diagnostic hematology: disorders of hemoglobin and gammopathies muhammad shoaib khan gm centre - 1
TRANSCRIPT

Diagnostic Hematology:Disorders of Hemoglobin andGammopathies
Muhammad Shoaib KhanGM Centre - 1

Hemoglobin structure
a globin globin
globin globin
Hgb A tetramer

Globin chain synthesis cluster - chromosome 16
cluster - chromosome 11
Gower 1
Portland Embryonic
Gower II
F Fetal <1%
A2 1.5-3.5%Adult
A >95%
Glo
bin
chai
n
com
pone
nt
Hgb n
ame
Devel
opm
ent
per
iod
% o
f adu
lt H
gb
G A

Thalassemia• Heterogenous group of disorders due to an imbalance of and
globin chain synthesis– thalssemia: -globin chain production decreased– thalassemia: globin chain production decreased
• The globin chains that are produced are normal
• Quantitative deficiency:– o thalassemia: No -globin chain is made– + thalassemia: decreased -globin chain is made
• With 4 genes and 2 genes there is wide phenotypic variation

Incidence of Thalassemia
• ~100,000 patients with homozygous -thalassemia world-wide
• Found in Mediterranean countries, South Asia and Far East
• Prevalence in the United Sates is increasing due to population migration

Alpha Thalassemia• Inadequate production of alpha chains• Hemoglobin analysis normal; can be detected by globin gene analysis• Absence of 1-2 alpha chains
– Common– Asymptomatic– Does not require therapy
• Absence of 3 alpha chains– Microcytic anemia (Hgb 7-10)– Splenomegaly
• Absence of 4 alpha chains– Hydrops fetalis (non-viable)

chains Hgb (g/dl) MCV (fl) RDW
/ Normal Normal Normal
/- 12-14 75-85 Normal
-/- or - -/ 11-13 70-75
- -/- 7-10 50-60
- -/- - - - -
Laboratory Findings in Alpha Thalassemia

Beta Thalassemia
Minor (Trait) / + or / ° 10-13
Intermedia +/+ 7-10
Major +/° or °/° < 7
ClinicalSyndrome Genotype Hemoglobin (g/dl)
Inadequate production of chains

Beta Thalassemia - Hgb analysis
Minor (Trait) / + or / ° 90-94 3.5-8 1-10
Intermedia +/+ 5-60 2-8 20-80%Major +/° 2-10 1-6 >85
°/° 0 1-6 >94
ClinicalSyndrome Genotype A A2 F
Hemoglobin analysis: Increased levels of Hgb A2 and Hgb F

Approach to Beta Thalassemia
• Screening/counseling
• RBC transfusion therapy
• Agents to increase hemoglobin F (Hydroxyurea)
• Bone marrow transplantation

Clinical Presentations of Abnormal Hemoglobins
• Sickling disorder
• Thalassemia or microcytic anemia
• Cyanosis
• Erythrocytosis
• Hemolytic anemia
• Asymptomatic (screening or family study)
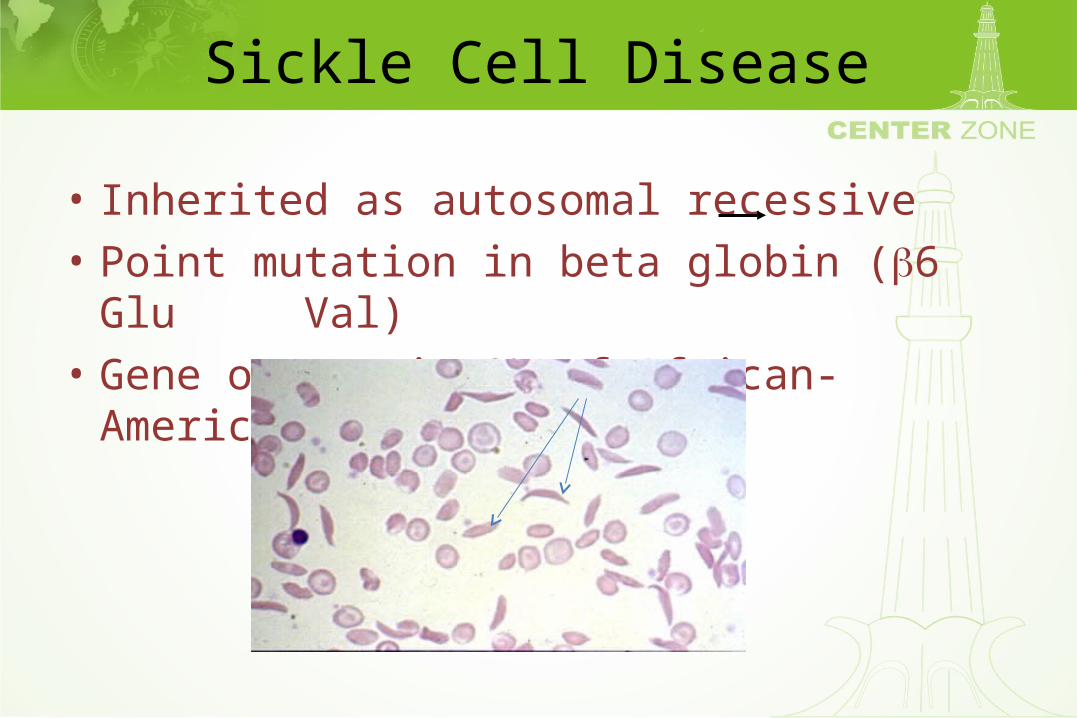
Sickle Cell Disease
• Inherited as autosomal recessive• Point mutation in beta globin (6 Glu Val)• Gene occurs in 8% of African-Americans
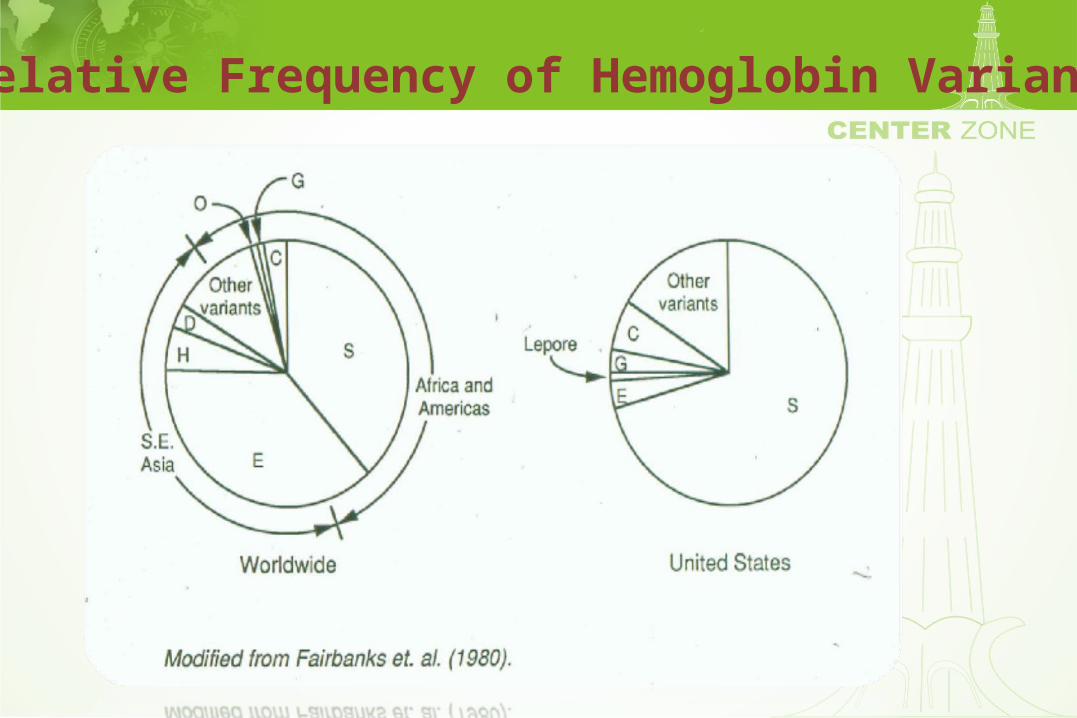
Relative Frequency of Hemoglobin Variants

Screening for Sickle Cell Trait and Disease
• RBC lysate with concentrated phosphate buffer and sodium hydrosulfite
• Incubate 10-20 min

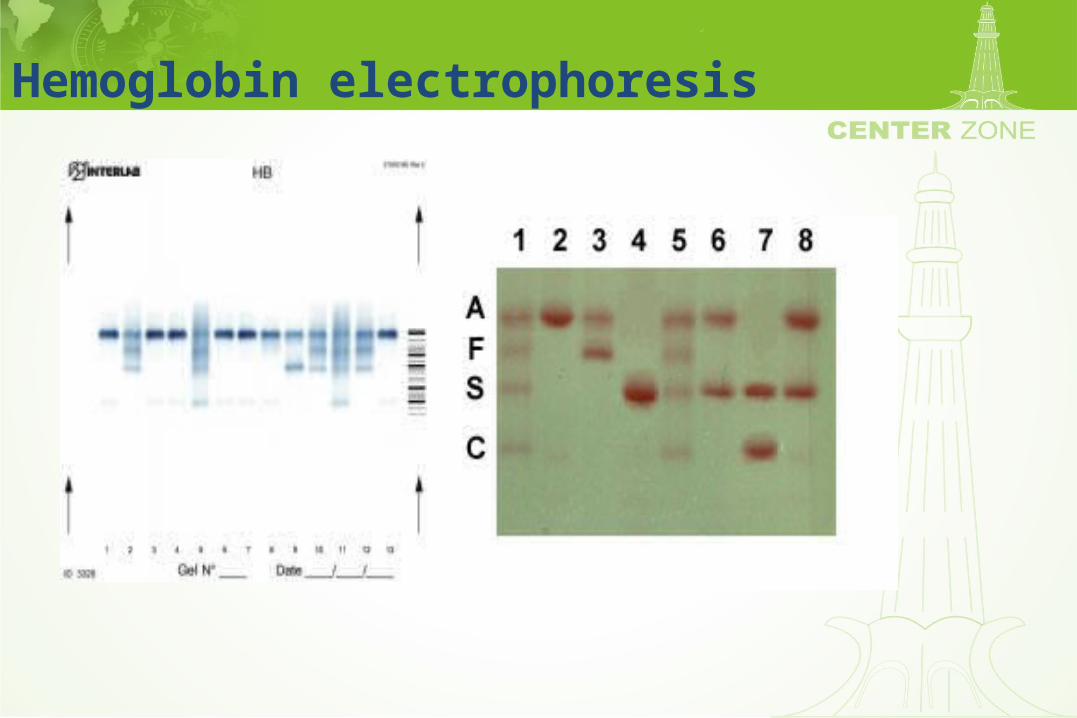
Hemoglobin Electrophoresis: Methodology
• Separates hemoglobins on solid support media– Cellulose acetate (Alkaline gel)– Citrate agar (Acid gel)
• Inexpensive and quickly prepared
• Sharp resolution of major hemoglobin bands
• Electrophoretic variability based on charge
QuickTime™ and aPhoto - JPEG decompressor
are needed to see this picture.
Hemoglobin electrophoresis

Hemoglobin electrophoresis:Variants of sickle cell anemia

Hemoglobin electrophoresis:Identification of abnormal hemoglobins

High Pressure Liquid Chromatography (HPLC)
• Separates hemoglobins by a cation exchange column
• Resolution of various hemoglobins including Hgb F is excellent
• Procedure can be automated leading to reliable interpretation
• Hemoglobin fractions can be quantified
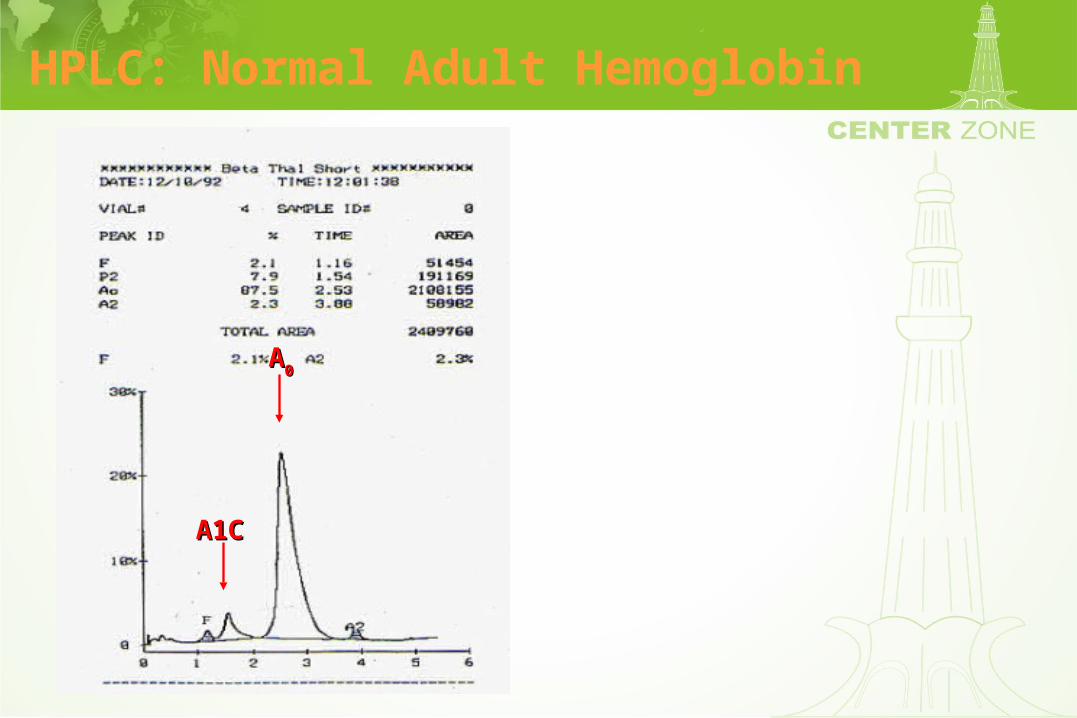
HPLC: Normal Adult Hemoglobin
A1CA1C
AA00
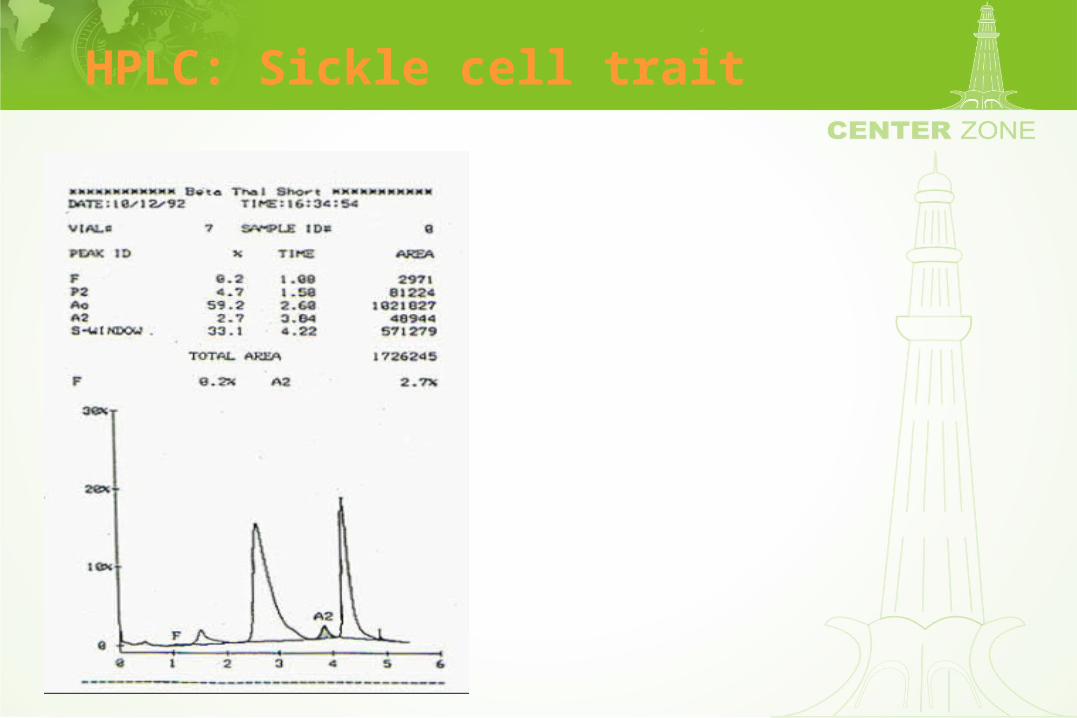
HPLC: Sickle cell trait
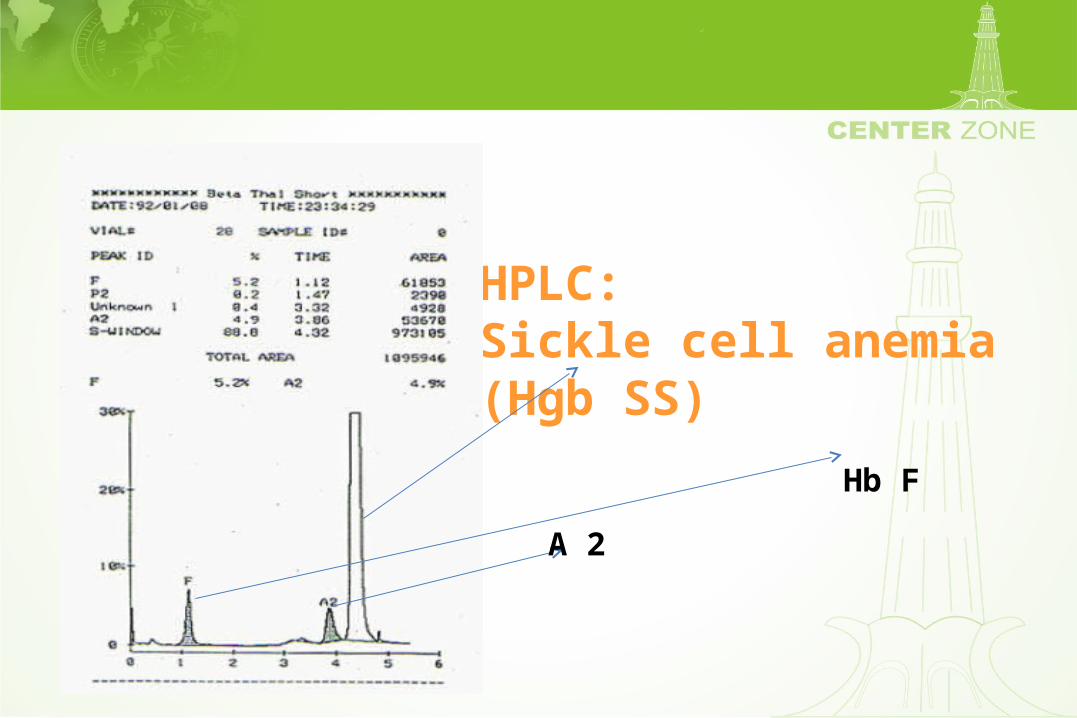
HPLC:Sickle cell anemia (Hgb SS)
A 2
Hb F

HPLC:Hgb SC disease

Monoclonal Gammopathies
• Laboratory evaluation of gammopathies
• Diseases associated with gammopathies
• Common clinical syndromes

Clinical indications for the evaluation of immunoglobulins
• Normochromic normocytic anemia
• Nephrotic syndrome in a non-diabetic patient
• Osteolytic lesions
• Lymphadenopathy
• Non-ischemic heart failure
• Elevated total serum protein
• Hypercalcemia

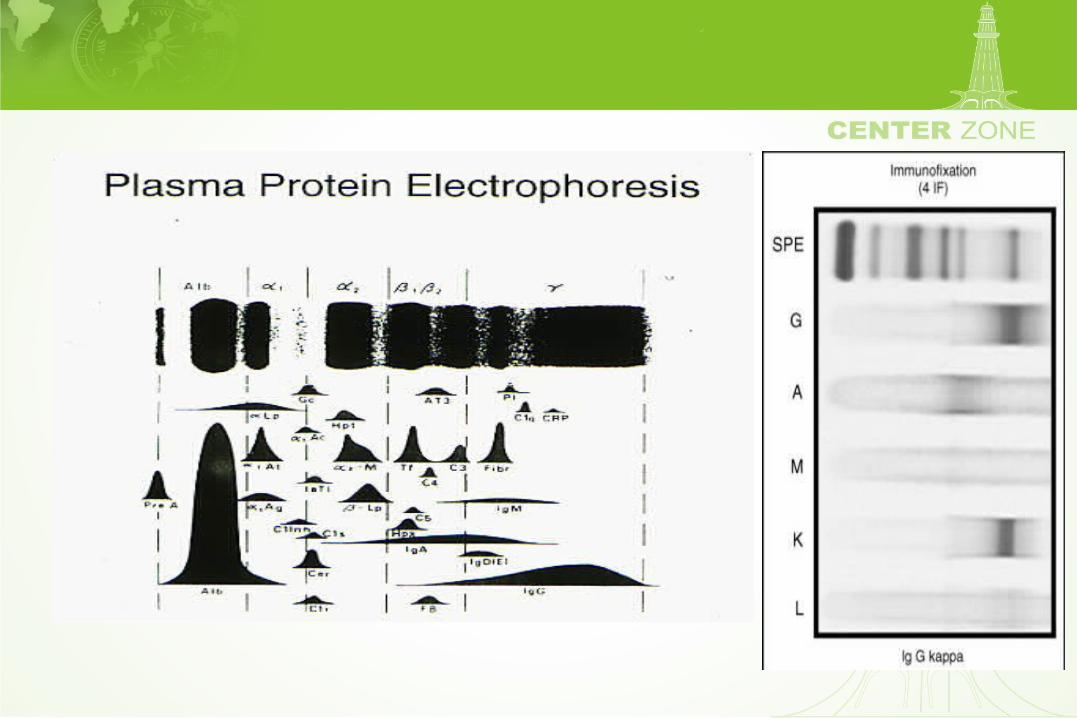

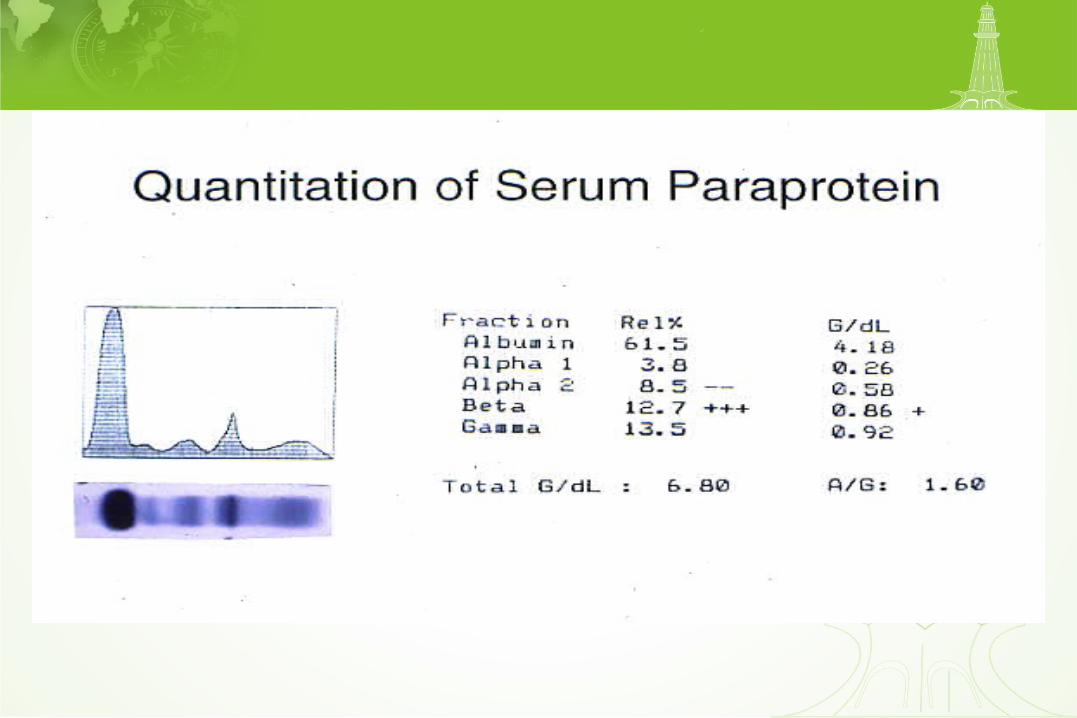


Free light chains
• Have been detected in urine for >50 years *• Polyclonal antibody against free LC• Purified so no cross-reactivity and does not bind to intact
immunoglobulin• Bound to latex beads - detected by a variety of techniques
(turbidity)
* Korngold and Lapiri Cancer: (1956) 9:262-272

Representative sensitivity levels
Kappa Lambda
SPEP 500-2000 mg/L 500-2000mg/L
IFE 150-500 mg/L 150-500 mg/L
Free light chains 1.5 mg/L 3.0 mg/L
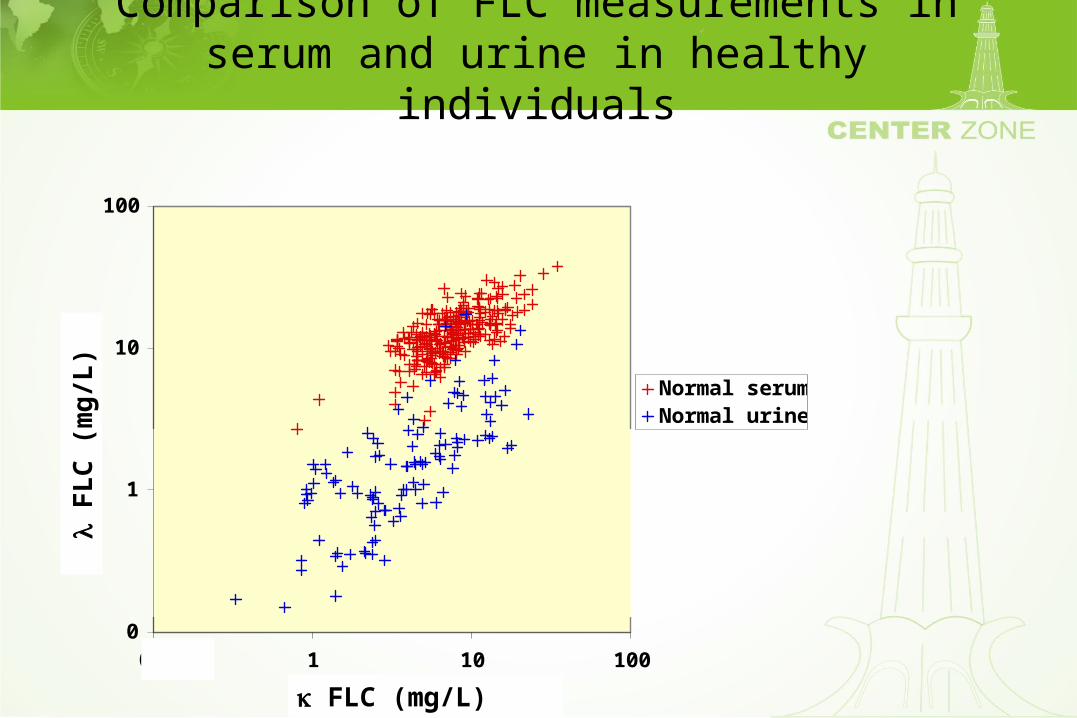
Comparison of FLC measurements in serum and urine in healthy individuals
0
1
10
100
0.1 1 10 100
FLC (mg/L)
_ F
LC
(m
g/L
)
Normal serumNormal urine
FLC (mg/L)
F
LC
(m
g/L
)

Serum free light chains
0.1
1
10
100
1000
10000
100000
0.1 1 10 100 1000 10000 100000
FLC (mg/L)
F
LC
(m
g/L
)
Normal sera
κ LCMM
λ LCMM
IIMM
High pIgG
AL Amyloidosis
Renal impairment
NSMM
IFE Sensitivity
SPE Sensitivity
F
LC
(m
g/L
)
Composite Figure of serum free light chain concentrations in various diseases

Potential uses of serum free light chains
• Sensitive marker for diagnosing monoclonal lymphoproliferative diseases
• ratio may be a prognostic marker for MGUS
• Useful marker in non-secretory myeloma or patients with only Bence-Jones proteinuria
• Marker to follow disease

Lymphoproliferative Disorders Commonly Associated with a Monoclonal Gammopathy
• Monoclonal gammopathy of undetermined significance (MGUS)
• Multiple myeloma• Waldenstroms macroglobulinemia• Amyloidosis

Monoclonal Gammopathies of Undetermined Significance (MGUS)
• Commonly found on serum protein electrophoresis• Occurs in ~2% of persons > 50 years of age• Characteristics
– Low serum monoclonal protein concentration (<3 g/dl)– Less than 5% plasma cells in bone marrow– Little or no monoclonal protein in urine– Absence of lytic bone lesions– No anemia, hypercalcemia, or renal insufficiency

“Benign Monoclonal Gammopathy” Course of MGUS in 241 Patients
Median follow-up 22 years
Group Description No. % 1 No substantial increase of serum 46 19 or urine monoclonal protein (benign) 2 Monoclonal protein ≥3.0g.dl but 23 10 no myeloma or related disease 3 Died of unrelated causes 113 47 4 Development of myeloma, 59 24 amyloidosis or related disease Total 241 100
N Engl J Med 2002;346:564-9 (Updated)Am J Med 1978; 64:814-26
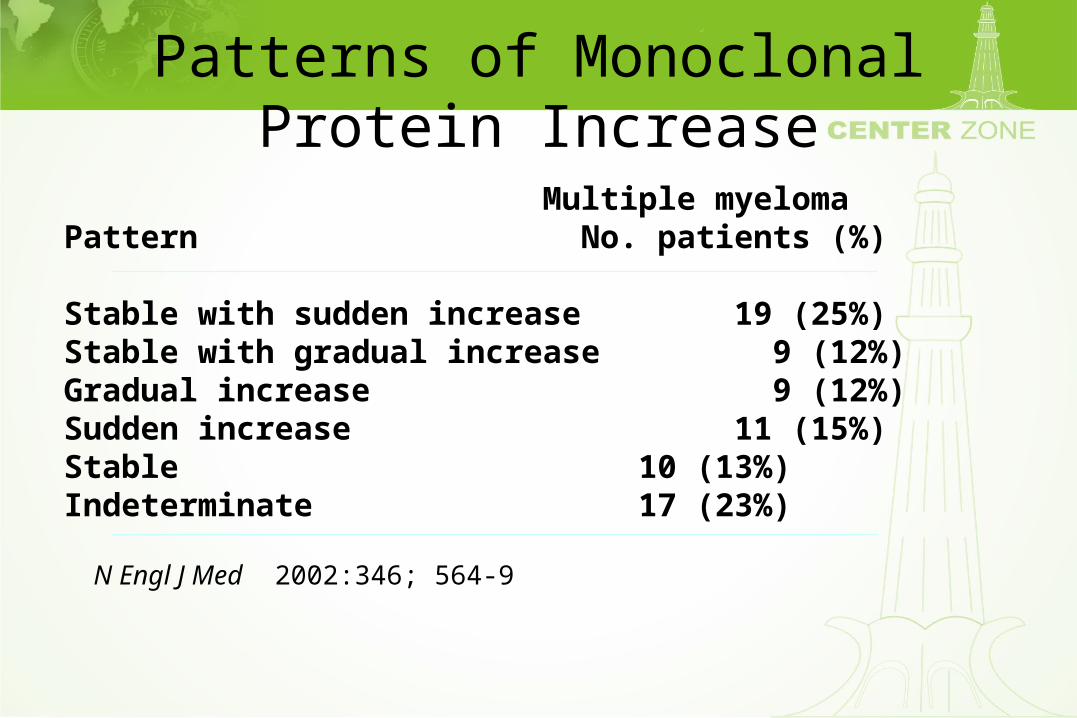
Patterns of Monoclonal Protein Increase
Multiple myelomaPattern No. patients (%)
Stable with sudden increase 19 (25%)Stable with gradual increase 9 (12%)Gradual increase 9 (12%)Sudden increase 11 (15%)Stable 10 (13%)Indeterminate 17 (23%)
N Engl J Med 2002:346; 564-9

Summary:(MGUS)
• Monoclonal proteins rarely disappear spontaneously (<5%)
• MGUS is a risk factor for multiple myeloma and related disorders
• Risk of progression to multiple myeloma or related disorders is increased with higher initial monoclonal protein levels
• Risk of progression is ~1 % per year

Multiple Myeloma: Incidence and Etiology
• 13,000 cases/year in USA• Median age - 65 yrs.• Incidence in African-Americans is two-fold other
ethnic groups• Familiar clusters are rare• Environmental/occupational exposures have been
implicated

Multiple Myeloma: Clinical Manifestations
• Bone pain/skeletal involvement• Fatigue/anemia• Renal insufficiency• Hypercalcemia• Neurologic symptoms• Infections

Laboratory evaluation
CBC with peripheral smear Chemistry panel (Include calcium and creatinine) SPEP/UPEP (immunofixation electrophoresis) Urinalysis/24 hr urine for protein Bone marrow exam Skeletal survey LDH and 2-microglobulin Serum viscosity

Peripheral smear: Plasma cell
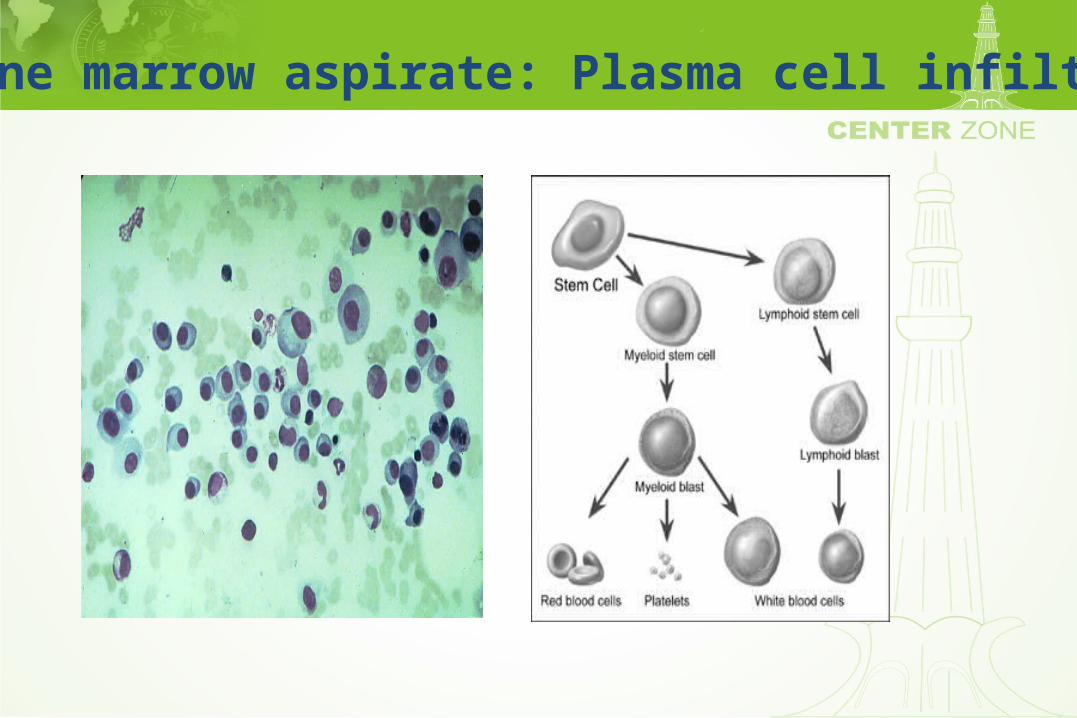
Bone marrow aspirate: Plasma cell infiltrate


Diagnostic Criteria for Multiple Myeloma
Major criteria I. Bone marrow plasmacytosis > 30% II. Histologic diagnosis of plasmacytoma III. Serum paraprotein IgG > 3.5 g/dl or IgA > 2.0 g/dl
Minor criteria a. Bone marrow plasmacytosis 10-30% b. Serum paraprotein less than major criteria c. Osteolytic lesion d. Hypogammaglobulinemia
One major criteria and one minor criteria Minor criteria a + b and one other

Waldenstroms MacroglobulinemiaIncidence and clinical features
• 1,500 cases/year in USA• Median age -, 63 yrs• Presenting symptoms
– Weakness and fatigue 44%
– Hemorrhagic manifestations 44%
– Weight loss 23%
– Neurologic symptoms 11%
– Visual disturbances 8%
– Raynauds phenomenon 3%

Waldenstroms Macroglobulinemia:Clinical Features
• Tumor infiltration– Bone marrow 90%– Splenomegaly 38%– Lymphadenopathy 30%
• Circulating IgM– Hyperviscosity syndrome 15-20%– Cryoglobulinemia 5-15%– Cold agglutinin disease 5-10%– Bleeding disorders 10%
• Tissue IgM– Neuropathy 10-20%


Amyloidosis: Classification and Biochemical Composition
• Primary amyloidosis– Immunoglobulin light chain (AL)
• Secondary amyloidosis– Amyloid A protein (AA)– Synthesized by liver as an acute phase reactant
• Hereditary amyloidosis– Transthyretin-derived amyloid (ATTR)

Primary Amyloidosis: Clinical Features• Nephropathy
– Renal function loss 80– Proteinuria 75
• Cardiomyopathy– Heart failure 40-50
• Neuropathy– Polyneuropathy 36– Orthostatic hypotension 26– Carpal tunnel syndrome 8
• Enteropathy– Hepatomegaly 57– Macroglossia 32– Diarrhea ± Malabsorption 8
% involved

Primary Amyloidosis: Histopathology
H&E Congo Red
Tongue(Macroglossia)

Primary amyloidosisKey points
1. Suspect amyloidosis when a patient has unexplained:Nephrotic range proteinuria with or without renal insufficiencyCardiomyopathy manifested by fatigue or CHFPeripheral neuropathyHepatomegaly
2. Pursue diagnosis if:A monoclonal protein is detected in serum or urine
3. Confirm diagnosis with Congo red stain of:Bone marrowSubcutaneous fatOther affected tissue
4. Perform echocardiogram to assess prognosis
5. Begin systemic treatment

Common clinical syndromesassociated with monoclonal gammopathies
• Bleeding disorders
• Hyperviscosity
• Cryoglobulinemia
• Peripheral neuropathy

Hemostatic defects associated withMonoclonal proteins
Effect on hemostasis Assay
Inhibition of platelet aggregation PFA; Bleeding time
Inhibition of fibrin polymerization Thrombin time
Acquired von Willebrand disease VWF activity and antigen
Acquired factor X deficiency Factor X activity

Acquired factor X deficiency
• Low factor X levels (<50%)• Severe bleeding with activity <10%• Associated with amyloidosis• Factor X binds to amyloid deposits in tissues• Treatment
– Underlying amyloidosis– Splenectomy– Large volumes of FFP/plasma exchange

Hyperviscosity syndrome
• Associated with Waldenstroms macroglobulinemia (15-20% of patients)• Measure serum viscosity (normal <1.8)• Clinical syndrome of hyperviscosity occurs >4.0• Symptoms
– Headaches
– Other neurologic symptoms (dizziness, mental status changes
– Blurry vision
– Easy bleeding


Cryoglobulinemia
• Type I (monoclonal) cryoglobulin• Associated with any lymphoproliferative disorder
– Waldenstroms macroglobulinemia 10-20%
• Symptoms– Raynaud phenomenon– Purpura– Renal insufficiency – Arthralgia
• Blood handling is difficult– Collect blood in 37° C tube– Transport and centrifuge at 37° C– Chill serum to 4° C for 48 hrs– Assay for cryoglobulin
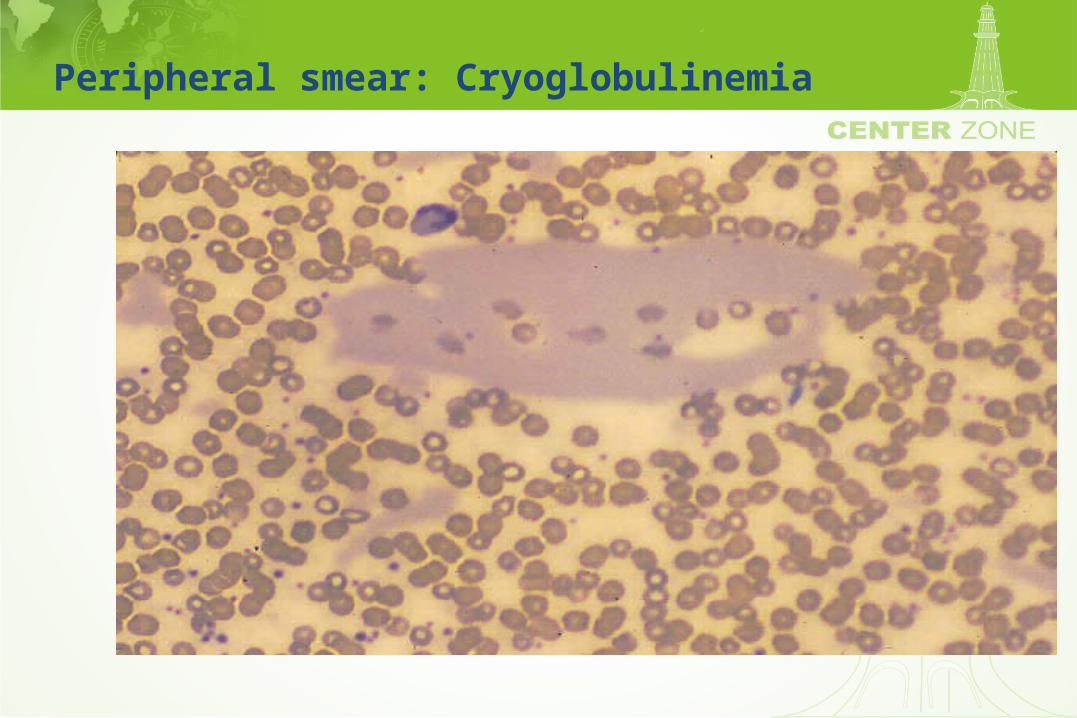
Peripheral smear: Cryoglobulinemia
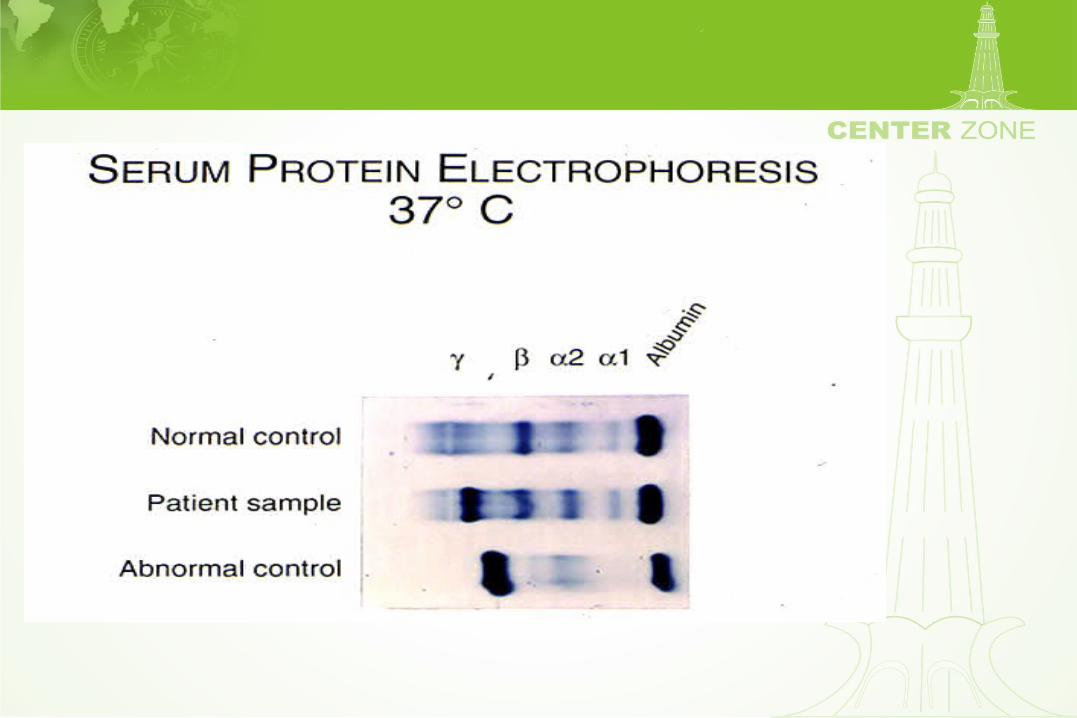



Neuropathies associated withmonoclonal protein disorders
• Associated with any lymphoproliferative disease
• Target antigens are occasionally identified (MAG; myelin associated glycoprotein)
• Symmetric, distal, sensory or sensorimotor
• May simulate CIDP (Chronic inflammatory demyelinating polyneuropathy)
• Associated with any class of monoclonal protein

Summary
• Lymphoproliferative disorders associated with monoclonal proteins are common
• Diagnosis may be difficult
• Treatment requires identification of underlying disease and any associated clinical syndromes