2-hydroxycinnamaldehyde inhibits the epithelial-mesenchymal transition in breast cancer cells
TRANSCRIPT

PRECLINICAL STUDY
2-Hydroxycinnamaldehyde inhibits the epithelial-mesenchymaltransition in breast cancer cells
Ismail Ahmed Ismail • Hye Sook Kang • Heon-Jin Lee • Hyeyoun Chang •
Jieun Yun • Chang Woo Lee • Nam Hee Kim • Hyun Sil Kim • Jong In Yook •
Su-Hyung Hong • Byoung-Mog Kwon
Received: 31 August 2012 / Accepted: 13 December 2012 / Published online: 3 January 2013
� Springer Science+Business Media New York 2012
Abstract Since epithelial-mesenchymal transition (EMT)
plays a critical role in cancer progression and in maintaining
cancer stem cell properties, EMT is emerging as a thera-
peutic target for inhibiting the metastatic progression of
cancer cells. 20-Hydroxycinnamaldehyde (HCA) and its
derivative, 20-benzoyloxycinnamaldehyde, have recently
been suggested as promising therapeutic candidates for
cancer treatment. The purpose of this study is to investigate
the anti-metastatic effect of HCA on breast cancer and
the molecular mechanisms by which HCA regulates the
transcriptional program during EMT. HCA induces epithe-
lial reversion at nanomolar concentrations by suppressing
Snail via the nuclear translocalization of GSK-3b, which
results in the transcriptional upregulation of E-cadherin.
HCA also activates the transcription factor KLF17, which
suppresses Id-1, indicating that HCA inhibits EMT by mul-
tiple transcriptional programs. Further, HCA treatment sig-
nificantly inhibits lung metastasis in a mouse orthotopic
breast cancer model. This study demonstrates the anti-met-
astatic effect of the non-toxic natural compound HCA
through attenuation of EMT in a breast cancer model.
Keywords 20-Hydroxycinnamaldehyde � Breast cancer
cells � Epithelial-mesenchymal transition (EMT) � Cell
invasion � Snail � KLF17
Introduction
Cinnamaldehyde is commonly used as a flavoring and
ingredient in food, beverages, medical products, cosmetics,
and perfumes. 20-Hydroxycinnamaeldehyde (HCA) is a
natural compound that is isolated from the bark of Cinna-
momum cassia Blume [1], and 20-benzoyloxycinnamalde-
hyde (BCA) is a well-known derivative of HCA. Both HCA
and BCA have been reported to have anti-tumor effects on
various types of cancer cells, inhibiting proliferation and
inducing apoptosis [2–5]. BCA was approved for clinical
tests by the Korean Food and Drug Administration on Jan-
uary 31, 2011. Although the cytotoxic or anti-angiogenic
effects of these cinnamaldehydes are exerted mainly at lm
to mM concentrations [2], the systemic administration
of cinnamaldehyde significantly suppressed tumor forma-
tion in vivo [5], suggesting that these compounds func-
tion to suppress tumorigenesis at physiological levels.
I. A. Ismail and H. S. Kang contributed equally to this work.
Electronic supplementary material The online version of thisarticle (doi:10.1007/s10549-012-2388-7) contains supplementarymaterial, which is available to authorized users.
I. A. Ismail � H. S. Kang � H.-J. Lee � S.-H. Hong (&)
Department of Oral Microbiology, School of Dentistry,
Kyungpook National University, Daegu, Republic of Korea
e-mail: [email protected]
I. A. Ismail
Laboratory of Molecular Cell Biology, Department of Zoology,
Faculty of Science, Assiut University, Assiut 71516, Egypt
H. Chang � B.-M. Kwon (&)
Laboratory of Chemical Biology and Genomics, Korea Research
Institute of Bioscience and Biotechnology, University of Science
and Technology, Daejon 305-806, Republic of Korea
e-mail: [email protected]
J. Yun � C. W. Lee
Bio-Evaluation Center, Korea Research Institute of Bioscience
and Biotechnology, Chungbuk, Republic of Korea
N. H. Kim � H. S. Kim � J. I. Yook
Department of Oral Pathology, Oral Cancer Research Institute,
College of Dentistry, Yonsei University, Seoul 120-752,
Republic of Korea
123
Breast Cancer Res Treat (2013) 137:697–708
DOI 10.1007/s10549-012-2388-7

However, the molecular mechanisms underlying the in vivo
effects of these compounds have not been clearly identified.
The pharmacokinetics and metabolism of HCA and BCA
were characterized in male Sprague–Dawley rats as part of a
preclinical evaluation. BCA was converted rapidly to HCA
in the rat serum after either intravenous or oral uptake; HCA
was subsequently converted to o-coumaric acid. The half-
lives of both HCA and BCA were *2 h [6].
Cancer metastases, rather than primary tumors, are
responsible for the most cancer deaths [7–9]. Epithelial-
mesenchymal transition (EMT) is a cellular program in which
polarized epithelial cells undergo complex biological changes
such that the epithelial cells express a mesenchymal pheno-
type, which induces enhanced migratory capacity, invasive-
ness, metastatic potential, and drug resistance [10]. Among
the number of molecular processes involved in EMT,
including the activation of transcription factors, regulation of
cellular adhesion, rearrangement of cytoskeletal proteins, and
changes in the expression of specific microRNAs [11], the
transcriptional programs of EMT are key targets for thera-
peutic intervention with natural compounds as well as for an
understanding of coordinated cellular program of EMT. For
example, members of the Snail superfamily of zinc finger
transcription factors bind directly to the proximal promoter
region of E-cadherin and induce an EMT phenotype in cancer
cells [12]. Interestingly, Snail expression is controlled by
GSK-3b-mediated phosphorylation, which is governed by
canonical Wnt signaling, allowing the coordinated transcrip-
tional regulation of b-catenin and Snail [13]. Importantly, the
p53 tumor suppressor directly regulates the Snail-mediated
EMT program via post-translational and post-transcriptional
mechanisms, suggesting that Snail may be an attractive ther-
apeutic target [14, 15] for the control of cancer metastasis.
The transcription factor Sp1 increases breast cancer cell
invasion and metastasis via the upregulation of urokinase
receptor or matrix metalloproteinase-2 expression [16, 17].
Furthermore, Sp1 binds to and activates Id-1 (inhibitor of
differentiation or DNA binding protein 1) promoter [18]. Id-
1 is overexpressed in highly invasive cancer cells, including
prostate [19], breast [20], cervical [21], and bladder cancers
[22]. Id-1 promotes metastasis in human breast cancer
in vivo via the upregulation of matrix metalloproteinase
MT1-MMP [20]. In human esophageal cancer, Id-1 activates
the PI3 K/AKT signaling pathway [23] or the N-cadherin-
RhoA axis [24], either of which can result in increased cancer
metastasis. Interestingly, KLF17 (Kruppel-like transcription
factor 17) is able to negatively regulate EMT and cell
invasion by directly binding to the Id-1 promoter to inhibit its
transcription in breast cancer cells [25].
Recently, the targeting of cancer stem cells has emerged
as a therapeutic strategy. EMT induced by Snail or TGF-bgenerates stem cell phenotype; the transformed cells exhibit
an increased ability to form mammospheres and resistance to
chemotherapy [26]. Selective inhibitors of cancer stem cells
have been identified through screening for compounds that
selectively kill mesenchymally transformed cells [27].
Although this has been a conceptual advance for cancer
therapeutics, several chemicals that target EMT, such as
salinomycin and etoposide, are of limited potential due to
systemic toxicity in mammals. Thus, natural compounds
with anti-EMT potential but without systemic adverse
effects may provide advanced therapeutic advantages. In this
study, we report that the natural food compound HCA blocks
a number of EMT transcriptional programs in breast cancer
cells, resulting in epithelial reversion. Specifically, we
examined the anti-EMT effect of HCA at non-toxic nano-
molar concentrations to explain the functional relevance of
anti-metastatic potential in vivo.
Materials and methods
Chemicals and reagents
MTT (3-[4, 5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetra-
zolium bromide) was purchased from Sigma (St. Louis, MO,
USA). DMEM, RPMI, FBS, and penicillin/streptomycin
antibiotics were purchased from Gibco (Invitrogen, CA,
USA). Qiazol was purchased from Qiagen (Valencia, CA,
USA), and 29 SYBR Green PCR master mix was purchased
from Takara Biotechnology (Dalian, Japan). The CytoSe-
lectTM 96-well cell invasion assay kit was purchased from
Cell Biolabs (San Diego, CA, USA). Rabbit polyclonal anti-
GSK3-b, anti-Snail, anti-KLF17, anti-Sp1, and rabbit
monoclonal anti-E-cadherin antibodies were purchased from
Abcam (Cambridge, UK). The mouse monoclonal anti-
vimentin antibody and anti-Id-1 antibody were purchased
from Lab Vision (Fremont, CA, USA) and Millipore
(Upstate Chemicon, Temecula, CA, USA), respectively.
Mouse monoclonal anti-HDAC1 and HRP-conjugated
mouse monoclonal IgG anti-b-actin antibody were pur-
chased from Santa Cruz Biotechnology (Santa Cruz, CA,
USA). HRP-conjugated goat anti-rabbit and anti-mouse
secondary antibodies were purchased from Pierce (Rock-
ford, IL, USA). Alexa Fluor goat anti-mouse and anti-rabbit
IgG fluorescent secondary antibodies were purchased from
Molecular Probes (Eugene, Oregon, USA). ECL Western
Blotting Detection Reagent was purchased from Neuronex
(Daegu, South Korea). All other reagents were obtained from
standard commercial sources.
Cell culture
The human breast cancer cell lines MCF-7, T47D, MDA-
MB-231, and MDA-MB-435 were obtained from the
American Type Culture Collection and maintained in
698 Breast Cancer Res Treat (2013) 137:697–708
123

DMEM (MCF-7 and MDA-MB-231) or RPMI (T47D and
MDA-MB-435) media containing 10 % FBS and
1 % penicillin/streptomycin solution. Cells were grown at
37 �C in a humidified atmosphere containing 5 % CO2.
Cell viability assay
Cells were seeded into 96-well plates (at a density of
5,000 cells/well). On the following day, cells were treated
with different concentrations of HCA (0–5 lm) in fresh
medium and incubated for another 24 h. Cell viability was
then assessed using the MTT assay, and the absorbance
was read at 570 nM using an ELISA microplate reader
(Molecular Devices, Downingtown, PA, USA).
Transwell migration assay
The effect of HCA on cell invasion was determined using a
CytoSelectTM 96-well cell invasion assay kit (Cell Biolabs, San
Diego, CA, USA) containing polycarbonate membrane inserts
(8 lm pore size) as indicated in the instruction manual and
previously described methods [28]. The invasion plate was
warmed for 10 min at room temperature. The basement
membrane layer was then rehydrated by adding 100 ll of
warm, serum-free medium to the inner compartment and
incubated for 1 h in a cell culture incubator. A cell suspension
(1 9 106 cells/ml) in serum-free medium (with or without
HCA) was cultured on the basement membrane after the
rehydration media was removed. A 150 ll aliquot of serum-
containing medium was added to the feeder tray. The basement
membrane chamber was inserted carefully to avoid air bubbles,
followed by the addition of 100 ll of cell suspension containing
the indicated doses of HCA. After 24 h of incubation, the
medium in the membrane chamber was transferred to a new
harvesting tray containing 150 ll of detachment solution for
30 min. The cells were dislodged completely from the under-
side of the membrane by gently tilting the membrane several
times. Then, 50 ll of 49 lysis buffer/CyQuant GR dye solution
was added to each sample and incubated for 20 min at room
temperature. Fluorescence measurements were performed in a
fluorescence plate reader at 480/520 nM.
Wound healing assay
The effect of HCA on breast cancer cell migration was
assessed using the wound healing assay as previously
described [29]. Briefly, cells were seeded in 100 mM cul-
ture plates and incubated for 16–24 h. When the cell
confluency reached approximately 80–90 %, a wound was
created manually by scratching the cell layer with a pipette
tip. Then, the cell medium was exchanged with new
medium containing either DMSO or 100 nM HCA. The
images were photographed immediately using phase-
contrast microscopy for the 0 h time point (control). After
24 h of incubation, the wounds were visualized and the
healed wound was compared with the control.
E-cadherin luciferase reporter assay
The E-cadherin reporter gene constructs E-cad (-108)-Luc
and E-cad (-108)-39Ebox. Mut-Luc and the pSV-gal (Pro-
mega) control vector have been described previously [30].
Cells were transfected with 0.75 lg E-cad (-108)-Luc or
E-cad (-108)-3xMut-Luc vector. Reporter gene activities
were measured with a luciferase assay system (Promega) at
48 h after transfection and normalized to the b-galactosidase
activities of co-transfected pSV-gal (0.25 g) measured with
a b-galactosidase enzyme assay system (Promega). Reporter
gene activities were reported as light units relative to those
obtained from mock (pCR3.1, Invitrogen)-transfected cells.
Quantitative real-time PCR
The effect of HCA (100 nM) on the expression of a panel of
genes involved in metastasis and EMT regulation in breast
cancer cells was investigated using real-time quantitative RT-
PCR analysis. In brief, 1 9 105 cells were plated in 60 mM
dishes and grown for 24 h, followed by treatment with the
indicated doses of HCA for 24 h. Total RNA was extracted
with Qiazol (Qiagen, Valencia, CA, USA) according to the
manufacturer’s instructions. Extracted RNA (5 lg) was
reverse transcribed into cDNA using a first-strand cDNA
synthesis kit (Applied Biosystems, Foster City, CA, USA),
and the resulting cDNA was diluted tenfold and kept at
-20 �C until use. The real-time qPCR primers were designed
using the Primer Express 1.5 software (Applied Biosystems)
as follows: E-cadherin forward, 50-CGACCCAACCCAA
GAATCTA-30; E-cadherin reverse, 50-CTCCAAGAATCC
CCAGAATG-30; Snail forward, 50-AGCTCTCTGAGGCCA
AG-GA TCT-30; Snail reverse, 50-TGTGGCTTCGGATGTG
CAT-30; KLF17 forward, 50-CTGCCTGAGCGTGGTATG
AG-30; KLF17 reverse, 50-TCATCCGGGAAGGAGTGA-G
A-30; Sp1 forward, 50-GGACTACCTGGAGTGATGCCT
AA-30; Sp1 reverse, 50-CCCATCAACGGTCTGGAACT-30;Id-1 forward, 50-CTCTACGACATGAACGGCTG -30; Id-1
reverse 50-TGCTCACCTTGCGGTTCTG-30; GAPDH
forward, 50-AGATCATCAGCAATGCCTCCTG-30 and GAP-
DH reverse, 50-ATGGCATGGACTGTGGTCATG-30. The
expressions of these genes were normalized to GAPDH. Real-
time qPCR was carried out using an ABI Prism 7500 sequence
detection system (Applied Biosystems, USA). Each 20 ll PCR
reaction contained 10 ll SYBR Green PCR master mix, 4 ll
diluted cDNA, and 200 nM primer set. All samples were
amplified in triplicate in a 96-well plate using the following
cycling conditions: 2 min at 50 �C, 10 min at 95 �C, and 40
cycles at 95 �C for 15 s followed by 1 min at 60 �C.
Breast Cancer Res Treat (2013) 137:697–708 699
123

Calculations were performed using the Dcycle threshold (DCt)
method, normalizing the average Ct value of each treatment
compared to its endogenous control (GAPDH), and then calcu-
lating the 2-DDCt for each treatment. Statistical analysis was
performed as described previously [31]. These experiments were
each repeated three times.
Western blot analyses
Cells were washed twice with cold PBS, after which
200 ll of PRO-PREP protein extraction solution (Intron,
Daejon, South Korea) was added. The cell lysates were
centrifuged, and protein concentrations were estimated
using the Coomassie protein assay reagent (Thermo Sci-
entific, Rockford, IL, USA). Protein samples (40 lg) were
electrophoresed on 8–15 % SDS-PAGE gels. Proteins
were transferred to nitrocellulose membranes, which were
blocked in 5 % skim milk in TBS (25 mM Tris base and
150 mM NaCl) for 2 h at room temperature, and then
incubated with primary antibody overnight at 4 �C. The
membranes were then incubated with horseradish peroxi-
dase-conjugated secondary antibodies at 1:5,000 dilutions
for 1 h at room temperature and then washed three times in
TBST (TBS and 0.1 % Tween 20). b-actin and HDAC-1
were used as reference proteins for the normalization of
cytosolic and nuclear protein contents, respectively. The
target proteins were detected with ECL detection reagents,
and the relative intensities of the bands were analyzed by
Image-J software.
Immunocytochemical analysis
The effect of HCA on GSK3-b and Snail localization in
breast cancer cells was demonstrated using immunofluo-
rescence staining as described [25]. Briefly, MDA-MB-231
and MDA-MB-435 cells were cultured in 6-well plates
containing cover slides. On the following day, the cells were
treated with 100 nM HCA for 24 h and subsequently washed
three times in PBS. Cells were immediately fixed in 4 %
paraformaldehyde for 1 h and then washed with PBS. Cells
were cleared in a buffer containing 0.1 % Triton X-100 and
0.1 % sodium citrate at pH 6 for 5 min, followed by three
washes in PBS. Cells were blocked by adding Tris-buffered
saline solution (TBS) containing BSA (0.05 M TBS ? 3
drops of albumin serum) for 1 h at room temperature. The
primary rabbit polyclonal GSK3b or Snail antibodies were
added to the TBS solution and incubated at room temperature
for 1 h. After three washes with PBS, the appropriate fluo-
rescent secondary antibodies were added for 40 min, fol-
lowed by three washes in PBS. Cells were then stained with
Hoechst nuclear counterstain for one minute, washed several
times with PBS, and then examined under fluorescence
microscopy (Olympus BX 51, Tokyo, Japan).
Subcellular protein fractionation
The effect of HCA on GSK3-b and Snail localization was
confirmed using subcellular fractionation followed by
western blot analysis, as described previously [32]. Cells
(1 9 106) were cultured in 100 mM plates overnight and
subsequently treated with 100 nM of HCA for 24 h. Cells
were washed in cold PBS and lysed in 300 ll of fraction-
ation buffer (10 mM Tris–HCl, 1 mM EDTA, 0.5 %
NP-40, and protease inhibitor cocktail). After incubation
on ice for 30 min, cells were centrifuged at 6009 g for
10 min. The supernatants and dissolved pellets were used
as cytoplasmic and nuclear fractions, respectively.
In vivo metastasis assay
All animal works were performed in accordance with a
protocol approved by the Institutional Animal Care and Use
Committee. To induce lung metastasis formation, mice were
anaesthetized, and a small incision was made to reveal the
mammary gland; 106 MDA-MB231 cells were injected
directly into the third mammary fat pad. The incision was
closed with wound clips, and the primary tumor was
removed at day 35. Mice were randomized into 3 groups
(n = 5): control (vehicle), HCA (50 mg/kg), and doxoru-
bicin (0.2 mg/kg) as a positive control. HCA was adminis-
tered daily by gavage, whereas doxorubicin was
administered by intraperitoneal injection once per 3 days.
The mouse lungs were harvested at day 35 after the removal
of the primary tumor, and the metastatic nodules in the lungs
were counted. For histological examination, the mouse lungs
were fixed into 10 % buffered formalin and subjected for
paraffin section with routine procedure. Microscopic
metastasis were examined and counted the number from the
H&E stained slide under light microscopy.
Statistical analysis
The differences in mean values among groups were eval-
uated and expressed as the mean ± SD. Averages were
drawn, and the statistical calculations were performed
using a student t test in Microsoft Excel 2007. For the
statistical difference of microscopic metastasis from H&E
sections, Mann–Whitney test in R (ver2.13.2) was used.
Results
HCA and BCA inhibit breast cancer cell migration
without affecting viability
To test the effect of HCA and BCA on EMT in breast
cancer cells, we examined the anti-migratory effect of
700 Breast Cancer Res Treat (2013) 137:697–708
123

these compounds at 100, 500, and 1,000 nM concentrations
by transwell migration assay kit. Under these conditions,
both HCA and BCA showed anti-migratory effects in a
panel of breast cancer cell lines (Fig. 1a) without affecting
cell viability (Fig. 1b). Anti-migratory effects for 100 nM
of HCA was also observed in wound healing assays using
MDA-MB-231 (Fig. 1c) and MDA-MB-435 (Fig. 1d)
breast cancer cells.
HCA suppresses Snail, resulting in increased
E-cadherin transcriptional activity
Since E-cadherin expression is a hallmark of EMT, and
Snail is a well-known EMT inducer that acts as a repressor
of E-cadherin transcription, we investigated the effect of
HCA on E-cadherin transcript expression in MDA-MB-231
and MDA-MB-435 cells. In both cell types, 100 nM HCA
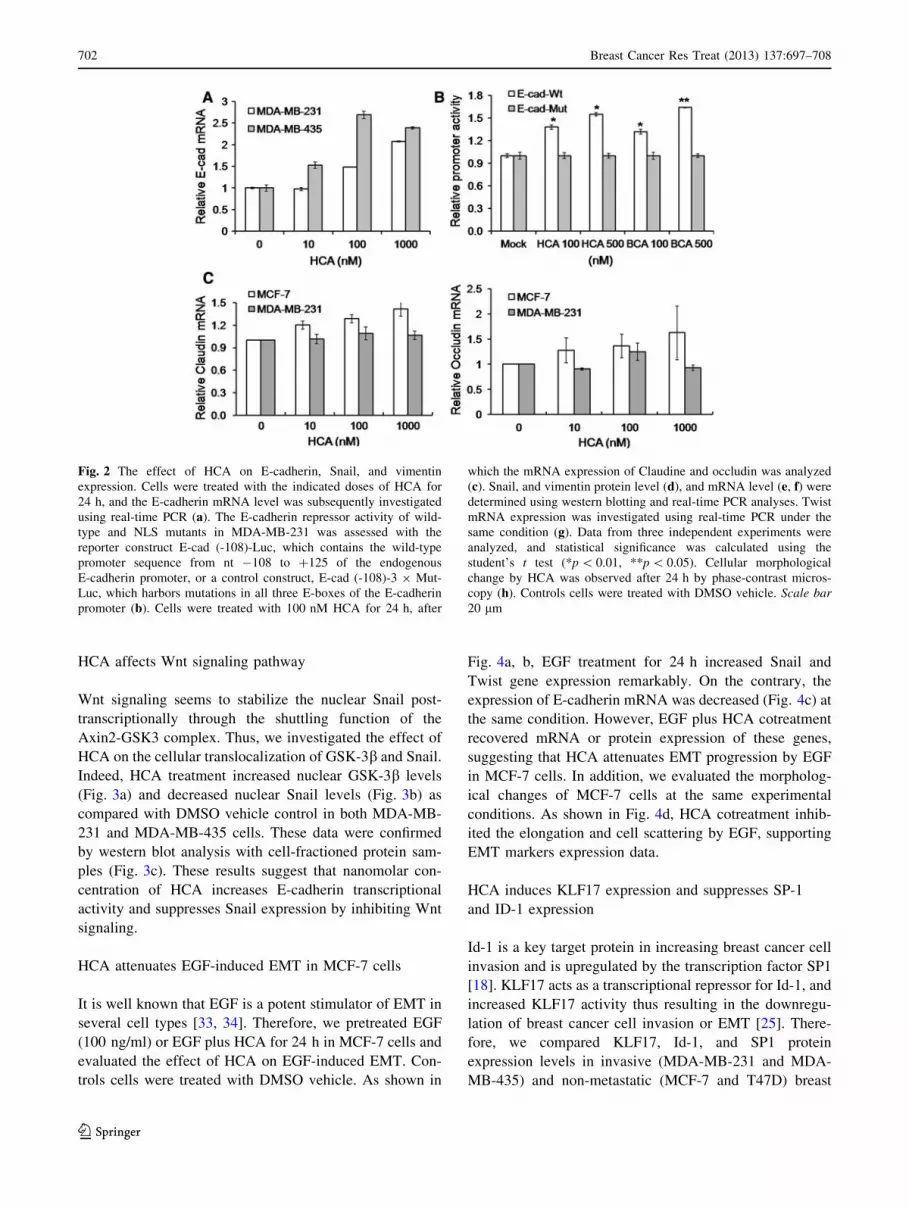
successfully increased E-cadherin transcription (Fig 2a),
revealing that nanomolar concentrations of HCA can
induce epithelial phenotypes in breast cancer cells. To test
whether this effect might be mediated by changes in the
transcriptional repressor that binds to the E-cadherin
proximal promoter, we next examined the E-cadherin
proximal promoter activity in response to HCA treatment.
HCA and BCA increased E-cadherin promoter activity
compared to the E-box mutant promoter (Fig. 2b). In
addition, HCA upregulated mRNA expression of claudin
and occludin which are representative epithelial markers
(Fig. 2c).
On the contrary, the endogenous Snail protein levels in
MDA-MB-231 and MDA-MB-435 cells were suppressed
by HCA (Fig. 2d), while the Snail transcript levels were
not affected significantly (Fig. 2e), suggesting that HCA
suppresses Snail expression via a post-translational mech-
anism. The expression of other mesenchymal markers such
as vimentin and Twist was also decreased by HCA
(Fig. 2d, f, g). Furthermore, cellular morphological change
by HCA was observed after 24 h by phase-contrast mi-
croscophy analysis. As shown in Fig. 2h, HCA treatment
decreased the elongation of cell shape in both MDA-MB-
231 and MDA-MB-435 cells, suggesting the epithelial-like
morphological changes.
Fig. 1 The effect of HCA on breast cancer cell migration and
invasion. Transwell cell invasion assay was performed with HCA and
its derivative BCA (100, 500, or 1,000 nM) in breast cancer cells (a).
To investigate the effect of HCA on cell viability, cells were
incubated with the indicated doses of HCA for 24 h, and then the cell
viability was assessed using MTT assay (b). These results are from
three independent experiments and each bar represents standard
deviation. Statistical significance was calculated using the student’s
t test (*p \ 0.01, **p \ 0.05). A wound healing assay was performed
to evaluate cell migration inhibition by HCA. MDA-MB-231 (c) and
MDA-MB-435 (d) cell monolayers were scratched, allowed to heal in
the presence of 100 nM of HCA for 24 h, and the images were
obtained by phase-contrast microscopy
Breast Cancer Res Treat (2013) 137:697–708 701
123
HCA affects Wnt signaling pathway
Wnt signaling seems to stabilize the nuclear Snail post-
transcriptionally through the shuttling function of the
Axin2-GSK3 complex. Thus, we investigated the effect of
HCA on the cellular translocalization of GSK-3b and Snail.
Indeed, HCA treatment increased nuclear GSK-3b levels
(Fig. 3a) and decreased nuclear Snail levels (Fig. 3b) as
compared with DMSO vehicle control in both MDA-MB-
231 and MDA-MB-435 cells. These data were confirmed
by western blot analysis with cell-fractioned protein sam-
ples (Fig. 3c). These results suggest that nanomolar con-
centration of HCA increases E-cadherin transcriptional
activity and suppresses Snail expression by inhibiting Wnt
signaling.
HCA attenuates EGF-induced EMT in MCF-7 cells
It is well known that EGF is a potent stimulator of EMT in
several cell types [33, 34]. Therefore, we pretreated EGF
(100 ng/ml) or EGF plus HCA for 24 h in MCF-7 cells and
evaluated the effect of HCA on EGF-induced EMT. Con-
trols cells were treated with DMSO vehicle. As shown in
Fig. 4a, b, EGF treatment for 24 h increased Snail and
Twist gene expression remarkably. On the contrary, the
expression of E-cadherin mRNA was decreased (Fig. 4c) at
the same condition. However, EGF plus HCA cotreatment
recovered mRNA or protein expression of these genes,
suggesting that HCA attenuates EMT progression by EGF
in MCF-7 cells. In addition, we evaluated the morpholog-
ical changes of MCF-7 cells at the same experimental
conditions. As shown in Fig. 4d, HCA cotreatment inhib-
ited the elongation and cell scattering by EGF, supporting
EMT markers expression data.
HCA induces KLF17 expression and suppresses SP-1
and ID-1 expression
Id-1 is a key target protein in increasing breast cancer cell
invasion and is upregulated by the transcription factor SP1
[18]. KLF17 acts as a transcriptional repressor for Id-1, and
increased KLF17 activity thus resulting in the downregu-
lation of breast cancer cell invasion or EMT [25]. There-
fore, we compared KLF17, Id-1, and SP1 protein
expression levels in invasive (MDA-MB-231 and MDA-
MB-435) and non-metastatic (MCF-7 and T47D) breast
Fig. 2 The effect of HCA on E-cadherin, Snail, and vimentin
expression. Cells were treated with the indicated doses of HCA for
24 h, and the E-cadherin mRNA level was subsequently investigated
using real-time PCR (a). The E-cadherin repressor activity of wild-
type and NLS mutants in MDA-MB-231 was assessed with the
reporter construct E-cad (-108)-Luc, which contains the wild-type
promoter sequence from nt -108 to ?125 of the endogenous
E-cadherin promoter, or a control construct, E-cad (-108)-3 9 Mut-
Luc, which harbors mutations in all three E-boxes of the E-cadherin
promoter (b). Cells were treated with 100 nM HCA for 24 h, after
which the mRNA expression of Claudine and occludin was analyzed
(c). Snail, and vimentin protein level (d), and mRNA level (e, f) were
determined using western blotting and real-time PCR analyses. Twist
mRNA expression was investigated using real-time PCR under the
same condition (g). Data from three independent experiments were
analyzed, and statistical significance was calculated using the
student’s t test (*p \ 0.01, **p \ 0.05). Cellular morphological
change by HCA was observed after 24 h by phase-contrast micros-
copy (h). Controls cells were treated with DMSO vehicle. Scale bar20 lm
702 Breast Cancer Res Treat (2013) 137:697–708
123

cancer cells. We found that KLF17 expression is signifi-
cantly higher in non-metastatic breast cancer cells, while
Sp1 and Id-1 are expressed at higher levels in metastatic
cells (Suppl. data Fig. 1).
We then investigated the effect of HCA on KLF17,
SP1, and Id-1 expression. KLF17 mRNA (Fig. 5a) and
protein (Fig. 5d) expression were significantly upregu-
lated by HCA treatment in MCF-7, MDA-MB-231, and
MDA-MB-435 breast cancer cells. Furthermore, HCA
treatment significantly decreased SP-1 mRNA (Fig. 5b)
and protein (Fig. 5d) expression levels. Together, these
data show that Id-1 expression is significantly inhibited
by HCA treatment in all three cell lines, as expected
(Fig. 5c, d).
HCA suppressed metastasis of MDA-MB-231 cells
in vivo
To test the effect of HCA on the in vivo metastatic capability
of breast cancer, we next treated mice that had been injected
orthotopically with MDA-MB-231 cells with HCA (50 mg/
kg), doxorubicin (2 mg/kg), or vehicle, administered daily.
After the removal of the primary tumors, the metastatic
potential of the tumors and the effect of HCA were evaluated
by lung autopsy. Indeed, the number of metastatic breast
cancer cell colonies in the lung parenchyma was significantly
reduced by HCA treatment. Metastasis in the HCA treatment
group was comparable to that in the doxorubicin treatment
group (Fig. 6a, b, p \ 0.05), while the body weight was not
Fig. 2 continued
Breast Cancer Res Treat (2013) 137:697–708 703
123
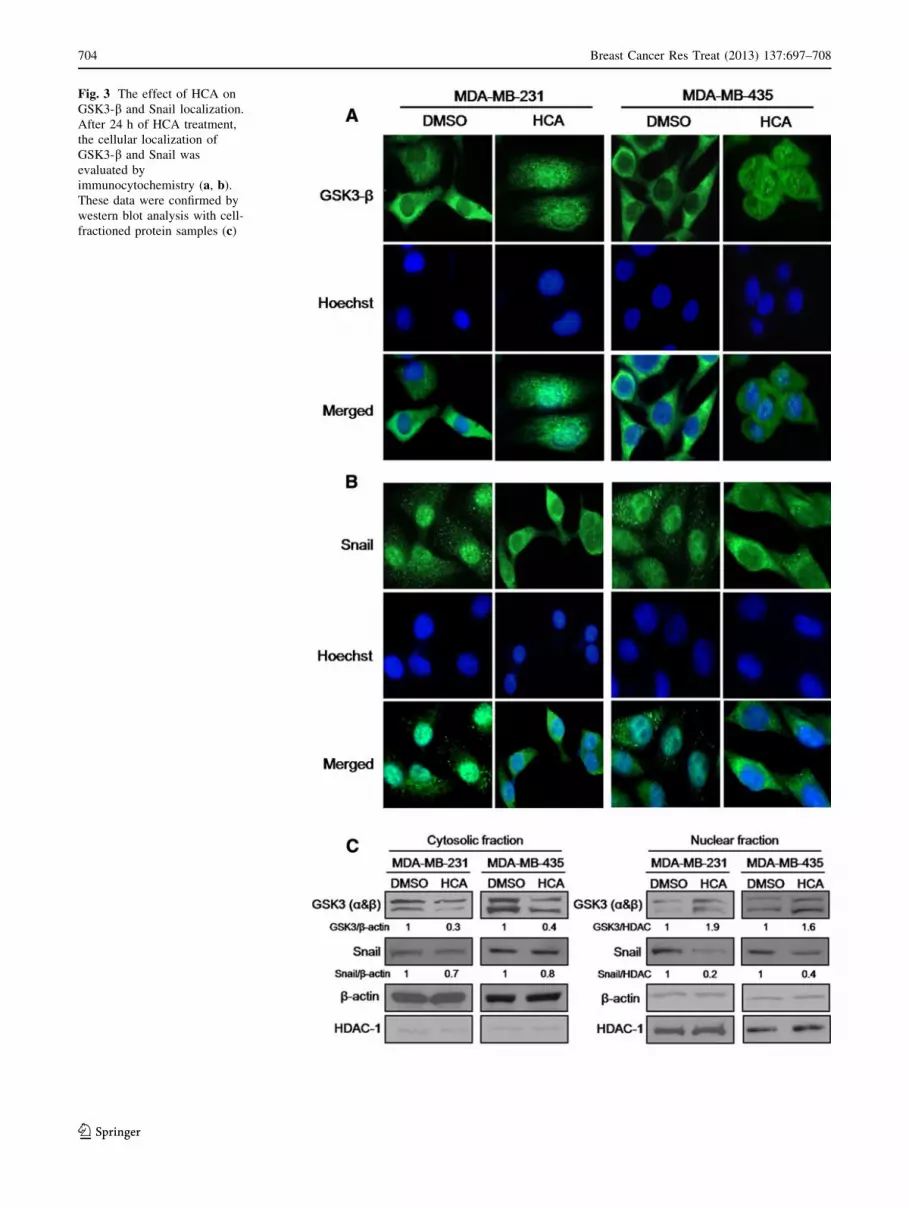
Fig. 3 The effect of HCA on
GSK3-b and Snail localization.
After 24 h of HCA treatment,
the cellular localization of
GSK3-b and Snail was
evaluated by
immunocytochemistry (a, b).
These data were confirmed by
western blot analysis with cell-
fractioned protein samples (c)
704 Breast Cancer Res Treat (2013) 137:697–708
123

Fig. 4 The effect of HCA on
EGF-induced EMT in MCF-7
cells. MCF-7 was treated with
EGF (50 ng/ml) and/or HCA
(100 nM). After 24 h, Snail (a),
Twist (b), and E-cadherin
(c) expression was analyzed.
Cellular morphological change
was observed after 24 h by
phase-contrast microscopy (d).
Controls cells were treated with
DMSO vehicle. These results
are from three independent
experiments and each bar
represents standard deviation.
Scale bar 20 lm
Fig. 5 mRNA (a, b, c) and
protein expression changes
(d) in KLF17, Sp1, and Id-1 in
breast cancer cells. After 24 h
of HCA (100 nM) treatment,
mRNA and protein expression
were evaluated using real-time
PCR and western blot analyses,
respectively. These results are
from three independent
experiments and each bar
represents standard deviation.
Statistical significance was
calculated using the student’s
t test (*p \ 0.01)
Breast Cancer Res Treat (2013) 137:697–708 705
123

affected by HCA (data not shown), demonstrating that HCA
administration effectively suppresses lung metastatic
potential without inducing systemic toxicity in vivo. Further,
HCA treatment was effective to reduce the micrometastasis
under histological examination (Fig. 6c, d, p \ 0.01). High
power examination of metastatic nodules showed similar
histologic findings among the group, suggesting that HCA
treatment prevents metastatic potential of cancer cells from
primary site rather than affect lung parenchyme of the
microenvironment.
Discussion
Breast cancer is the most common malignant disease in
women and is responsible for 23 % of all female cancers
worldwide. The main cause of cancer death is not the
primary tumor but its metastases at distal sites [35]. EMT is
critical for the development of cells with a mesenchymal
phenotype from the epithelial phenotype, which is neces-
sary to induce highly aggressive cancers [36]. Cinnamal-
dehyde and its derivatives inhibit cancer cell proliferation
and have toxic effects in several cancer cells [28, 37–40].
One recent study showed that cinnamon extract reduced
cell migration in human cervical cancer cell via the
downregulation of MMP-2 mRNA [41] or by blocking
VEGFR2 kinase and its downstream signaling [42]. Fur-
thermore, cinnamic acid was reported to reduce the inva-
sive capacity of melanoma cells via downregulation of
MMP-2 [43]. In contrast, it has been reported that cinna-
maldehyde increases Langerhans cell migration via the
upregulation of CXCL12 [44].
We investigated the effect of HCA on breast cancer cell
invasion and metastasis and found that, at subtoxic doses
(100 nM), both HCA and BCA inhibit breast cancer cell
invasion or EMT by upregulating the expression of E-cad-
herin. We investigated the molecular mechanism(s) by
which HCA inhibits breast cancer EMT and cell invasion.
HCA downregulates the Snail protein, which acts as a
transcriptional repressor of E-cadherin, and thus upregulates
E-cadherin promoter activity. HCA-induced Snail protein
downregulation seems to be associated with GSK3-bnuclear localization and Snail nuclear exclusion. This is
consistent with the fact that nuclear GSK3-b phosphorylates
Snail and thereby induces its nuclear export, which is fol-
lowed by Snail protein degradation [45–47].
Fig. 6 The effect of HCA on lung metastasis of breast cancer tissue
transplanted into the MPF of nude mice. Mice that had been injected
orthotopically with MDA-MB-231 cells received daily treatment with
HCA (50 mg/kg) or vehicle for 35 days. Doxorubicin (2 mg/kg) was
administered by intraperitoneal injection once per 3 days for the
same period. After the primary tumors were removed, the number
of metastases was evaluated by lung autopsy (a, b), (**p \ 0.01).
A representative histological finding of metastatic lung nodules
(c) under low magnification (upper panels) and high magnification
(lower panels). Small rectangular squares indicate micrometastatic
tumor nodules in lung parenchyme Scale bar 50 lm. Number of
micrometastsis in mouse lung in each groups (d) revealed HCA also
inhibits metastasis of the breast cancer cells (*p \ 0.05)
706 Breast Cancer Res Treat (2013) 137:697–708
123

Previous studies implicated that EGF can mediate EMT
in breast cancer cells [48] and other carcinomas [49, 50]. In
the present study, we treated EGF and/or HCA, and eval-
uated EMT markers expression and morphological change
to confirm the effect of HCA on the EMT of MCF-7 cell.
EGF treatment induced a mesenchymal phenotypes such
as increased motility and upregulation of mesenchymal
markers. Interestingly, EGF and HCA cotreatment restored
the epithelial phenotype from EGF-induced mesenchymal
cell shape. In addition, HCA cotreatment attenuated
upregulation of mesenchymal markers, supporting the
inhibitory effect of HCA on EMT in breast cancer cells.
Id-1 is one of the key target proteins that increases breast
cancer cell invasion [20], and it is upregulated by the tran-
scription factor SP1 [18]. Meanwhile, KLF17 acts as a
transcriptional repressor for Id-1, and its activity results in
the downregulation of breast cancer cell invasion or EMT
[25]. Therefore, we investigated the effect of HCA on Id-1,
KLF17, and SP1 expression. Interestingly, HCA induces a
significant increase in KLF17 mRNA and protein expression
in breast cancer cells. At the same time, HCA effectively
decreased SP1 and Id-1 transcription and protein levels,
suggesting that HCA inhibits breast cancer invasion via the
inhibition of Id-1 through the upregulation of KLF17 and the
downregulation of SP1. It has been reported that both Id-1
and Snail are overexpressed in several types of cancers,
particularly in highly invasive cells and tissues [18, 51].
Furthermore, Id-1 mRNA and protein expression are
increased in Snail-overexpressing MDCK cells, suggesting
that Id-1 is a downstream target of Snail [18].
In conclusion, we report for the first time that sub-toxic
doses of HCA have anti-metastastic effects on breast can-
cer and inhibit EMT. HCA exerts its EMT inhibitory
activity by downregulating Snail protein activity, which in
turn promotes the upregulation of E-cadherin promoter
activity. Furthermore, HCA inhibits breast cancer invasion
in part via the inhibition of the Sp1/Id-1 signaling pathway
by KLF17 upregulation.
Acknowledgments This work was supported by the Basic Science
Research Program through the National Research Foundation of
Korea Grant funded by the Korean Government (2009-0070462). This
research was supported by the Bio & Medical Technology Devel-
opment Program of the National Research Foundation funded by the
Korean government (2012M3A9C404877).
Disclosures None.
References
1. Kwon BM, Cho YK, Lee SH, Nam JY, Bok SH, Chun SK et al
(1996) 20-Hydroxycinnamaldehyde from stem bark of cinnamo-
mum cassia. Planta Med 62:183–184
2. Han DC, Lee MY, Shin KD, Jeon SB, Kim JM, Son KH et al (2004)
20-benzoyloxycinnamaldehyde induces apoptosis in human carci-
noma via reactive oxygen species. J Biol Chem 279:6911–6920
3. Kwon BM, Lee SH, Choi SU, Park SH, Lee CO, Cho YK et al
(1998) Synthesis and in vitro cytotoxicity of cinnamaldehydes to
human solid tumor cells. Arch Pharm Res 21:147–152
4. Lee CW, Hong DH, Han SB, Park SH, Kim HK, Kwon BM et al
(1999) Inhibition of human tumor growth by 20-hydroxy- and 20-benzoyloxycinnamaldehydes. Planta Med 65:263–266
5. Moon EY, Lee MR, Wang AG, Lee JH, Kim HC, Kim HM et al
(2006) Delayed occurrence of H-ras12V-induced hepatocellular
carcinoma with long-term treatment with cinnamaldehydes. Eur J
Pharmacol 530:270–275
6. Lee K, Kwon BM, Kim K, Ryu J, Oh SJ, Lee KS et al (2009)
Plasma pharmacokinetics and metabolism of the antitumour drug
candidate 20-benzoyloxycinnamaldehyde in rats. Xenobiotica 39:
255–265
7. Steeg PS (2006) Tumor metastasis: mechanistic insights and
clinical challenges. Nat Med 12:895–904
8. Steeg PS (2007) Cancer: micromanagement of metastasis. Nature
449:671–673
9. Eccles SA, Welch DR (2007) Metastasis: recent discoveries and
novel treatment strategies. Lancet 369:1742–1757
10. Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks
PB (2009) Tumour-initiating cells: challenges and opportunities
for anticancer drug discovery. Nat Rev Drug Discov 8:806–823
11. Sreekumar R, Sayan BS, Mirnezami AH, Sayan AE (2011)
MicroRNA Control of Invasion and Metastasis Pathways. Front
Genet 2:58
12. Nieto MA (2002) The snail superfamily of zinc-finger tran-
scription factors. Nat Rev Mol Cell Biol 3:155–166
13. Xu C, Kim NG, Gumbiner BM (2009) Regulation of protein stability
by GSK3 mediated phosphorylation. Cell Cycle 8:4032–4039
14. Lim SO, Kim H, Jung G (2010) p53 Inhibits tumor cell invasion
via the degradation of snail protein in hepatocellular carcinoma.
FEBS Lett 584:2231–2236
15. Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE et al (2011)
A p53/miRNA-34 axis regulates Snail1-dependent cancer cell
epithelial-mesenchymal transition. J Cell Biol 195:417–433
16. Zannetti A, Del Vecchio S, Romanelli A, Scala S, Saviano M, Cali
G et al (2005) Inhibition of Sp1 activity by a decoy PNA-DNA
chimera prevents urokinase receptor expression and migration of
breast cancer cells. Biochem Pharmacol 70:1277–1287
17. Hung WC, Chang HC (2009) Indole-3-carbinol inhibits Sp1-
induced matrix metalloproteinase-2 expression to attenuate
migration and invasion of breast cancer cells. J Agric Food Chem
57:76–82
18. Jorda M, Vinyals A, Marazuela A, Cubillo E, Olmeda D, Valero
E et al (2007) Id-1 is induced in MDCK epithelial cells by
activated Erk/MAPK pathway in response to expression of the
Snail and E47 transcription factors. Exp Cell Res 313:2389–2403
19. Zhang X, Ling MT, Wang Q, Lau CK, Leung SC, Lee TK et al
(2007) Identification of a novel inhibitor of differentiation-1 (ID-
1) binding partner, caveolin-1, and its role in epithelial-mesen-
chymal transition and resistance to apoptosis in prostate cancer
cells. J Biol Chem 282:33284–33294
20. Fong S, Itahana Y, Sumida T, Singh J, Coppe JP, Liu Y et al (2003)
Id-1 as a molecular target in therapy for breast cancer cell invasion
and metastasis. Proc Natl Acad Sci USA 100:13543–13548
21. Darnel AD, Wang D, Ghabreau L, Yasmeen A, Sami S, Akil N
et al (2010) Correlation between the presence of high-risk human
papillomaviruses and Id gene expression in Syrian women with
cervical cancer. Clin Microbiol Infect 16:262–266
22. Ding Y, Wang G, Ling MT, Wong YC, Li X, Na Y et al (2006)
Significance of Id-1 up-regulation and its association with EGFR
in bladder cancer cell invasion. Int J Oncol 28:847–854
Breast Cancer Res Treat (2013) 137:697–708 707
123

23. Li B, Tsao SW, Li YY, Wang X, Ling MT, Wong YC et al (2009)
Id-1 promotes tumorigenicity and metastasis of human esopha-
geal cancer cells through activation of PI3K/AKT signaling
pathway. Int J Cancer 125:2576–2585
24. Cheung PY, Yip YL, Tsao SW, Ching YP, Cheung AL (2011) Id-
1 induces cell invasiveness in immortalized epithelial cells by
regulating cadherin switching and Rho GTPases. J Cell Biochem
112:157–168
25. Gumireddy K, Li A, Gimotty PA, Klein-Szanto AJ, Showe LC,
Katsaros D et al (2009) KLF17 is a negative regulator of epi-
thelial-mesenchymal transition and metastasis in breast cancer.
Nat Cell Biol 11:1297–1304
26. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY
et al (2008) The epithelial-mesenchymal transition generates cells
with properties of stem cells. Cell 133:704–715
27. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg
RA et al (2009) Identification of selective inhibitors of cancer
stem cells by high-throughput screening. Cell 138:645–659
28. Cabello CM, Bair WB 3rd, Lamore SD, Ley S, Bause AS, Azi-
mian S et al (2009) The cinnamon-derived Michael acceptor
cinnamic aldehyde impairs melanoma cell proliferation, inva-
siveness, and tumor growth. Free Radic Biol Med 46:220–231
29. Liang CC, Park AY, Guan JL (2007) In vitro scratch assay: a
convenient and inexpensive method for analysis of cell migration
in vitro. Nat Protoc 2:329–333
30. Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK,
Choi YJ, Kim J, Fearon ER, Weiss SJ (2006) A Wnt-Axin2-
GSK3beta cascade regulates Snail1 activity in breast cancer cells.
Nat Cell Biol 8:1398–1406
31. Livak KJ, Schmittgen TD (2001) Analysis of relative gene
expression data using real-time quantitative PCR and the 2(-Delta
Delta C(T)) method. Methods 25:402–408
32. Fan J, Ren H, Fei E, Jia N, Ying Z, Jiang P et al (2008) Su-
moylation is critical for DJ-1 to repress p53 transcriptional
activity. FEBS Lett 582:1151–1161
33. Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL,
Hortobagyi GN, Hung MC (2007) Epidermal growth factor
receptor cooperates with signal transducer and activator of tran-
scription 3 to induce epithelial-mesenchymal transition in cancer
cells via up-regulation of TWIST gene expression. Cancer Res
67:9066–9076
34. Vergara D, Valente CM, Tinelli A, Siciliano C, Lorusso V,
Acierno R, Giovinazzo G, Santino A, Storelli C, Maffia M (2011)
Resveratrol inhibits the epidermal growth factor-induced epithe-
lial mesenchymal transition in MCF-7 cells. Cancer Lett 310:1–8
35. Weigelt B, Peterse JL, van ‘t Veer LJ (2005) Breast cancer
metastasis: markers and models. Nat Rev Cancer 5:591–602
36. Arias AM (2001) Epithelial mesenchymal interactions in cancer
and development. Cell 105:425–431
37. Ren Z, Kang YH, Shi ZY, Huang-Fu CS, Hu GQ, Liu B (2010)
Cinnamaldehyde of loxacin-3-ylhydrazone induces apoptosis of
human hepatocarcinoma SMMC-7721 cells. Yao Xue Xue Bao
45:1109–1115
38. Kwon HK, Hwang JS, So JS, Lee CG, Sahoo A, Ryu JH et al
(2010) Cinnamon extract induces tumor cell death through inhi-
bition of NFkappaB and AP1. BMC Cancer 10:392
39. Lee CW, Lee SH, Lee JW, Ban JO, Lee SY, Yoo HS et al (2007)
2-Hydroxycinnamaldehyde inhibits SW620 colon cancer cell
growth through AP-1 inactivation. J Pharmacol Sci 104:19–28
40. Ka H, Park HJ, Jung HJ, Choi JW, Cho KS, Ha J et al (2003)
Cinnamaldehyde induces apoptosis by ROS-mediated mito-
chondrial permeability transition in human promyelocytic leu-
kemia HL-60 cells. Cancer Lett 196:143–152
41. Koppikar SJ, Choudhari AS, Suryavanshi SA, Kumari S,
Chattopadhyay S, Kaul-Ghanekar R (2010) Aqueous cinnamon
extract (ACE-c) from the bark of Cinnamomum cassia causes
apoptosis in human cervical cancer cell line (SiHa) through loss
of mitochondrial membrane potential. BMC Cancer 10:210
42. Lu J, Zhang K, Nam S, Anderson RA, Jove R, Wen W (2010)
Novel angiogenesis inhibitory activity in cinnamon extract blocks
VEGFR2 kinase and downstream signaling. Carcinogenesis
31:481–488
43. Liu L, Hudgins WR, Shack S, Yin MQ, Samid D (1995) Cin-
namic acid: a natural product with potential use in cancer inter-
vention. Int J Cancer 62:345–350
44. Ouwehand K, Santegoets SJ, Bruynzeel DP, Scheper RJ, de
Gruijl TD, Gibbs S (2008) CXCL12 is essential for migration of
activated Langerhans cells from epidermis to dermis. Eur J
Immunol 38:3050–3059
45. Ko H, Kim HS, Kim NH, Lee SH, Kim KH, Hong SH et al (2007)
Nuclear localization signals of the E-cadherin transcriptional
repressor Snail. Cells Tissues Organs 185:66–72
46. Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ (2005) Wnt-
dependent regulation of the E cadherin repressor snail. J Biol
Chem 280:11740–11748
47. Yang Z, Rayala S, Nguyen D, Vadlamudi RK, Chen S, Kumar R
(2005) Pak1 phosphorylation of snail, a master regulator of epi-
thelial-to-mesenchyme transition, modulates snail’s subcellular
localization and functions. Cancer Res 65:3179–3184
48. Hardy KM, Booth BW, Hendrix MJ, Salomon DS, Strizzi L
(2010) ErbB/EGF signaling and EMT in mammary development
and breast cancer. J Mammary Gland Biol Neoplasia 15:191–199
49. Gan Y, Shi C, Inge L, Hibner M, Balducci J, Huang Y (2010)
Differential roles of ERK and Akt pathways in regulation of
EGFR-mediated signaling and motility in prostate cancer cells.
Oncogene 29:4947–4958
50. Cai Z, Zhou Y, Lei T, Chiu JF, He QY (2009) Mammary serine
protease inhibitor inhibits epithelial growth factor-induced epi-
thelial-mesenchymal transition of esophageal carcinoma cells.
Cancer 115:36–48
51. Li J, Jia H, Xie L, Wang X, He H, Lin Y, Hu L (2009) Correlation
of inhibitor of differentiation 1 expression to tumor progression,
poor differentiation and aggressive behaviors in cervical carci-
noma. Gynecol Oncol 114:89–93
708 Breast Cancer Res Treat (2013) 137:697–708
123