activation of retinoid receptors rara and rxra induces differentiation and apoptosis, respectively,...
TRANSCRIPT

Vol. 7, 179-186, February 1996 Cell Growth & Differentiation 179
Activation of Retinoid Receptors RARa and RXRa InducesDifferentiation and Apoptosis, Respectively, in HL-60 Cells’
Kapil Mehta,2 Teresa McQueen, Nouri Neamati,Steven Collins, and Michael AndreeffDepartments of Bioimmunotherapy [K. M., T. M., N. N.] andHematology [M. A.], The University of Texas M. D. Anderson CancerCenter, Houston, Texas 77030, and Fred Hutchinson Cancer ResearchCenter, Seattle, Washington 98125 [S. C.]
AbstractInduction of granulocytic differentiation in HL-60myeloid leukemia cells by retinoids is followed by theirdeath via apoptosis. Retinoids are known to mediatetheir biological effects through at least two distinct
types of nuclear receptors, the retinoic acid receptorsand retinoid X receptors. We undertook to characterizethe potential role of these receptors in inducingdifferentiation and apoptosis by retinoids. For this, weused a previously described variant of an HL-60 cellline (HL-60R) in which retinoid receptor function has
been abrogated due to a trans-dominant negativemutation. Retroviral vector-mediated gene transfer wasused to introduce the normal retinoic acid receptor(RARa) or retinoid X receptor (RXRa) into HL-60R cells.Our results suggest that ligand-Induced activation ofRARa is sufficient to induce differentiation in HL-60cells, whereas activation of RXRa can induce directapoptosis of these cells without their priorcommitment to differentiate.
IntroductionNormal myelopoiesis involves a series of events that leads tothe differentiation of very primitive hematopoietic precursorstem cells into terminally differentiated granulocytes (1). Ma-ture granulocytes represent the most abundant and shortest
lived of all the hematopoietic cell populations. The averagehalf-life of normal granulocytes in the circulating blood,where they function as specific effector cells in host-defensemechanisms, is about 1 2 h. After spending relatively short
periods in a functional state, granulocytes are eliminatedfrom the body through a genetically regulated mechanismcalled apoptosis or programmed cell death (2-4). A critical
Received 9/25/95; revised 1 1/6/95; accepted 11/17/95.The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to mdi-cate this fact.I This work is supported in part by USPHS Grant FDR000923 from theFood and Drug Administration and by a grant from Argus Pharmaceuti-cels, Inc. (The Woodlands, TX).2 To whom requests for reprints should be addressed, at Department ofBioimmunotherapy, Box 60, The University of Texas M. D. AndersonCancer Center, I 51 5 Holcombe Boulevard, Houston, TX 77030. Phone:(713) 792-2649; Fax: (713) 796-1731.
balance between the life span of these cells and their regu-lated death is important for normal homeostasis.
The human myeloblast cell line HL-60 provides a conve-nient model for studying the regulatory mechanisms of my-
eloid cell differentiation. This cell line can be induced todifferentiate into a granulocytic lineage following ATRA3treatment (5). Retinoid-differentiated HL-60 cells, like normalgranulocytes, have a limited in vitro life span that could befurther induced to undergo apoptosis in response to certainstimuli (6). This suggests that HL-60 cells could also serve asa model to study the mechanisms of apoptosis in terminallydifferentiated hematopoietic cells. However, the interpreta-tion of retinoid effects on cell growth, differentiation, or ap-optosis is difficult because retinoids mediate these effects bybinding and activating two different types of nuclear recep-tors: the RARs and RXRs, which differ in their sequences andexhibit distinct ligand-binding properties (7-9). The RARsbind both ATRA and its 9-cis stereoisomer (9-cis RA),whereas the RXRs bind only 9-cis RA (1 0-1 2). Since most ofthe blood cells, including HL-60, express both types of re-ceptors (1 3, 1 4), retinoid-induced effects in these cells maybe a result of activation of either RARs, RXRs, or both typesof receptors.
We directly addressed this problem in a mutant subcloneof the HL-6O cell line (HL-60R) in which retinoid receptorfunction has been abrogated as a result of a trans-dominantnegative regulatory point mutation in the ligand-binding do-main of the receptor (1 5). HL-60R subclones expressingspecific receptors were generated by retrovirus-mediatedtransduction of RARa- or RXRa-specific coding sequences(1 6, 17). Our results suggest that the introduction of RARainto HL-60R cells completely restored their sensitivity toATRA-induced granulocytic differentiation. In contrast, theintroduction of RXRa cDNA in HL-60R cells rendered themmarkedly sensitive to apoptosis in response to the RXR-specific ligand, 9-cis RA. Our observations provide directevidence that ATRA-induced terminal differentiation ofHL-60 cells is mediated by activation of RARa, whereasligand activation of RXRa receptors triggers a signal thatleads to programmed death, or apoptosis, of these cells.
Results
Receptor-specific Regulation of CD38 and TGase byRetinoids. Treatment of HL-60 cells with retinoids has beenshown to induce the rapid and acute expression of a cell
3 The abbreviations used are: ATRA, all-trans retinoic acid; RA, retinoicacid; AAR, retinoic acid receptor; RXA, retinoid X receptor; TGase, trans-glutaminase; S:N ratio, signal:noise ratio; TdT, terminal deoxynucleotidyltransferase; MiT, 3-(4,5-dimethyfthiazol-2-yl)-2,5-diphenyl tetrazoliumbromide.
A. B.
. control
U0 +9-cisRA
100
75
50
25
0
C’)0C.)
L’;�HL-GOR HL-60R-RARz HL�60R�RXRz
A B
11$.
t
S
HL-60R HL-60R-RARi HL-60R-RXAz
surface glycoprotein, CD38 (1 7-1 9), as well as of a protein
cross-linking enzyme, TGase (20). In an initial experiment, we
studied the effects of ATRA and 9-cis RA on CD38 and
TGase expression in the HL-60R cell line, as well as itssublines, that had been retrovirally transfected with full-
length RARa (HL-60R-RARa) or RXRa (HL-60R-RXRa)
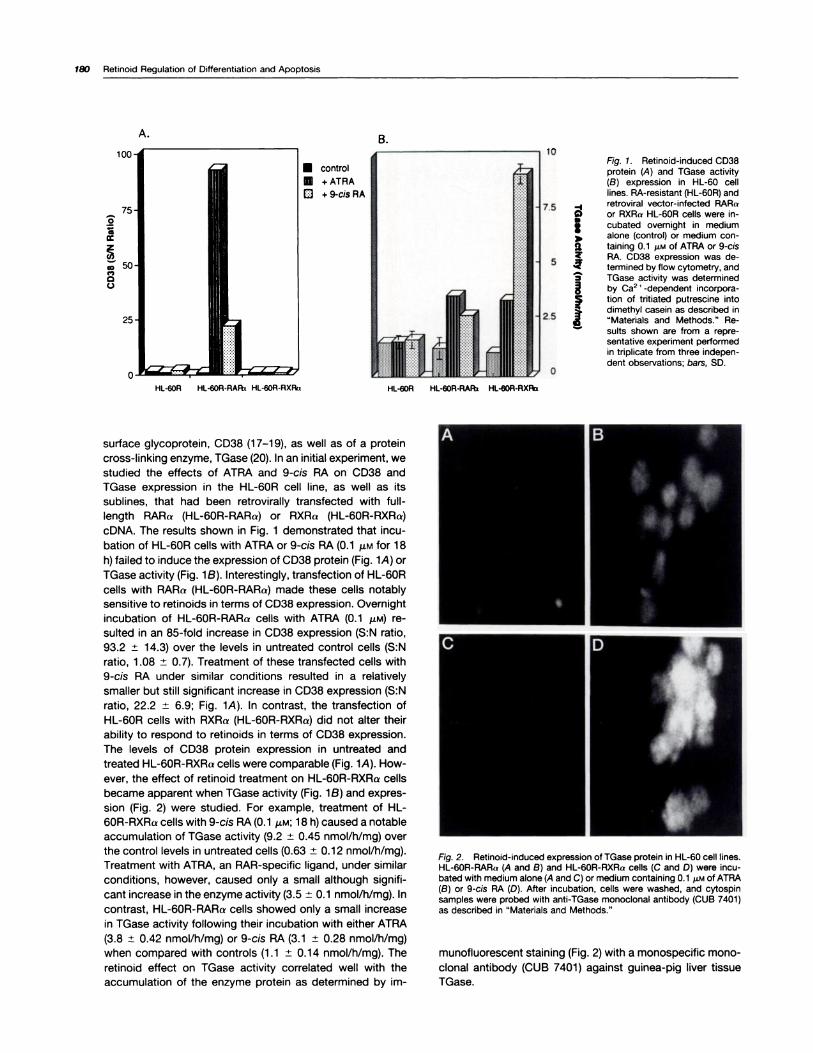
cDNA. The results shown in Fig. 1 demonstrated that incu-
bation of HL-60R cells with ATRA or 9-cis RA (0.1 �M for 18
h) failed to induce the expression of CD38 protein (Fig. 1A) or
TGase activity (Fig. 1B). Interestingly, transfection of HL-60R
cells with RARa (HL-60R-RARa) made these cells notablysensitive to retinoids in terms of CD38 expression. Overnight
incubation of HL-60R-RARa cells with ATRA (0.1 p.M) re-
suIted in an 85-fold increase in CD38 expression (S:N ratio,
93.2 ± 14.3) over the levels in untreated control cells (S:N
ratio, 1 .08 ± 0.7). Treatment of these transfected cells with
9-cis RA under similar conditions resulted in a relatively
smaller but still significant increase in CD38 expression (S:N
ratio, 22.2 ± 6.9; Fig. 1A). In contrast, the transfection of
HL-60R cells with RXRa (HL-60R-RXRa) did not alter their
ability to respond to retinoids in terms of CD38 expression.
The levels of CD38 protein expression in untreated andtreated HL-60R-RXRa cells were comparable (Fig. 1A). How-ever, the effect of retinoid treatment on HL-60R-RXRa cells
became apparent when TGase activity (Fig. 1B) and expres-
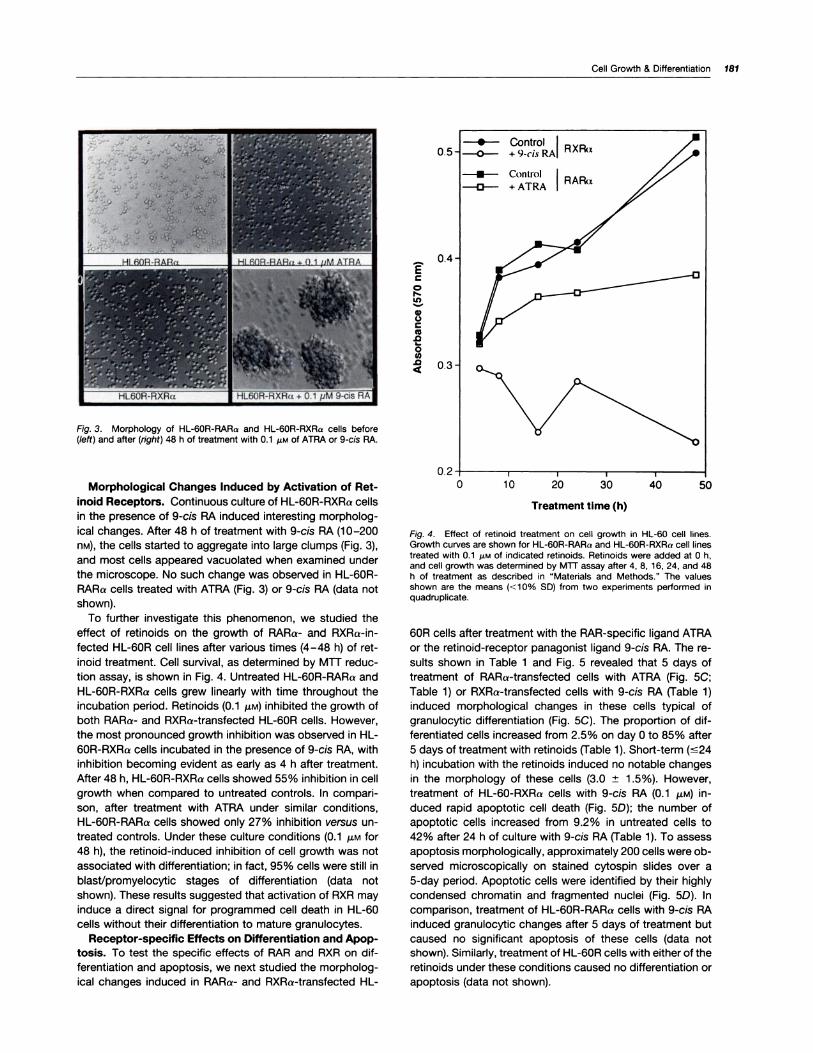
sion (Fig. 2) were studied. For example, treatment of HL-
60R-RXRa cells with 9-cis RA (0.1 �.LM; 18 h) caused a notable
accumulation of TGase activity (9.2 ± 0.45 nmol/h/mg) over
the control levels in untreated cells (0.63 ± 0.12 nmol/h/mg).
Treatment with ATRA, an RAR-specific ligand, under similar
conditions, however, caused only a small although signifi-
cant increase in the enzyme activity (3.5 ± 0.1 nmol/h/mg). In
contrast, HL-60R-RARa cells showed only a small increase
in TGase activity following their incubation with either ATRA
(3.8 ± 0.42 nmol/h/mg) or 9-cis RA (3.1 ± 0.28 nmol/h/mg)
when compared with controls (1 .1 ± 0.14 nmol/h/mg). The
retinoid effect on TGase activity correlated well with the
accumulation of the enzyme protein as determined by im-
(0�
V
Fig. 2. Retinoid-induced expression of TGase protein in HL-60 cell lines.HL-60R-RAAa (A and B) and HL-60A-RXRa cells (C and D) were incu-bated with medium alone (A and C) or medium containing 0.1 �M of ATRA(B) or 9-cis RA (D). After incubation, cells were washed, and cytospinsamples were probed with anti-TGase monoclonal antibody (CUB 7401)as described in “Materials and Methods.”
munofluorescent staining (Fig. 2) with a monospecific mono-
clonal antibody (CUB 7401) against guinea-pig liver tissue
180 Retinoid Regulation of Differentiation and Apoptosis
TGase.
Fig. 1. Aetinoid-induced CD38protein (A) and TGase activity(B) expression in HL-60 celllines. RA-resistant (HL-60R) and
-I retroviral vector-infected RARa�) or RXRa HL-60R cells were in-
I cubated ovemight in medium� alone (control) or medium con-
a taming o.i �LM of ATRA or 9-cis�. RA. CD38 expression was de-
termined by flow cytometry, and3 TGase activity was determined� by Ca2 � -dependent incorpora-� tion of tntiated putrescine into
� dimethyl casein as described in“Materials and Methods.” Re-
� suits shown are from a repre-sentative experiment performedin triplicate from three indepen-dent observations; bars, SD.
�‘- �
:Jr;T�1:�:r�:r. ___________________________________
F
- :Iv�’:;�:4;t. _________________________
Treatment time (h)
Cell Growth & Differentiation 181
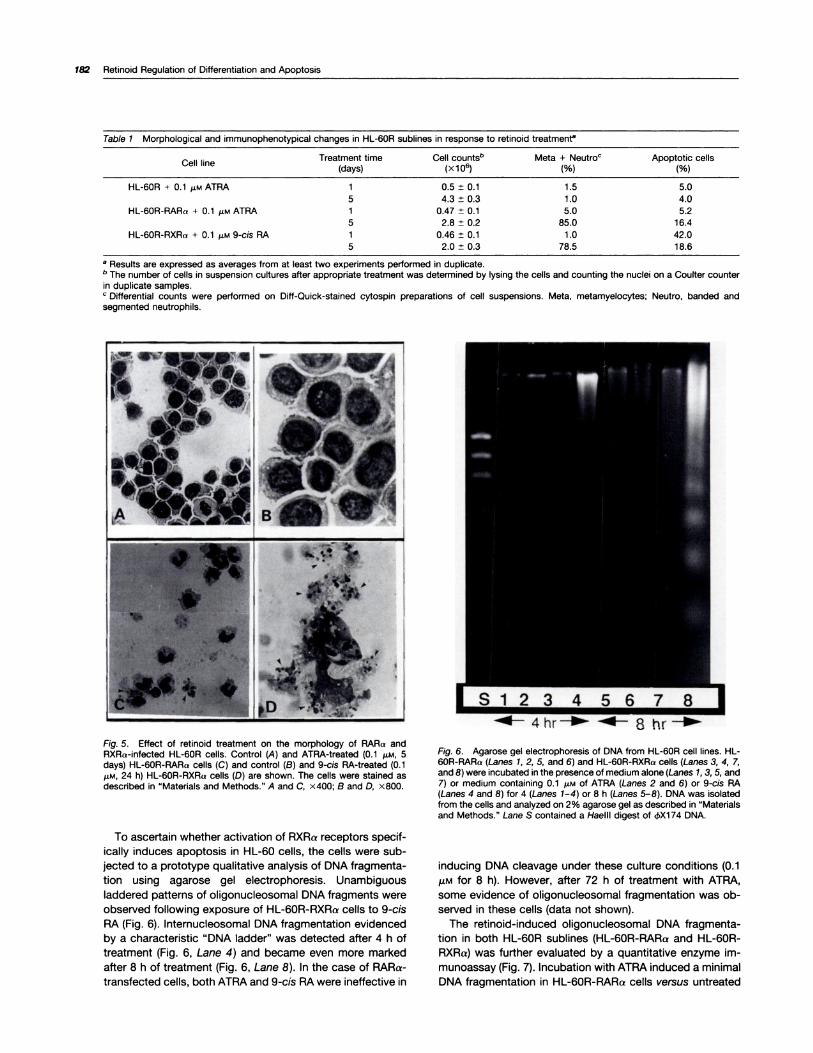
Fig. 3. Morphology of HL-60R-RARa and HL-60R-RXRo cells before(left) and after (right) 48 h of treatment with 0.1 j�M of ATRA or 9-cis RA.
Morphological Changes Induced by Activation of Ret-
inoid Receptors. Continuous culture of HL-60R-RXRa cells
in the presence of 9-cis RA induced interesting morpholog-ical changes. After 48 h of treatment with 9-cis RA (10-200
nM), the cells started to aggregate into large clumps (Fig. 3),
and most cells appeared vacuolated when examined under
the microscope. No such change was observed in HL-60R-
RARa cells treated with ATRA (Fig. 3) or 9-cis RA (data not
shown).
To further investigate this phenomenon, we studied the
effect of retinoids on the growth of RARa- and RXRa-in-
fected HL-60R cell lines after various times (4-48 h) of ret-
inoid treatment. Cell survival, as determined by MiT reduc-
tion assay, is shown in Fig. 4. Untreated HL-60R-RARa and
HL-60R-RXRa cells grew linearly with time throughout the
incubation period. Retinoids (0.1 f.LM) inhibited the growth of
both RARr- and RXRa-transfected HL-60R cells. However,
the most pronounced growth inhibition was observed in HL-
60R-RXRa cells incubated in the presence of 9-cis RA, withinhibition becoming evident as early as 4 h after treatment.
After 48 h, HL-60R-RXRa cells showed 55% inhibition in cell
growth when compared to untreated controls. In compari-
son, after treatment with ATRA under similar conditions,
HL-60R-RARa cells showed only 27% inhibition versus un-
treated controls. Under these culture conditions (0.1 j.�M for
48 h), the retinoid-induced inhibition of cell growth was not
associated with differentiation; in fact, 95% cells were still in
blast/promyelocytic stages of differentiation (data not
shown). These results suggested that activation of RXR may
induce a direct signal for programmed cell death in HL-60
cells without their differentiation to mature granulocytes.
Receptor-specific Effects on Differentiation and Apop-
tosis. To test the specific effects of RAR and RXR on dif-
ferentiation and apoptosis, we next studied the morpholog-
ical changes induced in RARa- and RXRa-transfected HL-
EC
0F,..In
0
C
.0
0(�)
.0
Fig. 4. Effect of retinoid treatment on cell growth in HL-60 cell lines.Growth curves are shown for HL-60R-RARa and HL-60R-RXRa cell linestreated with 0.1 �LM of indicated retinoids. Retinoids were added at 0 h,and cell growth was determined by MiT assay after 4, 8, 1 6, 24, and 48h of treatment as described in “Materials and Methods.” The valuesshown are the means (< 1 0% SD) from two experiments performed inquadruplicate.
60R cells after treatment with the RAR-specific ligand ATRA
or the retinoid-receptor panagonist ligand 9-cis RA. The re-
suIts shown in Table 1 and Fig. 5 revealed that 5 days of
treatment of RARa-transfected cells with ATRA (Fig. SC;
Table 1) or RXRa-transfected cells with 9-cis RA (Table 1)
induced morphological changes in these cells typical of
granulocytic differentiation (Fig. SC). The proportion of dif-
ferentiated cells increased from 2.5% on day 0 to 85% after
S days of treatment with retinoids (Table 1). Short-term (s24
h) incubation with the retinoids induced no notable changes
in the morphology of these cells (3.0 ± 1 .5%). However,
treatment of HL-60-RXRa cells with 9-cis RA (0.1 .LM) in-
duced rapid apoptotic cell death (Fig. SD); the number of
apoptotic cells increased from 9.2% in untreated cells to
42% after 24 h of culture with 9-cis RA (Table 1). To assess
apoptosis morphologically, approximately 200 cells were ob-
served microscopically on stained cytospin slides over a
S-day period. Apoptotic cells were identified by their highly
condensed chromatin and fragmented nuclei (Fig. SD). In
comparison, treatment of HL-60R-RARa cells with 9-cis RA
induced granulocytic changes after S days of treatment but
caused no significant apoptosis of these cells (data not
shown). Similarly, treatment of HL-60R cells with either of the
retinoids under these conditions caused no differentiation or
apoptosis (data not shown).
182 Retinoid Regulation of Differentiation and Apoptosis
Table 1 Morphological and immunophenotyp ical changes in HL-60R su blines in response to ret inoid treatment”
C II Ie ne Treatment time(days)
Cell countsb(x106)
Meta + Neutroc(%)
Apoptotic cells(%)
HL-60R + 0.1 ,.LM ATRA 1 0.5 ± 0.1 1.5 5.0
5 4.3 ± 0.3 1.0 4.0HL-60R-RARa + 0.1 �M ATRA 1 0.47 ± 0.1 5.0 5.2
5 2.8 ± 0.2 85.0 16.4
HL-60R-RXRr + 0.1 �M 9-cis RA 1 0.46 ± 0.1 1 .0 42.05 2.0 ± 0.3 78.5 18.6
a Results are expressed as averages from at least two experiments performed in duplicate.b The number of cells in suspension cultures after appropriate treatment was determined by lysing the cells and counting the nuclei on a Coulter counter
in duplicate samples.C Differential counts were performed on Duff-Quick-stained cytospin preparations of cell suspensions. Meta, metamyelocytes; Neutro, banded and
segmented neutrophils.
Fig. 5. Effect of retinoid treatment on the morphology of RARo andRXRa-infected HL-60R cells. Control (A) and ATRA-treated (0.1 �M, 5days) HL-60R-AARa cells (C) and control (B) and 9-cis RA-treated (0.1�LM, 24 h) HL-60R-RXRa cells (D) are shown. The cells were stained asdescribed in “Materials and Methods.” A and C, x400; B and D, x800.
To ascertain whether activation of RXRa receptors specif-
ically induces apoptosis in HL-60 cells, the cells were sub-
jected to a prototype qualitative analysis of DNA fragmenta-
tion using agarose gel electrophoresis. Unambiguous
laddered patterns of oligonucleosomal DNA fragments were
observed following exposure of HL-60R-RXRa cells to 9-cis
RA (Fig. 6). Internucleosomal DNA fragmentation evidenced
by a characteristic “DNA ladder” was detected after 4 h of
treatment (Fig. 6, Lane 4) and became even more marked
after 8 h of treatment (Fig. 6, Lane 8). In the case of RARa-
transfected cells, both ATRA and 9-cis RA were ineffective in
Fig. 6. Agarose gel electrophoresis of DNA from HL-60R cell lines. HL-60R-RARr (Lanes 1, 2, 5, and 6) and HL-60R-RXRa cells (Lanes 3, 4, 7,and 8) were incubated in the presence of medium alone (Lanes 1, 3, 5, and7) or medium containing 0.1 MM of ATRA (Lanes 2 and 6) or 9-cis RA(Lanes 4 and 8) for 4 (Lanes 1-4) or 8 h (Lanes 5-8). DNA was isolatedfrom the cells and analyzed on 2% agarose gel as described in “Materialsand Methods.” Lane S contained a Haelll digest of 4X174 DNA.
inducing DNA cleavage under these culture conditions (0.1
�M for 8 h). However, after 72 h of treatment with ATRA,some evidence of oligonucleosomal fragmentation was ob-
served in these cells (data not shown).
The retinoid-induced oligonucleosomal DNA fragmenta-
tion in both HL-60R sublines (HL-60R-RARa and HL-60R-
RXRa) was further evaluated by a quantitative enzyme im-
munoassay (Fig. 7). Incubation with ATRA induced a minimalDNA fragmentation in HL-60R-RARa cells versus untreated
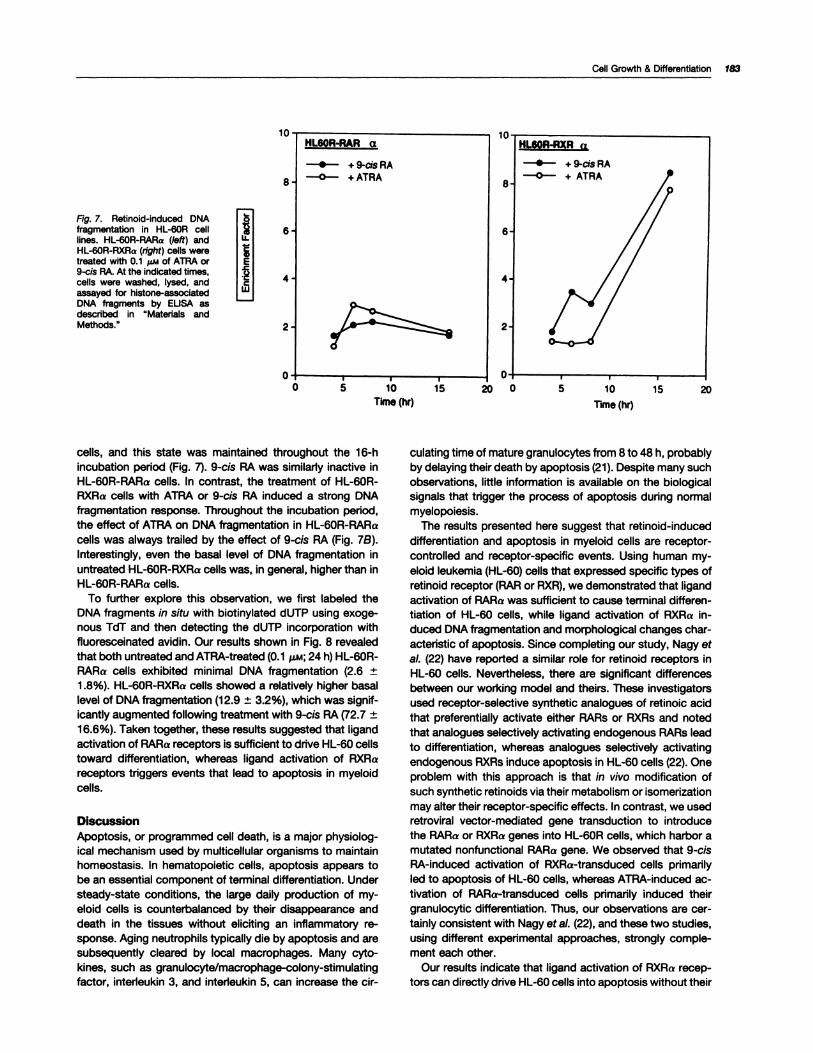
Fig. 7. Aetinoid-induced DNAfragmentation in HL-60A celllines. HL-60A-AARa (left) andHL-60A-AXAa (right) cells weretreated with 0.1 �i,,i of ATRA or9-cis RA. At the indicated times,cells were washed, lysed, andassayed for histone-associatedDNA fragments by EUSA asdescribed in “Materials andMethods.”
I�II El
L�j
HLSOR-RAR a
.- +9-cisRA
-0- +ATRA8
6
4
2
00 1’O 1’5 20 0
Time (hr) Time (hr)5 10 15 20
Cell Growth & Differentiation 183
cells, and this state was maintained throughout the 16-hincubation period (Fig. 7). 9-cis RA was similarly inactive in
HL-60R-RARa cells. In contrast, the treatment of HL-60R-RXRa cells with ATRA or 9-cis RA induced a strong DNAfragmentation response. Throughout the incubation period,the effect of ATRA on DNA fragmentation in HL-60R-RARacells was always trailed by the effect of 9-cis RA (Fig. 7B).
Interestingly, even the basal level of DNA fragmentation inuntreated HL-60R-RXRa cells was, in general, higher than inHL-60R-RARa cells.
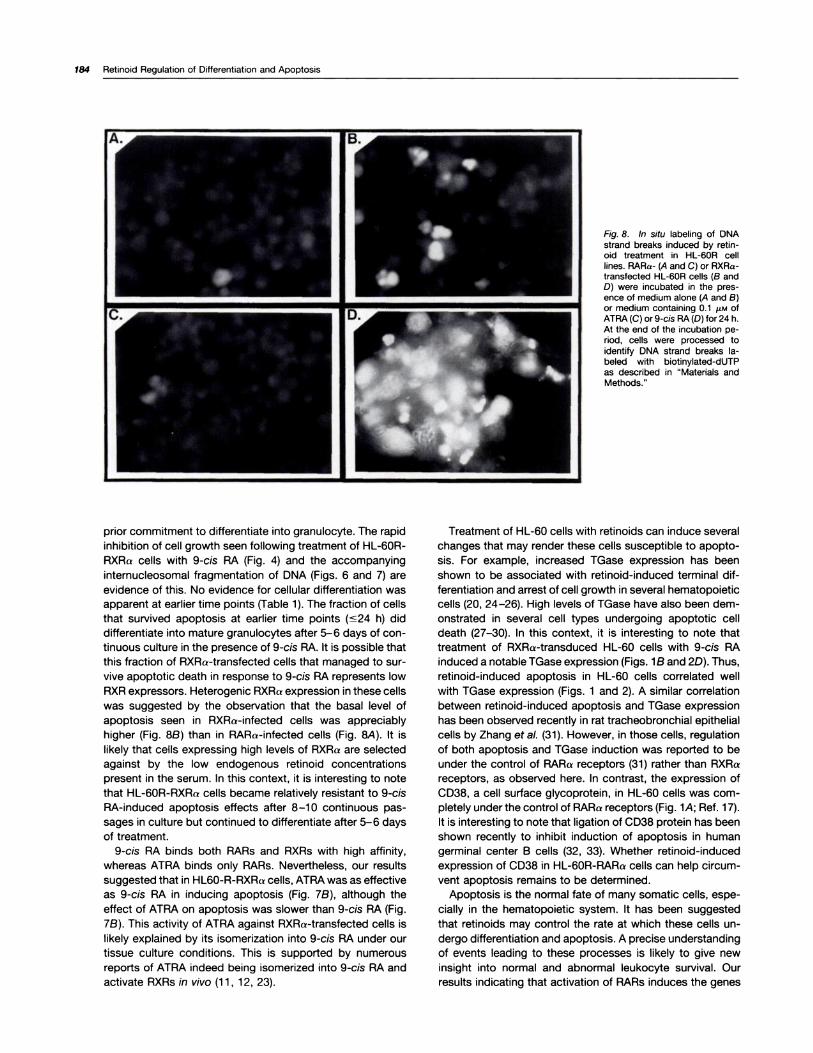
To further explore this observation, we first labeled theDNA fragments in situ with biotinylated dUTP using exoge-nous TdT and then detecting the dUTP incorporation withfluoresceinated avidin. Our results shown in Fig. 8 revealedthat both untreated and ATRA-treated (0.1 �i; 24 h) HL-60R-RARa cells exhibited minimal DNA fragmentation (2.6 ±1 .8%). HL-60R-RXRa cells showed a relatively higher basallevel of DNA fragmentation (12.9 ± 3.2%), which was signif-icantly augmented following treatment with 9-cis RA (72.7 ±16.6%). Taken together, these results suggested that ligandactivation of RARa receptors is sufficient to drive HL-60 cellstoward differentiation, whereas ligand activation of RXRa
receptors triggers events that lead to apoptosis in myeloidcells.
Discussion
Apoptosis, or programmed cell death, is a major physiolog-ical mechanism used by multicellular organisms to maintainhomeostasis. In hematopoietic cells, apoptosis appears to
be an essential component of terminal differentiation. Understeady-state conditions, the large daily production of my-
eloid cells is counterbalanced by their disappearance anddeath in the tissues without eliciting an inflammatory re-sponse. Aging neutrophils typically die by apoptosis and are
subsequently cleared by local macrophages. Many cyto-kines, such as granulocyte/macrophage-colony-stimulatingfactor, interleukin 3, and interleukin 5, can increase the cir-
culating time of mature granulocytes from 8 to 48 h, probablyby delaying their death by apoptosis (21). Despite many suchobservations, little information is available on the biologicalsignals that trigger the process of apoptosis during normalmyelopoiesis.
The results presented here suggest that retinoid-induceddifferentiation and apoptosis in myeloid cells are receptor-controlled and receptor-specific events. Using human my-eloid leukemia (HL-60) cells that expressed specific types of
retinoid receptor (RAR or RXR), we demonstrated that ligandactivation of RARa was sufficient to cause terminal differen-tiation of HL-60 cells, while ligand activation of RXRa in-
duced DNA fragmentation and morphological changes char-acteristic of apoptosis. Since completing our study, Nagy et
a!. (22) have reported a similar role for retinoid receptors inHL-60 cells. Nevertheless, there are significant differencesbetween our working model and theirs. These investigators
used receptor-selective synthetic analogues of retinoic acidthat preferentially activate either RARs or RXRs and notedthat analogues selectively activating endogenous RARs leadto differentiation, whereas analogues selectively activatingendogenous RXRs induce apoptosis in HL-60 cells (22). Oneproblem with this approach is that in vivo modification of
such synthetic retinoids via their metabolism or isomerizationmay alter their receptor-specific effects. In contrast, we usedretroviral vector-mediated gene transduction to introducethe RARa or RXRa genes into HL-60R cells, which harbor amutated nonfunctional RARa gene. We observed that 9-cis
RA-induced activation of RXRa-transduced cells primarilyled to apoptosis of HL-60 cells, whereas ATRA-induced ac-tivation of RARa-transduced cells primarily induced their
granulocytic differentiation. Thus, our observations are car-
tainly consistent with Nagy et aL (22), and these two studies,using different experimental approaches, strongly comple-ment each other.
Our results indicate that ligand activation of RXRa recep-
tors can directly drive HL-60 cells into apoptosis without their
184 Retinoid Regulation of Differentiation and Apoptosis
Fig. 8. In situ labeling of DNAstrand breaks induced by retin-oid treatment in HL-60R celllines. RARa- (A and C) or RXRa-transfected HL-60R cells (B andD) were incubated in the pres-ence of medium alone (A and B)or medium containing 0.1 �LM ofATRA(C) or 9-cis RA(D)for 24 h.At the end of the incubation pe-nod, cells were processed toidentify DNA strand breaks Ia-beled with biotinylated-dUTPas described in “Materials andMethods.”
prior commitment to differentiate into granulocyte. The rapid
inhibition of cell growth seen following treatment of HL-60R-
RXRa cells with 9-cis RA (Fig. 4) and the accompanyinginternucleosomal fragmentation of DNA (Figs. 6 and 7) are
evidence of this. No evidence for cellular differentiation was
apparent at earlier time points (Table 1). The fraction of cells
that survived apoptosis at earlier time points �s24 h) did
differentiate into mature granulocytes after 5-6 days of con-
tinuous culture in the presence of 9-cis RA. It is possible that
this fraction of RXRa-transfected cells that managed to sur-
vive apoptotic death in response to 9-cis RA represents low
RXR expressors. Heterogenic RXRa expression in these cells
was suggested by the observation that the basal level ofapoptosis seen in RXRa-infected cells was appreciably
higher (Fig. 8B) than in RARa-infected cells (Fig. 8A). It is
likely that cells expressing high levels of RXRa are selected
against by the low endogenous retinoid concentrations
present in the serum. In this context, it is interesting to note
that HL-60R-RXRa cells became relatively resistant to 9-cis
RA-induced apoptosis effects after 8-1 0 continuous pas-
sages in culture but continued to differentiate after 5-6 daysof treatment.
9-cis RA binds both RARs and RXRs with high affinity,
whereas ATRA binds only RARs. Nevertheless, our results
suggested that in HL6O-R-RXRa cells, ATRA was as effective
as 9-cis RA in inducing apoptosis (Fig. 7B), although the
effect of ATRA on apoptosis was slower than 9-cis RA (Fig.
7B). This activity of ATRA against RXRa-transfected cells is
likely explained by its isomerization into 9-cis RA under our
tissue culture conditions. This is supported by numerous
reports of ATRA indeed being isomerized into 9-cis RA and
activate RXRs in vivo (1 1 , 12, 23).
Treatment of HL-60 cells with retinoids can induce several
changes that may render these cells susceptible to apopto-
sis. For example, increased TGase expression has been
shown to be associated with retinoid-induced terminal dif-
ferentiation and arrest of cell growth in several hematopoietic
cells (20, 24-26). High levels of TGase have also been dem-
onstrated in several cell types undergoing apoptotic cell
death (27-30). In this context, it is interesting to note that
treatment of RXRa-transduced HL-60 cells with 9-cis RA
induced a notable TGase expression (Figs. 1B and 2D). Thus,
retinoid-induced apoptosis in HL-60 cells correlated well
with TGase expression (Figs. 1 and 2). A similar correlation
between retinoid-induced apoptosis and TGase expression
has been observed recently in rat tracheobronchial epithelial
cells by Zhang et a!. (31). However, in those cells, regulation
of both apoptosis and TGase induction was reported to be
under the control of RARa receptors (31) rather than RXRa
receptors, as observed here. In contrast, the expression of
CD38, a cell surface glycoprotein, in HL-60 cells was corn-
pletely under the control of RARa receptors (Fig. 1A; Ref. 17).
It is interesting to note that ligation of CD38 protein has been
shown recently to inhibit induction of apoptosis in human
germinal center B cells (32, 33). Whether retinoid-induced
expression of CD38 in HL-60R-RARa cells can help circum-
vent apoptosis remains to be determined.
Apoptosis is the normal fate of many somatic cells, espe-
cially in the hematopoietic system. It has been suggested
that retinoids may control the rate at which these cells un-
dergo differentiation and apoptosis. A precise understanding
of events leading to these processes is likely to give new
insight into normal and abnormal leukocyte survival. Our
results indicating that activation of RARs induces the genes

Cell Growth & Differentiation 185
linked to cellular differentiation while activation of RXRs in-duces genes linked to apoptosis may suggest ways to con-trol cell survival in diseases ranging from aplastic anemia to
leukemia.
Materials and Methods
Cells, Cell Growth, and Cellular Morphology. The human myeloid eu-kemia cell line HL-60 was cloned to produce a line resistant to the
differentiating effects of ATRA (designated HL-60R). This was done by
continuous culture of the parental HL-60 cell line in the presence of ATRA(Sigma Chemical Co., St. Louis, MO) as described earlier (34). Two addi-tional subclones from HL-60A cells HL-60R-RARa and HL-60A-AXRa
were developed by retroviral vector (LXSN)-mediated transduction ofcDNA fragments harboring the complete coding sequences of RAAa andAXAa, respectively (34). All three cloned cell lines were maintained inRPMI 1640 supplemented with 5% FCS, 2 mu L-glutamine, 100 units/mI
penicillin, and 1 00 �g/ml streptomycin. In addition, the two retinoid re-ceptor-transfected cell lines (HL-60R-RARa and HL-60R-RXAa) were
continuously cultured in the presence of neomycin (1 mg/mI) to preventselection against retrovirus-negative clones.
Cells were resuspended at a concentration of 0.5-5 x 1 0� cells/mi ingrowth medium containing 0.1 pi�i ATRA or 9-cis RA (kindly provided by
Dr. Richard Heyman, LJgand Pharmaceuticals, San Diego, CA). Cellsmaintained in medium or in DMSO at concentrations used for delivering
the retinoids served as controls. Results were obtained from at least three
separate experiments performed in duplicate.The number of viable cells after indicated times of retinoid treatment
was determined by thelr ability to reduce MU to insoluble formazan (35).Alternatively, the cell number was determined by counting the nuclel on a
Couitercounter(Coulter Electronics, Inc., Hialeah, FL)afterlysing the cellswith Zap-Oglobin (Coulter Diagnostics). Cell morphology was studied bylight microscopy. In brief, after appropriate treatment, cytospin prepara-tions of cells were fixed in 100% methanol and stained with Diff-Quick
stain (Dade Diagnostic, Aguada, Puerto Rico). Cells (�200 cells/sample)
were scored as differentiated if they showed metamyelocytic or moremature forms and as apoptotic if they contained fragmented nuclei.
Determination of CD38 Expression and TGase ACtIVItY. Expressionof CD38 protein was determined as described earlier (1 7). Briefly, the cellswere stained with Leu-17 (PE-conjugated; Becton Dickinson, San Jose,CA) anti-CD38 monoclonal antibody and analyzed on a FACScan flowcytometer using LYSIS II Research software (Becton Dickinson). Thepercentage of CD38� cells was determined over a background of cellsstained with an isotypic control antibody (PE-labeled IgGi ; Becton Dick-
inson). The results were expressed in a S:N ratio defined as the mean
fluorescence of CD38� cells divided by the mean fluorescence of cellsstained with the isotypic control antibody.
TGase activity in cell lysates was determined by enzymatic assay asdescribed earlier (35). The enzyme activity was expressed as nmols pu-
trescine incorporated/h/mg cell protein. Expression of TGase protein inretinoid-treated and untreated cells was also determined by immunoflu-
orescence staining. In brief, cytospin preparations of cells were fixed in
absolute methanol, blocked with human serum albumin and normal hu-man serum, and probed with a monoclonal antibody to tissue TGase (CUB7401 ; kindly provided by Dr. Paul Birckbichler, Medical Research Foun-
dation, Oklahoma City, OK). The secondary antibody was 1 :1000 dilutedfluorescein-conjugated goat antimouse immunoglobulin G (rchaln spe-
cific; Sigma).Analysis of DNA FragmentatIon. After appropriate treatment, cells
(2 x 106) were centrifuged (400 x g for 10 mm) into a pellet and lysed at
4#{176}Cfor 15 mm in 400 �I of2O mu Tris-HCI(pH 8.0)containing 10 m� EDTAand 0.2% Triton X-1 00. The cell lysates were then transferred to a mi-crofuge tube and centrifuged at 12,000 x g for 15 mm to separate soluble(fragmented) DNA from pellet (intact genomic) DNA. Supematants con-
taming soluble DNA were collected and resuspended in 1 M NaCI and 500
�tI isopropanol. After overnight incubation (at -20#{176}C),samples were cen-trifuged (12,000 x g for 15 mm), and the pellets were resuspended in abuffer containing 10 mM Tns-HCI (pH 7.4) and 1 m� EDTA. The sampleswere treated with ANase (50 pg/mI) for 1 h at 50#{176}C.After mixing with 5x
gel loading buffer (Sigma), samples were electrophoresed on a 2% aga-
rose gel and stained with ethidium bromide. The gels were then visualizedin the dark under UV illumination.
Determination of DNA Fragments by EUSA. After treatment withretinoids, cells were centrifuged (200 x g for 5 mm) and washed once inmedium. The resulting pellets were then lysed. The presence of histone-
associated DNA fragments in cell lysates, a sign of internucleosomaldegradation of genomic DNA during apoptosis, was determined using anELISA kit (Boehringer Mannheim, Indianapolis, IN). The specific level ofDNA fragmentation in retinoid-treated HL-60 cells was expressed as the
enrichment factor, defined as follows: milliunits of the sample/milliunits ofthe corresponding control, where milliunits is absorbance at 405 nm.
Labeling of DNA Strand Breaks by TdT Assay. Following retinoidtreatment, the cells were fixed in 1% formaldehyde for 15 mm on ice,washed with PBS, resuspended in 70% ethanol, and stored at -20#{176}Cfor
up to 3 days. Prior to use, the cells were centrifuged and resuspended inPBS. DNA strand breaks were labeled with biotinylated-dUTP at 37”C for1 h in a reaction catalyzed byTdT(Boehringer Mannheim)and stained withfluoresceinated avidin (Boohringer Mannheim). Finally, the cells werewashed and examined for fluorescence under a UV microscope.
AcknowledgmentsWe thank Julie Chen for excellent technical assistance, Jude Richard for
editorial assistance, and Dr. Paul Birckbichler for providing the monoclo-nal anti-TGase antibody.
References1 . Metcalf, D. The molecular control of division, differentiation commit-
ment, and maturation in hematopoietic cells. Nature (Lond.), 339: 27-30,1989.
2. Marx, J. Cell death studies yield cancer clues. Science (Washington
DC), 259: 760-761, 1993.
3. Stewart, B. W. Mechanisms of apoptosis: integration of genetic, bio-chemical, and cellular indicators. J. NatI. Cancer Inst., 86: 1286-1295,1994.
4. Steller, H. Mechanisms and genes of cellular suicide. Science (Wash-ington DC), 267: 1445-1449, 1995.
5. Breitman, T. A., Selonick, S. E., and Collins, S. J. Induction of differ-entiation of the human promyelocytic leukemia cell line (HL-60) by retinoicacid. Proc. NatI. Aced. Sci. USA, 77: 2936-2940, 1980.
6. Martin, S. J., Bradley, J. G., and Cotter, T. G. HL-60 cells induced todifferentiate towards neutrophils subsequently die via apoptosis. Clin.
Exp. Immunol., 79: 448-453, 1990.
7. Pfahl, M., Apfel, A., Bendik, I., Fanjul, A., Graupner, G., Lee, M.,La-vista, N., Lu, X., Piedrafita, J., Ortiz, M. A., Salbert, G., and Zhang, X.Nuclear retinoid receptors and their mechanisms of action. Vitam. Horm.,
49: 327-381 , 1994.
8. Mangelsdorf, D. J., Umesono, K., and Evans, A. M. The retinoid re-ceptors. In: M. B. Spom, A. B. Roberts, and D. S. Goodman (eds.), TheRetinoids, pp. 319-349. New York: Raven Press, 1994.
9. Chambon, P. The retinoids signaling pathway: molecular and geneticanalysis. Semin. Cell Biol., 5: 1 15-125, 1994.
10. Giguere, V., Ong, E. S., Seul, P., and Evans, A. M. identification of areceptor for the morphogen retinoic acid. Nature (Lond.), 330: 624-629,1987.
1 1 . Levin, A. A., Sturzenbecker, L J., Kazmer, S., Bosalkowski, T., Husel-ton, C., Allenby, G., Speck, G., Kratzeisen, C. I., Rosenberger, M., Lovey,A., and Grippe, J. F. 9-cis Aetinoic acid stereoisomer binds and activatesthe nuclear receptor AXRa. Nature (Lond.), 355: 359-361 , 1992.
12. Heyman, A. A., Mangelsdorf, D. J., Dyck, J. A., Stein, A. B., Eichele,
G., Evans, A. M., and Thaller, C. 9-cis Aetinoic acid is a high affinity ligand
for the retinoid X receptor. Cell, 68: 397-406, 1992.
13. Rees, J. The molecular biology of retinoic acid receptors: orphan from
good family seeks home. Br. J. Dermatol., 126: 97-104, 1992.
14. Hashimoto, V., Kagechika, H., and Shudo, K. Expression of retinoicacid receptor genes and ligand-binding selectivity of retinoic acid recep-tors. Biochem. Biophys. Res. Commun., 166: 1300-1307, 1990.

185 Aetinoid Regulation of Differentiationand Apoptosis
15. Robertson, K. A., Emami, B., and Collins, S. J. Retinoic acid-resistantHL-60A cells harbor a point mutation in the retinoic acid receptor ligand-binding domain that confers dominant negative activity. Blood, 80: 1885-1889, 1992.
16. Robertson, K. A., Emami, B., Lueller, L, and Collins, S. J. Multiplemembers of the retinoic acid receptor family are capable of mediating thegranulocytic differentiation of HL-60 cells. Mol. Cell. Biol., 12: 3743-3749,
1992.
17. Drach, J., McOueen, T., Engel, H., Andreeff, M., Robertson, K. A.,
Collins, S. J., Malavasi, F., and Mehta, K. Aetinoic acid-induced expres-
sion of CD38 antigen in myeloid cells is mediated through retinoic acidreceptor-a. Cancer Res., 54: 1746-1752, 1994.
18. Drach, J., Zhao, S., Malavasi, F., and Mehta, K. Rapid induction of
CD38 antigen on myeloid leukemia cells by retinoic acid. Biochem. Bio-phys. Res. Commun., 195: 545-550, 1993.
19. Kontanio, K., Nishina, H., Ohoka, V., Takahashi, K., and Katada, D.
NAD glycohydrolase specifically induced by retinoic acid in human leu-
kemic HL-60 cells. J. Biol. Chem., 268: 16895-16898, 1993.
20. Davies, P. J., Murtaugh, M. P., Moore, W. T., Johnson, G. S., andLucas, D. Retinoic acid-induced expression of tissue transglutaminase inhuman promyelocytic leukemia (HL-60) cells. J. Biol. Chem., 260: 5166-5174, 1985.
21 . Suuier, M. K. T., Sehnert, A. J., and Cohen, J. J. Apoptosis in leuko-
cytes. J. Leukocyte Biol., 57: 2-10, 1995.
22. Nagy, L, Thomazy, V. A., Shipley, G. L, Fesus, L, Lamph, W.,Heyman, A. A., Chandraratna, A. A. S., and Davies, P. J. A. Activation of
retinoid X receptors induces apoptosis in HL-60 cell lines. Mol. Cell. Biol.,15: 3540-3551, 1995.
23. Mangelsdorf, D. J., Ong, E. S., Dyck, J. A., and Evans, A. M. A nuclearreceptor that identifies a novel retinoic acid response pathway. Nature(Lond.), 345: 224-229, 1990.
24. Mehta, K., and Berestein, G. L Expression of tissue transglutaminase
in cultured monocytic leukemia (THP-1) cells during differentiation. Can-
cer Res., 46: 1388-1394, 1986.
25. Murtaugh, M. P., Arend, W. P., and Davies, P. J. A. Induction of tissuetransglutaminase in human peripheral blood monocytes. J. Exp. Med.,
159: 114-125, 1984.
26. Chiocca, E. A., Davies, P. J. A., and Stein, J. P. Regulation of tissuetransglutaminase gene expression as a molecular model for retinoid ef-fects on proliferation and differentiation. J. Cell. Biochem., 39: 293-304,
1989.
27. Fesus, L, Davies, P. J. A., and Piacentini, M. Apoptosis: molecularmechanisms in programmed cell death. Eur. J. Cell Biol., 56: 170-177,1991.
28. Fesus, L Biochemical events in naturally occurring forms of cell
death. FEBS Lett., 328: 1-5, 1993.
29. Jiang, H., and Kochhar, D. M. Induction of tissue transglutaminase
and apoptosis by retinoic acid in the limb bud. Teratology, 46: 333-340,
1992.
30. Piacentini, M., Petruzzelli, M. A., Oliverio, S., Piredda, L, Biedler, J.,and Melino, G. Phenotypic-specific tissue transglutaminase regulation in
human neuroblastoma cells in response to retinoic acid: correlation with
cell death by apoptosis. Int. J. Cancer, 52: 271-278, 1992.
31 . Zhang, L-X., Mills, K. J., Dawson, M. I., Collins, S. J., and Jetten, A.
M. Evidence for the involvement of retinoic acid receptor a-dependent
signaling pathway in the induction of tissue transglutaminase and apop-tosis by retinoids. J. Biol. Chem., 270: 6022-6029, 1995.
32. Kumagai, M., Smith, E. C., Murray, D. J., Silvennoinen, 0., Murti, K.
G., Evans, W. E., Malavasi, F., and Campana, D. Ligation of CD38 sup-
presses human B lymphopoiesis. J. Exp. Med., 181: 1101-1 110, 1995.
33. Zupo, S., Rugan, E., Dono, M., Taborelli, G., Malavasi, F., and Ferra-mi, M. CD38 signaling by agonistic monoclonal antibody prevents apop-tosis of human germinal center B cells. Eur. J. Immunol., 24: 1218-1222,1995.
34. Collins, S., Robertson, K. A., and Mueller, L Retinoic acid-inducedgranulocytic differentiation of HL-60 myeloid leukemia cells is mediated
directly through the retinoic acid receptor (RARa). Mol. Cell. Biol., 10:2154-2163, 1990.
35. Mehta, K. High levels of transglutaminase expression in doxorubicin-resistant human breast carcinoma cells. Int. J. Cancer, 58: 400-406,1994.