subsequent testing.
Additional violations related to laboratory records were found at the Piparia
Silvassa drug-product manufacturing facility where the firm failed to ensure
that laboratory records included complete data derived from all tests necessary
to assure compliance with established specifications and standards.
—Rita Peters
FDA Warns Cannabidiol Oil Sellers
FDA issued eight warning letters on Feb. 4, 2016 to various makers of a dietary
supplement called cannabidiol, which is derived from the cannabis plant. Can-
nabidiol, which is also known as CBD oil, is described as a nonpsychoactive (no
THC) dietary supplement that is purported to have numerous benefits, includ-
ing anti-inflammatory and antiemetic properties. The oils are also advertised
as being able to reduce depression, combat cancer, shrink tumors, facilitate
wound healing, and treat other serious, chronic conditions, such as rheumatoid
arthritis, schizophrenia, diabetes, lupus, and Crohn’s disease, among others.
The companies that received warning letters about the claims associated
with cannabidiol include Sana Te, PainBomb, Morguetorium, Michigan Herbal
Remedies, HealthyHempOil.com, Dose of Nature, Green Garden Gold, and ABC
Productions. FDA reviewed the websites, Etsy stores, and Facebook pages of
the companies, and concluded the companies were selling products marketed
as new drugs. FDA said the CBD oil products are being marketed as medications
and have not been adequately tested to prove they are safe and effective. The
agency wrote, “drugs may not be legally introduced or delivered for introduc-
tion into interstate commerce without prior approval from the FDA.”
To FDA, CBD products are excluded from the dietary supplement definition,
as CBD oil is already being investigated for its medicinal benefits in clinical trials.
The agency pointed out that CBD oil is currently undergoing clinical investiga-
tion, citing GW Pharmaceuticals’ studies in the United States of the products
Sativex and Epidiolex for the treatment of cancer pain and a form of epilepsy
called Dravet Syndrome, respectively. The agency says that unless a clinical in-
vestigation meets the limited criteria in that particular regulation, an investiga-
tional new drug (IND) application is required for all other clinical investigations
of products. Thus, any manufacturers of CBD oil making claims about clinical
efficacy for the treatment of disease must file an IND with the agency and have
scientific data to back up their claims.
—Randi Hernandez

Pharmaceutical Technology MARCH 2016 35
API Synthesis & Manufacturing
rather than biopharmaceutics proper-ties. “Only a few larger pharmaceutical companies and small CROs continue to employ high-throughput screening as a first pass strategy,” says Selbo.
What is important, according to Cowans, is how the results of high-throughput screening can contribute to better understanding of the proper-ties of potential solid-form candidates.
“As a screening tool, high-throughput technology provides a platform to evaluate the potential for crystallizing new solid forms under certain con-ditions, albeit with some limitations. The information obtained solely from high-throughput technology is not suf-ficient to select a solid-form candidate for development, however,” he says. Consequently, rational experimental design with fit-for-purpose screening strategies based on rational screening or mixed screening approaches have become the norm for the industry, ac-cording to Selbo.
Large-scale screening is still em-ployed as a part of intellectual prop-erty (IP) strategies for later-stage pro-grams where there is more certainty of compound survival. “High-through-put screening is today an essential technology for mapping polymorphic forms, generating IP, and broadening the protected space in terms of poly-morphs, and helping to avoid surprises as much as possible in advanced de-velopment phases,” Gil comments. For these programs, mixed screening services with rational, mid-, and high-throughput screens may be employed in an attempt to cover all bases, ac-cording to Cowans.
Sharp patternsWith respect to advances in technol-ogy for polymorph screening, Cow-ans points to improvements in x-ray powder diffraction (XRPD), which he refers to as the “quintessential tool for distinguishing different crystalline forms.” Most importantly, current-generation instruments are capable of providing the sharp, high-quality pat-terns that are necessary for identifying whether a material exists as a single
crystalline phase and are amenable to indexing algorithms.
The combination of indexing soft-ware and advanced XRPD technology improves the ability to identify crys-talline phases or mixtures, increases screening efficiency, and provides critical information about the various crystalline forms obtained, according to Cowans. SSCI has developed and patented its own indexing software
specifically targeting organic mol-ecules and integrated this tool into its routine screening activities.
Another important advance for polymorph screening is the reduc-tion in the quantity of API required for analytical techniques (e.g., XRPD), according to Gil. Automation has also increased the efficiency of these analyses and accelerated sample pre-characterization. PT
Request Your 2016 USP Catalog!
1,200+ USP Chemicals Bulk Prices Included Most Complete
Label Information
Serving Science and Industry since 1971
DEA Schedules I-V Controlled Substances
800.772.8786SpectrumChemical.com
du esSubstances
m

36 Pharmaceutical Technology MARCH 2016 PharmTech .com
PEER-REVIEWED
Statistical Tools to Aid
in the Assessment of
Critical Process ParametersKe Wang, Nathan D. Ide, Olivier Dirat, Ann K. Subashi, Nicholas M. Thomson, Kim E. Vukovinsky, and Timothy J. Watson
There are many different approaches for assessing
process parameter criticality, and assessing which
process parameters have a significant impact on
critical quality attributes (CQAs) is a particular
challenge. Including an unimportant process parameter
as a critical process parameter (CPP) in a control
strategy can be detrimental. The authors present a
statistical approach to determine when a statistically
significant relationship between a process parameter
and a CQA is large enough to make a practically
meaningful impact (i.e., practical significance).
The assessment of critical quality attributes (CQAs) and
the control of critical process parameters (CPPs) that
affect these attributes are important components of the
overall control strategy for drug substance and drug product
manufacturing. There are many different approaches for
assessing process parameter criticality, and statistics can
play an important role in these evaluations. One particular
challenge involves assessing when a relationship between
a process parameter and a CQA represents a significant
impact on that CQA. Assessing impact based solely on sta-
tistical significance (p-value) is not appropriate, because sta-
tistical significance does not take into account the strength
of the relationship relative to the relevant quality require-
ments and can lead to the inclusion of relatively unimpor-
tant process parameters as critical elements of the control
strategy. Including these unimportant process parameters
as CPPs is undesirable as it effectively dilutes the focus on
process parameters that are truly important for ensuring
product quality. The excessive assignment of criticality to
unimportant process parameters can also place an unneces-
sary burden on manufacturing operations.
This article introduces a statistical approach to help
determine when a statistically significant relationship
between a process parameter and a CQA is large enough
to make a practical meaningful impact (i.e., practical
significance). The assessment of practical significance can
then be used to determine if a parameter has a significant
impact on a CQA, thus helping to assess the criticality of
process parameters. The described statistical methodology
is intended to provide a consistent framework for
discussions on process parameter criticality and should be
used to support but not replace scientific judgment.
The statistical approach that has been developed takes
into account the process risk (Z score) and the parameter
effect size (20% rule). This article discusses some background
on criticality assessments, the motivation behind the
development of a new strategy for these assessments, and
examples of the implementation of this approach.
Background on criticality assessments
The control of CQAs, critical material attributes (CMAs), and
CPPs is an integral component of the overall control strategy
Ke Wang, PhD*, is associate director, Pharm Sci & Manufacturing
Statistics, Pfizer, MS 8220-4438, Eastern Point Road, Groton, CT 06340,
Tel.: +1.860.686.2888, [email protected]; Nathan D. Ide, PhD, is
principal research scientist, Process Research and Development, AbbVie,
1401 Sheridan Road, North Chicago, IL 60064; Olivier Dirat, PhD, is
associate research fellow, Chemical Research and Development, Pfizer,
Discovery Park House Ramsgate Road, Sandwich, CT13 9NJ, United
Kingdom; Ann Kathryn Subashi is director, Global CMC; Nicolas
M. Thomson, PhD, is director, Chemical Research and Development;
Kim Erland Vukovinsky is senior director, Pharm Sci & Manufacturing
Statistics; and Timothy J.N. Watson, PhD, is research fellow, CMC
Advisory Office, all four at Pfizer, Eastern Point Road, Groton, CT 06340.
*To whom correspondence should be addressed.
Submitted: May 19, 2015. Accepted: Aug. 14, 2015.
GE
OP
AU
L/G
ET
TY
IM
AG
ES

Focused on Your Success
Pharma&Biotech
Committed to Global Innovation for Human Health
Lonza has been a reliable partner in the life sciences industry for
over 30 years. Our experience in biological and chemical develop-
ment and manufacturing has allowed us to create a broad platform
of technologies and services for fine chemicals, advanced inter-
mediates, active pharma ceutical ingredients (APIs), functional
ingredients, biologics, cell and viral therapies.
We are committed to continued innovation with a focus on future
scale-up technologies and emerging markets. Whether you are
an established pharmaceutical company or an emerging biotech,
Lonza is prepared to meet your outsourcing needs at any scale.
Why Outsource with Lonza?
– Full range of services from preclinical risk assessment to
full-scale commercial manufacturing
– Advanced technologies and optimized processes to streamline
your product pipeline
– 10 contract development and manufacturing sites worldwide
– Experience with worldwide regulatory authorities
– Track record in meeting accelerated timelines associated with
breakthrough therapy designated products
– Dedicated project teams committed to comprehensive and
timely communications
– Lean, sustainable processes that minimize waste and
environmental risk
www.lonza.com/oursites
For more information, contact us at:
North America: +1 201 316 9200
Europe and Rest of World: +41 61 316 81 11

38 Pharmaceutical Technology MARCH 2016 PharmTech .com
Criticality Assessment
for drug substance and drug
product manufacturing. The
International Conference on
Harmonization (ICH) Q8(R2)
(1) provides the following def-
initions:
t� CQA is defined as a phys-
ical, chemical, biological. or
microbiological property or
characteristic that should
be within an appropriate
limit, range, or distribution to
ensure the desired product
quality.
t� CPP is a process parameter
whose variability has an
impact on a critical quality
attribute and therefore
should be monitored or con-
trolled to ensure the process
produces the desired quality.
ICH Q11 (2) states that:
“A control strategy should
e n s u r e t h a t e a c h d r u g
substance CQA is within the
appropriate range, limit, or
distribution to assure drug
substance quality. The drug
substance specification is
one part of a total control
strategy and not all CQAs
need to be included in the
drug substance specification.
CQAs can be (i ) included
on the specif ication and
confirmed through testing
the final drug substance, or (ii)
included on the specification
and con f i rmed th rough
upstream controls (e.g., as
in real-time release testing
[RTRT]), or (iii) not included on
the specification but ensured
through upstream controls.”
In addition, ICH Q11 states
that : “ Impurit ies are an
important class of potential
drug substance CQAs because of their potential impact on drug
product safety. For chemical entities, impurities can include
organic impurities (including potentially mutagenic impurities),
inorganic impurities (e.g., metal residues), and residual solvents.”
The term CMA is not defined in the glossary of ICH
Q8, but ICH Q11 clarifies that material attributes can be
intermediate, reagent, solvent, or starting material
attributes. It is appropriate to use the term CMA for any of
the starting material or intermediate attributes that have
an impact on a drug substance CQA (particularly in the case
where a specific CQA/impurity in the drug substance is
derived from a different chemical species upstream).
Based on the above definitions and ICH Q8/Q11 guidance,
the criticality of process parameters should be assessed for
their impact on the CQAs and CMAs. Upstream/intermediate
specifications can include quality attributes that are not
part of the control strategy for CQAs but are included for
monitoring and trending purposes only.
Design and run experiments
based on output from initial risk
assesments
Would considering the CPP enhance the control strategy?
Could the PP beexpected to be
critical based on established
science?
Holistic reviewof criticality
assessment andcontrol strategy
Non-criticalprocess
parameter
Criticalprocess
parameter(CPP)
Assess criticality of an individual
processparameter (PP)
Statisticallysignificant
relationshipbetween PP and
a CQA
Is therelationshippractically
significant?
Yes
Yes
Yes
Yes
No
No
No
No
Figure 1: Decision tree for assessment of process parameter criticality. CQA is critical quality
attribute.
Response
Target or specification
Explored space
Figure 2: Illustration of statistical significance and practical significance. The green
and blue lines represent two statistically significant relationships between a
process parameter and a response. The red dashed line indicates the acceptable
limit for this response. The relationship represented by the blue line is practically
significant, but the relationship represented by the green line is not, because the
effect size is small and all of the results are far below the limit.
AL
L F
IGU
RE
S A
RE
CO
UR
TE
SY
OF
TH
E A
UT
HO
RS
.

Unistick® single unit dose liquid stick packs are user-friendly, convenient,
and affordable. They help patients take their medicine on-time and in the
right amount, and can reduce the need for artificial preservatives.
Speak to Unither Pharmaceuticals today to differentiate your products
and improve your patient’s experience without increasing costs.
Unither is a global development and manufacturing partner for pharmaceutical
dosage forms, with facilities in Europe and North America.
Visit us at Interphex booth #1318 www.unither-pharma.com
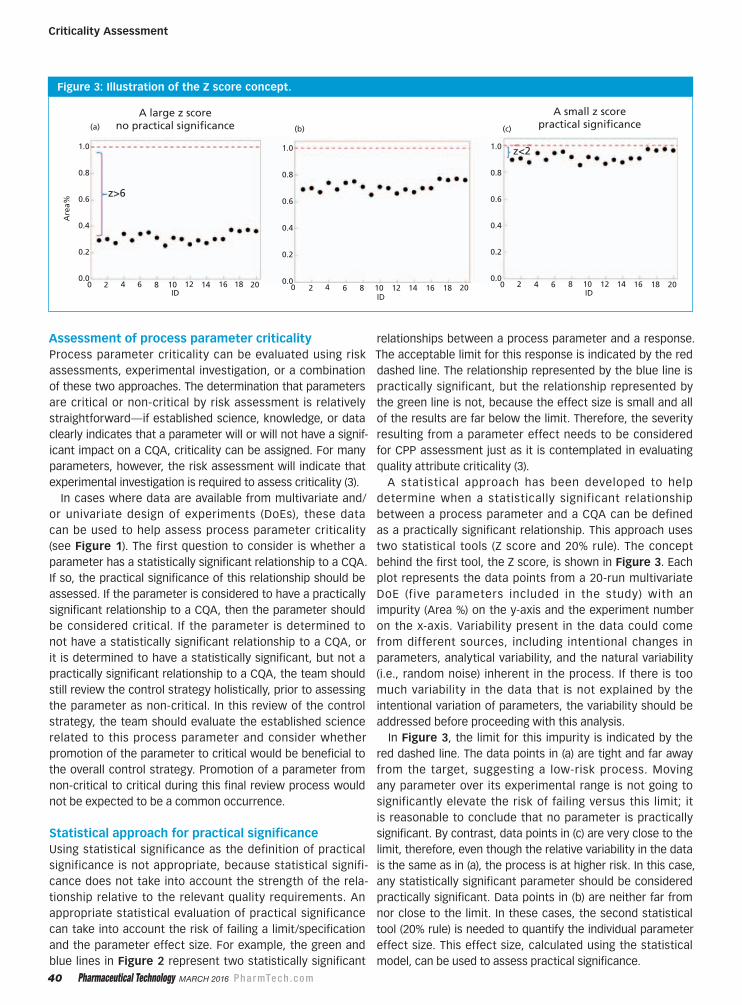
40 Pharmaceutical Technology MARCH 2016 PharmTech .com
Criticality Assessment
Assessment of process parameter criticality
Process parameter criticality can be evaluated using risk
assessments, experimental investigation, or a combination
of these two approaches. The determination that parameters
are critical or non-critical by risk assessment is relatively
straightforward—if established science, knowledge, or data
clearly indicates that a parameter will or will not have a signif-
icant impact on a CQA, criticality can be assigned. For many
parameters, however, the risk assessment will indicate that
experimental investigation is required to assess criticality (3).
In cases where data are available from multivariate and/
or univariate design of experiments (DoEs), these data
can be used to help assess process parameter criticality
(see Figure 1). The first question to consider is whether a
parameter has a statistically significant relationship to a CQA.
If so, the practical significance of this relationship should be
assessed. If the parameter is considered to have a practically
significant relationship to a CQA, then the parameter should
be considered critical. If the parameter is determined to
not have a statistically significant relationship to a CQA, or
it is determined to have a statistically significant, but not a
practically significant relationship to a CQA, the team should
still review the control strategy holistically, prior to assessing
the parameter as non-critical. In this review of the control
strategy, the team should evaluate the established science
related to this process parameter and consider whether
promotion of the parameter to critical would be beneficial to
the overall control strategy. Promotion of a parameter from
non-critical to critical during this final review process would
not be expected to be a common occurrence.
Statistical approach for practical significance
Using statistical significance as the definition of practical
significance is not appropriate, because statistical signifi-
cance does not take into account the strength of the rela-
tionship relative to the relevant quality requirements. An
appropriate statistical evaluation of practical significance
can take into account the risk of failing a limit/specification
and the parameter effect size. For example, the green and
blue lines in Figure 2 represent two statistically significant
relationships between a process parameter and a response.
The acceptable limit for this response is indicated by the red
dashed line. The relationship represented by the blue line is
practically significant, but the relationship represented by
the green line is not, because the effect size is small and all
of the results are far below the limit. Therefore, the severity
resulting from a parameter effect needs to be considered
for CPP assessment just as it is contemplated in evaluating
quality attribute criticality (3).
A statistical approach has been developed to help
determine when a statistically significant relationship
between a process parameter and a CQA can be defined
as a practically significant relationship. This approach uses
two statistical tools (Z score and 20% rule). The concept
behind the first tool, the Z score, is shown in Figure 3. Each
plot represents the data points from a 20-run multivariate
DoE (f ive parameters included in the study) with an
impurity (Area %) on the y-axis and the experiment number
on the x-axis. Variability present in the data could come
from different sources, including intentional changes in
parameters, analytical variability, and the natural variability
(i.e., random noise) inherent in the process. If there is too
much variability in the data that is not explained by the
intentional variation of parameters, the variability should be
addressed before proceeding with this analysis.
In Figure 3, the limit for this impurity is indicated by the
red dashed line. The data points in (a) are tight and far away
from the target, suggesting a low-risk process. Moving
any parameter over its experimental range is not going to
significantly elevate the risk of failing versus this limit; it
is reasonable to conclude that no parameter is practically
significant. By contrast, data points in (c) are very close to the
limit, therefore, even though the relative variability in the data
is the same as in (a), the process is at higher risk. In this case,
any statistically significant parameter should be considered
practically significant. Data points in (b) are neither far from
nor close to the limit. In these cases, the second statistical
tool (20% rule) is needed to quantify the individual parameter
effect size. This effect size, calculated using the statistical
model, can be used to assess practical significance.
Are
a%
A large z score
no practical significance
A small z score
practical significance
z>6
IDID
ID
z<21.0
0.8
0.6
0.4
0.2
0.00 2 4 6 8 10 12 14 16 18 20
1.0
0.8
0.6
0.4
0.2
0.00 2 4 6 8 10 12 14 16 18 20
1.0
0.8
0.6
0.4
0.2
0.00 2 4 6 8 10 12 14 16 18 20
(a) (b) (c)
Figure 3: Illustration of the Z score concept.

www.EurofinsLancasterLabs.com
Winner of the CRO Leadership Award for
quality, reliability, productivity and innovation.
Leading experts in:
Chemistry
Biochemistry
Microbiology
Molecular &Cell Biology
Virology
Global Services:
Method Development/Optimization
Validation/Qualification/Transfer
Product Release Testing
Stability Storage & Testing
Raw Materials Testing
Impurities & Residuals Testing
Characterization
Cell Banking
Cell Line Characterization
Viral Clearance
Bioassays
Professional Scientific Services
Perfect timing.
Every time.
The art of mastering cell bank production requires
expertise, innovation and exquisite attention to detail
where every minute counts. With more than 50 years
of combined experience, our passionate, and often
nocturnal, scientists obsess 24/7 to ensure your cell
banks become masterpieces. To that, we offer you:
t� Direct access to your scientist for any question, any time.
� t� Constant oversight and monitoring of your cell banks.
� t� Continual troubleshooting and status updates during
expansion.
For the most accomplished service in the industry,
trust our cell bank experts to be on your watch.
Cell banking
done right.

42 Pharmaceutical Technology MARCH 2016 PharmTech .com
Criticality Assessment
As introduced conceptually in Figure 3, the distance
between the data and the limit plays an important role
in process risk evaluation, and the Z score can be used
to measure this distance. If ϰ and s are the average and
standard deviation of the data respectively, and U is the
upper limit of the specif ication, the Z score can be
calculated using Equation 1:
Z = (U- ϰ)/s [Eq. 1]
The Z score evaluates how close the entire dataset is
to the limit, without focusing on any individual parameter
effects. The value for Z effectively indicates how far the
data is from the limit/specification; a large Z score indicates
that the data are far from the limit/specification, while a
small Z score indicates that the data are close to the limit/
specification. Conceptually, the Z score is similar to a process
capability index (Cpk)(4); a large Z score indicates a low-risk
process and a small Z Score indicates a high-risk process.
In this approach, Z score values of two and six are used as
the cut-off values for assessing practical significance. In cases
where a Z score is larger than six, as illustrated conceptually
in Figure 3(a), there are no practically significant parameters.
In cases where a Z score is less than two as illustrated
conceptually in Figure 3(c), it is generally appropriate to
conclude that every statistically significant parameter is
practically significant. If a Z score is between two and six,
as illustrated conceptually in Figure 3(b), the individual
effect size of statistically significant parameters needs to be
quantified and compared to the limit/specification. Here, 20%
of the limit/specification range (20% rule) is chosen as the
threshold for practical significance. If a parameter effect size is
greater than 20% of the limit/specification range, it should be
considered practically significant. If the parameter effect size
is less than 20% of the limit/specification range, then it is not
practically significant. The selection of 20% as the threshold
for practical significance is similar to the conventional criteria
(0.2–0.3) for a small effect size, using Cohan’s d (5). This
procedure for assessing practical significance is outlined in
Figure 4. The described approach allows for the rapid and
consistent assessment of process parameter criticality.
For simplicity, the above discussion of Z scores is focused
on one-sided, upper-limit specifications (U). It is worth noting
that the approach can be easily extended to one-sided, lower-
limit specifications and also to two-sided specifications.
Examples
Two examples are included to illustrate the implementation
of the described approach. The first example shows the criti-
cality assessment for six crystallization process parameters
in a drug substance intermediate step. The studied parame-
ters were seed temperature, temperature ramp rate 1, tem-
perature ramp 1 end temperature, temperature ramp rate
2, temperature ramp 2 end temperature, and seed loading.
These parameters were incorporated into a 26-2 fractional
factorial with four replicates, at the target conditions, for
a total of 20 experiments. Shown in Figure 5 are the bar
charts for one process-related impurity (impurity 1) with a
limit of not more than 1.0% at the intermediate step. This
impurity is a CMA, as it tracks forward to an impurity (CQA)
in the drug substance. The limit was set based on down-
stream fate and purge data. Statistical analysis revealed that
ramp rate 1 and seed loading had statistically significant
relationships with impurity 1. Given the calculated average
Statisticallysignificant
relationshipbetween a PP
and CQA
Assess practicalsignificance
Target is > 6sfrom average
(Z>6)
Target isbetween 2s and6s from average
(6≥Z≥2)
Is the effect size> 20% of targetor specification
value?
Relationship isnot practically
significant
No YesRelationship is
practicallysignificant
Target is < 2s from average
(Z < 2)
Calculateaverage and
standarddeviation (s) forresponse data
Calculatedistance from
average totarget
in units of s
Figure 4: Decision tree for practical significance. PP is process parameter. CQA is critical quality attribute.

8 weeks.;OH[»Z�^OH[�[OL�OHUK�V�Z��
ramp-up and rework
from First in Human to Proof of Concept
take from you.
��^LLRZ�`V\�JHU»[�H�VYK�[V�^HZ[L�
��^LLRZ�WH[PLU[Z�JHU»[�H�VYK�[V�^HP[�
But when you partner with
7H[OLVU�6UL:V\YJL�™ you learn how
you can get your 8 weeks back.*
And maybe even more.
We approach drug development
H�M\UKHTLU[HSS`�KP�LYLU[�^H �̀
Our way accelerates every step.
(UK��TVYL�PTWVY[HU[S �̀�LSPTPUH[LZ�
the spaces between them.
;VNL[OLY��^L»SS�NL[�`V\Y�TVSLJ\SL�
to Proof of Concept faster — and
better prepared for what comes next.
We’ve got your weeks. Come get them.
A HEALTHIER WORLD. DELIVERED.
3LHYU�HSS�[OL�ILULÄ[Z�VM�ZPUNSL�ZV\YJL�V\[ZV\YJPUN�at 7H[OLVU�JVT�6UL:V\YJL
* 8-week time savings estimate based on applying Patheon OneSource™ optimization
processes for typical multi-vendor Phase I – Phase IIb drug development program.
©2016 Patheon®. All rights reserved.
OneSource™
44 Pharmaceutical Technology MARCH 2016 PharmTech .com
Criticality Assessment
(0.31%), the standard deviation (0.037%), and the limit of
1.0%, a Z score of 18 is obtained. The Z score of 18 is greater
than six, and therefore none of the studied parameters have
a practically significant relationship with impurity 1.
A second example is shown in Figure 6. This example
is based on a measurement of tablet assay, in a 17-run
experimental study for a drug product process, with a two-
sided specification of 95%–105%. The study was designed
to explore the impact of excipients (magnesium stearate
quantity, dicalcium phosphate [DCP] source) and process
parameters (Comil shear force, pre-blending) on tablet
assay. Statistical analysis revealed a significant interaction
between DCP source and Comil shear force. The fitted
regression model is shown in Equation 2 as follows:
���������������������ComilShearForce – 0.70 �
����� �����������ComilShearForce * DCP [Eq. 2]
For a two-sided specification, two Z scores are calculated,
and the lower of the two values is used to assess practical
significance. Given the calculated average (99.24%) and
standard deviation (1.64%) for tablet assay, the upper limit
and lower limit had Z scores of 3.5 and 2.6, respectively. Both
Z scores require additional assessment via the 20% rule to
quantify individual parameter effect sizes. For a model with
a significant interaction between two parameters, each
calculated parameter effect needs to take into account both
the main effect and the interaction effect. The statistical model
in Equation 2 predicts that the maximum change in assay
is 3.36% for Comil shear force and 3.20% for DCP. Given the
specification range of 10% (105%–95%) for tablet assay, the
20% threshold is 2.0%. As both effect sizes are greater than
2.0%, Comil shear force and DCP are practically significant.
Summary
In general, it is expected that the evaluation of practical sig-
nificance presented in the two examples discussed would
encompass all relevant experiments that assess a given
parameter (i.e., the entire explored range). Given the expec-
tation that criticality assessments should be performed by
evaluating effects across the entire explored range, bounda-
ries of the explored ranges should be set in a pragmatic
fashion. These explored ranges need to effectively support
the development of process understanding and the need for
usable ranges in manufacturing, while avoiding unnecessary
expansion of the investigation into ranges that would never
be considered for manufacturing.
Conclusion
Process parameter criticality can be determined using risk
assessments, experimental investigation, established sci-
ence, or a combination of these approaches. The proposed
statistical methodology is intended to provide guidance and
a common language to facilitate discussions on process
parameter criticality. Criticality is assessed by determining
when a statistically significant relationship should be consid-
ered a practically significant relationship, which is then used
to aid the overall criticality assessment.
Acknowledgements
The authors would like to thank Brad Evans, Greg Steeno, Leslie
Van Alstine, Brian P. Chekal, Mark T. Maloney, Shengquan Duan,
Steven Guinness, and Ken Ryan for their helpful discussion and
feedback during the development of this approach.
References
1. ICH, Q8(R2) Pharmaceutical Development (2009).
2. ICH, Q11 Development and Manufacture of Drug Substances (2012).
3. L.X. Yu et al., The AAPS Journal, 16 (4) 771-783 (2014).
4. D. Montgomery, “Process Capability Analysis”, in Introduction to
Statistical Quality Control (John Wiley & Sons, Inc., New York, 3rd ed.,
1997), pp. 430-470.
5. J. Cohen, “Analysis of Variance”, in Statistical Power Analysis for the
Behavioral Sciences (Lawrence Erlbaum Associates, 2nd ed., 1988), pp.
273-406. PT
Target
Are
a%
Average
>6s
1.0
0.8
0.6
0.4
0.20 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
ID
Z=3.5
Z=2.6
Target
Target
105.0
102.5
100.0
97.5
95.00 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
ID
Figure 5: Bar chart for impurity 1 (y-axis) in Example 1
design of experiment (DoE) (x-axis). Black dashed line
is the average of impurity 1 and red dashed line is the
target. Z = 18.
Figure 6: Scatter plot of tablet assay (y-axis) in Example
2 design of experiment (DoE) (x-axis). Black dashed
line is the average of tablet assay and red dashed lines
represent the two-sided specification. Z1 = 3.5 and
Z2 = 2.6.

The Aenova Group’s services cover the entire value chain for the development and production of all the main dosage forms and product groups in pharmaceuticals and dietary supplements. With more than 4,000 employees and over 21 sites, Aenova is one of the leading companies in the pharmaceutical and healthcare industries...and the right choice for your next project.
Global Reach With A Breadth of Services
4����%�-,�
4���+����),.%�,�
4��( -!�%���),.%�,�
4��+�'.%�,�
4���%%�-,
Solids Liquids & Semi-solids
4��.+��(+��(%%(#��%��(%.-#(',�
4��&.%,#(',���.,)�',#(',�
4��#'-&�'-,�
4���%,
Specialty
4��2-(-(1#�,�
4��(+&('�,�
4� �-�����-�&��
����'-#�#(-#�,�
4���)"�%(,)(+#',
Injectables
4� �2()"#%#3�-#('�
4���,�)-#����'. ��-.+#'!�
4���+&#'�%��-�+#%#3�-#('
Members of the Aenova Group
��������4���+�!�'()"�+&��4�������4����.)-��"�+&���4���0#,,���),��4���0#,,�(��4����&&%�+
www.aenova-group.com » [email protected]
Packaging
4� %#,-�+,�
4� (--%�,� ��(%#�,����#*.#�,�
4���+-(',�
4� ����%#'!4��(.�"�,

46 Pharmaceutical Technology MARCH 2016 PharmTech .com
Single-use systems (SUS) featuring plastic components and materials are widely used in bio/pharmaceu-
tical development and clinical-trial ma-terial manufacturing, and are expected to become more common in commer-cial manufacturing. Drug manufactur-ers and contract manufacturers favor SUS for the benefits of fast changeover, reduced cross contamination, and man-ufacturing system flexibility.
Concern about the potential for con-taminants from the plastic materials interacting with the drug, unacceptable extractables data, and a lack of standards for evaluating SUS materials could, how-ever, slow adoption.
Nearly three-quarters of biophar-maceutical manufacturers surveyed for the 2015 BioPlan Associates industry study (1) identified extractables and leachables as a concern that may limit further use of SUS. The only greater concern was breakage of bags and loss of production material. Concerns about extractables and leachables have been steady during recent years, par-tially due to the increased use of dis-posables and increased awareness of the uncertainties about related regula-tory issues, according to the report.
Current good manufacturing practices (CGMPs) require that manufacturing equipment must be constructed so the surfaces that contact components, in-pro-
cess materials, or drug products “must not be reactive, additive or absorptive so as to alter the safety, identity, strength, quality, or purity of the drug product” (2). For bio-logic products, “All surfaces that come in contact with products shall be clean and free of surface solids, leachable contami-nants, and other materials that will hasten the deterioration of the product or other-wise render it less suitable for the intended use” (3). Similar requirements are in place for the European Union (4).
Therefore, bio/pharma and contract manufacturers must—using scientific- and risk-based approaches—test the materials and surfaces that come in direct or indirect contact with the API, ingredients, and drug product to demon-strate equipment and process suitability for regulatory filings.
Extractables studies identify chemi-cals that migrate from a material when exposed to a solvent at an elevated tem-perature, generating a worst-case test for contaminants. The data are used to assess toxicities of the extracted chemicals and to assess the potential for leachables—com-pounds that migrate into a specific drug product or process under normal condi-tions during the drug product’s lifecycle.
Some suppliers of manufacturing components and packaging materials provide extractables data that can be used for preliminary screening and risk assess-ment; however, the information often is
not available or sufficient. A complex supply chain, where different parts of an SUS technology may be manufactured or processed by multiple subcontractors at multiple locations, adds uncertainty to the quality and completeness of the data.
In the BioPlan study, more than 80% of the respondents “agreed” or “strongly agreed” that vendors of SUS should gen-erate and validate extractables and leach-ables data. The study authors concluded that drug owners believe SUS vendors need to do more testing and analysis of materials used in SUS devices, or they are not comfortable dealing with extract-ables and leachables issues and defer to suppliers for the testing (1).
The cost of acquiring data was also considered. Nearly one-third of the re-spondents said they would not pay SUS suppliers more for extractables and leachables data; 22% said they would pay up to 25% more. The estimated average upcharge was 13.4%. The study authors attributed the drug manufacturers’ price sensitivity to SUS-supplier upcharges for data to general cost-consciousness, in-creased knowledge about regulatory is-sues, a better assessment of developing the data internally, and an increasing number of contract service providers offering test-ing services of extractables and leachables.
Willingness to pay a markup for data has largely stabilized, the study concludes, although a growing number of drug owners are willing to do their own testing for early-phase development, where data requirements are less stringent.
Industry effortsThe lack of sufficient extractables data from suppliers of SUS has spurred activ-ity for standardized testing protocols for these technologies. Representatives of drug manufacturers, testing laboratories, SUS manufacturers and suppliers, inde-pendent consultants, and regulatory au-thorities have participated in discussions, authored position papers, proposed best practices and standards, and developed databases. Often, the proposals conflict on key points; however, the groups are reporting efforts to build consensus.
The Product Quality Research In-stitute (PQRI), a collaborative effort of
Building Consensus forE&L Testing StandardsRita Peters
Extractables and Leachables
Standardized testing protocols are crucial for acceptance of single-use systems.
TE
EK
ID/G
ET
TY
IM
AG
ES

Benef it from the focused expertise gained from working on 869
diverse products, collaborating with 436 companies over 24 years.
Talk with the people who can provide you the right answers
D e v e l o p m e n t S c i e n c e s C l i n i c a l M a n u f a c t u r i n g Te c h n i c a l S e r v i c e s
3 0 I N D I A N D R , �9 �( � � � , �9 �< �/ �$�1 �' � �PA 1 8 9 74 U S A P H O N E + 1 ( 2 1 5 ) 3 9 � � � � � � � � � � ) D [ � � � � � � � 5 ) 3 9 6 - 8 3 7 5w w w . l y o t e c h n o l o�J�\���F�R�P����� L�Q�T�X� L�U�\�#� O�\�o - t . c o m I n t e g r a t i n g S c i e n c e a n d T e c h n o l o g y
T E C H N O L O G Y, I N C.
LYOPHILIZATION

48 Pharmaceutical Technology MARCH 2016 PharmTech .com
Extractables and Leachables
FDA’s Center for Drug Evaluation and Research (CDER), industry, and aca-demia, has published several documents including safety thresholds and best practices for extractables and leachables in orally inhaled and nasal products (5), and a similar publication for parenterals and ophthalmic drug products (6).
Bio/pharma and medical-device com-panies formed the Extractables and Leach-ables Safety Information Exchange (ELSIE) Consortium to compile toxicological data on leachables and extractables to study their impact on drug product packaging, delivery, and manufacturing systems. As of October 2015, the consortium had devel-oped a database of nearly 400 compounds listing chemical, chronic toxicity, muta-genicity/carcinogenicity, reproductive/developmental toxicity, and absorption, distribution, metabolism, and excretion information (7).
An ELSIE materials working group also piloted an extraction study protocol on polyethylene and polyvinyl chloride that included extraction solvents, extrac-tion techniques, and a range of analytical techniques. The study investigated whether a reduced number of extraction techniques or a reduced number of solvents could be used to obtain information useful for mate-rial selection, regardless of product type (8).
Trade associations, including the Par-enteral Drug Association and the Inter-national Society of Pharmaceutical Engi-neers, also have been active in sponsoring presentations and publishing papers about extractables and leachables testing.
Proposals and consensusThe Bio-Process Systems Alliance (BPSA), a corporate member trade asso-ciation of component suppliers, systems integrators, users, and testing laborato-ries, was founded in 2005 to accelerate the adoption of single-use manufactur-ing technologies. Representing the in-dustry supplier market segment, BPSA has published recommended practices for extractables and leachables testing and has initiated efforts to standardize testing procedures for other aspects of SUS materials (9).
The BioPhorum Operations Group (BPOG), which represents the drug-owner segment of the industry, defined a standardized extractables testing proto-col for SUS manufacturing systems that covers methods for extractables testing studies, including sample preparation, extraction conditions, and reporting data. The authors, members of the BPOG extractables working group, wrote that a testing protocol, with agreed-upon test methods, would establish common ex-pectations among suppliers, users, and regulators on the type of testing data to be generated. The enhanced data would help users compare components from different suppliers, would assist suppliers in selecting materials that end users need, and would help in controlling product variability (10).
Following publication of the BPOG paper, BPSA published a perspective arti-cle noting a difference of opinions among end-user companies, suppliers, testing laboratories, and standards-writing bod-ies that must be resolved to achieve a full industry consensus standard (11).
Representatives of BPSA and BPOG are participating in an ASTM Interna-tional working group developing prac-tice documents for extractables studies. The extractables test solutions practice will define a standard method to create extraction samples from single-use bio-
process systems using model bioprocess extraction solutions. Data generated from these studies could be used to make risk-based decisions about the potential im-pact on the API or drug product and the selection of equipment or components. Analysis of extractables test solution will be covered in a separate practice (12).
ASTM is also developing a practice for testing of leachables for single-use mate-rials that contact APIs, intermediates, or the final drug product. The studies are designed to provide a leachables profile based on testing methodology compa-rable to extractables studies (13).
References 1. BioPlan Associates, 12th Annual Report
and Survey of Biopharmaceutical Manufac-turing Capacity and Production (Rockville, MD, April 2015), www.bioplanassociates.com/12th
2. Code of Federal Regulations, Title 21, Food and Drugs (Government Printing Office, Washington, DC), Part 211.65(a).
3. Code of Federal Regulations, Title 21, Food and Drugs (Government Printing Office, Washington, DC), Part 600.11 (b).
4. EudraLex, EU Guidelines for Good Manu-facturing Practice for Medicinal Products for Human and Veterinary Use (Brussels, Belgium, August 2014).
5. PQRI, Safety Thresholds and Best Practices for Extractables and Leachables In Orally Inhaled and Nasal Drug Products (September 2006).
6. D. Paskiet, PDA J. Pharm. Sci. Technol, 67 (5) 430-47 (2013).
7. Extractables and Leachables Safety Infor-mation Exchange, online www.elsiedata.org/elsie-database/, accessed Feb. 26, 2016.
8. A. Teasdale, et al., AAPS PharmSciTech, 16 (3) (June 2015).
9. BPSA, Technical Guides, Extractables and Leachables, online www.bpsalliance.org, accessed Feb. 26, 2016.
10. W. Ding, et al, Pharm. Eng, 34 (6) (2014). 11. BPSA, Toward Industry Standardization
of Extractables Testing for Single-Use Systems: A Collective BPSA Perspective, (March 10, 2015) online, www.biopro-cessintl.com, accessed Feb. 21, 2016.
12. ASTM WK43975, “New Practice for De-termining and Characterizing BioProcess Extractables from Components, Subas-semblies, and Assemblies Used in Single-Use Applications,” ASTM International, www.astm.org (West Conshohocken, PA), accessed Feb. 22, 2016.
13. ASTM WK48084, “New Practice for De-termining and Characterizing Leachables released from Materials used in Single-use Systems under bioprocess operating conditions,” ASTM International, www.astm.org (West Conshohocken, PA), accessed Feb 22, 2016. PT
The US Pharmacopeial Convention (USP) will pub-
lish a revision to the United States Pharmacopia
Chapter <661> on Plastic Containers in May 2016,
renaming the chapter Plastic Packaging Systems
and their Materials of Construction. New General
Chapter <1663> Assessment of Extractables As-
sociated with Pharmaceutical Packaging/Delivery
Systems describes scientific practices for accom-
plishing an extractables assessment. New General
Chapter <1664> Assessment of Drug Product
Leachables Associated with Pharmaceutical Pack-
aging/Delivery Systems outlines a framework for
the design, justification, and implementation of
assessments for drug-product leachables derived
from pharmaceutical packaging and delivery sys-
tems. General Chapter <1664.1> addresses spe-
cific considerations for leachables in metered-dose
inhalers, nasal sprays, dry-powder inhalers and
inhalation solutions, suspension, and sprays.
USP updates standards
for packaging plastics

May 16-18, 2016Sheraton Boston Hotelton Boston HotelBShera
SVISIT: www.aaps.org/nationalbiotechFOR MORE INFORMATION!

50 Pharmaceutical Technology MARCH 2016 PharmTech .com
Data integrity is a major regulatory topic in GMP-regulated laborato-ries. The problem is widespread,
as cases of non-compliance have also been observed in laboratories regulated by good laboratory practice (GLP) and good clinical practice (GCP), not just GMP. Non-compliances from the European Medicines Agency (EMA) or warning letters from FDA show ex-amples from companies in the United States, Canada, the United Kingdom, and Italy, as well as China and India. So, it is not just a problem for Asia—it is a global issue.
The non-compliances are not con-fined to falsification and fraud. In
fact, in the majority of labs, the main data integrity issues concern poor data management, which account for 95% of non-compliance cases; only 5% are a re-sult of falsification or fraud. Problems arise from basic errors such as relying on paper as raw data and failing to protect electronic records, rather than deliberate manipulation of data. It is the latter, however, that gets the major headlines. In July 2014, FDA issued a stern warning that data integrity was a key focus of its enforcement efforts (1).
Regulatory guidance on data integrityIn January 2015, the UK Medicines and Healthcare products Regulatory Agency (MHRA) went further and is-sued Guidance for Industry on Data In-tegrity (revised in March 2015) (2). This guidance consists of descriptions, defi-nitions, and expectations based around
those definitions, which include raw data, original records, file structures, and audit trail. The regulatory expec-tations presented in this guidance are useful. However, the definitions are simply presented as a shopping list; a diagram explaining and linking key definitions would be more beneficial.
The guidance presents data integ-rity as the extent to which all data are
“complete, consistent, and accurate” throughout the data lifecycle, which covers the period from data acquisi-tion through to interpretation, report-ing, and archiving and then destruc-tion after the record retention period. There are similarities in the US regula-tions; a 20-year-old FDA definition of data integrity describes the degree to which a collection of data is complete, consistent, and accurate (3).
In European GMP regulations, doc-umentation constitutes a key part of quality assurance and, therefore, com-pliance with GMP (4). An organization must have good documentation, follow standard operating procedures (SOPs), and demonstrate compliance with the applicable regulations. Several require-ments focus on data integrity for com-puterized systems (5). If critical data are entered into a data system, a second check is required, which can either be a second person or can be automated using the computer system itself.
In the US, the regulations for labo-ratory records focus on the concept of complete data (6).
A review of data integrity warning letters reveals several citations for fail-ure to have complete data including a failure to have a complete procedure (7), a failure to fully document the work that is carried out (8), or being selective in re-porting data (e.g., using test samples to
“check” if an instrument is working cor-rectly) (9). Complete data includes the actual observation, which can be visual or by computerized system. Contextual metadata, which puts the data or result in context, is also required. Examples of metadata include operator ID, units of measurement, sample information, identity, batch number, instrument suit-ability, and readiness. Audit trail events
Data Integrity
Data integrity is a widespread, global problem that must be addressed.
D3D
AM
ON
/GE
TT
Y I
MA
GE
S
Dr. Bob McDowall is director of R D
McDowall Ltd., [email protected].
Dr. Joanne Ratcliff is communications
project manager at Mettler Toledo GmbH,
How Important is Data Integrity to Regulatory Bodies? Bob McDowall and Joanne Ratcliff

Pharmaceutical Technology MARCH 2016 51
are also part of the metadata. If a hy-brid system or a fully electronic system is used, FDA and European regulatory agencies require companies to review and evaluate audit trail events to see if data have been manipulated without authorization.
FDA guidance comprises a list of com-monly asked questions and answers that are crucial for maintaining data integrity (10). Detailed explanations are given for:t� Why paper records from a hybrid
system should not be defined as raw data—instead it should be the
underlying electronic records; Al-though the reasoning focuses on chromatography, it is applicable to any computerized system.
t� Why sharing of user identities in a computerized system is not al-lowed—as it makes it impossible
Table I: Good Automated Manufacturing Practice (GAMP) criteria for data integrity—ALCOA+.
ALCOA Term Criteria Definition
A Attributable Who performed the action and when? If a record is changed, who did it and why? Link to the
source data.
L Legible Data must be recorded permanently in a durable medium and be readable.
C Contemporaneous The data should be recorded at the time the work is performed and date/time stamps should
follow in order.
O Original The information must be the original record or a certified true copy.
A Accurate No errors or editing performed without documented amendments.
+ Complete All data including any test, repeat, or reanalysis performed on the sample.
+ Consistent Consistent generation of records and application of date time stamps in the expected sequence.
+ Enduring Data should be recorded on controlled worksheets, in laboratory notebooks or in validated
electronic systems.
+ Available Data needs to be available and accessible for review, audit, or inspection over the lifetime of
the record.
www.rommelag.com
rommelag agP.O. Box · CH-5033 Buchs, SwitzerlandPhone: +41 62 834 55 55 · Fax: +41 62 8345500E-mail: [email protected]
rommelag Kunststoff-MaschinenVertriebsgesellschaft mbHP.O. Box 1611 · D-71306 Waiblingen, GermanyPhone: +49 7151 95811-0 · Fax: +49 7151 15526E-mail: [email protected]
rommelag USA, Inc.27905 Meadow Drive, Suite 9Evergreen CO 80439, USAPhone: +1.303. 674.8333 · Fax: +1.303.670.2666E-Mail: [email protected]
rommelag Trading (Shanghai) Co., Ltd. Room 1501 Xinyin BuildingNo. 888 Yishan Road · 200233 Shanghai, P.R.ChinaPhone: +86 21 6432 0166 · Fax: +86 21 6432 0266E-mail: [email protected]
Advanced aseptic packaging in one operation cycle
Reliable – Simple – Cost-Effective
bottelpack® Technology:- Integrated clean room US-class 100- Recognized by GMP, FDA, JP …- Aseptic packaging of liquids,
creams, ointments …- Endless container designs in PE, PP…
Your benefits:- Tamper-proof packaging- Easy to open- Simple to use- Shatter-proof, no splinter hazard
VISIT US ATINTERPHEX 2016New York – April 26-28Booth 3516

52 Pharmaceutical Technology MARCH 2016 PharmTech .com
to attribute the work carried out to a single individual.
t� Why actual samples should not be used as system suitability tests to see whether a batch passes or not. FDA warning letters reveal many citations for this transgression.
Data integrity criteriaThe criteria for data integrity are de-fined by the acronyms ALCOA and ALCOA+. ALCOA, which stands for attributable, legible, contemporane-ous, original, and accurate, was de-veloped by an FDA GMP inspector in the late 1980s. ALCOA has been used in many areas that are regulated both by FDA and other regulatory agencies worldwide. In 2010, EMA published a paper on electronic source data for clinical records, in which another four requirements relating to electronic data were added: complete, consistent, enduring, and available (11). The Good Automated Manufacturing Practice (GAMP) Data Integrity Special Inter-est Group (SIG) now refers to ALCOA+,
which includes the nine attributes or criteria for data integrity (see Table I).
Inspection trends In the past, an inspector would review piles of paper; to view a computer sys-tem, he would be shown print-outs of the screen. Now, the focus is on the computerized systems and the elec-tronic records within; the paper output is secondary. Inspectors will focus on the electronic records, looking at how they were generated and manipulated within the application. In the light of these changes, consideration needs to be given to the person who will manage the system during an inspection. How is the system configured to protect the electronic records? Are electronic sig-natures being used in the application, ensuring that the configuration of the application is documented and reflects the settings within the software?
Annex 11 now requires audit trail en-tries to be reviewed, and FDA considers these entries as part of the complete data. Many findings of non-compliance dur-
ing inspection have been discovered by looking at audit trail entries, therefore, an approach to reviewing audit trail events is needed. Citations have noted when audit trails were been turned off, the audit trail had not been reviewed, or user identities were shared, which is not allowed under the regulations. Ad-ditional citations can be found in the September 2014 issue of LCGC (12).
The ten compliance commandmentsThe 10 compliance commandments for computerized laboratory systems described in Table II should be consid-ered (12).
Where should the electronic data transfer begin?Let’s consider preparation of standards by way of example. In an analytical lab, standards are usually prepared using an analytical balance. The actual weight is typically printed on a strip printer and pasted into a lab journal. Afterwards, the standards are widely
Data Integrity
Table II: The 10 compliance commandments.
Commandment Comment
1 Management is responsible. Management must take the lead in making certain that the integrity of
data in the lab is managed and maintained.
2 Use a networked system, ideally with
a database.
Stand-alone systems should not be used in a regulated environment.
3 Document the system configuration
and manage all changes to it.
You must document and ensure the configuration protects the records.
4 Work electronically and use electronic
signatures.
Try to work electronically wherever possible. The advantage is that the
data are maintained within the system. Don’t use a hybrid because you
have two incompatible formats (worst possible situation).
5 Allocate each user a unique identity
and use adequate password strength.
Ensure that you have unique user identities, so that you can attribute the
work to a single individual.
6 Separate roles to avoid conflict of
interest.
Separate the roles within any computerized system. Typically “Admin” should only
be accessible by IT or a small group of people, not standard laboratory workers.
Exception if there are only 1–2 users (in which case it is necessary to share the
roles).
7 Define methods that can and cannot
be adjusted.
Consider which methods within a system can actually be changed and
which cannot.
8 Have a standard operating procedure
for data manipulation.
For chromatography data systems, a standard operating procedure
(SOP) for both automatic and manual integration is necessary.
9 Ensure staff are trained and
competent.
The need for an SOP is clear, but do people understand it and are they
competent to use it? Ensure the people are trained, both in data integrity
and the instrumental techniques they are using.
10 Carry out effective self-inspections or
internal audits.
Make certain that internal audits don’t just focus on paper. Instead, they
should go deeper and look at things within the computerized system.

Pharmaceutical Technology MARCH 2016 53
used for analytical methods, such as high-performance liquid chromatog-raphy (HPLC), gas chromatography (GC), titration, etc. Data management and documentation for analytical in-struments are usually managed by dedicated software or a laboratory in-formation management system (LIMS). This procedure is currently allowed by regulatory bodies, which state that print-outs representing original data for simple devices (such as balances) are acceptable (2) but is not the case for more complex devices. Nevertheless, it is important that reference standards are accurate and traceable because they represent the starting point of many analyses. Warning letters have cited
“no details available on the preparation of standards or solutions, especially of analytical reference standards” (13, 14). Independent of process, it is important to ensure that all the data are available.
This then begs the question: Where should the electronic transfer begin? In
the process described above, there is a gap in the data transfer between the “simple instrument” (the balance) and the “com-plex instrument” (e.g., the HPLC). Clearly, this is not recommended because it in-troduces an additional level of risk; it is obvious why no gaps should occur.
Capturing the data at the point of origin and transferring the data elec-tronically throughout the whole work-flow is a much lower risk approach and reduces the risk of errors during the early stages of a process, giving addi-tional confidence in the compliance of a workflow or laboratory.
What data should be transferred?The next question then becomes: What data need to be transferred? It is essen-tial to associate results with metadata to build context around the values collected. Although integrating even a simple in-strument can be tricky, the advantages of an integrated solution are obvious.
When electronic data transfer starts from the beginning of the process, each piece of information needs to be input only once for it to be available through-out the whole system. This allows seam-less movement of data and other infor-mation from the start of a process to the end, without the need for manual effort, such as manual transcription, creating an efficient work environment.
This transfer of data is achievable using a laboratory execution system (LES), such as LabX, which enables a variety of instruments to be directly connected (balances, titrators, density meters, refractometers, thermal analy-sis instruments, and pH meters). Use-ful features such as SOP-user guidance on the instrument terminal, automatic results capture in a database, and real-time data access support traceability of data and compliance with the GAMP data integrity criteria in ALCOA+.
biopuretech.com800-282-8823
BioPure fluid path components, connect with confidence
) ot numbers molded in for full traceability
) Emulate over-molded components -
full through bore diameter, no obstructions
) ower cGMP manufacturing costs
through simplified production operations
) �educe process validation
) �anufactured and packed in an
ISO Class 7 clean room
Specialists in the design and manufacture
of single-use bioprocessing components.
Contin. on page 76

54 Pharmaceutical Technology MARCH 2016 PharmTech .com
Process Control
OP
EN
ING
AR
T I
MA
GE
CO
UR
TE
SY
OF
SIE
ME
NS
In the past, process control data were used simply to control the process. With the explosion of data collection
and analysis capabilities, however, data can be harnessed to do much more. Pharmaceutical Technology spoke with Dr. Hartmut Klocker, vice-president of the Pharma Market Development Board at Siemens, about how data from process control systems can be used to optimize manufacturing processes.
Integrating dataPharmTech: What can be accomplished by more effectively using process con-trol data?
Klocker: In the past, process control data were collected in the distributed control system archives and dedicated plant information systems, and their only purposes were to control the process and visualize the status of the
process in real-time and historically. Today, however, pharma companies try to gain additional information by bringing together all different types of data from many sources including control systems, programmable logic controllers (PLCs), analyzers, building management systems, and laboratories.
The analysis of key performance indicators (KPIs) using all these data will open new insights into manufac-turing processes and provide the basis for process optimization while keep-ing the quality within tight specifica-tions. In addition, new technologies, such as continuous processes for tab-let manufacturing or the production of personalized medicine, would not work without integrating process and analyzer data and even parameters of the scheduling and planning tools. In the future, the industry will focus on
using process data to predict the end-product quality and thus the expected therapeutic effect using model predic-tive control.
Some data generated within PLCs, smart sensors, and complex analyz-ers are not collected and not used at all. Collecting these data, transferring them to a database, and applying tools to generate valuable information out of the pile of data are key challenges. A cloud solution may provide suffi-cient storage capacity and calculation power. The data analysis can then be performed by the customer or even outsourced to the suppliers of the automation systems or the machine manufacturers by giving them access to the relevant cloud data. An example, available today, is equipment monitor-ing, with the purpose of getting a bet-ter understanding of the condition of the machines and the overall produc-tion process. Data could be then used for energy management and condition-based maintenance.
PharmTech: How can data from dif-ferent sources be integrated and pre-sented to users?
Klocker: Data integration platforms are key to bringing together these data. The networking capabilities of devices are increasing steadily in line with the general industry trends of digitaliza-tion. Key technologies that allow this integration are the open standards Ole for Process Control, Unified Architec-ture (OPC UA) and OPC Analyzer-Device Integration (OPC UA ADI), which is for complex analyzers with large quantities of multivariate data.
Cloud technologies can provide a basis for such data platforms. Different experts can then be granted access to a subset of these data to provide data an-alytical services. For example, equip-ment experts can be shown data for predictive maintenance, and process engineers can be shown data to sup-port continuous process improvement. In addition, the common problem of overloading plant operators with con-trol data that are not directly relevant to monitor the process can be avoided. Data can be hidden on the operators’
Gaining Insight from Process Control Data Jennifer Markarian
Integrated data and cloud-based solutions can be used for process optimization.

Pharmaceutical Technology MARCH 2016 55
screens but transferred into the cloud, where other experts can look at various issues, such as machine health and per-formance or the relationship between climate and process quality parameters. The traditional operator screens will change, and instead of colorful graph-ics of process f low diagrams, process parameters and alarm messages with more focused views will prevail. The intent is to give a fast and clear picture of what is really going on, while hid-ing irrelevant details. Another tool is smart-phone apps that can be used to present the information needed to deal with specific tasks.
PharmTech: What are the security con-cerns with storing data in the cloud?
Klocker: Cyber-attacks are a chal-lenge that is mentioned in every in-dustry, especially when storing pro-cess data and accessing it broadly. To create the highest level of security, it is important to modernize and integrate the automation environment, with security built natively into the system from the field-level up. Siemens uses a ‘defense-in-depth’ concept that covers plant and network security as well as system integrity.
Using dataPharmTech: How can the collected data be used for determining KPIs, such as over-all equipment effectiveness (OEE)?
Klocker: KPIs, such as OEE and oth-ers, must use the whole range of data sources from automation systems, planning tools, building management systems, and laboratory management. There are two principal methods for achieving this. The first strategy is to collect all data in one large database and apply the KPI calculations to this database. This strategy is straightfor-ward as long as the intelligent device can be easily integrated into the plant information database using an open communication standard. In the alter-native strategy, the data are not copied into a database, but accessed from all the distributed databases on request. The databases accessed might be plant information systems and historical data from control systems, recipe-
control systems, enterprise resource planning data, and others. Intelligent caching of data to allow quick access using internet browsers is important. The advantage of this concept is to avoid copying of the original data, which might make the proof of data consistency (according to GMP regu-lations) cumbersome.
PharmTech: What are some best prac-tices for using process data to control the manufacturing process?
Klocker: Critical quality attributes (CQAs) determine the final product quality and are the basis for predict-ing therapeutic effect. Today, many CQAs can be measured in-line/on-line by applying process analytical technol-ogy (PAT). Data collected from experi-ments using the in-line/on-line mea-surement of the CQAs in the different unit operations of a production chain are the basis for process data model-ing. Correlation of the data provides a basis for understanding the process
and determining the critical process parameter (CPP) settings to obtain the right quality. Furthermore, the ap-plication of PAT, combined with model predictive control in the process con-trol system, allows us to adapt CPP set-tings dynamically and produce within specification all the time.
A good example of the need to combine data from several sources is personalized medicine, which is of increasing importance to the pharma-ceutical industry. The same principles in terms of manufacturing, quality control, and release of the drug apply to personalized medicine as to con-ventional drugs, but instead of large batches of active ingredients, a batch size becomes the drug product for a single patient. Without tight data in-tegration and a fully paperless pro-duction process, manufacturing costs would exceed any acceptable limit. To
Contin. on page 76

56 Pharmaceutical Technology MARCH 2016 PharmTech .com
In the past decade, there was a move-ment within the pharmaceutical in-dustry to benchmark drug manu-
facturers’ performance against that of manufacturers in other industries, to improve such areas as process robust-ness, speed and consistency, and to reduce waste (1). Proponents of lean manufacturing urged pharmaceutical companies to adopt best practices from other industries.
Thought leaders began to look more closely at manufacturing, as well as such standard industry benchmarks as changeover times and inventory turns, and asked whether pharmaceu-tical companies could improve per-formance and become more agile. To see how closely the industry has been examining its approach to inventory management and supply chain agil-ity, Pharmaceutical Technology asked Lora Cecere, founder of Supply Chain Insights, for her opinion on progress.
“Pharma is stuck in the mud,” notes Cecere, who writes the Supply Chain Shaman blog (2) and has more than 40 years of experience managing major corporate supply chains, consulting, and analyzing industrial performance.
As a 2012 Supply Chain Insights re-port (3) noted, pharma is immature, compared to other industries, in its supply-chain management practices. It still has higher operating margins, but faces more complexity, commoditiza-tion, and globalization than it did in the past. Mergers and acquisitions have also had an impact, Cecere says. Today, most pharmaceutical companies still generally take a conservative, func-tional approach to their supply chains, and focus more on the demand cycle than on channel data, says Cecere, who suggests that more industry executives follow the advice of supply-chain ex-pert Carol Ptak, a partner at the De-mand Driven Institute. In a blog post
on SupplyChainInsights.com (4), Ptak suggests that companies focus, first, on return on investment and cash flow, and secondarily on profit and loss, so that they can sense changes in demand and adapt planning and production dynamically.
Cecere and her collaborators have developed a supply chain index to measure how individual companies and industries are improving their supply chain agility. Considering such big issues as overall growth, profitabil-ity and complexity, the index tracks industries’ and companies’ improve-ment by averaging metrics over differ-ent time periods.
Each industry is analyzed individu-ally, based on its unique characteris-tics, but then companies are ranked, with those that have made the greatest gains recognized as “Supply Chains to Admire” (5) within those industries, based on improvements in inventory turns, return on capital investment, and operating margins.
Companies in industries with lower operating margins tend to manage their supply chains better than those with higher margins, so Cecere was surprised to find that generic pharma-ceutical manufacturers were not more agile than Big Pharma. In 2015, no ge-neric or over-the-counter drug compa-nies made the cut, while Biogen-Idec was the sole pharmaceutical company to make the list of 26 Supply Chains to Admire.
Biogen-Idec listed on the 2015 “Supply Chains to Admire” ListBiogen-Idec improved its overall sup-ply chain index ranking from 6 (when considering the period from 2006 to 2014), to 2 (when considering 2009 to 2014), the study found. The company increased return on capital from 13% to 21%, respectively, during these same time intervals, and inventory turns, re-spectively, from 9.99 to 14.55. Table I,
shows performance for the seven top-ranking pharmaceutical companies from 2011 through 2014, comparing them with metrics for a manufacturer from a different industry: Apple, Inc.
Supply Chain
DIE
TE
R S
PA
NN
KN
EB
EL/G
ET
TY
IM
AG
ES
Redefining Pharma Agility Agnes Shanley
Supply-chain success is measured by how effectively new medications reach patients, and how swiftly manufacturers can react to internal and external changes.

Pharmaceutical Technology MARCH 2016 57
If traditional supply chain manage-ment is not a top priority, how is the industry becoming more agile, and what metrics best reflect its progress? Pharmaceutical Technology asked Greg Anthos, senior managing consultant at Tunnell Consulting, for his thoughts. Anthos says that pharmaceutical manufacturers are focusing more of their attention upstream, to improve the research and development process, and, in particular, the way they handle clinical trials.
More pharmaceutical manufactur-ers are moving to adaptive trial de-signs, he says, and a growing number of them are working to be able to bring life-saving products to market under accelerated approval in Phase II. This is a major feat, considering the Phase III failure rate for new drugs (6).
At a time when some commodity ge-nerics continue to be in short supply, the emphasis is not on reducing inventories but on ensuring adequate supplies, An-thos says. Companies are also address-ing the challenges brought by mergers and acquisitions, which can temporarily distract staff, at all levels, from opera-tional and agility goals. McKinsey ana-lysts argue for the need for greater CEO involvement, standardized processes, and greater communication and feed-back, the hallmarks of any good lean or operational excellence program, when a merger or acquisition is underway (7).
The following are some opinions on agile manufacturing that Anthos shared in an interview with Pharma-ceutical Technology.
Fear of stockouts and tendency to play it safe with inventoryPharmTech: It was one of Tunnell’s ana-lysts who first drew industry’s atten-tion to pharma’s inventory turns, com-pared to those in other industries, as a measure of its agility. Why is it that the industry appears to have made so little progress in improving its perfor-mance in this area?
Anthos (Tunnell Consulting): Not a lot has changed if you analyze the indus-try based on the classic supply-chain measures. Some manufacturers are
probably carrying well over a year’s worth of inventory. Today, however, supply chain agility has to mean get-ting medicines to the patient more reli-ably and efficiently.
You’ll see some progress in inventory management, but it has not been the primary focus. Today, the industry’s first goal is ensuring supply to market so that there will not be any stock-
outs. A growing number of processes, especially in biotech, can be complex and fickle, so there is a tendency to play it safe with inventory. In the end, pharma companies will be measured based on ensuring supply, but also on revenue and profitability and cash flow, so inventory turns are largely issues for manufacturing departments, the CFO, and the guys in the warehouse.
%�� ��������� �� � ���������� �$�������!��� #
Thermo ScientificTM TruScanTM analyzers are fully compliant with the revised
standards in the European Pharmacopoeia Supplement 8.7, effective April
1, 2016. Unlike other handheld Raman analyzers that require either factory
recalibration or frequent field recalibration, TruScan is ready to meet your
EP 2.2.48 compliance challenges out of the box.
Worry-free compliance with TruScan
Are you ready for EP compliance?
&��
����
����
��
�������
���
� �$�
�������
������� �
������"
�����
��� �
����
���
��
���
� ����
�����
#���� ����
��
�������
���
� �$�
������
���
!����
�������
58 Pharmaceutical Technology MARCH 2016 PharmTech .com
PharmTech: So how is agility being defined in pharma today?
Anthos (Tunnell Consulting): Agility is much more about, in the short term, getting new products to market quickly. It’s about launching adaptive trials for breakthrough medications and having a sufficiently strong foundation to be able to launch a new product based on Phase II data.
In addition, agility means adapting immediately to changes in the market. These can be changes in demand, for example, when a competitor fails to get approval for a new drug and drops out of the market. But agility also means reacting to changes in internal pro-cesses or challenges within the com-pany that must be overcome.
In the end, agility is getting to mar-ket faster and better managing the supply chain, and overcoming, more dynamically, any issues, whether exter-nal or internal, that have disrupted the supply chain. It’s all about being more responsive to change.
The downsized challenge: doing more with lessPharmTech: How can companies be agile when they are dealing with more, and more complex processes, as well as with reductions in staff and resources?
Anthos (Tunnell Consulting): As I see it, the most progressive companies are focusing on three areas:
t� *ODSFBTJOH�SJTL�BTTFTTNFOUT �PO�
CPUI�UIF�TVQQMZ�DIBJO�BOE�UIF�FO-
UFSQSJTF��They are becoming more proactive about determining the risks that are out there, prioritiz-ing them, and ensuring that their resources are focused more effec-tively to address them.
t� 5IJOLJOH�UISPVHI�QPUFOUJBM�GV-
UVSF�DIBOHFT�BT�UIFZ�EFWFMPQ�BOZ�
OFX�QSPEVDU �i.e., how the process could be simplified to improve ro-bustness and process understand-ing. They are also taking steps to simplify their supply chains.
t� &OTVSJOH�UIF�QPUFOUJBM�PG�UIFJS�
QSPDFTTFT� UP� BEESFTT� GVUVSF�
DIBOHFT�� �Recently, I had a con-versation with someone at a phar-maceutical company who had been in discussions with FDA regarding process changes. He told me that a number of people within FDA have become frustrated by the fact that they are spending most of their time addressing process changes (e.g., changes that incor-porate the use of more up-to-date equipment, or even simple changes in the location of manufacture).
As a result, less of their time is being spent on their primary mission: ensur-ing that safe and efficacious medicines get to the public. For drug develop-ers and manufacturers today, the key question is: How do we develop new products so that, in the future, no or
only minimal regulatory changes will be required, except maybe filing for new drug approval in a new geographi-cal market. It’s a great stretch concept, but it takes a fair amount of work to get to a point where you can think about, and address, all the issues involved.
Certainly some of FDA’s and the industry’s early work with quality by design (QbD) had that goal in mind. The idea was to better understand your product performance ranges and the design space, file for approval in a wider range that would allow you to make adjustments in that range.
Today, complexity, both of the prod-ucts and of the regulatory landscape, has boxed us in. Significant numbers of supplements must now be filed for individual products. That’s a challenge that the industry still has to surmount if it is to develop more robust supply chains and more robust processes.
PharmTech: How are manufacturers doing this?
Anthos (Tunnell Consulting): It requires figuring out design choices. For exam-ple, a company might choose to launch a new process using a stable platform that is already fairly well understood and characterized, and that has al-ready been scaled up. It also means doing more of the classic QbD work, such as design of experiments (DoE) to really understand how the product behaves, where it works well, and what the process’ fault lines are. Simplifica-
Table I: Supply chain rankings (2011–2014), pharma and biopharma vs. Apple, Inc. (data from Supply Chain Insights).
Company and ranking for its industry (2006–2014 vs. 2009–2014) Growth (%)Operating
margin
Inventory
turns
Return on
capital (%)
Revenue per
employee ($/year)
Apple (16, 16) 0.32 0.31 136.6 25 2,077,891
Novartis (1, 1) 0.01 0.30 8.44 11 436,307
Biogen Idec (2, 6) 0.20 0.36 14.55 21 1,115,679
Merck (3,8) -0.02 0.16 7.22 11 556,423
Teva (4, 2) 0.06 0.14 4 7 444,822
Novo Nordisk (5,5) 0.10 0.37 8.16 60 412,580
Abbott Labs (6,3) 0.10 0.12 8.11 10 297,141
Eli Lilly (7,7) -0.04 0.21 8.40 18 585,537
Source: Supply Chains to Admire, 2015, excerpted with permission. Biogen-Idec was listed on the Supply Chains to Admire list for
2015, representing the pharmaceutical industry. Apple was not on the list and was ranked 16 on the consumer electronics list, but
is included for comparison. The full report may be accessed via Supply Chain Insight’s community site, www.beetfusion.com.
Supply Chain

Pharmaceutical Technology MARCH 2016 59
tion is key, so you lean out the process to reduce unnecessary complexity.
PharmTech: But how can companies do that effectively with biologics, and how can they do it when they are also rushing to get product to market faster?
Anthos (Tunnell Consulting): With biolog-ics, there is inherent complexity, so the key is designing in better controls or at least monitoring each process so that you better understand its behavior. It’s not easy. The industry is trying to move for-ward with greater process understanding while trying to get new products to mar-ket faster. These are competing forces.
PharmTech: How can all of this be done when international mergers are going on and day-to-day conditions are in such constant shift?
Anthos (Tunnell Consulting): Mergers distract staff. It’s not only a question of pondering who will get to keep his or her job, but such questions as which processes to follow, whether in manu-facturing or development. You want to create the best lean practices that are most effective, but you’re often in a rush to meet some cost reduction syn-ergy or regulatory target that a large consulting firm has told you to meet.
As a result, many companies reduce staff before they’ve figured out how to simplify and improve their processes.
Knowledge management is another major issue, because mergers and ac-quisitions drive a significant amount of employee turnover, both planned and unplanned. Knowledge often re-sides in individuals, rather than within the organization, and that jeopardizes business continuity. The way to avoid that is by understanding that you want to harmonize processes first, and en-gaging the folks who are already in the process of doing that to come up with the best work practices.
References 1. J. Macher and J. Nickerson, Pharmaceu-
tical Research Manufacturing Project,
Final Benchmarking Report, 2006, site
accessed February 20, 2016. http://apps.
olin.wustl.edu/faculty/nickerson/results/
PMRPFinalReportSept2006.pdf
2. L. Cecere, The Supply Shaman blog,
www.supplychainshaman.com/
3. A. Mayer, Supply Chain Metrics That
Matter: A Focus on the Pharmaceutical
Industry,” Dec. 3, 2012, Supply Chain
Insights, www.beetfusion.com (site reg-
istration required)
4. C. Ptak, What is Supply Chain Excel-
lence? Feb. 23, 2016, Supply Chain In-
sights, www.beetfusion.com/blogs/carol-
ptak/what-supply-chain-excellence
5. L. Cecere and R. Denman, Supply Chains
to Admire, 2015, Supply Chain Insights,
excerpted with permission, Sept. 8, 2015,
Accessible via www.beetfusion.com (site
registration and free membership re-
quired).
6. A. Shanley, Pharm Technol, 40 (13)(Feb-
ruary, 2016), pp 24-27.
7. A. Agrawal et al, Pharma M&A: Agile
Shouldn’t Mean Ad Hoc, McKinsey
and Co., site accessed February 19, 2016.
www.mckinsey.com/business-functions/
strategy-and-corporate-finance/our-in-
sights/pharma-m-and-a-agile-shouldnt-
mean-ad-hoc PT
� �����(+��- &���-&2�3�6��0$'�$)"������6��0$/ �����/'�)/�������������6��� �������6�$)!*�" (0��*(
www.gemu.com
1436 Intelligent Positioner and Process Controller
�# ���MÜ ������*. $.��)� ' �/-*)$��+*.$/$*) - 2$/#��)�*+/$*)�'�+-*� ..� �*)/-*'' -�/#�/��*)/-*'.� +- ..0- ��5*2�ra/ � *-�/ (+ -�/0- �$) �*)%0)�/$*)�2$/#���+) 0(�/$��''3���/0�/ � +-*� ..��*)/-*'�va'1 �
6� �. �2$/#�.$)"' �)���*0�'e ��/$)" '$) �r �)��,0�-/ -�/0-) va'1 .
6� �$- �/�1�'1e (*0)/$)" *- - (*/ �(*0)/$)"
6� �0'/$+' fie'���0.�$)/ -!�� .� $)�'0�$)"�
Pr*4�0.��)��De1$�eNe/
6� Oper�/*-�$)/ -!�� �0.$)"����0$'/�$) we��+�"
6� Pr*"-�((��' ��'�-(�- '�y *0/+0/.
6� �$(+' ��*(($..$*)$)" �)��*+ -�/$*)�1$a keyp�� �0//*). *-�/#-*0"h �*(+0/ -��*((0)$��/$*)�
6� � �0-$/3�' 1 '.�/*��*)/-*'�0.er ��� .. -$"#/.
3800 Camp Creek Parkway • Building 2600 • Suite 120
Atlanta, GA 30331 • 678-553-3400 • [email protected]
www.gemu.com
1436 Intelligent Positioner and Process Controller
The GEMÜ 1436 cPos is an electronic positioner with an optional process controller that controls pressure, flow rate or temperature in conjunction with a
pneumatically actuated process control valve.
• Use with single and double acting linear and quarter turn valves
• Direct valve mounting or remote mounting
• Multiple field bus interfaces, including
Profibus and DeviceNet
• Operator interface using a built-in web page
• Programmable alarm relay outputs
• Simple commissioning and operation via keypad buttons or through computer communication
• Security levels to control user access rights

60 Pharmaceutical Technology MARCH 2016 PharmTech .com
Facility Design: Right-Sizing
DA
NIE
L G
RIL
L/G
ET
TY
IM
AG
ES
Right-sizing implies operational ef-fectiveness and efficiency. Since the 1980s, the phrase has been
used in management circles as a euphe-mism for layoffs, but in facility design, right-sizing can be a euphemism for minimal cost. Flexibility, on the other hand, is about having the capacity to adapt to future changes quickly and easily. Flexibility, however, typically re-quires an investment in infrastructure. At their most extreme, these two ideas are at opposite ends of a facility-design continuum.
Anticipating the futureThe idea of “right-sizing,” when applied to design, is to create an optimal facility. An example is found in the best practice recommendation of designing for actual equipment loads instead of the traditional method of estimated loads (1). Without the data of real loads, the practice of esti-mating results in oversized systems that increase energy and operational cost. But at what point is the glove’s fit tailored too tight? Experience has established that—in the pharmaceutical industry—change is inevitable, making flexibility a necessary facility consideration. When building a facility, it is wise to build in some accom-modation for future modifications. An-ticipating the future can be daunting, but experience proves that it is prudent to try.
To effectively tailor a design for current needs, while at the same time planning for future potential, the different types of change that may occur must be consid-ered. The most obvious potential change is increasing the manufacturing capac-ity to meet demand for the current drug portfolio. This change will require more and/or larger equipment of the same type as already exists. Another consideration is how new products will be introduced to the facility. Over the long haul, the ability of a facility to adapt to changing market and business conditions ensures its value far into the future. Inevitably, this ability will require more than just adding similar equipment, and new and different tech-nologies will need to be accommodated.
Making spaceIn all cases, at a minimum, drug man-ufacturing facility owners need to allow for additional space. Planning for more space can be as simple as lay-ing out a facility so that the manufac-turing area has a direction in which to expand, such as into a warehouse or to the exterior into a future building addition. Key to effective growth, how-ever, is having a strategy for internal circulation, with a sound way for ma-terials and personnel to access the fu-ture area. Because continuity is harder to achieve if it has not been planned, a good place to start is ensuring that the current manufacturing circulation (i.e., the flow of materials and person-nel) can be extended directly into the future area. While the exact use of the future space may be unknown, the need to get personnel and materials in and out in a way that is operationally effective and compliant is certain.
Over time, the drive for greater manufacturing efficiencies and capa-bilities leads to new equipment. All equipment has a limited useful lifes-pan. In addition, drug reformulation can demand additional or modified equipment, and new products and their processes may be added to an existing portfolio. A facility design should allow for the removal of exist-ing equipment and the installation of new models. Dimensional clearances
Flexibility vs. Right-Sizing:
Determining the Right Facility Size
Eric Bohn
Eric Bohn is partner at Jacobs
Wyper Architects, 1232 Chancellor St.,
Philadelphia, PA 19107, tel: 215.985.0400,
www.jacobswyper.com.
Choosing the right facility size requires tailoring the design for current needs as well as anticipating the future.

Pharmaceutical Technology MARCH 2016 61
and movement pathways need to be integrated into the design. Without such a mindful approach, equipment may become inaccessible, and changes may require major interruption of on-going operations.
Planning for utilities and servicesPlanning for additional space and new equipment is relatively easy to imple-ment. A greater challenge, with greater cost burden, is planning for future utili-ties and services. Again, smart strate-gies need to be applied. Near-term flex-ibility can be provided by a thoughtful approach to system distribution. For example, service mains can be installed and sized so that local distribution can be easily added in the future. If judi-ciously done, this oversizing of a system can, for little upfront cost, provide for relatively easy and efficient growth in the future. In some situations, however, extending the current system may not be the right approach. If not, a strat-egy for future utilities still needs to be developed. One strategy is to plan a pathway and space for new mains and distribution to be installed later. Another strategy is to make a decision to develop a separate system in the fu-ture. In any case, it is better to build in a strategy at the beginning than to be without options later.
The greatest cost potential is in the gen-eration of overall utility capacity, which is typically a longer-term consideration related to an expanded manufacturing footprint. Using modular equipment that can provide phased growth is a strategy that has found much success. Admittedly, this will not work for all systems, but it can work for many. Such an approach enables spending only what is required and delaying future capital expenditures until they are actually needed. Another option is purchasing extra capacity up-front by way of oversized equipment, thus anticipating the future need for increased capacity. You can ignore this possibility by rationalizing, “if we are that success-ful, we’ll deal with it then,” or you can develop a strategy that affords flexibility and a relatively quick and easy response when needed.
ConclusionRight-sizing is more than responding to the immediate, foreseeable requirements of a facility. To optimize a design, there needs to be a prudent investigation of the potential for growth. With right-sizing, as the phrase implies, one size does not fit all. An approach needs to be crafted appropriate for the situation and the po-tentials that exist for a facility. Right-siz-ing is about finding value, the sweet spot
where design, management, and opera-tional considerations are balanced and deliver a quality facility for an appropri-ate cost. Finding the “right size,” involves the creation of an efficient design with an appropriate amount of flexibility.
Reference 1. US Environmental Protection Agency, Lab-
oratories for the 21st Century Best Practice Guide: Right-sizing Laboratory Equipment Loads (Washington, DC, August 2005). PT
SANITARY/USDA-AC ������9�������TIC CONVEYING C��������S & SYS�����9���TCH & CONTINUOUS OPERATION
��������������������������SYS�����9���T �������������� ������SYS�����9���-C���������� �������PACKAGES
FDA-APPROVED/ ���������������������� ONS��� �����9��� ����������� ������� �
�������VA ���� ONVEYING SYS�����9� ����������
�&3�42�2/,5&�8/41�0.&4-"3*$�$/.5&8*.(�
$)",,&.(&2���*2*3�5"$�4�-"7�$/-�0.&4-"3*$
/1 $",, 800-VAC-U-�� �
More than 60 years of custom application
experience in the pharmaceutical industry.
Over 10,000 different powder and bulk materials
handled—including Free-Flowing and Non-Free
Flowing Powders. Technical expertise including
FAT and SAT validation. And an airtight perfor-
mance guarantee. It’s what makes VAC-U-MAX
as unique and trusted as the pharmaceutical
process automation solutions we design.
�1/-�2".*3"18�$/-0/.&.32�3/�'4,,8�"43/-"3&d
#"3$)�/1�$/.3*.4/42�2823&-2��6&�$1&"3&
0)"1-"$&43*$",�01/$&22*.(�2/,43*/.2�
&.3*1&,8�"1/4.%�8/41�.&&%2�".%�(/",2��
VAC����� ��&,� "0�� "024,&��".%� /"3&%
T"#,&3� /.5&8/1�'/1��&.3,&��&,*5&18�/'��1/%4$3
3/� "024,&��*,,*.(�/1��"$+"(*.(��"$)*.&2

62 Pharmaceutical Technology MARCH 2016 PharmTech .com
TROUBLESHOOTING Equipment and Processing
DA
NL
EA
P/G
ET
TY
IM
AG
ES
PharmTech.com/Troubleshooting
The benefits of adopting single-use technologies in the production of biopharmaceuticals, such as lower
capital investment and increased flex-ibility, are now well documented and widely recognized in the industry. But when building a new facility based on single-use technologies, or incorporat-ing single-use into an existing facility,how do companies ensure they fully realize the benefits?
Facility design is a complex, multi-faceted, multi-step process, and early de-cisions can cause unforeseen limitations as the project progresses or, later, when further development of the facility is re-quired. Asking the right questions at the outset and having the depth of experi-ence and knowledge to understand theconsequences of the answers are vital to establishing the right specifications dur-ing the design phase.
Identifying a partner or partners tosupport the design and build of a facility and the process that sits within it is the first key decision. Traditionally, an archi-tectural and engineering firm and one, or possibly multiple, single-use process-equipment supply partners are selected. Working with a single external point of contact can help drive efficiencies in project-management and delivery. To be successful, however, the lead partner will need an understanding of biomanufac-turing facility design, engineering, qual-ification, and validation, as well as the
operational aspects of combining process hardware, single-use consumables, and automation platforms.
Overall, there are four sets of re-quirements to consider: product(s) to be made, process technologies, facility design, and supporting services. In eachcase, a series of questions will help iden-tify objectives, design specifications, and potential constraints.
Considering the productProduct class. The first element that de-fines any biopharmaceutical manufac-turing facility is the product itself. Will the facility be manufacturing monoclo-nal antibodies, recombinant proteins,vaccines, antibody-drug conjugates, or fragment antibodies? Also, will theproducts be mammalian cell-derived or microbial cell-derived? While thesequestions are most pertinent for the se-lection of the bioprocessing technolo-gies required, they are also importantfor the design of the facility itself.
The promise of f lexibility and sim-plification are often major deciding fac-tors for choosing single-use technology. Removing the need for cleaning and sanitization, for example, means that switching between one product and an-other becomes quicker and easier. One way to take advantage of this flexibility is by making the facility multi-purpose(i.e., the manufacture of two or more products) to drive greater facility utiliza-tion. Deciding between a single- or multi-product facility impacts facility design considerations. Factors such as avoiding cross-contamination between products and ensuring that process-specific equip-
ment can be moved around efficiently or housed nearby for rapid changeover need to be built into the design.
Regulations. With the plethora of regulatory guidelines and associated compliance requirements to adhere to when building a facility, it must be clear whether the product is for re-search and development purposes (pre-clinical), clinical trials, or commercial scale, as this will define the relevantGMP requirements. Also, if producing at commercial scale, which regulatory standard is needed? Is the product ap-proved by FDA, the European Medi-cines Agency (EMA), the China FDA, Brazil’s National Health Surveillance Agency (ANVISA), or other agencies? In some cases, local requirements go be-yond global ones. For example, Chinese fire regulations demand a greater levelof fire resistance than is typical glob-ally, and in countries such as Korea and Japan earthquake-proofing measures may have to be implemented.
Capacity. To define the necessary ca-pacity of the facility, the primary ques-tion is how many batches per productper year are needed? However, thisnumber has not always been defined when the facility design stage is reached. Alternatively, it should be possible to consider what quantity (in kilograms) of the bulk API needs to be produced for each product within the facility per year to meet clinical trial or commercial market requirements, and then work back to the number of batches.
For example, one can consider 2 x 2000-L bioreactors running a typical 14-day incubation period staggered a
Peter Genest is global operations manager,
FlexFactory, tel: 1.860.670.3014, pete.genest@
ge.com, and John Joseph is engineering
project leader, both at GE Healthcare’s Life
Sciences business.
Asking the right questions is crucial.
Peter Genest and John Joseph
Designing a Biomanufacturing Facility Incorporating Single-Use Technologies

April 26-28, 2016Javits Center | New York City
COST EFFECTIVE SOLUTIONS TO DEVELOP AND
MANUFACTURE PRODUCT
STATE-OF-THE-ART
TECHNOLOGIES LAUNCHES, from Industry Leaders
“INNOVATION IN
MANUFACTURING SCIENCES”
WIth PDA
CUTTING-EDGE Innovation & Technologies
REGISTER TODAY AT WWW.INTERPHEX.COM/ADVANSTAR
INTERPHEX was a great experience this year! Sat in on multiple presentations
and was very impressed with content, material presented, and the turnout.
I also thought the layout of the show this year was great. Finally, the mobile
app was awesome! Very helpful in locating companies, saving vendors I
wished to visit, and much more. - Lisa Birmingham
PREMIER SPONSOR:

64 Pharmaceutical Technology MARCH 2016 PharmTech .com
Troubleshooting
week apart, which equates to one batch produced each week. A typical batch at 2 g/L with a 70% overall yield in down-stream processing and a 95% production success rate will therefore yield 138 kg/yr in total. The final yield here is deter-mined by the product titer of the pro-duction bioreactor, combined with the efficiency of the downstream purifica-tion steps, both of which will be driven by the details of the bioprocess itself.
Selecting process technologiesThe next step is to drill down into the discrete unit operations of the biomanu-facturing workflow. If the production pro-cess is already defined, it should be listed out, but if not, then the contracted partner may be able to provide an equipment list with flexible process capability. Figure 1 shows an example of a production process from cell culture to bulk drug substance.
Starting with upstream, the status of the cell line and whether the process should be batch, fed-batch, or perfusion needs to be decided. Details about the nature of the process also need to be captured, including the bio-safety level and lengths of culture time for the seed and production bioreactors.
Moving to downstream, the overall yield of the purification process from
post-cell culture harvest through to purified bulk API should be provided, along with an estimate of the step yield of each unit operation. If chromatog-raphy columns are used in the process f low, also specify the column volume and diameter required along with the desired number of cycles for each step.
Many single-use consumable supply partners now offer large customized sys-tem designs that can be tailored exactly to a specific workflow. Having an all-en-compassing single-use system for a unit operation may seem to be the most effi-cient option. However, manufacturing a large single-use system comes with chal-lenges. Packaging size and transportation integrity, sterility validation, component supply, handling and staging, installa-tion, and operational use can all become more difficult and lead to greater risk lev-els. In some cases, defining and selecting smaller and simpler single-use systems to function in a modular workflow can be beneficial for minimizing risks.
Another important consideration in selecting single-use consumables is ensur-ing the supply chain is robust. Switching out any element of a validated process re-quires significant additional work. There-fore, make sure the supply partner has a proven track record, a materials policy
in place, transparency on how they work with raw material suppliers, and a proac-tive communication program, and that they can provide examples of how they have dealt with previous situations of raw material changes. Also check the robust-ness of the qualification and validation package supplied, and make sure it meets all relevant regulatory requirements.
Breaking new ground or renovating?The crucial point in designing a new facil-ity is whether it will be a brownfield/reno-vation or a greenfield site. If it is brown-field, then designers and engineers will need to know if the footprint is fixed and whether there are any restrictions on the space, such as floor strength, ceiling height, or door and elevator sizes. When thinking about the layout, are there existing person-nel, product, or material flows already in place? Also, is there existing support infra-structure, such as utilities, warehousing, or laboratory space, that can be accessed? If possible, plans for future plant expansion at the site, or at other sites, should be taken into account, particularly if they will have an impact on the product requirements of the facility being built now.
If it is a greenfield site, then there is increased flexibility in what can be built. However, sourcing an engineering firm
Project management
Centralized monitoring and control
Vial
Cell culture seed train
Purification Bulk formulation
Production bioreactor Harvest and viral inactivation
HyClone media
and supplements
WAVE
Bioreactor
Xcellerex XDR
bioreactor
UniFlux cross
flow filtration
Normal flow
filtration
Viral
inactivation
ÄKTA chromatography
system and
AxiChrom columns
ÄKTA chromatography
system and
AxiChrom columns
Nano-
filtration
Ultrafiltration/
diafiltration
conditioning
Bulk drug
substance
Sterile
filtration
Single-use tubing sets/assemblies
Figure 1: An example of a production process from cell culture to bulk drug substance.
FIG
UR
E I
S C
OU
RT
ES
Y O
F G
E H
EA
LT
HC
AR
E


66 Pharmaceutical Technology MARCH 2016 PharmTech .com
Troubleshooting
with the relevant experience for a stick-built biopharmaceutical facility design can be challenging in some parts of the world. In response to this, another op-tion that has emerged is the modular facility, made from standardized pre-built units delivered to the greenfield site. This approach can have benefits in ensuring consistent standards of quality and reduction of time to first batch. This modular approach to building allows site excavation to run in parallel with module construction and validation of unit operations to begin offsite.
For those on a brownfield site or those building a new facility adjacent to an existing one, any current cen-tralized automation platform for data archiving and process monitoring may need to be linked to the new facility. In other cases, a standalone automation platform will be appropriate.
Finally, the need for any additional support functions or buildings should be decided (e.g., fill and finish building, a black utility generation building, a ware-house, quality control [QC] laboratories, or a waste treatment plant).
The needs here can sometimes run counter to expectations. For example, when embarking on a first foray into single-use, many presume that the re-moval of the hard piping and utilities needed for clean in place of stainless steel will result in a reduced footprint require-ment. What is not always anticipated is the warehousing requirements for the stock of single-use consumables, which also need to be unpacked and prepared in a staging area. While having adjacent warehousing on a site may fulfill this need, more efficient tracking, set-up, and speed of changeover will be achieved if some consumables staging and storage sits within the facility itself, in close prox-imity to, or as part of the cleanroom en-vironment. In total, the footprint is likely to be reduced in switching from stainless steel to single-use, but the change is not always as significant as expected.
When adding a single-use train to complement existing stainless-steel production facilities, the flexibility of single-use can help reduce the need for additional utilities. In one case, when
designers and engineers looked at which existing underutilized utilities could be shared with a new single-use set-up, it turned out to be a significant amount. For example, the flexibility of single-use meant that single-use unit operations requiring a water supply could be sched-uled for the downtime or periods of low water consumption of the stainless-steel process. The reduced consumption of utilities required to operate the single-use process allowed for easier integration of additional capacity into the existing in-frastructure of a production site.
Safety and time considerationsThe ability of operators to safely work with biologic and potentially hazardous materials at any stage during the pro-cess is a key facility design consideration. Knowing where to place biosafety cabi-nets, if aseptic connections are required, and knowing any special design modi-fications to the single-use system (e.g., extra clamps, material selection, han-dling of highly toxic excipients) is vital.
Next, if known, specify the buffer and media requirements of each unit opera-tion step in the production process, in-cluding whether any solutions require special handling (e.g., 70% ethanol), if steps are time-constrained (e.g., a highly-labile product that must be processed in a specified period), or if temperatures other than room temperature are required (e.g., temperature-sensitive media for upstream or cold purification processing).
The buffer preparation schedule for downstream purification can have a sig-nificant impact on facility design. Whether it is just-in-time preparation, one day in advance of use, or before any purification is started, will influence how much space is required for buffer storage or whether a system of built-in piping is required.
Planning for the futureFacility design is a multifaceted, inter-locking web of needs, wants, and risks, and it must be properly managed from the outset to accommodate and account for all requests. Management includes being able to step back and take a holis-tic view. The prime driver and desired outcome, whether it is shortest time to
market, lowest overall cost, or capital preservation, will significantly direct the decisions made at all stages of the design and building process.
For example, for a small biotech that was particularly concerned about reduc-ing capital expenditure, the ultimate rec-ommendation was to buy-in ready-made buffer and media in single-use liquid delivery bags. The overall scale and out-put of the facility was relatively low, and therefore the additional infrastructure re-quired for in-house preparation was not going to drive significant savings in the longer term. This change in processing methodology minimized both footprint and utility needs.
Another element to consider at this point is how much “future flexibility” to account for during the design and build phase. Do you want to allow for the possi-bility of adding more production bioreac-tors to expand manufacturing capacity? Do you want to add 10% more communi-cation drops for the integration of future equipment? The balance to be struck is between too much and not enough.
One reason such flexibility is impor-tant is that future manufacturing needs are always uncertain. Factors such as in-creased productivity and titer, coupled with increased market competition due to products coming off patent, has led to some stainless-steel facilities becom-ing underutilized and ending up shut down or sold.
The facility itself is only the beginning. Operational training will be required, as a minimum, but many supply partners can offer a much wider range of services. Vali-dation requires significant experience and know-how and has the potential to con-sume significant internal resources. Out-sourcing this element to an experienced partner can be a cost-effective option.
Ultimately, of course, budget is a cru-cial factor, along with when production needs to commence. But these should be considered alongside a close appraisal of the experience and depth of knowledge of the team that will be delivering the project. By mapping skills against re-quirements, it is possible to identify key attributes external partners need to have to make a project a success, first time. PT

Sponsored by
Presented by
Engineering the Mechanical
Properties of Amorphous
Spray-Dried Dispersions
EVENT OVERVIEW:
Drug candidates with low oral absorption potential in the crystalline state are
frequently converted to the amorphous form to increase solubility, prevent
precipitation, and increase dissolution rate, thereby improving the extent of
absorption and bioavailability. Spray drying drug from a solution containing an
amphiphilic polymer is one of the most common and scalable methods used to
achieve these enabling properties. The resulting powder contains the amorphous
drug molecularly suspended or solubilized within the polymer matrix. Typical
crystalline drug properties, often unfavorable for downstream processing, are
masked by the amorphous state of the drug and matrix polymer. In addition,
unlike many crystalline drug substances, spray-dried dispersion (SDD) particles
are tunable, even within the spray drying process. Given this tunability, SDD
composition and physical properties can be co-optimized for absorption potential,
physical and chemical-state stability, mechanical and powder flow properties, and
spray drying throughput. This is a departure from traditional development, where
drug substance and drug product are progressed somewhat independently, with
drug substance form and morphology often changing during the development
and scaling processes. This often results in constraining the oral dosage formulation
and process to overcome undesirable crystalline drug properties.
In this webinar, experts will discuss:
■ How particle engineering by spray drying can be used to co-optimize several
facets of SDD development with a specific focus on optimization of SDD
mechanical properties
■ Reduction of SDD tableting scale up risks and pill burden through identification
of the primary mechanismof compaction and rational formulation design
■ Development case studies highlighting SDDs whose primary mechanism of
compaction is plastic flow versus brittle fracture.
Key Learning Objectives:
■ The mechanical properties of SDDs can be tuned by changing the degree of
atomization and drying rate in the spray dryer.
■ A review of typical SDD mechanical properties, including stress stain
behaviors, and how these can be exploited to optimize tableting.
■ Engineering SDD particles provides opportunities to reduce pill burden
by maximizing SDD loading in the dosage form through rational excipient
selection and SDD physical property design.
ON-DEMAND WEBCASTOriginally aired Feb. 23, 2016
For questions, contact Daniel Graves at [email protected]
Presenter
Aaron Goodwin, Ph.D.
Principal Investigator,
Research & Development
Bend Research
Bend Research is a division of
Capsugel Dosage Form Solutions
Moderator
Rita Peters
Editorial Director
Pharmaceutical
Technology
Register for free at www.pharmtech.com/pt/spray_drying
Who Should Attend:
■ Large pharma, mid-size pharma, biotech
companies
■ Product development group leaders and
formulators, technical, and engineering
positions involved in scale-up and
manufacturing of oral solid dosage forms

68 Pharmaceutical Technology MARCH 2016 PharmTech .com
OUTSOURCING OUTLOOK
SV
ETA
DE
MID
OF
F/G
ET
TY
IM
AG
ES
SV
ETA
DE
MID
OF
F/G
ET
TY
IM
AG
ES
PharmTech.com/ptoutsource
Pharmaceutical and biopharmaceuti-cal companies will continue to use outsourcing services, especially drug
manufacturing, due to cost concerns and mergers and acquisitions, according to a market survey of drug manufacturers by ISR Reports. The survey showed that the top five activities that drug companies outsource are drug product manufactur-ing, packaging and labeling, distribu-tion, small-molecule manufacturing, and holding and storage, according to Kate Hammeke of ISR Reports (1).
The ISR survey suggests that the majority of companies are outsourcing their small-molecule manufacturing. ISR’s 2016 small-molecule outsourcing survey also showed that other popular outsourced activities include packaging and labeling, distribution, and holding and storage. Respondents to ISR’s survey indicated resources, an increased product demand, cost, and mergers and acquisi-tions as some of the reasons small-mole-cule companies outsource these activities.
For large-molecule companies, distri-bution was the number one outsourced activity indicated by ISR’s survey, with packaging and labeling coming second. Cost was the number one reason large-molecule companies indicated as the reason for outsourcing.
Service providers are expanding their services and capabilities to keep up with the high demand. The following are some examples of the growth the outsourcing industry is experiencing.
Company developments, expansions, and acquisitionsThe first few months of 2016 has seen an array of investments, expansions, and acquisitions in the pharmaceutical
outsourcing market. Outsourcing com-panies appear to be looking to the early-phase development and clinical trial mar-kets to increase their portfolios.
MPI Research, an early-stage drug de-velopment contract research organization (CRO) based in Michigan, is investing more than $5 million in a facility renova-tion to begin in 2016. The company plans to renovate more than 55,000 square feet of facility space.
Ed Amat, executive vice-president of global sales and marketing, said in a press release that the expansion reflects the in-creasing demand for investigational and preclinical pharmaceutical and medical device studies (2). In the upcoming year, MPI hopes to increase the company’s workforce by approximately 10%.
LabConnect, a Seattle-based provider of laboratory services to biopharmaceu-tical, medical device, and contract re-search firms, has built a new 5000-sq-ft biorepository in Johnson City, TN, that includes space for ambient, refrigerated, cold (-20 °C), and ultra-low temperature (-70 to -80 °C) storage as well as liquid nitrogen vapor phase storage (-190 °C) (3).
The facility includes storage capac-ity for more than eight million samples, validated and mapped backup freezers and generators, redundant HVAC sys-tems, building and biorepository security systems, and a temperature monitoring system for freezers and refrigerators with a 21 Code of Federal Regulations Part 11 compliant audit trail. LabConnect also tracks sample locations and consolidates data within a centralized database.
Catalent Pharma Solutions announced on Feb. 2, 2016 (4) an investment of $4.6 million to expand its Singapore clinical supply facility by building GMP space
for secondary packaging. The invest-ment doubles the ambient storage space and quadruples cold-storage capacity, the company reports. The site provides clini-cal supply services including project and supply-chain management, comparator sourcing, clinical label printing, second-ary packaging, clinical storage, import/export management, importer of record service, and returns and destruction management services. It has served as a regional hub for studies in Australia, Singapore, Korea, Hong Kong, and other countries in Southeast Asia.
Onyx Scientific, a UK-based CRO and small-scale API manufacturer, has announced an investment in an addi-tional site located adjacent to its existing facility in North-East England to increase its laboratory facilities, GMP suites, and storage of GMP materials (5). In addition, the company has recruited several more chemists to support clients’ pre-clinical, development, and early-stage API manu-facturing projects. In 2015, the company grew its GMP space following an increase in demand for its small-scale API manu-facturing services.
Vetter announced on Jan. 28, 2016 that the company’s Schuetzenstrasse multi-functional building in Ravens-burg, Germany, has been completed on schedule and departments critical to its operation have started to move in (6). The $32 million (€29 million) investment is part of a $331 million (€300 million) total investment strategy announced by the company in September 2015 for further development to its manufacturing sites.
The continued demand by large and small customers for enhanced drug de-velopment services, as well as the need for ever-more future-oriented sophisticated
Pharma Outsourcing Market Expands
The pharma outsourcing market starts 2016
with expansions, acquisitions, and new offerings.
Susan Haigney


70 Pharmaceutical Technology MARCH 2016 PharmTech .com
Outsourcing Outlook
IT systems to protect their data, created the need for the new facility, the company reported in a press statement.
The 91,500-sq-ft, six-story building contains non-cGMP laboratories for development support, laboratory space for mi-crobiological analysis, office workplaces for Vetter Development Service and IT, and a data processing center with enhanced security systems, including a safety cell that protects technol-ogy and data from external physical hazards in the event of an emergency.
Novasep is building a new synthesis laboratory and adding kilogram-scale production at its existing US facility in Booth-wyn, PA (7). This extension will allow Novasep to offer both chemistry and purification services and to produce the initial kilogram-scale batches of synthetic molecules that are needed for biological testing and preclinical trials.
The investment is in response to increasing demand from US customers for closer proximity to Novasep’s con-tract manufacturing services for early-stage development and production scale-up. Novasep has provided purifica-tion development services to North American customers for more than 15 years at this location. The new laboratory, equipped with reactors up to 50 L in size, will start operation in May 2016. It will offer cryogenic capacities and standard chemistry, as well as preparative purification chromatog-raphy processes.
PBOA expands membershipThe Pharma & Biopharma Outsourcing Association (PBOA), founded in 2014, has been advocating for the pharma outsourc-ing industry as the global market changes and expands. “We’re focused on working on the reauthorization of the Generic Drug User Fee Amendment (GDUFA), while keeping an eye on FDA’s Quality Metrics initiative, and helping make sure that CMO/CDMOs [contract manufacturing organizations/contract de-velopment and manufacturing organizations] are prepared for track-and-track/serialization regulations as they roll out in the US and the EU in the next few years,” says PBOA President Gil Roth.
In February 2016, PBOA expanded its membership (8). IDT Bi-ologika and Ei SolutionWorks joined the PBOA as general mem-bers; 3M Drug Delivery Systems (DDS) joined as a sustaining member. Diego Romeu, manufacturing and supply chain director at 3M DDS, was also voted to a three-year term on the board of trustees, along with Rajan Puri, director of business development at Therapure, and Lee Karras, CEO of Halo Pharmaceutical.
“As we continue our mission to represent the CMO/CDMO industry before FDA, Congress and other stakeholders, it’s criti-cal that we increase our membership and provide a true voice for our industry,” said Roth. “We’ve been successful in bringing the CMO/CDMO perspective to issues such as GDUFA, quality metrics, and serialization, and we’re delighted to bring in new member companies and add fresh points of view to our Board of Trustees.”
References 1. A. Shanley, “Surveys Examine Outsourcing Trend,” Pharmaceutical
Technology, Supplement: Partnerships in Outsourcing, 40 (13), 32-33, www.pharmtech.com/surveys-examine-outsourcing-trend
2. PharmTech Editors, “MPI Research Invests in Upgrades and Depart-ment Expansions,” PharmTech.com, Feb. 11, 2016, www.pharmtech.com/mpi-reasearch-invests-upgrades-and-department-expansions
3. LabConnect, “LabConnect Builds New Biorepository, Expands Ser-vices, Offers Absolute Sample Protection,” Press Release, Feb. 3, 2016, www.labconnectllc.com/Documents/New%20Biorepository%20PR%2022016%20-%20New%20Biorepository%20Expands%20Ser-vices%20Offers%20Absolute%20Sample%20Protection.pdf, accessed Feb. 16, 2016.
4. Catalent, “Catalent Invests $4.6M to Further Expand Asia-Pacific Clinical Trials Hub in Singapore,” Press Release, Feb. 2, 2016, www.catalent.com/index.php/news-events/news/Catalent-Invests-4.6M-To-Further-Expand-Asia-Pacific-Clinical-Trials-Hub-In-Singapore, accessed Feb. 16, 2016.
5. Onyx Scientific, “Facility Expansion at CRO Following Record Year,”Press Release, Jan. 29, 2016, www.onyxipca.com/facility-ex-pansion-cro-record-year/, accessed Feb. 16, 2016.
6. Vetter, “Vetter Announces Completion of Multi-Functional Building for Development Service and State-of-the-Art IT,” Press Release, Jan. 28, 2016, www.vetter-pharma.com/en/newsroom/vetter-news/news-l-vetter-announces-completion-of-multi-functional-building-for-development-service-and-state-of-the-art-it, accessed Feb. 16, 2016.
7. Novasep, “Novasep Adds Synthesis and Kilo Lab Extensions at US Facility,” Press Release, www.novasep.com/home/about-novasep/media-events/press-release/novasep-adds-synthesis-and-kilo-lab-extensions-to-us-facility.html, accessed Feb. 16, 2016.
8. PBOA, “PBOA Welcomes New Members and Trustees,” Press Re-lease, Feb. 10, 2016, www.pharma-bio.org/news/pboa-welcomes-new-members-and-trustees/, accessed Feb. 16, 2013. PT
Find out why 7 of the 10 largest CMOs rely on
W«�ÙÃ^ÊçÙ���^ãÙ�ã�¦®����ò�Äã�¦��
Focused. Timely. Accurate.
This is an invaluable service
that’s instrumental to our
ĨŽƌĞĐĂƐƟ�ŶŐ��ƉůĂŶŶŝŶŐ�ĂŶĚ�
ŐƌŽǁƚŚ�
–Business Intelligence Director,
Top-10 Global CMO
“
нϭ�ϳϬϯ�ϯဒϯ�ϰဓϬϯ�Direct
ϭ�ဒဒဒ�ϳϳϳ�ဓဓϰϬ�Toll-free in USA
[email protected] | www.pharmsource.com
X �Proprietary databases�ϐ������
����� ���������������������Ǧ
��������������������Ǥ
X Expert reports ����������Ǧ������
�� ���Ȁ���������������ǡ�����
����������ǡ����Ǧ����������Ǥ
Market intelligence
you can rely on
from the source
you can trust.
Used and respected by the top CMOs, CDMOs and CROs around the world.

EVENT OVERVIEW:
The use of hot melt extrusion (HME) for formulating active
pharmaceutical ingredients with low solubility continues to
advance as interest in continuous manufacturing increases.
An understanding of the variable process parameters that
have a significant impact on the final pharmaceutical product
is necessary to successfully manage an HME process when
using twin-screw extrusion. In this webcast, experts will discuss
residence time, mechanical-energy consumption, and other
critical parameters, as well as their effects on a scientific
approach to process scale-up.
Key Learning Objectives:
■ How HME parameters affect final solid dosage forms
■ How to scale-up a continuous process from research to
production while minimizing process optimization
■ How to optimize parameters to meet product quality
Who Should Attend:
■ Scientists, engineers and specialists in formulation
research, process development and production; scientists
actively using twin-screw extruders in the pharmaceutical
industry who want to learn a systematic approach to HME
process scale-up.
For questions contact
Kristen Moore at [email protected]
Presenter:
DIRK LEISTER
Technical Marketing,
Pharmaceutical
Extrusion Applications
Thermo Fisher Scientific
Karlsruhe, Germany
Moderator:
RITA PETERS
Editorial Director
Pharmaceutical
Technology
Sponsored by
Presented by
LIVE WEBCAST Thursday, April 14, 2016 at 8 am PDT | 11 am EDT | 4 pm BST | 5 pm CEST
Register for free at
www.pharmtech.com/pt/hot_melt
Continuous Manufacturing of Pharmaceuticals :
Scale-up of a Hot Melt Extrusion Process

Stay Current on Drug Development
and Manufacturing Trends
Attend panel discussions and presentations,
developed by the editors of Pharmaceutical
Technology and BioPharm International, on
critical industry topics.
PharmTech/BioPharm Keynote Sessions in the
Exhibit Hall at
2016Free admission with Exhibit Hall Pass
Tuesday, April 26
10:15 AM – 11:45 AM: Overcoming Bottlenecks in
Biopharmaceutical Development
1:00 PM – 1:30 PM: Innovations in Solid Dosage Development and
Manufacturing: Drop-On-Demand Manufacturing;
Polymer Thin Films; AND Minitablets
Wednesday, April 27
10:30 AM – 11:30 AM: The Contract Services Market: 2016 Update
1:30 PM – 3:15 PM: Strategies and Innovations to Reduce Drug
Shortages and Improve Availability of Medicines
3:30 PM – 4:30 PM: Panel Discussion: Addressing
Sterile Manufacturing Challenges
For details: www.PharmTech.com/INTERPHEX2016
The Contract Services Market: 2016 Update
How will consolidation in the bio/pharmaceutical and contract services market, a changing financial market, and an active politi-cal and regulatory year shape the fortunes of the contract servic-es market? In his annual presentation, industry expert Jim Miller will offer his perspectives on the contract services landscape for the next few years.
Presenter: Jim Miller, president of PharmSource Information Services, Inc.
For details: www.PharmTech.com/INTERPHEX2016
SPECIAL SESSION
Wednesday, April 27
10:30 – 11:30 AM
INTERPHEX 2016 PLANNING GUIDE AND INDUSTRY PIPELINE
VISIT PHARMTECH AT BOOTH 1225
t� .FFU�UIF�FEJUPST
t� �3FWJFX�DVSSFOU�JTTVFT�PG�Pharmaceutical Technology
and BioPharm International
t� "OTXFS�TVSWFZT
t� .FFU�DPMMFBHVFT�BOE�QFFST
STAY CURRENT ON TECHNOLOGIES AND SERVICES
Visit Pharmaceutical Technology sponsors that are
FYIJCJUJOH�BU�*/5&31)&9�������4FF�EFTDSJQUJPOT�BOE�
CPPUI�JOGPSNBUJPO�PO�UIF�GPMMPXJOH�QBHFT�
Plan Your Visit to
2016April 26–28
Javits Center–New York City
INTERPHEX 2016
PLANNING GUIDE
PH
OT
OG
RA
PH
Y B
Y S
TE
VE
KE
LLE
Y A
KA
MU
DP
IG/
GE
TT
Y I
MA
GE
SIM
AG
ES

Pharmaceutical Technology MARCH 2016 73
VISIT US AT INTERPHEX 2016
VISIT US AT INTERPHEX 2016
VISIT US AT INTERPHEX 2016
INTERPHEX 2016 PLANNING GUIDE AND INDUSTRY PIPELINE
Admix sanitary mixing equipment complies
with strict cGMP requirements and is
constructed with FDA-approved materials. It
can handle simple liquid blending, stabilizer
dispersions, and complex emulsions.
Our innovative technology, applications
expertise, and understanding of APIs equate
to improved production times, reduced
horsepower, and optimized capital costs.
Factory acceptance testing, QA, and
validation packages are available.
Admix t�144 Harvey Road Londonderry,
NH 03053 t��www.admix.com/biotech t
tel. 800.466.2369
INTERPHEX Booth #1050
Eppendorf is a leading life-science company
that develops and supplies lab instruments
and bioprocess systems for microbial and cell-
culture applications. The Eppendorf Bioprocess
portfolio includes scalable platforms in stand-
alone, parallel, and single-use systems with
working volumes from 60mL to 2400L. Lab
products include liquid handling, centrifuges,
shakers, incubators, sample prep, and detection
all from Eppendorf.
Eppendorf, 102 Motor Parkway, Hauppauge,
NY 11788 t�www.eppendorf.com t [email protected] t�tel. 800.645.3050
INTERPHEX Booth #3432
Fette Compacting
America’s New FE75
Tablet Press
The FE75 double-sided rotary
tablet press is equipped with up
to 115 punch stations to produce more than 1.6
million tablets/hr. Ideal for large batches, the
FE75’s four compression rollers feature a special
control system for direct compression, enabling
operation with two intermediate pressures. The
FE75 shares several technologies with fellow
FE Series presses, including a patent-pending
conical filling unit, highly accurate manually
adjustable filling table and trouble-free tablet
discharge through the column. Fette Compacting
America, 400 Forge Way, Rockaway, NJ 07866 t
www.fetteamerica.com t�tel. 73.586.8722
INTERPHEX Booth # 2505C
Fluid Metering
Inc. Dispensers
& Pumps
Fluid Metering
has 55+ Years of
experience meet-
ing the demand-
ing needs of the
medical, analytical, process, and industrial
markets for ultra-precise fluid control
from microliters to liters. Fluid Metering’s
dependability has been demonstrated by
more than 250,000 OEM pumps in service.
Fluid Metering Inc t 5 Aerial Way #500 t
Syosset, NY 11791 t www.fmipump.com. t
tel. 800.223.3388 or 516.922.6050
INTERPHEX Booth #1468
GEMÜ Valves manufactures valves, mea-
surement and control systems for all sectors
of industry such as basic and fine chemical,
acid handling, mining, general manufac-
turing, pharmaceutical, biotechnological,
and food industries, as well as, microchip
production.
Gemu Valves t�3800 Camp Creek Parkway t
Building 2600 Suite 120 t Atlanta, GA
30331 t www.gemu.com t�[email protected] t�
tel. 678.553.3400 t
INTERPHEX Booth #2663
Jubilant HollisterStier Contract Manufacturing
is an integrated contract manufacturer, able
to manufacture sterile injectable, ointment,
cream, and liquid dosage forms. Our facilities
across North America provide specialized
manufacturing services for the pharmaceuti-
cal and biopharmaceutical industries. Fill/
Finish; Lyophilization; Multiple Dosage Forms;
Certified Project Managers
Jubilant HollisterStier Contract Manufacturing t
A Jubilant Pharma Company t 3525 N. Regal
St. t Spokane, WA 99207 t www.jublHS.com t
[email protected] t tel. 509.489.5656
INTERPHEX Booth #1830
MG America is the US sub-
sidiary of MG2 of Bologna,
Italy and a leading supplier
of processing and packag-
ing equipment. The
PLANETA 200 Capsule Filler is the latest evolu-
tion in a line of well-established continuous
motion machines. The PLANETA 200 produces
up to 200,000 capsules per hour, but its primary
advantage is its premium flexibility: its modular
design results in a highly configurable platform
suitable for a wide array of production require-
ments, including an ability to fit several dosing
units simultaneously, enabling one capsule to
be filled with differing products.
MG America, 31 Kulick Road, Fairfield, NJ 07004.
t www.mgamerica.com. t�tel. 973.808.8185 t
INTERPHEX Booth #2221
Meissner enables pharma-
ceutical and biopharma-
ceutical manufacturing
via advanced process
solutions encompassing
a wide variety of filtration
media available in an un-
paralleled range of form
factors, innovative single-use systems, and an
expanding portfolio of automation platforms.
From design engineering to implementation
and validation services, our team of experts
provides support throughout the lifecycle of
your process, including logistics assistance.
Meissner Filtration Products t 1001 Flynn Road
Camarillo, CA 93012 t�www.meissner.com t�[email protected] t�tel. +1-805.388.9911 t�fax +1-805.388.5948
INTERPHEX Booth #2341
Catalent’s ADVASEPT® Technology is an ad-
vanced aseptic filling solution for clinical or
commercial supply of biologics, parenteral,
respiratory, and other products.
ADVASEPT utilizes Blow-Fill-Seal manufactur-
ing to produce a glass-free primary packaging
container that reduces, and even eliminates,
the main concerns associated with glass vials.
ADVASEPT Lock incorporates a needle-
free luer lock connection to increase prac-
titioner safety when handling parenteral
drug products.
Catalent Pharma Solutions, 14 Schoolhouse
Rd, Somerset, NJ 08873, t�www.catalent.com
t tel. +1.888.SOLUTION (765.8846)
INTERPHEX Booth # 1546

INDUSTRY PIPELINE
74 Pharmaceutical Technology MARCH 2016 PharmTech .com
INTERPHEX 2016 PLANNING GUIDE AND INDUSTRY PIPELINE
VISIT US AT INTERPHEX 2016
VISIT US AT INTERPHEX 2016
VISIT US AT INTERPHEX 2016
One 2 One
One 2 One™ is a global leader in sterile
injectables contract manufacturing. We
carefully guide your medicine from devel-
opment through launch, harnessing 25+
years of expertise in biopharmaceutical
fill and finish. Organizational strengths
include in-depth experience with complex
biologics and a lyophilization center of
excellence. One 2 One t 275 North Field Drive,
Lake Forest, IL 60045 t�www.one2onecmo.com
t tel. 224.212.2267
INTERPHEX Booth #1235
Watson-Marlow Fluid Technology Group
(WMFTG) is the world leader in peristaltic
pumps and associated fluid path technologies
for the pharmaceutical market. Comprising
eight established brands, each with their
own area of expertise, but together offering
our customers unrivaled solutions for their
pumping and fluid transfer applications.
Watson-Marlow Fluid Technology Group t 37 Upton
Drive, Wilmington, MA 01887 t
www.wmftg.com t [email protected]
INTERPHEX Booth #2833
Company Services
Patheon, a business unit of DPx Holdings B.V.,
is a leading provider of contract development
and commercial manufacturing services to the
pharmaceutical and biotechnology sectors.
The company offers one of the broadest sets of
solutions to customers including commercial
manufacturing, drug product services, biolog-
ics, pharmaceutical development services, and
active pharmaceutical ingredients.
Patheon, 4815 Emperor Blvd. Suite 300,
Durham, NC 27703 t www.patheon.com t��
tel. +1 919.226.3200
INTERPHEX Booth # 1424
VAI’s manufacturing and testing operations
mirror current GMP/GLP standards. With
over 35 years of contamination control ex-
perience, our products and services include
a comprehensive line of disinfectants, spo-
ricides, residue removers, process cleaners,
quality water, pre-saturated and dry wipes,
disposable garments, cleanroom paper,
documentation materials, printing systems,
viable monitoring, cleaning equipment,
consulting and laboratory services.
Veltek Associates, Inc. t��www.sterile.com
INTERPHEX Booth # 2527
Company
Services
SMI, founded in 1982,
brings over 30 years of
engineering excellence to the industry. Initially,
SMI pioneered instrumentation systems for
pharmaceutical equipment.
SMI introduced a tablet press of its own, the
“Piccola”. The Piccola was the first bench-top
tablet press designed for R&D. Today, SMI
provides a complete line of solid dosage equip-
ment ranging from the single station MiniPress,
to bench top rotary, to the 21 station pilot scale
Nova, designed for mono and multi-layered
tablets. Specialty Measurements Inc. (SMI), P.O. Box
356, 1309 US Highway 22 East, Lebanon, NJ
08833 t�www.smitmc.com t tel. 908.534.1500
INTERPHEX Booth # 3153
rommelag has been the driving force be-
hind advanced aseptic Blow/Fill/Seal (BFS)
filing technology for over 50 years. Rom-
melag provides services for container and
closure development, clinical trials and
aseptic production. The bottelpack 430®
closed parison BFS machine with disposable
fill system, designed to provide advanced
aseptic fill/finish for injectable drug and
biotech products, represents the newest
application into the BFS market.
Rommelag USA, Inc. t 27905 Meadow Dr.
Suite 9, Evergreen, CO 80439 t
www.rommelag.com t 303.674.8333
INTERPHEX Booth # 3516
Ropack Pharma Solutions supports solid
oral dosage projects from early-stage
formulation through clinical and
commercial manufacturing, packaging,
and distribution. We’re not just another
CMO. We’re a trusted partner. Never
losing sight that it’s our clients’ vision
we’re bringing to market, we collaborate,
communicate, and work together toward a
common goal—a successful outcome.
Ropack Pharma Solutions t 49 Mall Drive,
Commack, NY 11725 t www.ropack.com
INTERPHEX Booth #1140
A Top 20 contract development and manu-
facturing organization (CDMO), Rottendorf
Pharma formulates, develops, manufac-
tures, and packages solid oral-dosage forms
for the global pharmaceutical industry. Cli-
ents choose Rottendorf for its broad-rang-
ing expertise, global regulatory capabilities,
commitment to quality, state-of-the-art
facilities, and exceptional customer service
exemplified by Total Process Ownership.
Rottendorf Pharma GmbH t 875 North Michigan
Avenue, 31st Floor, Chicago, IL 60611 t
www.rottendorf.com t�tel. 312.794.7836
INTERPHEX Booth # 1541
The ASEP-TECH® Blow/Fill/Seal (BFS) system
has been marketed by Weiler Engineering,
Inc. for over 45 years to the world’s leading
pharmaceutical and healthcare companies.
This advanced aseptic technology com-
bines blow molding of plastic bottles, ster-
ile filling of liquid products and hermetic
sealing on one compact machine frame in
bottle sizes ranging from 0.2mL to 1000mL.
Weiler Engineering, Inc. t�1395 Gateway Drive
t�Phone: 847.697.4900 t�Fax: 847.697.4915 t�
www.weilerengineering.com t solutions@
weilerengineering.com
INTERPHEX Booth #1224

Pharmaceutical Technology MARCH 2016 75
Dispersers
Ross Bow Tie Dispersers,
or High Viscosity
Dispersers, are heavy-
duty mixers designed
for heavy pastes and
viscous liquids up
to several hundred
thousand centipoise,
with stainless steel 304 wetted parts. An
explosion-proof inverter-duty motor helps
the mixers incorporate solids into liquid
vehicles in a consistent manner without
running at high speeds.
Ross, Charles & Son Company t Hauppauge, NY t�
www.mixers.com t tel. 800.243.ROSS
INTERPHEX 2016 PLANNING GUIDE AND INDUSTRY PIPELINE
NEW PRODUCTS AND SERVICES
NEW PRODUCTS AND SERVICES
NEW PRODUCTS AND SERVICES
Mikart specializes in the development,
manufacturing, and packaging of solid-dose
and liquid-oral dose products. The company’s
services include formulation development;
analytical, manufacturing, packaging, and
regulatory services; and complete project
management. Mikart offers clients more than
40 years of experience, a responsive working
relationship, and the ability to take products
from formulation development through full-
scale commercial production.
Mikart, Inc., t www.mikart.com t tel. 404.351.4510
Company Services
EtQ is the leading FDA Compliance, Quality,
EHS, and Operational Risk Management
software provider for identifying, mitigating,
and preventing high-risk events through
integration, automation, and collaboration.
EtQ’s modules are tightly integrated to
deliver a leading FDA compliance solution.
Key modules include Corrective Action,
Audits, Risk Management, Complaint
Handling, and more. EtQ t�www.etq.com
Clinical Material Manufacturing
Lyophilization Technology, Inc. is a Contract
Development and Manufacturing Organiza-
tion providing development, clinical manu-
facturing, and technical services focused on
lyophilized products. The comprehensive
range of services includes thermal analysis,
formulation development, process engineer-
ing, cytotoxic/high potent APIs, pre-clinical to
Phase II clinical material preparation for freeze
dried pharmaceuticals, biologics, and biophar-
maceuticals. Technical services encompass
consulting, compliance support and training.
Lyophilization Technology, Inc., 30 Indian Drive,
Ivyland, PA 18974-1431 USA, t�
www.lyotechnology.com t tel. 215.396.8373
Shimadzu is a world leader in the analytical
instruments industry. Instruments include
UHPLC, SFE/SFC, LC-MS/MS, GC-MS, UV-Vis,
FTIR, MALDI-TOF, EDXRF, thermal analyzers, and
TOC analyzers. They are used throughout the
pharmaceutical pipeline, from proteomics and
metabolomics research and drug discovery/
development to QA/QC and manufacturing,
providing a total solution to researchers working
within the pharmaceutical/biopharma industry.
Shimadzu Scientific Instruments, 7102 Riverwood
Drive, Columbia, MD 21046 t www.ssi.shimadzu.com
t [email protected] t tel. 800.477.1227
t fax. 410.381.1222
Featured
Product
VAC-U-MAX
manufactures
Vacuum Conveying
Equipment and
Systems designed
for the Pharmaceutical Industry. Construction
of equipment is USDA Accepted. The vacuum
conveyors range in convey rates from 500
pounds per hour to 5,000 pounds per hour
and beyond. Designed for Free and Non Free
Flowing powders they can be equipped for
use with any application. Loading tablet press
machines, mix tanks, blenders.
VAC-U-MAX, Belleville, NJ t
www.vac-u-max.com t tel. 973.759.4600
You deserve a strategic, responsive, and effi-
cient partner for your early-stage drug devel-
opment. MPI Research offers that and more.
With an impressive breadth of discovery,
preclinical, and clinical scientific knowledge
and services, our team of highly trained scien-
tists and world-class facilities offer the insights
to see your project through. We do every-
thing we can to make your vision a reality.
MPI Research, 54943 North Main Street, Mattawan,
MI 49071 USA, t�www.mpiresearch.com t tel. +1.269.668.3336
PYRAMID Laboratories, Inc. is located in
Southern California, United States. Our fa-
cilities are housed in three buildings cover-
ings more than 70,000 ft. The combination
of our manufacturing facilities and state-of-
the-art laboratory allows PYRAMID to offer
the pharmaceutical and biotech industry
both analytical and manufacturing support
capabilities. PYRAMID Laboratories, Inc., 3598
Cadillac Avenue, Costa Mesa, CA 92626 t www.pyramidlabs.com t [email protected]
t tel. 714.435.9800 t fax 714.435.9585
TruTag Technologies offers the most advanced
product authentication and brand protection
solutions for pharmaceutical, nutritional, food
and food packaging, electronics, industrial, and
consumer goods industries. The TruTag solu-
tion addresses the $1 trillion global counterfeit
and diversion problem with a breakthrough
technology using microtags made of the high-
est purity silica that serve as “covert on-product
codes.” TruTag® microtags are biologically inert,
edible, virtually invisible, and can integrate into
the very fabric of a product, independent of
packaging and labels. TruTag t�2045 Lauwiliwili
Street, Unit 301, Kapolei, HI t�www.TruTags.com
t�tel. 808.878.8247

76 Pharmaceutical Technology MARCH 2016 PharmTech .com
overcome this challenge, process automation data and data from the laboratory information system have to be shared with the information technology (IT) systems for batch re-cording, reviewing, and releasing. Furthermore, scheduling systems are a key part aligning all production steps and ensuring planning principles like first-in/first-out priori-tization rules. Extensive integration of these data sources by IT and automation systems will allow production costs to be lowered by at least an order of magnitude.
Another case that presents some challenges is transform-ing a batch pharmaceutical process into a continuous pro-cess. A continuous process requires a robust process design and continual monitoring, typically using online analyz-ers. Multivariate analyzer data and other process param-eters from simple sensors must be combined and treated by statistical engines to give the operators a clear view of the process so that they can ensure that the process always stays within the quality boundaries. In the next genera-tion of continuous production processes, it will become important to use these quality parameters to automatically adjust the process, thus closing the loop. The core of such an advanced process-control system is a data platform han-dling a large quantity of multivariate and univariate data in real time and applying statistical methods like principle
component analysis. The easy integration of multivariate analyzer data (e.g., spectral data) following open standards is an important requirement. An equally important feature is the real-time integration to the control system for adjust-ing the set-point dynamically. Advanced process control methods, especially model predictive control strategies, can add the benefit of controlling the overall quality at the low-est variance within the lower and upper limits. PT
Process Control
Visit PharmTech.com for more articles on process control, such as:
t� Reducing False Out-of-Control Signals, PharmTech.com/reducing-false-
out-control-signals
t� Model-Predictive Design, Control, and Optimization, PharmTech.com/
model-predictive-design-control-and-optimization
t� Flowsheet Models Modernize Pharmaceutical Manufacturing Design
and Risk Assessment, PharmTech.com/flowsheet-models-modernize-
pharmaceutical-manufacturing-design-and-risk-assessment
t� Incorporating Process Analytical Technology Data into Process Control,
PharmTech.com/incorporating-process-analytical-technology-data-
process-control.
More on process control
Process Control — contin. from page 55
Data Integrity — contin. from page 53
As regulators continue to tighten their inspection ap-proaches, it is crucial for managers and scientists in regu-lated GXP laboratories to understand these criteria for data integrity and to assess and improve laboratory data manage-ment processes to ensure compliance with current regula-tions. Only after all these points have been addressed can data integrity truly be achieved.
References 1. C. Rosa, “Current Regulatory/Inspection Issues Related to Supply
Chain,” Food and Drug Law Institute (FDLI), Conference Un-derstanding cGMPS–What Attorneys Need to Know, Washing-ton DC, July 15, 2014, www.fdli.org/docs/cgmps/carmelo-rosa.pdf?sfvrsn=0
2. MHRA, Guidance for Industry on Data Integrity (MHRA, March 2015), www.gov.uk/government/uploads/system/uploads/attach-ment_data/file/412735/Data_integrity_definitions_and_guid-ance_v2.pdf
3. FDA, “Glossary of Computer System Software Development Terminology,” 1995, www.fda.gov/iceci/inspections/inspection-guides/ucm074875.htm
4. European Commission, EudraLex, The Rules Governing Medicinal Products in the European Union, Volume 4, Good Manufacturing Practice, Chapter 4 Documentation (June 2011), http://ec.europa.eu/health/files/eudralex/vol-4/chapter4_01-2011_en.pdf
5. European Commission, EudraLex, The Rules Governing Medici-nal Products in the European Union, Volume 4, Good Manufac-turing Practice, Annex 11 Computerised Systems (January 2011),
http://ec.europa.eu/health/files/eudralex/vol-4/annex11_01-2011_en.pdf
6. US Electronic Code of Federal Regulations, 21 CFR 211.194(a). 7. FDA, FDA Warning Letter to Trifarma S.p.A, July 2014, www.fda.gov/
ICECI/enforcementactions/warningletters/2014/ucm404316.htm 8. FDA, FDA Warning Letter to Ipca Laboratories Ltd., January
2016, www.fda.gov/ICECI/enforcementactions/warningletters/ucm484910.htm
9. FDA, FDA Warning Letter to Micro Laboratories Ltd., Janu-ary 2015, www.fda.gov/iceci/enforcementactions/warninglet-ters/2015/ucm431456.htm
10. FDA, Questions and Answers on Current Good Manufacturing Practices, Good Guidance Practices, Level 2 Guidance–Records and Reports, www.fda.gov/Drugs/GuidanceComplianceRegula-toryInformation/Guidances/ucm124787.htm
11. EMA, GCP Inspectors Working Group publication, Ref lec-tion paper on expectations for electronic source data and data transcribed to electronic data collection tools in clinical trials (London, June 2010), www.ema.europa.eu/docs/en_GB/docu-ment_library/Regulatory_and_procedural_guideline/2010/08/WC500095754.pdf
12. R.D. McDowall, LCGC 27 (9) (September 2014), www.chromatog-raphyonline.com/role-chromatography-data-systems-fraud-and-falsification
13. FDA, FDA Warning Letter to RPG Life Sciences Ltd., May 2013, www.fda.gov/ICECI/EnforcementActions/WarningLetters/2013/ucm355294.htm
14. FDA, FDA Warning Letter to Wockhardt Ltd., July 2013, www.fda.gov/ICECI/EnforcementActions/WarningLetters/2013/ucm361928.htm PT

Pharmaceutical Technology MARCH 2016 77
MANUFACTURING
STERILE MANUFACTURING
Mixing/Blending/Drying
MANUFACTURING/PROCESSING EQUIPMENT
Purity.Eriez.com � 888-300-3743
Quick ShipMetal Detector Program
In Stock� 8 Xtreme® Metal
Detector aperture heights
� Available in 12, 18 and 24-inch widths
� Sanitary-grade polypropylene belt
+]ZM�NWZ�\PM�+WUUWV�2WJ�Proven relief from the signs and symptoms of Chronic Job Search (CJS)
Joel Kern - East Coast Sales Manager
�������������t�KLFSO!BEWBOTUBS�DPN
Irene Onesto - MiE-West Sales Manager
�������������t�JPOFTUP!BEWBOTUar�DoN
Paul Milazzo - Director of Sales
�������������t�QNJMB[[P!BEWBOTUBS�DPN
Leverage branded content from
Pharmaceutical Technology to create a more
powerful and sophisticated statement about
your product, service, or company in your next
marketing campaign. Contact Wright’s Media to
fi nd out more about how we can customize your
acknowledgements and recognitions to enhance
your marketing strategies.
Marketing solutions fi t for:
Outdoor | Direct Mail | Print Advertising
Tradeshow/POP Displays | Social Media | Radio & TV
Content Licensing for Every
Marketing Strategy
For information, call Wright’s Media at 877.652.5295
or visit our website at www.wrightsmedia.com
SANITARYRIBBON
BLENDERS
1-800-243-ROSS www.ribbonblenders.com
Scan to learn more. Try our mobile app:mixers.com/web-app

78 Pharmaceutical Technology MARCH 2016 PharmTech .com
ASK THE EXPERT
Q: Our company covers the entire lifecycle for our drugs, from
R&D through clinical trials to commercial product. The quality
management system covers good clinical practice (GCP) and cur-
rent good manufacturing practice (cGMP). In a recent client audit,
we received an observation: “You do not apply the same level of
cGMP to clinical-trial material manufacture as you apply to com-
mercial product.” Is this a new regulatory expectation?
A:It is not a regulatory expectation to apply the same level of
cGMP to investigational new drug (IND) and marketed prod-
ucts. Although it is correct that you need to apply cGMP to all your
clinical-trial supplies (referred to as investigational medicinal prod-
ucts [IMP] in the European Union [EU] and IND in the United States)
and to commercial operations, the level of cGMP differs. The rea-
sons for this are practical constraints as well as regulatory require-
ments. Under FDA regulations, the manufacture of most INDs used
in Phase I clinical trials is exempt from cGMPs (1) (i.e., 21 Code of
Federal Regulations Part 211 is not applicable). For INDs for clinical
Phases II and III and for commercial product, cGMP applies (2, 3).
In the EU, the relevant regulations are in EudraLex Vol. 4, Part x
I (drug product) and Part II (drug substance/API), and IMPs are
covered specifically in Annex 13 “Manufacture of Investigational
Medicinal Products,” which was last updated in 2010 (4).
EudraLex, and any laws and regulations laid down by EU memberx
states, are based on Commission Directive 2003/94/EC, which
applies to both IMP and marketed products (5).
Paragraph 17 in Annex 13 summarizes the differences in
cGMP for IMP and marketed product: “Production processes
for investigational medicinal products are not expected to be
validated to the extent necessary for routine production but
premises and equipment are expected to be qualified. For
sterile products, the validation of sterilizing processes should be
of the same standard as for products authorized for marketing.”
Though these guidances are useful, they will not necessarily
answer all questions. The common adage supported by the
majority of regulatory agencies is to apply a scientifically sound
risk-based approach to compliance. More information can be
found in several publications, including a widely publicized
technical report by the Parenteral Drug Association (6).
The regulators do understand that product and process
knowledge will continue to grow during the lifecycle from
clinical Phase I to launch and beyond. With increased
understanding, companies can and need to apply more
controls and move towards a fully validated process. You may
wish to discuss the observation with the auditor in light of the
regulations and guidance documents mentioned above.
References
1. FDA, Guidance for Industry: cGMP for Phase 1 Investigational Drugs (Rockville, MD, July 2008).
2. FDA, Draft Guidance for Industry: INDs for Phase 2 and 3 Stud-ies of Drugs, Including Specified and Therapeutic Biotechnology Derived Products (Rockville, MD, February 1999).
3. 21 CFR Parts 210 and 211, www.ecfr.gov. 4. European Commission, EudraLex-Volume 4 Good manufactur-
ing practice (GMP) Guidelines, http://ec.europa.eu/health/docu-ments/eudralex/vol-4/
5. Commission Directive 2003/94/EC on the principles and guide-lines of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human use, http://ec.europa.eu/health/files/eudralex/vol-1/dir_2003_94/dir_2003_94_en.pdf
6. PDA Technical Report 56 Application of Phase-Appropriate Qual-ity System and cGMP to the Development of Therapeutic Protein Drug Substance (PDA, 2012). PT
Ad Index
Siegfried Schmitt, principal consultant, PAREXEL, discusses the regulatory
requirements for cGMPs in the different phases of drug development and manufacture.
Phase-Appropriate GMP
AAPS ...........................................49Admix Inc ....................................55Aenova ........................................45AirBridge Cargo Airlines............ 16AMRI ............................................31Capsugel .....................................67Catalent Pharma Solutions.......80Emergent Biosolutions .............. 15Eppendorf North America ..........3ETQ Inc ........................................ 17Eurofins LancasterLaboratories ............................... 41Excipient Fest .............................65Fette Compacting America Inc ...............................9Fluid Metering Inc ........................6Gemu Valves Inc ........................59GlobePharma .............................20INTERPHEX .................................63Jubilant HollisterStier .................27Lonza ...........................................37Lyophilization Technology Inc ..47Meissner Filtration Products ......2MG America Inc ......................... 11Mikart ..........................................25MPI Research .............................21One 2 One .....................................7Parenteral Drug Association ....23
Patheon Pharmaceutical
Svc Inc ......................................43
Pharmacore ..................................6
PharmSource Information
Services Inc .............................70
Pyramid Laboratories ................ 19
Rommelag USA Inc .................... 51
Ropack ........................................69
Rottendorf Pharmaceuticals ....33
SMI ............................................... 13
Spectrum Chemical
Mfg Corp ..................................35
Suheung-America
Corporation ...............................4
Thermo Fisher Scientific ..... 57, 71
Trutag Technologies, Inc ...........29
Unither Pharmaceuticals ..........39
University Of Tennessee .............4
Vac U Max ................................... 61
Veltek Associates ........................5
Watson Marlow Fluid Technology
Group ...........................................53
Weiler Engineering Inc .............79
COMPANY PAGE COMPANY PAGE

To see our hands-free, sterile manufacturing process in action,
visit www.ASEP-TECH.com/ptus or call 847-697-4900.
Safeguarding solutions through innovative packaging
Some things are better in ...safer inside
At Weiler Engineering, Inc. our ASEP-TECH® Blow/Fill/Seal
machines produce shatterproof plastic, aseptically packaged products
MR�E�GPSWIH�IRZMVSRQIRX°E�FIXXIV�EPXIVREXMZI�XS�GSRZIRXMSREP�½PPMRK�
of glass vials for parenteral, ophthalmic, biologics, vaccines and
respiratory products.
ADVANCED ASEPTIC TECHNOLOGY ADVANTAGES:
) Increased safety for both the patient and healthcare provider
)� Lower product to market cost with streamlined packaging process
)� Improved product quality with no aluminum leaching issues
)��Less material handling reduces risk of product contamination
©20
15 W
eile
r En
gine
erin
g, In
c.

˝ 2
016
Cata
len
t P
harm
a S
olu
tio
ns.
All r
igh
ts r
ese
rve
d.
Catalent. More products. Better treatments. Reliably supplied.™
us + 1 888 SOLUTION eu + 800 8855 6178 catalent.com/potent
POTENT HANDLING EXPERTISE
Proven track record with 20+ years of potent
handling experience and 80+ years providing
innovative pharmaceutical delivery solutions.
300+ potent and highly-potent compounds,
as well as hormones and cytotoxics.
INTEGRATED SERVICES
Integrated analytical, development and
supply solutions with recent investments
in high potency clinical packaging, Micron
Technologies particle size engineering, and
expanded manufacturing services for oral solids.
SAFELY & RELIABLY SUPPLIED
Robust engineering and PPE controls.
Comprehensive risk assessment processes.
Highly-trained, experienced team with deep
expertise in special handling protocols
in 10+ sites across our global network.
potent compounds. integrated solutions. reliably supplied.
SITES IN THE GLOBAL NETWORKIntegrated solutions from
optimization through supply
300+POTENT COMPOUNDS HANDLED
Potent, hghly potent, hormones
and cytotoxics
YEARS EXPERTISE IN HIGH POTENT HANDLING 80+ years providing drug delivery solutions20+
10+