a new high-throughput aflp approach for identification of new genetic polymorphism in the genome of...
TRANSCRIPT

www.elsevier.com/locate/jmicmeth
Journal of Microbiological Methods 56 (2004) 49–62
A new high-throughput AFLP approach for identification of
new genetic polymorphism in the genome of the clonal
microorganism Mycobacterium tuberculosis
Nicole van den Braaka, Guus Simonsb, Roy Gorkinkb, Martin Reijansb,Kimberly Eadiea, Kristin Kremersc, Dick van Soolingenc, Paul Savelkould,
Henri Verbrugha, Alex van Belkuma,*
aDepartment of Medical Microbiology and Infectious Diseases, Erasmus MC, Dr. Molewaterplein 40, 3015 GD Rotterdam, The NetherlandsbDepartment of Microbial Genomics, Keygene BV, Agro Businesspark 90, 6708 PW Wageningen, The Netherlands
cDiagnostic Laboratory for Infectious Diseases and Perinatal Screening, National Institute of Public Health and the Environment,
P.O. Box 1, 3720 BA Bilthoven, The NetherlandsdDepartment of Medical Microbiology, Free University of Amsterdam, De Boelelaan, Amsterdam, The Netherlands
Received 21 August 2003; received in revised form 13 September 2003; accepted 15 September 2003
Abstract
We have here applied high-throughput amplified fragment length polymorphism (htAFLP) analysis to strains belonging to the
five classical species of the Mycobacterium tuberculosis complex. Using 20 strains, three enzyme combinations and eight
selective amplification primer pairs, 24 AFLP reactions were performed per strain. Overall, this resulted in 480 DNA fingerprints
and more than 1200 htAFLP-amplified PCR fragments were visualised per strain. The cumulative dendrogram correctly
clustered strains from the various species, albeit within a distance of 6.5% for most of them. The single isolate ofMycobacterium
canettii presented separately at 19% distance. All over, 169 fragments (14%) appeared to be polymorphic. Sixty-eight were
specific forM. canetti and forty-five forMycobacterium bovis. For the 10 differentM. tuberculosis strains included in the present
analysis, 56 polymorphic markers were identified. Upon sequencing 20 of these marker regions and comparisons with the H37Rv
genome sequence, 25% appeared to share homology to members of the antigenically variable PE/PPE surface protein encoding
gene family confirming previous findings on the genetic heterogeneity within these genes. In addition, homologues for phage
genes and insertion element-encoded genes were detected. Forty-five percent of the sequences derived from ORFs with a
currently unknown function, which was corroborated by genome sequence comparison for the clinical M. tuberculosis CD 1551
isolate. Sequence variation in M. tuberculosis was assessed in more detail for a subset of these loci by newly designed PCR
restriction fragment length polymorphism (RFLP) tests and direct sequencing. Fourteen novel PCR RFLP tests were developed
and twelve novel single nucleotide polymorphisms (SNPs) were identified, all suited for epidemiological analysis of M.
tuberculosis. The tests allowed for identification of the major Mycobacterium species and M. tuberculosis variants and clones.
D 2003 Elsevier B.V. All rights reserved.
Keywords: Mycobacterium tuberculosis; Genetic variation; High-throughput AFLP; PCR RFLP; Single nucleotide polymorphism
0167-7012/$ - see front matter D 2003 Elsevier B.V. All rights reserved.
doi:10.1016/j.mimet.2003.09.018
* Corresponding author. Tel.: +31-10-4635813; fax: +31-10-4633875.
E-mail address: [email protected] (A. van Belkum).

N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–6250
1. Introduction
Mycobacterium tuberculosis, a cause of severe
infectious morbidity and mortality among humans, is
a clonally reproducing infectious microorganism. The
most likely explanation for its genetic homogeneity is
a short evolutionary history (Kapur et al., 1994). This
hypothesis was verified by high-throughput sequenc-
ing of housekeeping genes, which demonstrated that,
despite normal mutation frequencies (David and New-
man, 1971), M. tuberculosis only accumulated limited
numbers of allelic variants in these genes (Sreevatsan
et al., 1997; Kapur et al., 1994; Musser et al., 2000). It
was shown that the largest proportion of mutations re-
corded was associated with selective pressure exerted
by antimicrobial agents, for instance. However, vari-
ous variable elements or loci were still identifiable in
theM. tuberculosis genome. The first in this series was
a set of insertion elements including IS1081 (Collins
and Stephens, 1991), IS1547 (Fang et al., 1998), an IS-
like element (Mariani et al., 1993) and the highly
polymorphic IS6110 (Thierry et al., 1990). Because
of its flexibility, both in copy number and chromo-
somal location, mapping of IS6110 restriction frag-
ment length polymorphism (RFLP) has become an
important tool for investigation of the epidemiology of
M. tuberculosis (Small et al., 1994; Van Embden et al.,
1993).
The nucleotide sequence for the entire genome of
M. tuberculosis H37Rv revealed the presence of
additional IS elements (Cole et al., 1998) and other
regions of putative variability, including minisatellite-
like elements. These mycobacterial interspersed re-
petitive units (MIRUs) were usually between 40 and
100 nucleotides in length and were found to be
dispersed across the chromosome (Supply et al.,
2000). Out of 31 candidate loci, 12 appeared to be
variable in both repeat copy number and primary
structure, a feature that is not uncommon among
microbes (for a review, see Van Belkum et al.,
1998). It was simultaneously documented that the
variable number of tandem repeats (VNTRs) in these
MIRUs seemed to be evolving slowly in mycobac-
terial populations. MIRU-VNTRs can be used for
assessing diversity among strains of M. tuberculosis
(Supply et al., 2001) and Mycobacterium bovis
(Roring et al., 2002). Another example of such a
repetitive domain, the direct repeat or DR locus, was
already discovered in the pre-genomics era (Hermans
et al., 1993; Kamerbeek et al., 1997). Within the DR
locus constant repeats are alternated with variable
sections. These latter elements have been used for the
development of a typing system suited for large-scale
mycobacterial epidemiology (Fang et al., 1998; Groe-
nen et al., 1993). Population genetic studies involv-
ing the DR element indicated that successive
deletions in the region rather than scrambling of the
variable units gave rise to significant levels of
diversity (Van Embden et al., 2000). In conclusion,
even after the elucidation of the first M. tuberculosis
genome sequence, genetic variation seemed to be
confined to a limited number of loci and frequently
based on repeat diversity or inherently dynamic
insertion elements. This implied that additional meth-
ods for defining genetic variability in M. tuberculosis
are still required prior to our full understanding of the
population genetics of this medically highly relevant
microorganism.
More recent developments in microbial genomics
and DNA array technology generated improvements in
our understanding of the biology of M. tuberculosis.
Initially, arrays of bacterial artificial chromosome
(BAC) clones were used to make inventories of
genomic differences between strains of M. tuberculo-
sis (Gordon et al., 1999). Especially mapping genomic
deletions revealed important details on the phylogeny
of M. tuberculosis and M. bovis. Similar but more
detailed data were obtained using whole genome
arrays manufactured by spotting 250–1000 nucleoti-
des long PCR products for all of the predicted open
reading frames in the genome of H37Rv on glass slides
(Behr et al., 1999). The large deletions are currently
supposed to involve genes whose functions are no
longer necessary for certain lineages of mycobacteria.
This was confirmed by the observation that strains
containing increasing numbers of deletions caused less
severe infections without pulmonary cavitation (Kato-
Maeda et al., 2001a,b). Most recently, a second M.
tuberculosis genome sequence became available for
the clinical isolate CD 1551 (Fleischmann et al., 2002).
This enabled the most detailed genetic comparison
between two strains of a single bacterial species
presented thus far. Various novel polymorphic sites
were identified, not only deletions and insertions but
also widespread single nucleotide polymorphism
(SNP). Apparently, genetic variation in M. tuberculo-

Table 1
Survey of strains used for htAFLP analysis
Numbers between brackets refer to numbering as given in Kremer et
al. (1999). Genuine duplicates are indicated by coloured shades, the
boxed region on the right indicates the strains also present in the
pilot collection.
N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–62 51
sis may be more widespread than anticipated. It has to
be realised, however, that determination of genetic
variability based on direct genome comparisons is
not always feasible: it is not (yet?) economically
possible to sequence the entire chromosome for two
or more representative strains of all bacterial species.
In addition, the variability between two members of a
species may not be representative for the general
genetic heterogeneity within the species as a whole.
This raises the need for alternative technology that
could be more generally applied to discover genetic
variation among all of the microbial pathogens.
We here demonstrate that high-throughput amplifi-
cation fragment length polymorphism (htAFLP) anal-
ysis is a likely choice for genome wide mutation
screening. AFLP as such is a restriction-amplification
method developed in the early 1990s (Vos et al.,
1995). After an additional digestion of genomic
DNA, by a combination of restriction enzymes, re-
striction site-specific extensions are ligated to the
multitude of DNA fragments. The attached linker
contains site-specific PCR priming sequences. When
targeted by PCR, the combination of sites at the
termini of an individual restriction fragment deter-
mines whether the fragment is amplified or not.
Usually, due to the complexity of the restriction digest,
one or two AFLP fingerprints suffice for obtaining a
reliable genetic signature for a microbial strain. In
principle, the method screens restriction site polymor-
phism and a clever extension of the primers used
during AFLP also facilitates monitoring of DNA
polymorphism in the restriction site-neighbouring re-
gion. This has resulted in the establishment of repro-
ducible and robust microbial typing strategies that do
not only provide genetic epidemiological information,
but which can also be used to identify new species,
even within the M. tuberculosis complex (Ahmed et
al., 2003). We here demonstrate that analysis of
various restriction enzyme combinations together with
differentially extended AFLP primers facilitates high-
density genomic screening, independent of knowledge
of mutation positions. Our current studies were aimed
at the detection of novel sites of genetic variation not
previously characterised in the genetically homoge-
neous species belonging to the M. tuberculosis com-
plex. This is the first model study on the value of
htAFLP for detecting genetic variants in an essentially
clonal bacterial species.
2. Materials and methods
2.1. Strains and DNA isolation
Strains used for the htAFLP analysis have been
described before (Kremer et al., 1999). A survey of
the isolates in the collection is given in Table 1. It is
important to emphasise that two separate but over-
lapping collections of strains were used. The first
collection was employed for the htAFLP, whereas the
second collection was used for the validation of
markers developed using the pilot collection. Both
collections were provided in a blinded fashion, the
receiving institution being unaware of the nature of
the strains before htAFLP was finished. In the
validation collection, three times three outbreak re-
lated isolates were included to confirm inter-strain
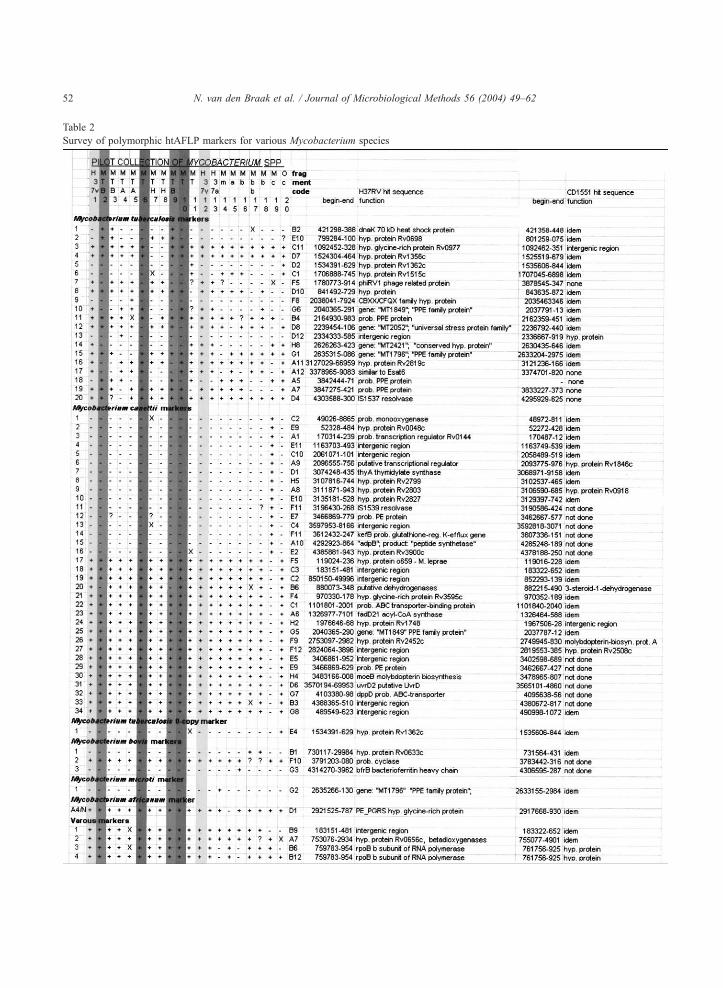
Table 2
Survey of polymorphic htAFLP markers for various Mycobacterium species
N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–6252
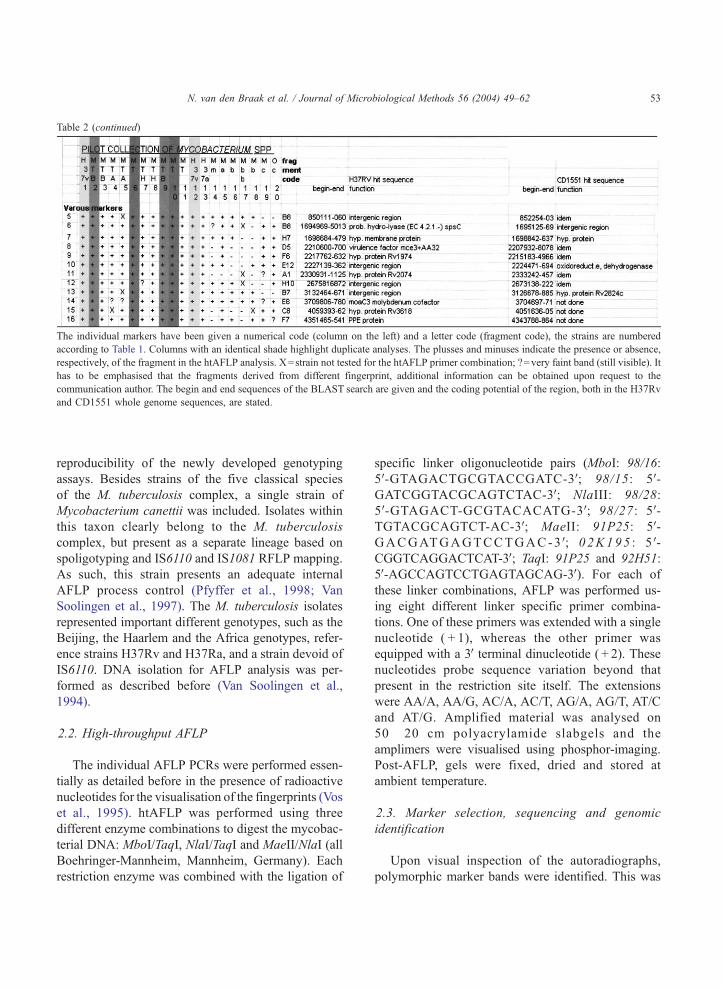
Table 2 (continued)
The individual markers have been given a numerical code (column on the left) and a letter code (fragment code), the strains are numbered
according to Table 1. Columns with an identical shade highlight duplicate analyses. The plusses and minuses indicate the presence or absence,
respectively, of the fragment in the htAFLP analysis. X=strain not tested for the htAFLP primer combination; ?=very faint band (still visible). It
has to be emphasised that the fragments derived from different fingerprint, additional information can be obtained upon request to the
communication author. The begin and end sequences of the BLAST search are given and the coding potential of the region, both in the H37Rv
and CD1551 whole genome sequences, are stated.
N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–62 53
reproducibility of the newly developed genotyping
assays. Besides strains of the five classical species
of the M. tuberculosis complex, a single strain of
Mycobacterium canettii was included. Isolates within
this taxon clearly belong to the M. tuberculosis
complex, but present as a separate lineage based on
spoligotyping and IS6110 and IS1081 RFLP mapping.
As such, this strain presents an adequate internal
AFLP process control (Pfyffer et al., 1998; Van
Soolingen et al., 1997). The M. tuberculosis isolates
represented important different genotypes, such as the
Beijing, the Haarlem and the Africa genotypes, refer-
ence strains H37Rv and H37Ra, and a strain devoid of
IS6110. DNA isolation for AFLP analysis was per-
formed as described before (Van Soolingen et al.,
1994).
2.2. High-throughput AFLP
The individual AFLP PCRs were performed essen-
tially as detailed before in the presence of radioactive
nucleotides for the visualisation of the fingerprints (Vos
et al., 1995). htAFLP was performed using three
different enzyme combinations to digest the mycobac-
terial DNA: MboI/TaqI, NlaI/TaqI and MaeII/NlaI (all
Boehringer-Mannheim, Mannheim, Germany). Each
restriction enzyme was combined with the ligation of
specific linker oligonucleotide pairs (MboI: 98/16:
5V-GTAGACTGCGTACCGATC-3V; 98/15: 5V-GATCGGTACGCAGTCTAC-3V; NlaIII: 98/28:
5V-GTAGACT-GCGTACACATG-3V; 98/27: 5V-TGTACGCAGTCT-AC-3V; MaeII: 91P25: 5V-GACGATGAGTCCTGAC-3V; 02K195 : 5V-CGGTCAGGACTCAT-3V; TaqI: 91P25 and 92H51:
5V-AGCCAGTCCTGAGTAGCAG-3V). For each of
these linker combinations, AFLP was performed us-
ing eight different linker specific primer combina-
tions. One of these primers was extended with a single
nucleotide ( + 1), whereas the other primer was
equipped with a 3Vterminal dinucleotide ( + 2). These
nucleotides probe sequence variation beyond that
present in the restriction site itself. The extensions
were AA/A, AA/G, AC/A, AC/T, AG/A, AG/T, AT/C
and AT/G. Amplified material was analysed on
50 � 20 cm polyacrylamide slabgels and the
amplimers were visualised using phosphor-imaging.
Post-AFLP, gels were fixed, dried and stored at
ambient temperature.
2.3. Marker selection, sequencing and genomic
identification
Upon visual inspection of the autoradiographs,
polymorphic marker bands were identified. This was

Table 3
Development of novel PCR RFLP tests for M. tuberculosis
The fragment codes are identical to those listed in Table 2. Shaded areas on the right identify those PCRs that did not result in amplification. Different numbers in the PCR RFLP
result sections indicate different RFLP patterns.
N.vanden
Braaket
al./JournalofMicro
biologica
lMeth
ods56(2004)49–62
54

N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–62 55
corroborated by indexing using the automated inter-
pretation software package AFLP QuattroPro (Key-
gene, Wageningen, The Netherlands). The fragments
were excised from the gels and re-amplified using their
matching AFLP consensus primer set without restric-
tion site-specific + 1/ + 2 extension sequences attached
(99G18: 5V-AGCGGATAACAATTTCACAGAGGA-CACACTGGTATA-GACTGCGT-ACCGAT-3V;99G22: 5V-AGCGGATAACAATTTCACACAGGA-CAC-ACTGGTATAGACTGCGTACA-CATG-3V).The amplimers were subjected to DNA sequencing
using a 96-well capillary sequencing machine (Meg-
aBace; ABI, Gouda, The Netherlands). We used the
nucleotide sequences of the fragments derived from
the htAFLP of the H37Rv strain in order to facil-
itate fragment identification using BLAST analysis
versus the genome sequence for this strain. Table 2
lists the fragments that were successfully subjected
to DNA sequencing and analysed by BLAST. Iden-
tification codes for the fragments are included as
well.
2.4. Development of PCR RFLP tests and directed
amplicon sequencing
In case an amplimer sequence matched a target
region in the M. tuberculosis H37Rv genome, a novel
PCR test was designed for probing the genetic poly-
morphism detected in more detail. This involved the
synthesis of forward and reverse primers located ap-
proximately 50 nucleotides upstream or downstream of
the region of homology, respectively, for all of the
validation strains (n = 26) (see Table 3), thereby ampli-
fying not only the differentially present AFLP fragment
itself but also neighbouring sequences. First, the
amplimers were digested with the restriction enzymes
used for the AFLP reaction. PCR RFLP digests were
analysed on agarose gels. In addition, some of the
fragments were amplified and completely sequenced.
This analysis revealed whether or not the variability
was due to variation in the restriction sites or in the
adjourning + 1/ + 2 nucleotides encoded by the AFLP
primers. DNA sequences were compared with the
genome sequence using the web-accessible version of
the Basic Local Alignment Search Tool (BLAST).
Additional comparison was performed by alignment
of all sequences using DNASTAR (Lasergene, Madi-
son WI, USA).
3. Results
3.1. htAFLP analysis and fragment analysis for
Mycobacterium strains
The first 20 Mycobacterium spp. strains (pilot col-
lection, see Table 1) were subjected to htAFLP. For
these strains, three enzyme combinations were com-
bined with eight different selective primer pairs; this
generated 480 different fingerprint types for all of the
strains. This resulted in an overall number of approx-
imately 1200 amplimers. Fig. 1 highlights the dendro-
gram as based on all of thesemarkers. It can be seen that
all of the different M. tuberculosis genotype strains
(Haarlem, Beijing and Africa) cluster closely. The M.
bovis strains cluster with the single M. bovis BCG and
the Mycobacterium microti isolates. Mycobacterium
africanum andM. canettii form quite distinct branches
in the phylogenetic tree, withM. canettii occupying the
most exceptional position. The genome strains were
analysed in duplicate with the genotyping results clus-
tering most closely. This difference is less than 0.2%,
indicative of the AFLP high signal-to-noise ratio.
Different numbers of selectively amplified marker
bands, not universally present for all strains in the pilot
collection, were identified per enzyme combination. In
case of MboI/TaqI, 58 useful markers were observed,
whereas NlaI/TaqI and MaeIII/NlaI digests rendered
64 and 47 differentiating markers, respectively. Over-
all, this amounted to 169 well-scored markers. Among
these, 31 were positive and 37 were negative for theM.
canettii strain only. Out of the remaining 101 markers,
45 were specific for M. bovis, leaving 56 polymorphic
markers for M. tuberculosis, essentially generated
using 10 different strains only.
3.2. DNA sequencing of M. tuberculosis specific
markers
Out of the 56 polymorphic markers identified forM.
tuberculosis, a randomly selected set of 20 was se-
quenced (Table 2), including a description of their
differential occurrence in the AFLP banding patterns.
For theM. tuberculosismarkers, the data obtained with
respect to the homology searches for the two currently
available genomes (H37Rv and CD 1551) are quite
similar: in 18 out of 20 comparisons, homologous
sequences were identified in both genome sequences.
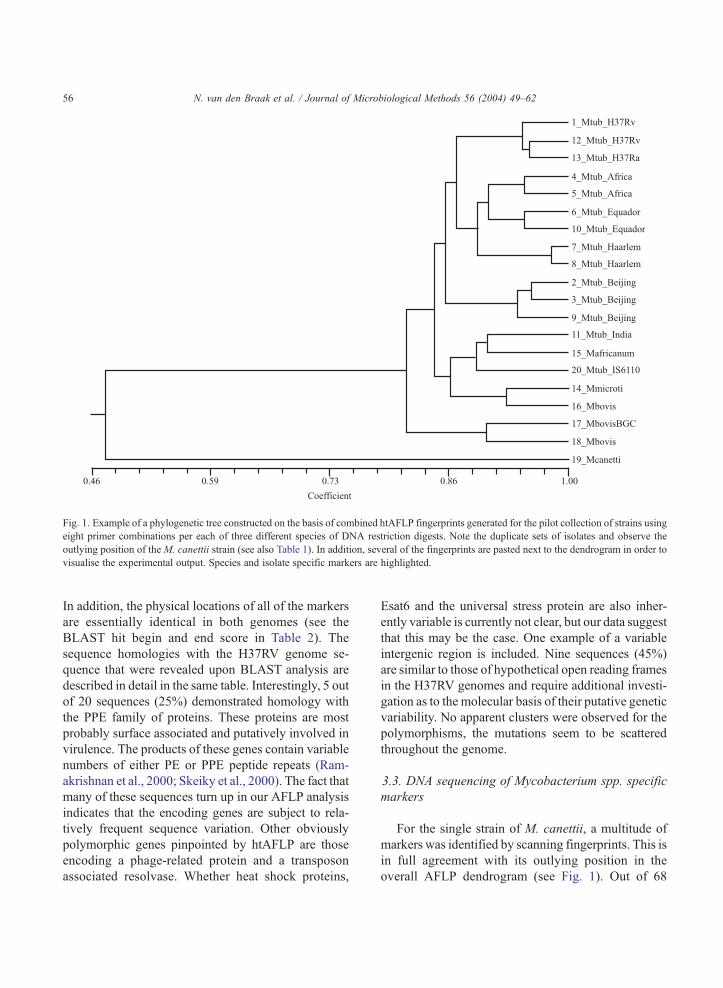
Fig. 1. Example of a phylogenetic tree constructed on the basis of combined htAFLP fingerprints generated for the pilot collection of strains using
eight primer combinations per each of three different species of DNA restriction digests. Note the duplicate sets of isolates and observe the
outlying position of the M. canettii strain (see also Table 1). In addition, several of the fingerprints are pasted next to the dendrogram in order to
visualise the experimental output. Species and isolate specific markers are highlighted.
N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–6256
In addition, the physical locations of all of the markers
are essentially identical in both genomes (see the
BLAST hit begin and end score in Table 2). The
sequence homologies with the H37RV genome se-
quence that were revealed upon BLAST analysis are
described in detail in the same table. Interestingly, 5 out
of 20 sequences (25%) demonstrated homology with
the PPE family of proteins. These proteins are most
probably surface associated and putatively involved in
virulence. The products of these genes contain variable
numbers of either PE or PPE peptide repeats (Ram-
akrishnan et al., 2000; Skeiky et al., 2000). The fact that
many of these sequences turn up in our AFLP analysis
indicates that the encoding genes are subject to rela-
tively frequent sequence variation. Other obviously
polymorphic genes pinpointed by htAFLP are those
encoding a phage-related protein and a transposon
associated resolvase. Whether heat shock proteins,
Esat6 and the universal stress protein are also inher-
ently variable is currently not clear, but our data suggest
that this may be the case. One example of a variable
intergenic region is included. Nine sequences (45%)
are similar to those of hypothetical open reading frames
in the H37RV genomes and require additional investi-
gation as to the molecular basis of their putative genetic
variability. No apparent clusters were observed for the
polymorphisms, the mutations seem to be scattered
throughout the genome.
3.3. DNA sequencing of Mycobacterium spp. specific
markers
For the single strain of M. canettii, a multitude of
markers was identified by scanning fingerprints. This is
in full agreement with its outlying position in the
overall AFLP dendrogram (see Fig. 1). Out of 68

N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–62 57
markers, a random set of 34 were successfully se-
quenced. It is interesting to note that all of the sequen-
ces showed significant homology scores with the
H37Rv and CD 1551 genome sequences, indicating
high levels of cross-species sequence identity and
suggesting that sequence variation rather than gene
gain or loss defines the species’ boundaries. Nine out of
thirty-four sequences matched with intergenic motifs
identified in the H37RV genome, suggesting mutations
seem to accumulate in intergenic regions (1/20 versus
9/34 inM. tuberculosis versusM. canettii, respectively;
p = 0.07). Eight sequences matched with open reading
frames for which no function has been proposed as yet.
For six of the hits, the BLAST analysis revealed a
match with a gene encoding a putative surface compo-
nent. Three of these matched again with the so-called
PE/PPE protein genes. The variability that was encoun-
tered using the htAFLP approach seems to be in
agreement with the expectations. The other surface
components appeared to be encoded by the ABC
transporter and potassium efflux genes.
Various other species-specific polymorphisms were
identified in addition. For the 21 markers that appeared
to separate either M. bovis, M. microti or M. africa-
num, 5 coincided with hypothetical protein genes, 5
with intergenic regions and, again, 5 identified surface-
associated protein genes. The comparison performed
with the genome sequence obtained for the clinical M.
tuberculosis isolate CD1551 largely confirmed the
findings listed above (see Table 2 for details).
3.4. Development of PCR RFLP tests for M. tuber-
culosis strains
Based on the sequences listed in Table 2, PCR tests
for amplifying the locus identified by htAFLP and its
surrounding regions were developed. The correct frag-
ments were generated for 17 out of 20 markers. In
these cases, the PCR facilitated the amplification of a
correctly sized fragment, albeit that for three tests the
PCRs failed to deliver sufficient quantities of the
amplicon. In addition, the PCRs were isolate-selective
for 5 out of 17 tests. Ultimately, 17 RFLP analyses
were performed. The results for the differential ampli-
fication and subsequent RFLP analysis are summar-
ised in Table 3. It is immediately obvious that the PCR
amplification in itself was already indicative for the
genetic heterogeneity among the strains. The grey
blocks in Table 3 reveal that PCRs turned out negative
quite regularly, but it has to be stated that this is
concordant with the epidemiological data: when one
of the strains belonging to either of the three clusters
included in the validation strains, the two other strains
were negative as well. In addition, negative amplifi-
cation was relatively often documented for the non-
tuberculosis species. One of the PCR data sets
appeared to be specifically negative for the M. bovis
strains (fragments C1 and F5). It is interesting to note
that the three epidemiologically related clusters of
three strains can be adequately discriminated on the
basis of PCR effectiveness and RFLP analysis. Com-
bining the data from 3 out of 14 tests accurately
discriminated the clusters (fragments A5, A11 and
F5). The results show that the PCR in itself already
corroborated the epidemiological relatedness between
the strains. The explanation could be that different
AFLP patterns are due to mutations in the primer
extension region rather than in the restriction sites
themselves. For this reason, we decided to completely
sequence the C11, H8 and D4 fragments generated for
all 26 strains from the validation collection.
3.5. Detection of single nucleotide polymorphism in
marker fragments for M. tuberculosis
Fragments C11, D4 and H8were amplified for all 26
strains in the validation collection. Amplification was
successful in all cases, but use of the corresponding
AFLP restriction enzymes did not result in distinguish-
able patterns. Sequencing these fragments in full,
however, revealed the presence of 12 single nucleotide
polymorphisms (SNPs) (see Table 4 for the precise
nature and position of the mutations). Out of these 12, 4
were physically linked and most likely the result of a
recombination event (see Table 4 for the position and
the nature of the SNPs). Two mutations appeared to be
insertions, the others were point mutations. The two
insertion sites were located within coding sequences:
whether the insertion causes abrogation of the gene
sequences is subject to current investigations. One of
the two genes encodes a member of the PE/PPE
proteins and it could well be that the insertion event
is part of the antigenic variation noted before for these
genes. When all mutation are accumulated, nine differ-
ent overall genotypes are found among the 26 strains.
WithinM. tuberculosis, five different types were found
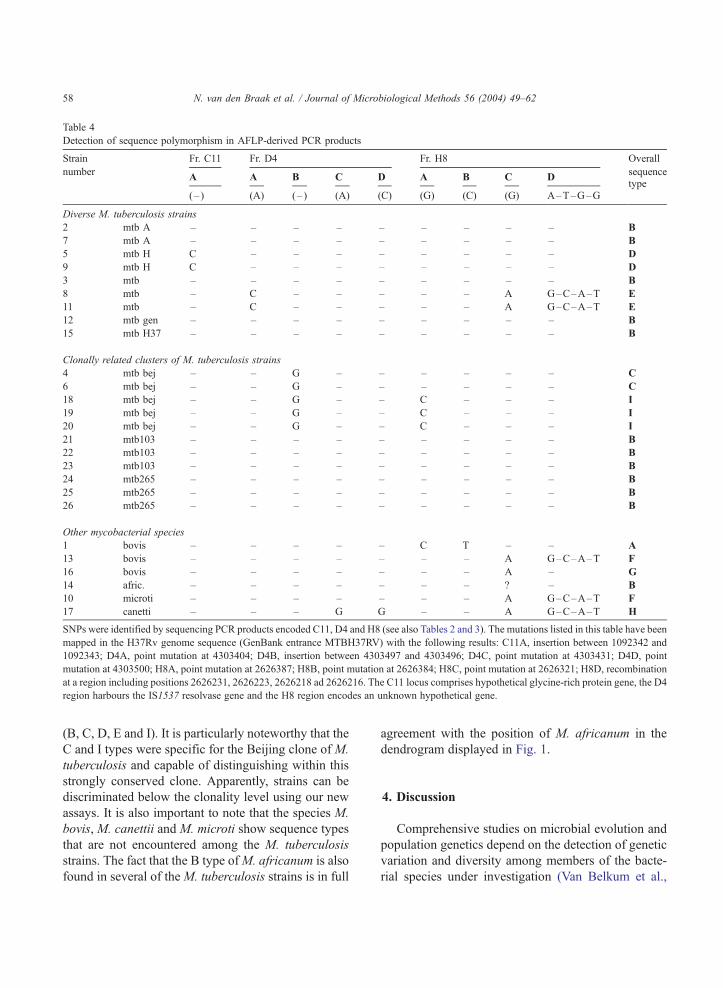
Table 4
Detection of sequence polymorphism in AFLP-derived PCR products
Strain Fr. C11 Fr. D4 Fr. H8 Overall
numberA A B C D A B C D
sequence
(– ) (A) (– ) (A) (C) (G) (C) (G) A–T–G–Gtype
Diverse M. tuberculosis strains
2 mtb A – – – – – – – – – B
7 mtb A – – – – – – – – – B
5 mtb H C – – – – – – – – D
9 mtb H C – – – – – – – – D
3 mtb – – – – – – – – – B
8 mtb – C – – – – – A G–C–A–T E
11 mtb – C – – – – – A G–C–A–T E
12 mtb gen – – – – – – – – – B
15 mtb H37 – – – – – – – – – B
Clonally related clusters of M. tuberculosis strains
4 mtb bej – – G – – – – – – C
6 mtb bej – – G – – – – – – C
18 mtb bej – – G – – C – – – I
19 mtb bej – – G – – C – – – I
20 mtb bej – – G – – C – – – I
21 mtb103 – – – – – – – – – B
22 mtb103 – – – – – – – – – B
23 mtb103 – – – – – – – – – B
24 mtb265 – – – – – – – – – B
25 mtb265 – – – – – – – – – B
26 mtb265 – – – – – – – – – B
Other mycobacterial species
1 bovis – – – – – C T – – A
13 bovis – – – – – – – A G–C–A–T F
16 bovis – – – – – – – A – G
14 afric. – – – – – – – ? – B
10 microti – – – – – – – A G–C–A–T F
17 canetti – – – G G – – A G–C–A–T H
SNPs were identified by sequencing PCR products encoded C11, D4 and H8 (see also Tables 2 and 3). The mutations listed in this table have been
mapped in the H37Rv genome sequence (GenBank entrance MTBH37RV) with the following results: C11A, insertion between 1092342 and
1092343; D4A, point mutation at 4303404; D4B, insertion between 4303497 and 4303496; D4C, point mutation at 4303431; D4D, point
mutation at 4303500; H8A, point mutation at 2626387; H8B, point mutation at 2626384; H8C, point mutation at 2626321; H8D, recombination
at a region including positions 2626231, 2626223, 2626218 ad 2626216. The C11 locus comprises hypothetical glycine-rich protein gene, the D4
region harbours the IS1537 resolvase gene and the H8 region encodes an unknown hypothetical gene.
N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–6258
(B, C, D, E and I). It is particularly noteworthy that the
C and I types were specific for the Beijing clone of M.
tuberculosis and capable of distinguishing within this
strongly conserved clone. Apparently, strains can be
discriminated below the clonality level using our new
assays. It is also important to note that the species M.
bovis, M. canettii and M. microti show sequence types
that are not encountered among the M. tuberculosis
strains. The fact that the B type ofM. africanum is also
found in several of theM. tuberculosis strains is in full
agreement with the position of M. africanum in the
dendrogram displayed in Fig. 1.
4. Discussion
Comprehensive studies on microbial evolution and
population genetics depend on the detection of genetic
variation and diversity among members of the bacte-
rial species under investigation (Van Belkum et al.,

N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–62 59
2001; Kato-Maeda et al., 2001a,b). Assessment of
panmicticism versus clonality is based on the degree
of genetic variability that can be addressed in the
microbial genetics laboratory. Consequently, tractabil-
ity of a bacterial genotype may be limited once only
small numbers of distinct genetic polymorphisms that
effectively separate strains are known. The mechanism
underlying the epidemic spread of organisms that are
largely clonal remains incompletely understood. M.
tuberculosis is an example of such a microorganism:
although major clones have been described and fol-
lowed globally (Bifani et al., 2002; Kato-Maeda et al.,
2001a,b), additional genotyping tools are required in
order to deepen our understanding of M. tuberculosis’
recent evolution and its mode of transmission (Brosch
et al., 2002; Mostowy et al., 2002). This is important
since control of mycobacterial diseases can only be
optimised once dissemination mechanisms have been
elucidated in full detail (Mathema and Kreiswirth,
2003). In addition, it has been demonstrated that single
mutations can change the pathogenicity of a M.
tuberculosis isolate (Collins et al., 1995) or its resis-
tance against some of the most commonly applied
antibiotics (Troesch et al., 1999; Upton et al., 2001;
Ramaswamy et al., 2003). This once more argues for
the need of identification of additional molecular
markers suited for identifying clinically relevant ge-
netic polymorphism for M. tuberculosis even beyond
the large deletions that were detected upon whole-
genome sequence comparison.
4.1. Distinguishing among M. tuberculosis strains
We here show that 10 strains of M. tuberculosis
suffice for the detection and identification of several
new genetic markers by htAFLP. Straightforward
application of htAFLP resulted in significant numbers
of markers for which sequence data could be
obtained. Further development of a small subset of
these marker molecules already resulted in several
novel and convenient PCR RFLP tests for mapping
genomic polymorphism that are epidemiologically
concordant among M. tuberculosis strains (see Table
3). In addition, based on sequencing of again a
limited set of htAFLP-identified loci, we identified
several new polymorphisms suited for distinguishing
strains in the M. tuberculosis complex including
members of the highly conserved Beijing family.
The universal spread of this strain and its frequent
association with outbreaks of disease indicate that
such additional molecular markers may be used to
further refine the ontogeny of this particularly path-
ogenic strain (Glynn et al., 2002). The mutation we
detected is located in a conserved hypothetical gene
for which further functional studies are certainly
warranted.
4.2. htAFLP and whole genome sequences
In principle, AFLP is a simple laboratory tech-
nology, requiring PCR machines and electrophoresis
equipment only. This renders the htAFLP easily
accessible to the microbiology research laboratory.
AFLP can also be used diagnostically and when
adequate software is available fingerprints can be
stored in exchangeable and expandable databases.
Informative evolutionary and epidemiological com-
parisons can be made (Kassama et al., 2002). In
addition, when whole genome sequences are avail-
able, AFLP fingerprints can be predicted on the
basis of computerised analyses. The number of frag-
ments generated by our htAFLP method is in rea-
sonable agreement with the expectations based on in
silico analyses (results not shown). This is in con-
trast with a previous study where the authors pre-
sented significant differences between theoretical and
practical outcomes (Sims et al., 2002). The data
presented by these authors were not supported by
sequencing of polymorphic fragments. It seems as if
the choice of the restriction enzyme is critical in this
respect, whereas also the methylation status of a
restriction site may be important (Hemavathy and
Nagaraja, 1995). Furthermore, it was recently dem-
onstrated for another clonal bacterial species that
even in the absence of a full genome sequence
AFLP can be used for the generation of informative
DNA probes. For Salmonella enterica serovar typhi-
murium, it was shown that probes specific for a
certain phage type could be readily developed (Hu et
al., 2002). Among 46 strains, 84 phage type-specific
fragments were identified on the basis of a single
restriction enzyme combination and all 16 different
+ 1 extensions for the primer pair used. This corrob-
orated the approach as sketched here, be it that in
case of an even more clonal organism such as M.
tuberculosis, the number of variant fragments is less.

N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–6260
Other procedures, such as for instance mapping IS
element insertion sites (Collins et al., 2002), are
clearly less efficient for mutation detection in my-
cobacterial species.
The availability of two genome sequences for M.
tuberculosis (Cole et al., 1998; Fleischmann et al.,
2002) facilitates initial functional analysis our specific
htAFLP markers. Mathematical studies performed by
Hughes et al. (2002) indicated that the rate of synon-
ymous mutations was found to be 0.000328F0.000022% when the two full genome sequences were
compared. More than 80% of all sites appeared to be
nonvariable. We here establish approximately 5%
polymorphism when screening 1200 loci by htAFLP.
Apparently, when more than two strains are used, the
relative easy with which mutation can be tracked down
increases, which does not support the claim by Hughes
et al. that ‘‘large numbers of loci need to be screened’’
before significant variability can be detected for my-
cobacterial strains. When assessing the nature of the
variable regions, the genomics approaches identified
phospholipase, membrane lipoproteins, the PE/PPE
surface proteins and certain cyclases (Fleischmann et
al., 2002). All of these elements can also be traced back
in Table 2, highlighting the BLAST searches for many
of the AFLP fragments. Additional genetic elements
such as the prophage phiRv1 and molybdopterin cofac-
tor biosynthesis genes were also identified by both
approaches. Based on their genomic comparison,
Fleischmann et al. (2002) literally conclude that they
were able to ‘‘. . . develop a set of markers that would be
valuable in studying the phylogenetics of theM. tuber-
culosis species and other tubercle bacilli’’. In view of
the overlapping outcome of our AFLP analysis, we feel
confident in repeating this statement for the AFLP
approach.
4.3. Concluding remarks
We have not yet reached the stage where for all
species of microorganisms two full genome sequences
are available. This calls for alternative strategies for
high-density assessment of informative genetic poly-
morphism. We here provide proof of principle for one
such method. htAFLP data as presented in this com-
munication are in adequate agreement with whole
genome comparisons. It is also shown that the avail-
ability of such markers can be helpful in the develop-
ment of simple tests for assessment of genetic
polymorphism between large number of microbial
strains. In conclusion, in the absence of multiple
genome sequences for a given microbial species,
htAFLP provides an attractive option for high-density
genotyping and the subsequent development of phy-
logenetically informative molecular variables.
Acknowledgements
The research described in this communication has
in part been facilitated by a grant provided by the
Dutch Ministry of Economic Affairs (BTS 00145).
References
Ahmed, N., Alam, M., Majeed, A.A., Rahman, S.A., Cataldi, A.,
Cousins, D., Hasnain, S.E., 2003. Genome sequence based, com-
parative analysis of the fluorescent amplified fragment length
polymorphisms (FAFLP) of tubercle bacilli from seals provides
molecular evidence for a new species within the Mycobacterium
tuberculosis complex. Infect. Genet. Evol. 2, 193–199.
Behr, M.A., Wilson, M.A., Gill, W.P., Salamon, H., Schoolnik,
G.K., Rane, S., Small, P.M., 1999. Comparative genomics of
BCG vaccines by whole genome DNA microarray. Science
284, 1520–1523.
Bifani, B.J., Mathema, B., Kurepina, N.E., Kreiswirth, B.N., 2002.
Global dissemination of the Mycobacterium tuberculosis W–
Beijing family strains. Trends Microbiol. 10, 45–52.
Brosch, R., Gordon, S.V., Marmiesse, M., Brodin, P., Buchrieser, C.,
Eiglmeier, K., Garnier, T., Gutierrez, C., Hewinson, G., Kremer,
K., Parsons, L.M., Pym, A.S., Samper, S., Van Soolingen, D.,
2002. A new evolutionary scenario for theMycobacterium tuber-
culosis complex. Proc. Natl. Acad. Sci. 99, 3684–3689.
Cole, S.T., Brosch, J., Parkhill, J., Garnier, T., Churcher, C., Harris,
D., Gordon, S.V., Eiglmeier, K., Gas, S., Barry, C.E., Tekaia,
K., Badcock, K., Baham, D., Brown, D., Chillingworth, T.,
Connor, R., Davies, R., Devlin, K., Feltwell, T., Gentles, S.,
Hamlin, N., Holroyd, S., Hornsby, T., Jagels, K., Krogh, A.,
McClean, J., Moule, S., Murphy, L., Oliver, K., Osborne, J.,
Quail, M.A., Rajandream, M.A., Rogers, R., Sutter, S., Seeger,
K., Skelton, J., Squares, R., Sulston, J.E., Taylor, K., White-
head, S., Barrell, B.G., 1998. Deciphering the biology of Myco-
bacterium tuberculosis from the complete genome sequence.
Nature 393, 537–544.
Collins, D.M., Stephens, D.M., 1991. Identification of insertion
sequence, IS1081, in Mycobacterium bovis. FEMS Lett. 83,
11–16.
Collins, D.M., Kawakami, R.P., De Lisle, G.W., Pascopella, L.,
Bloom, B.R., Jacobs, W.R., 1995. Mutation of the principal
sigma factor causes loss of virulence in a strain of the Myco-

N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–62 61
bacterium tuberculosis complex. Proc. Natl. Acad. Sci U. S. A.
92, 8036–8040.
Collins, D.M., De Zoete, M., Cavaignac, S.M., 2002. Mycobacte-
rium avium subsp. paratuberculosis strains from cattle and
sheep can be distinguished by a PCR test based on a novel
DNA sequence difference. J. Clin. Microbiol. 40, 4760–4762.
David, H.L., Newman, C.M., 1971. Some observations on the ge-
netics of isoniazid resistance in the tubercle bacilli. Am. Rev.
Respir. Dis. 104, 508–515.
Fang, Z., Morrison, N., Watt, B., Doig, C., Forbes, K.J., 1998.
IS6110 transposition and evolutionary scenario of the direct
repeat locus in a group of closely related Mycobacterium tuber-
culosis strains. J. Bacteriol. 180, 2102–2109.
Fleischmann, R.D., Alland, D., Eisen, J.A., Carpenter, L., White,
O., Peterson, J., DeBoy, R., Dodson, R., Gwinn, M., Haft, D.,
Hickey, E., Kolonay, J.F., Nelson, W.C., Umayam, L.A., Ermo-
laeva, M., Salzberg, S.L., Delcher, A., Utterback, T., Weidman,
J., Khouri, H., Gill, J., Mikula, A., Bishai, W., Jacobs, W.R.,
Venter, J.C., Fraser, C.M., 2002. Whole genome comparison
of Mycobacterium tuberculosis clinical and laboratory strains.
J. Bacteriol. 184, 5479–5490.
Glynn, J.R., Whiteley, J., Bifani, P.J., Kremer, K., Van Soolingen,
D., 2002. Worldwide occurrence of Beijing/W strains of Myco-
bacterium tuberculosis: a systematic review. Emerg. Infect. Dis.
8, 843–849.
Gordon, S.V., Brosch, R., Billault, A., Garnier, T., Eiglmeier, K.,
Cole, S.T., 1999. Identification of variable regions in the ge-
nomes of tubercle bacilli using bacterial artificial chromosome
arrays. Mol. Microbiol. 32, 643–655.
Groenen, P.M.A., Bunschoten, A.E., Van Soolingen, D., Van
Embden, J.D.A., 1993. Nature of DNA polymorphism in the
direct repeat cluster of Mycobacterium tuberculosis: application
for strain differentiation by a novel typing method. Mol. Micro-
biol. 10, 1057–1085.
Hemavathy, K.C., Nagaraja, V., 1995. DNA methylation in myco-
bacteria: absence of methylation at GATC (Dam) and CCA/
TGG (Dcm) sequences. FEMS Immunol. Med. Microbiol. 11,
291–296.
Hermans, P.W.M., Van Soolingen, D., Bik, E.M., De Haas, P.E.W.,
Dale, J.W., Van Embden, J.D.A., 1993. The insertion element
IS987 from Mycobacterium bovis BCG is located in a hot spot
integration region for insertion elements in Mycobacterium
tuberculosis complex strains. Infect. Immun. 59, 2695–2705.
Hu, H., Lan, R., Reeves, P.R., 2002. Fluorescent amplified fragment
length polymorphism analysis of Salmonella enterica serovar
typhimurium reveals phage-type specific markers and potential
for microarray typing. J. Clin. Microbiol. 40, 3406–3415.
Hughes, A.L., Friedman, R., Murray, M., 2002. GenomEwide pat-
tern of synonymous nucleotide substitution in two complete
genomes of Mycobacterium tuberculosis. Emerg. Infect. Dis.
8, 1342–1345.
Kamerbeek, J., Schouls, L.M., Kolk, A., Van Agterveld, M., Van
Soolingen, D., Kuijper, S., Bunschoten, J.E., Molhuizen, H.,
Shaw, R., Goyal, M., Van Embden, J.D.A., 1997. Simultaneous
detection and strains differentiation of Mycobacterium tubercu-
losis for diagnosis and epidemiology. J. Clin. Microbiol. 35,
907–914.
Kapur, V., Whittam, T.S., Musser, J.M., 1994. Is Mycobacterium
tuberculosis 15,000 years old? J. Infect. Dis. 170, 1348–1349.
Kassama, Y., Rooney, P.J., Goodacrs, R., 2002. Fluorescent ampli-
fied fragment length polymorphisms probabilistic database for
identification of bacterial isolates from uninary tract infections.
J. Clin. Microbiol. 40, 2795–2800.
Kato-Maeda, M., Bifani, P.J., Kreiswirth, B.N., Small, P.M., 2001a.
The nature and consequence of genetic variability within Myco-
bacterium tuberculosis. J. Clin. Invest. 107, 533–537.
Kato-Maeda, M., Rhee, J.T., Gingeras, T.R., Salamon, H., Dren-
kow, J., Smittipat, N., Small, P.M., 2001b. Comparing genomes
within the species Mycobacterium tuberculosis. Genome Res.
11, 547–554.
Kremer, K., Van Soolingen, D., Frothingham, R., Haas, W.H.,
Hermans, P.W.M., Martin, C., Palittapongearnpin, P., Plikaytis,
P.P., Riley, L.W., Yakrus, M.A., Musser, J.M., Van Embden,
J.D.A., 1999. Comparison of methods based on different mo-
lecular epidemiological markers for typing of Mycobacterium
tuberculosis complex strains: interlaboratory study of discrim-
inatory power and reproducibility. J. Clin. Microbiol. 37,
2607–2618.
Mariani, F., Piccolella, E., Collizzi, V., Rappuoli, R., Gross, R.,
1993. Characterization of an IS-like element from Mycobacte-
rium tuberculosis. J. Gen. Microbiol. 139, 1767–1772.
Mathema, B., Kreiswirth, B.N., 2003. Rethinking tuberculosis epi-
demiology: the utility of molecular methods. ASM News 69,
80–85.
Mostowy, S., Cousins, D., Brinkman, J., Aranaz, A., Behr, M.A.,
2002. Genomic deletions suggest a phylogeny for the Mycobac-
terium tuberculosis complex. J. Infect. Dis. 186, 74–80.
Musser, J.M., Amin, A., Ramaswamy, S., 2000. Negligible genetic
diversity of Mycobacterium tuberculosis host immune system
protein targets. Evidence of limited selective pressure. Genetics
155, 7–16.
Pfyffer, G.E., Auckenthaler, R., Van Embden, J.D.A., Van Soolingen,
D., 1998. Mycobacterium canettii, the smooth variant of M.
tuberculosis, isolated from a Swiss patient exposed in Africa.
Emerg. Infect. Dis. 4, 631–634.
Ramakrishnan, L., Federspiel, N.A., Falkow, S., 2000. Granuloma-
specific expression of Mycobacterium virulence proteins from
the glycine-rich PE-PGRS family. Science 288, 1436–1439.
Ramaswamy, S.V., Reich, R., Dou, S.J., Jasperse, L., Pan, X.,
Wanger, A., Quitugua, T., Graviss, E.A., 2003. Single nucleo-
tide polymorphisms in genes associated with isoniazid resist-
ance in Mycobacterium tuberculosis. Antimicrob. Agents
Chemother. 47, 1241–1250.
Roring, S., Scott, A., Brittain, D., Walker, I., Hewison, G., Neill, S.,
Skuce, R., 2002. Development of variable number of tandem
repeat typing of Mycobacterium bovis: comparison of results
with those obtained by using existing exact tandem repeats
and spoligotyping. J. Clin. Microbiol. 40, 2126–2133.
Sims, E.J., Goyal, M., Arnold, C., 2002. Experimental versus in
silico fluorescent amplified fragment length polymorphism anal-
ysis of Mycobacterium tuberculosis: improved typing with and
extended fragment range. J. Clin. Microbiol. 40, 4072–4076.
Skeiky, Y.A., Ovendale, P.J., Jen, S., Alderson, M.R., Dillon, D.C.,
Smith, S., Wilson, C.B., Orme, I.M., Reed, S.G., Campos-Neto,

N. van den Braak et al. / Journal of Microbiological Methods 56 (2004) 49–6262
A., 2000. T cell expression cloning of a Mycobacterium tuber-
culosis gene encoding a protective antigen associated with the
early control of infection. J. Immunol. 165, 7140–7149.
Small, P.M., Hopewell, P.C., Singh, S.P., Paz, A., Parsonnet, J.,
Ruston, D.C., Schecter, G.F., Daley, C.L., Schoolnik, G.A.,
1994. The epidemiology of tuberculosis in San Francisco. A
population based study using conventional and molecular
methods. N. Engl. J. Med. 330, 1703–1709.
Sreevatsan, S., Pan, X., Stockbauer, K.E., Connell, N.D., Kreis-
wirth, B.N., Whittam, T.S., Musser, J.M., 1997. Restricted struc-
tural gene polymorphism in the Mycobacterium tuberculosis
complex indicates evolutionary recent global dissemination.
Proc. Natl. Acad. Sci. U. S. A. 94, 9869–9874.
Supply, P., Lesjean, S., Savine, E., Kremer, K., Van Soolingen, D.,
Locht, C., 2001. Automated high throughput genotyping for
study of global epidemiology of Mycobacterium tuberculosis
based on mycobacterial interspersed repetitive units. J. Clin.
Microbiol. 39, 3563–3571.
Supply, P., Mazars, E., Lesjean, S., Vincent, V., Gicquel, B., Locht,
C., 2000. Variable human minisatellite-like regions in the Myco-
bacterium tuberculosis genome. Mol. Microbiol. 36, 762–771.
Thierry, D., Brisson Noel, A., Vincent-Levy-Frebault, V., Nguyen,
S., Guesdon, J., Gicquel, B., 1990. Characterization of a Myco-
bacterium tuberculosis insertion sequence, IS6110, and its ap-
plication in diagnosis. J. Clin. Microbiol. 28, 2668–2673.
Troesch, A., Nguyen, H., Miyada, C.G., Desvarenne, S., Gingeras,
T.R., Kaplan, P.M., Cros, P., Mabilat, C., 1999. Mycobacterium
species identification and rifampin resistance testing with high-
density DNA probe arrays. J. Clin. Microbiol. 37, 49–55.
Upton, A.M., Mushtaq, A., Victor, T.C., Sampson, S.L., Sandy, J.,
Smith, D.M., Van Helden, P.V., Sim, E., 2001. Arylamine N-
acetyltransferase of Mycobacterium tuberculosis is a polymor-
phic enzyme and a site of isoniazid metabolism. Mol. Microbiol.
42, 309–317.
Van Belkum, A., Scherer, S., Van Alphen, L., Verbrugh, H., 1998.
Short sequence DNA repeats in prokaryotic genomes. Microbiol
Mol. Biol. Rev. 62, 275–293.
Van Belkum, A., Struelens, M., De Visser, A., Verbrugh, H.,
Tibayrenc, M., 2001. Role of genomic typing in taxonomy, evo-
lutionary genetics, and microbial epidemiology. Clin. Microbiol.
Rev. 14, 547–560.
Van Embden, J.D.A., Crawford, J.T., Dale, J.W., Gicquel, B., Her-
mans, P.W.A., McAdam, R., Shinnick, T., Small, P.M., 1993.
Strain identification ofMycobacterium tuberculosis by DNA fin-
gerprinting: recommendations for a standardized method. J. Clin.
Microbiol. 31, 406–409.
Van Embden, J.D.A., Van Gorkom, T., Kremer, K., Jansen, R.,
Van der Zeijst, B.A.M., Schouls, L.M., 2000. Genetic varia-
tion and evolutionary origin of the direct repeat locus of
Mycobacterium tuberculosis complex bacteria. J. Bacteriol.
182, 2393–2401.
Van Soolingen, D., De Haas, P.E.W., Hermans, P.W.M., Van
Embden, J.D.A., 1994. DNA fingerprinting of Mycobacterium
tuberculosis. Methods Enzymol. 235, 196–205.
Van Soolingen, D., Hoogenboezem, T., De Haas, P.E.W., Hermans,
P.W.M., Koedam, M.A., Teppema, K.S., Brennan, P.J., Besra,
G.S., Portaels, F., Top, J., Schouls, L.M., Van Embden, J.D.,
1997. A novel pathogenic taxon of the Mycobacterium tuber-
culosis complex, Canettii: characterization of an exceptional
isolate from Africa. Int. J. Syst. Bacteriol. 47, 1236–1245.
Vos, P., Hogers, R., Bleeker, M., Reijans, M., Van de Lee, T.,
Hornes, M., Frijters, A., Pot, J., Peleman, J., Kuiper, M., Za-
beau, M., 1995. AFLP: a new technique for DNA fingerprinting.
Nucleic Acids Res. 23, 4407–4414.