a polyvalent aptamer system for targeted drug delivery
TRANSCRIPT

lable at ScienceDirect
Biomaterials xxx (2013) 1e8
Contents lists avai
Biomaterials
journal homepage: www.elsevier .com/locate/biomater ia ls
A polyvalent aptamer system for targeted drug delivery
Zhiqing Zhang a, b, c, M. Monsur Ali b, c, Mark A. Eckert b, c, Dong-Ku Kang b, c,Yih Yang Chen b, c, Leonard S. Sender d, e, David A. Fruman f, Weian Zhao b, c, *
a State Key Laboratory of Heavy Oil Processing, College of Science, China University of Petroleum, Qingdao 266580, People’s Republic of Chinab Department of Pharmaceutical Sciences, Sue and Bill Gross Stem Cell Research Center and Chao Family Comprehensive Cancer Center, University ofCalifornia, Irvine, 845 Health Sciences Road, Irvine, CA 92697, USAc Department of Biomedical Engineering, and Edwards Lifesciences Center for Advanced Cardiovascular Technology, University of California, Irvine, 845Health Sciences Road, Irvine, CA 92697, USAd Department of Medicine, University of California, Irvine, Irvine, CA, USAe Hyundai Cancer Institute, CHOC Children’s Hospital, Orange, CA, USAf Department of Molecular Biology & Biochemistry, University of California, Irvine, CA, USA
a r t i c l e i n f o
Article history:Received 19 July 2013Accepted 27 August 2013Available online xxx
Keywords:AptamerRolling circle amplificationMultivalencyDrug deliveryLeukemiaCancer
* Corresponding author. Department of PharmaceGross Stem Cell Research Center and Chao Family CoUniversity of California, Irvine, 845 Health Sciences R
E-mail address: [email protected] (W. Zhao).
0142-9612/$ e see front matter � 2013 Elsevier Ltd.http://dx.doi.org/10.1016/j.biomaterials.2013.08.079
Please cite this article in press as: Zhang Z, et10.1016/j.biomaterials.2013.08.079
a b s t r a c t
Poor efficacy and off-target systemic toxicity are major problems associated with current chemothera-peutic approaches to treat cancer. We developed a new form of polyvalent therapeutics that is composedof multiple aptamer units synthesized by rolling circle amplification and physically intercalatedchemotherapy agents (termed as “Poly-Aptamer-Drug”). Using a leukemia cell-binding aptamer anddoxorubicin as a model system, we have successfully constructed Poly-Aptamer-Drug systems anddemonstrated that the Poly-Aptamer-Drug is significantly more effective than its monovalent counter-part in targeting and killing leukemia cells due to enhanced binding affinity (w40 fold greater) and cellinternalization via multivalent effects. We anticipate that our Poly-Aptamer-Drug approach will yieldnew classes of tunable therapeutics that can be utilized to effectively target and treat cancers whileminimizing the side effects of chemotherapy.
� 2013 Elsevier Ltd. All rights reserved.
1. Introduction
Targeted cancer therapy is still a major unmet need. Currentchemotherapeutic drugs lack selectivity and are associated withmajor side-effects and lack of efficacy in many patients. Recenteffort has therefore focused on the development of targeted ap-proaches (e.g., antibodyedrug conjugates, targeted drug delivery(TDD) systems with nanoparticles) to deliver drugs selectively tocancerous cells, thus enhancing drug efficacy and reducing non-specific toxicity [1e6]. However, current targeted cancer therapysystems, which typically utilize monovalent molecular recognition[7e11] between targeting molecules (e.g., antibody and aptamers)and receptors on cancer cells, still suffer from poor targeting andcellular internalization efficiency, selectivity, and overall killingefficacy.
In nature, biological systems often use multivalent, cooperativeinteractions where multiple ligands on one biological entity
utical Sciences, Sue and Billmprehensive Cancer Center,oad, Irvine, CA 92697, USA.
All rights reserved.
al., A polyvalent aptamer sys
simultaneously bind to receptors on another to achieve highbinding affinity and selectivity [12]. Inspired by nature, engineeredmultivalency has become an emerging and powerful strategy toimprove targeting efficacy and selectivity in drug delivery [12e17].For example, cooperative, multivalent binding between targetingmolecules immobilized on a polymer scaffold or nanoparticle andreceptors at target sites can improve not only the affinity but alsothe specificity of molecular interactions involved in drug delivery[2,12e14]. Intriguingly, multivalent ligandereceptor binding at thecell membrane can promote cellular internalization, likely throughenergy-dependent endocytic pathways [2,12e14]. While these ex-amples demonstrate the advantages of multivalency in drug tar-geting and delivery, current methods to prepare multivalent TDDsystems are complex and often involve chemistries that are noteasily modified [2,12e14].
We have recently exploited a simple, powerful isothermalenzymatic reaction called rolling circle amplification (RCA) tosynthesize multivalent scaffolds [18e20]. In RCA, DNA polymerase(e.g., phi29 DNA polymerase) extends DNA from a primer byreplicating a circular DNA template many times to yield a single-stranded (ss) DNA product that is typically tens of thousands ofnucleotides long (Fig.1a) [21e28]. Note also that RCA products
tem for targeted drug delivery, Biomaterials (2013), http://dx.doi.org/
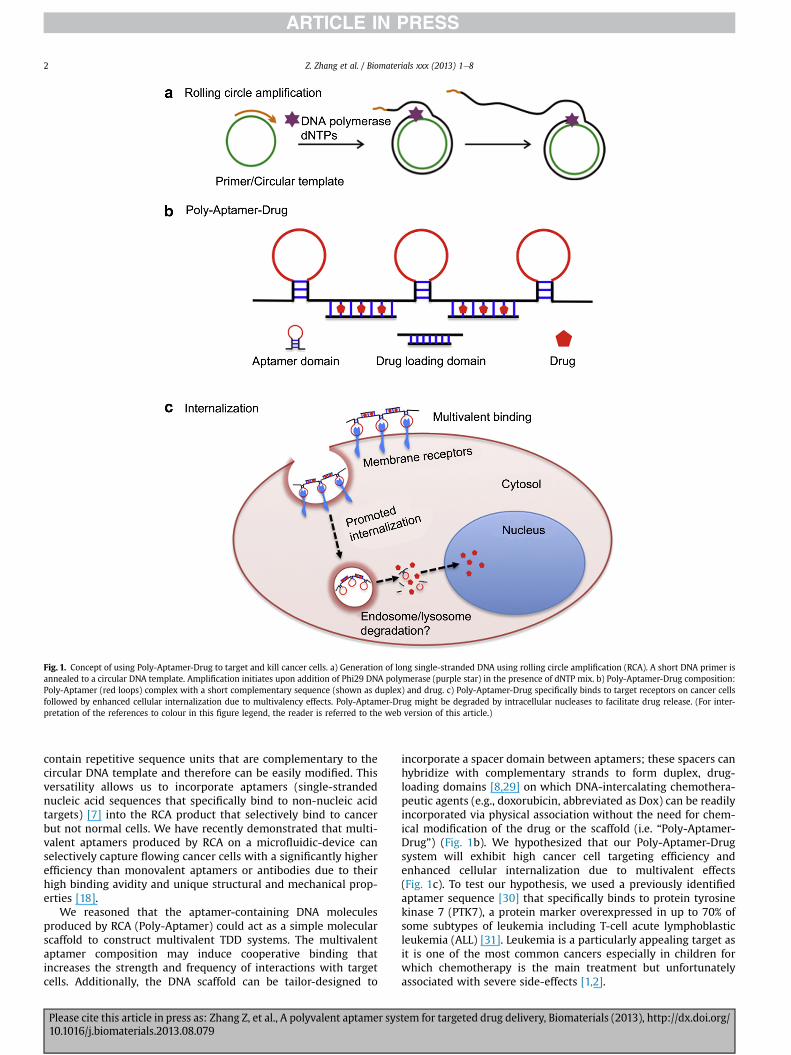
Fig. 1. Concept of using Poly-Aptamer-Drug to target and kill cancer cells. a) Generation of long single-stranded DNA using rolling circle amplification (RCA). A short DNA primer isannealed to a circular DNA template. Amplification initiates upon addition of Phi29 DNA polymerase (purple star) in the presence of dNTP mix. b) Poly-Aptamer-Drug composition:Poly-Aptamer (red loops) complex with a short complementary sequence (shown as duplex) and drug. c) Poly-Aptamer-Drug specifically binds to target receptors on cancer cellsfollowed by enhanced cellular internalization due to multivalency effects. Poly-Aptamer-Drug might be degraded by intracellular nucleases to facilitate drug release. (For inter-pretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Z. Zhang et al. / Biomaterials xxx (2013) 1e82
contain repetitive sequence units that are complementary to thecircular DNA template and therefore can be easily modified. Thisversatility allows us to incorporate aptamers (single-strandednucleic acid sequences that specifically bind to non-nucleic acidtargets) [7] into the RCA product that selectively bind to cancerbut not normal cells. We have recently demonstrated that multi-valent aptamers produced by RCA on a microfluidic-device canselectively capture flowing cancer cells with a significantly higherefficiency than monovalent aptamers or antibodies due to theirhigh binding avidity and unique structural and mechanical prop-erties [18].
We reasoned that the aptamer-containing DNA moleculesproduced by RCA (Poly-Aptamer) could act as a simple molecularscaffold to construct multivalent TDD systems. The multivalentaptamer composition may induce cooperative binding thatincreases the strength and frequency of interactions with targetcells. Additionally, the DNA scaffold can be tailor-designed to
Please cite this article in press as: Zhang Z, et al., A polyvalent aptamer sys10.1016/j.biomaterials.2013.08.079
incorporate a spacer domain between aptamers; these spacers canhybridize with complementary strands to form duplex, drug-loading domains [8,29] on which DNA-intercalating chemothera-peutic agents (e.g., doxorubicin, abbreviated as Dox) can be readilyincorporated via physical association without the need for chem-ical modification of the drug or the scaffold (i.e. “Poly-Aptamer-Drug”) (Fig. 1b). We hypothesized that our Poly-Aptamer-Drugsystem will exhibit high cancer cell targeting efficiency andenhanced cellular internalization due to multivalent effects(Fig. 1c). To test our hypothesis, we used a previously identifiedaptamer sequence [30] that specifically binds to protein tyrosinekinase 7 (PTK7), a protein marker overexpressed in up to 70% ofsome subtypes of leukemia including T-cell acute lymphoblasticleukemia (ALL) [31]. Leukemia is a particularly appealing target asit is one of the most common cancers especially in children forwhich chemotherapy is the main treatment but unfortunatelyassociated with severe side-effects [1,2].
tem for targeted drug delivery, Biomaterials (2013), http://dx.doi.org/
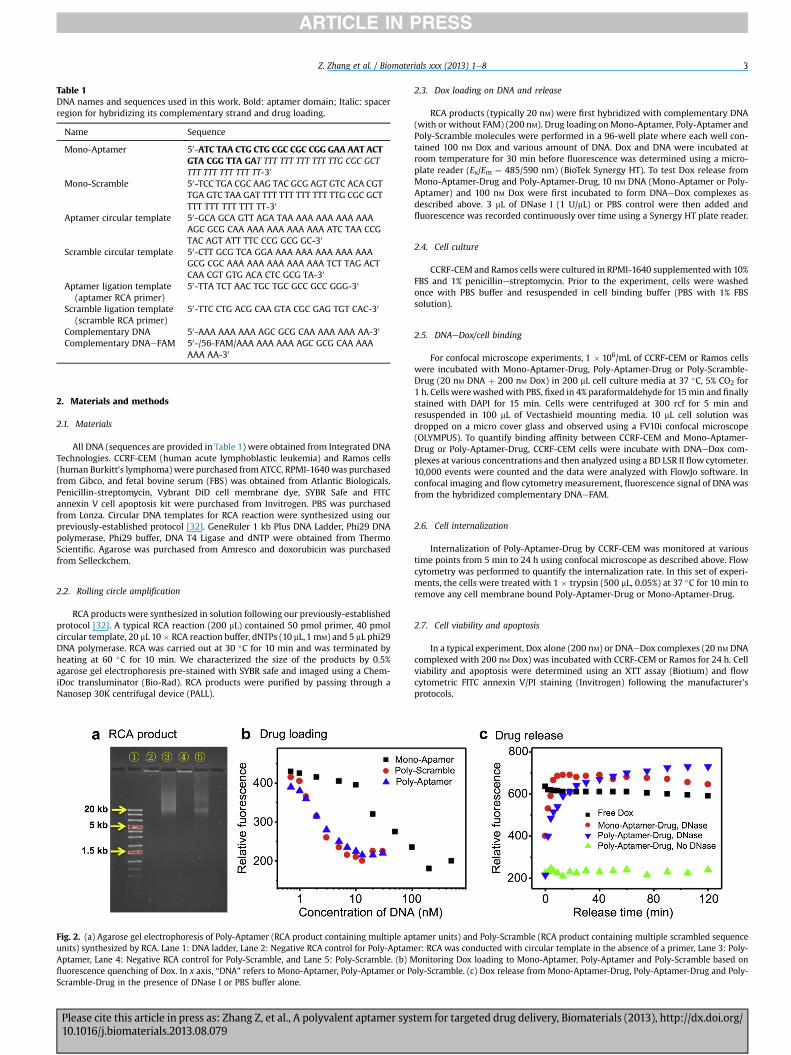
Table 1DNA names and sequences used in this work. Bold: aptamer domain; Italic: spacerregion for hybridizing its complementary strand and drug loading.
Name Sequence
Mono-Aptamer 50-ATC TAA CTG CTG CGC CGC CGG GAA AAT ACTGTA CGG TTA GAT TTT TTT TTT TTT TTG CGC GCTTTT TTT TTT TTT TT-30
Mono-Scramble 50-TCC TGA CGC AAG TAC GCG AGT GTC ACA CGTTGA GTC TAA GAT TTT TTT TTT TTT TTG CGC GCTTTT TTT TTT TTT TT-30
Aptamer circular template 50-GCA GCA GTT AGA TAA AAA AAA AAA AAAAGC GCG CAA AAA AAA AAA AAA ATC TAA CCGTAC AGT ATT TTC CCG GCG GC-30
Scramble circular template 50-CTT GCG TCA GGA AAA AAA AAA AAA AAAGCG CGC AAA AAA AAA AAA AAA TCT TAG ACTCAA CGT GTG ACA CTC GCG TA-30
Aptamer ligation template(aptamer RCA primer)
50-TTA TCT AAC TGC TGC GCC GCC GGG-30
Scramble ligation template(scramble RCA primer)
50-TTC CTG ACG CAA GTA CGC GAG TGT CAC-30
Complementary DNA 50-AAA AAA AAA AGC GCG CAA AAA AAA AA-30
Complementary DNAeFAM 50-/56-FAM/AAA AAA AAA AGC GCG CAA AAAAAA AA-30
Z. Zhang et al. / Biomaterials xxx (2013) 1e8 3
2. Materials and methods
2.1. Materials
All DNA (sequences are provided in Table 1) were obtained from Integrated DNATechnologies. CCRF-CEM (human acute lymphoblastic leukemia) and Ramos cells(human Burkitt’s lymphoma) were purchased fromATCC. RPMI-1640 was purchasedfrom Gibco, and fetal bovine serum (FBS) was obtained from Atlantic Biologicals.Penicillin-streptomycin, Vybrant DiD cell membrane dye, SYBR Safe and FITCannexin V cell apoptosis kit were purchased from Invitrogen. PBS was purchasedfrom Lonza. Circular DNA templates for RCA reaction were synthesized using ourpreviously-established protocol [32]. GeneRuler 1 kb Plus DNA Ladder, Phi29 DNApolymerase, Phi29 buffer, DNA T4 Ligase and dNTP were obtained from ThermoScientific. Agarose was purchased from Amresco and doxorubicin was purchasedfrom Selleckchem.
2.2. Rolling circle amplification
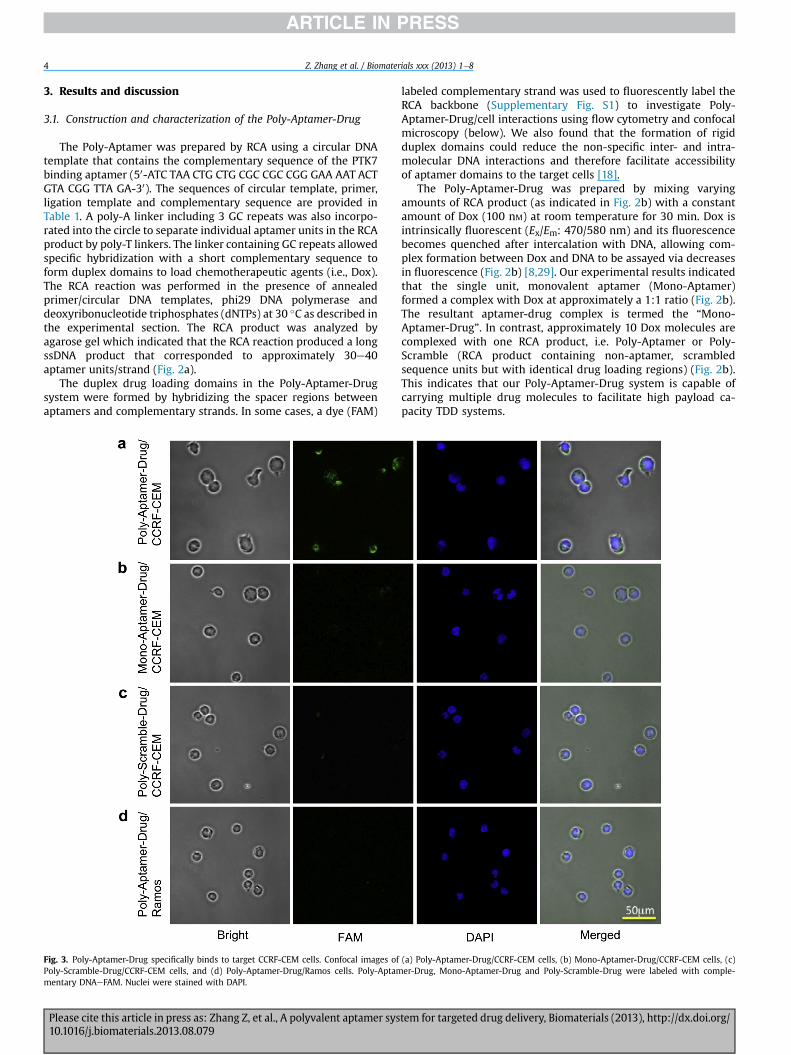
RCA products were synthesized in solution following our previously-establishedprotocol [32]. A typical RCA reaction (200 mL) contained 50 pmol primer, 40 pmolcircular template, 20 mL 10� RCA reaction buffer, dNTPs (10 mL,1 mM) and 5 mL phi29DNA polymerase. RCA was carried out at 30 �C for 10 min and was terminated byheating at 60 �C for 10 min. We characterized the size of the products by 0.5%agarose gel electrophoresis pre-stained with SYBR safe and imaged using a Chem-iDoc transluminator (Bio-Rad). RCA products were purified by passing through aNanosep 30K centrifugal device (PALL).
Fig. 2. (a) Agarose gel electrophoresis of Poly-Aptamer (RCA product containing multiple apunits) synthesized by RCA. Lane 1: DNA ladder, Lane 2: Negative RCA control for Poly-AptamAptamer, Lane 4: Negative RCA control for Poly-Scramble, and Lane 5: Poly-Scramble. (b) Mfluorescence quenching of Dox. In x axis, “DNA” refers to Mono-Aptamer, Poly-Aptamer or PScramble-Drug in the presence of DNase I or PBS buffer alone.
Please cite this article in press as: Zhang Z, et al., A polyvalent aptamer sys10.1016/j.biomaterials.2013.08.079
2.3. Dox loading on DNA and release
RCA products (typically 20 nM) were first hybridized with complementary DNA(with or without FAM) (200 nM). Drug loading onMono-Aptamer, Poly-Aptamer andPoly-Scramble molecules were performed in a 96-well plate where each well con-tained 100 nM Dox and various amount of DNA. Dox and DNA were incubated atroom temperature for 30 min before fluorescence was determined using a micro-plate reader (Ex/Em ¼ 485/590 nm) (BioTek Synergy HT). To test Dox release fromMono-Aptamer-Drug and Poly-Aptamer-Drug, 10 nM DNA (Mono-Aptamer or Poly-Aptamer) and 100 nM Dox were first incubated to form DNAeDox complexes asdescribed above. 3 mL of DNase I (1 U/mL) or PBS control were then added andfluorescence was recorded continuously over time using a Synergy HT plate reader.
2.4. Cell culture
CCRF-CEM and Ramos cells were cultured in RPMI-1640 supplementedwith 10%FBS and 1% penicillinestreptomycin. Prior to the experiment, cells were washedonce with PBS buffer and resuspended in cell binding buffer (PBS with 1% FBSsolution).
2.5. DNAeDox/cell binding
For confocal microscope experiments, 1 � 106/mL of CCRF-CEM or Ramos cellswere incubated with Mono-Aptamer-Drug, Poly-Aptamer-Drug or Poly-Scramble-Drug (20 nM DNA þ 200 nM Dox) in 200 mL cell culture media at 37 �C, 5% CO2 for1 h. Cells werewashedwith PBS, fixed in 4% paraformaldehyde for 15min and finallystained with DAPI for 15 min. Cells were centrifuged at 300 rcf for 5 min andresuspended in 100 mL of Vectashield mounting media. 10 mL cell solution wasdropped on a micro cover glass and observed using a FV10i confocal microscope(OLYMPUS). To quantify binding affinity between CCRF-CEM and Mono-Aptamer-Drug or Poly-Aptamer-Drug, CCRF-CEM cells were incubate with DNAeDox com-plexes at various concentrations and then analyzed using a BD LSR II flow cytometer.10,000 events were counted and the data were analyzed with FlowJo software. Inconfocal imaging and flow cytometry measurement, fluorescence signal of DNAwasfrom the hybridized complementary DNAeFAM.
2.6. Cell internalization
Internalization of Poly-Aptamer-Drug by CCRF-CEM was monitored at varioustime points from 5 min to 24 h using confocal microscope as described above. Flowcytometry was performed to quantify the internalization rate. In this set of experi-ments, the cells were treated with 1 � trypsin (500 mL, 0.05%) at 37 �C for 10 min toremove any cell membrane bound Poly-Aptamer-Drug or Mono-Aptamer-Drug.
2.7. Cell viability and apoptosis
In a typical experiment, Dox alone (200 nM) or DNAeDox complexes (20 nM DNAcomplexed with 200 nM Dox) was incubated with CCRF-CEM or Ramos for 24 h. Cellviability and apoptosis were determined using an XTT assay (Biotium) and flowcytometric FITC annexin V/PI staining (Invitrogen) following the manufacturer’sprotocols.
tamer units) and Poly-Scramble (RCA product containing multiple scrambled sequenceer: RCA was conducted with circular template in the absence of a primer, Lane 3: Poly-onitoring Dox loading to Mono-Aptamer, Poly-Aptamer and Poly-Scramble based on
oly-Scramble. (c) Dox release from Mono-Aptamer-Drug, Poly-Aptamer-Drug and Poly-
tem for targeted drug delivery, Biomaterials (2013), http://dx.doi.org/
Z. Zhang et al. / Biomaterials xxx (2013) 1e84
3. Results and discussion
3.1. Construction and characterization of the Poly-Aptamer-Drug
The Poly-Aptamer was prepared by RCA using a circular DNAtemplate that contains the complementary sequence of the PTK7binding aptamer (50-ATC TAA CTG CTG CGC CGC CGG GAA AAT ACTGTA CGG TTA GA-30). The sequences of circular template, primer,ligation template and complementary sequence are provided inTable 1. A poly-A linker including 3 GC repeats was also incorpo-rated into the circle to separate individual aptamer units in the RCAproduct by poly-T linkers. The linker containing GC repeats allowedspecific hybridization with a short complementary sequence toform duplex domains to load chemotherapeutic agents (i.e., Dox).The RCA reaction was performed in the presence of annealedprimer/circular DNA templates, phi29 DNA polymerase anddeoxyribonucleotide triphosphates (dNTPs) at 30 �C as described inthe experimental section. The RCA product was analyzed byagarose gel which indicated that the RCA reaction produced a longssDNA product that corresponded to approximately 30e40aptamer units/strand (Fig. 2a).
The duplex drug loading domains in the Poly-Aptamer-Drugsystem were formed by hybridizing the spacer regions betweenaptamers and complementary strands. In some cases, a dye (FAM)
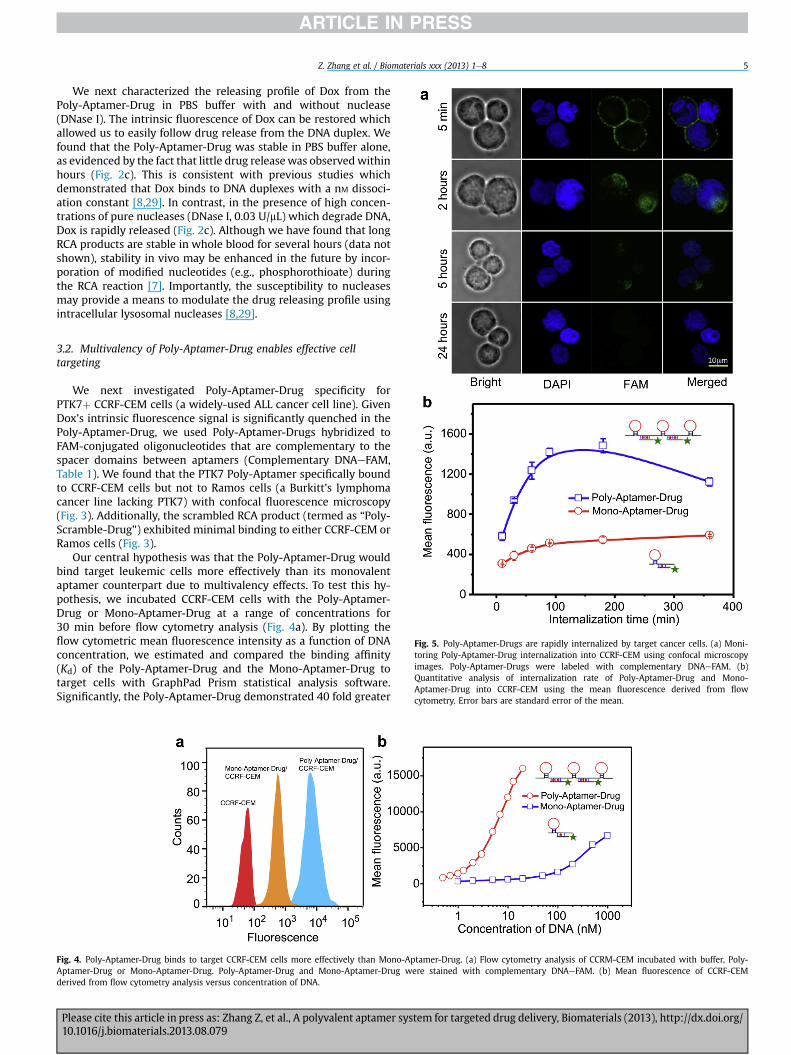
Fig. 3. Poly-Aptamer-Drug specifically binds to target CCRF-CEM cells. Confocal images ofPoly-Scramble-Drug/CCRF-CEM cells, and (d) Poly-Aptamer-Drug/Ramos cells. Poly-Aptammentary DNAeFAM. Nuclei were stained with DAPI.
Please cite this article in press as: Zhang Z, et al., A polyvalent aptamer sys10.1016/j.biomaterials.2013.08.079
labeled complementary strand was used to fluorescently label theRCA backbone (Supplementary Fig. S1) to investigate Poly-Aptamer-Drug/cell interactions using flow cytometry and confocalmicroscopy (below). We also found that the formation of rigidduplex domains could reduce the non-specific inter- and intra-molecular DNA interactions and therefore facilitate accessibilityof aptamer domains to the target cells [18].
The Poly-Aptamer-Drug was prepared by mixing varyingamounts of RCA product (as indicated in Fig. 2b) with a constantamount of Dox (100 nM) at room temperature for 30 min. Dox isintrinsically fluorescent (Ex/Em: 470/580 nm) and its fluorescencebecomes quenched after intercalation with DNA, allowing com-plex formation between Dox and DNA to be assayed via decreasesin fluorescence (Fig. 2b) [8,29]. Our experimental results indicatedthat the single unit, monovalent aptamer (Mono-Aptamer)formed a complex with Dox at approximately a 1:1 ratio (Fig. 2b).The resultant aptamer-drug complex is termed the “Mono-Aptamer-Drug”. In contrast, approximately 10 Dox molecules arecomplexed with one RCA product, i.e. Poly-Aptamer or Poly-Scramble (RCA product containing non-aptamer, scrambledsequence units but with identical drug loading regions) (Fig. 2b).This indicates that our Poly-Aptamer-Drug system is capable ofcarrying multiple drug molecules to facilitate high payload ca-pacity TDD systems.
(a) Poly-Aptamer-Drug/CCRF-CEM cells, (b) Mono-Aptamer-Drug/CCRF-CEM cells, (c)er-Drug, Mono-Aptamer-Drug and Poly-Scramble-Drug were labeled with comple-
tem for targeted drug delivery, Biomaterials (2013), http://dx.doi.org/
Z. Zhang et al. / Biomaterials xxx (2013) 1e8 5
We next characterized the releasing profile of Dox from thePoly-Aptamer-Drug in PBS buffer with and without nuclease(DNase I). The intrinsic fluorescence of Dox can be restored whichallowed us to easily follow drug release from the DNA duplex. Wefound that the Poly-Aptamer-Drug was stable in PBS buffer alone,as evidenced by the fact that little drug releasewas observedwithinhours (Fig. 2c). This is consistent with previous studies whichdemonstrated that Dox binds to DNA duplexes with a nM dissoci-ation constant [8,29]. In contrast, in the presence of high concen-trations of pure nucleases (DNase I, 0.03 U/mL) which degrade DNA,Dox is rapidly released (Fig. 2c). Although we have found that longRCA products are stable in whole blood for several hours (data notshown), stability in vivo may be enhanced in the future by incor-poration of modified nucleotides (e.g., phosphorothioate) duringthe RCA reaction [7]. Importantly, the susceptibility to nucleasesmay provide a means to modulate the drug releasing profile usingintracellular lysosomal nucleases [8,29].
Fig. 5. Poly-Aptamer-Drugs are rapidly internalized by target cancer cells. (a) Moni-toring Poly-Aptamer-Drug internalization into CCRF-CEM using confocal microscopyimages. Poly-Aptamer-Drugs were labeled with complementary DNAeFAM. (b)Quantitative analysis of internalization rate of Poly-Aptamer-Drug and Mono-Aptamer-Drug into CCRF-CEM using the mean fluorescence derived from flowcytometry. Error bars are standard error of the mean.
3.2. Multivalency of Poly-Aptamer-Drug enables effective celltargeting
We next investigated Poly-Aptamer-Drug specificity forPTK7þ CCRF-CEM cells (a widely-used ALL cancer cell line). GivenDox’s intrinsic fluorescence signal is significantly quenched in thePoly-Aptamer-Drug, we used Poly-Aptamer-Drugs hybridized toFAM-conjugated oligonucleotides that are complementary to thespacer domains between aptamers (Complementary DNAeFAM,Table 1). We found that the PTK7 Poly-Aptamer specifically boundto CCRF-CEM cells but not to Ramos cells (a Burkitt’s lymphomacancer line lacking PTK7) with confocal fluorescence microscopy(Fig. 3). Additionally, the scrambled RCA product (termed as “Poly-Scramble-Drug”) exhibited minimal binding to either CCRF-CEM orRamos cells (Fig. 3).
Our central hypothesis was that the Poly-Aptamer-Drug wouldbind target leukemic cells more effectively than its monovalentaptamer counterpart due to multivalency effects. To test this hy-pothesis, we incubated CCRF-CEM cells with the Poly-Aptamer-Drug or Mono-Aptamer-Drug at a range of concentrations for30 min before flow cytometry analysis (Fig. 4a). By plotting theflow cytometric mean fluorescence intensity as a function of DNAconcentration, we estimated and compared the binding affinity(Kd) of the Poly-Aptamer-Drug and the Mono-Aptamer-Drug totarget cells with GraphPad Prism statistical analysis software.Significantly, the Poly-Aptamer-Drug demonstrated 40 fold greater
Fig. 4. Poly-Aptamer-Drug binds to target CCRF-CEM cells more effectively than Mono-Aptamer-Drug. (a) Flow cytometry analysis of CCRM-CEM incubated with buffer, Poly-Aptamer-Drug or Mono-Aptamer-Drug. Poly-Aptamer-Drug and Mono-Aptamer-Drug were stained with complementary DNAeFAM. (b) Mean fluorescence of CCRF-CEMderived from flow cytometry analysis versus concentration of DNA.
Please cite this article in press as: Zhang Z, et al., A polyvalent aptamer system for targeted drug delivery, Biomaterials (2013), http://dx.doi.org/10.1016/j.biomaterials.2013.08.079
Z. Zhang et al. / Biomaterials xxx (2013) 1e86
affinity (Kd ¼ 6.5 nM) for target cells than the Mono-Aptamer-Drug(Kd ¼ 260 nM) (Fig. 4b).
3.3. Multivalency of Poly-Aptamer-Drug facilitates cellularinternalization
Bypassing the cell membrane barrier is a prerequisite fordelivering chemotherapeutics into cancer cells to effectively killcancer cells. We therefore investigated whether the Poly-Aptamer-Drug could be efficiently internalized and whether multivalencyfacilitates cellular internalization. Despite their negative charge,aptamer-modified nanoparticles, and DNA nanostructures canbe efficiently internalized by cells [33e42]. Importantly, multiva-lent binding on the cell membrane could lead to enhanced endo-cytosis compared to monovalent binding [12,17]. We first utilizedconfocal microscopy to examine Poly-Aptamer-Drug internaliza-tion following binding at different time points (5 min, 2 h, 5 h and24 h). Significantly, we observed that Poly-Aptamer-Drugs wererapidly (<1e2 h) internalized into intracellular domains (Fig. 5a,Supplementary information Fig. S2, and Supplementary Videos forreconstructed 3D confocal images). Intriguingly, the fluorescencesignals from complementary DNAeFAM that were hybridized ontoPoly-Aptamer-Drugs gradually diminished inside the cells over thecourse of several hours. We do not yet know whether this isbecause of the degradation of the dye itself or of the Poly-Aptamer-Drug backbone which led to dissociation of dyes and thereforedilution of the fluorescence signal. Work to understand the un-derlying mechanisms of intracellular trafficking and drug release ofthe Poly-Aptamer-Drug is ongoing.
Supplementary data related to this article can be found online athttp://dx.doi.org/10.1016/j.biomaterials.2013.08.079.
To investigate the effects of multivalency on cellular internali-zation, we compared the internalization rates between the Poly-Aptamer-Drug and the Mono-Aptamer-Drug following binding. Inthis set of experiments, the cells were treated with trypsin, anestablished procedure to remove any cell membrane bound DNAbefore they were analyzed by flow cytometry [2,9]. Importantly, wefound that the internalization half-time (defined as half of the timeto reach the plateau) of Poly-Aptamer-Drug is approximately 1 hwhile the internalization of Mono-Aptamer-Drug did not reach the
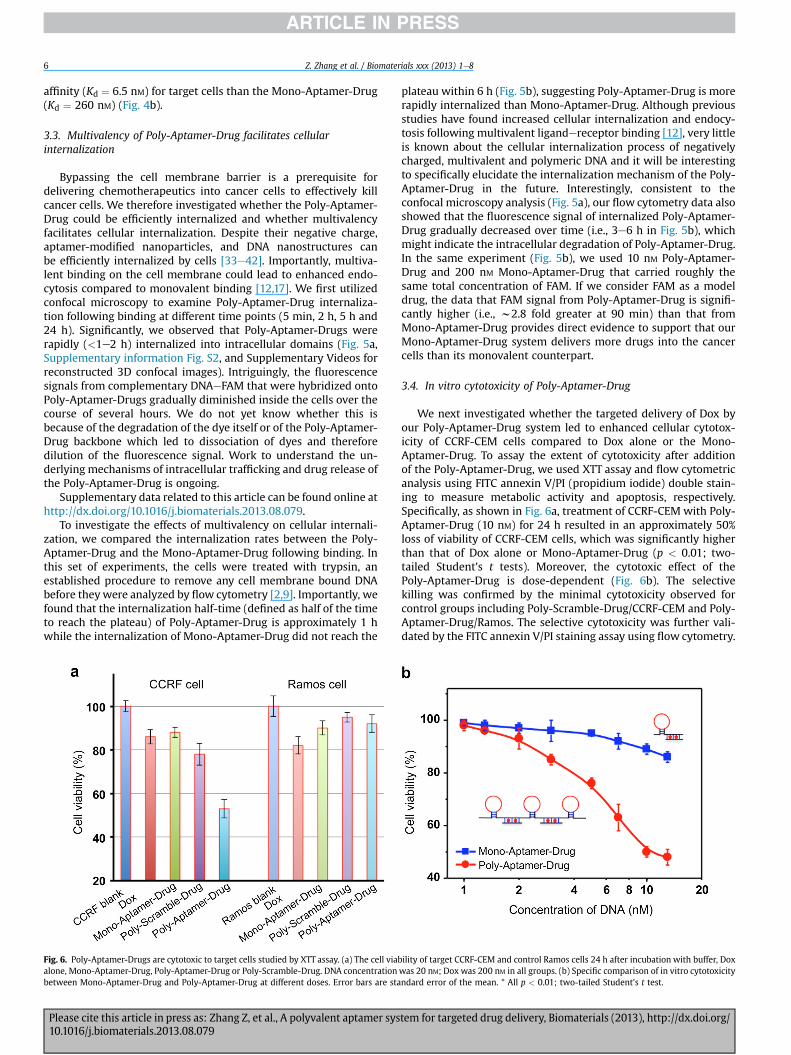
Fig. 6. Poly-Aptamer-Drugs are cytotoxic to target cells studied by XTT assay. (a) The cell viabalone, Mono-Aptamer-Drug, Poly-Aptamer-Drug or Poly-Scramble-Drug. DNA concentrationbetween Mono-Aptamer-Drug and Poly-Aptamer-Drug at different doses. Error bars are sta
Please cite this article in press as: Zhang Z, et al., A polyvalent aptamer sys10.1016/j.biomaterials.2013.08.079
plateau within 6 h (Fig. 5b), suggesting Poly-Aptamer-Drug is morerapidly internalized than Mono-Aptamer-Drug. Although previousstudies have found increased cellular internalization and endocy-tosis following multivalent ligandereceptor binding [12], very littleis known about the cellular internalization process of negativelycharged, multivalent and polymeric DNA and it will be interestingto specifically elucidate the internalization mechanism of the Poly-Aptamer-Drug in the future. Interestingly, consistent to theconfocal microscopy analysis (Fig. 5a), our flow cytometry data alsoshowed that the fluorescence signal of internalized Poly-Aptamer-Drug gradually decreased over time (i.e., 3e6 h in Fig. 5b), whichmight indicate the intracellular degradation of Poly-Aptamer-Drug.In the same experiment (Fig. 5b), we used 10 nM Poly-Aptamer-Drug and 200 nM Mono-Aptamer-Drug that carried roughly thesame total concentration of FAM. If we consider FAM as a modeldrug, the data that FAM signal from Poly-Aptamer-Drug is signifi-cantly higher (i.e., w2.8 fold greater at 90 min) than that fromMono-Aptamer-Drug provides direct evidence to support that ourMono-Aptamer-Drug system delivers more drugs into the cancercells than its monovalent counterpart.
3.4. In vitro cytotoxicity of Poly-Aptamer-Drug
We next investigated whether the targeted delivery of Dox byour Poly-Aptamer-Drug system led to enhanced cellular cytotox-icity of CCRF-CEM cells compared to Dox alone or the Mono-Aptamer-Drug. To assay the extent of cytotoxicity after additionof the Poly-Aptamer-Drug, we used XTT assay and flow cytometricanalysis using FITC annexin V/PI (propidium iodide) double stain-ing to measure metabolic activity and apoptosis, respectively.Specifically, as shown in Fig. 6a, treatment of CCRF-CEM with Poly-Aptamer-Drug (10 nM) for 24 h resulted in an approximately 50%loss of viability of CCRF-CEM cells, which was significantly higherthan that of Dox alone or Mono-Aptamer-Drug (p < 0.01; two-tailed Student’s t tests). Moreover, the cytotoxic effect of thePoly-Aptamer-Drug is dose-dependent (Fig. 6b). The selectivekilling was confirmed by the minimal cytotoxicity observed forcontrol groups including Poly-Scramble-Drug/CCRF-CEM and Poly-Aptamer-Drug/Ramos. The selective cytotoxicity was further vali-dated by the FITC annexin V/PI staining assay using flow cytometry.
ility of target CCRF-CEM and control Ramos cells 24 h after incubation with buffer, Doxwas 20 nM; Dox was 200 nM in all groups. (b) Specific comparison of in vitro cytotoxicityndard error of the mean. * All p < 0.01; two-tailed Student’s t test.
tem for targeted drug delivery, Biomaterials (2013), http://dx.doi.org/
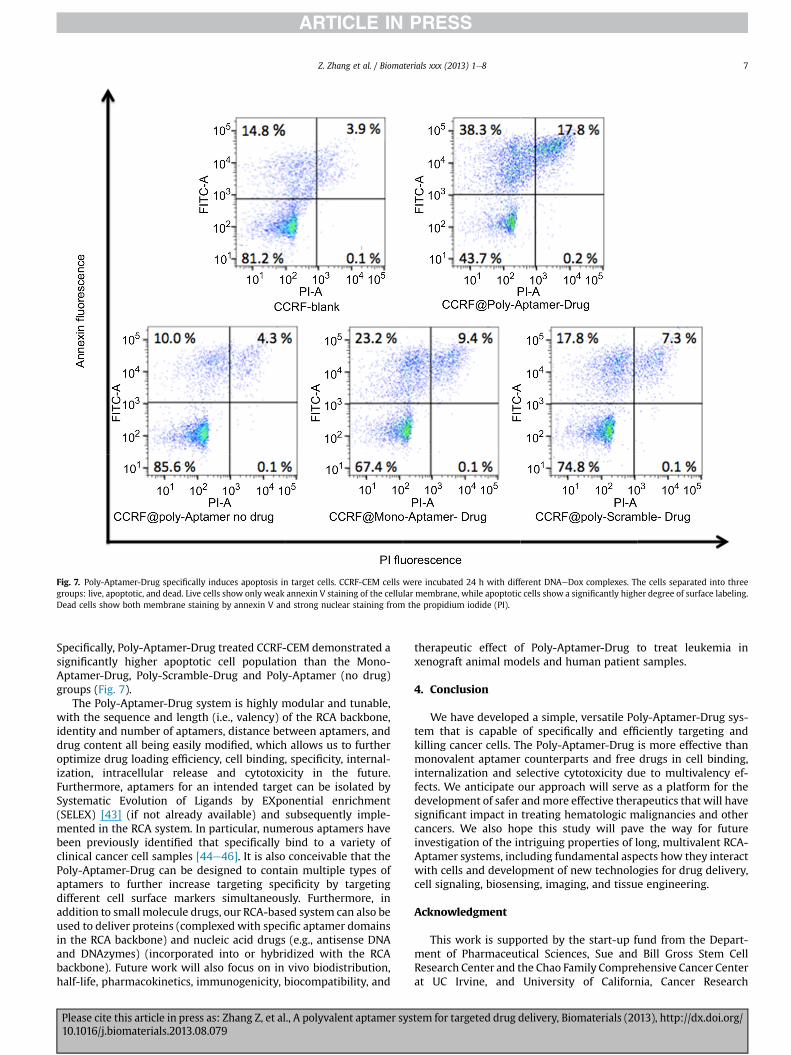
Fig. 7. Poly-Aptamer-Drug specifically induces apoptosis in target cells. CCRF-CEM cells were incubated 24 h with different DNAeDox complexes. The cells separated into threegroups: live, apoptotic, and dead. Live cells show only weak annexin V staining of the cellular membrane, while apoptotic cells show a significantly higher degree of surface labeling.Dead cells show both membrane staining by annexin V and strong nuclear staining from the propidium iodide (PI).
Z. Zhang et al. / Biomaterials xxx (2013) 1e8 7
Specifically, Poly-Aptamer-Drug treated CCRF-CEM demonstrated asignificantly higher apoptotic cell population than the Mono-Aptamer-Drug, Poly-Scramble-Drug and Poly-Aptamer (no drug)groups (Fig. 7).
The Poly-Aptamer-Drug system is highly modular and tunable,with the sequence and length (i.e., valency) of the RCA backbone,identity and number of aptamers, distance between aptamers, anddrug content all being easily modified, which allows us to furtheroptimize drug loading efficiency, cell binding, specificity, internal-ization, intracellular release and cytotoxicity in the future.Furthermore, aptamers for an intended target can be isolated bySystematic Evolution of Ligands by EXponential enrichment(SELEX) [43] (if not already available) and subsequently imple-mented in the RCA system. In particular, numerous aptamers havebeen previously identified that specifically bind to a variety ofclinical cancer cell samples [44e46]. It is also conceivable that thePoly-Aptamer-Drug can be designed to contain multiple types ofaptamers to further increase targeting specificity by targetingdifferent cell surface markers simultaneously. Furthermore, inaddition to small molecule drugs, our RCA-based system can also beused to deliver proteins (complexed with specific aptamer domainsin the RCA backbone) and nucleic acid drugs (e.g., antisense DNAand DNAzymes) (incorporated into or hybridized with the RCAbackbone). Future work will also focus on in vivo biodistribution,half-life, pharmacokinetics, immunogenicity, biocompatibility, and
Please cite this article in press as: Zhang Z, et al., A polyvalent aptamer sys10.1016/j.biomaterials.2013.08.079
therapeutic effect of Poly-Aptamer-Drug to treat leukemia inxenograft animal models and human patient samples.
4. Conclusion
We have developed a simple, versatile Poly-Aptamer-Drug sys-tem that is capable of specifically and efficiently targeting andkilling cancer cells. The Poly-Aptamer-Drug is more effective thanmonovalent aptamer counterparts and free drugs in cell binding,internalization and selective cytotoxicity due to multivalency ef-fects. We anticipate our approach will serve as a platform for thedevelopment of safer andmore effective therapeutics that will havesignificant impact in treating hematologic malignancies and othercancers. We also hope this study will pave the way for futureinvestigation of the intriguing properties of long, multivalent RCA-Aptamer systems, including fundamental aspects how they interactwith cells and development of new technologies for drug delivery,cell signaling, biosensing, imaging, and tissue engineering.
Acknowledgment
This work is supported by the start-up fund from the Depart-ment of Pharmaceutical Sciences, Sue and Bill Gross Stem CellResearch Center and the Chao Family Comprehensive Cancer Centerat UC Irvine, and University of California, Cancer Research
tem for targeted drug delivery, Biomaterials (2013), http://dx.doi.org/

Z. Zhang et al. / Biomaterials xxx (2013) 1e88
Coordinating Committee. Z.Z. is supported by the National NaturalScience Foundation of China (NSFC, No. 51103179), the NaturalScience Foundation of Shandong Province, China (Grant No.ZR2011BL017), the Fundamental Research Funds for the CentralUniversities and the scholarship of China Scholarship Council (CSC201206455009).
Appendix A. Supplementary information
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials.2013.08.079.
References
[1] Mallikaratchy PR, Ruggiero A, Gardner JR, Kuryavyi V, Maguire WF,Heaney ML, et al. A multivalent DNA aptamer specific for the B-cell receptoron human lymphoma and leukemia. Nucleic Acids Res 2011;39:2458e69.
[2] Yang L, Meng L, Zhang X, Chen Y, Zhu G, Liu H, et al. Engineering polymericaptamers for selective cytotoxicity. J Am Chem Soc 2011;133:13380e6.
[3] Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers asan emerging platform for cancer therapy. Nat Nanotechnol 2007;2:751e60.
[4] Liu Z, Duan JH, Song YM, Ma J, Wang FD, Lu X, et al. Novel HER2 aptamerselectively delivers cytotoxic drug to HER2-positive breast cancer cellsin vitro. J Transl Med 2012;10:148e57.
[5] Xu W, Siddiqui IA, Nihal M, Pilla S, Rosenthal K, Mukhtar H, et al. Aptamer-conjugated and doxorubicin-loaded unimolecular micelles for targeted ther-apy of prostate cancer. Biomaterials 2013;34:5244e53.
[6] Hubbell JA, Chilkoti A. Nanomaterials for drug delivery. Science 2012;337:303e5.
[7] Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov2010;9:537e50.
[8] Bagalkot V, Farokhzad OC, Langer R, Jon S. An aptameredoxorubicin physicalconjugate as a novel targeted drug-delivery platform. Angew Chem Int Ed2006;45:8149e52.
[9] Huang YF, Shangguan D, Liu H, Phillips JA, Zhang X, Chen Y, et al. Molecularassembly of an aptameredrug conjugate for targeted drug delivery to tumorcells. Chembiochem 2009;10:862e8.
[10] Ray P, White RR. Aptamers for targeted drug delivery. Pharmaceuticals2010;3:1761e78.
[11] Thiel K. Oligo oligarchy-the surprisingly small world of aptamers. Nat Bio-technol 2004;22:649e51.
[12] Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biologicalsystems: implications for design and use of multivalent ligands and inhibitors.Angew Chem Int Ed 1998;37:2754e94.
[13] Kolonko EM, Kiessling LL. A polymeric domain that promotes cellular inter-nalization. J Am Chem Soc 2008;130:5626e7.
[14] Kiessling LL, Gestwichi JE, Strong LE. Synthetic multivalent ligands in theexploration of cell-surface interactions. Curr Opin Chem Biol 2000;4:696e703.
[15] Kibria G, Hatakeyama H, Ohga N, Hida K, Harashima H. The effect of liposomalsize on the targeted delivery of doxorubicin to integrin avb3-expressing tu-mor endothelial cells. Biomaterials 2013;34:5617e27.
[16] Gorityala BK, Lu Z, Leow ML, Ma J, Liu XW. Design of a “turn-off/turn-on”biosensor: understanding carbohydrate-lectin interactions for use in non-covalent drug delivery. J Am Chem Soc 2012;134:15229e32.
[17] Tong GJ, Hsiao SC, Carrico ZM, Francis MB. Viral capsid DNA aptamer conjugatesas multivalent cell-targeting vehicles. J Am Chem Soc 2009;131:11174e8.
[18] Zhao W, Cui CH, Bose S, Guo D, Shen C, Wong WP, et al. Bioinspired multi-valent DNA network for capture and release of cells. Proc Natl Acad Sci U S A2012;109:19626e31.
[19] Zhao W, Ali MM, Brook MA, Li Y. Rolling circle amplification: applications innanotechnology and biodetection with functional nucleic acids. Angew ChemInt Ed 2008;47:6330e7.
[20] Tang L, Liu Y, Ali MM, Kang DK, Zhao W, Li J. Colorimetric and ultrasensitivebioassay based on a dual-amplification system using aptamer and DNAzyme.Anal Chem 2012;84:4711e7.
Please cite this article in press as: Zhang Z, et al., A polyvalent aptamer sys10.1016/j.biomaterials.2013.08.079
[21] Hamblin GD, Carneiro KMM, Fakhoury JF, Bujold KE, Sleiman HF. Rolling circleamplification-templated DNA nanotubes show increased stability and cellpenetration ability. J Am Chem Soc 2012;134:2888e91.
[22] Li Z, Wei B, Nangreave J, Lin C, Liu Y, Mi Y, et al. A replicable tetrahedralnanostructure self-assembled from a single DNA strand. J Am Chem Soc2009;131:13093e8.
[23] Amiram M, Quiroz FG, Callahan DJ, Chilkoti A. A highly parallel method forsynthesizing DNA repeats enables the discovery of ‘smart’ protein polymers.Nat Mater 2011;10:141e8.
[24] Larsson C, Koch J, Nygren A, Janssen G, Raap AK, Landegren U, et al. In situgenotyping individual DNA molecules by target-primed rolling-circle ampli-fication of padlock probes. Nat Methods 2004;1:227e32.
[25] Lee JB, Peng S, Yang D, Roh YH, Funabashi H, Park N, et al. A mechanicalmetamaterial made from a DNA hydrogel. Nat Nanotechnol 2012;7:816e20.
[26] Tian Y, He Y, Mao C. Cascade signal amplification for DNA detection. Chem-biochem 2006;7:1862e4.
[27] Cheglakov Z, Weizmann Y, Braunschweig AB, Wilner OI, Willner I. Increasingthe complexity of periodic protein nanostructures by the rolling circleamplified synthesis of aptamers. Angew Chem Int Ed 2008;47:126e30.
[28] Cheglakov Z, Weizmann Y, Basnar B, Willner I. Diagnosing viruses by therolling circle amplified synthesis of DNAzymes. Org Biomol Chem 2007;5:223e5.
[29] Zhu G, Zheng J, Song E, Donovan M, Zhang K, Liu C, et al. Self-assembled,aptamer-tethered DNA nanotrains for targeted transport of molecular drugsin cancer theranostics. Proc Natl Acad Sci U S A 2013;110:7998e8003.
[30] Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P, et al. Aptamersevolved from live cells as effective molecular probes for cancer study. ProcNatl Acad Sci U S A 2006;103:11838e43.
[31] Shangguan D, Cao Z, Meng L, Mallikaratchy P, Sefah K, Wang H, et al. Cell-specific aptamer probes for membrane protein elucidation in cancer cells.J Proteome Res 2008;7:2133e9.
[32] Zhao W, Brook MA, Li Y. Periodic assembly of nanospecies on repetitive DNAsequences generated on gold nanoparticles by rolling circle amplification.Methods Mol Biol 2008;474:79e90.
[33] Bagalkot V, Zhang L, Levy-Nissenbaum E, Jon S, Kantoff PW, Langer R, et al.Quantum dot�aptamer conjugates for synchronous cancer imaging, therapy,and sensing of drug delivery based on bi-fluorescence resonance energytransfer. Nano Lett 2007;7:3065e70.
[34] Xiao Z, Levy-Nissenbaum E, Alexis F, Luptak A, Teply BA, Chan JM, et al. En-gineering of targeted nanoparticles for cancer therapy using internalizingaptamers isolated by cell-uptake selection. ACS Nano 2012;6:696e704.
[35] Liu X, Yan H, Liu Y, Chang Y. Targeted cell-cell interactions by DNAnanoscaffold-templated multivalent bispecific aptamers. Small 2011;7:1673e82.
[36] Min K, Jo H, Song K, Cho M, Chun YS, Jon S, et al. Dual-aptamer-based deliveryvehicle of doxorubicin to both PSMA (þ) and PSMA (�) prostate cancers.Biomaterials 2011;32:2124e32.
[37] Liu J, Cao Z, Lu Y. Functional nucleic acid sensors. Chem Rev 2009;109:1948e98.
[38] Zhang H, Li F, Dever B, Li XF, Chris Le X. DNA-mediated homogeneous bindingassays for nucleic acids and proteins. Chem Rev 2013;113:2812e41.
[39] Douglas SM, Bachelet I, Church GM. A logic-gated nanorobot for targetedtransport of molecular payloads. Science 2012;335:831e4.
[40] Zhu G, Zhang S, Song E, Zheng J, Hu R, Fang X, et al. Building fluorescent DNAnanodevices on target living cell surfaces. AngewChem Int Ed 2013;52:5490e6.
[41] Cheng Z, Zaki AA, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nano-particles: cost versus benefit of adding targeting and imaging capabilities.Science 2012;338:903e10.
[42] Wang K, You M, Chen Y, Han D, Zhu Z, Huang J, et al. Self-assembly of abifunctional DNA carrier for drug delivery. Angew Chem Int Ed 2011;50:6098e101.
[43] Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment:RNA ligands to bacteriophage T4 DNA polymerase. Science 1990;249:505e10.
[44] Kim E, Jung Y, Choi H, Yang J, Suh JS, Huh YM, et al. Prostate cancer cell deathproduced by the co-delivery of Bcl-xL shRNA and doxorubicin using anaptamer-conjugated polyplex. Biomaterials 2010;31:4592e9.
[45] Mi J, Zhang X, Rabbani ZN, Liu Y, Reddy SK, Su Z, et al. RNA aptamer-targetedinhibition of NF-kB suppresses non-small cell lung cancer resistance todoxorubicin. Mol Ther 2007;16:66e73.
[46] Cerchia L, Esposito CL, Camorani S, Rienzo A, Stasio L, Insabato L, et al.Targeting Axl with an high-affinity inhibitory aptamer. Mol Ther 2012;20:2291e303.
tem for targeted drug delivery, Biomaterials (2013), http://dx.doi.org/