discrete rna libraries from pseudo-torsional space
TRANSCRIPT

doi:10.1016/j.jmb.2012.03.002 J. Mol. Biol. (2012) 421, 6–26
Contents lists available at www.sciencedirect.com
Journal of Molecular Biologyj ourna l homepage: ht tp : / /ees .e lsev ie r.com. jmb
Discrete RNA Libraries from Pseudo-Torsional Space
Elisabeth Humphris-Narayanan 1 and Anna Marie Pyle 1, 2⁎1Department of Molecular, Cellular and Developmental Biology and Department of Chemistry, Yale University,New Haven, CT 06520, USA2Howard Hughes Medical Institute, Chevy Chase, MD 20815, USA
Received 20 December 2011;received in revised form28 February 2012;accepted 6 March 2012Available online13 March 2012
Edited by M. F. Summers
Keywords:RNA structure;RNA backbone conformation;RNA fragment library;RNA modeling
*Corresponding author. DepartmenCellular and Developmental BiologyChemistry, Yale University, New HAbbreviations used: 2-D, two-dim
deformation index.
0022-2836/$ - see front matter © 2012 E
The discovery that RNA molecules can fold into complex structures andcarry out diverse cellular roles has led to interest in developing tools formodeling RNA tertiary structure. While significant progress has been madein establishing that the RNA backbone is rotameric, few libraries of discreteconformations specifically for use in RNA modeling have been validated.Here, we present six libraries of discrete RNA conformations based on asimplified pseudo-torsional notation of the RNA backbone, comparable tophi and psi in the protein backbone. We evaluate the ability of each libraryto represent single nucleotide backbone conformations, and we show howindividual library fragments can be assembled into dinucleotides that areconsistent with established RNA backbone descriptors spanning from sugarto sugar. We then use each library to build all-atom models of 20 test folds,and we show how the composition of a fragment library can limit modelquality. Despite the limitations inherent in using discretized libraries, wefind that several hundred discrete fragments can rebuild RNA folds up to174 nucleotides in length with atomic-level accuracy (b1.5 Å RMSD). Weanticipate that the libraries presented here could easily be incorporated intoRNA structural modeling, analysis, or refinement tools.
© 2012 Elsevier Ltd. All rights reserved.
Introduction
The cellular role of RNA is now known to extendfar beyond simple transfer of genetic information toinclude catalysis, molecular recognition, and geneticcontrol.1 Thus, RNA can act as a folded macromol-ecule with striking parallels to proteins.2 Knowl-edge of RNA three-dimensional structure cantherefore be critical for understanding the structuralmechanisms involved in RNA conformationalchanges, ligand and protein binding, and catalysis.Despite the growing interest in RNA tertiarystructure, the development and success of compu-tational tools for RNA structural modeling have
t of Molecular,and Department of
aven, CT 06520, USA.ensional; DI,
lsevier Ltd. All rights reserve
lagged behind the counterpart tools for proteins.This is in part due to the difficulty in determiningexperimental RNA structures with sufficiently highresolution and is in part due to the complexityinherent in the six torsional degrees of freedomwithin the RNA backbone of each nucleotide (Fig.1a). In the last two decades, the number, diversity,and quality of solved RNA structures have growntremendously, and this has allowed the structuralfeatures of the RNA backbone3–8 and bases9,10 to beanalyzed in great detail.Recently, several groups identified rotameric back-
bone conformations that occur repeatedly withinRNA structures.5–8 Rotameric conformations havelong been observed for the torsion angles of smallmolecules, as well as for the torsions of the proteinbackbone11 and side chains.12 The discovery thatprotein side chains have strong torsional preferencesled to the development of protein rotamer librariesthat have been used with great success in molecularmodeling for prediction and design13,14 or structuralvalidation.15 Initially, protein rotamer libraries
d.

C4’i+1
Pi+1
C4’i-1C3’i-1O3’i-1
C5’i+1
O5’i+1
(a)
(b)
C4’iPi
C4’i+1
Pi+1
C4’i-1θ
SuiteConformer
Fig. 1. Using pseudo-torsions to reduce RNA backbonedimensionality. (a) A nucleotide, with its six standardbackbone torsions labeled, is depicted in black stick atoms.A suite, which spans from sugar to sugar and comprisesseven torsions, is also denoted. Atoms defined to be partof a filtered fragment, which include the O3′, C3′, and C4′atoms of the preceding nucleotide and the P, O5′, C5′, andC4′ atoms of the following nucleotide, are also shown instick. (b) Two pseudo-torsions (black arrows) per filteredfragment are created by forming pseudo-bonds betweenconsecutive C4' and phosphorus atoms along the RNAbackbone (black lines and spheres, respectively). The tworesulting pseudo-torsions are named eta, η [C4′i− 1, Pi,C4′i, Pi+1 ], and theta, θ, [Pi, C4′i, Pi+1, C4′i+1 ].
7Discrete RNA libraries from pseudo-torsional space
consisted of a limited number of idealized side-chainconformations that were understood to representlocal minima on the potential energy surface.16,17
However, several studies suggested that early rota-mer libraries were incomplete, and as a result,expanded protein rotamer libraries were developedthat consisted of hundreds, or even thousands, ofside-chain conformations.18–21 The conformationswithin these libraries typically consisted of side chainstaken directly from high-resolution crystal structuresand therefore did not always correspond to localenergy minima. Nevertheless, the larger rotamerlibraries were shown to be superior to earlier librariesin achieving accuracy in protein modeling.18–21
A consensus RNA backbone rotamer library of 46conformations was recently published8 that incor-porates and builds upon several earlier RNArotamer libraries.5–7 While the consensus library
represents a significant achievement in terms ofquantitatively describing RNA backbone structure,incorporating the consensus library into modelingtools that build RNA structure may present a uniquechallenge. This is because each rotameric backbonestate is defined in terms of a new unit of RNAstructure, termed a “suite” (Fig. 1a).7,8 A suiteconsists of seven backbone torsions (δ, ɛ, ζ, α, β, γ,and δ) and spans two nucleotide sugars (Fig. 1a).The suite notation is straightforward to use forstructure quality assessment.22 However, becauseeach suite both begins and ends with a sugar ring,assembling individual suites into larger RNAstructures can be difficult. Whereas two traditionalnucleotides can be joined at a single phosphateatom, joining two suites requires that they overlapcompletely by one sugar ring. Therefore, a minimi-zation protocol would need to resolve any poten-tially differing sugar conformations resulting fromoverlapping suites. While a local minimization stepcould be incorporated into rotamer-based modelingtools, doing so could undercut advantages incomputational speed that normally would be gainedfrom using a purely rotamer-based approach.To date, no finite list of representatives of the 46
suites within the consensus rotamer set has beenpublished. Thus, to use the suite notation duringmodeling building, a protocol is needed to selectamong the many different possible conformationsthat could simultaneously satisfy the ranges of sevenbackbone torsions involved in each of the consensussuites. Keating and Pyle recently illustrated the onlytechnique available thus far to combine suitesduring model building. Their protocol requiresthat a backbone trace is known in advance andthen uses coordinate minimization to generate suiteconformations compatible with the preexistingbackbone trace.23 In this work, we provide analternative approach to using the consensus setduring model building. Specifically, we presentseveral discrete libraries of RNA conformationsthat are easy to combine and do not requirecoordinate minimization or a preexisting backbonetrace. The libraries we present should be ideal foruse in de novo modeling tools that employ pairwisedecomposable energy functions or require discreterotamers. However, they could also be useful as astarting point for modeling approaches that employconformational minimization.Instead of a rotamer set,most tools thatmodel RNA
either employ a coarse-grainedmodeling approach ormake use of large databases of RNA fragments (for areview, see Ref. 24). In coarse-grainedmodeling, low-resolution models are generated by representing eachnucleotide in a highly reduced form, typically as oneor more spheres.25–27 Coarse-grained modeling canafford large advantages in speed, especially formodeling larger RNA folds.25–27 However, a secondround of computational prediction is required to

8 Discrete RNA libraries from pseudo-torsional space
produce all-atom models from coarse-grainedtraces.28 In contrast, all-atom RNA models are oftenbuilt using either groups of base pairs29,30 or three-nucleotide-long fragments taken from a single ribo-somal subunit structure.31 Fragment-based structureassembly has successfully generated models of small-and medium-sized RNA molecules with backboneaccuracies of 2–10 Å.29–31 However, it is currently
(a)
0 60 120 180 240 300 360
0
60
120
180
240
300
360
0
60
120
180
240
300
360
0 60 120 180 240 300 360
Helical
Non-Helical
Helical
(e)
(f)
θ
θ
(b)
0 60 120
180
240
300
0
60
120
180
240
300
3600 60 120
180
240
300
0
60
120
180
240
300
360
C3 Hel
C3Non-H
C2
Total LLibrary S
Tottal Bins
I II
III
IV
Fig. 2. Using pseudo-torsions to generate filtered fragmentof quality-filtered RNA nucleotides (see Materials and Methotorsions are shown for C3′-endo (top) and C2-endo (bottom)depict ranges of eta (150bηb190) and theta (190bθb260) assoof nucleotides previously associated with kink-turn and π-turare denoted by I, II, and III, respectively (bottom).29 The clusadenosine platforms as well as the second position of π-turnswere generated by binning pseudo-torsional space, separated bRNA extended nucleotide with pseudo-torsions closest to the c(c) (green dots), and 10° (d) (brown dots) libraries is shown. (e)10° library. All fragments have a helical η torsion but differ in wbase placement of the central nucleotide is often similar for all frequired to define each fragment's pseudo-torsions are depictefragment pseudo-bonds are shown as spheres. (f) The total sibinning and the relative number of fragments helical in η or θbins created by each grid is also given.
unknown what limitations these fragment librariescurrently have. It is possible that some fragmentlibraries may overrepresent certain RNA structuralfeatures, such as helical regions, but completely lackappropriate representatives for others.In this work, we aim to develop libraries of
discrete conformations (“filtered fragment librar-ies”) that exhaustively span RNA conformational
(c) (d)
0 60 120
180
240
300
360
0
60
120
180
240
300
360
0 60 120
180
240
300
360
0
60
120
180
240
300
3600 60 120
180
240
300
360
0
60
120
180
240
300
360
0 60 120
180
240
300
360
0
60
120
180
240
300
360
360
360
60° 30° 20° 15° 10° 5°
ical 11 40 74 103 170 304
elical 23 39 53 57 63 73
33 81 115 136 169 200
ize 67 160 242 296 402 577
72 228 648 1152 2592 10,368
libraries. (a) Pseudo-torsions were measured for a data setds) and plotted in a Ramachandran-like manner. Pseudo-nucleotides separately. Horizontal and vertical gray barsciated with nucleotides in a helical conformation. Clustersn motifs, asymmetrical internal loops, or S1 and S2 motifster of nucleotides denoted as IV includes the 5′-halves ofand Ω-turns (bottom).29 (b–d) Filtered fragment librariesy sugar pucker, at varying degrees and selecting the singleenter of each bin. Construction of the 60° (b) (blue dots), 30°Example C3′-endo fragment representatives, taken from thehether their θ torsion is helical or non-helical. Note that theragments shown, regardless of pseudo-torsions. All atomsd in gray stick form, and the C4′ and P atoms defining theze of each of the six libraries created by pseudo-torsionalare given. For reference, the number of pseudo-torsional

9Discrete RNA libraries from pseudo-torsional space
space, are easy to assemble without implementationof minimization protocols, and are consistent withthose already identified using the more comprehen-sive suite notation. To generate the filtered fragmentsets, we use a pseudo-torsional notation that mimicsthe phi–psi notation of the protein backbone.32–34 Toform the pseudo-torsions, consecutive RNA back-bone C4′ and phosphate atoms are linked withvirtual bonds (Fig. 1b). This creates two pseudo-torsions per RNA nucleotide: η [C4′i− 1, Pi, C4′i,Pi+1] and θ [Pi, C4′i, Pi+1, C4′i+1] (arrows in Fig. 1b).The RNA pseudo-torsion nomenclature might beideal for generating libraries of RNA conformationsfor several reasons. First, nucleotides with similar ηand θ values are often found within the same unitsof tertiary structure,32 and these values can be usedto identify known or novel structural motifs withinexisting RNA structures.35,36 Second, small motifs ofRNA, such as the GNRA tetra-loop, can be rebuiltwith high accuracy by replacing native nucleotidesin silico with non-tetra-loop nucleotides that havesimilar pseudo-torsions.33 Finally, when nucleotidepseudo-torsions are plotted in two-dimensional(2-D) space, their associated RNA backbone confor-mations appear to cluster.32,33 Importantly, theclustering of nucleotide pseudo-torsions in two-dimensions has recently been shown to correspondto the clusters of the RNA backbone suites observedin seven-dimensions.23
The accuracy of protein modeling had generallyimproved after expanded rotamer libraries wereintroduced.18–21 Thus, we created six libraries ofRNA filtered fragments that varied in size fromsmall (∼70 fragments) to large (∼500 fragments),and then we examined how the accuracy of RNAmodeling depended on the choice of library used.The various libraries were constructed by using thepseudo-torsional notation to select representativesdirectly from a data set of high-quality crystallo-graphic structures with η and θ values spaced every60°, 30°, 20°, 15°, 10°, or 5°. While each library wascreated using a coarse-grained approach based ononly two atoms per nucleotide (C4′ and P, seeFig. 1), each individual library conformationretained all-atom detail. As the word rotamer istypically reserved for ideal conformations located atthe bottom of a local energy minimum, we refer tothe members of each of the libraries of discrete RNAconformations as “filtered fragments.”Here, we first briefly describe the features of the
six libraries, and then we present methodologicalrules for connecting the fragments into dinucleo-tides that are consistent with the previously pub-lished suite nomenclature. We evaluate theperformance of each library at modeling singlenucleotides, dinucleotides, and entire RNA folds,and we find that the modeling accuracy at allstructural levels is dependent on the filteredfragment library used. Importantly, we find that
fewer than several hundred well-chosen fragmentsare sufficient to build models of RNA folds withatomic-level accuracy. These sets of pseudo-torsion-based libraries are small enough to ensure speed andefficiency for modeling tools but large enough tomodel RNA folds with high accuracy. Thus, weanticipate that the pseudo-torsion-based librarieswill be of use in the future for a wide variety ofmodeling applications.
Results
Generation of filtered libraries ofpseudo-torsional fragments
The same data set of 171 high-quality crystallo-graphic structures that was used to identify clustersof RNA backbone torsions in seven-dimensions7
(“RNA05”; see Materials and Methods) was alsoused to generate the pseudo-torsion-based libraries.As in Ref. 7, we eliminated from the data setnucleotides with high atomic B-factors or residueswith steric clashes (see Materials and Methods).Further, to ensure that only the highest-qualitynucleotides were included within the libraries, weremoved nucleotides that had poorly defined sugarpuckers or that lacked all necessary pseudo-torsion-al atoms (see Fig. 1a and Materials and Methods).Once the data set of high-quality filtered nucleo-
tides was created, we first surveyed the range ofpseudo-torsions present by measuring the η and θvalues of each filtered nucleotide (see Materials andMethods and Fig. 1b) and plotting these valuesagainst each other in a 2-D, Ramachandran-likescatter plot (Fig. 2a). In keeping with precedent setin other studies,32,33 nucleotides were first groupedby sugar pucker (see Materials and Methods), andtwo η–θ plots were created: one η–θ plot fornucleotides with C3′-endo sugar pucker (Fig. 2a,top) and a second plot for nucleotides with C2′-endo(Fig. 2a, bottom) sugar pucker.Not surprisingly, the η–θ plots of the filtered
RNA05 data set were remarkably similar to η–θplots observed in an early analysis of pseudo-torsions within a small set of 52 structures.32 Mostnotably, a large number of C3′-endo nucleotides hadη–θ values within a very narrow range that waspreviously associated with the helical A-form ofRNA32,33 (Fig. 2a, gray regions; 150bηb190;190bθb240). The η–θ plots of the filtered RNA05data set were also roughly the same as η–θ plotsgenerated after applying an automated clusteringalgorithm to a larger set of approximately 7000unfiltered crystallographic nucleotides.33 Interest-ingly, the regions of scatter removed by automati-cally clustering nucleotides based on the similarityof their η–θ values33 appeared similar to the regions
RMSD(Angst
Den
sity
0.0 0.5
01
23
45
6
Den
sity
0.0 0.5
01
23
45
6
RMSD(Angstroms)
Den
sity
0.0 0.5 1.0 1.5
01
23
45
6
.5
.5
RMSD(Angstroms)
Den
sity
0.0 0.5 1.0 1.5
01
23
(a)
ms
ms
02,1174,2336,3508,466
<0
02,1174,2336,3508,466
<0
02,117
<0.5 Angstroms0
2,1174,2336,3508,466
<0.5 Angstroms
(b)
RMSD(Angst
Den
sity
0.0 0.5
01
23
45
6
RMSD(Angst
Den
sity
0.0 0.5
01
23
45
6
RMSD(Angstroms)D
ensi
ty
0.0 0.5 1.0 1.5
01
23
45
6
.5
.5
RMSD(Angstroms)
Den
sity
0.0 0.5 1.0 1.5
01
23
ms
ms
02,1174,2336,3508,466
<0
02,1174,2336,3508,466
<0
02,117
<0.5 Angstroms0
2,1174,2336,3508,466
<0.5 Angstroms
6453
6633
4349
5773
67206331
6255
6440
4106
5557
65865995
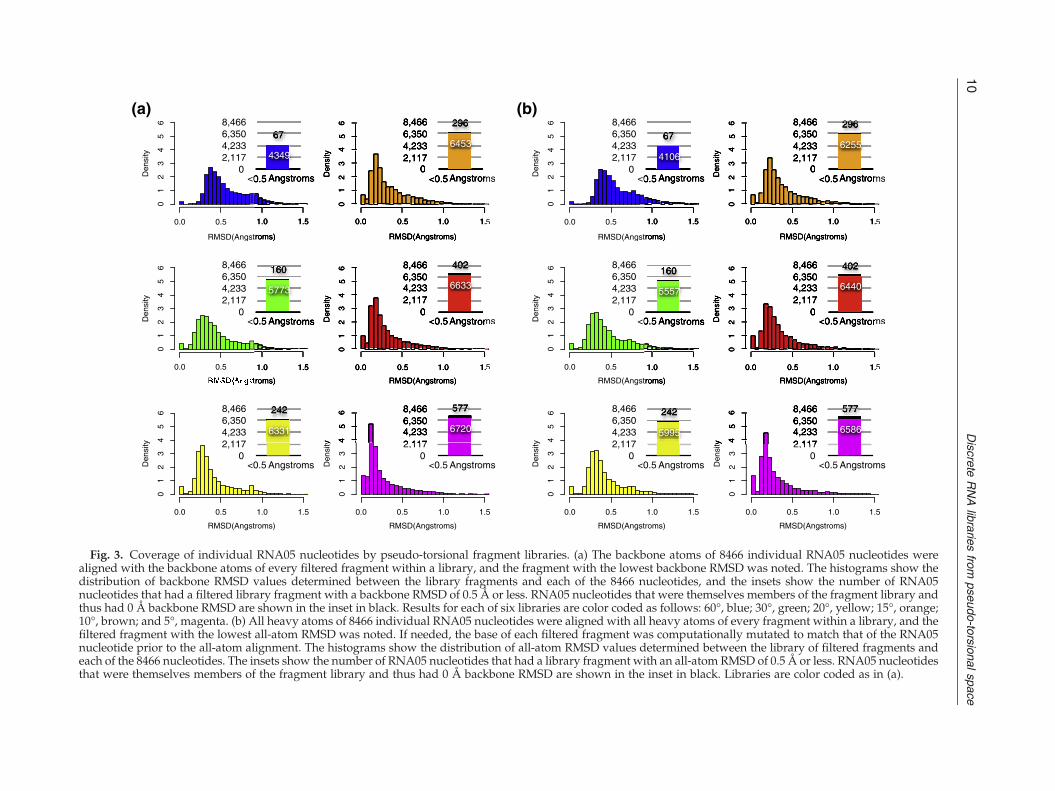
Fig. 3. Coverage of individual RNA05 nucleotides by pseudo-torsional fragment libraries. (a) The backbone atoms of 8466 individual RNA05 nucleotides werealigned with the backbone atoms of every filtered fragment within a library, and the fragment with the lowest backbone RMSD was noted. The histograms show thedistribution of backbone RMSD values determined between the library fragments and each of the 8466 nucleotides, and the insets show the number of RNA05nucleotides that had a filtered library fragment with a backbone RMSD of 0.5 Å or less. RNA05 nucleotides that were themselves members of the fragment library andthus had 0 Å backbone RMSD are shown in the inset in black. Results for each of six libraries are color coded as follows: 60°, blue; 30°, green; 20°, yellow; 15°, orange;10°, brown; and 5°, magenta. (b) All heavy atoms of 8466 individual RNA05 nucleotides were aligned with all heavy atoms of every fragment within a library, and thefiltered fragment with the lowest all-atom RMSD was noted. If needed, the base of each filtered fragment was computationally mutated to match that of the RNA05nucleotide prior to the all-atom alignment. The histograms show the distribution of all-atom RMSD values determined between the library of filtered fragments andeach of the 8466 nucleotides. The insets show the number of RNA05 nucleotides that had a library fragment with an all-atom RMSD of 0.5 Å or less. RNA05 nucleotidesthat were themselves members of the fragment library and thus had 0 Å backbone RMSD are shown in the inset in black. Libraries are color coded as in (a).
10Discrete
RNA
librariesfrom
pseudo-torsionalspace

C4’i C4’j-1
Pi+1
Pj
C4’i+1
C4’j
C4’i-1
Pj+1
C4’j+1
C4’i
Pi+1
C4’i+1
C4’j-1
Pj
C4’j
+Pj+1
C4’j+1
C4’i-11
θ1
2θ2
C4’i
Pj
C4’jPi
θ1 2
(a) (d)(c)(b)
0
200
400
600
800
1000
50
100
150
200
250
300
350
50
100
150
200
250
300
350
Theta
Eta
0
2
4
6
8
10
50 100
150
200
250
300
350
50
100
150
200
250
300
350
Theta
Eta
0
1
2
3
4
50 100
150
200
250
300
350
50
100
150
200
250
300
350
Theta
Eta
0
2
4
6
8
10
12
14
50
100
150
200
250
300
350
50
100
150
200
250
300
350
Theta
Eta
C3’-endo to C3’-endo C3’-endo to C3’-endo
C3’-endo to C2’-endo C3’-endo to C2’-endo
C3’-endo to C3’-endo
C3’-endo to C2’-endo
C2’-endo to C3’-endo C2’-endo to C3’-endo
C2’-endo to C2’-endo C2’-endo to C2’-endo
C2’-endo to C3’-endo
C2’-endo to C2’-endo
0
50
100
150
200
250
0 50 100 150 200 250 300 350
0
50
100
150
200
250
300
350
Theta
Eta
0
50
100
150
200
250
300
350
0 50 100 150 200 250 300 350
0
50
100
150
200
250
300
350
Theta
Eta
0
50
100
150
200
250
0 50 100 150 200 250 300 350
0
50
100
150
200
250
300
350
Theta
Eta
0
50
100
150
200
250
0 50 100 150 200 250 300 350
0
50
100
150
200
250
300
350
Theta
Eta
0
20
40
60
80
100
120
0 50 100 150 200 250 300 350
0
50
100
150
200
250
300
350
Theta
Eta
0
20
40
60
80
100
120
0 50 100 150 200 250 300 350
0
50
100
150
200
250
300
350
Theta
Eta
0
200
400
600
800
1000
0 50 100 150 200 250 300 350
0
50
100
150
200
250
300
350
Theta
Eta
0
200
400
600
800
1000
0 50 100 150 200 250 300 350
0
50
100
150
200
250
300
350
Theta
Eta
Library Total DinucleotidesSterically Tolerated
DinucleotidesIdentified as
Suites# Suites Identified
Most frequently Identified Suites
60° 4,489 90% 53% 51 2a, 7d, 1a
30° 25,600 90% 53% 52 2[, 1a, 2a
20° 58,564 89% 58% 54 1a, 2a, 7d
15° 72,361 88% 60% 54 1a, 2a, 7d
10° 161,604 88% 63% 54 1a, 2a, 7d
5° 332,929 88% 69% 54 1a, 2a, 1b
RNA05 8,466 N/A 86% 54 1a, 1c, 1b
(e)
Fig. 4. Assembly of pseudo-torsional fragments into dinucleotides. (a) The nucleotides (black atoms) of any two filteredfragments can be connected into dinucleotides by using the extended pseudo-torsional atoms (gray atoms) to guideassembly by orienting one nucleotide relative to another (top). The last three atoms involved in the θ torsion of the firstfragment [C4′i, Pi+1, C4′i+1] are aligned with the first three atoms involved in the η torsion of the second fragment [C4′j− 1,Pj, C4′j] (middle). To connect the two fragments at the adjoining phosphate, a small translation was performed such thatthe overlapping phosphate atoms of the two fragments had identical coordinates. After attachment, the overlappingatoms used in the alignment (gray atoms) are removed and discarded. The connectivity of a dinucleotide can berepresented in shorthand by the combination of θ–η torsions formed (bottom). If a longer stretch of RNA is desired, thelast three extended atoms of the end fragment can be retained and used to add an additional fragment. (b and c) Thefrequency of θ–η torsions within two-nucleotide stretches of the RNA05 data set (b) and the frequency of θ–η torsionswithin in silico dinucleotides assembled from the 10° library (c) are shown, color coded to the scales, in (b) and (c),respectively. Dinucleotides from the 10° library determined to have steric clashes via overlap of van der Waals radii(scaled by 60%, see Materials and Methods) are excluded from the plots in (d). (e) For each filtered fragment library(column 1), the total number of dinucleotides generated (column 2), the percentage of dinucleotides determined to be freeof serious atomic overlaps (column 3), and the percentage of dinucleotides identified by Suitename as a rotameric suite(column 4) are given. The total number, out of 54, of suites identified within the dinucleotides generated from each library(column 5) and the most frequently identified suites (column 6) are also given.
11Discrete RNA libraries from pseudo-torsional space

12 Discrete RNA libraries from pseudo-torsional space
of scatter removed by quality filtering the RNA05nucleotides to eliminate steric clashes, to have lowB-factors, and to have well-defined pseudo-torsionsand sugar pucker (see Materials and Methods andSupplementary Fig. 1).We next sought to create libraries of nucleotides
with pseudo-torsions that spanned, or completelycovered, the range of observed η–θ values.A previousstudy identified 11 pseudo-torsion-based clusters byusing standard clustering techniques.33 However, weemployed a non-clustering-based methodology thatallowed us to systematically generate libraries thatvaried in size but also ensured that representatives ofall 11 previously observed clusters were included ineach filtered fragment library. Specifically, we choseto bin the two filtered η–θ plots at one of six differentresolutions (60°, 30°, 20°, 15°, 10°, or 5°) and selectedfrom each resolution of bins the single nucleotideclosest to the center of each bin (Fig. 2b–d; see alsoMaterials andMethods). If a binwas unpopulated, nonucleotide was selected. In such a manner, one“ideal” representative was chosen from each binand taken to be representative of all other nucleotideswithin the same bin.This binning process created six libraries that
ranged in size from 67 to 577 (Fig. 2f). Becausealmost every 60° and 30° bin was populated (Fig. 2band c), the 60° and 30° libraries contained filteredfragments with fairly uniformly spaced η–θ values(Fig. 2f). In contrast, when the η–θ plots were binnedat 20° or finer, many bins were located within theempty regions of the η–θ plots, which wereunpopulated (Fig. 2d). As a result, the 20°, 15°, 10°,and 5° libraries were significantly smaller in sizethan expected from the total number of bins (Fig. 2f).Further, a large number of the nucleotides within the20°, 15°, 10°, or 5° binswere locatedwithin the helicalη–θ region (Fig. 2d). As a result, the largest fourlibraries did not have evenly spaced η–θ values butinstead were biased toward helical conformations(Fig. 2d and f). As an example, Fig. 2d illustrates theselection of nucleotides using a 10° bin size, and Fig.2e shows 10 filtered fragments, each with helical ηbut varying θ, selected after binning at 10°. Note thateach fragment consists of the backbone and basecoordinates of a single selected nucleotide (Fig. 2e,wheat atoms), as well as the coordinates of all theatoms involved in defining the selected nucleotide'spseudo-torsions (Fig. 2e, gray atoms). By saving theatoms that define the η and θ values of each libraryfragment, the pseudo-torsions of each fragment canbe directly used during model building.
Filtered fragment library accuracy: Modeling thebackbone and bases of individual nucleotides
To build accurate models of RNA folds, a filteredfragment library must reproduce structural featuresthat are found within individual nucleotides. As a
first test of each library, we thus asked howaccurately the filtered fragments within each librarycould reproduce the backbone coordinates of each ofthe 8466 individual nucleotide conformations withinthe original unfiltered RNA05 data set (see Materialsand Methods). To do so, we aligned the backboneatoms of every fragment within each library to thecorresponding backbone atoms of every nucleotidein the RNA05 data set and noted the RMSD over allthe backbone fragment atoms, including thosedefining its η and θ values. We then used thebackbone RMSD calculations to determine which ofthe library fragments was the most structurallysimilar to each individual RNA05 nucleotide.We evaluated the ability of each library to
represent the diversity of backbone conformationswithin the RNA05 data set by counting how manynucleotides had a library fragment with a backboneRMSD within 1 Å or 0.5 Å. Regardless of whichfiltered fragment library was examined, the majorityof RNA05 nucleotides had a library fragment within1 Å backbone RMSD (Fig. 3a). However, the sixlibraries differed in the number of nucleotides with alibrary fragment within 0.5 Å backbone RMSD (Fig.3a, inset). For example, the 60° library modeled thebackbone coordinates of approximately 50% (4349/8466) of the nucleotides to within an accuracy of0.5 Å, while the 30° library reproduced the backbonecoordinates of 68% (5773/8466) of the nucleotides towithin 0.5 Å (Fig. 3a, blue and green). In this case, asmall increase in library size of only approximately100 fragments resulted in a large increase in thenumber of nucleotides modeled to within 0.5 Åaccuracy. The shift toward modeling more RNA05nucleotides with increased backbone accuracy con-tinued for the remaining libraries. Impressively, allfour libraries binned at 20° or finer were able tocover or “mimic” the backbone structure of 75–80%of the RNA05 nucleotides to within 0.5 Å (Fig. 3,yellow, orange, brown, and magenta). This level ofstructural accuracy in modeling individual nucleo-tides is comparable to that typically calculated formany protein side-chain rotamer libraries.18,19
We next evaluated how accurately each librarycould reproduce the full coordinates of all of theunfiltered RNA05 nucleotides, including eachnucleotide's base. To compute all-atom RMSDs, wecomputationally mutated the base of each filteredfragment to match that of each RNA05 nucleotideprior to aligning all heavy atoms (see Materials andMethods). The library fragment with the minimumall-atom RMSD to each nucleotide was then noted.Surprisingly, the accuracy of the library fragmentsin modeling the data set of nucleotides in all-atomdetail was very similar to the accuracy previouslyobserved for modeling only the backbone atoms ofeach nucleotide. Even when the base atoms wereincluded, each of the filtered fragment librariesmodeled the majority of nucleotides to within 1 Å

13Discrete RNA libraries from pseudo-torsional space
accuracy. Further, the largest four libraries modeled70–78% of nucleotides to within 0.5 Å accuracy (Fig.3b). Often, the filtered fragment that “best fit” thecoordinates of an entire nucleotide when calculatingall-atom RMSD was the same library fragment thatbest fit the nucleotide when only backbone RMSDcoordinates had been considered (SupplementaryFig. 2). These results suggest that when a libraryfragment accurately models the backbone atoms of anucleotide, the base atoms of the nucleotide willoften be modeled accurately as well.
Pseudo-torsion-guided assembly of filteredfragments into in silico dinucleotides
We next asked whether the filtered fragmentswithin each library couldbe assembled into physicallyrealistic dinucleotides. Assembling two single nucle-otides into a dinucleotide requires choosing how toplace one nucleotide with respect to another. While a
Backbone RMSD (Angstroms)
Den
sity
Den
sity
Den
sity
Den
sity
(a)
0 1 2 3 4 5 6 7
01
23
45
0 1 2 3 4 5 6 7
01
23
45
0 1 2 3 4 5 6 7
01
23
45
0 1 2 3 4 5 6 7
01
23
45
(b) (cHairpin2ANN23 nts
SAM-III Riboswitch 3E5C52nts
Hammerhead Ribozyme2QUS68 nts
Lysine Riboswitch3DIL
174 nts
Fig. 5. Assembly of pseudo-torsional filtered fragments intoobserved for 1000 models assembled from each of six filteredThe folds shown range in size from 27 to 158 nucleotides, andgreen; 20°, yellow; 15°, orange; 10°, brown; and 5°, magenta. (bcoloring), as well as for the best model observed for each target5° (f) (magenta) libraries, is shown in cartoon format. BackbonFig. 6, and all-atom RMSD values are given in Fig. 7 and Sup
large number of dinucleotide conformations couldtheoretically be formed from a pair of individualnucleotides, we chose to orient and assemble individ-ual library fragments into dinucleotides by using theirpseudo-torsions as a guide (Fig. 4a).Specifically, dinucleotides were formed from two
individual fragments by aligning three of the atomsinvolved in defining the θ torsion of one fragmentwith three of the atoms involved in defining the ηtorsion of a second fragment (see Fig. 4a andMaterials and Methods). In order to form acontiguous dinucleotide, the aligned pseudo-torsionatoms were joined at the phosphate atom, and allpseudo-torsional atoms were removed (see Fig. 4band Materials and Methods). However, for buildingstructures longer than dinucleotides (see Deriving a“lower-limit” estimate of model quality: ModelingRNA folds), the pseudo-torsional atoms at the endsof a joined dinucleotide can remain and be used toguide attachment of the next incoming fragment. As
) (d) (e) (f)
RNA folds. (a) The distribution of backbone RMSD valuesfragment libraries is shown for the four RNA target folds.the distributions are color coded as follows: 60°, blue; 30°,–f) The native fold for each of the four targets (b) (rainbowfrom the 60° (c) (blue), 30° (d) (green), 15° (e) (orange), ande RMSD values for each model to the targets are given inplementary Table 3.

14 Discrete RNA libraries from pseudo-torsional space
a shorthand, we refer to each assembled dinucleo-tide by its θ–η value (red arrows in Fig. 4a, bottom).We evaluated whether assembling filtered frag-
ments into dinucleotides based on their pseudo-torsions built realistic two-nucleotide conformationsin the following manner. To begin, we created alibrary of dinucleotides from each fragment library byconnecting, pairwise, every combination of individu-al fragments in silico using the three-step assemblyprotocol. We then noted the θ–η value of everydinucleotide assembled in silico. To survey theconnectivity of the in silico dinucleotides, we separat-ed the dinucleotides by sugar pucker and plotted howfrequently each pair of θ–η values occurredwithin theset of in silicodinucleotides (Fig. 4c).We observed thata large number of dinucleotides with helical confor-mations connecting individual C3′-endo nucleotideshad been formed in silico (Fig. 4c; note C3′-endo sugarswith 190bθb240 or 150bηb190). This trend wasespecially prevalent for the dinucleotides assembledfrom the 20°, 15°, 10°, and 5° libraries (SupplementaryFig. 3). As these four libraries contained a relativelylarge number of individual fragments with C3′-endohelical conformations (Fig. 2f), this bias toward helicaldinucleotide connectivities was not too surprising.We then compared the frequency of the in silicodinucleotide θ–η values (Fig. 4c) to the frequency ofthe θ–η values calculated for two-nucleotide stretcheswithin the data set of RNA05 structures (Fig. 4b). Asimilar strong bias toward connecting nucleotideswith helical C3′-endopseudo-torsions occurredwithinthe experimental structures. We note that there isintrinsically no reason for this bias toward C3′-endoconnectivity. Rather, it is just a consequence of thepopulation of filtered fragments selected to be withineach library. Nevertheless, we conclude that assem-bling individual library fragments by using theirpseudo-torsions as a guide results in dinucleotideorientations that largely mimic those observedwithincrystallographic structures.While we observed that the in silico dinucleotides
had orientations that largely mimic those seenexperimentally, there was no guarantee per se thatthe in silico dinucleotide conformations were physi-cally realistic and free of steric overlaps. To addressthis, we next checked to see whether each in silicodinucleotide contained steric clashes by measuringoverlap of van der Waals radii for each pair of atoms(seeMaterials andMethods; van derWaals radii werescaled by 60% due to the discrete nature of thefragments being assembled). After the dinucleotidesidentified to have serious clashes were removed fromthe exhaustive set (approximately 10–12%; Fig. 4e)and the combinations of θ–η torsions of the remainingdinucleotides were replotted (Fig. 4d), we observedthat the pattern of θ–η frequencies appeared virtuallyunchanged (compare Fig. 4c andd).We thus concludethat the majority of the time, when two arbitraryfragments are assembled into a dinucleotide using
their respective pseudo-torsions, the conformationthat results will be physically realistic.
Comparison of in silico dinucleotides androtameric suites
Ideally, any library of discrete RNA conforma-tions should include representatives of each of thepreviously identified backbone rotameric states.8
The two sugars within each in silico dinucleotideconstitute one RNA suite (see Fig. 1a). Based on thisinformation, we were able to determine whethereach set of in silico dinucleotides contained all of thepreviously published rotamer suites. To do so, weused the program Suitename8 to calculate whichsuite, in seven-dimensional space, was most closelyidentified with each dinucleotide assembled in silico.We performed this calculation only for dinucleo-tides that had already been determined by van derWaals overlap to be free of steric clashes.Most, but not all, of the consensus rotamer suites
were identified within the dinucleotides generatedin silico from the 60° and 30° libraries (46 publishedsuites+8 “wannabe” suites; see Fig. 4e and Supple-mentary Fig. 4). In contrast, all consensus suiteconformations were observed repeatedly within thedinucleotides assembled from the libraries binned atfiner resolution (Fig. 4e; Supplementary Fig. 4).Unsurprisingly, the most frequently generated suitetype from the 20°, 15°, 10°, and 5° libraries was suite1a, which is the suite most closely associated withthe helical A-form of RNA (Fig. 4e; SupplementaryFig. 4). While most in silico dinucleotides hadtorsions consistent with one of the establishedrotamer suites, each library also generated dinucle-otide conformations that were considered non-rotamer outliers (Fig. 4e). This trend occurred lessoften for the larger, more extensive filtered fragmentlibraries. Backbone conformations identified as out-liers by Suitename also occur within crystallograph-ic structures and within the original, unfilteredRNA05 data set; approximately 14% of the RNAconformations could not be identified by Suitenameto be associated with any consensus rotamer suite(Fig. 4e). Thus, while the majority of dinucleotideconformations generated from the filtered fragmentlibraries are suite like, other dinucleotide fragmentconformations may represent previously unidenti-fied suites or contain torsional values that lie justoutside a traditional suite.
Deriving a “lower-limit” estimate of modelquality: Modeling RNA folds
Thus, far we have found the libraries binned at 20°or finer to be superior in reproducing the coordinatesof individual nucleotides and generating dinucleo-tides compatible with the rotameric suites. We nextsubjected the filtered fragment libraries to a more
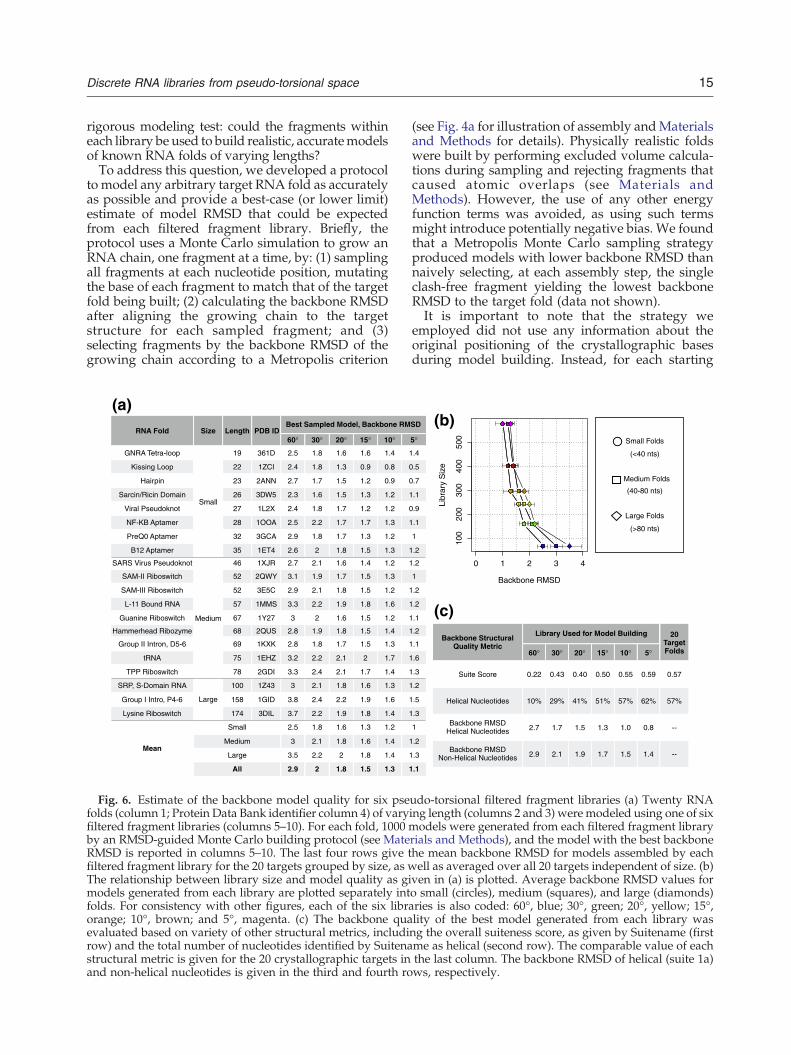
15Discrete RNA libraries from pseudo-torsional space
rigorous modeling test: could the fragments withineach library be used to build realistic, accuratemodelsof known RNA folds of varying lengths?To address this question, we developed a protocol
to model any arbitrary target RNA fold as accuratelyas possible and provide a best-case (or lower limit)estimate of model RMSD that could be expectedfrom each filtered fragment library. Briefly, theprotocol uses a Monte Carlo simulation to grow anRNA chain, one fragment at a time, by: (1) samplingall fragments at each nucleotide position, mutatingthe base of each fragment to match that of the targetfold being built; (2) calculating the backbone RMSDafter aligning the growing chain to the targetstructure for each sampled fragment; and (3)selecting fragments by the backbone RMSD of thegrowing chain according to a Metropolis criterion
RNA Fold Size Length PDB ID Best SSampleed Modeel, Backkbone RRM
g60° 30° 20° 15° 10°
GNRA Tetra-loop 19 361D 2.5 1.8 1.6 1.6 1.4
Kissing Loop 22 1ZCI 2.4 1.8 1.3 0.9 0.8
Hairpin 23 2ANN 2.7 1.7 1.5 1.2 0.9
Sarcin/Ricin DomainSmall
26 3DW5 2.3 1.6 1.5 1.3 1.2
Viral PseudoknotSmall
27 1L2X 2.4 1.8 1.7 1.2 1.2
NF-KB Aptamer 28 1OOA 2.5 2.2 1.7 1.7 1.3
PreQ0 Aptamer 32 3GCA 2.9 1.8 1.7 1.3 1.2
B12 Aptamer 35 1ET4 2.6 2 1.8 1.5 1.3
SARS Virus Pseudoknot 46 1XJR 2.7 2.1 1.6 1.4 1.2
SAM-II Riboswitch 52 2QWY 3.1 1.9 1.7 1.5 1.3
SAM-III Riboswitch 52 3E5C 2.9 2.1 1.8 1.5 1.2
L-11 Bound RNA 57 1MMS 3.3 2.2 1.9 1.8 1.6
Guanine Riboswitch Medium 67 1Y27 3 2 1.6 1.5 1.2
Hammerhead Ribozyme 68 2QUS 2.8 1.9 1.8 1.5 1.4
Group II Intron, D5-6 69 1KXK 2.8 1.8 1.7 1.5 1.3
tRNA 75 1EHZ 3.2 2.2 2.1 2 1.7
TPP Riboswitch 78 2GDI 3.3 2.4 2.1 1.7 1.4
SRP, S-Domain RNA 100 1Z43 3 2.1 1.8 1.6 1.3
Group I Intro, P4-6 Large 158 1GID 3.8 2.4 2.2 1.9 1.6
Lysine Riboswitch 174 3DIL 3.7 2.2 1.9 1.8 1.4
Small 2.5 1.8 1.6 1.3 1.2
MeanMedium 3 2.1 1.8 1.6 1.4
MeanLarge 3.5 2.2 2 1.8 1.4
All 2.9 2 1.8 1.5 1.3
(a)
Fig. 6. Estimate of the backbone model quality for six psefolds (column 1; Protein Data Bank identifier column 4) of varyfiltered fragment libraries (columns 5–10). For each fold, 1000by an RMSD-guided Monte Carlo building protocol (see MateRMSD is reported in columns 5–10. The last four rows give tfiltered fragment library for the 20 targets grouped by size, as wThe relationship between library size and model quality as gimodels generated from each library are plotted separately intfolds. For consistency with other figures, each of the six libraorange; 10°, brown; and 5°, magenta. (c) The backbone quaevaluated based on variety of other structural metrics, includirow) and the total number of nucleotides identified by Suitenastructural metric is given for the 20 crystallographic targets inand non-helical nucleotides is given in the third and fourth ro
(see Fig. 4a for illustration of assembly andMaterialsand Methods for details). Physically realistic foldswere built by performing excluded volume calcula-tions during sampling and rejecting fragments thatcaused atomic overlaps (see Materials andMethods). However, the use of any other energyfunction terms was avoided, as using such termsmight introduce potentially negative bias. We foundthat a Metropolis Monte Carlo sampling strategyproduced models with lower backbone RMSD thannaively selecting, at each assembly step, the singleclash-free fragment yielding the lowest backboneRMSD to the target fold (data not shown).It is important to note that the strategy we
employed did not use any information about theoriginal positioning of the crystallographic basesduring model building. Instead, for each starting
SD
5°
1.4
0.5
0.7
1.1
0.9
1.1
1
1.2
1.2
1
1.2
1.2
1.1
1.2
1.1
1.6
1.3
1.2
1.5
1.3
1
1.2
1.3
1.1
0 1 2 3 4
100
200
300
400
500
Backbone RMSD
Libr
ary
Siz
eSmall Folds
(<40 nts)
Medium Folds
(40-80 nts)
Large Folds
(>80 nts)
(b)
(c)
Backbone Structural Q lit M t i
Libbrary Ussed forr Modell Buildiing 20 TargetQuality Metric
60° 30° 20° 15° 10° 5°Target Folds
Suite Score 0.22 0.43 0.40 0.50 0.55 0.59 0.57
Helical Nucleotides 10% 29% 41% 51% 57% 62% 57%
Backbone RMSD Helical Nucleotides 2.7 1.7 1.5 1.3 1.0 0.8 --
Backbone RMSD Non-Helical Nucleotides 2.9 2.1 1.9 1.7 1.5 1.4 --
udo-torsional filtered fragment libraries (a) Twenty RNAing length (columns 2 and 3) were modeled using one of sixmodels were generated from each filtered fragment libraryrials and Methods), and the model with the best backbonehe mean backbone RMSD for models assembled by eachell as averaged over all 20 targets independent of size. (b)
ven in (a) is plotted. Average backbone RMSD values foro small (circles), medium (squares), and large (diamonds)ries is also coded: 60°, blue; 30°, green; 20°, yellow; 15°,lity of the best model generated from each library wasng the overall suiteness score, as given by Suitename (firstme as helical (second row). The comparable value of eachthe last column. The backbone RMSD of helical (suite 1a)ws, respectively.

16 Discrete RNA libraries from pseudo-torsional space
crystal structure, the bases were removed, and onlythe coordinates of crystallographic backbone wereused to guide rebuilding the fold from each set ofdiscrete library fragments.
Pseudo-torsional libraries can model RNA foldswith atomic-level accuracy
We selected 20 RNA folds that ranged in size andcomplexity from simple hairpins to complex ribo-zymes (Fig. 6a) and used the lower-limit protocol inconjunction with each filtered fragment library torebuild each fold 1000 times (Fig. 5a). For thelibraries binned every 60° or 30°, sampling of lowRMSD structures was often poor (2–6 Å; Fig. 5a, blueand green curves). Further, even though every stepof model assembly was directly guided by backboneRMSD, the best models generated by the 60° and 30°libraries were only in the 2- to 4-Å range (Fig. 6a).This agrees with the overall poor coverage found forthese libraries at the nucleotide level. The other fourlibraries consistently sampled low backbone RMSDmodels (1–4 Å; Fig. 5a, yellow, orange, brown, andmagenta curves). The backbone RMSD of the best-sampled models improved as library binning be-came finer and finer (Fig. 6a), and the 15°, 10°, and 5°libraries consistently produced models with atomic-level accuracy (b1.5 Å backbone RMSD; Figs. 6a and5c–f). As each of the filtered fragment libraries variedin its ability to accurately model the crystallographictarget folds, we conclude that the quality offragments used in RNA modeling can limit theaccuracy with which RNA models can be built.Surprisingly, we found no large differences in the
accuracy with which each library was able to modelsmall, medium, and large RNA folds (Fig. 6a and b).The one exception to this finding was the 60° library,which generated relatively poor quality modelsoverall, regardless of size (see standard deviationbars in Fig. 6b). While the filtered fragment librariesmodeled large and small RNA folds with approxi-mately the same accuracy, there was often a signifi-cant variation in the RMSD of the models producedby the Monte Carlo protocol for large folds (Fig. 5a,3DIL). This was reflected in both a significantbroadening of the 1000 RMSD values sampled bythe Monte Carlo protocol (see Fig. 5a, 3DIL) and thespeed with which the best-observed fold among the1000 was sampled (data not shown). Because of this,we conclude that even if a library contains fragmentscapable of generating a high-qualitymodel, increasedstructural sampling may be needed to produceaccurate models of longer RNA folds.
Evaluating model folds using other backbonemetrics: Suiteness and helicity
The backbone quality of the models generatedfrom each filtered fragment library was also
evaluated by two other non-RMSD-based metrics.First, we used Suitename to calculate the overall“suite score” for the original 20 targets as well as forthe models of each target fold (Fig. 6c, first row).Briefly, the suite score reflects how many suiteswithin a structure have backbone torsions consistentwith one of the previously identified rotamericsuites. Again, models built from the 60°, 30°, and20° libraries performed poorly, with their averagesuite score indicating only 22–40% of the modelnucleotides to be suite like (Fig. 6c, first row). Incontrast, the average suite score of models generatedfrom all other libraries was almost identical withthat of the original data set (Fig. 6c, first row). In afew cases, the suite score of a crystallographic targetwas dramatically improved when the fold wasrebuilt using library fragments (see SupplementaryTable 2 and Discussion).Perhaps the most simple and defining character-
istic of RNA folds is that they contain a highpercentage of helical nucleotides. Thus, we alsodeterminedwhether the models and target folds hada similar number of helical nucleotides. To do so, weagain used the program Suitename8 and determinedhowmany nucleotides within each target andmodelwere identified as the helical 1a suite. Only 10–40%of the nucleotides within the 20 best models builtfrom the 60°, 30°, and 20° libraries were identified ashelical (Fig. 6c, second row). These percentages werefar less than the 57% of nucleotides identified ashelical within the 20 crystallographic folds (Fig. 6c,second row). In contrast, the 15°, 10°, and 5° librariesconsistently rebuilt the target folds into RNAmodels with an average percent of helical nucleo-tides close to the original data set (51–62% ascompared to 57%; Fig. 6c, second row). Afterexamining the helical 1a suite, we asked whethercorresponding nucleotide positions for models andtargets had identical suite conformations over theentire set of 54 conformers (see Materials andMethods). Suites within the 60° models rarelymatched that of the target fold (out of 966 suites,115 suites had identical conformers and 29 werenear identical). In contrast, N80% of suites within the5° models were identical with that of their targetfolds (out of 966 suites, 727 suites had identicalconformers and 72 suites were near identical). Thesefindings are in general agreement with the resultspreviously described for evaluating modeling accu-racy for each library based on backbone RMSD.
Evaluating the RNA models in all-atom detail:All-atom RMSD
Base pairing and positioning often play a funda-mental role in most computational tools that modelRNA de novo. However, the lower-limit protocolselects fragments during model building using onlybackbone RMSD and ignores the location of all base

17Discrete RNA libraries from pseudo-torsional space
atoms, except to disallow fragments whose baseatoms result in steric clashes. Thus, it was possiblethat the backbone coordinates of lower-limit modelswere accurate but that the individual bases co-ordinates were not.To check whether the models built from each
library using a backbone-based RMSD approach hadaccurate base placement, we first calculated the all-atom RMSD of the 20 best models from each of thelibraries to their targets (Fig. 7a, first row). All-atomRMSD values correlated strongly with the backboneRMSD values and, in most cases, were approximate-ly 0.6–0.7 Å greater (Supplementary Table 3). The 60°library produced structures with relatively poor all-atom RMSD values (4.6 Å; Fig. 7a, first row), whilethe all-atommodels generated by the 5° library weresurprisingly accurate (1.7 Å; Fig. 7a, first row). Wealso examined whether the models had any system-atic differences in structural quality at helical andnon-helical regions. To do so, we aligned each of thebest models to its target using all backbone atoms,and then, using this fixed alignment, we calculatedthe all-atom RMSD overall helical (e.g., suite 1a) andnon-helical nucleotides separately.We observed thathelical regions were modeled more accurately thanthe non-helical regions (Fig. 7a, second and fourthrows; 5° model mean accuracy, 1.2 Å and 2.3 Å,respectively; see also Supplementary Table 4). Thisdifference in accuracy appeared largely due to baseplacement: base atoms within helical regions of the5° models were located, over average, 1.5 Å awayfrom their position in the target fold, while the baseatoms within non-helical regions of the samemodelswere located much farther away on average (3.2 Å;Fig. 7a, third and fifth rows). The lower-limitprotocol had ensured that fragments with near-ideal backbone RMSD had been selected duringmodel building for both helical and non-helicalregions alike. Thus, the RMSD of base atoms withinnon-helical regions such as loops and junctions maybe somewhat limited by the current base conforma-tions within the 5° library.
Evaluating the RNA models in all-atom detail:Base orientation and base pairing
In addition to RMSD, we also evaluated theaccuracy of base positioning within the modelsbuilt by each library using two other metrics. First,we identified the number of nucleotides within eachof the models that had chi angles within 20° of thenative nucleotide at the same chain position. Correctbase placement, as measured by chi angle, showedsteady improvements as the filtered fragmentlibraries grew larger and the bin resolution grewfiner. Using this metric, the 60° library performedpoorly and positioned only approximately 40%(468/1207) of bases positioned within 20° of theirtargets (Fig. 7a, sixth row). In contrast, the models
generated using the 5° library had almost 80% (959/1207) of nucleotides placed in a correct baseorientation (Fig. 7a, sixth row). Achieving such ahigh level of accurate base placement, despite thelack of enforcing any criteria to favor base orienta-tion during model building other than sterics, mightbe surprising. However, accurate base placementbased on pseudo-torsional information alone hasbeen observed before.23,33,36
Placement of side chains within 20° is a standardoften used for protein side-chain modeling.20
However, it is not clear whether this level ofaccuracy would be sufficient to observe hydrogenbonding patterns among RNA base pairs. We thusused two freely available annotation programs(RNAView and MC-annotate37) (see Materials andMethods) to calculate, for each target and each 5°model, how many canonical Watson–Crick pairs(G-C and A-U) and how many other “non-Watson–Crick” hydrogen bond pairs9 were present (Fig. 7cand d; Supplementary Table 5). As in Ref. 37, weused the intersection of the paired interactionsreported by both annotation tools.RNAView and MC-annotate both found instances
of all 12 combinations of orientations between theWatson–Crick, Hoogsteen, and sugar “faces” ofnucleotide bases9 within the 5° models (data notshown). Importantly, whenever the two annotationtools agreed that a base pair was present within the5° models, the identical base pairing was almostfound within the target (high sensitivity, as reportedby PPV values in Fig. 7b). In contrast, many pairingsfound within the target fold were not detected in the5° models (low specificity, as reported by STYvalues in Fig. 7b). Upon examining the annotationresults in greater detail, we observed that the twotools often found widely differing sets of hydrogenbonding interactions within the 5° models. Forinstance, within the 5° models, almost 65% of thebase pairing identified by MC-annotate and almost45% of base pairings identified by RNAView weredisregarded because they did not intersect (data notshown). Figure 7c and d demonstrate one examplewhere both annotation tools failed to agree on basepairings within a model, even though the baseatoms of the model were located very close to thebase atoms of the target (model and target coloredmagenta and gray, respectively).This failure todetect hydrogen bonding within helical regions ofthe 5° models was a common occurrence, despite thefact that the RMSD of the base atoms within suchregions was typically low (1.5 Å on average; Fig. 7a,third row). Thus, while the criteria commonlyemployed by tools such as RNAView and MC-annotate may be reliable for detecting hydrogenbonding patterns in crystal structures, the samecriteria may also fail to detect pairings in modelsthat contain bases with close to, but not ideal,geometry.

RNA FoldInteraction Network
Fidelity AnalysisInteraction Network
Fidelity AnalysisInteraction Network
Fidelity AnalysisRNA Fold(Best 5° Model)
PPV STY DI
Small 1 0.4 1.8
Medium 0.9 0.2 3.3
Large 0.8 0.3 2.9
All 0.9 0.3 2.6
(a)
A9
A17
A16
A15A14
A13
A12
A11
A10
(c)
A26
A25
A24
A23
A22
A4
A3
A1
A2
(d)
False Positives: 0
Nuc #1 Nuc #2 Relation
True Positives: 2
Nuc #1 Nuc #2 Relation
A4 A22 W/WA12 A15 S/H
False Negatives: 8
Nuc #1 Nuc #2 Relation
A1 A25 W/WA2 A24 W/WA3 A23 W/WA5 A21 W/WA8 A9 S/HA9 A18 W/HA10 A17 H/SA11 A16 W/W
Base Structural Quality Metric
Library Used for Model BuildingLibrary Used for Model BuildingLibrary Used for Model BuildingLibrary Used for Model BuildingLibrary Used for Model BuildingLibrary Used for Model BuildingBase Structural Quality Metric
60° 30° 20° 15° 10° 5°
All-Atom RMSD 4.6 3.0 2.7 2.3 2.0 1.7
Base RMSD Helical Nucleotides
5.9 3.3 3.1 2.3 1.7 1.5
Base RMSD Non-Helical Nucleotides
6.5 4.7 4.0 3.9 3.3 3.2
Chi +-20deg 39% 62% 69% 74% 78% 79%
(b)A5
A6
A7A8
A21
A20
A19
A18
A17
Base Structural Quality Metric
Best 5° Model of 3DW5
All-Atom RMSD (26 NTs)
2.0Å
Base RMSD Helical Nucleotides
(18 NTs)1.0Å
Base RMSD Non-Helical Nucleotides
(8 NTs)4.6Å
PPV=1.0 STY=0.2 DI=2.05
Fig. 7. Estimate of the base model quality for six pseudo-torsional filtered fragment libraries: All-atom RMSD and hydrogen bond network fidelity. (a) The averageall-atom RMSD over the best 20 models generated from each library is given in the first row. The average all-atom RMSD values, after finding the optimal alignmentbetween each model and target based on backbone atoms, is given separately for helical (second row) and non-helical (fourth row) nucleotides. The average all-atomRMSD values over only base atoms are given in the third and fifth rows for helical and non-helical nucleotides, respectively. The last row gives the percent of allnucleotides within the models determined to have chi torsions within 20° of their targets. (b) Interaction network fidelity (INF) analysis was performed between each ofthe best 5° models of the 20 RNA test folds. Results for specificity, PPV=tp/(tp+fp), and sensitivity, STY=tp/(tp+fn), are given in the second and third columns,respectively. The last column reports the DI, or DI=RMSD/INF. Interactions were calculated as the intersection of pairings detected by RNAView and MC-annotate,and results were averaged over all folds based on their size (e.g., small, medium, or large). (c) The crystal structure of test fold 3DW5 (gray) is shown aligned with thebackbone of its best 5° model (magenta). All 26 nucleotides are shown within the three panels. (d) The all-atom RMSD and the RMSD of the helical and non-helical baseatoms of 3DW5 aligned to its best 5° model are given in rows 1–3 of the table. The box regions denote the INF analysis. Base pairs are denoted using numbering identicalwith (c).
18Discrete
RNA
librariesfrom
pseudo-torsionalspace

19Discrete RNA libraries from pseudo-torsional space
Despite the large number of false negatives withinthe 5° models, we nevertheless calculated thedeformation index (DI) for all 20 targets. The DI isa measure that accounts for both base-pairinginteractions and RMSD (defined as √(PPV×STV)/RMSD37). Over all 20 targets, the average DI valuewas 2.6 (Fig. 7b, last column). As a comparison,several of the structures within the test set (1KXK,1XJR, and 2QUS) were recently modeled using bothMC-Fold and FARNA.38 Despite having poorRMSD overall (ranging from 9 to 15 Å), both theMC-Fold and FARNA models nevertheless hadnotably high specificity and sensitivity values(PPVN0.8 and STYN0.6).38 Thus, DI values inthese three modeling cases were far higher thanthose reported here for the lower-limit models andranged from approximately 14 to 20.
Discussion
In this study, we used a pseudo-torsionalnotation of the RNA backbone to generate sixfiltered libraries of discrete fragments. We alsopresented a methodology for assembling theindividual filtered fragments into larger structuresusing each fragment's pseudo-torsional values as aguide. We found that accuracy in modelingindividual nucleotides, dinucleotides, and entireRNA folds consistently improved as the librariesgrew in size and more thoroughly covered pseudo-torsional space. The largest four libraries modeledmost individual nucleotides to within 0.5 Å, repro-duced all the previously described rotamer suites,and built RNA folds with subatomic accuracy.Consequently, these libraries should be useful fornumerous modeling applications including de novostructural modeling, structure analysis, or crystal-lographic refinement.
Lessons learned from lower-limit modelassembly
Use of discrete libraries inherently limits modelingaccuracy
Building all-atom models of RNA folds usingbackbone RMSD as a guide is not an approach thatcan be directly incorporated into modeling foldsde novo. Nevertheless, certain lessons can be learnedthat are applicable to de novo modeling strategies.First, almost all RNA tertiary modeling tools buildmodels out of discrete pieces of RNA structure,most commonly either RNA fragments31 or cyclicnucleotides.29 However, the extent to which usingdifferent sets of RNA pieces limits modelingaccuracy is not tested explicitly. Twenty yearsago, an early test of nucleotide-based samplingdetermined that approximately 30 discrete confor-
mations could rebuild tRNA to an accuracy of3.1 Å.39,40 This result is consistent with our findingthat the 60° library of 67 filtered fragments buildsmodels with an accuracy of 2–3 Å. However, sincethis initial work,39 few or no tests have beenperformed to indicate the range of modelingaccuracy that can be expected from any givenlibrary of RNA conformations.The model building protocol we used here is
guided by RMSD and lacks energetic terms. Thus,we could assume that when models were poorlybuilt or had high RMSD to the target fold, the errorswere not due to scoring. Further, the libraries wetested were small and this allowed us to use a modelbuilding protocol to perform exhaustive sampling ateach point in the assembly protocol (e.g., at eachstep of the assembly protocol, every fragment in thelibrary was tested and scored based on its RMSD tothe target). Under these conditions, we were able todirectly study how library quality can affectmodeling accuracy. While we found that the use ofdiscrete fragments during modeling especiallylimits accuracy when libraries are small, we foundthat even the largest libraries we tested imposedsome limitations on modeling accuracy.
Filtered fragment libraries can build large and smallfolds with comparable accuracy
Despite the limitations inherent in using discretefragments, the RMSD-based building protocolshowed that discrete libraries are capable ofrebuilding both small and large RNA folds withapproximately equal accuracy. Our lower-limitprotocol builds models using an RMSD-basedapproach and thus eliminates any errors thatmight be introduced by use of a scoring function.Thus, we conclude that if an appropriate library ofRNA conformations is used with near-perfectsampling, there should be no inherent difference inmodeling large and small folds. In contrast, largeRNA folds are often modeled with far worseaccuracy than small hairpins and folds in de novomodeling.31,38 A similar phenomenon is observedwhen building random models of RNA: the meanRMSD of a random model has been shown toincrease with RNA chain length.41 We were unableto use our building protocol to directly test theaccuracy of other published RNA fragment librariesfor building larger RNA folds. However, most of thefragment libraries currently in use are quite largeand likely contain a large diversity of RNAconformations. Thus, the difficulties in de novomodeling of larger folds, as compared with smallerfolds, likely result from insufficiencies in eithersampling or scoring and not the quality of the RNAfragment libraries being used. With respect tosampling, we observed that even when using

20 Discrete RNA libraries from pseudo-torsional space
RMSD to the target as a guide to sample fragments,rebuilding larger RNA folds to the same modelingaccuracy as smaller RNA folds often requiredincreased sampling. Small structural differences inthe fragments selected when building larger RNAfolds may more easily propagate through an entirestructure, causing the models generated for largerRNA folds to vary more widely in their overallbackbone RMSDs. We conclude that a similarincreased sampling of larger RNA folds might alsobe necessary for accurately modeling large RNAfolds de novo.
The backbone conformations and base orientationsof filtered library fragments are linked
Perhaps the most striking result of this study wasthat we observed a strong correlation between thebackbone orientations of the library fragments andtheir base orientations. The result that correct baseorientation can be ascertained from backbone co-ordinates is not new but has also been observedduring semiautomated crystallographic modelbuilding using pseudo-torsions.23 Here, we showthat selecting a library of fragments based on theirbackbone η and θ values and assembling thesefragments based on their backbone RMSD to a targetfold can generate models that accurately reproduceboth the all-atom coordinates of individual nucleo-tides (Fig. 3b) and entire RNA folds (Figs. 6 and 7;Supplementary Table 3).The all-atom models built in this study were not
sampled using a base-centric approach with strin-gent hydrogen bonding criteria. As a result, thehydrogen bonding network analysis did not detectall native base-pairing interactions within themodels (Fig. 7c and d). However, we found thatweakening the structural constraints by introducingan increased kT value produced models with higher-quality backbones overall. Indeed, enforcing perfectbase planarity or strict hydrogen bonding at an earlystage of modeling is likely to limit overall de novomodeling accuracy. This may be especially true incases where slight deviations from strict hydrogenbonding criteria could result in the correct place-ment of the correlated backbone atoms. Certainly atlater stages of modeling, one would fix inaccuraciesintroduced by using a discrete set of fragments andcorrect base placement to conform to stricterhydrogen boding criteria.Finally, we note that a correlation between base
orientation and the η and θ backbone torsions hasbeen observed before.23,33,36 In contrast, a similarcorrelation between base orientation and the stan-dard six backbone torsions was not observed.33
Thus, the strong correlation we observe betweencorrect backbone conformation and base orientationmay be a property unique to using pseudo-torsion-based fragments.
Advantages of using pseudo-torsionfragment libraries
Selecting libraries for modeling accuracy
One advantage of the methodology presented hereis that a library of appropriate size and structuralresolution can be selected for the modeling task athand. Many classification schemes have producedsmall sets of less than 100 RNA conformations.3,8,39,42
Our results show that using rigid sets of this sizeshould be appropriate for building RNA models inthe range of 2–4 ÅRMSD. For instance, the 60° librarydeveloped in this work contains approximately 70fragments and was able to build models withaccuracies of 2.5–4 Å backbone RMSD. This result isconsistent with the 3.1 Å accuracy noted for buildingtRNA with 30 discrete conformations.39,40 However,as illustrated by thework of Keating andPyle, atomic-level accuracies (e.g., b1.5 Å) may be obtained fromlibraries of this size if a coordinate minimization stepis included into the building process.23
In agreement with this idea, many all-atomstructural modeling tools use large libraries ofRNA structural fragments that can contain hundredsor even thousands of conformations.29,31 However,our results suggest that 300–500 well-chosen frag-ments are sufficient to build RNA models withaccuracies of 1.5 Å backbone RMSD or better. Thus,tools using libraries significantly larger than thiscould gain an increase in modeling speed withoutmaking a large sacrifice in modeling accuracy byselecting an appropriately sized fragment set.
Focused library sampling usingpseudo-torsion-based fragments
Several tools for modeling RNA incorporate exper-imental data or secondary structure predictions.27,30
Such tools might enjoy an additional advantage byusing pseudo-torsion filtered fragment libraries.Nucleotides involved in helical regions, tetra-loops,pi loops, and other diverse structural motifs havebeen shown to have η and θ values within well-defined ranges.33,36 Thus, only the subset of libraryfragments within these pseudo-torsion ranges mayneed to be sampled in order to model such regions.Such a strategy of focused sampling could biassimulations toward favorable conformations while,at the same time, increasing computational speed.While some tools, such as MC-Sym, currently catalogstructural pieces of RNA as belonging to particularstructuralmotifs,29 the pseudo-torsion-based librarieswe present here could extend this idea to the singlenucleotide level.Likewise, generating all-atom models of medium-
and large-sized RNAs still remains a computationalchallenge, in part due to limitations imposed byconformational sampling. As a result, coarse-grained

21Discrete RNA libraries from pseudo-torsional space
models are often first produced for larger RNAs and,if desired, all-atom detail is added later in a separateprediction step using the coarse-grained backbonetrace as a guide.28 Fragment libraries have alreadybeen employed in generating all-atom models fromcoarse-grained backbone traces with good success.28
However, the pseudo-torsion-based filtered frag-ment libraries are smaller in overall size relative tofragment libraries, and they provide the advantagesof focused sampling based on structural motifsmentioned above. Additionally, if the pseudo-torsions of a coarse-grained model can be directlymeasured, then these values could be used to directlyguide fragment selection. A similar approach, inwhich pseudo-torsions are measured from an elec-tron density backbone trace and used to guide all-atom crystallographic model building, has recentlybeen published.23
Rebuilding models with increased rotamericity
One final advantage of using the pseudo-torsionlibraries we present is that they were generated fromthe same high-quality data set, RNA05, that wasoriginally used to determine the consensus set ofrotamer suites.8 As a result, crystallographic foldsthat contained poor suite conformations or overallpoor suite scores could be rebuilt using libraryfragments into almost identical folds with improvedscores. For example, two crystallographic folds inthe rebuilding test set, 361D and 1Z43, originallycontained a large number of dinucleotide suitesidentified as outliers, or non-rotameric (11/19 and53/112 suites, respectively). When each of thesefolds was rebuilt using the filtered fragments fromthe 5° library, the new models contained notablyfewer non-rotameric suites (3/19 and 4/112, respec-tively, for 361D and 1Z43). We thus anticipate thatusing the pseudo-torsional fragment libraries incrystallographic or de novo modeling applicationscould improve the quality of the modeled backbone.Finally, we note that the quality of the filtered
fragment libraries we present here is dependent onthe quality of the initial data set, RNA05, fromwhich they were generated. Thus, as the quality ofthe data set gets better, the quality of the fragmentlibraries will likely also improve. A new data set ofhigh-quality RNA structures, RNA09, has recentlybeen made available†, and it would be of interest tocompare fragment libraries generated from this dataset with those published here using RNA05.Preliminary results suggest that libraries generatedfrom the newer RNA09 data set would be slightlylarger but largely overlap with the RNA05 libraries(Supplementary Fig. 6).
†http://kinemage.biochem.duke.edu/databases/rnadb.php
Comparison of pseudo-torsion fragmentlibraries to semiautomated model buildingwith consensus conformers
Importantly, the libraries we have presented arenot the only approach to incorporating the structur-al diversity of the consensus conformers into modelbuilding. For building RNA folds de novo, thediscrete sets presented in this work can be easilyassembled and do not require coordinate minimi-zation. However, if a backbone trace has beenalready been generated, the semiautomated ap-proach of Keating and Pyle can use the consensussuites and coordinate minimization to build an all-atom model.23 For the one test case that overlappedbetween the two methodologies (the guanineriboswitch), the accuracy between the two methodsappeared to be comparable (1.1 Å backbone RMSDfor the 5° library, as reported in this work; mostbackbone atoms to within 0.9 Å of their crystallo-graphic coordinates, as reported in Ref. 23). Thus,both approaches appear suitable for rebuildingknown backbones, including rebuilding those back-bones with increased rotamericity.
Conclusions
To summarize, we have presented six filteredlibraries of pseudo-torsional fragments and validatedtheir ability to reproduce the structural features ofRNA at the level of individual nucleotides anddinucleotides and in the building of entire RNAfolds. Importantly, the fragments are easy to assembleand can be classified, using their pseudo-torsions, intohelical and non-helical RNA conformations. Becausewe have shown that the filtered fragment libraries arecapable of building high-quality, all-atommodels, weanticipate that they should be useful for a variety ofmodeling applications including de novo RNA struc-ture prediction and design, as well as in RNAstructure analysis and refinement.
Materials and Methods
Selection of RNA structural data set
Filtered fragment libraries were generated by takingcoordinates directly from the RNA Database 2005(RNA052).7 The RNA05 data set is hand-curated andconsists of 171 RNA coordinate files (9482 nucleotidestotal) of resolution ≤3.0 Å.
Application of quality filters
Prior to selection of fragments, quality filters wereapplied to each coordinate file in RNA05 on a nucleotide-

22 Discrete RNA libraries from pseudo-torsional space
by-nucleotide basis as follows. First, the tool PROBE43 wasused to check each RNA structure for steric clashes byflagging nucleotides containing any single atom withgreater than 0.4 Å van der Waals radii overlap with anyother atom. In order to be as strict as possible, both intra-and inter-chain overlaps of N0.4 Å were taken intoaccount. Nucleotides passing the steric clash quality filterwere then subjected to a second round of quality filteringand excluded from consideration if any heavy atom(backbone or base) within the nucleotide contained a B-factorN 60. Finally, nucleotides containing alternativeconformations were excluded. Quality filtering removed6018 nucleotides from the starting data set, leaving a totalof 3464.
Preparation of RNA structural data set
Only nucleotides containing a 2′-hydroxyl and baseidentity of A, C, G, or U were considered, and modifiedbases were not used for this analysis. All non-RNAmolecules, waters, heteroatoms and duplicate copies ofRNA had been already removed within the previouslypublished data set. Hydrogens, which had previouslybeen added to each structure, were removed from theRNA05 data set.
Measurement of nucleotide sugar pucker andpseudo-torsions
Sugar pucker was determined for each RNA05 nucle-otide by using a combination of two separate criteria. First,the standard backbone torsion delta (C5′, C4′, C3′, O3′)was calculated for each nucleotide using DANGLE.43
Next, the perpendicular distance between the glycosidicbond of each nucleotide and the following phosphate wascalculated using a perl script (e.g., the base–phosphateperpendicular distance).23,44 Nucleotides were then de-fined as having a C3′-endo sugar pucker if their deltavalues were 84±30° and their base–phosphate perpendic-ular values were N2.9 Å. Likewise, nucleotides weredefined to have C2′-endo sugar pucker if their delta valueswere 147±30° and their base–phosphate perpendiculardistances were≤2.9 Å. Of the RNA05 nucleotides, 838 haddelta values or base–phosphate perpendicular distancesoutside of these ranges and were discarded.The backbone pseudo-torsions eta [η: C4′i− 1, Pi, C4′i,
Pi+1] and theta [θ: Pi, C4′i, Pi+1, C4′i+1] were measured foreach quality-filtered RNA05 nucleotide determined tohave a well-defined sugar pucker using the programDANGLE.43 Nucleotides at the beginning or end of astructure, as well as nucleotides directly preceding orfollowing a chain break, were excluded from analysisbecause both pseudo-torsions could not be measured.Nucleotides were also excluded from analysis if thenucleotide immediately preceding (used in defining η) orfollowing (used in defining θ) failed to meet all filteringcriteria. In all, pseudo-torsions were recorded for 1780nucleotides (1562 and 218 for C3′-endo and C2′-endo,respectively).In developing a semiautomated approach for crystallo-
graphic model building,23 a new pseudo-torsional nota-tion of C1′-P and η′/θ′ was introduced. While this newnotation has some advantages in generating all-atom
detail from backbone traces of electron density, definedstructural motifs have not yet been correlated with η′/θ′values. In contrast, structural motifs have been wellcharacterized using the original η–θ notation, allowingfor the possibility to bias library sampling towarddesired motifs (see Focused library sampling usingpseudo-torsion-based fragments above). Thus, in thiswork, we have chosen to generate discrete fragment setsusing the original C4′-P and η–θ notation.
Selection of filtered fragments
To create filtered fragment libraries, individual RNA05nucleotides were selected based on their measuredpseudo-torsions as follows. First, 2-D pseudo-torsionalspace was partitioned uniformly at six varying degrees,60°, 30°, 20°, 15°, 10°, and 5°. The partitioningwas repeatedindependently for each C2′- and C3′-endo sugar pucker.Next, for each partitioning, we calculated the η–θ valuesfor the center of each (ηbin_center and θbin_center). Finally, aperl script was used to search the list of quality-filteredRNA05 nucleotides for the single instance of correct sugarpucker with pseudo-torsional values closest (as measuredby Euclidean distance, d) to the bin center. If the bin wasempty and did not contain a quality-filtered nucleotide, nofragment was added to the library. The Euclidean distance(d) between the pseudo-torsion values of the bin center(ηbin_center, θbin_center) and the pseudo-torsion values ofevery nucleotide within the bin (η,θ) was calculated as
follows: d =ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi½ ηbin center−ηð Þ2 + θbin center−θð Þ2�
q.
The crystallographic coordinates of each RNA05 nucleo-tide that best represented a pseudo-torsional bin wererecorded and included in the appropriate filtered fragmentlibrary. The backbone coordinates defining theη and θ of theselected RNA05 nucleotides (gray atoms, Fig. 1a) were alsorecorded and added to the library. Fragment bond lengthsand angles were assumed rigid, and neither the backbonenor the glycosidic bond lengths or angles were modifiedfrom those found in the original RNA05 nucleotide. Thedistribution of standard torsions (α, β, γ, δ, ɛ, ζ, and Χ) foreach of the six libraries mimicked that of the entire RNA05data set (Supplementary Fig. 5).
Fragment attachment
Two fragments (i and j) can be attached at a singlephosphate using pseudo-torsions as follows. First, thelast three atoms involved in defining the θ pseudo-torsion of fragment i, (C4′i, Pi+1, C4′i+1; Fig. 4a) arealigned to the first three atoms defining the η pseudo-torsion of fragment j (C4′j− 1, Pj, C4′j; Fig. 4a). Thisalignment will bring the phosphates Pi+1and Pj into veryclose proximity. In order to ensure direct connectivitybetween the two fragments, the coordinates of thephosphate atom of fragment j were then translated, ifnecessary, to overlap exactly the coordinates of thephosphate of fragment i. To form a dinucleotide, alloverlapping and nonoverlapping atoms involved indefining the pseudo-torsions of both fragments can beremoved (Fig. 4a). If attachment of additional fragmentsis desired, as is the case when building an entire RNAfold, the atoms involved in defining the θ pseudo-torsionof fragment j can be retained and used to guide the

23Discrete RNA libraries from pseudo-torsional space
attachment of an additional fragment to the end of agrowing RNA chain. All alignments and translationswere performed using the Biopython SVDSuperimposer,and all fragment backbone and base bond lengths andangles remained fixed during fragment attachment (seealso RMSD calculations).
Steric exclusion calculations
In order to build physically realistic dinucleotides andfolds, excluded volume calculations were performed afterattaching one fragment to another. Attachments thatresulted in steric overlaps were rejected. For generationof dinucleotides, excluded volume calculations werecomputed pairwise over all atoms (backbone and base)between the two joined nucleotides except the atomsdirectly connected by the intervening phosphate (e.g., theO3′ of the first nucleotide with the O1P, O2P, and O5′atoms of the second nucleotide). For building of RNAfolds, excluded volume calculation were also performedpairwise over all the atoms (backbone and base) ofnucleotides that “neighbored” the added fragment,where neighboring nucleotides were defined to be thosehaving a phosphate–phosphate distance to the addedfragment of less than 20 Å.For each set of atom pairs, the distance between the two
atoms, in angstroms, was compared to the sum of the vander Walls radii of the same two atoms. If the distancebetween any two atoms was found to be less than theirsummed van der Waals radii, scaled by 60%, then a stericoverlap was considered to occur. The scaling of radii by60% was used to allow for the discrete nature of thefragments being assembled. The van der Waals radii usedfor calculations were as follows: carbon, 1.7 Å; oxygen,1.52 Å; phosphorous, 1.8 Å; and nitrogen, 1.55 Å.For building of dinucleotides, if a steric clash occurred
due to the presence of a purine base, the purine base wasmutated in silico to a pyrimidine, and the excluded volumecalculations were repeated to check whether the clash hadbeen resolved. During the mutation process, the glysco-sidic bond angle of the base was left unchanged (seeSequence mutation protocol). For rebuilding target RNAfolds, the base of each fragment was computationallymutated, if needed, to the sequence identical with thetarget fold being modeled prior to excluded volumecalculations.
Sequence mutation protocol
A reference file, consisting of a single representative setof atomic coordinates for each RNA base type (ProteinData Bank ID 1ET4: A203, G207, C211, and U215), wasused to mutate a fragment base to match any arbitrarytarget sequence. The mutation protocol left all fragmentsugar atoms fixed but replaced the original fragment basecoordinates with the desired mutant base coordinateswithin the reference file as follows. First, BioPython'sSVD-based Superimposer (see RMSD calculations) wasused to align the reference base with the fragment baseusing three of the four atoms involved in the chi torsion ofeach base: purine [C1′, N9, C4] or pyrimidine [C1′, N1,C2]. Next, the reference base coordinates were translated,if needed, such that the reference base N1/N9 atom
coordinates exactly matched those of the original fragmentbase N1/N9 atom coordinates. The old base coordinateswere then replaced with the reference base coordinates.The chi angles of reference bases attached via the abovemethod of superposition followed by translation weretypically within 1° of those measured for the originalfragment base.
RMSD calculations
All superpositions and RMSD calculations were calcu-lated using the Bio.SVDSuperimposer module of BioPy-thon, which implements a singular value decompositionsuperposition algorithm based on Ref. 45. BackboneRMSD values were calculated over all heavy atoms inthe sugar phosphate backbone (e.g., P, O1P, O2P, O5′,C5′, C4′, O4′, C3′, O3′, C2′, O2′, C1′). All-atom RMSDvalues were calculated (after mutation of fragment basesto match target structure, as necessary) over all non-heavy atoms.
Model building protocol
We developed a model building protocol that aims torebuild a “target” RNA fold with the lowest possiblebackbone RMSD. The protocol uses filtered libraryfragments and builds a model of a target fold in astepwise fashion. The first fragment in the model is thesingle fragment within a library having the smallestheavy-atom backbone RMSD to the first nucleotide inthe target fold. The base of this first fragment iscomputationally mutated to match the target sequence(see Sequence mutation protocol), and all atoms definingthe fragment's η pseudo-torsion are then removed (grayatoms in Fig. 1a). To grow the chain by one nucleotide,every fragment in the library is attached to the first, one ata time in a randomly selected order. After attachment, thebackbone RMSD of the entire chain built thus far to thetarget fold is calculated and compared with the chainbackbone RMSD calculated for the last attached fragment.If the backbone RMSD of the most recently attachedfragment is lower than previously observed or ifthe score passes the Metropolis criterion [Paccept=min(1,e−ΔRMSD/KT)], the fragment nucleotide base was mu-tated to match the target sequence (see Sequence mutationprotocol) and checked for steric clashes with other atomsof growing chain (see Steric exclusion calculations). If thefragment is found to be clash free, its chain RMSD value isrecorded as the new best observed so far and used forcomparison with later fragments.Chain growth continued in this manner until all but the
last nucleotide in the target RNA fold had been modeled.The last fragment, as with the first fragment, is selectedwithout the Monte Carlo criterion (e.g., the single clash-free fragment with lowest backbone RMSD was chosen),and the atoms defining the θ pseudo-torsion of the lastadded central nucleotide (gray atoms, Fig. 1a) areremoved. Occasionally, after attempting to attach allfragments in a library to the end of a growing chain, noclash-free fragment was found. In such cases, chaingrowth was stopped, and a new Monte Carlo chainbuilding simulation was begun. This phenomenon wasinitially observed when we attempted to build models

24 Discrete RNA libraries from pseudo-torsional space
without using theMonte Carlo criterion (e.g., at each chaingrowth step, the clash-free fragment with best score wasselected deterministically). For each target fold, we usedthis protocol to build 500 model folds using a KT=0.001and to build 500 model folds using a KT=0.005. EachMonte Carlo simulation was performed on a singlecomputing node of the Yale computing cluster, whichhad eight 2. 66-GHz Intel Xenon cores and 16- GB RAM.The computational time required to build each target folddepended on the size of the target, as well as the size ofthe library (Supplementary Fig. 7). Small folds (b40 NTs)were typically built in less than 15 min, while mediumfolds (40–80 NTs) were built in 15–45 min. In contrast,large structures (N80 NTs) were built in 1–3 h (Supple-mentary Fig. 7).
Model building test set
To test model building accuracy using each filteredfragment library, 20 RNA crystallographic structures wereselected (Figs. 6 and 7). Each of the 20 structures wascontiguous over its entire length (e.g., structures withchain breaks were not considered), and the meanstructural resolution over all structures was 2.3 Å (resolu-tions varied from 1.0 to 3.0 Å). Each fold was classifiedbased on its chain length as small (19–35 nucleotides),medium (46–78 nucleotides), or large (100 nucleotidesor more).The structures within the test set were diverse and
included the tRNA fold, aptamers, riboswitches, and theP4–P6 domain of the group I intron. The majority of testset either was solved post-2005 and had no comparablerepresentative within the RNA05 data set (SupplementaryTable 1; 3DIL, 2GDI, 3E5C, 2QWY, 3GCA, 2ANN, 1ZCI,and 361D) or did not contribute fragments to any of the sixlibraries (Supplementary Table 1; 1OOA, 1XJR, and1KXK). During analysis of the test set (Figs. 5–7), nosignificant differences were found between the modelingaccuracy of the structures that had originally beencontained within RNA05 and those that were unique.
Evaluation of modeled suite conformations
We examined 20 targets that contained a total of 1207suites. For each target, the percentage of suites that weremodeled correctly was determined as follows. First, theprogram Suitename8 was used to classify all conformerswithin a target and model. The classifications were thencompared on a suite-by-suite basis. If a correspondingnucleotide position was classified as having the same suitein both the target and model, then it was labeled as“identical.” Corresponding nucleotide positions classifiedas having helical (1a) or helical-like (1 m, 1 L, &a, 1c, or 1f)conformers in both the target and model were labeled as“near-identical.” Nucleotides that Suitename triaged (106suites over all 20 targets) or determined to be outliers (135suites over all 20 targets) were disregarded.
‡http://www.major.iric.ca/MC-Pipeline/§http://www.pylelab.org/software/index.html
Evaluation of base pairing
The RNA interaction network fidelity between modelsand target folds was calculated using two freely available
annotation tools from MC-Pipeline‡: RNAView andMC-Sym.37,38 For both the model and the target structure,the network fidelity calculation was calculated bydetermining the intersection of the base-pairing typesdetailed in Ref. 9. Stacking was not considered in thenetwork fidelity calculation.
Availability
All six filtered fragment libraries, as well as code tobuild dinucleotides, are freely available§.
Acknowledgements
We thank Kevin Keating (Yale) and Laura Murray(Yale) for early discussions of the project andproviding critical comments on the manuscript. Wealso thank Kevin Keating for suggesting the term“filtered fragment library.” We thank the Yale HighPerformance Computing Center for use of the HighPerformance Computing computational cluster. Thiswork was supported by National Institutes of Healthgrant (F32GM096516) to E.H.N. A.M.P. is an inves-tigator of the Howard Hughes Medical Institute.
Supplementary Data
Supplementary data associated with this articlecan be found, in the online version, at doi:10.1016/j.jmb.2012.03.002
References
1. Gesteland, R. F., Cech, T. & Atkins, J. F. (2006). TheRNA world: the nature of modern RNA suggests aprebiotic RNA. 3rd edit. Cold Spring Harbor Mono-graph Series 43, Cold Spring Harbor Laboratory Press,Cold Spring Harbor, NY.
2. Tinoco, J. I. & Bustamante, C. (1999). How RNA folds.J. Mol. Biol. 293, 271–281.
3. Kim, S. H., Berman, H. M., Seeman, N. C. & Newton,M. D. (1973). Seven basic conformations of nucleicacid structural units. Acta Crystallogr., Sect. B: Struct.Crystallogr. Cryst. Chem. 29, 703–710.
4. Murthy, V. L., Srinivasan, R., Draper, D. E. & Rose,G. D. (1999). A complete conformational map forRNA. J. Mol. Biol. 291, 313–327.
5. Hershkovitz, E., Sapiro, G., Tannenbaum, A. &Williams, L. D. (2006). Statistical analysis of RNAbackbone. IEEE/ACM Trans. Comput. Biol. Bioinform. 3,33–46.
6. Schneider, B., Morávek, Z. & Berman, H. M. (2004).RNA conformational classes. Nucleic Acids Res. 32,1666–1677.

25Discrete RNA libraries from pseudo-torsional space
7. Murray, L. J. W., Arendall, r., Bryan, W., Richardson,D. C. & Richardson, J. S. (2003). RNA backbone isrotameric. Proc. Natl Acad. Sci. USA, 100, 13904–13909.
8. Richardson, J. S., Schneider, B., Murray, L. W., Kapral,G. J., Immormino, R. M., Headd, J. J. et al. RNAOntology Consortium(2008). RNA backbone: consen-sus all-angle conformers and modular string nomen-clature (an RNA Ontology Consortium contribution).RNA, 14, 465–481.
9. Leontis, N. B. & Westhof, E. (2001). Geometricnomenclature and classification of RNA base pairs.RNA, 7, 499–512.
10. Lescoute, A., Leontis, N. B., Massire, C. & Westhof, E.(2005). Recurrent structural RNA motifs, IsostericityMatrices and sequence alignments. Nucleic Acids Res.33, 2395–2409.
11. Ramachandran,G.N., Ramakrishnan,C.&Sasisekharan,V. (1963). Stereochemistry of polypeptide chainconfigurations. J. Mol. Biol. 7, 95.
12. Chandrasekaran, R. & Ramachandran, G. N. (1970).Studies on the conformation of amino acids. XI.Analysis of the observed side group conformation inproteins. Int. J. Protein Res. 2, 223–233.
13. Dunbrack, R. L., Jr & Karplus, M. (1993). Backbone-dependent rotamer library for proteins. Application toside-chain prediction. J. Mol. Biol. 230, 543–574.
14. Dunbrack, R. L., Jr &Karplus,M. (1994). Conformationalanalysis of the backbone-dependent rotamer preferencesof protein sidechains. Nat. Struct. Biol. 1, 334–340.
15. Lovell, S. C., Word, J. M., Richardson, J. S. &Richardson, D. C. (2000). The penultimate rotamerlibrary. Proteins: Struct., Funct., Genet. 40, 389–408.
16. Ponder, J. W. & Richards, F. M. (1987). Tertiarytemplates for proteins. Use of packing criteria in theenumeration of allowed sequences for differentstructural classes. J. Mol. Biol. 193, 775–791.
17. Dunbrack, R. L., Jr (2002). Rotamer libraries in the 21stcentury. Curr. Opin. Struct. Biol. 12, 431–440.
18. Peterson, R. W., Dutton, P. L. & Wand, A. J. (2004).Improved side-chain prediction accuracy using an abinitio potential energy function and a very largerotamer library. Protein Sci. 13, 735–751.
19. Xiang, Z. & Honig, B. (2001). Extending the accuracylimits of prediction for side-chain conformations.J. Mol. Biol. 311, 421–430.
20. Schrauber, H., Eisenhaber, F. & Argos, P. (1993).Rotamers: to be or not to be? An analysis of aminoacid side-chain conformations in globular proteins.J. Mol. Biol. 230, 592–612.
21. De Maeyer, M., Desmet, J. & Lasters, I. (1997). All inone: a highly detailed rotamer library improves bothaccuracy and speed in the modelling of sidechains bydead-end elimination. Folding Des. 2, 53–66.
22. Davis, I. W., Murray, L. W., Richardson, J. S. &Richardson, D. C. (2004). MOLPROBITY: structure vali-dation and all-atom contact analysis for nucleic acidsand their complexes. Nucleic Acids Res. 32, W615–619.
23. Keating, K. S. & Pyle, A. M. (2010). Semiautomatedmodel building for RNA crystallography using adirected rotameric approach. Proc. Natl Acad. Sci.USA, 107, 8177–8182.
24. Shapiro, B. A., Yingling, Y. G., Kasprzak, W. &Bindewald, E. (2007). Bridging the gap in RNA structureprediction. Curr. Opin. Struct. Biol. 17, 157–165.
25. Ding, F., Sharma, S., Chalasani, P., Demidov, V. V.,Broude, N. E. & Dokholyan, N. V. (2008). Ab initioRNA folding by discrete molecular dynamics: fromstructure prediction to folding mechanisms. RNA, 14,1164–1173.
26. Sharma, S., Ding, F. & Dokholyan, N. V. (2008).iFoldRNA: three-dimensional RNA structure predic-tion and folding. Bioinformatics, 24, 1951–1952.
27. Jonikas, M. A., Radmer, R. J., Laederach, A., Das, R.,Pearlman, S., Herschlag, D. & Altman, R. B. (2009).Coarse-grained modeling of large RNA moleculeswith knowledge-based potentials and structural fil-ters. RNA, 15, 189–199.
28. Jonikas, M. A., Radmer, R. J. & Altman, R. B. (2009).Knowledge-based instantiation of full atomic detailinto coarse-grain RNA 3D structural models. Bioinfor-matics, 25, 3259–3266.
29. Major, F., Turcotte, M., Gautheret, D., Lapalme, G.,Fillion, E. & Cedergren, R. (1991). The combination ofsymbolic and numerical computation for three-dimensional modeling of RNA. Science, 253,1255–1260.
30. Parisien, M. &Major, F. (2008). The MC-Fold andMC-Sym pipeline infers RNA structure from sequencedata. Nature, 452, 51–55.
31. Das, R. & Baker, D. (2007). Automated de novoprediction of native-like RNA tertiary structures. Proc.Natl Acad. Sci. USA, 104, 14664–14669.
32. Duarte, C. M. & Pyle, A. M. (1998). Stepping throughan RNA structure: a novel approach to conformationalanalysis. J. Mol. Biol. 284, 1465–1478.
33. Wadley, L. M., Keating, K. S., Duarte, C. M. & Pyle,A. M. (2007). Evaluating and learning from RNApseudotorsional space: quantitative validation of areduced representation for RNA structure. J. Mol. Biol.372, 942–957.
34. Keating, K. S., Humphris, E. L. & Pyle, A. M. (2011). Anew way to see RNA. Q. Rev. Biophys., 1–34.
35. Duarte, C. M., Wadley, L. M. & Pyle, A. M. (2003).RNA structure comparison, motif search and discov-ery using a reduced representation of RNA conforma-tional space. Nucleic Acids Res. 31, 4755–4761.
36. Wadley, L. M. & Pyle, A. M. (2004). The identificationof novel RNA structural motifs using COMPADRES:an automated approach to structural discovery.Nucleic Acids Res. 32, 6650–6659.
37. Parisien, M., Cruz, J. A., Westhof, E. & Major, F.(2009). New metrics for comparing and assessingdiscrepancies between RNA 3D structures andmodels. RNA, 15, 1875–1885.
38. Laing, C. & Schlick, T. (2010). Computational ap-proaches to 3D modeling of RNA. J. Phys. Condens.Matter. 22, 283101.
39. Gautheret, D., Major, F. & Cedergren, R. (1993).Modeling the three-dimensional structure of RNAusing discrete nucleotide conformational sets. J. Mol.Biol. 229, 1049–1064.
40. Major, F., Gautheret, D. & Cedergren, R. (1993).Reproducing the three-dimensional structure of atRNA molecule from structural constraints. Proc.Natl Acad. Sci. USA, 90, 9408–9412.
41. Hajdin, C. E., Ding, F., Dokholyan, N. V. & Weeks,K. M. (2010). On the significance of an RNA tertiarystructure prediction. RNA, 16, 1340–1349.

26 Discrete RNA libraries from pseudo-torsional space
42. Sykes, M. T. & Levitt, M. (2005). Describing RNAstructure by libraries of clustered nucleotide doublets.J. Mol. Biol. 351, 26–38.
43. Word, J. M., Lovell, S. C., LaBean, T. H., Taylor, H. C.,Zalis, M. E., Presley, B. K. et al. (1999). Visualizing andquantifying molecular goodness-of-fit: small-probecontact dots with explicit hydrogen atoms. J. Mol.Biol. 285, 1711–1733.
44. Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N.,Kapral, G. J., Wang, X. et al. (2007). MolProbity: all-atom contacts and structure validation for proteinsand nucleic acids. Nucleic Acids Res. 35, W375–W383.
45. Golub, G. H. & Van Loan, C. F. (1989). MatrixComputations (Johns Hopkins Series in the MathematicalSciences 3), 2nd edit. Johns Hopkins University Press,Baltimore, MD.