abnormalities in intracellular ca2+ regulation in fetal vascular smooth muscle in pre-eclampsia:...
TRANSCRIPT

The FASEB Journal express article 10.1096/fj.02-0507fje. Published online December 17, 2002.
Abnormalities in intracellular Ca2+ regulation in fetal vascular smooth muscle in pre-eclampsia: enhanced sensitivity to arachidonic acid Joern R. Steinert, Lucilla Poston, Giovanni E. Mann, and Ron Jacob�
Centre for Cardiovascular Biology and Medicine, GKT Schools of Biomedical Sciences and Medicine, King�s College London, Guy�s Campus, London SE1 1UL, UK �Address for correspondence: Ron Jacob, PhD, Center for Cardiovascular Biology and Medicine, GKT School of Biomedical Sciences, King�s College London, Guy's Campus, London SE1 1UL, UK. E-mail: [email protected] ABSTRACT Pre-eclampsia (PE) is a leading cause of maternal and fetal mortality and morbidity. As free fatty acid metabolism is abnormally regulated in PE, we investigated the intracellular Ca2+ ([Ca2+]i) response to arachidonic acid (AA) in primary cultures of human umbilical artery smooth muscle cells (HUASMC). AA (50 µM) caused a significantly greater [Ca2+]i elevation in PE than in normal HUASMC, with many cells displaying a delayed secondary increase. The nonmetabolizable AA analog ETYA did not induce a response, suggesting that the augmented PE response depends on an AA metabolite. Inhibition of the AA metabolizing cyclooxygenase or lipoxygenase pathways did not affect the AA response of PE HUASMC but induced in normal cells the secondary rise of [Ca2+]i observed in PE cells. This potentiated response and the response in PE cells were blocked by inhibitors of the monooxygenase pathway, a third AA metabolizing pathway. We conclude that the [Ca2+]i response of HUASMC is elevated in PE because of an increased level of a monooxygenase metabolite that stimulates Ca2+ influx and that this can be mimicked in normal cells by blocking cyclooxygenase or lipoxygenase to divert AA to the monooxygenase. This and our work with fetal endothelial cells (FASEB J. 10.1096/fj.01-0916fje) demonstrate phenotypic changes in the fetal vasculature in PE. Key Words: calcium • arachidonic acid • pre-eclampsia • cyclooxygenase • smooth muscle cell
P re-eclampsia (PE) remains a potentially life threatening complication of pregnancy and is a leading cause of maternal and fetal morbidity and mortality. Premature delivery is often indicated, contributing to the high prenatal mortality associated with this disease. The diagnostic criteria are gestational hypertension and significant
proteinuria. Maternal endothelial dysfunction is now considered to play a pivotal role. Potential causes include elevation of free fatty acid (FFA) (1), lipoproteins, oxidized lipoproteins, or lipid peroxides (2) and deportation of syncytiotrophoblastic microvillous

fragments from the placenta (3). We previously found disturbances in PE of [Ca2+]i regulation in human umbilical vein endothelial cells (4) and now examine whether the same is true for human umbilical artery smooth muscle cells (HUASMC). A feature of PE is abnormal activation of the maternal coagulation cascade (5) with an imbalance between production of the arachidonic acid metabolites thromboxane A2 (TXA2) and prostaglandin I2 (PGI2), favoring TXA2, compared to normotensive pregnancies where an eightfold increase in PGI2 production normally dominates a small increase in TXA2 synthesis (6-8). Coupled with the observed elevation of maternal circulating arachidonic acid (AA) concentrations (9), this implies a substantial abnormality in AA metabolism. AA and its metabolites are implicated in the control of [Ca2+]i homeostasis, including modulation of Ca2+ release and Ca2+ entry via voltage-gated and nonvoltage-gated pathways by AA itself or by metabolites synthesised via cyclooxygenase (COX), lipoxygenase (LOX), or monooxygenase (cytochrome P450, MOX) pathways. In general, AA and its metabolites stimulate Ca2+ mobilization, although there are reports of inhibition of voltage-gated Ca2+ channels (10). AA stimulates nonvoltage-gated Ca2+ entry in several cell types and by a variety of mechanisms. In some instances this appears to be a direct effect of AA itself, since responses are mimicked by the nonmetabolizable AA analog ETYA (11, 12). A recent report demonstrates AA-mediated activation of the human Trp4 expressed in HEK293 cells (13). In other cases, AA metabolites such as the MOX metabolite 5,6-epoxyeicosatrienoic acid (5,6 EET) have been proposed to be the signal (14) for activating capacitative Ca2+ entry after Ca2+ store depletion (15). In addition to changes in the maternal vasculature (16-20), there are numerous reports of fetal vascular abnormalities (21-24) in PE pregnancies. The noninvasive characterization of the fetal vasculature using Doppler analysis provides the clearest evidence of worsening fetal outcome (22). Several studies characterizing maternal and fetal circulations using Doppler waveforms indicate that in cases of severe maternal hypertension, the presence of abnormal uterine artery waveforms increases the likelihood of abnormal umbilical artery waveforms (22). Given the evidence for altered AA metabolism and the involvement of AA in Ca2+ mobilization, we have examined the Ca2+ response to AA to further characterize potential abnormalities related to fetal vascular dysfunction in PE. METHODS Isolation and primary culture of human umbilical artery smooth muscle cells Umbilical cords were obtained with consent from normal term (26 cords) and pre-eclamptic deliveries (38 cords). PE was defined as a gestational hypertension (blood pressure >140/90 mm Hg measured on two occasions more than 4 h apart) in association with proteinuria (>300 mg/24 h). Cords were stored for up to 1 day at 4°C in Hanks' basic salt solution (BSS) supplemented with 150 µg mL-1 gentamycin, 10 mmol l-1 HEPES, and 10 mmol l-1 NaHCO3. HUASMC were cultured from explants in endothelial basal medium (EBM) supplemented with 10% fetal calf serum, 3.2 mM L-glutamine, penicillin (100 units mL-1), and streptomycin (100 units mL-1) at 37oC in a 5% CO2 atmosphere. The medium was

changed every 2 days. Cells were grown in T75 flasks until they reached confluence, then seeded onto 24 mm glass coverslips (#1, BDH, Poole, Dorset, UK). All experiments were performed using cells in passages 1-3. Fluorescence measurements of [Ca2+]i in HUASMC For [Ca2+]i determinations, subconfluent HUASMC were loaded for 1 h at room temperature with 1 µM fura 2-AM in HEPES-buffered Dulbecco's modified Eagle�s medium containing 20% fetal calf serum. After loading, cells were kept in a balanced salt solution (BSS (mM): 145 NaCl, 5 KCl, 1 MgCl2, 10 HEPES, 1 CaCl2, 10 D-glucose, pH 7.4) containing 1% (w/v) bovine serum albumin (BSA) for up to 1 h. Fluorescence was measured as described previously (25) using a rotating wheel spectrophotometer (Cairn, Sittingbourne, Kent, UK) with excitation at 340, 360, and 380 nm and emission detected at >505 nm. Coverslips were mounted on a thermostatted stage of an inverted epifluorescence microscope equipped with a 40× objective (N.A. 1.3) and superfused with BSS by gravity feed. The entire field of view was measured, usually containing several cells. For Ca2+ imaging experiments, excitation at 350 and 380 nm was via an LEP dual filter wheel system (Ludl, Hawthorne, NY, USA) equipped with various neutral density and interference filters. Emission was measured >400 nm and captured with a 14 bit cooled CCD camera (Hamamatsu C4880-80) controlled by Openlab software (Improvision, Coventry UK). An image pair was captured every 1-3 s. The ratio of fluorescence at 340 or 350 and 380 nm excitation was used as a measure of [Ca2+]i. Autofluorescence was estimated by addition of 2 mM Mn2+. MATERIALS Indomethacin, nordihydroguaiaretic acid (NDGA), eicosatetraynoic acid (ETYA), 17-octadecynoic acid (17-ODYA), isoniazid (isonicotinic acid hydrazide), octadecanoic acid, arachidonic, and oleic and linoleic acid were obtained from Sigma Chemical Co. (Poole, Dorset, UK); metyrapone (2-methyl-1,2-di-3-pyridyl-1-propanone, MPP) was from Aldrich (Poole, Dorset, UK) and fura 2-AM from Calbiochem (Nottingham, UK). Arachidonic acid, indomethacin, NDGA, ETYA, 17-ODYA, isoniazid, octadecanoic acid, and fura 2-AM were dissolved in dimethylsulphoxide (DMSO) and MPP in methanol. All drugs were stored as frozen stocks at �20°C. All drug concentrations are expressed as final molar concentrations in the superfusate (BSS (mM): 145 NaCl, 5 KCl, 1 MgCl2, 10 HEPES, 10 D-glucose, 1 CaCl2, pH 7.4). Ca2+-free solutions were nominally so with omission of CaCl2. Statistics Results are expressed as means ± SE of measurements in at least 3 different cell cultures (n=number of umbilical cords). Statistical significance was evaluated using an ANOVA or Student�s t test for unpaired observations with P < 0.05 considered significant (*).

RESULTS Arachidonic acid-induced increases in [Ca2+]i Figure 1A shows ratio traces from experiments where in the presence of 1 mM [Ca2+]o application of 50 µΜ AA evoked a sustained [Ca2+]i plateau phase in normal cells, whereas pre-eclamptic HUASMC responded with a significantly greater increase in [Ca2+]i [∆ ratio, normal: 1.44 ± 0.28 (16) vs. PE: 2.99 ± 0.47 (19), P<0.009]. The nature of the rise in [Ca2+]i was also different for PE cells with 39 of 59 experiments displaying two separate phases with a secondary plateau phase occurring a few minutes after the initial [Ca2+]i increase. We did not detect any difference in the basal [Ca2+]i [ratio, normal: 0.43 ± 0.01 (5) vs. PE: 0.46 ± 0.01 (7)] (Fig. 1B). The initial responses to AA in PE cells [∆ ratio, 0.97 ± 0.04 (19)] were not different from those observed in normal cells [∆ ratio, 1.02 ± 0.03, (16)] but the secondary plateau phase, which was absent in normal cells, was responsible for the significantly greater Ca2+ responses. Data in Fig. 1C summarize the [Ca2+]i plateau responses after 600-800 s exposure to AA. The increase in [Ca2+]i in both types of cells returned to baseline after removing AA and addition of 0.1% (w/v) BSA-containing solution. BSA was used to aid the complete removal of AA, since it has high-affinity binding sites for fatty acids (26). As pre-eclampsia often necessitates a premature delivery, the larger AA responses observed in PE cells could have been due to the shorter gestational period rather than the PE per se. However, there was no correlation between gestational age and [Ca2+]i response to AA (Fig. 2). Moreover, for those pregnancies that lay within the normal gestational period of 38 to 40 wk, the [Ca2+]i responses were still significantly larger in PE (normal: 0.96 ± 0.20 (3) vs. PE: 3.60 ± 0.98 (6), P<0.02, one-tailed t test assuming unequal variance). To investigate whether AA-induced increases in [Ca2+]i involved Ca2+ release from internal stores, we tested the response in nominally Ca2+-free BSS. As shown in Fig. 3A, normal and PE cells both responded to AA [∆ ratio, normal: 0.27 ± 0.08 (16) vs. PE: 0.50 ± 0.09 (19)] although in both cases the response was less than that seen in the presence of extracellular Ca2+ (normal: P<0.01; PE: P=0.005). As was the case in the presence of Ca2+
o, the response was larger in PE than in normal cells (P<0.05). Data from 16 normal and 19 pre-eclamptic cell cultures are summarized in Fig. 3B. Effect of other free fatty acids To determine whether the observed effects of AA on HUASMC were specific to AA, we examined the effects of other unsaturated fatty acids on normal and PE HUASMC. As summarized in Fig. 4A, when HUASMC were challenged with either 50 µM AA, oleic (OA), or linoleic acid (LA), cells responded in the presence of Ca2+
o with a significantly lower sustained [Ca2+]i plateau than that elicited by AA in both normal cells [OA: 3.0 ± 2.7%; LA: 31.9 ± 15.6% (3), expressed as a percentage of the AA response] and PE cells [OA: 8.3 ± 3.2%; LA: 34.3 ± 7.7% (6)] (ANOVA, P<0.0001 for the type of fatty acid). Moreover, a biphasic Ca2+ increase with a secondary plateau phase was not observed with

oleic or linoleic acid. These data were obtained using imaging; hence, the ratios could not be compared directly with those obtained using the spectrophotometer. The lack of a significant difference between the response of normal and PE cells to AA is most likely due to the small number of cell cultures used in this set of experiments. Experiments in the absence of Ca2+
o (Fig. 4B) produced smaller responses to all three fatty acids, with no significant variation between them (ANOVA, P>0.1). We also tested the response to the nonmetabolizable AA analog eicosatetraynoic acid (ETYA, 50 µM) in normal HUASMC, but did not observe a Ca2+ response (∆ ratio, 0.01±0.01, n=3; data not shown), suggesting that one of the metabolites of AA was responsible for the observed increases in [Ca2+]i. Modulation of AA-induced Ca2+ signaling by inhibitors of AA metabolism AA is metabolized via cyclooxygenases (COX-1/2), lipoxygenases (LOX), or monooxygenases (cytochrome P450); inhibition of any of these pathways will affect the cellular distribution of its metabolites. We determined the effect of inhibiting COX with 10 µM indomethacin and LOX with 10 µM nordihydroguaiaretic acid (NDGA) in PE cells to investigate the role of metabolites from these two pathways in generating the secondary plateau phase of the Ca2+ response. However, as shown in Fig. 5A, B and summarized in Fig. 5C, PE HUASMC responses to 50 µM AA were not significantly altered by indomethacin or NDGA [∆ ratio, PE: 2.35 ± 0.31 (8) vs. Indo: 2.97 ± 0.93 (8) vs. NDGA: 3.80 ± 1.33 (8)], even though these cells were preincubated with the inhibitors for 20 min. In control experiments, the response of normal cells was markedly potentiated by either indomethacin or NDGA, or a combination of the two, resulting in a secondary [Ca2+]i plateau phase and a response not significantly different from that of PE cells [∆ ratio, normal: 0.83 ± 0.12 (3); Indo: 3.12 ± 0.87 (3), P<0.04; NDGA: 8.05 ± 1.71 (3); NDGA +Indo: 5.09 ± 1.27 (3), P<0.02] (Fig. 5C). The potentiating effect of COX and LOX inhibition on the response of normal HUASMC to AA could be due to potentiation of the effective AA concentration by inhibition of its metabolism; the intracellular accumulation of AA could then potentiate the Ca2+ response directly or via a metabolite generated as a result of AA diversion to the monooxygenase pathway. We suspected the latter because of the lack of effect of ETYA. To test this hypothesis, we tested three inhibitors of the monooxygenase pathway: metyrapone (MPP, 50 µM), isoniazid (200 µM), and 17-octadecynoic acid (17-ODYA, 10 µΜ). The first two have some structural similarity, each containing a pyridine ring with a carbonyl group; however, the carbonyl group is in the meta position in metyrapone and in the para position in isoniazid. 17-ODYA is completely different structurally, being an alkyne carboxylate. As a control we also examined the effect of 17-octadecanoic acid (17-ODA), which is an alkane carboxylate lacking the terminal methyne. All three inhibitors reduced the response of PE cells to AA [∆ ratio, 5.11 ± 1.05 (100%) vs. MPP: 37.8 ± 11.8%; isoniazid: 55.9 ± 6.6%; 17-ODYA: 7.6 ± 2.2%, n=3 for each inhibitor, ANOVA, P<0.001, Fig. 6]: these are quoted as percentages because of the small number of cultures used. Moreover, all three MOX inhibitors reduced the indomethacin-augmented AA response in normal cells [∆ ratio, 4.55 ± 1.4 (100%) vs. MPP: 26.3 ± 6.2%; isoniazid: 18.9 ± 6.9% and 17-ODYA: 13.7 ± 8.1%, n=3 for each inhibitor, ANOVA, P<0.01, Fig. 6]. The native AA response in normal

cells (without indomethacin augmentation) was also inhibited by 17-ODYA [∆ ratio, 1.02 ± 0.13 (100%) vs. 17-ODYA: 43.9 ± 10.5% (3), P<0.05, Fig. 6]; metyrapone caused a slight inhibition that was not significant [MPP: 81.1 ± 16.9% (3), P>0.05]; isoniazid was not tested. Furthermore, 10 µM 17-ODA did not affect the response of indomethacin-pretreated normal cells to AA (122 ± 24%, Fig. 6). Calcium and smooth muscle cell contraction The delay before the onset of the secondary [Ca2+]i plateau phase could be due to the time required to accumulate a metabolite. Another possibility is that in PE (or the presence of COX or LOX inhibition), [Ca2+]i rises to a higher level in a monophasic manner, but then causes a contraction sufficient to generate an artefactual increase in fluorescence ratio. To rule out this possibility, we recorded responses of indomethacin pretreated normal HUASMC to 50 µM AA using ratio imaging to examine the relationship between the second phase of the fluorescence ratio increase and contraction. Of the 18 cells imaged, 3 cells did not show a clear distinction between the initial and later increases in [Ca2+]i; in the remaining 15 cells there was a distinct secondary phase rise in Ca2+, and in 11 of these cells this occurred before contraction started (Fig. 7). These results show that the second plateau phase is not an artefact of contraction. DISCUSSION The present study provides insight into possible abnormalities of fetal human artery smooth muscle cells isolated from pre-eclamptic pregnancies. Our findings provide the first evidence that AA-stimulated Ca2+ signaling is modulated differently in fetal smooth muscle cells in PE and that this alteration is linked to differences in AA metabolism in PE. We have shown that HUASMC have an elevated [Ca2+]i response to AA in PE and that this is not due to a shorter gestational period for PE pregnancies. The main mechanism for this increased response is a potentiated Ca2+ influx, since responses were significantly larger in the presence than in the absence of Ca2+
o. This response could be a direct action of AA itself or a response to one of its metabolites. AA is also known to stimulate Ca2+ influx nonspecifically by increasing membrane fluidity, as reported in HEK293 cells (27). This is unlikely here, since oleic acid has been reported to increase membrane fluidity to a similar extent as AA (28) and ETYA to a greater extent than AA (29) and yet oleic acid induced a smaller [Ca2+]i response than AA and ETYA induced no response at all (30). The lack of any effect of the nonmetabolizable analog ETYA and the considerably smaller responses to oleic and linoleic acid in the presence of Ca2+
o strongly suggest that AA is not activating Ca2+ influx directly, but rather via a metabolite. The lack of any significant difference between the responses to the oleic, linoleic, and arachidonic acids in normal and PE HUASMC in the absence of Ca2+
o may indicate that the ability of AA to release Ca2+ from internal stores is a relatively nonspecific effect compared to its effect on Ca2+ influx. The intermediate-sized response to linoleic acid could be a consequence of linoleic acid being a precursor for AA synthesis.

In contrast, the response to AA in the presence of Ca2+o clearly involves AA metabolism
since, in normal HUASMC, the response was potentiated by inhibition of either the COX or LOX signaling pathways. Moreover, the potentiation of Ca2+ influx is due to a MOX metabolite, as inhibition of the MOX pathway by structurally dissimilar compounds inhibited the Ca2+ response in normal cells (either the native response or the potentiated response after COX or LOX inhibition) as well as the large response seen in PE cells. The ability of MOX inhibitors to inhibit even the nonpotentiated response in normal cells suggests that a MOX metabolite may be involved in the control of Ca2+ influx even in normal HUASMC. Our data show that the MOX pathway can be stimulated by the increased availability of AA achieved by inhibiting AA metabolism via alternative pathways. This may seem surprising when there is an essentially unlimited provision of extracellular AA, and suggests that the diffusion of extracellular AA to the MOX enzyme is a rate-limiting step. A similar mechanism could be responsible for the potentiating effect of indomethacin on the AA-induced [Ca2+]i increases in somatotropes (31). Another indication of AA accumulation and diversion for use as a substrate for MOX metabolism is the frequently observed substantial delay in the onset of the secondary phase, perhaps reflecting a gradual rise in AA and attainment of a critical substrate concentration for MOX activation. Although it is apparent that the increased response in PE is due to a greater degree of metabolism of AA via MOX, it is not clear whether this is due to increased MOX activity or to decreased activity by COX and/or LOX pathways. The latter is more probable since the PE response was exactly mimicked in normal cells by inhibiting COX or LOX. Moreover, COX or LOX inhibitors did not affect the AA response of PE cells, suggesting that the combined activity of these pathways is already reduced; however, the ability of the MOX pathway to metabolize AA in PE cells may already be fully saturated. PE-associated disturbances of Ca2+ regulation have been reported in endothelial cells and erythrocytes (4, 32-34). There have also been reports of altered maternal and fetal fatty acid metabolism usually reflected in altered levels of various fatty acids, including AA (9, 35-37). Raised maternal plasma phospholipase A2 has been reported in PE (38, 39), and this could be responsible for raised concentrations of AA in the maternal circulation. Various AA metabolites have been reported to stimulate Ca2+ influx, including epoxyeicosatrienoic acids (EETs). Of particular relevance here are reports of this effect in vascular smooth muscle cells (40). Urinary levels of EETs and their hydroxylated derivatives are elevated in pregnant women and even more so in those with pregnancy-induced hypertension (42), so it is tempting to argue that this may contribute to the hypertension. However, EETs have been canvassed as a candidate for EDHF (42, 43), and blocking their further metabolism by inhibiting their conversion by soluble epoxide hydrolase to the corresponding dihydroxy epoxyeicosatrienoic acids has been reported to lower blood pressure in rats with hypertension induced by angiotensin II (44). One possible resolution of these contradictions is that the effect of EETs may depend on the state of the muscle, since 14,15-EET produced both contraction and relaxation in porcine coronary artery rings depending on whether the rings were precontracted with acetylcholine or the thromboxane mimetic U46619, respectively (40).

Exogenously applied or endogenously generated AA leads to vasorelaxation, which is variously reported to be insensitive (45) or sensitive (46) to MOX inhibitors. In the intact vessel, this is due to release of endothelial-derived prostacyclin and EDHF: prostacyclin is a COX metabolite, and there is evidence that EDHF may be an AA MOX metabolite (42, 43). However, it is quite clear that, in these HUASMC, AA causes a rise in [Ca2+]i, which is not only likely to cause a contraction, but is observed to do so in normal cells when the response is potentiated by COX inhibition (Fig. 5). It is therefore likely that in PE, AA increases production of MOX metabolites such as 5,6 EET in vascular smooth muscle, and this will mitigate or even predominate over any relaxing effect of prostacyclin or EDHF released from the endothelium. This may be especially likely in the presence of endothelial dysfunction, described in the maternal circulation (17) and sometimes in the fetoplacental circulation (47). Our data suggest that in PE there could be increased fetal vascular tone due to the increased sensitivity of vascular smooth muscle to AA. Increased fetoplacental vascular resistance is detectable by alterations in Doppler profiles and provide a prediction of worsening fetal outcome (22). Another important issue relating to the fetal circulation is the current substantial interest in the field of fetal programming of the cardiovascular system (48). Our previous results with fetal endothelial cells (4) and current results with vascular smooth muscle cells demonstrate phenotypic changes that are persistent in culture, suggesting the possibility of adding PE to the list of intrauterine factors implicated in fetal programming. Should the altered AA metabolism that we report here extend to the maternal circulation, it may also contribute to the pathogenesis of the hypertension, one of the hallmarks of PE. ACKNOWLEDGEMENTS We gratefully acknowledge the generous support from Tommy�s the Baby Charity, UK, and the Community Fund and are indebted to Julie Adams and the midwives of St. Thomas� and Guy�s Hospital for collecting normal and PE umbilical cords. We thank Simon Bartlett, Anthony Morgan, and John Mitchell for helpful discussions and Sara Cato for her expert assistance with cell culture. REFERENCES 1. Endresen, M. J., Tosti, E., Heimli, H., Lorentzen, B., Henriksen, T. (1994) Effects of
free fatty acids found increased in women who develop pre-eclampsia on the ability of endothelial cells to produce prostacyclin, cGMP and inhibit platelet aggregation. Scand. J. Clin. Lab. Invest. 54, 549-557
2. Hubel, C. A., McLaughlin, M. K., Evans, R. W., Hauth, B. A., Sims, C. J., Roberts, J. M. (1996) Fasting serum triglycerides, free fatty-acids, and malondialdehyde are increased in preeclampsia, are positively correlated, and decrease within 48 hours post-partum. Am. J. Obstet. Gynecol. 174, 975-982

3. Cockell, A. P., Learmont, J. G., Smarason, A. K., Redman, C. W., Sargent, I. L., Poston, L. (1997) Human placental syncytiotrophoblast microvillous membranes impair maternal vascular endothelial function. Br. J. Obstet. Gynaecol. 104, 235-240
4. Steinert, J. R., Wyatt, A. W., Poston, L., Jacob, R., Mann, G. E. (2002) Preeclampsia is associated with altered Ca2+ regulation and nitric oxide production in human fetal venous endothelial cells. FASEB J. 10.1096/fj.01-0916fje,
5. Lyall, F., Greer, I. A. (1994) Preeclampsia---a multifaceted vascular disorder of pregnancy. J. Hypertens. 12, 1339-1345
6. Hsu, C. D., Copel, J. A., Hong, S. F., Chan, D. W. (1995) Thrombomodulin levels in preeclampsia, gestational hypertension, and chronic hypertension. Obstet. Gynecol. 86, 897-899
7. Walsh, S. W., Wang, Y., Jesse, R. (1996) Placental production of lipid peroxides, thromboxane, and prostacyclin in preeclampsia. Hypertens. Pregnancy 15, 101-111
8. Hayman, R., Brockelsby, J., Kenny, L., Baker, P. (1999) Preeclampsia: the endothelium, circulating factor(s) and vascular endothelial growth factor. J. Soc. Gynecol. Invest. 6, 3-10
9. Shouk, T. A., Omar, M. N., Fayed, S. T. (1999) Essential fatty acids profile and lipid peroxides in severe pre-eclampsia. Ann. Clin. Biochem. 36, 62-65
10. Liu, L., Rittenhouse, A. R. (2000) Effects of arachidonic acid on unitary calcium currents in rat sympathetic neurons. J. Physiol. (London) 525, 391-404
11. Gailly, P. (1998) Ca2+ entry in CHO cells, after Ca2+ stores depletion, is mediated by arachidonic acid. Cell Calcium 24, 293-304
12. Munaron, L., Antoniotti, S., Distasi, C., Lovisolo, D. (1997) Arachidonic acid mediates calcium influx induced by basic fibroblast growth factor in Balb-c 3T3 fibroblasts. Cell Calcium 22, 179-188
13. Wu, X., Babnigg, G., Zagranichnaya, T., Villereal, M. L. (2002) The role of endogenous human Trp4 in regulating carbachol-induced calcium oscillations in HEK-293 cells. J. Biol. Chem. 277, 13597-13608
14. Alvarez, J., Montero, M., García-Sancho, J. (1992) Cytochrome P450 may regulate plasma membrane Ca2+ permeability according to the filling state of the intracellular Ca2+ stores. FASEB J. 6, 786-792
15. Graier, W. F., Simecek, S., Sturek, M. (1995) Cytochrome P450 mono-oxygenase-regulated signalling of Ca2+ entry in human and bovine endothelial cells. J. Physiol. (London) 482, 259-274

16. Pascoal, I. F., Lindheimer, M. D., Nalbantian-Brandt, C., Umans, J. G. (1998) Preeclampsia selectively impairs endothelium-dependent relaxation and leads to oscillatory activity in small omental arteries. J. Clin. Invest. 101, 464-470
17. McCarthy, A. L., Woolfson, R. G., Raju, S. K., Poston, L. (1993) Abnormal endothelial-cell function of resistance arteries from women with preeclampsia. Am. J. Obstet. Gynecol. 168, 1323-1330
18. Ashworth, J. R., Warren, A. Y., Baker, P. N., Johnson, I. R. (1997) Loss of endothelium-dependent relaxation in myometrial resistance arteries in pre-eclampsia. Br. J. Obstet. Gynaecol. 104, 1152-1158
19. Kublickiene, K. R., Lindblom, B., Kruger, K., Nisell, H. (2000) Preeclampsia: evidence for impaired shear stress-mediated nitric oxide release in uterine circulation. Am. J. Obstet. Gynecol. 183, 160-166
20. Suzuki, Y., Saitoh, M., Suzumori, K., Kajikuri, J., Itoh, T. (2000) Characterization of changes in mechanical responses to histamine in omental resistance arteries in pre-eclampsia. Br. J. Pharmacol. 131, 37-42
21. Bodelsson, G., Marsal, K., Stjernquist, M. (1995) Reduced contractile effect of endothelin-1 and noradrenaline in human umbilical artery from pregnancies with abnormal umbilical artery flow velocity waveforms. Early Hum. Dev. 42, 15-28
22. Harrington, K., Thompson, M. O., Carpenter, R. G., Nguyen, M., Campbell, S. (1999) Doppler fetal circulation in pregnancies complicated by pre-eclampsia or delivery of a small for gestational age baby: 2. Longitudinal analysis. Br. J. Obstet. Gynaecol. 106, 453-466
23. Saijo, Y., Maeda, K., Nakaya, Y., Kamada, M., Mitani, R., Endo, S., Irahara, M., Yamano, S., Aono, T. (2001) Altered sensitivity to a novel vasoconstrictor endothelin-1 (1-31) in myometrium and umbilical artery of women with severe preeclampsia. Biochem. Biophys. Res. Commun. 286, 964-967
24. Akar, F., Ark, M., Uydes, B. S., Soysal, M. E., Saracoglu, F., Abacioglu, N., van der Vliet, Kanzik, I. (1994) Nitric oxide production by human umbilical vessels in severe pre-eclampsia. J. Hypertens. 12, 1235-1241
25. Morgan, A. J., Jacob, R. (1996) Ca2+ influx does more than provide releasable Ca2+ to maintain repetitive spiking in human umbilical vein endothelial cells. Biochem. J. 320, 505-517
26. Ek, B. A., Cistola, D. P., Hamilton, J. A., Kaduce, T. L., Spector, A. A. (1997) Fatty acid binding proteins reduce 15-lipoxygenase-induced oxygenation of linoleic acid and arachidonic acid. BBA-Lipid Lipid Metab. 1346, 75-85

27. Mignen, O., Shuttleworth, T. J. (2000) IARC, a novel arachidonate-regulated, noncapacitative Ca2+ entry channel. J. Biol. Chem. 275, 9114-9119
28. Badea, M., Jinga, V., Horer, O. (1984) Experimental modulation of the plasmalemmal microfluidity. Studies on endothelial and aortic smooth muscle cells. Physiologie 21, 39-44
29. Brown, M., Anderson, K. M., Patel, H., Hopfinger, A. J., Harris, J. E. (1992) Eicosatetraynoic and arachidonic acid-induced changes in cell membrane fluidity consonant with differences in computer-aided design-structures. Biochem. Biophys. Acta 1105, 285-290
30. Beck, R., Abbot, S. E., Aaronson, P. I., Smirnov, S. V. (1998) Effect of temperature and albumin on membrane fluidity in vascular smooth muscle and endothelial cells. J. Physiol. (London) 507P, P71
31. Roudbaraki, M. M., Drouhault, R., Bacquart, T., Vacher, P. (1996) Arachidonic acid-induced hormone release in somatotropes: involvement of calcium. Neuroendocrinol. 63, 244-256
32. Matteo, R., Proverbio, T., Cordova, K., Proverbio, F., Marin, R. (1998) Preeclampsia, lipid peroxidation, and calcium adenosine triphosphatase activity of red blood cell ghosts. Am. J. Obstet. Gynecol. 178, 402-408
33. Mahdy, Z., Otun, H. A., Dunlop, W., Gillespie, J. I. (1998) The responsiveness of isolated human hand vein endothelial cells in normal pregnancy and in pre-eclampsia. J. Physiol. (London) 508, 609-617
34. AbdAlla, S., Lother, H., el Massiery, A., Quitterer, U. (2001) Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nature Med. 7, 1003-1009
35. Velzing-Aarts, F. V., van der Klis, F. R., van der Dijs, F. P., Muskiet, F. A. (1999) Umbilical vessels of preeclamptic women have low contents of both n-3 and n-6 long-chain polyunsaturated fatty acids. Am. J. Clin. Nutr. 69, 293-298
36. Lorentzen, B., Drevon, C. A., Endresen, M. J., Henriksen, T. (1995) Fatty acid pattern of esterified and free fatty acids in sera of women with normal and pre-eclamptic pregnancy. Br. J. Obstet. Gynaecol. 102, 530-537
37. Ogburn, P. L., Williams, P. P., Johnson, S. B., Holman, R. T. (1984) Serum arachidonic acid levels in normal and preeclamptic pregnancies. Am. J. Obstet. Gynecol. 148, 5-9
38. Jendryczko, A., Drozdz, M. (1990) Increased placental phospholipase A2 activities in pre-eclampsia. Zbl. Gynacol. 112, 889-891

39. Lim, K. H., Rice, G. E., de Groot, C. J., Taylor, R. N. (1995) Plasma type II phospholipase A2 levels are elevated in severe preeclampsia. Am. J. Obstet. Gynecol. 172, 998-1002
40. Fang, X., Weintraub, N. L., Stoll, L. L., Spector, A. A. (1999) Epoxyeicosatrienoic acids increase intracellular calcium concentration in vascular smooth muscle cells. Hypertension 34, 1242-1246
41. Catella, F., Lawson, J. A., Fitzgerald, D. J., FitzGerald, G. A. (1990) Endogenous biosynthesis of arachidonic acid epoxides in humans: increased formation in pregnancy-induced hypertension. Proc. Natl. Acad. Sci. USA 87, 5893-5897
42. Campbell, W. B., Gebremedhin, D., Pratt, P. F., Harder, D. R. (1996) Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 78, 415-423
43. Fisslthaler, B., Popp, R., Kiss, L., Potente, M., Harder, D. R., Fleming, I., Busse, R. (1999) Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature (London) 401, 493-497
44. Imig, J. D., Zhao, X., Capdevila, J. H., Morisseau, C., Hammock, B. D. (2002) Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 39, 690-694
45. Fukao, M., Hattori, Y., Kanno, M., Sakuma, I., Kitabatake, A. (1997) Evidence against a role of cytochrome P450-derived arachidonic acid metabolites in endothelium-dependent hyperpolarization by acetylcholine in rat isolated mesenteric artery. Br. J. Pharmacol. 120, 439-446
46. Singer, H. A., Saye, J. A., Peach, M. J. (1984) Effects of cytochrome P-450 inhibitors on endothelium-dependent relaxation in rabbit aorta. Blood Vessels 21, 223-230
47. Barber, A., Robson, S. C., Myatt, L., Bulmer, J. N., Lyall, F. (2001) Heme oxygenase expression in human placenta and placental bed: reduced expression of placenta endothelial HO-2 in preeclampsia and fetal growth restriction. FASEB J. 15, 1158-1168
48. Godfrey, K. M., Barker, D. J. (2001) Fetal programming and adult health. Pub. Health Nutr. 4, 611-624
Received June 25, 2002; accepted October 23, 2002.
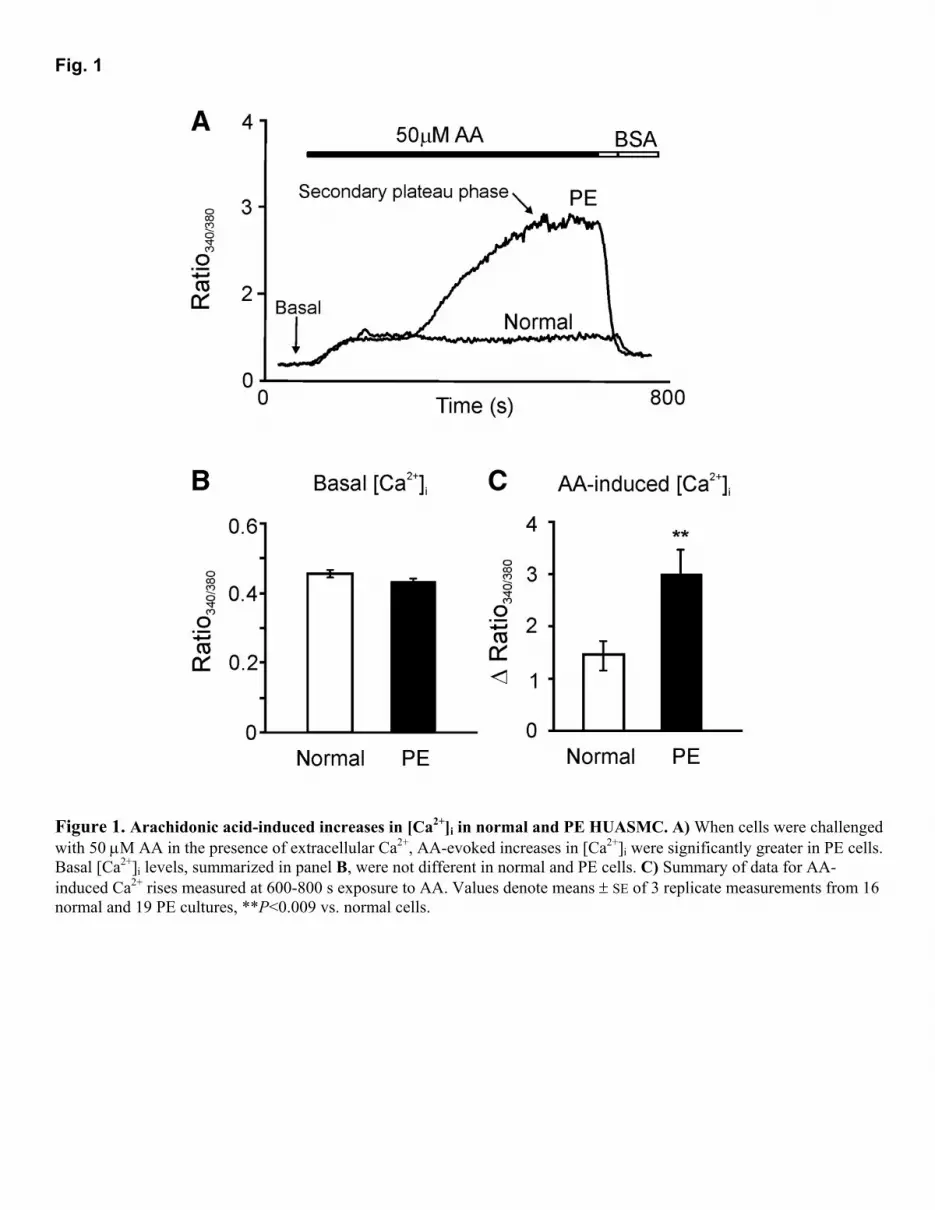
Fig. 1
Figure 1. Arachidonic acid-induced increases in [Ca2+]i in normal and PE HUASMC. A) When cells were challenged with 50 µM AA in the presence of extracellular Ca2+, AA-evoked increases in [Ca2+]i were significantly greater in PE cells. Basal [Ca2+]i levels, summarized in panel B, were not different in normal and PE cells. C) Summary of data for AA-induced Ca2+ rises measured at 600-800 s exposure to AA. Values denote means ± SE of 3 replicate measurements from 16 normal and 19 PE cultures, **P<0.009 vs. normal cells.
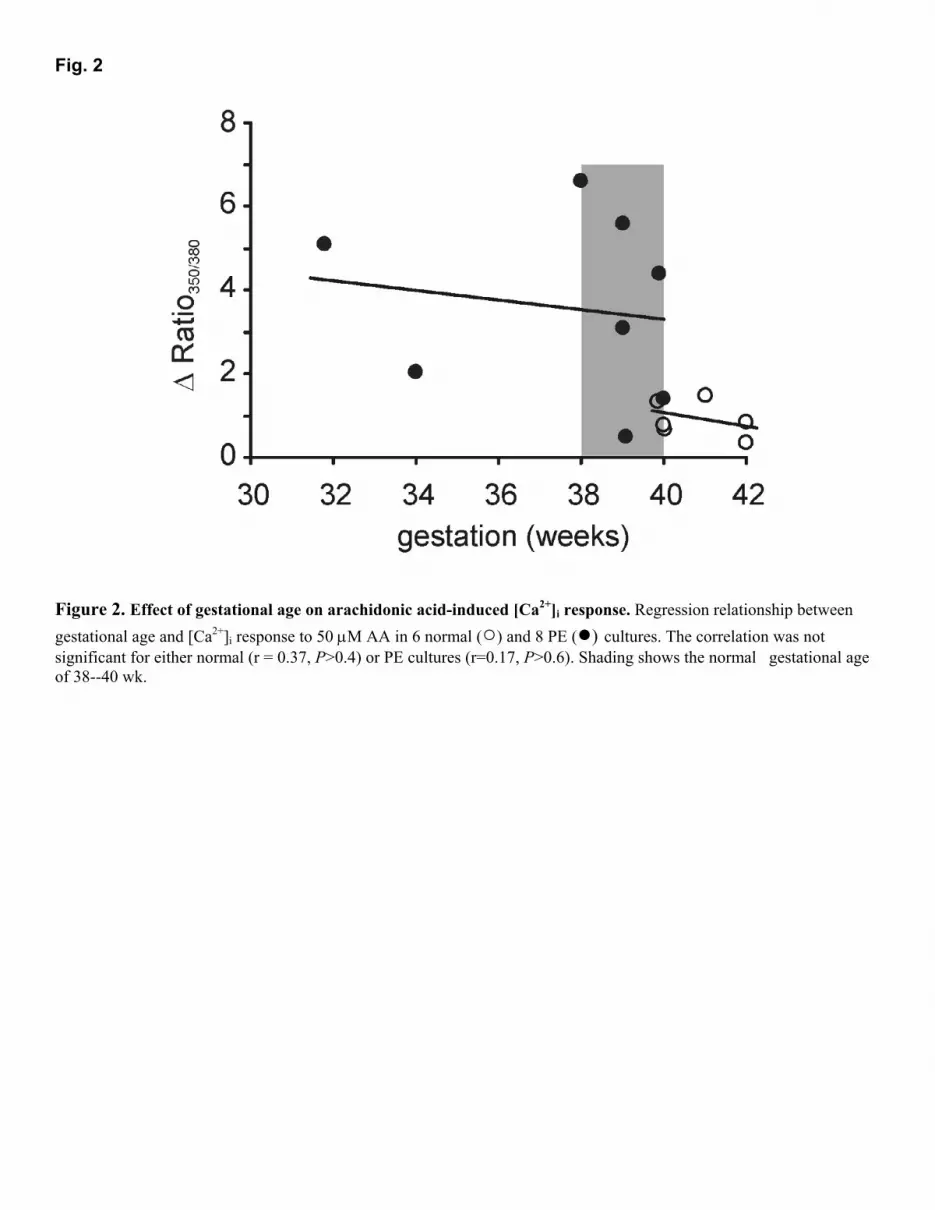
Fig. 2
Figure 2. Effect of gestational age on arachidonic acid-induced [Ca2+]i response. Regression relationship between gestational age and [Ca2+]i response to 50 µM AA in 6 normal (○) and 8 PE (●) cultures. The correlation was not significant for either normal (r = 0.37, P>0.4) or PE cultures (r=0.17, P>0.6). Shading shows the normal gestational age of 38--40 wk.
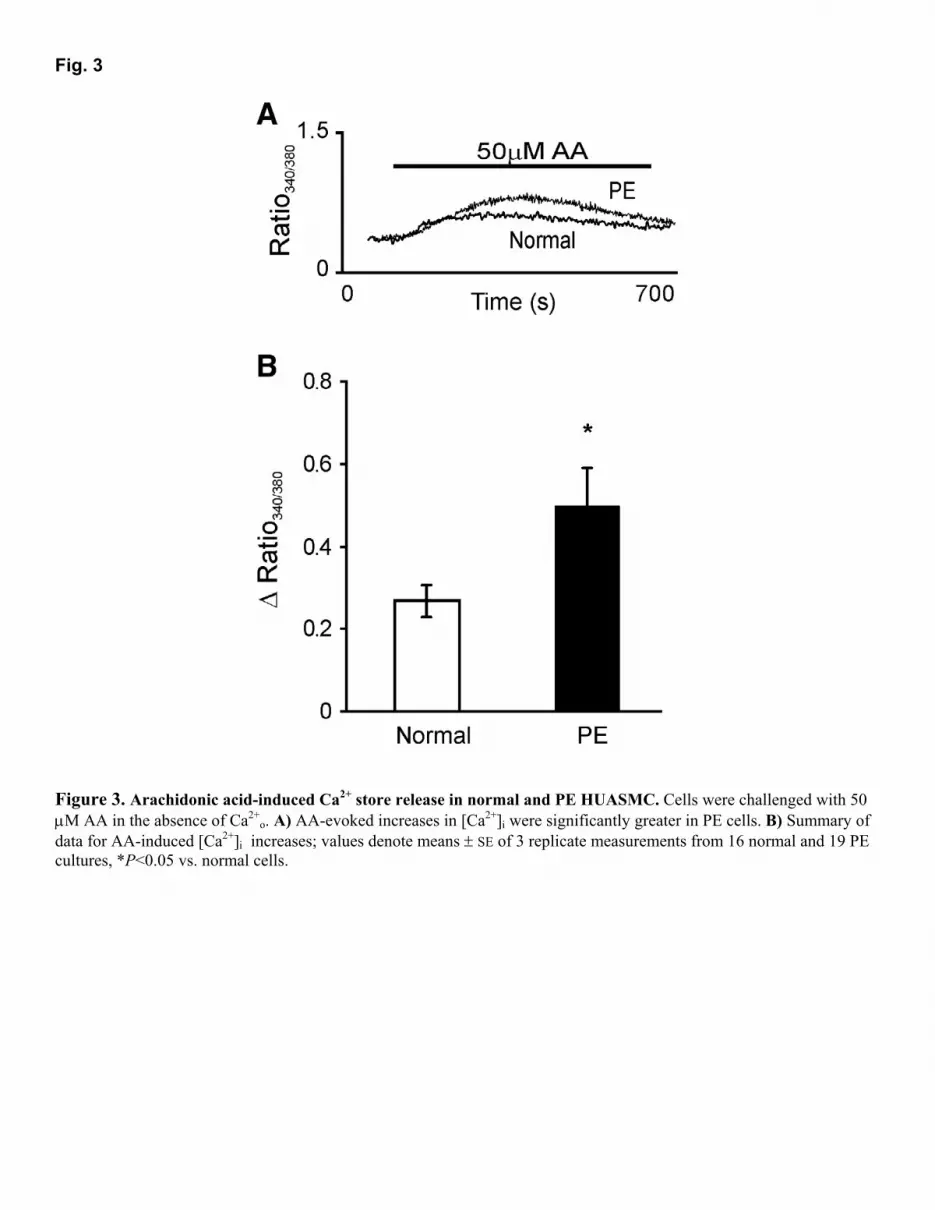
Fig. 3
Figure 3. Arachidonic acid-induced Ca2+ store release in normal and PE HUASMC. Cells were challenged with 50 µM AA in the absence of Ca2+
o. A) AA-evoked increases in [Ca2+]i were significantly greater in PE cells. B) Summary of data for AA-induced [Ca2+]i increases; values denote means ± SE of 3 replicate measurements from 16 normal and 19 PE cultures, *P<0.05 vs. normal cells.

Fig. 4
Figure 4. [Ca2+]i responses of normal and PE HUASMC to fatty acids. Normal (□) and PE (■) HUASMC were challenged with 50 µM arachidonic acid (AA), 50 µM oleic acid, and 50 µM linoleic acid in Ca2+-containing solution (A) or nominally Ca2+-free solution (B). Data denote means ± SE of replicate measurements in 3 different normal and 6 PE cell cultures, **P<0.0004 vs. AA in normal cells, �P<0.02 vs. AA in PE cells. Data obtained by imaging, so fluorescence ratios are not directly comparable with other data.
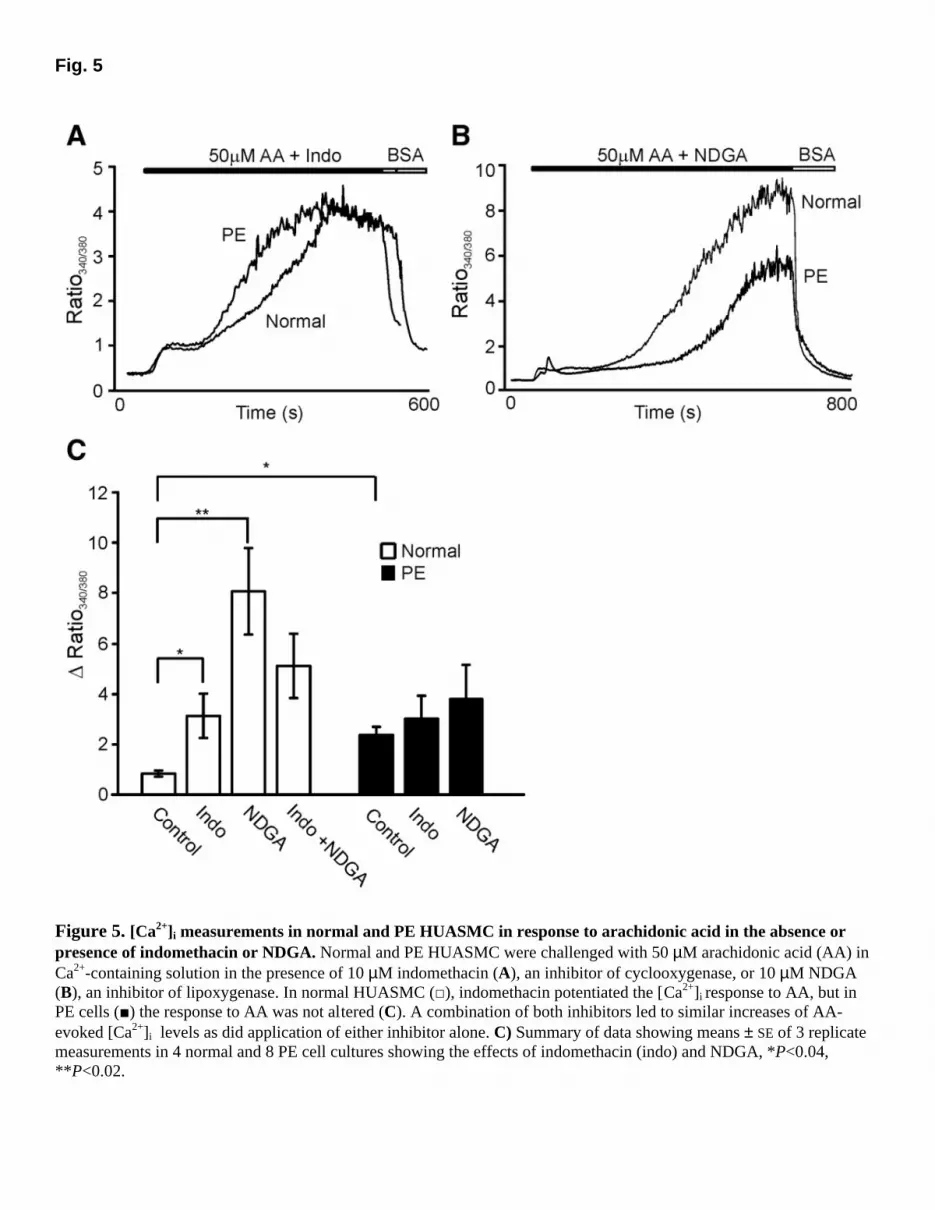
Fig. 5
Figure 5. [Ca2+]i measurements in normal and PE HUASMC in response to arachidonic acid in the absence or presence of indomethacin or NDGA. Normal and PE HUASMC were challenged with 50 µM arachidonic acid (AA) in Ca2+-containing solution in the presence of 10 µM indomethacin (A), an inhibitor of cyclooxygenase, or 10 µM NDGA (B), an inhibitor of lipoxygenase. In normal HUASMC (□), indomethacin potentiated the [Ca2+]i response to AA, but in PE cells (■) the response to AA was not altered (C). A combination of both inhibitors led to similar increases of AA-evoked [Ca2+]i levels as did application of either inhibitor alone. C) Summary of data showing means ± SE of 3 replicate measurements in 4 normal and 8 PE cell cultures showing the effects of indomethacin (indo) and NDGA, *P<0.04, **P<0.02.
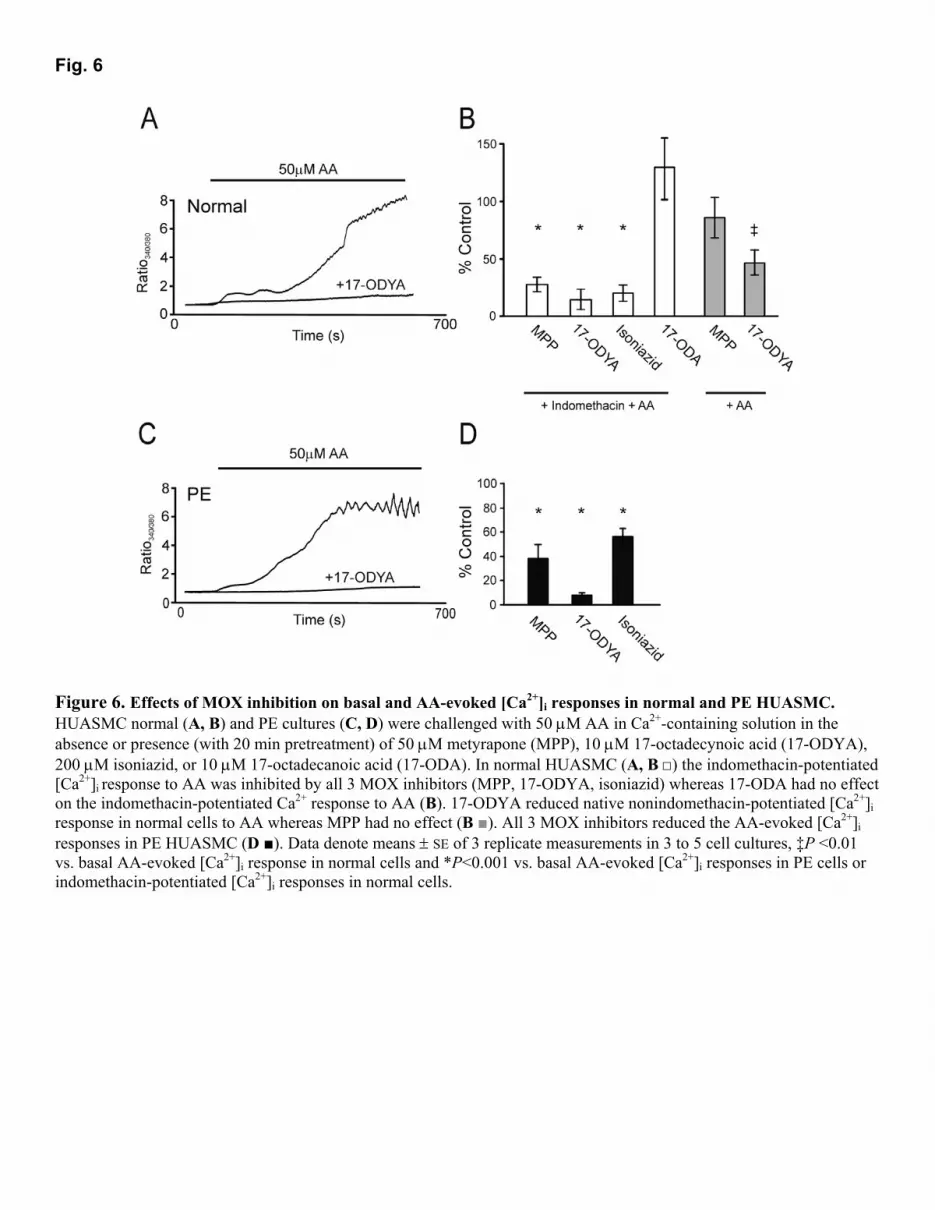
Fig. 6
Figure 6. Effects of MOX inhibition on basal and AA-evoked [Ca2+]i responses in normal and PE HUASMC. HUASMC normal (A, B) and PE cultures (C, D) were challenged with 50 µM AA in Ca2+-containing solution in the absence or presence (with 20 min pretreatment) of 50 µM metyrapone (MPP), 10 µM 17-octadecynoic acid (17-ODYA), 200 µM isoniazid, or 10 µM 17-octadecanoic acid (17-ODA). In normal HUASMC (A, B □) the indomethacin-potentiated [Ca2+]i response to AA was inhibited by all 3 MOX inhibitors (MPP, 17-ODYA, isoniazid) whereas 17-ODA had no effect on the indomethacin-potentiated Ca2+ response to AA (B). 17-ODYA reduced native nonindomethacin-potentiated [Ca2+]i response in normal cells to AA whereas MPP had no effect (B ■). All 3 MOX inhibitors reduced the AA-evoked [Ca2+]i responses in PE HUASMC (D ■). Data denote means ± SE of 3 replicate measurements in 3 to 5 cell cultures, �P <0.01 vs. basal AA-evoked [Ca2+]i response in normal cells and *P<0.001 vs. basal AA-evoked [Ca2+]i responses in PE cells or indomethacin-potentiated [Ca2+]i responses in normal cells.
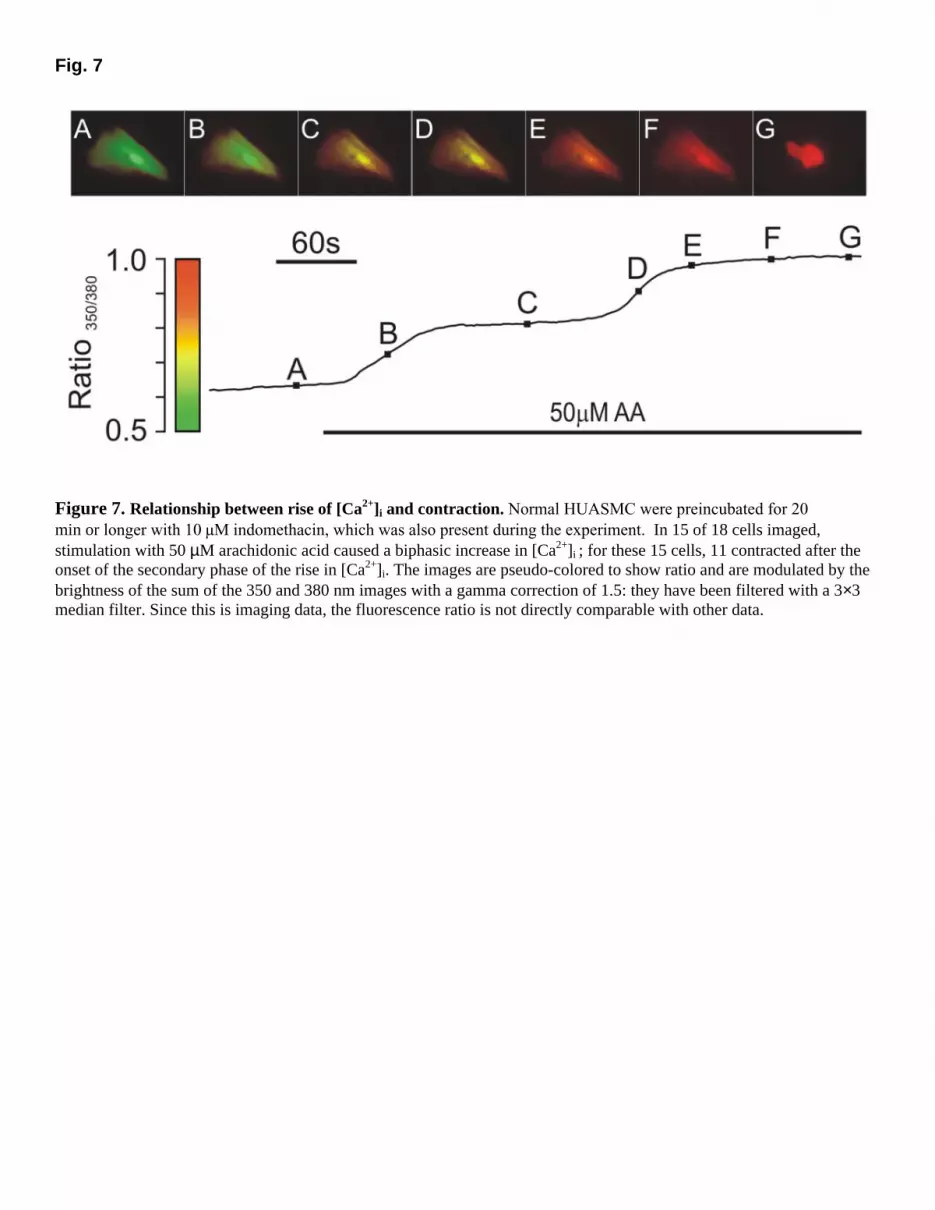
Fig. 7
Figure 7. Relationship between rise of [Ca2+]i and contraction. Normal HUASMC were preincubated for 20 min or longer with 10 µM indomethacin, which was also present during the experiment. In 15 of 18 cells imaged, stimulation with 50 µM arachidonic acid caused a biphasic increase in [Ca2+]i ; for these 15 cells, 11 contracted after the onset of the secondary phase of the rise in [Ca2+]i. The images are pseudo-colored to show ratio and are modulated by the brightness of the sum of the 350 and 380 nm images with a gamma correction of 1.5: they have been filtered with a 3×3 median filter. Since this is imaging data, the fluorescence ratio is not directly comparable with other data.