activation of wnt signaling rescues neurodegeneration and behavioral impairments induced by...
TRANSCRIPT

IMMEDIATE COMMUNICATION
Activation of Wnt signaling rescues neurodegenerationand behavioral impairments induced by b-amyloid fibrilsGV De Ferrari, MA Chacon, MI Barrıa, JL Garrido, JA Godoy, G Olivares, AE Reyes, A Alvarez,
M Bronfman and NC Inestrosa
Centro de Regulacion Celular y Patologıa, MIFAB, Facultad de Ciencias Biologicas, P Universidad Catolica de Chile, Santiago,Chile
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder, which is probablycaused by the cytotoxic effect of the amyloid b-peptide (Ab). We report here molecular changesinduced by Ab, both in neuronal cells in culture and in rats injected in the dorsal hippocampuswith preformed Ab fibrils, as an in vivo model of the disease. Results indicate that in bothsystems, Ab neurotoxicity resulted in the destabilization of endogenous levels of b-catenin, akey transducer of the Wnt signaling pathway. Lithium chloride, which mimics Wnt signaling byinhibiting glycogen synthase kinase-3b promoted the survival of post-mitotic neurons againstAb neurotoxicity and recovered cytosolic b-catenin to control levels. Moreover, the neurotoxiceffect of Ab fibrils was also modulated with protein kinase C agonists/inhibitors and reversedwith conditioned medium containing the Wnt-3a ligand. We also examined the spatial memoryperformance of rats injected with preformed Ab fibrils in the Morris water maze paradigm, andfound that chronic lithium treatment protected neurodegeneration by rescuing b-catenin levelsand improved the deficit in spatial learning induced by Ab. Our results are consistent with theidea that Ab-dependent neurotoxicity induces a loss of function of Wnt signaling componentsand indicate that lithium or compounds that mimic this signaling cascade may be putativecandidates for therapeutic intervention in Alzheimer’s patients.Molecular Psychiatry (2003) 8, 195–208. doi:10.1038/sj.mp.4001208
Keywords: Alzheimer’s disease; amyloid b-peptide; neurodegeneration; behavior; lithium;Wnt signaling
Introduction
Alzheimer’s disease (AD) is an age-related neuro-degenerative disease characterized by two types oflesions in the brain: neuritic plaques composedmainly of the amyloid b-peptide (Ab) and neuro-fibrillary tangles (NFTs) composed mainly of hyper-phosphorylated forms of the protein tau, and by aselective neuronal cell death.1 Ab neurotoxicity isassociated with morphological alterations in rat andin human neurons in cultureFincluding neuronalshrinkage plus severe axonal and dendritic dystrophyFand similar dystrophic changes are seen in ADneurons that develop neurofibrillar pathology.2 Re-markably, it has been observed that primary culturesof fetal rat hippocampal and human cortical neuronsexposed to Ab induced the activation of glycogensynthase kinase-3b (GSK-3b), the hyperphosphoryla-tion of tau proteins, and the loss of the microtubularnetwork.3–5
GSK-3b is a key modulator of the Wnt signaltransduction pathway.6–8 According to the classicalview of Wnt signaling,9,10 in the presence of anextracellular Wnt ligand, membrane-anchored recep-tors of the Frizzled protein family transduce its signalto the intracellular space activating Dishevelledprotein. Dishevelled, in turn, inactivates GSK-3bactivity, through the formation of a multiproteincomplex. As a result of GSK-3b inactivation, intra-cellular levels of b-catenin increase, allowing itsbinding to components of the high mobility groupfamily of transcription factors T-cell factor/lymphoidenhancer-binding factor (Tcf/LEF). Finally, b-catenin–Tcf/LEF complexes enter the nucleus and activate theexpression of Wnt-target genes. Alternatively, in theabsence of a Wnt ligand, the activity of GSK-3b isswitched on and thus it phosphorylates b-catenin forubiquitin-proteosome-mediated degradation.11,12 As anet result, b-catenin levels are diminished within thecytosol and therefore the expression of the Wnt-targetgenes is switched off.
Wnt signaling is essential in developmental andoncogenic processes.13,14 More recently, it has beenimplicated in neurodegenerative disorders such asautism,15,16 schizophrenia,17,18 and AD.19,20 Indeed,several studies have shown that the familial AD-linked presenilin-1 proteins form high molecular
Received 26 July 2001; revised 7 April 2002; accepted 23 April2002
Correspondence: Prof NC Inestrosa, Molecular NeurobiologyUnit, P Catholic University of Chile, PO Box 114-D, Santiago,Chile. E-mail: [email protected]
Molecular Psychiatry (2003) 8, 195–208& 2003 Nature Publishing Group All rights reserved 1359-4184/03 $25.00
www.nature.com/mp

weight multiprotein complexes with the b-cateninprotein.21–23 Further studies showed that b-cateninlevels were markedly reduced in AD patients carryingpresenilin-1-inherited mutations,24 and it has beensuggested that similar mutations may disturb b-catenin translocation to the nuclei,25,26 likely affectingWnt activity. Likewise, presenilin-1–b-catenin com-plexes also contain GSK-3b27,28 and its substrate tau,27
and it was suggested that presenilin-1-inheritedmutations altered the affinity and thus the activityof GSK-3b.29,30
Considering that lithium mimics Wnt signaling byreversibly inactivating GSK-3b,31,32 here we examinedwhether lithium could act as a neuroprotective factoragainst Ab-dependent damage. Since it has beensuggested that Wnt signaling inactivates GSK-3b viaan intracellular pathway that involves the proteinkinase C (PKC) enzyme,7,8,33 we also studied theability of PKC agonists/inhibitors to modulate Abneurotoxicity. We report here that Ab fibrils inducethe destabilization of endogenous levels of b-catenin,and that activation of Wnt signaling by inhibitingGSK-3b activity either with lithium, the PKC activatorphorbol 12-myristate 13-acetate (PMA) or with con-ditioned medium containing the Wnt-3a ligandprotected post-mitotic neurons from Ab neurotoxi-city. Furthermore, lithium reduced hippocampaldegeneration, recovered b-catenin levels, and sub-stantially improved spatial memory deficits of ratsinjected with Ab fibrils. A preliminary account of thiswork has been presented elsewhere.34
Materials and methods
Ab fibril formationHuman wild-type Ab1–40 peptide (Bachem, CA, USA)was aggregated as described previously.35 PreformedAb fibrils were washed several times in either sterilephosphate-buffered saline (PBS) (pH 7.4), or sterileartificial cerebrospinal fluid (aCSF; 130 mM NaCl,2.6 mM KCl, 4.3 mM MgCl2, 1.8 mM CaCl2), and thenpelleted by centrifugation (30 min at 14 000 rpm in anEppendorf microcentrifuge). Ab concentrations weredetermined from denaturing Tris-Tricine SDS-PAGEgels subjected to densitometric scanning and the datawere processed using the GS365W program (HoeferScientific Instruments, CA, USA). The final pelletwas resuspended at a concentration of 2.5 mg/ml.
Primary cultures of rat hippocampal neuronsCultures were prepared from 18-day-old Sprague–Dawley rat fetuses.36 The hippocampi were dissectedunder a stereomicroscope and individual cells wereprepared by trypsinization (0.1%) in Hank’s balancedsalt solution. Cells were seeded for 2 h onto 24- to 96-well culture dishes or cover glasses coated with poly-D-lysine (50mg/ml) and adhesion medium: Dulbecco’smodified Eagle’s medium (DMEM, Gibco/BRL), 10%horse serum, 100 U/ml penicillin/streptomycin. Theculture medium was then substituted with Neuro-basal media supplemented with B27, 100mg/ml
streptomycin and 100 U/ml penicillin, and cells wereincubated at 371C and 8% CO2. For experiments,6 to 8-day-old hippocampal cell cultures were shiftedto B27-free media.
Neuro 2A cellsNeuro 2A cells37 were grown in DMEM supplementedwith 5% fetal bovine serum (FBS), 100mg/ml strepto-mycin, 100 U/ml penicillin and 25mg/ml fungizone,and maintained at 371C, in 5% CO2 and saturatedhumidity. Cells were then induced to differentiate48 h after plating by applying 5 mM dibutyryl cAMP.
HEK-293 cell line and conditioned medium containingthe Wnt-3a ligandHEK-293 cells were grown essentially as described.38
Cells were transiently transfected with the expressionvector containing HA-tagged mouse Wnt-3a underthe control of a CMV promoter or with its controlplasmid (Upstate Biotechnology, NY, USA), by usingLipofectamine-plus (Gibco/BRL) according to instruc-tions from the manufacturer. For conditioned media,cells were grown to 95% confluence, washed withPBS, and maintained in serum-free DMEM over-night. The conditioned media were filtered through0.22mM filter units, aliquoted, and stored at –801Cuntil use.
In vitro cytotoxicity of Ab fibrilsHippocampal neurons or Neuro 2A cells (3� 103/100ml per well) were assayed for the cytotoxic effectsof Ab (0.1–10mM) by measuring the reduction of3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) to a color compound.35 The cytotoxiceffects of Ab were assayed in the presence ofincreasing concentrations of LiCl (1 mM–10 mM),PMA (50 nM–2.5 mM), POC-16 (1–100mM) and AM-44 (1–100 mM)39 or with 0.2 ml/cm2 conditionedmedia containing Wnt-3a.38 MTT reduction wasdetermined in a Labsystem Uniskam I spectrophot-ometer (Finland) at 540 and 650 nm. MTT values (intriplicate) correspond to 4–6 separate experimentsand are expressed as the percentage of control(untreated) cells.
Immunocytochemistry and cell fractionation ofhippocampal neuronsHippocampal neurons grown on cover slides exposedto different treatments were washed once with PBS at371C, fixed with 4% paraformaldehyde/PBS for 4 h,extracted using 0.1%. Triton X-100/PBS, and immuno-stained using primary monoclonal antibodies (diluted1:300 in PBS) for b-catenin (Santa Cruz Biotech., CA,USA). The secondary antibody was a fluorescein-conjugated polyclonal antibody. Cells were viewed byoptic fluorescence using a ZEISS LSM-400 confocalmicroscope (63�oil immersion objectives) and re-corded at 520�520 pixels/image. Cellular morphol-ogy was evaluated using phase-contrast microscopy(Optiphot-NIKON, Japan). Cytosolic fractions of trea-ted cells (1.2�105 cells in 24-well culture dishes)
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
196
Molecular Psychiatry

were harvested by scraping and homogenized inbuffer A (10 mM HEPES, 1.5 mM MgCl, 10 mM KCl,1 mM EDTA, 1 mM DTT, 10% glycerol, pH 7.5)containing a cocktail of protease inhibitors (2 mMphenylmethylfluorosulfonate, 10 mg/ml aprotinin,10 mg/ml leupeptin, and 10 mg/ml trypsin inhibitor).The resulting pellet was resuspended in buffer B(20 mM HEPES, 1.5 mM MgCl, 0.4 mM NaCl, 0.5 mMDTT, 0.4 mM EDTA, 25% glycerol, pH 7.9) plusprotease inhibitors, and centrifuged at 13 000 g for30 min (41C). Supernatants were separated and keptas cytosol. Fractions were loaded onto 10% SDS-PAGE gels and electrophoretically transferred ontonitrocellulose membranes at 1.5 mA (41C for 2 h), in25 mM Tris-HCl, 192 mM glycine (pH 8.2) and 20%methanol. The membranes were blocked with TBS/non-fat dried milk (5%) for 2 h and incubated over-night (41C) with the anti-b-catenin (Santa CruzBiotech., CA, USA), the anti-Phospho-GSK-3b (Ser9)(Upstate Biotechnology, NY, USA), or the anti-GSK-3b(Santa Cruz Biotech., CA, USA) antibodies diluted1 : 1000 in TBS/non-fat dried milk 5%. The secondaryantibody was labeled with alkaline phosphatase anddetected using NBT and BCIP. The anti-tubulinantibody (Santa Cruz Biotech., CA, USA) was usedas a load control.
Intrahippocampal injection with preformed Ab fibrilsMale Sprague–Dawley rats (200 g) were randomlydivided into two groups: the saline-treated group(control) and the chronic LiCl-treated group. LiCl wasdissolved in physiological saline (0.9% NaCl) andwas administered intraperitoneally once a day with aload dose of 3 meq/kg for 2 weeks prior to surgery. Amaintenance dose of 2 meq/kg was given during thefollowing 2 weeks. Control rats were injected withphysiological saline alone. Plasma LiCl concentra-tions were measured in a separate group of ratstreated with chronic lithium doses, at the end of thepre- and post-treatment period (data not shown).After 2 weeks of lithium treatment, rats wereanesthetized using Equitesin (2.5 ml/kg i.p.) andinjected bilaterally into the dorsal hippocampus(�3.5 mm AP, 72.0 mm ML and �2.7 DV, accordingto Bregma) stereotaxically with a 10ml Hamiltonsyringe. In total, 3.0 ml of Ab fibrils (7.5 mg/ml),ibotenic acid (3.0 mg/3ml), or aCSF were administeredat a rate of 0.5 ml/min. On the second day aftersurgery, the animals were post-treated with LiCl orsaline as control for 2 weeks before the Morris watermaze task40 was applied.
Perfusion and fixation of rat brainsAnimals were anesthetized using Equitesin (2.5 ml/kgi.p.) and injected with heparin (4 USP/kg i.p.) beforeperfusion. Rats were perfused through the heart withbuffer containing 0.1% sodium nitrite, followed byfixation with 4% paraformaldehyde in 0.1 M phos-phate buffer (PB) for 30 min. Brains were removedfrom their skulls and post-fixed in the same fixativesolution for 3 h at room temperature, followed by 10%
sucrose in PB overnight at 41C. After fixation, brainswere coded to ensure unbiased processing andanalysis. Finally, the brains were cut into 40 mmcoronal sections with a cryostat (Leitz 1900) at �201C.Sections from the same brain were divided intogroups for analysis by Nissl staining (0.3% cresylviolet), as previously described,41 and for immuno-histochemical staining (see below).
Immunohistochemical staining of hippocampalsectionsAll sections were mounted on gelatin-coated slides,air dried, dehydrated by serial rinses in gradedethanol solutions, cleared with xylene and cover-slipped with Canada (Merck, Damstadt, Germany).Free floating immunohistochemical procedures wereperformed as described.41 Throughout experiments,washing and dilution of immunoreagents was carriedout using 0.01 M PBS and 0.2% Triton X-100 (PBS+T);two PBS+T washes were performed following eachantibody incubation. Immunodetection of Ab wasperformed using rabbit anti-Ab (1 : 500) polyclonalantibody (Sigma Co, St Louis) incubated overnight at41C. GSK-3b was detected with a goat polyclonalantibody (Santa Cruz Biotech., CA, USA) diluted1 : 300 in PBS+T. A horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG was used as a secondantibody (1 : 600) and incubated for 1 h at roomtemperature. After two washes, staining was gener-ated by incubating in 0.6% DAB for 15 min, addingH2O2 at a final concentration of 0.01% and incubatingfor 4 min. Hippocampal sections were pre-treatedwith 0.5% H2O2 for 30 min to reduce endogenousperoxidase activity, followed by treatment with 5%normal goat serum (DAKO) at room temperature for1 h to avoid non-specific binding. b-catenin wasimmunodetected with the anti b-catenin antibody(1 : 200, Santa Cruz Biotech., CA, USA) and asecondary antibody conjugated to FITC (1 : 300).Sections were observed under a fluorescent micro-scope. Finally, amyloid deposits were also visualizedby Thioflavin-S (Th-S) staining, as described pre-viously.35 To quantify b-catenin-positive cells, fluor-escence microscopy images were placed under atransparency sheet divided into nine equal squares.Eight different persons, without knowing the corre-sponding treatments, counted b-catenin-positive cellsin each picture by this method. Then, the valuesobtained were graphed as a vertical point scatter plot.The mean of the eight observations is shown in eachcase.
TUNEL assayIn order to detect the onset of apoptotic processes, weused the In Situ Cell Death Detection Kit, TMR redTUNEL assay (Roche Molecular Biochemicals, Mann-heim, Germany). Rats injected either with Ab fibrilsor with ibotenic acid were fixed by perfusion 24 hafter injection and the brains were cut into 30 mmsections. To reduce the fluorescent backgroundinduced by paraformaldehyde fixation, sections
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
197
Molecular Psychiatry
mounted in gelatin-coated slides were treated with0.15 M glycine pH 7.4 at RT for 15 min followingincubation with NaBH4 for 15 min at RT. The sectionswere permeabilized with 0.1% TX-100 in 0.1%sodium citrate at 41C. Then, brain sections wereincubated with the TUNEL reaction mixture for60 min at 371C in a humidified atmosphere in thedark. After brain sections were washed twice withblocking buffer (PBS, 0.1% TX-100, 0.5% BSA),coverslips were mounted with fluorescent mediumand directly analysed under a fluorescent microscopeand by confocal microscopy. As a negative control,brain sections were incubated with Label Solution(without terminal transferase) instead of TUNELreaction mixture. As a positive control, brain sectionswere incubated with DNase I to induce DNA strandbreaks. Negative and positive controls are not shown.
Behavioral testsAll animals were trained in a circular water maze40
(1.6 m diameter, 75 cm deep and painted black) usinga two trial per day regime. Rats were trained for 5consecutive days, followed by 2 days off, and thentrained for 4 additional days. Each trial began whenrats were allowed to swim, always starting at the sameeastern point (see below) and ended when the animalresided on a platform for 5 s. Rats were then removed
from the maze, dried and returned to their cages. Fordescriptive data collection, imaginary lines sub-divided the pool into four equal quadrants. Theselines intersected the edge of the pool at the arbitrarycardinal start locations called north, south, east andwest. The platform (9 cm in diameter) was located inthe center of the northwest quadrant (hidden quad-rant) and the pool was divided into three equidistantconcentric annuli. Trial data were gathered using awater maze video tracking system (HVS Imagem,Hampton, UK). Water depth and temperature weremaintained at 50 cm and 19–211C.
Results
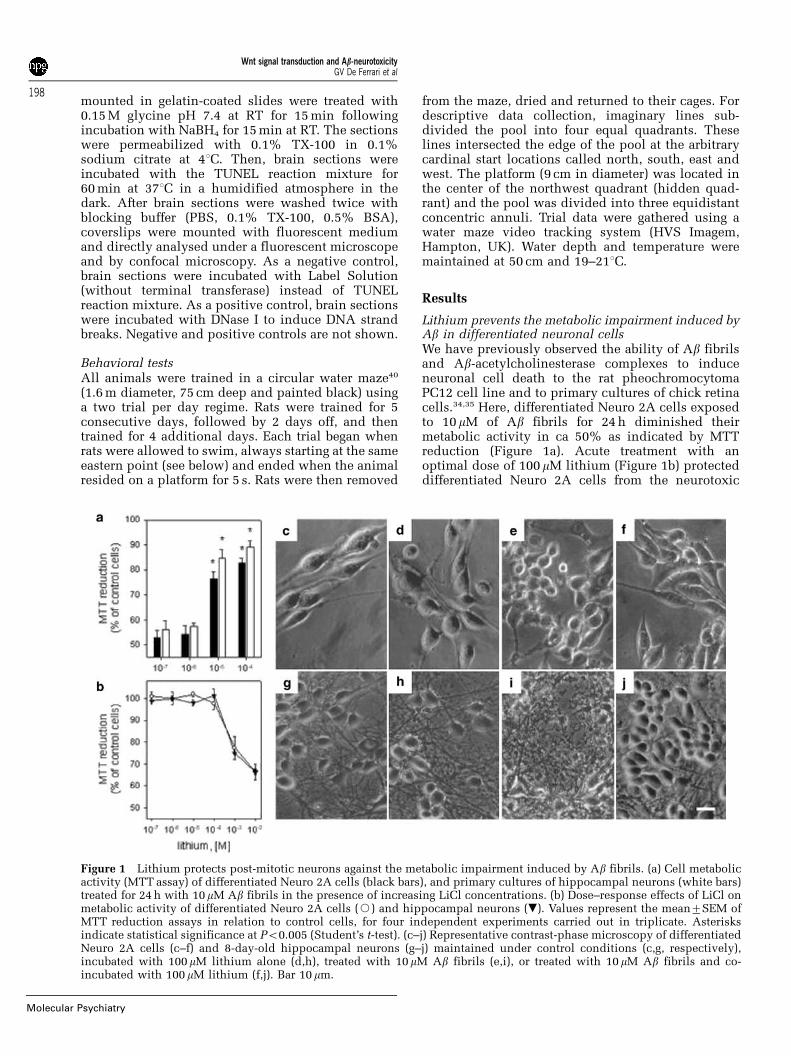
Lithium prevents the metabolic impairment induced byAb in differentiated neuronal cellsWe have previously observed the ability of Ab fibrilsand Ab-acetylcholinesterase complexes to induceneuronal cell death to the rat pheochromocytomaPC12 cell line and to primary cultures of chick retinacells.34,35 Here, differentiated Neuro 2A cells exposedto 10mM of Ab fibrils for 24 h diminished theirmetabolic activity in ca 50% as indicated by MTTreduction (Figure 1a). Acute treatment with anoptimal dose of 100mM lithium (Figure 1b) protecteddifferentiated Neuro 2A cells from the neurotoxic
Figure 1 Lithium protects post-mitotic neurons against the metabolic impairment induced by Ab fibrils. (a) Cell metabolicactivity (MTT assay) of differentiated Neuro 2A cells (black bars), and primary cultures of hippocampal neurons (white bars)treated for 24 h with 10mM Ab fibrils in the presence of increasing LiCl concentrations. (b) Dose–response effects of LiCl onmetabolic activity of differentiated Neuro 2A cells (J) and hippocampal neurons (.). Values represent the mean7SEM ofMTT reduction assays in relation to control cells, for four independent experiments carried out in triplicate. Asterisksindicate statistical significance at Po0.005 (Student’s t-test). (c–j) Representative contrast-phase microscopy of differentiatedNeuro 2A cells (c–f) and 8-day-old hippocampal neurons (g–j) maintained under control conditions (c,g, respectively),incubated with 100mM lithium alone (d,h), treated with 10mM Ab fibrils (e,i), or treated with 10 mM Ab fibrils and co-incubated with 100 mM lithium (f,j). Bar 10 mm.
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
198
Molecular Psychiatry
effects of Ab and determined a significant increase incellular activity for those cells (480% MTT reduc-tion). Primary cultures of hippocampal neurons,which represent a population of mature excitatoryneurons maintained throughout development andadulthood, showed a significantly impaired cellularmetabolism when exposed to Ab fibrils (ca 45% MTTreduction), although this was not so when co-incubated with 100mM lithium (Figure 1a). Underlight microscopy, differentiated Neuro 2A (Figures1c–f), as well as hippocampal cells (Figures 1g–j),showed clear morphological alterations upon expo-sure to 10mM Ab fibrils. In particular, differentiatedNeuro 2A and hippocampal neurons, which undercontrol conditions formed an extensive network ofwell-spread shapes and a profusion of long neuritesfibrils showed somatic shrinkage plus severe axonaland dendritic dystrophy (Figures 1e and i). A total of
100mM lithium protected post-mitotic neurons byway of preserving their normal shape, axonal pro-cesses, and neurites (Figures 1f and j). Finally,neurons incubated only with 100mM lithium didnot display altered morphologies (Figures 1d and h).
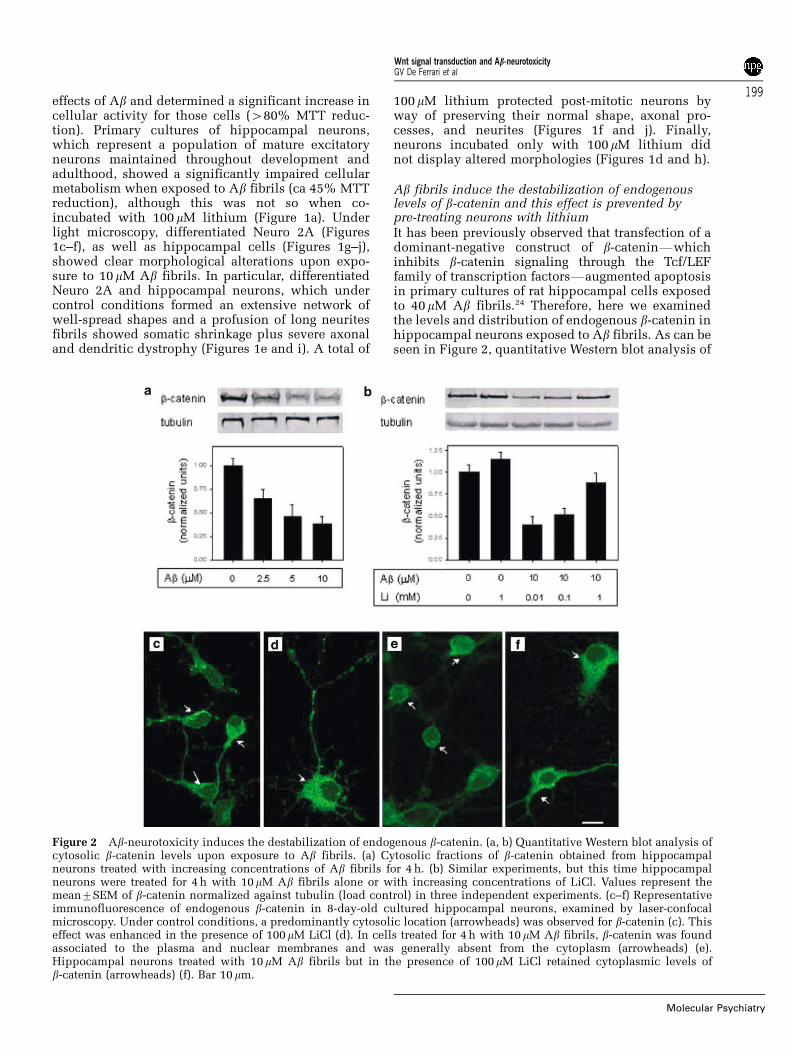
Ab fibrils induce the destabilization of endogenouslevels of b-catenin and this effect is prevented bypre-treating neurons with lithiumIt has been previously observed that transfection of adominant-negative construct of b-cateninFwhichinhibits b-catenin signaling through the Tcf/LEFfamily of transcription factorsFaugmented apoptosisin primary cultures of rat hippocampal cells exposedto 40mM Ab fibrils.24 Therefore, here we examinedthe levels and distribution of endogenous b-catenin inhippocampal neurons exposed to Ab fibrils. As can beseen in Figure 2, quantitative Western blot analysis of
Figure 2 Ab-neurotoxicity induces the destabilization of endogenous b-catenin. (a, b) Quantitative Western blot analysis ofcytosolic b-catenin levels upon exposure to Ab fibrils. (a) Cytosolic fractions of b-catenin obtained from hippocampalneurons treated with increasing concentrations of Ab fibrils for 4 h. (b) Similar experiments, but this time hippocampalneurons were treated for 4 h with 10mM Ab fibrils alone or with increasing concentrations of LiCl. Values represent themean7SEM of b-catenin normalized against tubulin (load control) in three independent experiments. (c–f) Representativeimmunofluorescence of endogenous b-catenin in 8-day-old cultured hippocampal neurons, examined by laser-confocalmicroscopy. Under control conditions, a predominantly cytosolic location (arrowheads) was observed for b-catenin (c). Thiseffect was enhanced in the presence of 100mM LiCl (d). In cells treated for 4 h with 10 mM Ab fibrils, b-catenin was foundassociated to the plasma and nuclear membranes and was generally absent from the cytoplasm (arrowheads) (e).Hippocampal neurons treated with 10mM Ab fibrils but in the presence of 100mM LiCl retained cytoplasmic levels ofb-catenin (arrowheads) (f). Bar 10mm.
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
199
Molecular Psychiatry
hippocampal neurons, incubated with increasingconcentrations of Ab fibrils (2.5–10 mM) for 4 h,revealed a significant dose-dependent decrease inthe levels of cytosolic b-catenin up to ca. 35%,compared to control hippocampal neurons (Figure2a). We next hypothesized that the neuroprotectiveeffect of lithium was achieved by way of b-cateninstabilization. Indeed, this effect was reversed in adose-dependent manner when cells were co-incu-bated with Ab fibrils (10mM) and increasing concen-trations of lithium (10mM–1 mM) (Figure 2b).Examined under the confocal microscope, b-catenindisplayed predominantly a cytoplasmic localizationin control hippocampal neurons (Figure 2c), and thisdistribution was enhanced to some extent when cellswere incubated with low doses of lithium (100mM)(Figure 2d). b-catenin was also seen associated toplasma and nuclear membranes and low levels of theprotein were also observed in patchy distributionswithin cell nuclei (Figures 2c and d), a fact that isconsistent with its normal role in cell adhesion andnuclear/transcriptional events. Conversely, b-cateninalmost disappeared from the cytoplasm of hippocam-pal cells when exposed to 10mM Ab fibrils (Figure2e). Remarkably, neurons similarly treated with Abfibrils, but this time in the presence of 100mMlithium, retained the distribution of b-catenin in thecytosol (Figure 2f), as it was observed for controlcells.
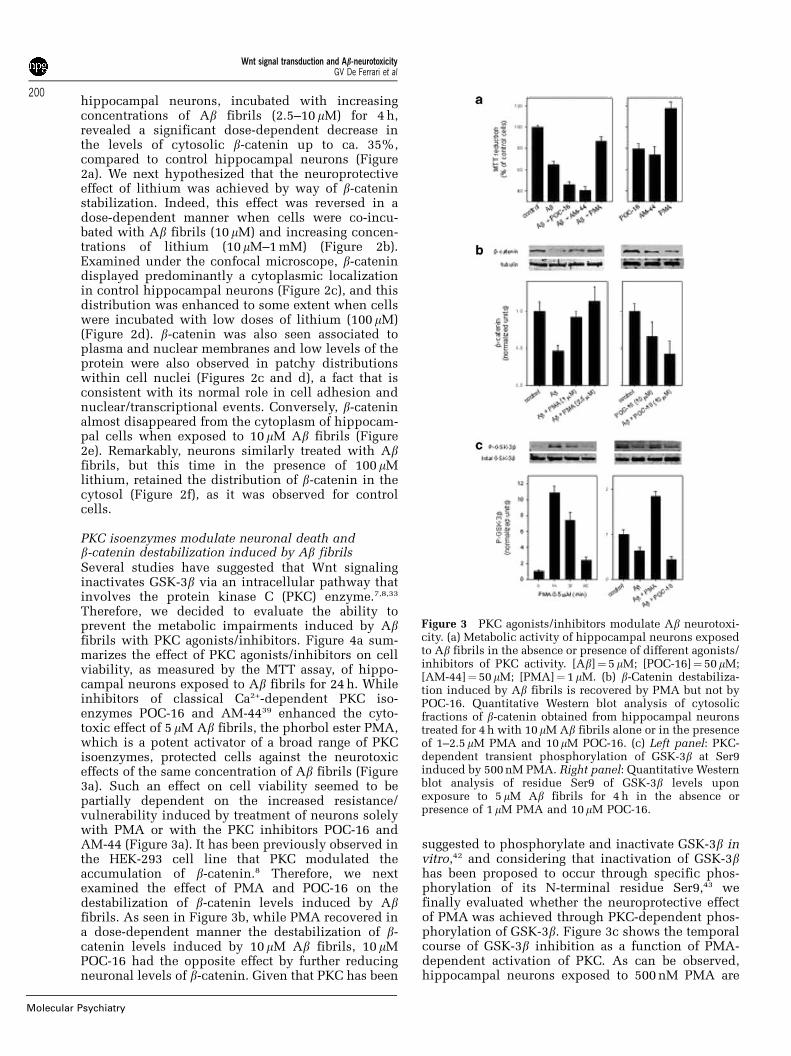
PKC isoenzymes modulate neuronal death andb-catenin destabilization induced by Ab fibrilsSeveral studies have suggested that Wnt signalinginactivates GSK-3b via an intracellular pathway thatinvolves the protein kinase C (PKC) enzyme.7,8,33
Therefore, we decided to evaluate the ability toprevent the metabolic impairments induced by Abfibrils with PKC agonists/inhibitors. Figure 4a sum-marizes the effect of PKC agonists/inhibitors on cellviability, as measured by the MTT assay, of hippo-campal neurons exposed to Ab fibrils for 24 h. Whileinhibitors of classical Ca2+-dependent PKC iso-enzymes POC-16 and AM-4439 enhanced the cyto-toxic effect of 5 mM Ab fibrils, the phorbol ester PMA,which is a potent activator of a broad range of PKCisoenzymes, protected cells against the neurotoxiceffects of the same concentration of Ab fibrils (Figure3a). Such an effect on cell viability seemed to bepartially dependent on the increased resistance/vulnerability induced by treatment of neurons solelywith PMA or with the PKC inhibitors POC-16 andAM-44 (Figure 3a). It has been previously observed inthe HEK-293 cell line that PKC modulated theaccumulation of b-catenin.8 Therefore, we nextexamined the effect of PMA and POC-16 on thedestabilization of b-catenin levels induced by Abfibrils. As seen in Figure 3b, while PMA recovered ina dose-dependent manner the destabilization of b-catenin levels induced by 10mM Ab fibrils, 10 mMPOC-16 had the opposite effect by further reducingneuronal levels of b-catenin. Given that PKC has been
suggested to phosphorylate and inactivate GSK-3b invitro,42 and considering that inactivation of GSK-3bhas been proposed to occur through specific phos-phorylation of its N-terminal residue Ser9,43 wefinally evaluated whether the neuroprotective effectof PMA was achieved through PKC-dependent phos-phorylation of GSK-3b. Figure 3c shows the temporalcourse of GSK-3b inhibition as a function of PMA-dependent activation of PKC. As can be observed,hippocampal neurons exposed to 500 nM PMA are
Figure 3 PKC agonists/inhibitors modulate Ab neurotoxi-city. (a) Metabolic activity of hippocampal neurons exposedto Ab fibrils in the absence or presence of different agonists/inhibitors of PKC activity. [Ab]¼ 5mM; [POC-16]¼ 50 mM;[AM-44]¼ 50 mM; [PMA]¼ 1mM. (b) b-Catenin destabiliza-tion induced by Ab fibrils is recovered by PMA but not byPOC-16. Quantitative Western blot analysis of cytosolicfractions of b-catenin obtained from hippocampal neuronstreated for 4 h with 10mM Ab fibrils alone or in the presenceof 1–2.5 mM PMA and 10 mM POC-16. (c) Left panel: PKC-dependent transient phosphorylation of GSK-3b at Ser9induced by 500 nM PMA. Right panel: Quantitative Westernblot analysis of residue Ser9 of GSK-3b levels uponexposure to 5mM Ab fibrils for 4 h in the absence orpresence of 1mM PMA and 10mM POC-16.
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
200
Molecular Psychiatry
induced to phosphorylate transiently the residue Ser9of GSK-3b (as recognized by a phospho-specificantibody against phospho-Ser9), which suggests thatinhibition of GSK-3b is an essential requisite forprotection against the cytotoxic effects of Ab fibrils.Indeed, hippocampal neurons exposed to 5 mM Abfibrils for 24 h were induced to decrease in ca. 30%the phosphorylation of residue Ser9 of GSK-3b, withrespect to control levels (Figure 3c). According towhat was expected, this cytotoxic effect induced byAb fibrils was reversed when neurons were treatedsimultaneously with 2.5 mM PMA, but not when cellswere co-incubated with 10 mM POC-16 (Figure 3c).
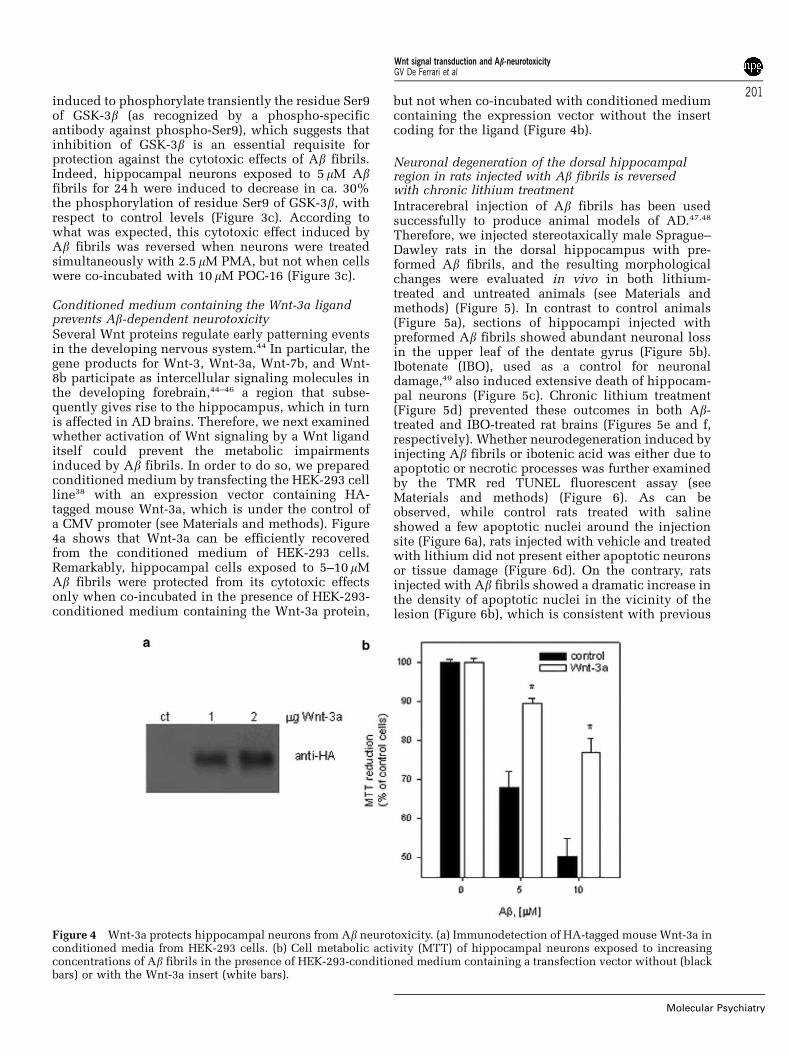
Conditioned medium containing the Wnt-3a ligandprevents Ab-dependent neurotoxicitySeveral Wnt proteins regulate early patterning eventsin the developing nervous system.44 In particular, thegene products for Wnt-3, Wnt-3a, Wnt-7b, and Wnt-8b participate as intercellular signaling molecules inthe developing forebrain,44–46 a region that subse-quently gives rise to the hippocampus, which in turnis affected in AD brains. Therefore, we next examinedwhether activation of Wnt signaling by a Wnt liganditself could prevent the metabolic impairmentsinduced by Ab fibrils. In order to do so, we preparedconditioned medium by transfecting the HEK-293 cellline38 with an expression vector containing HA-tagged mouse Wnt-3a, which is under the control ofa CMV promoter (see Materials and methods). Figure4a shows that Wnt-3a can be efficiently recoveredfrom the conditioned medium of HEK-293 cells.Remarkably, hippocampal cells exposed to 5–10 mMAb fibrils were protected from its cytotoxic effectsonly when co-incubated in the presence of HEK-293-conditioned medium containing the Wnt-3a protein,
but not when co-incubated with conditioned mediumcontaining the expression vector without the insertcoding for the ligand (Figure 4b).
Neuronal degeneration of the dorsal hippocampalregion in rats injected with Ab fibrils is reversedwith chronic lithium treatment
Intracerebral injection of Ab fibrils has been usedsuccessfully to produce animal models of AD.47,48
Therefore, we injected stereotaxically male Sprague–Dawley rats in the dorsal hippocampus with pre-formed Ab fibrils, and the resulting morphologicalchanges were evaluated in vivo in both lithium-treated and untreated animals (see Materials andmethods) (Figure 5). In contrast to control animals(Figure 5a), sections of hippocampi injected withpreformed Ab fibrils showed abundant neuronal lossin the upper leaf of the dentate gyrus (Figure 5b).Ibotenate (IBO), used as a control for neuronaldamage,49 also induced extensive death of hippocam-pal neurons (Figure 5c). Chronic lithium treatment(Figure 5d) prevented these outcomes in both Ab-treated and IBO-treated rat brains (Figures 5e and f,respectively). Whether neurodegeneration induced byinjecting Ab fibrils or ibotenic acid was either due toapoptotic or necrotic processes was further examinedby the TMR red TUNEL fluorescent assay (seeMaterials and methods) (Figure 6). As can beobserved, while control rats treated with salineshowed a few apoptotic nuclei around the injectionsite (Figure 6a), rats injected with vehicle and treatedwith lithium did not present either apoptotic neuronsor tissue damage (Figure 6d). On the contrary, ratsinjected with Ab fibrils showed a dramatic increase inthe density of apoptotic nuclei in the vicinity of thelesion (Figure 6b), which is consistent with previous
Figure 4 Wnt-3a protects hippocampal neurons from Ab neurotoxicity. (a) Immunodetection of HA-tagged mouse Wnt-3a inconditioned media from HEK-293 cells. (b) Cell metabolic activity (MTT) of hippocampal neurons exposed to increasingconcentrations of Ab fibrils in the presence of HEK-293-conditioned medium containing a transfection vector without (blackbars) or with the Wnt-3a insert (white bars).
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
201
Molecular Psychiatry
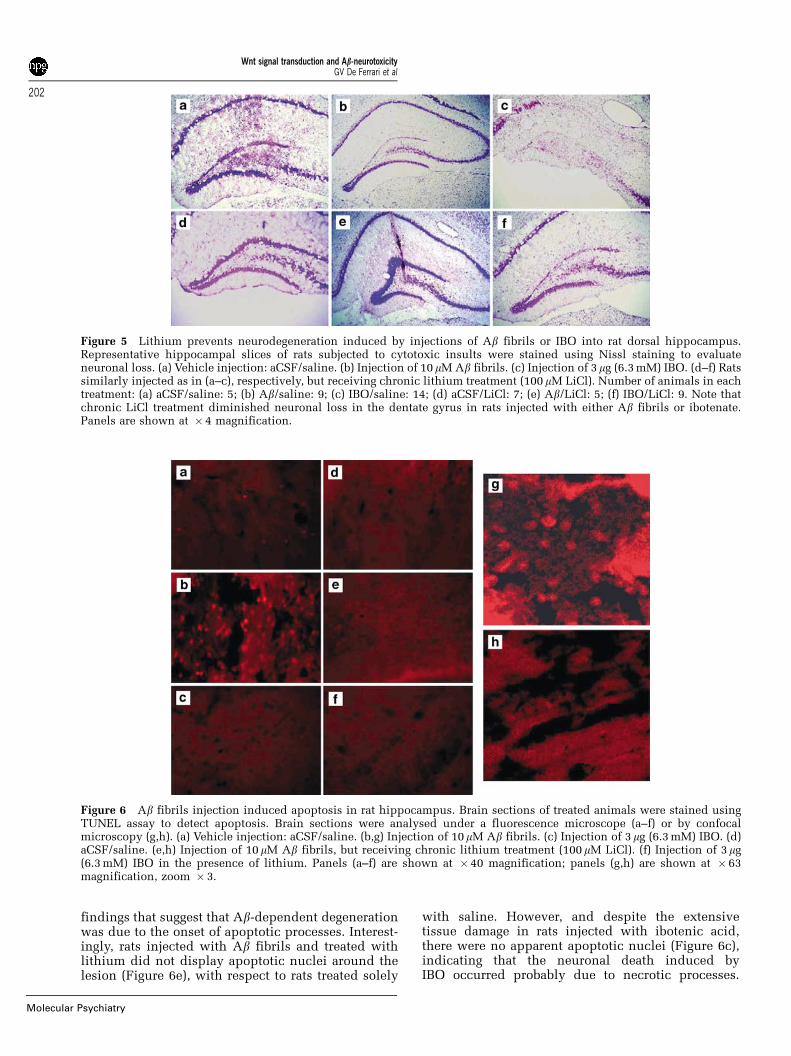
findings that suggest that Ab-dependent degenerationwas due to the onset of apoptotic processes. Interest-ingly, rats injected with Ab fibrils and treated withlithium did not display apoptotic nuclei around thelesion (Figure 6e), with respect to rats treated solely
with saline. However, and despite the extensivetissue damage in rats injected with ibotenic acid,there were no apparent apoptotic nuclei (Figure 6c),indicating that the neuronal death induced byIBO occurred probably due to necrotic processes.
Figure 5 Lithium prevents neurodegeneration induced by injections of Ab fibrils or IBO into rat dorsal hippocampus.Representative hippocampal slices of rats subjected to cytotoxic insults were stained using Nissl staining to evaluateneuronal loss. (a) Vehicle injection: aCSF/saline. (b) Injection of 10 mM Ab fibrils. (c) Injection of 3mg (6.3 mM) IBO. (d–f) Ratssimilarly injected as in (a–c), respectively, but receiving chronic lithium treatment (100 mM LiCl). Number of animals in eachtreatment: (a) aCSF/saline: 5; (b) Ab/saline: 9; (c) IBO/saline: 14; (d) aCSF/LiCl: 7; (e) Ab/LiCl: 5; (f) IBO/LiCl: 9. Note thatchronic LiCl treatment diminished neuronal loss in the dentate gyrus in rats injected with either Ab fibrils or ibotenate.Panels are shown at � 4 magnification.
Figure 6 Ab fibrils injection induced apoptosis in rat hippocampus. Brain sections of treated animals were stained usingTUNEL assay to detect apoptosis. Brain sections were analysed under a fluorescence microscope (a–f) or by confocalmicroscopy (g,h). (a) Vehicle injection: aCSF/saline. (b,g) Injection of 10mM Ab fibrils. (c) Injection of 3mg (6.3 mM) IBO. (d)aCSF/saline. (e,h) Injection of 10 mM Ab fibrils, but receiving chronic lithium treatment (100 mM LiCl). (f) Injection of 3mg(6.3 mM) IBO in the presence of lithium. Panels (a–f) are shown at � 40 magnification; panels (g,h) are shown at � 63magnification, zoom � 3.
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
202
Molecular Psychiatry
Moreover, although rats injected with IBO and treatedwith lithium did not show apoptotic neurons, thetissue in the vicinity of the injection site was never-theless protected (Figure 6f). Micrographs obtained athigher magnification by confocal microscopy (63� ,zoom 3� ) also revealed the presence of apoptoticnuclei in saline-treated rats injected with Ab fibrils incontrast with lithium-treated rats injected with Abfibrils (Figures 6g and h, respectively).
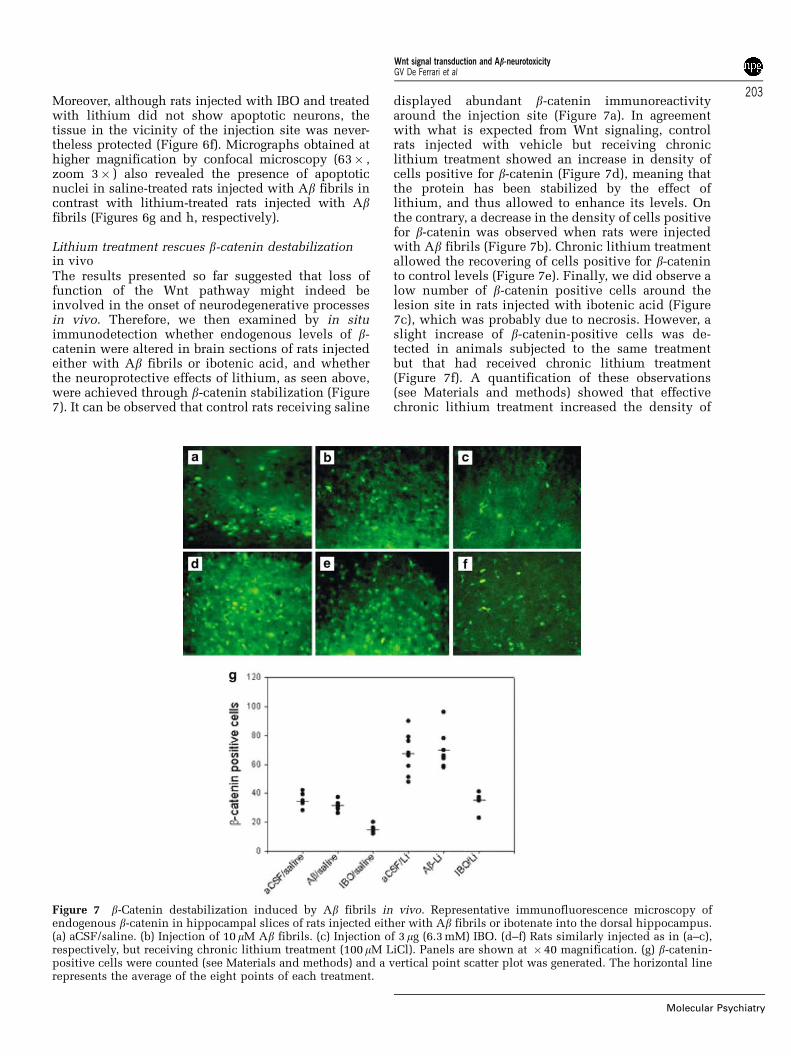
Lithium treatment rescues b-catenin destabilizationin vivoThe results presented so far suggested that loss offunction of the Wnt pathway might indeed beinvolved in the onset of neurodegenerative processesin vivo. Therefore, we then examined by in situimmunodetection whether endogenous levels of b-catenin were altered in brain sections of rats injectedeither with Ab fibrils or ibotenic acid, and whetherthe neuroprotective effects of lithium, as seen above,were achieved through b-catenin stabilization (Figure7). It can be observed that control rats receiving saline
displayed abundant b-catenin immunoreactivityaround the injection site (Figure 7a). In agreementwith what is expected from Wnt signaling, controlrats injected with vehicle but receiving chroniclithium treatment showed an increase in density ofcells positive for b-catenin (Figure 7d), meaning thatthe protein has been stabilized by the effect oflithium, and thus allowed to enhance its levels. Onthe contrary, a decrease in the density of cells positivefor b-catenin was observed when rats were injectedwith Ab fibrils (Figure 7b). Chronic lithium treatmentallowed the recovering of cells positive for b-cateninto control levels (Figure 7e). Finally, we did observe alow number of b-catenin positive cells around thelesion site in rats injected with ibotenic acid (Figure7c), which was probably due to necrosis. However, aslight increase of b-catenin-positive cells was de-tected in animals subjected to the same treatmentbut that had received chronic lithium treatment(Figure 7f). A quantification of these observations(see Materials and methods) showed that effectivechronic lithium treatment increased the density of
Figure 7 b-Catenin destabilization induced by Ab fibrils in vivo. Representative immunofluorescence microscopy ofendogenous b-catenin in hippocampal slices of rats injected either with Ab fibrils or ibotenate into the dorsal hippocampus.(a) aCSF/saline. (b) Injection of 10 mM Ab fibrils. (c) Injection of 3mg (6.3 mM) IBO. (d–f) Rats similarly injected as in (a–c),respectively, but receiving chronic lithium treatment (100mM LiCl). Panels are shown at � 40 magnification. (g) b-catenin-positive cells were counted (see Materials and methods) and a vertical point scatter plot was generated. The horizontal linerepresents the average of the eight points of each treatment.
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
203
Molecular Psychiatry

b-catenin-positive cells in comparison with animalstreated with saline (Figure 7g). Therefore, our resultsdemonstrate that Ab-dependent neurotoxicity targetsb-catenin-responsive neurons for degeneration andthat chronic lithium treatment protects these cellsagainst such cytotoxic insults probably by directinhibition of GSK-3b. A fact consistent with such apossibility is that in the hippocampal region of Ab-treated rats, we observed a strong GSK-3b immunor-eactivity (Figure 8d) around deposits of Ab fibrils,visualized either with the amyloid dye Thioflavine-Sor with an anti-Ab antibody (Figures 8e and f,respectively). Control animals showed neither GSK-3b immunoreactivity nor Ab deposits (Figures 8a–c).
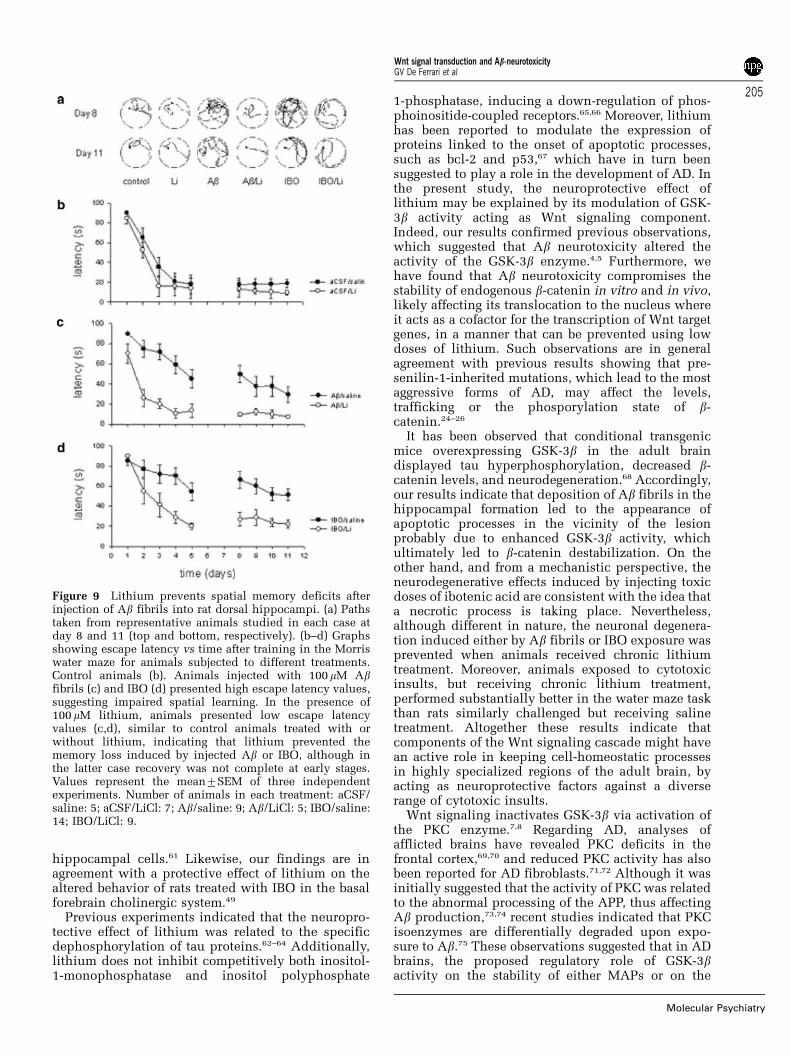
Lithium averts the behavioral impairments induced byhippocampal injection of Ab fibrils
Severe behavioral deficits have been observed inanimals exposed to Ab47,50 and in transgenic miceoverexpressing mutant forms of the hAPP.51,52 There-fore, the possibility to ameliorate with chroniclithium treatment the spatial memory deficits in-duced by intrahippocampal administrations either ofAb fibrils or ibotenic acid was further investigated.Treated rats were trained for 2 weeks in the Morriswater maze paradigm with the hidden platform andthe learning score was recorded (see Materials andmethods). Representative paths taken by control andtreated animals are shown in Figure 9a. It can beobserved that during the first week of maze trials andcompared to control rats injected solely with vehicle(Figure 9b), animals injected with Ab fibrils or IBObut receiving chronic lithium treatment (Figures 9cand d, respectively) showed a faster learning responsethan rats lacking lithium. In the second week, allgroups treated with lithium displayed similar andreduced escape latency values with respect to controlanimals. Therefore, our results suggest that chroniclithium treatment heightened spatial memory andthus prevented the behavioral impairment induced
either by Ab fibrils or IBO, although in the latter caseto a lesser extent.
Discussion
In this study, we have found that Ab-dependentneurotoxicity alters the activity of key components ofthe Wnt signal transduction pathway and that activa-tion of this signaling cascade is sufficient to preventsuch cytotoxic effects. Previously, Ab neurotoxicityhas been linked to the abnormal phosphorylationstate of tau, as is observed in AD brains.3–5 Given thathyperphosphorylation of tau occurs primarily at (Seror Thr)-Pro motifs, it was suggested that proline-directed kinases were responsible for this activity.Among this group of kinases, it is presently acceptedthat GSK-3b plays a major role in the stability ofMAPs such as tau,4,53–55 and the responsive target forWnt signaling MAP-1B.34,56 These observations sug-gested that the activity of the GSK-3b enzyme shouldbe kept tightly regulated in mature neurons. Consis-tent with such idea is the observation in AD brains insitu that neurons containing tangle-like inclusionsthat parallel the sequence of the development ofneurofibrillary changes are positive for active, but notinactive, GSK-3b.57
Here we have observed that lithium, which in-activates GSK-3b and thus acts as a positive regulatorin the Wnt signaling pathway,31,32 prevents thecytotoxic effects of Ab fibrils to post-mitotic neuronsFviz. differentiated Neuro 2A cells and primarycultures of hippocampal neuronsFand recoversneurodegeneration and behavioral impairments inanimals injected either with preformed Ab fibrils orIBO in the dorsal hippocampus in vivo. Our findingsexpand on previous studies in vitro, which showedthat lithium protected rat cortical neurons, cerebellargranule cells and the PC12 cell line from Ab-inducedcytotoxic stresses,58,59 and with experiments in vivo,which suggested that chronic lithium prevented focalcerebral ischemia60 and increased neurogenesis of
Figure 8 Ab deposition in the dorsal hippocampus induces GSK-3b immunoreactivity. Representative hippocampal slicesof control animals (a–c) and rats injected with 10 mM Ab fibrils (d–f). (a,d) Anti-GSK-3b antibody. (b,e) Amyloid staining withThioflavine-S. (c,f) Anti-Ab antibody. Panels are shown at � 10 magnification.
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
204
Molecular Psychiatry
hippocampal cells.61 Likewise, our findings are inagreement with a protective effect of lithium on thealtered behavior of rats treated with IBO in the basalforebrain cholinergic system.49
Previous experiments indicated that the neuropro-tective effect of lithium was related to the specificdephosphorylation of tau proteins.62–64 Additionally,lithium does not inhibit competitively both inositol-1-monophosphatase and inositol polyphosphate
1-phosphatase, inducing a down-regulation of phos-phoinositide-coupled receptors.65,66 Moreover, lithiumhas been reported to modulate the expression ofproteins linked to the onset of apoptotic processes,such as bcl-2 and p53,67 which have in turn beensuggested to play a role in the development of AD. Inthe present study, the neuroprotective effect oflithium may be explained by its modulation of GSK-3b activity acting as Wnt signaling component.Indeed, our results confirmed previous observations,which suggested that Ab neurotoxicity altered theactivity of the GSK-3b enzyme.4,5 Furthermore, wehave found that Ab neurotoxicity compromises thestability of endogenous b-catenin in vitro and in vivo,likely affecting its translocation to the nucleus whereit acts as a cofactor for the transcription of Wnt targetgenes, in a manner that can be prevented using lowdoses of lithium. Such observations are in generalagreement with previous results showing that pre-senilin-1-inherited mutations, which lead to the mostaggressive forms of AD, may affect the levels,trafficking or the phosporylation state of b-catenin.24–26
It has been observed that conditional transgenicmice overexpressing GSK-3b in the adult braindisplayed tau hyperphosphorylation, decreased b-catenin levels, and neurodegeneration.68 Accordingly,our results indicate that deposition of Ab fibrils in thehippocampal formation led to the appearance ofapoptotic processes in the vicinity of the lesionprobably due to enhanced GSK-3b activity, whichultimately led to b-catenin destabilization. On theother hand, and from a mechanistic perspective, theneurodegenerative effects induced by injecting toxicdoses of ibotenic acid are consistent with the idea thata necrotic process is taking place. Nevertheless,although different in nature, the neuronal degenera-tion induced either by Ab fibrils or IBO exposure wasprevented when animals received chronic lithiumtreatment. Moreover, animals exposed to cytotoxicinsults, but receiving chronic lithium treatment,performed substantially better in the water maze taskthan rats similarly challenged but receiving salinetreatment. Altogether these results indicate thatcomponents of the Wnt signaling cascade might havean active role in keeping cell-homeostatic processesin highly specialized regions of the adult brain, byacting as neuroprotective factors against a diverserange of cytotoxic insults.
Wnt signaling inactivates GSK-3b via activation ofthe PKC enzyme.7,8 Regarding AD, analyses ofafflicted brains have revealed PKC deficits in thefrontal cortex,69,70 and reduced PKC activity has alsobeen reported for AD fibroblasts.71,72 Although it wasinitially suggested that the activity of PKC was relatedto the abnormal processing of the APP, thus affectingAb production,73,74 recent studies indicated that PKCisoenzymes are differentially degraded upon expo-sure to Ab.75 These observations suggested that in ADbrains, the proposed regulatory role of GSK-3bactivity on the stability of either MAPs or on the
Figure 9 Lithium prevents spatial memory deficits afterinjection of Ab fibrils into rat dorsal hippocampi. (a) Pathstaken from representative animals studied in each case atday 8 and 11 (top and bottom, respectively). (b–d) Graphsshowing escape latency vs time after training in the Morriswater maze for animals subjected to different treatments.Control animals (b). Animals injected with 100 mM Abfibrils (c) and IBO (d) presented high escape latency values,suggesting impaired spatial learning. In the presence of100 mM lithium, animals presented low escape latencyvalues (c,d), similar to control animals treated with orwithout lithium, indicating that lithium prevented thememory loss induced by injected Ab or IBO, although inthe latter case recovery was not complete at early stages.Values represent the mean7SEM of three independentexperiments. Number of animals in each treatment: aCSF/saline: 5; aCSF/LiCl: 7; Ab/saline: 9; Ab/LiCl: 5; IBO/saline:14; IBO/LiCl: 9.
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
205
Molecular Psychiatry

b-catenin molecule may well relate to deficits in PKCactivity acting as a Wnt signaling component. Indeed,here we have found that while POC-16 and AM-44,which are inhibitors of classical Ca2+-dependent PKCisoenzymes,39 enhanced the metabolic impairmentsinduced by Ab fibrils (namely cell viability, b-catenindestabilization, and GSK-3b activation), the phorbol-ester PMA prevented such neurotoxic effects. Ourresults are in general agreement with previousobservations in PC12 cells and dorsal root ganglioncultures,76,77 which support the notion of a protectiverole for PKC upon Ab insults.
Wnt signaling is revealing itself to be increasinglycomplex and is only now beginning to be under-stood.9,10 Although new modulators of this signalingcascade are being identified and their functionsdiscerned at a rapid pace, there is a major need toestablish whether the function of Wnt components issustained throughout neurodegeneration of the adulthuman brain, as occurs in oncogenic processes.13,14 Inthis respect, we have found that the metabolicimpairments induced by Ab fibrils in hippocampalneurons were reversed selectively when these cellswere co-incubated with conditioned medium con-taining Wnt-3a. Although current experiments are
being undertaken in our lab to establish properly therole that Wnt ligands play in Ab neurotoxicity, ormolecules that help to present its signal to its receptor,our results are suggestive of a major protectivefunction of such molecules during the onset ofneurodegenerative processes (Figure 10). RegardingAD, several genes coding for components of Wntsignaling are located in close proximity to geneticmarkers that account for putative locus for late-onsetAD pedigrees (GV De Ferrari and NC Inestrosa,unpublished observations). Indeed, Wnt8B45 andsecreted Frizzled related protein-5 (SFRP-5)78 arelocated around marker D10S1671 in the long arm ofchromosome 10.79 Likewise, Wnt180 and Wnt10B81 arelocated around markers D12S390 and D12S96 in thelong arm of chromosome 12.82,83 Finally, the genecoding for the low-density lipoprotein receptor-related protein-6 (LRP-6),84 which functions as a co-receptor of Wnt signaling,85 is located close to markerD12S1623 in the short arm of chromosome 12.86
In conclusion, our results indicate that Ab-depen-dent neurotoxicity leads to a loss of function of Wntsignaling components,19 opening the possibility thatlithium or compounds that mimic Wnt signaling maybe used as putative candidates for therapeutic inter-vention in AD patients.
Acknowledgements
We thank Dr FJ Munoz and S Lienlaff for helping usin the early stages of this project. This work wassupported by grants FONDAP Biomedicine No.13980001 and Millenium Institute for Fundamentaland Applied Biology (MIFAB) No. P 99-007-F. NCIand MB are recipients of a Presidential Chair inScience from the Chilean government. NCI is a JohnSimon Guggenheim Memorial Foundation Fellow.
References
1 Selkoe DJ. The origins of Alzheimer disease: A is for amyloid.JAMA 2000; 283: 1615–1617.
2 Mandelkow E-M, Mandelkow E. t in Alzheimer’s disease. TrendsCell Biol 1998; 8: 425–427.
3 Busciglio J, Lorenzo A, Yeh J, Yankner BA. b-amyloid fibrilsinduce t phosphorylation and loss of microtubule binding.Neuron 1995; 14: 879–888.
4 Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Tauprotein kinase I is essential for amyloid b-protein-inducedneurotoxicity. Proc Natl Acad Sci USA 1993; 90: 7789–7793.
5 Takashima A, Honda T, Yasutake K, Michel G, Murayama O,Murayama M et al. Activation of t protein kinase I/glycogensynthase kinase-3b by amyloid b-peptide (25–35) enhancesphosphorylation of t in hippocampal neurons. Neurosci Res1998; 31: 317–323.
6 Siegfried E, Chou TB, Perrimon N. Wingless signaling acts throughzeste-white 3, the Drosophila homolog of glycogen synthasekinase-3, to regulate engrailed and establish cell fate. Cell 1992;71: 1167–1179.
7 Cook D, Fry MJ, Hughes K, Sumathipala R, Woodget JR, Dale TC.Wingless inactivates glycogen synthase kinase-3 via an intracel-lular signaling which involves a protein kinase C. EMBO J 1996;15: 4526–4536.
8 Chen RH, Ding WV, McCormick F. Wnt signaling to b-catenininvolves two interactive components. Glycogen synthase
Figure 10 Ab neurotoxicity induces loss of function ofWnt signaling components. Abnormal processing of APPleads to increased deposition/toxicity of the Ab peptide.Increased levels of Ab alter the normal activity of Ca2+-dependent PKC isoenzymes and enhance the activity ofGSK-3b. Activated GSK-3b targets b-catenin for its degrada-tion and tau proteins (t) for hyperphosphorylation. De-graded b-catenin no longer can act as a cofactor for thetranscription of Wnt target genes (eg C-jun); thus loss offunction of Wnt signaling has occurred. Wnt signaling istissue specific; therefore, survival of neurons that dependon its trophic support in selected regions of the brain iscompromised (eg hippocampus), leading to cognitivedecline as is observed in AD brains.
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
206
Molecular Psychiatry

kinase-3b inhibition and activation of protein kinase C. J BiolChem 2000; 275: 17 894–17 899.
9 Wodarz A, Nusse R. Mechanisms of Wnt signaling in develop-ment. Annu Rev Cell Dev Biol 1998; 14: 59–88.
10 Kuhl M, Sheldahl LC, Park M, Miller JL, Moon RT. The Wnt/Ca2+pathway: a new vertebrate Wnt signaling pathway takes shape.Trends Genet 2000; 16: 279–283.
11 Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. Theaxis-inducing activity, stability, and subcellular distribution ofb-catenin is regulated in Xenopus embryos by glycogen synthasekinase 3. Genes Dev 1996; 10: 1443–1454.
12 Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. b-catenin is atarget for the ubiquitin-proteasome pathway. EMBO J 1997; 16:3797–3804.
13 Nusse R, Varmus HE. Wnt genes. Cell 1992; 69: 1073–1087.14 Polakis P. Wnt signaling and cancer. Genes Dev 2000; 14: 1837–
1851.15 Wassink TH, Piven J, Vieland VJ, Huang J, Swiderski E, Pietila J
et al. Evidence supporting WNT2 as an autism susceptibility gene.Am J Med Genet 2001; 105: 406–413.
16 Andres C. Molecular genetics and animal models in autisticdisorder. Brain Res Bull 2002; 57: 109–119.
17 Cotter D, Kerwin R, al-Sarraji S, Brion JP, Chadwich A, LovestoneS et al. Abnormalities of Wnt signalling in schizophreniaFevi-dence for neurodevelopmental abnormality. Neuroreport 1998; 9:1379–1383.
18 Miyaoka T, Seno T, Ishino H. Increased expression of Wnt-1 inschizophrenic brains. Schizophr Res 1999; 38: 1–6.
19 De Ferrari GV, Inestrosa NC. Wnt signaling function in Alzhei-mer’s disease. Brain Res Brain Res Rev 2000; 33: 1–12.
20 Anderton BH, Dayanandan R, Killick R, Lovestone S. Doesdysregulation of the Notch and wingless/Wnt pathways underliethe pathogenesis of Alzheimer’s disease? Mol Med Today 2000; 6:54–59.
21 Zhou J, Liyanage U, Medina M, Ho C, Simmons AD, Lovett M et al.Presenilin 1 interaction in the brain with a novel member of theArmadillo family. NeuroReport 1997; 8: 2085–2090.
22 Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque Let al. The presenilin 1 protein is a component of a high molecularweight intracellular complex that contains b-catenin. J Biol Chem1998; 273: 16 470–16 475.
23 Tesco G, Kim TW, Diehlmann A, Beyreuther K, Tanzi RE.Abrogation of the presenilin 1/b-catenin interaction and preserva-tion of the heterodimeric presenilin 1 complex following caspaseactivation. J Biol Chem 1998; 273: 33 909–33 914.
24 Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C,Staufenbiel M et al. Destabilization of b-catenin by mutations inpresenilin-1 potentiates neuronal apoptosis. Nature 1998; 395:698–702.
25 Nishimura M, Yu G, Levesque G, Zhang DM, Ruel L, Chen F et al.Presenilin mutations associated with Alzheimer disease causedefective intracellular trafficking of b-catenin, a component of thepresenilin protein. Nat Med 1999; 5: 164–169.
26 Kawamura Y, Kikuchi A, Takada R, Takada S, Sudoh S, ShibamotoS et al. Inhibitory effect of a presenilin 1 mutation on the Wntsignalling pathway by enhancement of b-catenin phosphorylation.Eur J Biochem 2001; 268: 3036–3041.
27 Takashima A, Murayama M, Murayama O, Kohno T, Honda T,Yasutake K et al. Presenilin 1 associates with glycogen synthasekinase-3b and its substrate t. Proc Natl Acad Sci USA 1998; 95:9637–9641.
28 Kang DE, Soriano S, Frosch MP, Collins T, Naruse S, Sisodia SSet al. Presenilin 1 facilitates the constitutive turnover ofb-catenin: differential activity of Alzheimer’s disease-linked PS1mutants in the b-catenin-signaling pathway. J Neurosci 1999; 19:4229–4237.
29 Tesco G, Tanzi RE. GSK3b forms a tetrameric complex withendogenous PS1-CTF/NTF and b-catenin. Effects of the D257/D385A and FAD-linked mutations. Ann NY Acad Sci 2000; 920:227–232.
30 Gantier R, Gilbert D, Dumanchin C, Campion D, Davoust D, TomaF et al. The pathogenic L392V mutation of presenilin 1 decreasesthe affinity to glycogen synthase kinase-3b. Neurosci Lett 2000;283: 217–220.
31 Klein PS, Melton DA. A molecular mechanism for the effectof lithium on development. Proc Natl Acad Sci USA 1996; 93:8455–8459.
32 Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogensynthase kinase-3 activity and mimics wingless signalling inintact cells. Curr Biol 1996; 6: 1664–1668.
33 Sheldahl LC, Park M, Malbon CC, Moon RT. Protein kinase C isdifferentially stimulated by Wnt and Frizzled homologs in aG-protein-dependent manner. Curr Biol 1999; 9: 695–698.
34 Inestrosa NC, Alvarez A, Godoy J, Reyes A, De Ferrari GV.Acetylcholinesterase-amyloid–b-peptide interaction and Wnt sig-naling involvement in Ab neurotoxicity. Acta Neurol Scand Suppl2000; 176: 53–59.
35 Alvarez A, Alarcon R, Opazo C, Campos EO, Munoz FJ, CalderonFH et al. Stable complexes involving acetylcholinesterase andamyloid-b peptide change the biochemical properties of theenzyme and increase the neurotoxicity of Alzheimer’s fibrils.J Neurosci 1998; 18: 3213–3223.
36 Banker GA, Cowan WM. Rat hippocampal neurons in dispersedcell culture. Brain Res 1977; 126: 397–442.
37 Calderon FH, von Bernhardi R, De Ferrari GV, Luza S, Aldunate R,Inestrosa NC. Toxic effects of acetylcholinesterase on neuronaland glial-like cells in vitro. Mol Psychiatry 1998; 3: 247–255.
38 Ding VW, Chen RH, McCormick F. Differential regulation ofglycogen synthase kinase 3b by insulin and Wnt signaling. J BiolChem 2000; 275: 32 475–32 481.
39 Garcıa-Huidobro T, Valenzuela E, Leisewitz AV, Valderrama J,Bronfman M. Anti-proliferative effect of two novel palmitoyl-carnitine analogs selective inhibitors of protein kinase C conven-tional isoenzymes. Eur J Biochem 1999; 266: 855–864.
40 Morris R. Developments of a water-maze procedure for studyingspatial learning in the rat. J Neurosci Meth 1984; 11: 47–60.
41 Cote SL, Ribeiro-Da-Silva A, Cuello AC. In: Cuello AC (ed).Immunocytochemistry II. John Wiley & Sons Ltd.: UK, 1993.
42 Goode N, Hughes K, Woodgett JR, Parker PJ. Differential regulationof glycogen synthase kinase-3b by protein kinase C isotypes. J BiolChem 1992; 267: 16 878–16 882.
43 Sutherland C, Leighton IA, Cohen P. Inactivation of glycogensynthase kinase-3b by phosphorylation: new kinase connectionsin insulin and growth-factor signalling. Biochem J 1993; 296:15–19.
44 Patapoutian A, Reichardt LF. Roles of Wnt proteins in neuraldevelopment and maintenance. Curr Opin Neurobiol 2000; 10:392–399.
45 Lako M, Lindsay S, Bullen P, Wilson DI, Robson SC, Strachan T. Anovel mammalian wnt gene WNT8B shows brain-restrictedexpression in early development, with sharply delimited expres-sion boundaries in the developing forebrain. Hum Mol Genet 1998;7: 813–822.
46 Lee SM, Tole S, Grove E, McMahon AP. A local Wnt-3a signal isrequired for development of the mammalian hippocampus.Development 2000; 127: 457–467.
47 O’Hare E, Weldon DT, Mantyh PW, Ghilardi JR, Finke MP,Kuskowski MA et al. Delayed behavioral effects followingintrahippocampal injection of aggregated Ab (1–42). Brain Res1999; 815: 1–10.
48 Chambers CB, Sigurdsson EM, Hejna MJ, Lorens SA, Lee JM,Muma NA. Amyloid-b injection in rat amygdala alters t proteinbut not mRNA expression. Exp Neurol 2000; 162: 158–170.
49 Pascual T, Gonzalez JL. A protective effect of lithium on ratbehaviour altered by ibotenic acid lesions of the basal forebraincholinergic system. Brain Res 1995; 695: 289–292.
50 Yamada K, Tanaka T, Han D, Senzaki K, Kameyama T, NabeshimaT. Protective effects of idebenone and a-tocopherol on b-amyloid-(1–42)-induced learning and memory deficits in rats: implicationof oxidative stress in b-amyloid-induced neurotoxicity in vivo. EurJ Neurosci 1999; 11: 83–90.
51 Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SDet al. Ab peptide immunization reduces behavioural impairmentand plaques in a model of Alzheimer’s disease. Nature 2000; 408:979–982.
52 Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, HardyJ et al. Ab peptide vaccination prevents memory loss in an animalmodel of Alzheimer’s disease. Nature 2000; 408: 982–985.
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
207
Molecular Psychiatry

53 Lovestone S, Reynolds CH, Latimer D, Davis DR, Anderton BH,Gallo JM et al. Alzheimer’s disease-like phosphorylation of themicrotubule-associated protein t by glycogen synthase kinase-3 intransfected mammalian cells. Curr Biol 1994; 4: 1077–1086.
54 Sperber BR, Leight S, Goedert M, Lee VM. Glycogen synthasekinase-3b phosphorylates t protein at multiple sites in intact cells.Neurosci Lett 1995; 197: 149–153.
55 Zheng-Fischhofer Q, Biernat J, Mandelkow EM, Illenberger S,Godemann R, Mandelkow E. Sequential phosphorylation of Tauby glycogen synthase kinase-3b and protein kinase A at Thr212and Ser214 generates the Alzheimer-specific epitope of antibodyAT100 and requires a paired-helical-filament-like conformation.Eur J Biochem 1998; 252: 542–552.
56 Lucas FR, Goold RG, Gordon-Weeks PR, Salinas PC. Inhibition ofGSK-3b leading to the loss of phosphorylated MAP-1B is an earlyevent in axonal remodelling induced by WNT-7a or lithium. J CellSci 1998; 111: 1351–1361.
57 Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B et al.Distribution of active glycogen synthase kinase 3b (GSK-3b) inbrains staged for Alzheimer disease neurofibrillary changes.J Neuropathol Exp Neurol 1999; 58: 1010–1019.
58 Alvarez G, Munoz-Montano JL, Satrustegui J, Avila J, Bogonez E,Diaz-Nido J. Lithium protects cultured neurons against b-amyloid-induced neurodegeneration. FEBS Lett 1999; 453: 260–264.
59 Wei H, Leeds PR, Qian Y, Wei W, Chen R, Chuang D. b-amyloidpeptide-induced death of PC12 cells and cerebellar granule cellneurons is inhibited by long-term lithium treatment. Eur JPharmacol 2000; 392: 117–123.
60 Nonaka S, Chuang DM. Neuroprotective effects of chronic lithiumon focal cerebral ischemia in rats. NeuroReport 1998; 9: 2081–2084.
61 Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK.Enhancement of hippocampal neurogenesis by lithium. J Neuro-chem 2000; 75: 1729–1734.
62 Munoz-Montano JR, Moreno FJ, Avila J, Diaz-Nido J. Lithiuminhibits Alzheimer’s disease-like t protein phosphorylation inneurons. FEBS Lett 1997; 411: 183–188.
63 Hong M, Chen DCR, Klein PS, Lee VM-Y. Lithium reduces tphosphorylation by inhibition of glycogen synthase kinase-3.J Biol Chem 1997; 272: 25 326–25 332.
64 Lovestone S, Davis DR, Webster MT, Kaech S, Brion JP, Matus A etal. Lithium reduces t phosphorylation: effects in living cells andin neurons at therapeutic concentrations. Biol Psychiatry 1999; 45:995–1003.
65 Berridge MJ. Cell signalling. A tale of two messengers. Nature1993; 365: 456–459.
66 Acharya JK, Labarca P, Delgado R, Jalink K, Zuker CS. Synapticdefects and compensatory regulation of inositol metabolism ininositol polyphosphate 1-phosphatase mutants. Neuron 1998; 20:1219–1229.
67 Chen R, Chuang D. Long term lithium treatment suppresses p53and Bax expression but increases Bcl-2 expression. A prominentrole in neuroprotection against excitotoxicity. J Biol Chem 1999;274: 6039–6042.
68 Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J.Decreased nuclear b-catenin t hyperphosphorylation and neuro-degeneration in GSK-3b conditional transgenic mice. EMBO J2001; 20: 27–39.
69 Cole G, Dobkins KR, Hansen LA, Terry RD, Saitoh T. Decreasedlevels of protein kinase C in Alzheimer brain. Brain Res 1988; 452:165–174.
70 Shimohama S, Narita M, Matsushima H, Kimura J, Kameyama M,Hagiwara M et al. Assessment of protein kinase C isozymes bytwo-site enzyme immunoassay in human brains and changes inAlzheimer’s disease. Neurology 1993; 43: 1407–1413.
71 Govoni S, Bergamaschi S, Racchi M, Battaini F, Binetti G,Bianchetti A et al. Cytosol protein kinase C down regulation infibroblasts from Alzheimer’s disease patients. Neurology 1993; 43:2581–2586.
72 Van Huynh T, Cole G, Katzman R, Huang KP, Saitoh T. Reducedprotein kinase C immunoreactivity and altered protein phosphor-ylation in Alzheimer’s disease fibroblasts. Arch Neurol 1989; 46:1195–1199.
73 Hung AY, Haass C, Nitsch RM, Qiu WQ, Citron M, Wurtman RJet al. Activation of protein kinase C inhibits cellular production ofthe amyloid b-protein. J Biol Chem 1993; 268: 22 959–22 962.
74 Gabuzda D, Busciglio J, Yankner BA. Inhibition of b-amyloidproduction by activation of protein kinase C. J Neurochem 1993;61: 2326–2329.
75 Favit A, Grimaldi M, Nelson TJ, Alkon DL. Alzheimer’s-specificeffects of soluble b-amyloid on protein kinase C-a and -gdegradation in human fibroblasts. Proc Natl Acad Sci USA 1998;95: 5562–5567.
76 Xie J, Guo Q, Zhu H, Wooten MW, Mattson MP. Protein kinase Ciota protects neural cells against apoptosis induced by amyloidb-peptide. Brain Res Mol Brain Res 2000; 82: 107–113.
77 Ma W, Zheng WH, Belanger S, Kar S, Quirion R. Effects of amyloidpeptides on cell viability and expression of neuropeptides incultured rat dorsal root ganglion neurons: a role for free radicalsand protein kinase C. Eur J Neurosci 2001; 13: 1125–1135.
78 Chang JT, Esumi N, Moore K, Li Y, Zhang S, Chew C et al. Cloningand characterization of a secreted frizzled-related protein that isexpressed by the retinal pigment epithelium. Hum Mol Genet1999; 8: 575–583.
79 Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S et al.Evidence for genetic linkage of Alzheimer’s disease to chromo-some 10q. Science 2000; 290: 2302–2303.
80 Arheden K, Mandahl N, Strombeck B, Isaksson M, Mitelman F.Chromosome localization of the human oncogene INT1 to 12q13by in situ hybridization. Cytogenet Cell Genet 1988; 47: 86–87.
81 Bui TD, Rankin J, Smith K, Huguet EL, Ruben S, Strachan T et al.A novel human Wnt gene, WNT10B, maps to 12q13 and isexpressed in human breast carcinomas. Oncogene 1997; 13: 1249–1253.
82 Pericak-Vance MA, Bass MP, Yamaoka LH, Gaskell PC, Scott WK,Terwedow HA et al. Complete genomic screen in late-onsetfamilial Alzheimer disease. Evidence for a new locus on chromo-some 12. JAMA 1997; 278: 1237–1241.
83 Rogaeva E, Premkumar S, Song Y, Sorbi S, Brindle N, Paterson Aet al. Evidence for an Alzheimer disease susceptibility locus onchromosome 12 and for further locus heterogeneity. JAMA 1998;280: 614–618.
84 Brown SD, Twells RC, Hey PJ, Cox RD, Levy ER, Soderman ARet al. Isolation and characterization of LRP6, a novel member ofthe low density lipoprotein receptor gene family. BiochemBiophys Res Commun 1998; 248: 879–888.
85 Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Yet al. LDL-receptor-related proteins in Wnt signal transduction.Nature 2000; 407: 530–535.
86 Mayeux R, Lee JH, Romas SN, Mayo D, Santana V, Williamson Jet al. Chromosome-12 mapping of late-onset Alzheimer diseaseamong Caribbean Hispanics. Am J Hum Genet 2002; 70: 237–243.
Wnt signal transduction and Ab-neurotoxicityGV De Ferrari et al
208
Molecular Psychiatry