activity reversal of tet repressor caused by single amino acid exchanges
TRANSCRIPT

Molecular Microbiology (2004)
53
(3), 777–789 doi:10.1111/j.1365-2958.2004.04159.x
© 2004 Blackwell Publishing Ltd
Blackwell Science, LtdOxford, UKMMIMolecular Microbiology0950-382XBlackwell Publishing Ltd, 2004
? 2004
53
3777789
Original Article
Reverse TetRO. Scholz
et al
.
Accepted 2 April, 2004. *For correspondence. E-mail [email protected]; Tel. (
+
49) 9131/85 28081; Fax (
+
49) 9131/85 28082.
Activity reversal of Tet repressor caused by single amino acid exchanges
Oliver Scholz,
1
Eva-Maria Henßler,
1
Johannes Bail,
1
Peter Schubert,
2
Joanna Bogdanska-Urbaniak,
1
Sabine Sopp,
1
Marco Reich,
1
Stefanie Wisshak,
1
Martin Köstner,
1
Ralph Bertram
1
and
Wolfgang Hillen
1
*
1
Lehrstuhl für Mikrobiologie, Institut für Mikrobiologie,
Biochemie und Genetik, Friedrich-Alexander Universität
Erlangen-Nürnberg, Staudtstraße 5, 91058 Erlangen,
Germany.
2
Biomedical Research Centre, Department of Medicine,
University of British Columbia, 2222 Health Sciences Mall,
Vancouver, BC, V6T 1Z3, Canada.
Summary
We explore by extensive mutagenesis regions in the
sequence allowing reversal of the allosteric response
of Tet repressor. The wild type requires anhydrotetra-
cycline for induction. About 100 mutants are pre-
sented, which, in contrast, require the drug for
repression. Their mutations are clustered at the inter-
face of the DNA- and inducer-binding domains. This
interface consists of a central hydrophobic region
surrounded by several hydrogen bonds. While most
of the mutants described here contain two to five
mutations, we found five positions in this region of
TetR, at which single amino acid exchanges lead to
activity reversal. They may disrupt the hydrogen-
bonding network bordering the domain interface. We
assume that the mutations cause a repositioning of
the DNA reading head with respect to the effector
binding core so that the same conformational change
can result in opposite activities.
Introduction
Ten years ago, Rose and Creamer (1994) proposed theso-called ‘Paracelsus challenge’ by asking what the mini-mum number of amino acids required to specify a proteinfold of the
a
-helix or the
b
-sheet-type would be. Subse-quently, three groups accomplished changing the foldingtype of a protein with retention of at least 50% of its aminoacids (Jones
et al
., 1996; Dalal
et al
., 1997; Yuan and
Clarke, 1998). Their results had a strong impact on ourknowledge of protein folding and especially for proteinsthat easily adopt alternative folds such as the amyloidprecursor protein (APP). As structure determines thefunction of proteins, the ‘functional Paracelsus question’would be how many mutations are required to change (notjust lose) the function of a given protein. We present herean example in which one single amino acid exchange issufficient for inversion of protein function. Moreover, thischange at one residue can take place at any of five aminoacids.
Tet repressor (TetR) is an allosteric DNA-binding protein(Hillen and Berens, 1994). It is of practical interestbecause of its applications for regulating genes in virtuallyall organisms (Gossen and Bujard, 2001; Berens andHillen, 2003). Crystal structures of the active forms andthe free protein have been elucidated (Hinrichs
et al
.,1994; Kisker
et al
., 1995; Orth
et al
., 1998; 2000), andpowerful screens have already led to many mutants withaltered activities (Helbl and Hillen, 1998; Helbl
et al
.,1998; Scholz
et al
., 2003). We report here an extensiveset of new mutations in TetR, which affect the allostery ofthat protein in a dramatic way, namely turning the induceranhydrotetracycline (atc) into a co-repressor.
TetR, a homodimeric protein built up of 10
a
-helices ineach subunit (see Fig. 1A), is one of the best investigatedprokaryotic effector-dependent regulatory proteins. Itbinds to
tet
operator with high specificity, while tetracycline(tc) binding leads to induction (Hinrichs
et al
., 1994; Kisker
et al
., 1995; Orth
et al
., 1998; 2000). Helices
a
1 to
a
3include a helix–turn–helix (HTH) motif and form the DNA-binding domains. The core domain consists of helices
a
5to
a
10 from both subunits and harbours two tc bindingpockets as well as the dimer interface. The two DNA-binding domains are each connected to the core domainvia helix
a
4. Based on the comparison of operator-boundand induced conformations of TetR, a model for the con-formational transition accompanying induction has beenproposed (Orth
et al
., 2000). The induction signal needsto be conducted over a 33 Å distance through the TetRprotein from the tc binding pocket to the
tetO
bindingsurface. The structural changes affect helices
a
4 and
a
6and result in a pendulum-like motion of the DNA-bindingdomain relative to the core domain. As a consequence,the
tetO
affinity drops about eight orders of magnitude(Lederer
et al
., 1996).

778
O. Scholz
et al.
© 2004 Blackwell Publishing Ltd,
Molecular Microbiology
,
53
, 777–789
A few mutants of TetR-based eukaryotic transactivatorshave been described for which the inducer tc has becomea co-repressor (Gossen
et al
., 1995; Urlinger
et al
., 2000).We explore here whether and how the unfused TetR pro-tein can be converted to revTetR by mutagenesis. Wehave obtained more than 100 TetR mutants that requireatc, a more potent effector than tc, for
tetO
binding. Theirlocations are discussed in view of the conformationalchange in TetR, and the derived hypotheses may be rel-evant for allosteric proteins in general.
Results
RevTetR mutations obtained from random mutagenesis
To explore the sequence space allowing reverse TetR
variants in
Escherichia coli
, we applied DNA shuffling(Stemmer, 1994a,b) to generate random mutations in thecore domain (codons 51–208). The purpose of thisapproach was to identify regions in the TetR sequencewhere mutants with this specialized phenotype are possi-ble. Mutant pools of TetR were screened for atc-depen-dent
b
-galactosidase (
b
-gal) expression. In addition tomany non-inducible TetR variants, 15 revTetR candidateswere found in 4000 mutants (see Table 1). They wereisolated, and
b
-gal assays with and without atc confirmedtheir phenotypes. The revTetR mutants were namedaccording to the location of the mutations as outlined in
Experimental procedures
. Their amino acid exchangesare shown in Fig. 2. The efficiencies of the revTetR vari-ants are given as induction factors (IF), which is the ratioof
b
-gal activity with and without atc. Mutant TetR r6.2
A
B
Fig. 1.
A. View of the structure of the TetR-[tc-mg]
+
complex. One TetR monomer is shown as light grey ribbon, the other in dark grey. Tc is drawn as a grey stick model. The helices
a
1,
a
4 and
a
6 are colour coded in blue, yellow and red, respectively, in one TetR monomer. Resi-dues within the three helices that are frequently mutated in revTetR variants are shown as stick models in the same colours in the other mono-mer. In detail, L17, N18, V20, I22 and E23 in
a
1 are shown in blue; D53, A56, I59 and A61 in
a
4 are shown in yellow, and D95, G96 and V99 in
a
6 are shown in redB. Stereoview of the interface of helices
a
1,
a
4 and
a
6. Residues L17 (blue), I59 (yellow) and V99 (red) are mutated in reverse single exchange mutants and point to the hydrophobic centre of the interface between the DNA read-ing head (represented by helix
a
1) and the core domain (represented by helices
a
4 and
a
6). The other positions where reverse single exchange mutants were found, G96 and A97, are marked in red. Hydrogen bonds between the DNA-binding head and the core domain involving D11, N18, I22, R62, R94 and D95 are indicated.
Reverse TetR
779
© 2004 Blackwell Publishing Ltd,
Molecular Microbiology
,
53
, 777–789
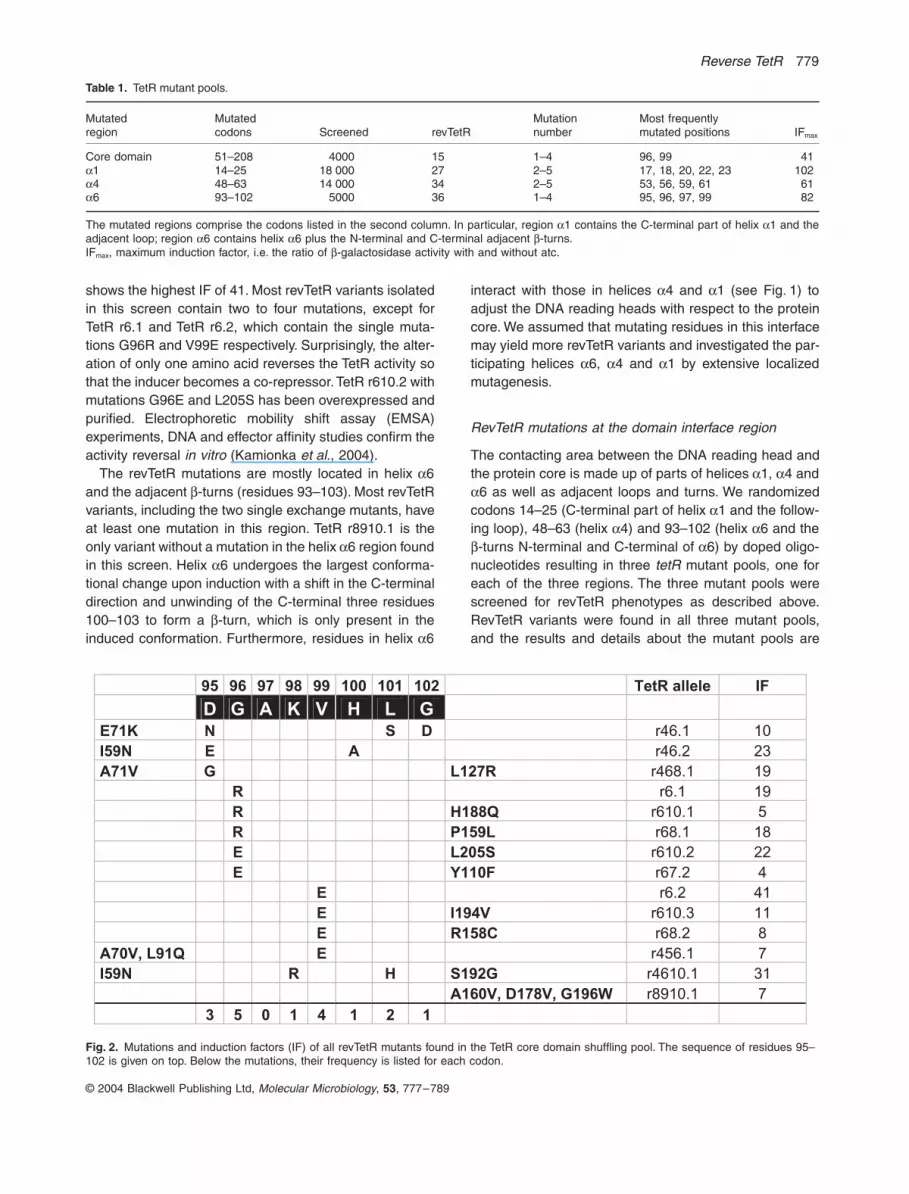
shows the highest IF of 41. Most revTetR variants isolatedin this screen contain two to four mutations, except forTetR r6.1 and TetR r6.2, which contain the single muta-tions G96R and V99E respectively. Surprisingly, the alter-ation of only one amino acid reverses the TetR activity sothat the inducer becomes a co-repressor. TetR r610.2 withmutations G96E and L205S has been overexpressed andpurified. Electrophoretic mobility shift assay (EMSA)experiments, DNA and effector affinity studies confirm theactivity reversal
in vitro
(Kamionka
et al
., 2004)
.
The revTetR mutations are mostly located in helix
a
6and the adjacent
b
-turns (residues 93–103). Most revTetRvariants, including the two single exchange mutants, haveat least one mutation in this region. TetR r8910.1 is theonly variant without a mutation in the helix
a
6 region foundin this screen. Helix
a
6 undergoes the largest conforma-tional change upon induction with a shift in the C-terminaldirection and unwinding of the C-terminal three residues100–103 to form a
b
-turn, which is only present in theinduced conformation. Furthermore, residues in helix
a
6
interact with those in helices
a
4 and
a
1 (see Fig. 1) toadjust the DNA reading heads with respect to the proteincore. We assumed that mutating residues in this interfacemay yield more revTetR variants and investigated the par-ticipating helices
a
6,
a
4 and
a
1 by extensive localizedmutagenesis.
RevTetR mutations at the domain interface region
The contacting area between the DNA reading head andthe protein core is made up of parts of helices
a
1,
a
4 and
a
6 as well as adjacent loops and turns. We randomizedcodons 14–25 (C-terminal part of helix
a
1 and the follow-ing loop), 48–63 (helix
a
4) and 93–102 (helix
a
6 and the
b
-turns N-terminal and C-terminal of
a
6) by doped oligo-nucleotides resulting in three
tetR
mutant pools, one foreach of the three regions. The three mutant pools werescreened for revTetR phenotypes as described above.RevTetR variants were found in all three mutant pools,and the results and details about the mutant pools are
Table 1.
TetR mutant pools.
Mutatedregion
Mutatedcodons Screened revTetR
Mutationnumber
Most frequently mutated positions IF
max
Core domain 51–208 4000 15 1–4 96, 99 41
a
1 14–25 18 000 27 2–5 17, 18, 20, 22, 23 102
a
4 48–63 14 000 34 2–5 53, 56, 59, 61 61
a
6 93–102 5000 36 1–4 95, 96, 97, 99 82
The mutated regions comprise the codons listed in the second column. In particular, region
a
1 contains the C-terminal part of helix
a
1 and theadjacent loop; region
a
6 contains helix
a
6 plus the N-terminal and C-terminal adjacent
b
-turns.IF
max
, maximum induction factor, i.e. the ratio of
b
-galactosidase activity with and without atc.
Fig. 2.
Mutations and induction factors (IF) of all revTetR mutants found in the TetR core domain shuffling pool. The sequence of residues 95–102 is given on top. Below the mutations, their frequency is listed for each codon.
95 96 97 98 99 100 101 102 TetR allele IF
D G A K V H L G
E71K N S D r46.1 10
I59N E A r46.2 23
A71V G L127R r468.1 19
R r6.1 19
R H188Q r610.1 5
R P159L r68.1 18
E L205S r610.2 22
E Y110F r67.2 4
E r6.2 41
E I194V r610.3 11
E R158C r68.2 8
A70V, L91Q E r456.1 7
I59N R H S192G r4610.1 31
A160V, D178V, G196W r8910.1 7
3 5 0 1 4 1 2 1

780
O. Scholz
et al.
© 2004 Blackwell Publishing Ltd,
Molecular Microbiology
,
53
, 777–789
summarized in Table 1. We sequenced at least 12unscreened mutants of each pool and verified their ran-dom distribution. Thus, the revTetR variants most proba-bly identify all residues with the potential to yield thisactivity upon mutation.
All revTetR obtained from the helix
a
1 pool are shownin Fig. 3A along with their mutations. A total of 27 revTetRvariants containing mutations at all randomized positionswere found in 18 000 screened candidates. The majority
of the mutations occur at residues L17, N18, V20, I22 andE23. Furthermore, three revTetR were identified that lackone, two or three residues in helix
a
1. The IF range fromthree to 100. Thus, the regulation efficiencies of the bestrevTetR are the same as for wild-type TetR.
RevTetR obtained from the helix
a
4 pool are shown inFig. 3B. Screening of 14 000 candidates yielded 34mutants with the desired phenotype (see Table 1). The IFof the best mutant is 60, somewhat less than that seen
Fig. 3.
Mutations and induction factors (IF) of all revTetR mutants found in the helix
a
1 (A), helix
a
4 (B) and helix
a
6 pool (C). The frequency of mutations is listed for each codon at the bottom. Deletions of complete codons are marked
∞
.
14 15 16 17 18 19 20 21 22 23 24 25 TetR allele IF
L E L L N E V G I E G L
I G G V r1.13 62
E G r1.20 29
G A G r1.11 15
F o r1.5 57
H F G r1.2 53
A T V r1.22 40
G M r1.10 56
A G V r1.7 102
V A D r1.4 95
S D r1.17 27
S K r1.1 86
G H V T W r1.25 25
Y D T L60S r14.21 86
V Y T r1.16 6
T Y F r1.9 23
Y E K r1.26 20
V F Y M K r1.18 10
S Y F r1.23 6
S Y F r1.6 26
S K V r1.8 31
V V K r1.21 14
K D L N A r1.3 72
D R N r1.14 3
G R N r1.12 90
D G96R r16.36 8
V E o o r1.24 46
o o o S r1.19 37
6 3 1 11 13 7 8 6 13 11 2 2
A

Reverse TetR
781
© 2004 Blackwell Publishing Ltd,
Molecular Microbiology
, 53, 777–789
for the best ones from the helix a1 pool. The most fre-quently mutated residues are D53, A56, I59 and A61.Alterations in the C-terminal six residues of helix a4 showthe highest frequency in this revTetR pool. All mutantsisolated from the helix a4 pool contain two to five muta-tions; no single exchange mutants appeared in thisregion.
RevTetR obtained from the helix a6 pool are shown inFig. 3C. Screening of 5000 candidates yielded 36 revTetR(see Table 1), in which the residues D95, G96, A97 andV99 were most frequently altered. The IF of the bestmutant from this pool is 80. RevTetR V99R represents thethird single exchange mutant in helix a6 besides revTetRG96R and V99E.
Fig. 3. Cont.
48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 TetR
allele IF
K R A L L D A L A V E I L A R H N F G r4.10 6
L V T F S r4.25 8
N T F Y r4.8 12
N G F S F r4.16 6
S S r4.4 2
F L S S r4.21 6
F T M S Q r4.36 15
F M S Q r4.37 16
G N L Q r4.2 2
F S Q A97T r46.15 8
Q V A Q Q r4.24 4
S F G I Q r4.1 33
S Y G I Q r4.29 61
Y G G r4.39 5
Y P r4.43 13
Y P L r4.6 6
P S r4.11 6
M P L V H188Y r410.1 51
H P L M r4.19 20
T M P S r4.41 10
P G r4.31 4
N S P L r4.35 7
E S R r4.42 5
E S P r4.30 4
E S M P r4.3 4
A P r4.40 2
Q K V P D r4.14 6
K V T G r4.20 6
V K V G r4.28 21
D V Q S r4.23 4
V V L T S r4.32 7
V H L r4.38 4
R E N r4.7 3
A N Y r4.34 5
0 3 3 1 3 17 6 3 13 9 10 14 7 15 10 11
B

782 O. Scholz et al.
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 53, 777–789
RevTetR with more than one essential mutation
The helices a1 and a4 mutants show that the reversephenotype can be obtained without mutations in the helixa6 region. Moreover, the mutation of only one residue
can be sufficient to reverse the effector response of TetR.Thus, only one of the exchanges in revTetR with multiplemutations may be sufficient for the phenotype. To clarifythis question, we separated the mutations occurring inthe three revTetR, r46.1, r4610.1 and r410.1 (Figs 2 and
Fig. 3. Cont.
93 94 95 96 97 98 99 100 101 102
TetR
allele IF
Y R D G A K V H L G
E T r6.34 3
E V113A r610.4 5
A71T E r46.16 2
R r6.1 19
R I r6.13 10
R L I r6.33 15
G R I r6.35 7
C R N r6.70 65
W P r6.36 3
G W r6.14 5
F W E N r6.17 8
P V T N r6.37 7
Q S r6.20 4
G P r6.12 3
T R G r6.19 19
L79V T R G r6.21 15
T G r6.29 8
V G r6.38 43
E r6.2 15
Y E r6.23 17
P E r6.39 82
C E r6.40 12
H N T L r6.41 17
R S L r6.18 2
P R D r6.30 12
F N F r6.31 7
R r6.8 16
P E D r6.7 3
E E r6.15 2
C E T r6.73 24
S E A r6.16 10
H H r6.45 2
N Q F r6.47 3
L A Q r6.22 2
H P Q r6.46 2
H R r6.44 2
5 9 11 14 13 6 15 6 5 3
C

Reverse TetR 783
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 53, 777–789
3B), by oligonucleotide-directed mutagenesis. The regu-latory activities of the resulting TetR variants are shownin Fig. 4A–C. One of the four mutations in revTetR r46.1and r4610.1 (E71K in TetR r46.1 and S192G in TetRr4610.1) can be removed without affecting the reversephenotype. Two mutations in revTetR r410.1 (L52M andH188Y) are not necessary. All possible combinations ofsingle and double exchanges of the remaining threemutations were then constructed for all three revTetRvariants. None of the single mutants shows a reversephenotype. Most of the single and double mutation vari-ants are either non-inducible or behave like wild-typeTetR. At least two mutations are required for reversebehaviour, as shown by the combinations A56P V57Land A56P E58V, which are derived from r410.1. Mutantsderived from r46.1 and r4610.1 require at least threemutations to show the reverse phenotype. This result,together with the three single mutation variants, indicatesthat revTetR behaviour can result from single or multipleexchanges.
RevTetR with single mutations
RevTetR variants with multiple exchanges were found inall three investigated regions, but those with single muta-tions only occurred in helix a6. The fact that multipleexchange revTetR contain non-essential alterations indi-cates that we have performed a saturating mutagenesisof the three regions. Thus, there may be more singleexchange mutants possible than we have found so far. Asthese are important for the mechanistic understanding ofthe reverse phenotype, we tried to find more singleexchange mutants in helices a1, a4 and a6 by randomiz-ing the 13 codons most frequently affected in the revTetRvariants (positions 17, 18, 20, 22, 23, 53, 56, 59, 61, 95,96, 97 and 99). Screening as described above revealedsingle exchange revTetR at positions 17, 59, 96, 97 and99. They fall into two groups according to the chemicalnature of the introduced amino acids (see Fig. 5A). Atposition 17, we found large to small exchanges (L17G),whereas charged amino acids are introduced at all otherpositions. To exclude the possibility that this observationoccurred by chance, we constructed all 19 single aminoacid exchanges of V99 and L17 (see Fig. 5B and C). Atposition 17, residues smaller than L, such as G, A and S,cause reverse behaviour. Most charged or larger polaramino acids at position 17 yield inactive proteins, whereasnon-polar residues yield wild type-like phenotypes, unlessthey are too bulky. A different result is obtained at position99: non-polar side-chains as in A, C, I, L or M have noinfluence on TetR activity. Bulky side-chains as in W, Y, For Q cause poorly inducible or only slightly reverse phe-notypes. Pronounced reverse phenotypes are obtained ifV99 is substituted by the charged residues E, K or R. All
reverse single mutants at positions 59, 96 and 97 intro-duce charged amino acids as well. Interestingly, positiveor negative charges lead to the same reverse phenotype.
Discussion
TetR mutants with new DNA sequence or inducer recog-nition specificities as well as non-inducible TetR mutantshave been obtained and analysed before (Hecht et al.,1993; Müller et al., 1995; Helbl and Hillen, 1998; Helblet al., 1998; Scholz et al., 2003). The set of mutantsdescribed by Müller et al. (1995) has defects in tc or atcresponse. They were grouped into several classes: oneclass is deficient in inducer binding, a second class isaffected in the allosteric conformational change, and athird class contains mutations in the TetR dimerizationdomain. Among them was the first revTetR, whichrepressed a little better in the presence of effector andwas subsequently used to construct the mammalian reg-ulator rtTA. This showed a much better reverse pheno-type in eukaryotes than in E. coli (Gossen et al., 1995).The widespread use of rtTA in many organisms providedthe motivation to develop a yeast-based screen ofmutated tTA, from which better rtTA variants were subse-quently obtained (Urlinger et al., 2000). The revTetR vari-ants presented here cover a much larger sequencespace and may lead to better rtTA variants when fused toeukaryotic readout domains. Furthermore, they shouldbe applicable to target validation in prokaryotes becausea target gene may now be switched on (using TetR) or off(using revTetR) upon administering the effector. Theaddition of effector often causes a faster and betterdefined expression response than its depletion. Thus,revTetR may be especially useful for bacteria in compli-cated environments such as model organisms for patho-genicity, e.g. mice.
This is the first study reporting an extensive set ofrevTetR variants in E. coli, represented by more than 100individual mutants, which most probably cover the entiresequence space of this phenotype. The regulatory effi-cacy of some revTetR mutants yields 100%, as measuredfor wild-type TetR in the same genetic background. Thus,the TetR fold is capable of totally turning the allostericresponse around from the wild type so that the induceratc becomes a co-repressor. We are able to define pre-cisely one region of TetR in which revTetR mutants occurwith high frequency: the contacting area of the DNA-binding domain with the core domain. It is built up by ahydrophobic interface surrounded by three hydrogenbonds (see Fig. 1B). The residues L17 (a1), I59 (a4), G96and V99 (both a6) are part of the interface between thethree helices (Fig. 1B), and all of them yield single muta-tion revTetR variants. A97 is the only position with reversesingle exchange mutations that is not part of the hydro-

784 O. Scholz et al.
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 53, 777–789
phobic interface, but is very close to that region. Introduc-tion of single charged residues at positions 59, 96, 97 or99 puts them in a hydrophobic region, where they are inan unfavourable state, undoubtedly leading to structuralalterations. In three of the four positions, a positive andnegative charge causes the same effect. Thus, there can-not be a neutralizing interaction partner in their new con-formation. It is conceivable that charged residues atposition 96 or 99 interfere with the H-bonding networkbetween a1 and a6 (N18 and R94; I22 and D95; Kisker
et al., 1995). Thus, they might cause a conformationalrearrangement of the two DNA-binding domains withrespect to the protein core. The volume reduction of theL17G exchange in the hydrophobic interface probably alsoresults in a changed conformation of the DNA-bindingdomain. We assume that these mutations alter the posi-tioning of the DNA reading heads with respect to theprotein core, although structural information on themutants is needed to describe the exact nature of theseconformational changes.
wt
r46
.1
D95
N L
10
1S
D9
5N
G1
02
D
L1
01
S G
10
2D
D9
5N
L1
01
S
G1
02
D
% b
-ga
lacto
sid
ase
activity
0
20
40
60
80
100
120
-
atc
D9
5N
L1
01
S G
10
2D
r461
0.1
I59N
K98
R L
10
1H
I59
N K
98
R
I59
N L
10
1H
K9
8R
L10
1H
I59
N
K9
8R
L1
01
H
S19
2G
% b
-ga
lacto
sid
ase
activity
0
20
40
60
80
100
120
-
atc
A
B
Fig. 4. In vivo data of single, double and three-fold mutants constructed from TetR r46.1 (A), TetR r4610.1 (B) and TetR r410.1 (C). Two (TetR r410.1) or three (TetR r46.1 and TetR r4610.1) mutations are required for the reverse phenotype in these alleles.

Reverse TetR 785
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 53, 777–789
We propose the following explanation for the reversephenotype: the conformational change leading to TetRinduction is a motion of the DNA-binding domains withrespect to the protein core. In the repressing conforma-tion, the DNA-binding heads fit into successive majorgrooves of tetO. A shift of both DNA-binding domainsrelative to each other leads to release of the DNA uponinducer binding. A putative trigger for this movement orig-inates at residue T103 and leads to formation of a b-turnC-terminal of helix a6, which is essential for induction(Orth et al., 1998; 1999; Scholz et al., 2000). As helicesa4 and a1 are connected to a6 via several interactions(Kisker et al., 1995; Orth et al., 2000), they follow the a6motion and take along the entire DNA-binding domain.The distances between residues L17, I59 and V99remain essentially unchanged during this motion in wild-type TetR. As the reverse mutations only occur in resi-dues that do not move with respect to each other, thedirection and scale of the motion should remainunchanged in revTetR, but the DNA-binding domains areprobably in a different position with respect to the proteincore, so that they cannot bind operator in the absence ofinducer. Binding of atc supposedly leads to the sameinternal motions in TetR and revTetR, but yields a DNA-binding conformation of the DNA reading heads inrevTetR. The effector binding signal would still be trans-mitted in the same way in this model, albeit with theinverse effect on DNA binding. The in vitro data obtainedwith TetR r610.2 confirm that atc binding increases the
tetO affinity of revTetR (Kamionka et al., 2004), which isconsistent with our model.
If this hypothesis is true, it would have interestingimplications for the flexibility of free revTetR. We assumethat an allosteric protein may interchange freely betweenits conformations in the absence of ligand. However, thiscannot be the case for revTetR because it can assumethe tetO binding conformation only in the presence of co-repressor. Thus, there must be a block in the conforma-tional transition that is overcome by co-repressor bind-ing. As revTetR and TetR are assumed to exhibitidentical allostery, this hypothesis would also hold truefor TetR.
A mechanistic interpretation of the revTetR variantsrequiring multiple mutations or the deletion mutants foundin the helix a1 region is much less straightforward.
Taken together, we assume for several mutants that themoving portions of the allosteric protein TetR can be struc-turally readjusted by single or multiple mutations, while theability to perform a conformational transition remainsintact. The mutant protein is still capable of adopting twodifferent conformations depending on the absence orpresence of the ligand atc, but both conformations aredifferent from the corresponding ones of the wild-typeprotein.
We have shown that one amino acid exchange is suffi-cient for reversal of TetR activity. It is conceivable thatanalogous metamorphoses might be possible for otherallosteric proteins, making them surprisingly versatile. By
r41
0.1
A56P
V57L E
58
V
A56P
E58
V
A56P
V5
7L
V57L E
58V
A56
P
V5
7L
E58
V
b-g
ala
cto
sid
ase a
ctivity
0
20
40
60
80
100
120
-
atc
L52M
A56P
V57L E
58V
A56P
V57L E
58V
H188Y
C Fig. 4. Cont.

786 O. Scholz et al.
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 53, 777–789
addressing this ‘functional Paracelsus challenge’ for otherallosteric responses, we should not only be able to gainnew properties but also expand our understanding of howproteins adopt and interchange between functionally dif-ferent states.
Experimental procedures
Materials and general methods
Atc was purchased from Acros and tc from Merck. MacCon-key agar base was purchased from Becton Dickinson. All
Fig. 5. In vivo data of single exchange mutants.A. revTetR variants with mutations at position 96 or 97.B and C. Phenotypes of all 19 single amino acid exchange mutants at position 17 (B) and 99 (C). Reverse phenotypes are caused by small residues such as G and S at position 17. Amino acid 99 needs a charged residue for reverse behaviour.
I59D
I59R
G9
6E
G9
6R
G9
6P
A9
7K
A9
7R
% b
-ga
lacto
sid
ase
activity
0
20
40
60
80
100
120
-
atc
L17 mutations
L(wt) C I V M F T Q Y H P W N D E K R G S A
% b
-gala
cto
sid
ase a
ctivity
0
20
40
60
80
100
120
-
atc
B
A

Reverse TetR 787
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 53, 777–789
other chemicals were from Merck, Roth or Sigma at thehighest available purity. Enzymes for DNA restriction andmodification were from New England Biolabs, Roche, Strat-agene or Pharmacia. Isolation and manipulation of DNA wasperformed as described previously (Sambrook and Russel,2001). Oligonucleotides were obtained from Applied Biosys-tems. Sequencing was carried out according to the protocolprovided by Applied Biosystems for cycle sequencing andanalysed with an ABI PRISM™ 310 genetic analyser(Applied Biosystems).
Construction of pWH1925
tetR(BD), which codes for a chimeric TetR containing aminoacids 1–50 from TetR(B) and amino acids 51–208 fromTetR(D), was chosen for this study because of its advanta-geous regulation properties (Schnappinger et al., 1998).tetR(BD) was cut out of pWH1411BD (Scholz et al., 2003)via HincII. Plasmid pUC19 was cut with PvuII. Both enzymescreate blunt ends. tetR was ligated non-directionally intopUC19, giving a pWH1925 precursor, in which tetR runs inthe opposite direction as the ¢lacZa remnant. This constructcontains two additional FspI sites to the FspI site in tetR,which is needed to be singular for further cloning purposes.The additional FspI sites were eliminated using the Strat-agene QuikChange™ mutagenesis kit according to the man-ufacturer’s recommendations and primers pUC_Fsp_del_1acttcccaacagttgcgtagcctgaatggc and pUC_Fsp_del_1b gccattcaggctacgcaactgttgggaag for elimination of FspI in the¢lacZa remnant, and pUC_Fsp_del_2c gttcgccagttaatagtttgcgtaacgttgttgcc and pUC_Fsp_del_2d ggcaacaacgttacgcaaactattaactggcgaac for silent mutation of the FspI sitein the bla gene. The underlined positions represent the alter-ations compared with the pUC19 sequence.
Designation of revTetR mutants
The revTetR variants have been named according to thelocation of their mutations. For this purpose, we used the 10a-helices of TetR to define 10 regions. Each region consistsof one helix and the adjacent residues in the C-terminaldirection. The names start with an ‘r’, which stands forreverse, followed by the number (from 1 to 10) for theregion(s) carrying mutations. Finally, an identification numberis added, separated by a dot. Example: r4610.1 is a revTetRmutant with mutations in regions 4, 6 and 10 and has theidentification number 1.
Construction of tetR mutant pools
DNA shuffling. DNA shuffling (Stemmer, 1994b) was used tointroduce random mutations in tetR. An error-prone poly-merase chain reaction (PCR) of the tetR gene with plasmidpWH1411BD was followed by a partial DNase I digest andrecovery of 10–70 bp fragments from SDS-PAGE. Themutated tetR genes were then reassembled in two furtherPCRs and cloned via ApaI and NcoI into pWH1411BD.
Oligonucleotide-directed randomization mutagenesis. Tointroduce random mutations into distinct regions of tetR, oli-gonucleotides with a defined amount of non-wild-type baseswere used. The sequences of the oligonucleotides are:RegHel1, 5¢-aagtaaagtgattaacagcgcattagagctgcttaatgagg
tcggaatcgaaggtttaacaacccgta-3¢; RgeHel4, 5¢-ataatcatgatg
acgcgccaagatctccaccgccagcgcatccagtagggcccgcttattttttac; RegHel6, 5¢-catcaggacgcgtgccgaggtgcacttttgccccgt
cacggtaacgcagcagcgcgcggc-3¢ The bold positions contained86% wild-type and 14% of the three non-wild-type bases
Fig. 5. Cont.
V99 mutations
V (wt) G A L I C M S H P T N Q W Y F D E K R
% b
-gala
cto
sid
ase a
ctivity
0
20
40
60
80
100
120
-
atc
C

788 O. Scholz et al.
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 53, 777–789
(91% and 9% for RegHel1 respectively). The expected pre-dominant frequency of mutations per oligonucleotide (e.g. pertetR after cloning) can be calculated according to a binomialdistribution and is three to four. The mutant tetR genes werethen assembled by PCR with the partially randomized oligo-nucleotides according to the three-primer method (Landtet al., 1990).
Single codon randomization
TetR mutant pools harbouring randomizations of single aminoacids were constructed according to the three-primer method(Landt et al., 1990). The mutagenesis primer was designedto contain the base sequence NNS at the correspondingposition.
Escherichia coli screening systems
Two different screening systems were used. In the first sys-tem, the mutant pools constructed in pWH1411BD (coredomain pool, helix a4 and a6 pool) were tested for inducibilitywith tc derivatives in a genetic screen as follows. E. coli strainWH207/lWH25 (Wissmann et al., 1991) with plasmidpWH414 was transformed with the pWH1411BD mutantpools. pWH414 contains a tetA–lacZ fusion expressing b-galunder tet control. pWH1411BD directs a constitutive expres-sion of tetR. The cells were plated on M9 minimal medium(Sambrook and Russel, 2001) containing 0.2% glucose ascarbon source and 0.004% Xgal. The colonies werescreened for their b-gal activities in the absence and pres-ence of 0.4 mM atc.
All other pools used in this study (helix a1 pool, singlecodon randomizations) were constructed in pWH1925 andanalysed in the second system. To screen for reversemutants, E. coli strain WH207/ltet50 was transformed withthe corresponding pools in pWH1925. E. coli WH207/ltet50has a chromosomal tetA–lacZ fusion under tet control so thatTetR represses the transcription of lacZ. lacZ expression wastested on MacConkey agar plates (59 g l-1 MacConkey agarbase and 14 g l-1 lactose). The colonies were screened fortheir b-gal activities in the absence and presence of 0.4 mMatc.
Quantification of repression and induction of TetR mutants
To determine the in vivo repression and induction of TetR, E.
coli WH207/ltet50 (Smith and Bertrand, 1988; Wissmannet al., 1991) was transformed with the tetR variants locatedon pWH853 (Wissmann et al., 1991; applies to data pre-sented in Fig. 4A), pWH1411BD (applies to data shown inFig. 4B) or pWH1925 (applies to all other data in this paper)and grown in LB with or without 0.4 mM atc at 37∞C. pWH853,pWH1411 and pWH1925 are expression vectors for TetR.The TetR expression levels are similar in pWH1411 andpWH1925, but clearly lower in pWH853, as derived fromWestern blot analysis (data not shown). Thus, pWH853 pro-vides an increased resolution for TetR variants with strong(co)-repression properties. b-Galactosidase activities weredetermined from log phase cultures of three independentclones at 37∞C as described previously (Miller, 1972) and
were repeated at least twice. The b-gal activity of E. coli
WH207/ltet50 without tetR ranged from 6500 to 9000 Millerunits. It was determined separately for samples with andwithout atc in each experiment and set to 100%.
Acknowledgements
This work was supported by the Deutsche Forschungsge-meinschaft through the SFB 473, the Graduiertenkolleg 805and the Fonds der Chemischen Industrie.
References
Berens, C., and Hillen, W. (2003) Gene regulation by tetra-cyclines. Constraints of resistance regulation in bacteriashape TetR for application in eukaryotes. Eur J Biochem
270: 3109–3121.Dalal, S., Balasubramanian, S., and Regan, L. (1997) Protein
alchemy: changing beta-sheet into alpha-helix. Nature
Struct Biol 4: 548–552.Gossen, M., and Bujard, H. (2001) Tetracyclines in the control
of gene expression in eukaryotes. In Tetracyclines in
Biology, Chemistry and Medicine. Nelson, M., Hillen, W.,and Greenwald, R.A. (eds). Basel: Birkhäuser, pp. 139–158.
Gossen, M., Freundlieb, S., Bender, G., Müller, G., Hillen,W., and Bujard, H. (1995) Transcriptional activation bytetracyclines in mammalian cells. Science 268: 1766–1769.
Hecht, B., Müller, G., and Hillen, W. (1993) Noninducible Tetrepressor mutations map from the operator binding motifto the C terminus. J Bacteriol 175: 1206–1210.
Helbl, V., and Hillen, W. (1998) Stepwise selection of TetRvariants recognizing tet operator 4C with high affinity andspecificity. J Mol Biol 276: 313–318.
Helbl, V., Tiebel, B., and Hillen, W. (1998) Stepwise selectionof TetR variants recognizing tet operator 6C with high affin-ity and specificity. J Mol Biol 276: 319–324.
Hillen, W., and Berens, C. (1994) Mechanisms underlyingexpression of Tn10 encoded tetracycline resistance. Annu
Rev Microbiol 48: 345–369.Hinrichs, W., Kisker, C., Duvel, M., Müller, A., Tovar, K.,
Hillen, W., and Saenger, W. (1994) Structure of the Tetrepressor-tetracycline complex and regulation of antibioticresistance. Science 264: 418–420.
Jones, D.T., Moody, C.M., Uppenbrink, J., Viles, J.H., Doyle,P.M., Harris, C.J., et al. (1996) Towards meeting theParacelsus challenge: the design, synthesis, and charac-terization of paracelsin-43, an alpha-helical protein withover 50% sequence identity to an all-beta protein. Proteins
24: 502–513.Kamionka, A., Bogdanska-Urbaniak, J., Scholz, O., and
Hillen, W. (2004) Two mutations in the tetracycline repres-sor change the inducer anhydrotetracycline to a corepres-sor. Nucleic Acids Res 32: 842–847.
Kisker, C., Hinrichs, W., Tovar, K., Hillen, W., and Saenger,W. (1995) The complex formed between Tet repressor andtetracycline-Mg2+ reveals mechanism of antibiotic resis-tance. J Mol Biol 247: 260–280.
Landt, O., Grunert, H.P., and Hahn, U. (1990) A general

Reverse TetR 789
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 53, 777–789
method for rapid site-directed mutagenesis using the poly-merase chain reaction. Gene 96: 125–128.
Lederer, T., Kintrup, M., Takahashi, M., Sum, P.E., Ellestad,G.A., and Hillen, W. (1996) Tetracycline analogs affectingbinding to Tn10-encoded Tet repressor trigger the samemechanism of induction. Biochemistry 35: 7439–7446.
Miller, J.H. (1972) Experiments in Molecular Genetics. ColdSpring Harbor, NY: Cold Spring Harbor Laboratory Press.
Müller, G., Hecht, B., Helbl, V., Hinrichs, W., Saenger, W.,and Hillen, W. (1995) Characterization of non-inducible Tetrepressor mutants suggests conformational changes nec-essary for induction. Nature Struct Biol 2: 693–703.
Orth, P., Cordes, F., Schnappinger, D., Hillen, W., Saenger,W., and Hinrichs, W. (1998) Conformational changes of theTet repressor induced by tetracycline trapping. J Mol Biol
279: 439–447.Orth, P., Saenger, W., and Hinrichs, W. (1999) Tetracycline-
chelated Mg2+ ion initiates helix unwinding in Tet repressorinduction. Biochemistry 38: 191–198.
Orth, P., Schnappinger, D., Hillen, W., Saenger, W., andHinrichs, W. (2000) Structural basis of gene regulation bythe tetracycline inducible Tet repressor-operator system.Nature Struct Biol 7: 215–219.
Rose, G.D., and Creamer, T.P. (1994) Protein folding: pre-dicting predicting. Proteins 19: 1–3.
Sambrook, J., and Russel, D. (2001) Molecular Cloning: a
Laboratory Manual. New York: Cold Spring Harbor Labo-ratory Press.
Schnappinger, D., Schubert, P., Pfleiderer, K., and Hillen, W.(1998) Determinants of protein–protein recognition by four
helix bundles: changing the dimerization specificity of Tetrepressor. EMBO J 17: 535–543.
Scholz, O., Schubert, P., Kintrup, M., and Hillen, W. (2000)Tet repressor induction without Mg2+. Biochemistry 39:
10914–10920.Scholz, O., Köstner, M., Reich, M., Gastiger, S., and Hillen,
W. (2003) Teaching TetR to recognize a new inducer. J Mol
Biol 329: 217–227.Smith, L.D., and Bertrand, K.P. (1988) Mutations in the Tn10
tet repressor that interfere with induction. Location of thetetracycline-binding domain. J Mol Biol 203: 949–959.
Stemmer, W.P. (1994a) Rapid evolution of a protein in vitro
by DNA shuffling. Nature 370: 389–391.Stemmer, W.P. (1994b) DNA shuffling by random fragmen-
tation and reassembly: in vitro recombination for molecularevolution. Proc Natl Acad Sci USA 91: 10747–10751.
Urlinger, S., Baron, U., Thellmann, M., Hasan, M.T., Bujard,H., and Hillen, W. (2000) Exploring the sequence spacefor tetracycline-dependent transcriptional activators: novelmutations yield expanded range and sensitivity. Proc Natl
Acad Sci USA 97: 7963–7968.Wissmann, A., Wray, L.V., Jr, Somaggio, U., Baumeister, R.,
Geissendorfer, M., and Hillen, W. (1991) Selection for Tn10tet repressor binding to tet operator in Escherichia coli:isolation of temperature-sensitive mutants and combinato-rial mutagenesis in the DNA binding motif. Genetics 128:
225–232.Yuan, S.M., and Clarke, N.D. (1998) A hybrid sequence
approach to the Paracelsus challenge. Proteins 30: 136–143.