adenovirus vai rna antagonizes the rna-editing activity of the adar adenosine deaminase
TRANSCRIPT

Adenovirus VAI RNA Antagonizes the RNA-Editing Activityof the ADAR Adenosine Deaminase
Ming Lei, Yong Liu, and Charles E. Samuel1
Department of Molecular, Cellular and Developmental Biology and Graduate Program of Biochemistry and Molecular Biology,University of California, Santa Barbara, California 93106
Received August 12, 1997; returned to author for revision September 9, 1997; accepted March 26, 1998
The virus-associated VAI RNA of adenovirus is a small highly structured RNA that is required for the efficient translationof cellular and viral mRNAs at late times after infection. VAI RNA antagonizes the activation of the interferon-inducibleRNA-dependent protein kinase, PKR, an important regulator of translation. The RNA-specific adenosine deaminase, ADAR, isan interferon-inducible RNA-editing enzyme that catalyzes the site-selective C-6 deamination of adenosine to inosine. ADARpossesses three copies of the highly conserved RNA-binding motif (dsRBM) that are similar to the two copies found in PKR,the enzyme in which the prototype dsRBM motif was discovered. We have examined the effect of VAI RNA on ADAR function.VAI RNA impairs the activity of ADAR deaminase. This inhibition can be observed in extracts prepared from interferon-treatedhuman cells and from monkey COS cells in which wild-type recombinant ADAR was expressed. Analysis of wild-type andmutant forms of VA RNA suggests that the central domain is important in the antagonism of ADAR activity. These resultssuggest that VAI RNA may modulate viral and cellular gene expression by modulating RNA editing as well as mRNAtranslation. © 1998 Academic Press
INTRODUCTION
RNA-specific adenosine deaminase (ADAR),2 alsoknown as dsRAD and DRADA, is an interferon-inducibleRNA-editing enzyme (Patterson and Samuel, 1995).ADAR catalyzes the C-6 deamination of adenosine toyield inosine in viral RNAs and cellular pre-mRNAs(Bass, 1997). For example, ADAR catalyzes the site-se-lective editing of hepatitis delta virus RNA (Polson et al.,1996) and the cellular pre-mRNAs encoding the gluta-mate-gated ion channel GluR receptor (Sommer et al.,1991; Lomeli et al., 1994) and the serotonin 5-HT2c recep-tor (Burns et al., 1997). In addition to selective RNAediting of structured single-stranded RNA substrates,ADAR can also deaminate adenosine-containing syn-thetic double-stranded RNA substrates (Liu and Samuel,1996; Nishikura et al., 1991; Polson and Bass, 1994). ThedsRNA deaminase activity is presumed to be responsi-ble for the biased hypermutations observed during thereplication of negative-stranded RNA viral genomes dur-ing lytic and persistent infections, as exemplified bymeasles virus (Cattaneo et al., 1988; Wong et al., 1989;Cattaneo, 1994). However, hypermutation deamination is
not limited to viruses with RNA genomes; extensiveadenosine modifications have also been identified in theearly transcripts of the DNA virus polyoma virus (Kumarand Carmichael, 1997). The A to I deaminations observedin both cellular pre-mRNA transcripts and viral RNAs aredependent upon double-stranded structures within thesubstrate RNA. The dsRNA binding properties of thepurified ADAR proteins (Hough and Bass, 1994; Kim etal., 1994a; Nishikura et al., 1991; O’Connell and Keller,1994) are similar to those of the IFN-inducible RNA-dependent protein kinase PKR (Samuel, 1979, 1993).
cDNA and genomic clones of the human ADAR deami-nase have been isolated and characterized. The humanADAR protein is a 1226-amino-acid protein as deducedfrom the cDNA sequence (Kim et al., 1994b; O’Connell etal., 1995; Patterson and Samuel, 1995) that is encoded byan IFN-inducible ;6.7-kb transcript (Patterson et al.,1995) that undergoes alternative splicing (Liu et al., 1997).The human ADAR gene maps to chromosome 1q21.1–21.2 (Weier et al., 1995). ADAR deaminase possessesthree copies of the highly conserved dsRNA bindingmotif dsRBM (Patterson and Samuel, 1995; Liu and Sam-uel, 1996) that was first identified in PKR kinase (McCor-mack et al., 1992; Green and Mathews, 1992) and Droso-philia Staufen protein (St. Johnston et al., 1992). Thecodon phasing of exons 3, 5, and 7 encoding the threedsRBM motifs in human ADAR (Liu et al., 1997) areexactly conserved and identical to the two dsRBM motif-containing exons of about 40 amino acids each presentin both human (Kuhen et al., 1996) and mouse (Tanaka
1 To whom reprint requests should be addressed. Fax: (805) 893-4724.2 The abbreviations used: Ad, adenovirus; ADAR, dsRNA-specific
adenosine deaminase; dsRNA, double-stranded RNA; dsRBM, dsRNAbinding motif previously designated R-motif; IFN, interferon; kb, kilo-base; nt, nucleotide; PKR, RNA-dependent protein kinase; VA, virus-associated.
VIROLOGY 245, 188–196 (1998)ARTICLE NO. VY989162
0042-6822/98 $25.00Copyright © 1998 by Academic PressAll rights of reproduction in any form reserved.
188

and Samuel, 1994) PKR. Three splice-site variants of theADAR protein have been identified. They all retain thedsRBM motif subdomains functionally intact, but pos-sess deletions of 19 and 26 amino acids between thedsRBMII and dsRBMIII subdomains and the dsRBMIII andcatalytic subdomains, respectively, which result in func-tionally distinct enzymes (Liu et al., 1997).
Adenovirus virus-associated (VA) RNAs are small reg-ulatory RNAs transcribed by RNA polymerase III(Mathews and Shenk, 1991). They are highly structuredRNAs, about 160 nt in size. Three structural features ofthe VAI RNA from Ad2 and Ad5 include two long imper-fectly base-paired stem regions, the terminal stem andthe apical stem-loop, which are linked by a central do-main (Ma and Mathews, 1996a,b). VAI RNA accumulatesto high concentrations in the cytoplasm at late times afteradenovirus infection (Soderlund et al., 1976), is requiredfor efficient protein synthesis (Schneider et al., 1984), andantagonizes the IFN-induced antiviral response in ade-novirus-infected cells (Kitajewski et al., 1986). One of theprinciple targets of VA RNA is the RNA-dependent pro-tein kinase PKR (Mathews and Shenk, 1991; Samuel,1991). PKR is an IFN-inducible cellular protein kinase thatplays a central role in the antiviral action of IFN and thecontrol of translation in virus-infected cells (Samuel,1993; Clemens, 1996). VAI RNA binds to the dsRNA bind-ing motifs of PKR (McCormack et al., 1992; McCormackand Samuel, 1995) and blocks the dsRNA-dependentactivation of PKR by autophosphorylation (Kitajewski etal., 1986; O’Malley et al., 1986). Activated PKR catalyzesthe phosphorylation of the a subunit of protein synthesisinitiation factor eIF-2 at serine 51 (Samuel, 1979; Pathaket al., 1988), which leads to an inhibition of translation(Clemens, 1996). In the case of adenovirus, VAI RNAcompetes with the dsRNA activator of PKR believed to besynthesized by symmetrical transcription of the viral ge-nome (Maran and Mathews, 1988). Because ADAR andPKR are both IFN-inducible proteins (Patterson and Sam-uel, 1995; Patterson et al., 1995; Meurs et al., 1990;Thomis et al., 1992), because ADAR and PKR are bothRNA-binding proteins that possess the same highly con-served dsRBM (Patterson and Samuel, 1995; McCor-mack et al., 1992; Liu et al., 1997), and because it iswell-established that the adenovirus VAI RNA antago-nizes the activation and function of the PKR kinase(Mathews and Shenk, 1991; Samuel, 1993), we consid-ered the possibility that VAI RNA may also antagonize theADAR RNA editing enzyme.
We report here the effect of adenovirus VAI RNA on thefunction of the ADAR deaminase. VAI RNA inhibitedADAR and was not a detectable substrate of ADAR.ADAR activity present in extracts prepared from eitherIFN-treated cells or from untreated cells transfected witha cDNA clone encoding wild-type ADAR was inhibited byVAI RNA. Analysis of VAI RNA mutants indicated that the
central domain region was important for the inhibition ofADAR as well as the antagonism of PKR.
RESULTS
Adenovirus VAI RNA inhibits RNA-specific adenosinedeaminase activity
The interferon inducible RNA-specific ADAR deami-nase contains three copies of the dsRNA-binding motiforiginally identified in the IFN-inducible RNA-dependentprotein kinase PKR (Patterson and Samuel, 1995; Liu etal., 1997). The RNA-dependent activation of the PKR ki-nase, an important regulator of translation, is selectivelyantagonized by adenovirus VAI RNA (Samuel, 1993;Mathews and Shenk, 1991). To explore the possible re-lationship between adenovirus VAI RNA and ADAR ac-tivity, cytoplasmic extracts prepared from IFN-treated Ucells were examined for their ability to catalyze thedeamination of 32P-labeled dsRNA substrate in the pres-ence of VAI RNA. As shown by the analysis of the ADARdeaminase activity by TLC (Fig. 1A), wild-type VAI RNAadded to the standard ADAR reaction mixture reducedthe amount of inosine formed. The inhibition of deami-nase activity was dependent upon the concentration ofadded VA RNA; maximal inhibition was observed be-tween 10 and 20 mg VAI RNA/ml (Fig. 1B). Bulk transferRNA did not significantly affect deaminase activity (Fig.1B). 32P-labeled VA RNA was also examined as a sub-strate in place of [32P]dsRNA in the standard ADARreaction mixture. As measured by the formation of ino-sine detected by TLC and autoradiography, wild-type VAIRNA was not a substrate for ADAR (data not shown). Theeffect of HIV TAR RNA on ADAR activity was also exam-ined. TAR(1-82) RNA did not antagonize ADAR activitymeasured with dsRNA substrate under conditions com-parable to those in which VA RNA did antagonize ADARactivity, and TAR RNA itself was not a detectable sub-strate of the deaminase (data not shown).
Inhibition of recombinant RNA-specific adenosinedeaminase by VAI RNA is independentof the N-terminus of ADAR
The ADAR cDNA expressed in transfected COS cells,either as the full-length protein (M1 ADAR) or as a trun-cated protein lacking the N-terminal 295 residues (M296ADAR), possesses dsRNA adenosine deaminase activity(Patterson and Samuel, 1995; Liu et al., 1997). The deami-nase activity associated with recombinant human ADARexpressed in monkey COS cells was inhibited by VAIRNA (Fig. 2). Both the M1 ADAR and the truncated M296ADAR enzymes were inhibited by the addition of VAI RNAto the reaction mixture (Fig. 2). The results shown in Fig.2 were obtained with the ADAR-b splice-site variant (Liuet al., 1997). We also found that VAI RNA inhibited thedeaminase activity of the recombinant ADAR-a and
189VA RNA ANTAGONIZES ADAR

ADAR-c variants, both as the M1 and M296 forms of theprotein (data not shown). Characterization of mutantADAR proteins possessing single amino acid substitu-tions in dsRBMI, dsRBMII, and dsRBMIII has establishedthat dsRBMIII is the most important for deaminase activ-ity and that dsRBMII is least important (Liu and Samuel,1996; Liu et al., 1997). Consistent with these earlier ob-servations, the dsRBMIII mutant of M1 ADAR did not
display significant enzymatic activity in either the ab-sence or the presence of added VAI RNA (Fig. 2). Bycontrast, the dsRBMII mutant of M296 ADAR, which dis-played significant deaminase activity in the absence ofVAI RNA, was inhibited by the addition of VAI RNA to thereaction mixture (Fig. 2). The low residual activity of thedsRBMI mutant was further reduced by addition of VAIRNA (Fig. 2).
FIG. 1. Effect of VAI RNA on ADAR deaminase activity present in extracts of interferon-treated human cells. The effect of adenovirus wild-type VAIRNA on double-stranded RNA-dependent adenosine deaminase (ADAR) activity was measured with cytoplasmic extracts prepared from interferon-treated human U cells. The indicated concentration of unlabeled Ad VAI RNA or transfer RNA was added to the reaction mixture before addition ofthe 32P-labeled dsRNA substrate. (A) Autoradiogram showing ADAR deaminase activity. Inosine formation was analyzed by thin-layer chromatography;the positions of AMP, IMP, and the origin are indicated. (B) Quantitation of ADAR deaminase activity observed in the standard reaction mixturecontaining U cell extract and either wild-type VAI RNA (open circles) or bulk tRNA (filled circles) as indicated.
FIG. 2. Effect of VAI RNA on the activity of recombinant wild-type and mutant human ADAR proteins expressed in transfected COS cells. Wild-typeand mutant forms of the human ADAR cDNA were expressed in transfected monkey COS cells. Cytoplasmic extracts were prepared and analyzedfor ADAR protein expression and enzyme activity with 32P-labeled dsRNA substrate. (A) Autoradiogram showing ADAR deaminase activity in theabsence (2) and the presence (1) of 12.5 mg/ml of VAI RNA. Inosine formation was analyzed by thin-layer chromatography; the positions of AMP, IMP,and the origin are indicated. (B) Quantitation of ADAR deaminase activity. Deaminase activity was calculated based on the percentage of A-to-Iconversion catalyzed per equivalent amount of expressed ADAR protein determined by Western immunoblot analysis (Liu et al., 1997). Two forms ofwild-type (WT) ADAR were expressed: M1, wild-type full-length protein; M296, N-terminal truncated protein beginning at methionine 296. ADARproteins possessing a substitution mutation in one of the three RNA-binding domains included RI (K554E), RII (K665E), and RIII (K776E), which wereexpressed as either the full-length or the M296 protein version (Liu et al., 1997).
190 LEI, LIU, AND SAMUEL
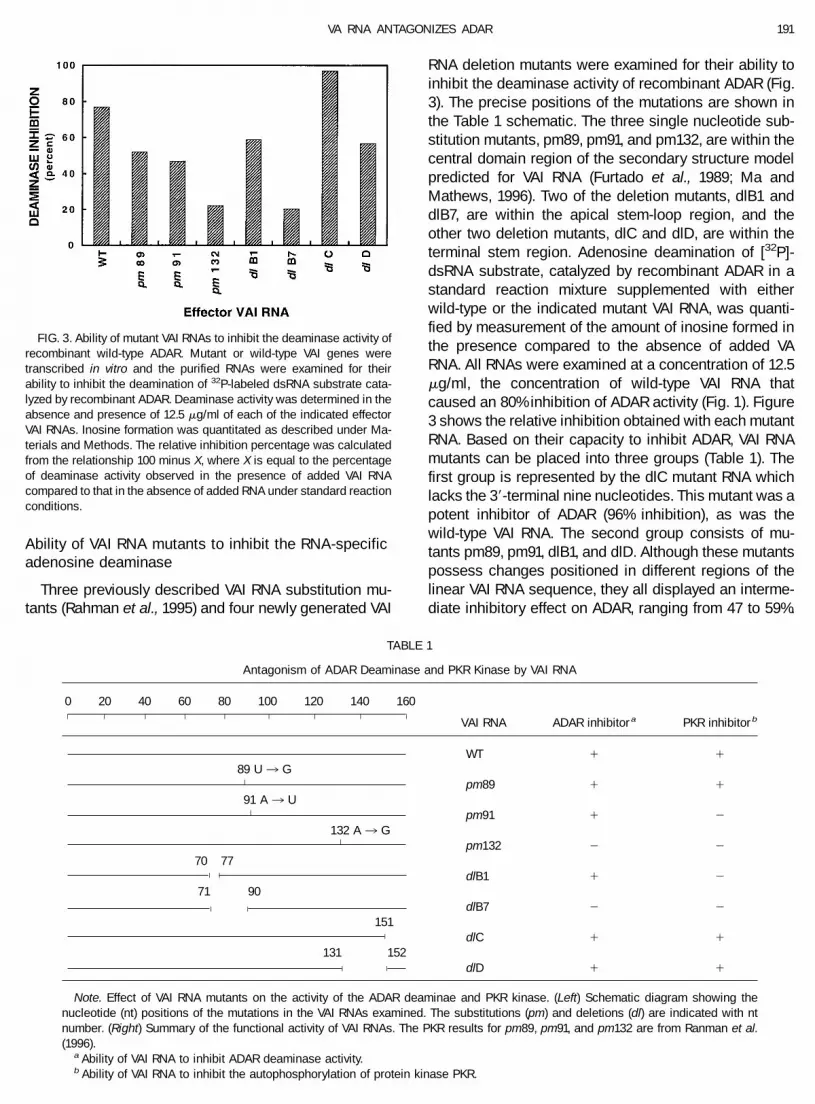
Ability of VAI RNA mutants to inhibit the RNA-specificadenosine deaminase
Three previously described VAI RNA substitution mu-tants (Rahman et al., 1995) and four newly generated VAI
RNA deletion mutants were examined for their ability toinhibit the deaminase activity of recombinant ADAR (Fig.3). The precise positions of the mutations are shown inthe Table 1 schematic. The three single nucleotide sub-stitution mutants, pm89, pm91, and pm132, are within thecentral domain region of the secondary structure modelpredicted for VAI RNA (Furtado et al., 1989; Ma andMathews, 1996). Two of the deletion mutants, dlB1 anddlB7, are within the apical stem-loop region, and theother two deletion mutants, dlC and dlD, are within theterminal stem region. Adenosine deamination of [32P]-dsRNA substrate, catalyzed by recombinant ADAR in astandard reaction mixture supplemented with eitherwild-type or the indicated mutant VAI RNA, was quanti-fied by measurement of the amount of inosine formed inthe presence compared to the absence of added VARNA. All RNAs were examined at a concentration of 12.5mg/ml, the concentration of wild-type VAI RNA thatcaused an 80% inhibition of ADAR activity (Fig. 1). Figure3 shows the relative inhibition obtained with each mutantRNA. Based on their capacity to inhibit ADAR, VAI RNAmutants can be placed into three groups (Table 1). Thefirst group is represented by the dlC mutant RNA whichlacks the 39-terminal nine nucleotides. This mutant was apotent inhibitor of ADAR (96% inhibition), as was thewild-type VAI RNA. The second group consists of mu-tants pm89, pm91, dlB1, and dlD. Although these mutantspossess changes positioned in different regions of thelinear VAI RNA sequence, they all displayed an interme-diate inhibitory effect on ADAR, ranging from 47 to 59%.
FIG. 3. Ability of mutant VAI RNAs to inhibit the deaminase activity ofrecombinant wild-type ADAR. Mutant or wild-type VAI genes weretranscribed in vitro and the purified RNAs were examined for theirability to inhibit the deamination of 32P-labeled dsRNA substrate cata-lyzed by recombinant ADAR. Deaminase activity was determined in theabsence and presence of 12.5 mg/ml of each of the indicated effectorVAI RNAs. Inosine formation was quantitated as described under Ma-terials and Methods. The relative inhibition percentage was calculatedfrom the relationship 100 minus X, where X is equal to the percentageof deaminase activity observed in the presence of added VAI RNAcompared to that in the absence of added RNA under standard reactionconditions.
TABLE 1
Antagonism of ADAR Deaminase and PKR Kinase by VAI RNA
0 20 40 60 80 100 120 140 160
VAI RNA ADAR inhibitor a PKR inhibitor b
WT 1 189 U 3 G
pm89 1 191 A 3 U
pm91 1 2132 A 3 G
pm132 2 270 77
dlB1 1 271 90
dlB7 2 2151
dlC 1 1131 152
dlD 1 1
Note. Effect of VAI RNA mutants on the activity of the ADAR deaminae and PKR kinase. (Left) Schematic diagram showing thenucleotide (nt) positions of the mutations in the VAI RNAs examined. The substitutions (pm) and deletions (dl) are indicated with ntnumber. (Right) Summary of the functional activity of VAI RNAs. The PKR results for pm89, pm91, and pm132 are from Ranman et al.(1996).
a Ability of VAI RNA to inhibit ADAR deaminase activity.b Ability of VAI RNA to inhibit the autophosphorylation of protein kinase PKR.
Ç Ç Ç Ç Ç Ç Ç Ç ÇÇ
ÇÇ
Ç ÇÇ Ç Ç
Ç Ç
191VA RNA ANTAGONIZES ADAR

The third group of mutant RNAs includes pm132 anddlB7. Their ability to inhibit ADAR was severely dimin-ished, causing only a 22% reduction or less in deaminaseactivity (Fig. 3).
Comparison of the effects of VAI RNA onRNA-specific adenosine deaminase ADARand RNA-dependent protein kinase PKR
Previous studies established that the central domainof VAI RNA is of critical importance in the antagonism ofactivation of the RNA-dependent protein kinase (PKR) byAd VAI RNA (Furtado et al., 1989; Clarke and Mathews,1995; Green et al., 1995; Rahman et al., 1995). Threesite-directed substitution mutations in the central domainregion of VAI RNA have differential effects on PKR acti-vation: point mutant pm89 inhibits PKR autophosphory-lation, whereas pm91 and pm132 do not antagonize PKRactivation (Rahman et al., 1995). Our results, summarizedby Table 1, reveal that these three VAI RNA point mutantsaffect ADAR function differently from how they affect PKRactivation. For example, pm89 can inhibit PKR activationas efficiently as WT VAI RNA (Rahman et al., 1995), butthe ability of pm89 to inhibit ADAR is reduced relative tothat of WT VAI RNA. Mutant pm91 cannot block PKR, butit retains its capacity to inhibit ADAR. Only pm132 losesits ability to block both PKR and ADAR, possibly becausethe VAI RNA secondary structure involving the centraldomain may be substantially altered by the A to G nu-cleotide substitution (Rahman et al., 1995).
We generated four deletion mutants of VAI RNA. Two ofthem are within the apical duplex region, and the othertwo are in the lower duplex region. Mutant dlB1 pos-sesses a six-nucleotide deletion within the apical stemnear to the upper loop; this mutant retains the capacity toefficiently inhibit ADAR activity, but it is a poor antagonistof PKR. Mutant dlB7 possesses a much larger deletion(18 nt) within the apical stem, extending from the apicalloop to the central domain; this mutation most likelyaffects the secondary structure of the apical stem as wellas the central domain structure. Mutant dlB7 does notinhibit either ADAR or PKR. Mutant dlC is a 39-truncationmutant in which the terminal nine nucleotides involved inthe base-paired terminal stem of VAI RNA are deleted.The dlC mutant blocks PKR as efficiently as WT VAI RNAand ADAR even somewhat more efficiently than WT VARNA. Mutant dlD possesses a large (20-nt) internal de-letion within the terminal stem region of VAI RNA, ex-tending from the central domain to the bottom of thestem. This mutant VAI RNA inhibited both ADAR and PKR.
DISCUSSION
The double-stranded RNA adenosine deaminase,ADAR, is an interferon-inducible RNA-editing enzyme(Patterson and Samuel, 1995; Patterson et al., 1995)which catalyzes the C-6 deamination of adenosine to
yield inosine within RNA (Bass, 1997). The resultsreported herein establish that adenovirus VAI RNAimpairs the deaminase activity of ADAR. This findinghas important implications related to the antiviral ac-tions of interferon and antisense-mediated inhibitionof gene expression.
Adenovirus VAI RNA is a ;160-nt regulatory RNAfound in abundant amounts in the cytoplasm of infectedcells at late times after infection (Mathews and Shenk,1991). Our results establish that VAI RNA can inhibitADAR deaminase activity. Both natural ADAR present inextracts from IFN-treated human U cells (Fig. 1) andrecombinant ADAR expressed in transfected monkeyCOS cells (Fig. 2) were inhibited by VAI RNA. An ;1000-fold molar excess of wild-type VA RNA over the syntheticdsRNA substrate was required to observe maximal inhi-bition of deaminase activity. Very little is known aboutnaturally occurring RNAs that may be targets for adeno-sine deamination by ADAR and thus in vivo candidatesfor impairment of the deamination by adenovirus VA RNAat late times in infection. Conceivably the relative differ-ence in concentration or binding affinity between targetsubstrates and VA RNA in vivo are less or, alternatively,additional interacting proteins may play a role in vivo inmodulating the relative affinity of the ADAR deaminasefor substrate RNAs versus effector RNAs.
The principle function previously elucidated for VAIRNA relates to its ability to antagonize the activation ofPKR, the RNA-dependent inhibitor of translation(Mathews and Shenk, 1991; Samuel, 1993). PKR plays akey role in the antiviral and antiproliferative actions ofinterferon (Samuel, 1991; Lengyel, 1993). PKR, like ADAR,is an IFN-inducible enzyme (Samuel, 1993; Clemens,1996). Indeed, VAI RNA is the best characterized RNAantagonist of the PKR kinase. It is well-established thatadenoviruses are relatively resistant to the antiviral ac-tions of interferon (Stewart, 1979). This resistance is due,at least in part, to the antagonism of PKR function by VAIRNA. PKR possesses two copies of the dsRBM motif thatpossesses RNA-binding activity (McCormack et al., 1992,1995; Green and Mathews, 1992). VAI RNA binds to PKRvia dsRBM motifs and prevents the subsequent auto-phosphorylation and activation of PKR kinase activity bydsRNA activators (Kitajewski et al., 1986; O’Malley et al.,1986; Mathews and Shenk, 1991; Samuel, 1993).
How could VAI RNA further antagonize the antiviralaction of interferon? ADAR possesses three copies ofthe highly conserved dsRBM motif which are responsiblefor binding of the substrate RNAs for deamination (Kimet al., 1994b; Patterson and Samuel, 1995; O’Connell etal., 1995; Liu et al., 1997). In addition to dsRNA sub-strates, ADAR also catalyzes the deamination of selec-tive adenosine residues in structured viral RNAs andcellular pre-mRNAs (Bass, 1997). VA RNA could impairADAR activity by either competing with the binding of thesubstrate RNA or alternatively by affecting the conforma-
192 LEI, LIU, AND SAMUEL

tion of ADAR in such a manner that abrogates deamina-tion of bound substrate RNA. Interestingly, a ribonucle-ase specific for inosine-containing RNA has been iden-tified in HeLa cells (Scadden and Smith, 1997). TheIFN-inducible ADAR deaminase (Patterson et al., 1995)may conceivably work in concert with the RNase-I spe-cific for inosine-containing RNAs (Scadden and Smith,1997), analogous to the manner in which the IFN-induc-ible 29,59-oligoadenylate synthetase functions to producethe 29,59-oligoadenylate activator of the constitutively ex-pressed ribonuclease L (Samuel, 1991; Zhou et al., 1993).The antagonism of the IFN-induced ADAR deaminase byVAI RNA would be expected to reduce the formation ofinosine-containing RNA transcripts and thus impair theformation of potential substrates for RNase-I.
From a comparison of our results for the VAI-mediatedantagonism of ADAR with those observed for PKR, twoimportant points emerge. First, our results obtained withmutants of VAI RNA suggest that the central domain of VAIRNA is essential for its ability to inhibit ADAR and that theapical duplex stem may be more important than the termi-nal stem. The central domain and an adjacent region in theapical stem are likewise important for the binding of VARNA to PKR and for the functional impairment of PKRactivity (Furtado et al., 1989; Clarke et al., 1994; Green et al.,1995; Rahman et al., 1995). Second, the interactions of VAIwith ADAR and PKR, while similar, are not equivalent. Forexample, pm91 and dlB1 do not significantly inhibit PKR, butboth of these mutants inhibit ADAR reasonably well (Table1). The basis for this difference is unknown, but may berelated to the fact that ADAR possesses three copies of thedsRBM motif, whereas PKR has only two copies. Althoughthe sequences of the dsRBM motifs are highly conserved,the individual motifs do display functional differences inADAR (Lai et al., 1995; Liu and Samuel, 1996) and PKR(McCormack et al., 1995; Green et al., 1995). Because thecentral domain of VAI RNA does not possess a double-stranded stem as found in perfectly paired dsRNA, but yetwe observe that the central domain of VAI RNA is importantfor the inhibition of ADAR deaminase activity, it is not un-expected that VAI RNA is not a substrate of ADAR.
We did not observe an inhibitory effect of TAR RNA onADAR activity, even though TAR RNA is bound by ADAR(Patterson and Samuel, 1995) and is reported to bedeaminated in a Tat-dependent manner (Sharmeen et al.,1991). The lack of inhibition of ADAR by TAR may relate tothe fact that ADAR possesses three copies of the dsRBM(Kim et al., 1994b; Patterson and Samuel, 1995). Mu-tagenesis studies have revealed that dsRBM3 is abso-lutely essential for deamination of a synthetic dsRNAsubstrate, whereas dsRBM1 and dsRBM2 are not (Liuand Samuel, 1996; Liu et al., 1997). For the PKR kinasewhich possesses two copies of the dsRBM, binding ofRNA does not necessarily correlate with function. Forexample, in vitro selection experiments have identifiedmultiple RNA sequences that bind to the dsRBM of PKR
with comparable Kd values; some selected RNAs func-tion as agonists or antagonists, whereas others do notsignificantly affect PKR activity (Bevilacqua et al., 1998).
The replicative form of single-stranded RNA virus ge-nomes (Cattaneo, 1994) or symmetrical transcripts ofdouble-stranded DNA virus genomes such as adenovi-rus (Maran and Mathews, 1988) may serve as potentialsources of double-stranded RNA in virus-infected cellsand thus targets for the biased hypermutation via deami-nation by ADAR. Antisense-induced regulation of tran-script levels via extensive adenosine deamination hasbeen reported for polyoma virus (Kumar and Carmichael,1997). Site-selective editing of adenovirus mRNA tran-scripts would provide another potential target for ADAR,similar to the highly selective editing established for theGluR and 5HT2c cellular pre-mRNAs (Lomeli et al., 1994;Burns et al., 1997). Although not yet established for ad-enovirus-infected cells, it is conceivable that either post-transcriptional selective editing of adenovirus mRNA oran antisense-mediated inhibition of gene expression, orboth, occurs during adenovirus multiplication. VAI RNA,which accumulates to high concentrations at late timesafter infection, would have the potential to modulate suchediting by ADAR.
Although extensive evidence from several laboratorieshas unequivocally established that VAI RNA functions toenhance protein synthesis, by a mechanism that involvesthe antagonism of the translation inhibitor PKR (Mathewsand Shenk, 1991), this does not preclude other additionalroles for VAI RNA. For example, it has been reported thatVAI RNA has a role in controlling mRNA stability (Strijkeret al., 1989). VAI RNA also has been found to increasecytoplasmic RNA abundance in an RNA length-depen-dent manner (Svensson and Akusjarvi, 1990). It is now ofutmost importance to establish the functional signifi-cance of the antagonism of the ADAR deaminase by VAIRNA, in the context of both the regulation of adenovirusgene expression and the antiviral actions of interferon.
EXPERIMENTAL PROCEDURES
Cell culture, interferon treatment, and transfection
Human amnion U cells, either untreated or treated withIFN-a (1000 units/ml), were maintained in Dulbecco’s mod-ified Eagle’s medium (DMEM) supplemented with 5% (v/v)fetal bovine serum (FBS, Hyclone) as previously described(Patterson and Samuel, 1995). Monkey kidney COS cellsmaintained in DMEM supplemented with 10% (v/v) FBSwere used to express recombinant ADAR proteins, eitherwild-type ADAR (Patterson and Samuel, 1995) or mutantADAR with substitutions in copy I, II, or III of the dsRBM(previously designated RI, RII, or RIII mutants) (Liu and Sam-uel, 1996). Transfections with the ADAR cDNAs using thepcDNA I/neo expression vector were by the DEAE–dextran/chloroquine phosphate method (Luthman and Magnusson,1983) using 5 mg of DNA/ml when the cultures in 60-mm
193VA RNA ANTAGONIZES ADAR

dishes were about 70% confluent. Cells were harvested 48to 60 h after transfection and extracts were prepared aspreviously described (Patterson and Samuel, 1995; Liu etal., 1997). ADAR protein expression was monitored by West-ern immunoblot analysis as described previously, usingantiserum generated against recombinant ADAR proteinexpressed in Escherichia coli (Patterson and Samuel, 1995).
Adenovirus VAI RNA plasmid constructions and invitro transcription
The pT7VA plasmid with the wild-type adenovirus type 2VAI RNA gene cloned downstream of the T7 promoter(Mellits et al., 1990) was generously provided by Dr. Mi-chael Mathews (Cold Spring Harbor Laboratory, New York).The pT7TAR plasmid was as previously described (McCor-mack et al., 1992). Single-base substitution mutants pm89,pm91, and pm132 of the Ad5 VAI RNA gene (Rahman et al.,1995) were kindly provided by Dr. Bayar Thimmapaya(Northwestern University Medical School). Deletion mu-tants dlB1, dlB7, dlC, and dlD were generated from plasmidpT7VA. All DNA manipulations were carried out by standardrecombinant DNA technology (Sambrook et al., 1989). Togenerate dlB1 and dlB7, pT7VA was linearized with BamHIand then treated with S1 nuclease. Recessed ends wereblunted with T4 DNA polymerase prior to plasmid closureusing T4 DNA ligase. dlC was generated by digestion ofpT7VA with Eco47III prior to in vitro transcription. dlD wasgenerated by digestion with AatII and EcoR47III to deletethe 20-nt AatII–EcoR47III fragment and treated with T4 DNApolymerase to remove the 39 overhangs, and then thegel-purified plasmid fragment was reclosed using T4 DNAligase. All VAI RNA mutations were confirmed by directsequence determination by the dideoxy chain-terminationmethod (Sanger et al., 1977). For in vitro transcription, plas-mid DNA constructions (5 mg) were linearized as follows:wild-type VA plasmid construction, with DraI or PstI; dlB1,dlB7, and dlD, with DraI; dlC, with Eco47III; and, pm89,pm91, and pm132 with EcoRI. Transcription was with T7RNA polymerase (Promega) according to the manufactur-er’s instructions. In vitro transcripts were subjected to or-ganic solvent extraction, ethanol precipitation, and gel elec-trophoresis, and they displayed the anticipated size char-acteristics (McCormack et al., 1992; Kitajewski et al., 1986).
Double-stranded RNA-specific adenosine deaminaseassay
The assay conditions for measurement of ADAR activ-ity using 32P-labeled dsRNA substrate were as describedpreviously (Patterson and Samuel, 1995; Liu and Samuel,1996). The standard reaction mixture (40 ml) containedabout 10 fmol of synthetic 32P-labeled dsRNA substrateand varying amounts of cytoplasmic extract protein asindicated in 50 mM Tris–HCl buffer, 100 mM KCl, 5 mMEDTA, 10% (v/v) glycerol, 1 mM DTT, and 0.5 mM phenyl-methylsulfonyl fluoride. Where noted, VAI RNA was
added to the reaction mixture and preincubated on icewith the ADAR-containing extract prior to addition of the32P-labeled dsRNA substrate. The complete reactionmixture was incubated at 30°C for 2 h and then extractedwith phenol–chloroform and chloroform. RNA was recov-ered by ethanol precipitation in the presence of 0.5 mg ofpoly(rI) (Sigma) carrier RNA, washed with 70% ethanol,dried, and suspended in 10 ml of nuclease digestionbuffer (30 mM KOAc, pH 5.3, 10 mM ZnSO4) containing1.5 mg of P1 nuclease (Pharmacia Biotech, Inc.). Incuba-tion was at 50°C for 2 h. IMP and AMP were resolvedfrom each other by thin-layer chromatography (TLC) oncellulose NM300 glass plates (Macherey and Nagel) in asolvent consisting of saturated (NH4)2SO4, 100 mM NaAc(pH 6.0), and isopropanol (79:19:2). Radioactivity corre-sponding to IMP and AMP was quantified using a Beck-man LS1801 liquid scintillation system. Quantitated val-ues from three independent experiments are shown withstandard error determinations.
Double-stranded RNA-dependent protein kinase PKRassay
Procedures for measurement of the VAI RNA-mediatedantagonism in vitro of the [g-32P]ATP-mediated autophos-phorylation of PKR were essentially as described beforein detail (Samuel et al., 1986; Kitajewski et al., 1986). Thestandard reaction mixture (25 ml) included HPLC-purifiedhuman PKR protein, eIF-2 (0.25 mg), and VAI RNA (0.5 mg)as indicated, which were preincubated prior to additionof synthetic poly(rI):poly(rC) dsRNA and [g-32P]ATP. Thecomplete reaction mixture was then incubated at 30°Cfor 15 min. 32P-labeled protein products were fraction-ated by SDS–polyacrylamide gel electrophoresis andquantified as previously described (Samuel et al., 1986).
ACKNOWLEDGMENT
This work was supported in part by Research Grant AI-12520 fromthe National Institute of Allergy and Infectious Diseases, NationalInstitutes of Health.
REFERENCES
Bass, B. L. (1997). RNA editing and hypermutation by adenosine deami-nation. Trends Biochem. Sci. 22, 157–162.
Bevilacqua, P. C., George, C. X., Samuel, C. E., and Cech, T. R. (1998).Binding of the protein kinase PKR to RNAs with secondary structuredefects: Role of the tandem A-G mismatch and noncontiguous he-lixes. Biochemistry 37, 6303–6316.
Burns, C. M., Chu, H., Rueter, S. M., Hutchinson, L. K., Canton, H.,Sanders-Bush, E., and Emeson, R. B. (1997). Regulation of seroto-nin-2C receptor G-protein coupling by RNA editing. Nature 387,303–308.
Cattaneo, R. (1994). Biased (A3 I) hypermutation of animal RNA virusgenomes. Curr. Opin. Genet. Dev. 4, 895–900.
Cattaneo, R., Schmid, A., Eschle, D., Baczko, K., ter Meulen, V., andBilleter, M. A. (1988). Biased hypermutation and other geneticchanges in defective measles viruses in human brain infections. Cell55, 255–265.
194 LEI, LIU, AND SAMUEL

Clarke, P. A., and Mathews, M. B. (1995). Interactions between thedouble-stranded RNA binding motif and RNA: definition of the bind-ing site for the interferon-induced protein kinase DAI (PKR) on ade-novirus VA RNA. RNA 1, 7–20.
Clemens, M. J. (1996). Protein kinases that phosphorylate eIF-2 andeIF-2B, and their role in eukaryotic cell translational control. In‘‘Translational Control,’’ pp. 139–172. Cold Spring Harbor LaboratoryPress, Cold Spring Harbor, NY.
Furtado, M. R., Subramanian, S., Bhat, R. A., Fowlkes, D. M., Safer, B.,and Thimmappaya, B. (1989). Functional dissection of adenovirus VAIRNA. J. Virol. 63, 3423–3434.
Green, S. R., and Mathews, M. B. (1992). Two RNA binding motifs in thedouble-stranded RNA activated protein kinase, DAI. Genes Dev. 6,2478–2490.
Green, S. R., Manche, L., and Mathews, M. B. (1995). Two functionallydistinct RNA binding motifs in the regulatory domain of the proteinkinase, DAI. Mol. Cell Biol. 15, 358–364.
Hough, R. F., and Bass, B. L. (1994). Purification of the Xenopus laevisdouble-stranded RNA adenosine deaminase. J. Biol. Chem. 269,9933–9939.
Kim, U., Garner, T. L., Sanford, T., Speicher, D., Murray, J. M., andNishikura, K. (1994a). Purification and characterization of double-stranded RNA adenosine deaminase from bovine nuclear extracts.J. Biol. Chem. 269, 13480–13489.
Kim, U., Wang, Y., Sanford, T., Zeng, Y., and Nishikura, K. (1994b).Molecular cloning of cDNA for double-stranded RNA adenosinedeaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl.Acad. Sci. USA 91, 11457–11461.
Kitajewski, J., Schneider, R. J., Safer, B., Munemitsu, S. M., Samuel, C. E.,Thimmappaya, B., and Shenk, T. (1986). Adenovirus VAI RNA antag-onizes the antiviral action of interferon by preventing activation of theinterferon-induced eIF-2 alpha kinase. Cell 45, 195–200.
Kuhen, K. L., Shen, X., Carlisle, E. R., Richardson, A. L., Weier, H. U. G.,Tanaka, H., and Samuel, C. E. (1996). Mechanism of interferon action.Structural organization of the human Pkr Gene encoding an interfer-on-inducible RNA-dependent protein kinase (PKR) and differencesfrom its mouse homolog. Genomics 36, 197–201.
Kumar, M., and Carmichael, G. G. (1997). Nuclear antisense RNAinduces extensive adenosine modifications and nuclear retention oftarget transcripts. Proc. Natl. Acad. Sci. USA 94, 3542–3547.
Lai, F., Drakas, R., and Nishikura, K. (1995). Mutagenic analysis ofdouble-stranded RNA adenosine deaminase, a candidate enzyme forRNA editing of glutamate-gated ion channel transcripts. J. Biol.Chem. 270, 17098–17105.
Lengyel, P. (1993). Tumor-suppressor genes: News about the interferonconnection. Proc. Natl. Acad. Sci. USA 90, 5893–5895.
Liu, Y., and Samuel, C. E. (1996). Mechanism of interferon action:functionally distinct RNA-binding and catalytic domains in the inter-feron-inducible, double-stranded RNA-specific adenosine deami-nase. J. Virol. 70, 1961–1968.
Liu, Y., George, C. X., Patterson, J. B., and Samuel, C. E. (1997). Func-tionally distinct double-stranded RNA-binding domains associatedwith alternative splice-site variants of the interferon-inducible dou-ble-stranded RNA-specific adenosine deaminase. J. Biol. Chem. 272,4419–4428.
Lomeli, H., Mosbacher, J., Melcher, T., Hoger, T., Geiger, J. R. P., Kuner,T., Monyer, H., Higuchi, M., Bach, A., and Seeburg, P. H. (1994).Control of kinetic properties of AMPA receptor channels by nuclearRNA editing. Science 266, 1709–1713.
Luthman, H., and Magnusson, G. (1983). High efficiency polyoma DNAtransfection of chloroquine-treated cells. Nucleic Acids Res. 11,1295–1308.
Ma, Y., and Mathews, M. B. (1996a). Structure, function, and evolution ofadenovirus-associated RNA: A phylogenetic approach. J. Virol. 70,5083–5099.
Ma, Y., and Mathews, M. B. (1996b). Secondary and tertiary structure inthe central domain of adenovirus type 2 VA RNA I. RNA 2, 937–951.
Maran, A., and Mathews, M. B. (1988). Characterization of the double-stranded RNA implicated in the inhibition of protein synthesis in cellsinfected with a mutant adenovirus defective for VA RNA. Virology 164,106–113.
Mathews, M. B., and Shenk, T. (1991). Adenovirus virus-associated RNAand translational control. J. Virol. 65, 5657–5662.
McCormack, S. J., and Samuel, C. E. (1995). Mechanism of interferonaction. RNA-binding activity of full-length and R-domain forms of theRNA-dependent protein kinase PKR—Determination of KD values forVAI and TAR RNAs. Virology 206, 511–519.
McCormack, S. J., Thomis, D. C., and Samuel, C. E. (1992). Mechanismof interferon action: Identification of a RNA binding domain within theN-terminal region of the human RNA-dependent P1/eIF-2a proteinkinase. Virology 188, 47–56.
Meurs, E., Chong, K., Galabru, J., Thomas, N. S., Kerr, I. M., Williams,B. R. G., and Hovanessian, A. G. (1990). Molecular cloning andcharacterization of the human double-stranded RNA-activated pro-tein kinase induced by interferon. Cell 62, 379–390.
Mellits, K. H., Kostura, M., and Mathews, M. B. (1990). Interaction ofadenovirus VA RNAI with the protein kinase DAI-nonequivalence ofbinding and function. Cell 61, 843–852.
Nishikura, K., Yoo, C., Kim, U., Murray, J. M., Estes, P. A., Cash, F. E., andLiebhaber, S. A. (1991). Substrate specificity of the dsRNA unwinding/modifying activity. EMBO J. 10, 3523–3532.
O’Connell, M. A., and Keller, W. (1994). Purification and properties ofdouble-stranded RNA-specific adenosine deaminase from calf thy-mus. Proc. Natl. Acad. Sci. USA 91, 10596–10600.
O’Connell, M. A., Krause, S., Higuchi, M., Hsuan, J. J., Totty, N. F., Jenny,A., and Keller, W. (1995). Cloning of cDNAs encoding mammaliandouble-stranded RNA-specific adenosine deaminase. Mol. Cell. Biol.15, 1389–1397.
O’Malley, R. P., Mariano, T. M., Siekierka, J., and Mathews, M. B. (1986).A mechanism for the control of protein synthesis by adenovirus VARNAI. Cell 44, 391–400.
Pathak, V. K., Schindler, D., and Hershey, J. W. B. (1988). Generation ofa mutant form of protein synthesis initiation factor eIF-2 lacking thesite of phosphorylation by eIF-2 kinases. Mol. Cell. Biol. 8, 993–995.
Patterson, J. B., Thomis, D. C., Hans, S. L., and Samuel, C. E. (1995).Mechanism of interferon action. Double-stranded RNA-specificadenosine deaminase from human cells is inducible by alpha andgamma interferons. Virology 210, 508–511.
Patterson, J. B., and Samuel, C. E. (1995). Expression and regulation byinterferon of a double-stranded RNA-specific adenosine deaminasefrom human cells: Evidence for two forms of the deaminase. Mol.Cell. Biol. 15, 5376–5388.
Polson, A. G., and Bass, B. L. (1994). Preferential selection of ad-enosines for modification by double-stranded RNA adenosinedeaminase. EMBO J. 13, 5701–5711.
Polson, A. G., Bass, B. L., and Casey, J. L. (1996). RNA editing ofhepatitis delta virus antigenome by dsRNA-adenosine deaminase.Nature 380, 454–456.
Rahman, A., Malhotra, P., Dhar, R., Kewalramani, T., and Thimmapaya,B. (1995). Effect of single-base substitutions in the central domain ofvirus-associated RNA I on its function. J. Virol. 69, 4299–4307.
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). ‘‘Molecular Cloning:A Laboratory Manual,’’ 2nd ed. Cold Spring Harbor Laboratory Press,Plainview, NY.
Samuel, C. E. (1979). Mechanism of Interferon Action. Phosphorylationof protein synthesis initiation factor eIF-2 in interferon-treated humancells by a ribosome-associated protein kinase possessing site-spec-ificity similar to hemin-regulated rabbit reticulocyte kinase. Proc.Natl. Acad. Sci. USA 76, 600–604.
Samuel, C. E. (1991). Antiviral actions of interferon. Interferon-regulatedcellular proteins and their surprisingly selective antiviral activities.Virology 183, 1–11.
Samuel, C. E. (1993). The eIF-2a protein kinases, regulators of trans-
195VA RNA ANTAGONIZES ADAR

lation in eukaryotes from yeasts to humans. J. Biol. Chem. 268,7603–7606.
Samuel, C. E., Knutson, G. S., Berry, M. J., Atwater, J. A., and Lasky, S. R.(1986). Purification of double stranded RNA-dependent protein ki-nase from mouse fibroblasts. Methods Enzymol. 119, 499–516.
Sanger, F., Nicklen, S., and Coulson, A. R. (1977). DNA sequencing withchain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74, 5463–5467.
Scadden, A. D. J., and Smith, C. W. J. (1997). A ribonuclease specific forinosine-containing RNA: A potential role in antiviral defence? EMBOJ. 16, 2140–2149.
Schneider, R. J., Weinberger, C., and Shenk, T. (1984). Adenovirus VAIRNA facilitates the initiation of translation in virus-infected cells. Cell37, 291–298.
Sharmeen, L., Bass, B., Sonenberg, N., Weintraub, H., and Groudine, M.(1991). Tat-dependent adenosine-to-inosine modification of wild-typetransactivation response RNA. Proc. Natl. Acad. Sci. USA 88, 8096–8100.
Soderlund, H., Pettersson, U., Vennstrom, B., Philipson, L., andMathews, M. B. (1976). A new species of virus-coded low molecularweight RNA from cells infected with adenovirus type 2. Cell 7,585–593.
Sommer, B., Kohler, M., Sprengel, R., and Seeburg, P. H. (1991). RNAediting in brain controls a determinant of ion flow in glutamate-gatedchannels. Cell 67, 11–19.
Stewart, W. E. (1979). ‘‘The Interferon System.’’ Springer-Verlag, Berlin/New York.
St. Johnston, D., Brown, N. H., Gall, J. G., and Jantsch, M. (1992). Aconserved double-stranded RNA-binding domain. Proc. Natl. Acad.Sci. USA 89, 10979–10983.
Strijker, R., Fritz, D. T., and Levinson, A. D. (1989). Adenovirus VAI-RNAregulates gene expression by controlling stability of ribosome-boundRNAs. EMBO J. 8, 2669–2675.
Svensson, C., and Akusjarvi, G. (1990). A novel effect of adenovirus VARNAI on cytoplasmic mRNA abundance. Virology 174, 613–617.
Tanaka, H., and Samuel, C. E. (1994). Mechanism of interferon action:Structure of the mouse PKR gene encoding the interferon-inducibleRNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 91, 7995–7999.
Thomis, D. C., Doohan, J. P., and Samuel, C. E. (1992). Mechanism ofinterferon action: cDNA structure, expression and regulation of theinterferon-induced, RNA-dependent Pl/eIF-2a protein kinase fromhuman cells. Virology 188, 33–46.
Weier, H-U. G., George, C. X., Greulich, K. M., and Samuel, C. E. (1995).The interferon-inducible, double-stranded RNA-specific adenosinedeaminase gene (DSRAD) maps to human chromosome 1q21.1–21.2.Genomics 30, 372–375.
Wong, T. C., Ayata, M., Hirano, A., Yoshikawa, Y., Tsuruoka H., andYamanouchi, K. (1989). Generalized and localized biased hypermu-tation affecting the matrix gene of a measles virus strain that causessubacute sclerosing panencephalitis. J. Virol. 63, 5464–5468.
Zhou, A., Hassel, B. A., and Silverman, R. H. (1993). Expression cloningof 2-5A-dependent RNAase: A uniquely regulated mediator of inter-feron action. Cell 72, 753–765.
196 LEI, LIU, AND SAMUEL