a study on the interactions between coenzyme q 0 and superoxide anion. could ubiquinones mimic...
TRANSCRIPT

A STUDY ON THE INTERACTIONS BETWEEN
COENZYME Q0 AND SUPEROXIDE ANION. COULD
UBIQUINONES MIMIC SUPEROXIDE DISMUTASE
(SOD)?
RITA PETRUCCI,a ELISABETTA GIORGINI,b ELISABETTA DAMIANI,b PATRICIA CARLONI,b GIANCARLO MARROSU,a ANTONIO TRAZZA,a GIAN
PAOLO LITTARRUc and LUCEDIO GRECI*,b
aDipartimento di Ingegneria Chimica, dei Materiali, delle Materie Prime e Metallurgia, Università "La Sapienza", via del Castro Laurenziano 7, 1-00161 Roma, ITALY; bDipartimento di Scienze dei Materiali e della Terra, Università, via Brecce Bianche, I-60131 Ancona, ITALY; cIstituto di Biochimica, Università, via Brecce Bianche, 1-60131 Ancona, ITALY.
Received 13 July 1999; accepted 12 August 1999
Abstract--An electrochemical study was carried out on 1,4-benzoquinone, duroquinone, coenzymes Q0 and Q10 in the absence and in the presence of molecular oxygen in aprotic (DMF) and protic (DMF/H2O 95:5 (v/v)) media. Water was added because the investigated reactions are deeply influenced by the presence of protons. Q0 and Q10 exhibited a similar electrochemical behaviour. Since
Q0 is more soluble in protic medium than the biologically more important analogue Q10, it was chosen as a model for a more detailed investigation. Voltammetric studies of Q0 carried out in aprotic and
protic media in the presence of oxygen showed that, besides simple O2- dismutation, theQ0 promoted dismutation of O2- should also be considered. Spectroelectrochemical experiments with the same
experimental conditions support the electrochemical results, showing that in the presence of superoxide and in aprotic medium semiquinone Q0- gives rise to a disproportionation equilibrium, while in the
presence of water it tends to be reoxidized to the starting Q0 by OOH. EPR measurements are also in
agreement with these results.
INTRODUCTION
In the last decades the inhibition of radical chain reactions involved in the oxidation
of numerous substrates has become of prime importance not only in the polymer and
food industries, but also in the field of biology and medicine [1]. In this context,
ubiquinones, and especially their reduced forms, are increasingly being used as
antioxidants for the treatment of a variety of diseases and the modulation of ageing
[2]. However, it should be stressed that the principal biological function of
ubiquinones remains their role as electron carriers in the mitochondrial respiratory

270
chain [3]. Apart from these classical functions, the intervention of ubiquinones has
been invoked to account for superoxide radical formation in normal cell respiration
[4], while their reaction with superoxide itself yields the corresponding radical anions
and oxygen [5-7].
Starting from a rejuvenated interest in ubiquinones, and in the wake of a
previous study in which we examined the mechanism of the dismutation of
superoxide radical in the presence of a series of aminoxyls [8], we sought to
reinvestigate, under the same experimental conditions, in aprotic and protic media the
electrochemical behaviour of 1,4-benzoquinone (1), duroquinone (2) and ubiquinones
Qo (3) and Qlo (4) toward superoxide radical. In fact, although the electrochemical
behaviour of quinones and ubiquinones has been previously and thoroughly
investigated [5, 9-10] the study of their interactions with superoxide [11] is still far
from being exhausted, especially because of the lack of comparable data.
RESULTS AND DISCUSSION
V oltammetric Results
Compounds 1-4 all exhibit, in anhydrous deoxygenated DMF, two well defined
monoelectronic reduction steps, a first one reversible and ranging between -0.53 V
and -0.86 V, and a quasi-reversible second one ranging between -1.31 V and -1.51 1
V. In protic medium [(DMF/H20 95:5 (v/v): the percentage of water was chosen in
order to obtain the same experimental conditions for (Qlo), the least soluble
compound] all compounds exhibit a positive shift of both the reduction steps, with a
much larger shift for the second than for the first (see Table 1).

271

272
The voltammetric study carried out with different concentrations of 1-4
showed that in all cases both the first and the second reduction potentials shift towards
more negative values on increasing the concentration. Representative voltammograms for compound 3 are shown in Figures 1 and 2 (solid line).
Figure 1 Cyclic voltammograms of Qo in anhydrous DMF 0.1 mol rl TEAP at a GC electrode, scan rate 0.200 V s-1, in the absence (solid line) and in the presence (dotted line) of oxygen.
It is worth noting that in anhydrous DMF, oxygen exhibits two
monoelectronic reduction steps (see Table): the first one (formation of 020- is a
reversible process, while the irreversibility of the second, i.e. the formation of O2==
may reflect the fast protonation of the dianion to give H02-, even in dry DMF
(promoted by traces of water in the oxygen flux) [8]. Furthermore, the second
reduction step generates a broadened anodic peak at a very positive potential ( Ep,, =
+1 V), previously attributed to the oxidation of H02 [8]. In protic medium
(DMF/HZO 95:5 (v/v)), both cathodic peaks shift toward more positive potentials, this
effect being much more pronounced for the second one; in addition a loss of
reversibility is observed even in the first step.
Compounds 1-4 all were also studied in the presence of oxygen in both
aprotic and protic media. The cyclic voltammograms of quinones 1-2 were difficult
to interpret because of the ovelap of their reduction peak with that of oxygen: in
particular, the first reduction peak of oxygen partly overlaps with the second reduction

273
peak of 1 and completely with the first reduction peak of 2. This overlap was not a
problem in the case of Qo (see Fig. 1 and 2) and QlO which showed a similar
electrochemical behaviour (see Epc values in Table). Qo was chosen as the substrate
for a more careful and thorough voltammetric study being more soluble in both media
(even at concentrations greater than 5 x 10-3 mol 1-'), with respect to its biologically more important analogue Qlo. Therefore, results and discussion will be referred to Qo
only.
Figure 2. Cyclic voltammograms of Qo in DMF/H20 95:5 (v/v) 0.1 mol 1-' TEAP at a GC electrode, scan rate 0.200 V s-', in the absence (solid line) and in the presence (dotted line) of oxygen.
In the literature it is often reported that quinone radical anions can be
oxidized in the presence of oxygen, with formation of superoxide anion and the
corresponding quinones: QO*- + Qo + 02'- this reaction is commonly
considered as an equilibrium, and some authors claim that this is thermodynamically
shifted to the left [6, 12]. The equilibrium seems to be shifted to the right only in the
presence of species that can promote superoxide dismutation [6,11,13,14] or of
protons. Although 02'- is a very weak base (pK, of the acid HOO' is 4.69) [15]
reaction (4) is very fast [16,17] and shifts the equilibrium of eqn. (2) to the right,
increasing the basicity of O2.- to an extent equivalent to that of a very strong base
[17]. The high rate constant of reaction (4) is justified by the high oxidising power
of HOO* [8,18] but the data reported in the literature cannot be used to evaluate the

274
interaction between these species because they lack homogeneity having been
measured under different experimental conditions [5,9,10].
When Qo and oxygen are reduced to their corresponding anions, a series of
reactions (eqn.s (1)-(6)) could occur, most of which involve radical anions: therefore
these reactions are seriously affected by the presence of a proton source [9, 14]. This
is why our experiments were carried out also in a well defined protic medium
(DMF/H20 95:5 (v/v)).
From the cyclic voltammogram of Qo in aprotic medium and in the presence of oxygen (Fig. 1, dotted line) it can be seen that the height of the first reduction peak
(a) remains unchanged, while the height of the second one (b) increases by about 20
% and the corresponding Epc shifts to a more negative value by 50 mV (as observed
for increasing concentrations of compounds 1-4 in aprotic medium). This suggests that the radical anion Qo*- is generated at first via electrochemical reduction and then,
when the superoxide anion is available, via electron transfer, according to reaction ( 1 ).
In addition, the second reduction peak of oxygen at about -2.0 V almost completely
disappears (y in Fig. 3) supporting the consumption of superoxide; furthermore, the
anodic peak corresponding to the process 02'- --. O2 + e (x') also decreases.
These observations were confirmed by applying the Marcus theory (see
experimental) to our electrochemical data in aprotic medium; the kl value ( 1 0 M-' s'')
calculated for reaction (1) matches well with that reported in the literature [19],
showing that the reaction is very fast. However, it must be considered that in
biological systems, the presence of the enzyme SOD (superoxide dismutase), which
dismutates 020. at a rate of 10 M-1 s' [20], could shift the equilibrium of eqn. (1) to
the left, above all under aerobic conditions [6]. In addition, even in aqueous medium
the equilibrium is shifted to the left due to protonation of superoxide anion (eqn. (2)) and its subsequent dismutation (eqn.s (4) and (5)).
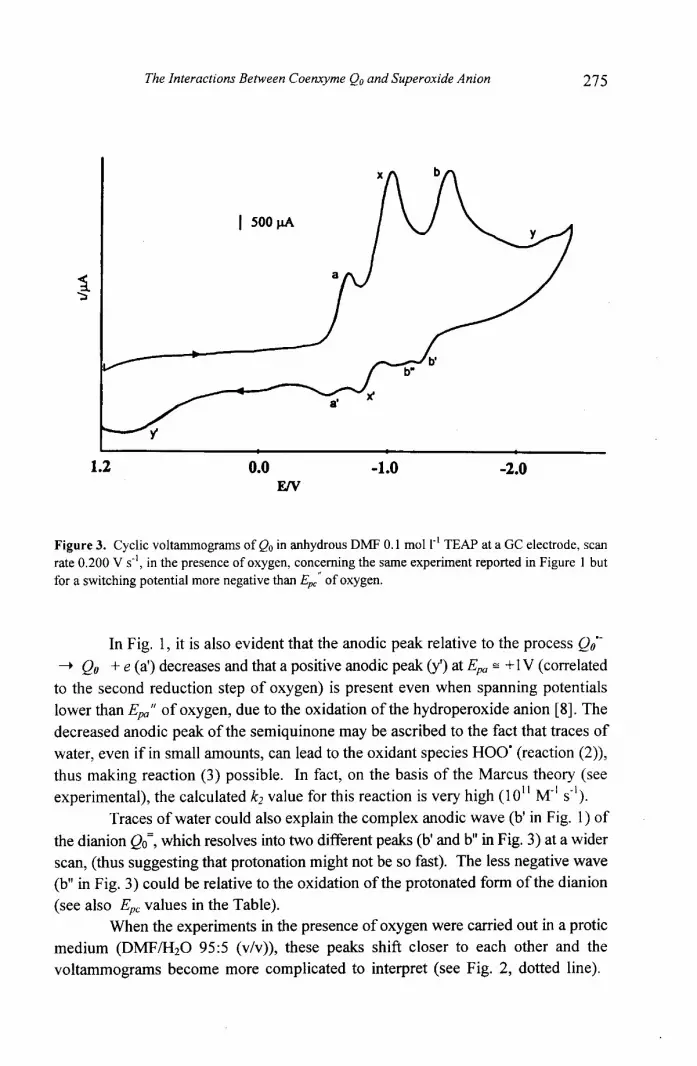
275
Figure 3. Cyclic voltammograms of Qo in anhydrous DMF 0.1 mol 1-' TEAP at a GC electrode, scan rate 0.200 V s', in the presence of oxygen, concerning the same experiment reported in Figure I but for a switching potential more negative than E,, of oxygen.
In Fig. 1, it is also evident that the anodic peak relative to the process Qoo- - -t Qo + e (a') decreases and that a positive anodic peak (y') at Epa = +1 V (correlated
to the second reduction step of oxygen) is present even when spanning potentials lower than Epp" of oxygen, due to the oxidation of the hydroperoxide anion [8]. The
decreased anodic peak of the semiquinone may be ascribed to the fact that traces of
water, even if in small amounts, can lead to the oxidant species HOO' (reaction (2)),
thus making reaction (3) possible. In fact, on the basis of the Marcus theory (see
experimental), the calculated k2 value for this reaction is very high ( 10" M-' 1 s- I).
Traces of water could also explain the complex anodic wave (b' in Fig. 1) of
the dianion which resolves into two different peaks (b' and b" in Fig. 3) at a wider
scan, (thus suggesting that protonation might not be so fast). The less negative wave
(b" in Fig. 3) could be relative to the oxidation of the protonated form of the dianion
(see also Epc values in the Table). When the experiments in the presence of oxygen were carried out in a protic
medium (DMF/H20 95:5 (v/v)), these peaks shift closer to each other and the
voltammograms become more complicated to interpret (see Fig. 2, dotted line).
276
However, it can be seen that the second reduction peak of 3 (b) substantially decreases
in height while the corresponding anodic peak (b') seems to be higher than the
cathodic one; the anodic peak relative to the process 02'- - 02+ e almost completely
disappears (compare x' in Fig. 1); the anodic peak relative to the process Qo'- Qo + e (a') also decreases; the positive anodic peak (y') at Epa Ë +1 V is still present. The
consumption of 02'- and Qo'- and the presence of the very positive anodic peak
suggests that besides dismutation of superoxide (reaction (4)), the reaction of HOO'
with Qo'- might also occur according to reaction (3), as already observed previously in the presence of only traces of water (y' in Figures 1 and 3).
Spectroelectrochemical Results
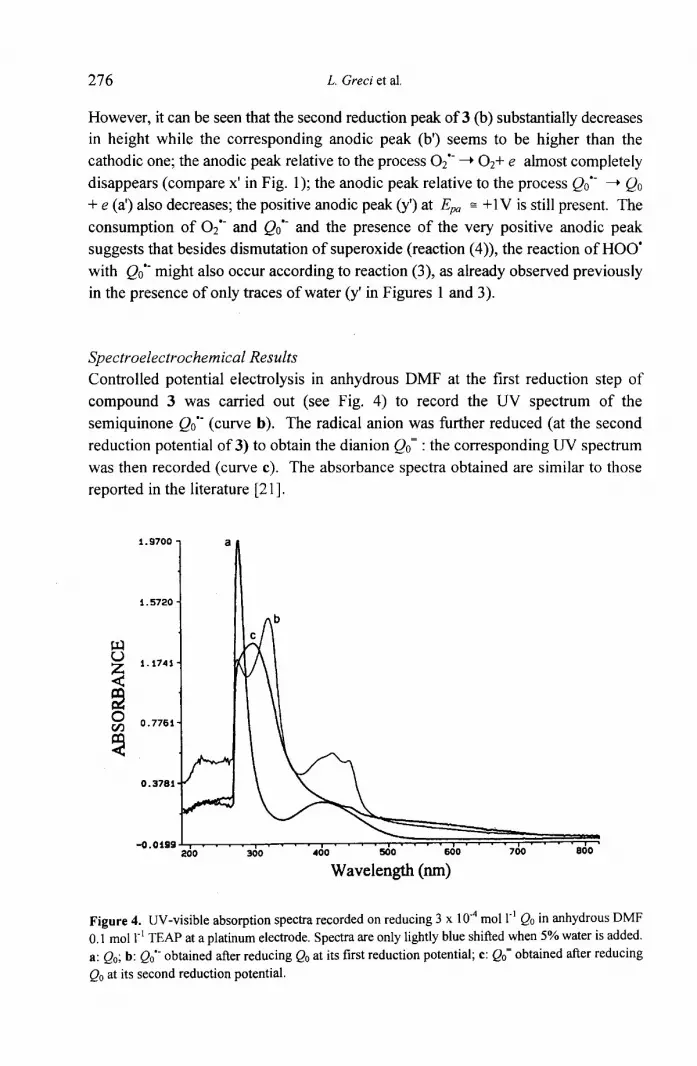
Controlled potential electrolysis in anhydrous DMF at the first reduction step of
compound 3 was carried out (see Fig. 4) to record the UV spectrum of the
semiquinone Qoo- (curve b). The radical anion was further reduced (at the second
reduction potential of 3) to obtain the dianion Qo- : the corresponding UV spectrum was then recorded (curve c). The absorbance spectra obtained are similar to those
reported in the literature [21 ].
Figure 4. UV-visible absorption spectra recorded on reducing 3 x 10 -4 mol 1-' Qo in anhydrous DMF
0.1 1 mol 1-1 TEAP at a platinum electrode. Spectra are only lightly blue shifted when 5% water is added.
a: Qo ; b: Qo°" obtained after reducing Qo at its first reduction potential; c: Qo obtained after reducing
Qo at its second reduction potential.

277
The reaction between Qo and 02'- was then studied in the UV cell, both in
aprotic and protic medium. Figures 5 and 6 show the significant spectra recorded
during the reaction, the results being independent of the method used to generate
superoxide anion.
Figure 5. UV-visible spectra recorded in anhydrous DMF during the reaction between Qo and
superoxide anion. a: Qo; b: after addition. of superoxide anion; c: after 30 minutes from start of the reaction.
In aprotic medium (Fig. 5), after the addition of superoxide to the solution
containing quinone 3, the typical absorbance peak at 320 nm of the semiquinone
appears at once (see spectrum b), indicating the occurrence of reaction ( 1 ); with time,
spectrum b evolves into c, which could be interpreted as the spectrum resulting from
a mixture of Qo, Qo*- the dianion Qo . This last result is in agreement with the
presence of the equilibrium described in eqn. (6).
Upon addition of superoxide in protic medium, the spectrum typical of the
semiquinone was also observed (Fig. 6), even if less pronounced (see the small
absorption band at 320 nm, Fig. 6 curve b): this suggests that superoxide reacts with
Qo affording Qo*- (reaction ( 1 )), even if to a lesser extent due to protonation of
superoxide; in any case its intensity gradually decreases and the spectrum with time
tends to assume the initial shape typical of Qo (Fig. 6 curve c). This could most likely
be interpreted by the fact that HOO' generated by protonation of 02" (eqn. (2)),
rapidly reoxidizes Qo'- to Qo (eqn. (3)).
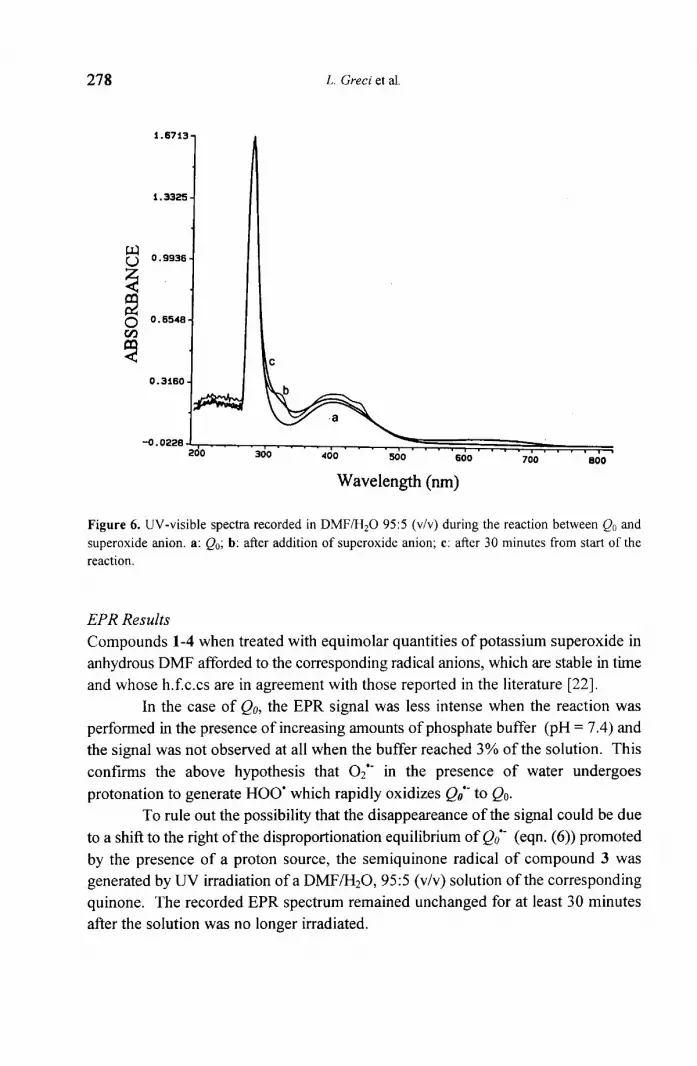
278
1.9
Figure 6. UV-visible spectra recorded in DMF/H20 95:5 (v/v) during the reaction between Qo and
superoxide anion. a: Qo; b: after addition of superoxide anion; c: after 30 minutes from start of the reaction.
EPR Results
Compounds 1-4 when treated with equimolar quantities of potassium superoxide in
anhydrous DMF afforded to the corresponding radical anions, which are stable in time
and whose h.f.c.cs are in agreement with those reported in the literature [22]. In the case of Qo, the EPR signal was less intense when the reaction was
performed in the presence of increasing amounts of phosphate buffer (pH = 7.4) and
the signal was not observed at all when the buffer reached 3% of the solution. This
confirms the above hypothesis that 02'- in the presence of water undergoes
protonation to generate HOO' which rapidly oxidizes Qoo. to Qo. To rule out the possibility that the disappeareance of the signal could be due
to a shift to the right of the disproportionation equilibrium of Qoo- (eqn. (6)) promoted
by the presence of a proton source, the semiquinone radical of compound 3 was
generated by UV irradiation of a DMF/H20, 95:5 (v/v) solution of the corresponding
quinone. The recorded EPR spectrum remained unchanged for at least 30 minutes
after the solution was no longer irradiated.

279
CONCLUSION
We believe that on the basis of the above results, a possible SOD mimic action for
ubiquinones may not be excluded. In fact, superoxide anion can be rapidly oxidized
by Qo to oxygen (reaction (1)) while Qo is reduced to the corresponding radical anion.
The protonated superoxide (reaction (2)) can reoxidize Qoo- to give the starting
quinone and the hydroperoxy anion (reaction 3). Reaction (1) is fast and shifted to
the right only in aprotic media; however it is an equilibrium and even in protic media
a low quantity of Qo*- may be formed. The high rate of reaction (3) can shift
equilibrium (1) to the right as much as (or more) reactions (2) and (4) shift it to the
left. In summary, Qo is recycled and superoxide anion is transformed into oxygen and
hydrogen peroxide. However, it is worth bearing in mind that because of the low
solubility of ubiquinones in water and their high solubility in lipids, the reactions
discussed above may be valid only at the interface of the lipid and aqueous phases. On the whole, it is evident that the reactivity of ubiquinones with superoxide
is complex, being strongly influenced by all the different parameters such as pH, the
presence of enzymes, lipo- or hydro-philicity, protic or aprotic media.
EXPERIMENTAL
Compounds 1-3 and all the solvents were purchased from Aldrich, while compound 4 was kindly donated by Kaneka. EPR spectra were recorded on a Varian E4
spectrometer interfaced with a PC, containing a ruby in the cavity as reference and
equipped with a medium pressure mercury lamp (250 W). A three-electrode
multipolarograph (AMEL 472) coupled with a digital x/y recorder (AMEL 863) was
employed for the voltammetric measurements, carried out at a pulsed (polarographic
measurements) or static (cyclic voltammetries) glassy-carbon electrode. Hg - Hg2C12 - NaCI (sat.aq.) - DMF - Et4NCl04 / sintered glass disk [23] was used as reference,
and a platinum wire as counter electrode. All the experiments were carried out using deionized water by Millipore Mill-Q Purification System. The spectroelectrochemical
study was carried out with a Diode Array Spectrophotometer HP 8452A and a
potentiostat (AMEL 552) coupled with an integrator (AMEL 731) and a recorder
(LINSEIS L250E) for controlled potential electrolyses, using an UV three-electrode
modified cell, a platinum wire as working electrode, an Ag/AgCI04 (0.1 mol 1-') - MeCN / fine-porosity fitted glass disk / DMF - Et4NC104 / sintered glass disk as
reference and a platinum wire (placed on the inner wall of a glass tube and connected
to the test solution via a sintered glass-disk) as auxiliary electrode.

280
Electrochemical and Spectroelectrochemical Measurements
Electrochemical measurements were carried out on nitrogen purged solutions of
quinones 1-4 at the concentration of about 1 x 10-3 mol 1-' in anhydrous DMF and
DMF/H20 95:5 (v/v), using TEAP (crystallised and dried under vacuum) 0.1 mol 1-'
as supporting electrolyte. The concentration of an oxygen saturated DMF solution at
25°C is about 4.8 x 10-3 mol 1-', therefore to have a substrate ratio 1:l, measurements in the presence of oxygen, in anhydrous DMF and DMF/H20 95:5 (v/v), were carried
out using a concentration of about 5 x 10-3 mol 1-' for compounds 1-4. In the case of
4 in DMF/H20, the solution at this concentration was opalescent. Throughout all the
measurements the temperature was kept at 25°C, constant within ± 0.1 °C. The
accuracy of the potentials reported in Tables is ± 5 mV.
Compound 3 was electrolysed in the UV cavity, at a concentration of about
3 x 104 mol 1-'. The solution was stirred by purging with a continuous nitrogen flow.
Experiments with potassium superoxide [24]. were carried out adding to the test
solution aliquots of an anhydrous DMF saturated solution of potassium superoxide. To prepare equimolar solutions of Potassium superoxide and Qo, dicyclohexane-18-
crown-6 was added to increase the solubility ofK02 in dry DMF. In this case a 0.05
mol r1 DMF solution of potassium superoxide containing 0.15 mol 1-' of crown was
used. Superoxide anion was also electrochemically generated [25] in the same aprotic medium by controlled potential electrolysis of an oxygen saturated solution, carried
out in anhydrous DMF 0.1 mol 1-' TEAP, using a pool of mercury as working
electrode, a platinum wire as counter and Hg - Hg2Clz - NaCI (sat.aq.) - DMF -
Et4NCl04 / sintered glass disk [23] as reference. Small amounts of the electrolysed
solution were rapidly transferred into the UV cell containing the Qo solution.
EPR Measurements
Dry DMF solutions of compounds 1-4 (0.02 mol 1'') were treated with saturated dry
DMF solutions of potassium superoxide and the EPR signals of the corresponding
semiquinone radical anions were recorded.
In order to find the equimolar ratio between Qo and superoxide, increasing
amounts of a saturated dry DMF solution of potassium superoxide were added to 0.1 1
ml of a solution of Qo (0.02 mol 1-') in anhydrous DMF, until the signal of the radical
anion increased no more (final volume 2 ml). Then, the signal of the radical anion
was recorded and compared with the signal of a ruby present in the cavity. The same
experiment was repeated using a solution containing increasing amounts of water
(phosphate buffer pH 7.4) until the signal completely disappeared. A DMF/H20 95:5 (v/v) solution (0.002 mol 1-1) of Qo, was UV-irradiated for
10 minutes until the signal of the semiquinone radical increased no more. The EPR
spectrum was checked for 30 minutes after having stopped irradiating.

281
Marcus Theory Treatment
The standard free energy change of the putative ET step was not corrected by an
electrostatic term. To compute the AG# by the Marcus expression [AG# = (1 +
where AG° ' = (E°°X - EOred) x 23.06 Kcal mol-' V-1] the following data were
used. Reorganization energy [26,27]. À(Oi02.-) = 49; À(Qo/QoO_) = 17.7;
À(HOOo/HOO-) = 30. Reduction potentials of Qo, oxygen and HOO' are reported in
the Table. For the calculation of the rate constant, the Arrhenius equation was used
with a preexponential factor of 1012 [26].
Acknowledgements We thank the Ministero dell'Universitd e della Ricerca Scientifica e Tecnologica
(MURST), the University of Ancona and the CNR for financial support and Angelo Alberti for helpful discussions.
REFERENCES
1. S. Al-Malaika, in C. Booth and C. Price (Eds.), Comprehensive Polymer Science, Pergamon Press, Oxford, New York, 1989, 2, 539; J. Pospisil and P.P. Klemchuk, Oxidation Inhibition
in Organic Materials, CRC Press Inc., Boca Raton, Florida, 1989, 1 and 2; B. Halliwell and J.M.C. Gutteridge in Lund (Ed.), Free Radicals in Biology and Medicine, , Clarendon Press, Oxford, 1993; C.E. Eriksson, and A. Na in F. Corongiu, S. Banni, M.A. Dessi and C. Rice- Evans (Eds.), Free Radicals and Antioxidants in Nutrition, Richelieu Press, 1993, 205-224.
2. S.A. Mortensen, Clin. Invest., , 71, 116 (1993); S.R. Thomas, J. Neuzil and R. Stocker, Arterioscler. Thromb. Vasc. Biol., 16, 687 (1996).
3. G. Lenaz, M. Battino, C. Castelluccio, R. Fato, M. Cavozzoni, H. Ranchova, C. Bovina, G.
Formiggini and G.P. Castelli, Free. Rad. Res. Comm., 8, 317 (1990). 4. E. Cadenas, A. Boveris, C.I. Ragan and A.O.M. Stoppani, Arch. Biochem. Biophys., 180, 248
(1977). 5. R.C. Prince, P.L. Dutton and J.M. Bruce, FEBS Lett., 160, 273 (1983). 6. K. Sugioka, M. Nakano, H. Totsune-Nakano, H. Minakami, S. Tero-Kubota and Y. Ikegami,
Biochim. Biophis. Acta., 936, 377 (1988). 7. H. Nohl and W. Jordan, Biochem. Biophys. Res. Commun., 138, 533 (1986). 8. P. Carloni, E. Damiani, L. Greci, P. Stipa, G. Marrosu, R. Petrucci and A. Trazza,
Tetrahedron, 52, 11257 (1996). 9. A. Brunmark and E. Cadenas, Free Rad. Biol. Med., 7, 435 91989). 10. Y.A. Ilan, G. Czapski and D. Meisel, Biochim. Bhiophys. Acta, 430(2), 209 91976); G.R.
Buettner, Arch. Biochem. Biophys., 300, 535 (1993). 11. T. Ozawa and A. Hanaki, Chem. Pharm. Bull., 31, 2535 (1983). 12. C.C. Winterbourn, Arch. Biochem. Biophys., 209, 159 (1981). 13. D.A. Stoyanovsky, A.N. Osipov, P.J. Quinn and V.E. Kagan, Arch. Biochem. Biophys., 323,
343 91995); H. Nohl, L. Gille and A. V. Kozlov, Free Rad. Biol. Med., 25, 666 (1988); K.
Schnurr, M. Hellwing, B. Seidemann, P. Jungblut, H. Kuhn, S.M. Rapoport and T. Schewe, Free Rad. Biol. Med., 20, 11 (1996).

282
14. H. Nohl, L. Gille, K. Schonheit and Y. Liu, Free Rad. Biol. Med., 20, 207 (1996). 15. J. Wilshire and D.T. Sawyer, Acc. Chem. Res., 12, 105 (1979). 16. D.T. Sawyer, M.J. Gibian, M.M. Morrison and T. Seo, J. Am. Chem. Soc., 100, 627 (1978). 17. A.A. Frimer Organic Reactions Involving the Superoxide Anion, in S. Patai (Ed.), The
Chemistry of Functional Groups. Peroxides, Wiley, New York, 1983, 429. 18. G. Merenyi, J. Lind and L. Engman, J. Chem. Soc. Perkin Trans. 2, 2551 (1994). 19. B. Kantilal, B. Patel and J.S. Willson, J. Chem. Soc. Faraday Trans. 1, 69, 814 (1973). 20. J.M. McCord and I. Fridovich, J. Biol. Chem., 243, 5753 (1968). 21. R.S.K.A. Gamage, S. Umapathy and A.J. McQuillan, J. Electroanal. Chem., 284, 229 (1990);
M. Degli Esposti, E. Ferri, G. Lenaz, Ital. J. Biochem., 30, 437 (1981). 22. K.B. Ulmschneider and H.B. Stegmann, Semiquinones and Related Species in H. Fischer and
K.-H. Hellwege (Eds.), Magnetic Properties of Free radicals. Part d1: Organic Anion
Radicals, Landolt-Börnstein Ser. II/9, , Springer-Verlag, Berlin-Heidelberg, 1980. 23. R. Andruzzi, A. Trazza, L. Greci and L. Marchetti, Ann. Chim. (Rome), 69, 583 (1979). 24. D.T. Sawyer and J.J. Gibian, Tetrahedron, 35, 1471 (1979). 25. D.T. Sawyer and J L. Roberts Jr., J Electroanal. Chem., 12, 90 (1969). 26. L. Eberson, Electron Transfer Reactions in Organic Chemistry, Springer-Verlag, Heidelberg,
1987. 27. E. Pellizzetti, E. Mentasti and E. Pramauro, Inorg. Chem., 17, 1988 (1978).