developmental disturbances of oral and para oral tissues fileetiology and pathogenesis: hereditary,...
TRANSCRIPT

Developmental Disturbances Of Oral And Para oral Tissues
BY
Dr: Hanan Eid Gamal

Definitions Developmental diseases: are those resulting from disturbance in normal development of an individual.
Congenital: developmental diseases manifested at birth.
Tarda: developmental diseases appear later in life.
Developmental diseases may be caused by hereditary or environmental (acquired) factors.
Hereditary diseases: Transmitted from parents to child.
Acquired diseases: Developed in reaction to environment.
Hamartoma: normal tissue in normal site but in excessive amount.
Ectopic: normal tissue in abnormal site.
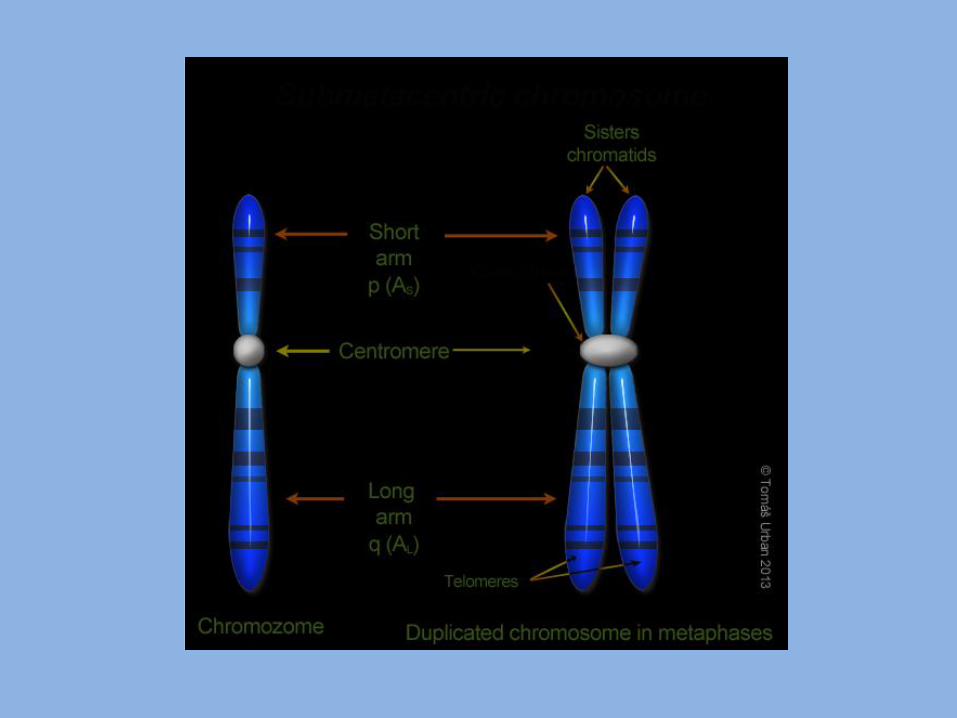

Classification of hereditary diseases: 1- chromosomal abnormalities: - Can affect autosomes or sex chromosomes. - May be numerical or structural. - Numerical such as trisomy and monosomy. - Structural such as deletion and translocation. 2- Single gene abnormalities: - May affect autosomes or sex chromosomes. - May be dominant, recessive. - May show varying degrees of expressivity and penetrance. 3- Multifactorial inheritance: - Result from the action of more than one gene and modified by environmental factors.

Environmental factors
Teratogens:
An agent believed to cause congenital abnormalities is referred to as teratogen, such as:
- Drugs and chemicals.
- Infections.
- Radiation.

Developmental Disturbances of Face 1- Treacher collins syndrome(mandibulo-facial dysostosis).
Definition: developmental malformation affecting the face, It constitutes one member of the first arch syndromes.
- The first arch syndromes includes in addition to treacher collins syndrome, pierre robin syndrome, mandibular dysostosis and cleft lip and palate.
Etiology and pathogenesis:
Hereditary, transmitted as autosomal dominant trait. The mutant gene located on the long arm of chromosome 5(5q32-33.1).
The mutant gene result in:

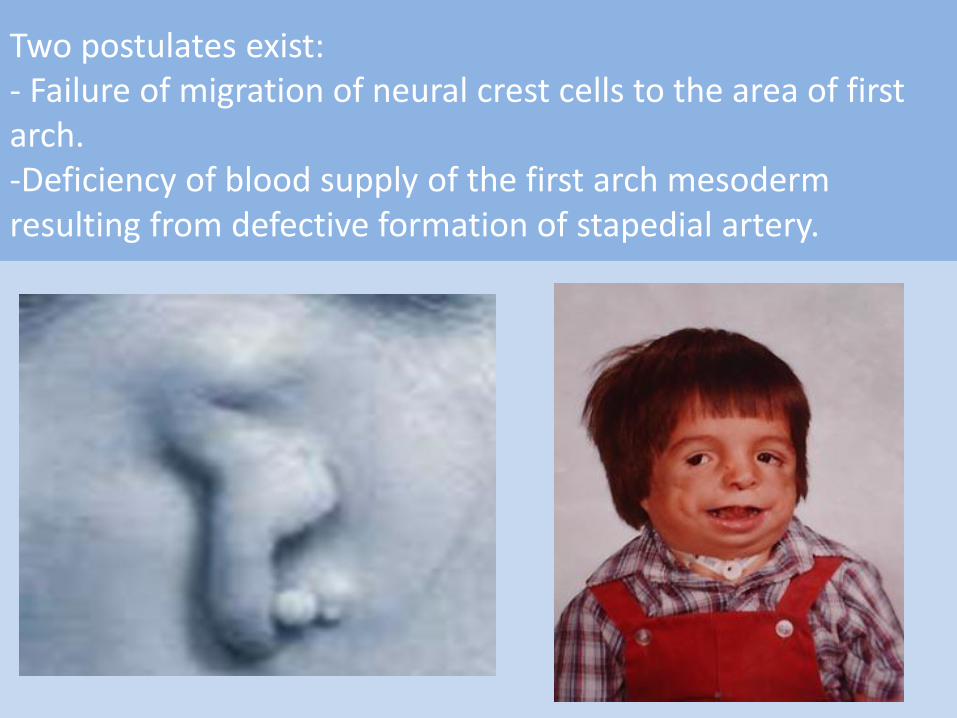
Two postulates exist: - Failure of migration of neural crest cells to the area of first arch. -Deficiency of blood supply of the first arch mesoderm resulting from defective formation of stapedial artery.
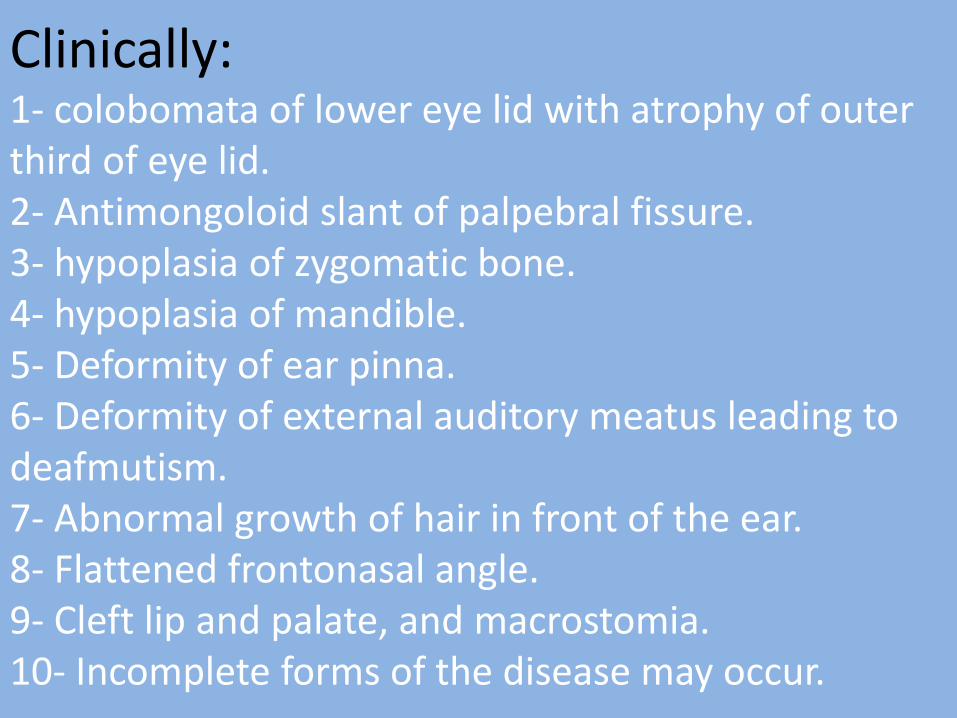
Clinically: 1- colobomata of lower eye lid with atrophy of outer third of eye lid. 2- Antimongoloid slant of palpebral fissure. 3- hypoplasia of zygomatic bone. 4- hypoplasia of mandible. 5- Deformity of ear pinna. 6- Deformity of external auditory meatus leading to deafmutism. 7- Abnormal growth of hair in front of the ear. 8- Flattened frontonasal angle. 9- Cleft lip and palate, and macrostomia. 10- Incomplete forms of the disease may occur.


2- Pierre Robin syndrome( Pierre Robin Sequence, mandibular dysostosis with glossoptosis)
Definition: developmental malformation characterized by mandibular hypoplasia, cleft palate and glossoptosis.
Syndrome: errors of morphogenesis with simultaneous presence of multiple anomalies.
Sequence: any condition that includes series of anomalies caused by cascade of events initiated by single malformation.
Etiology: two postulates:
1- Hereditary, autosomal recessive or x- linked inheritance.
2- Mechanical theory, oligohydramnios theory. Supported by presence of shoulder impression on the body of mandible and the characteristic u-shaped cleft palate.

Arrest of mandibular development may prevent descent of the tongue and failure of palatal shelves elevation and fusion.

Clinically:
1- Hypoplasia of mandible
2- Glossoptosis(ptosis in Greek means downward displacement.
3- Cleft palate.

3- Crouzon Syndrome (cranio- facial dysostosis)
Definition: developmental disorder characterized by craniocynostosis and cranio- facial malformation.
- Craniocynostosis: means early ossification of one or more of the cranial sutures resulting in abnormal head shape.
- Skull deformity due to retarded growth perpendicular to the affected suture with increased growth along the same direction that the suture follow.
- Types: primary and secondary.
- Primary (syndromic): occur as a result of many syndromes, the major ones are: Crouzon syndrome, Apert syndrome, Pfeiffer syndrome, and Carpenter syndrome.
- Secondary: show normal suture biology but there is abnormal internal or external forces resulting in early closure.
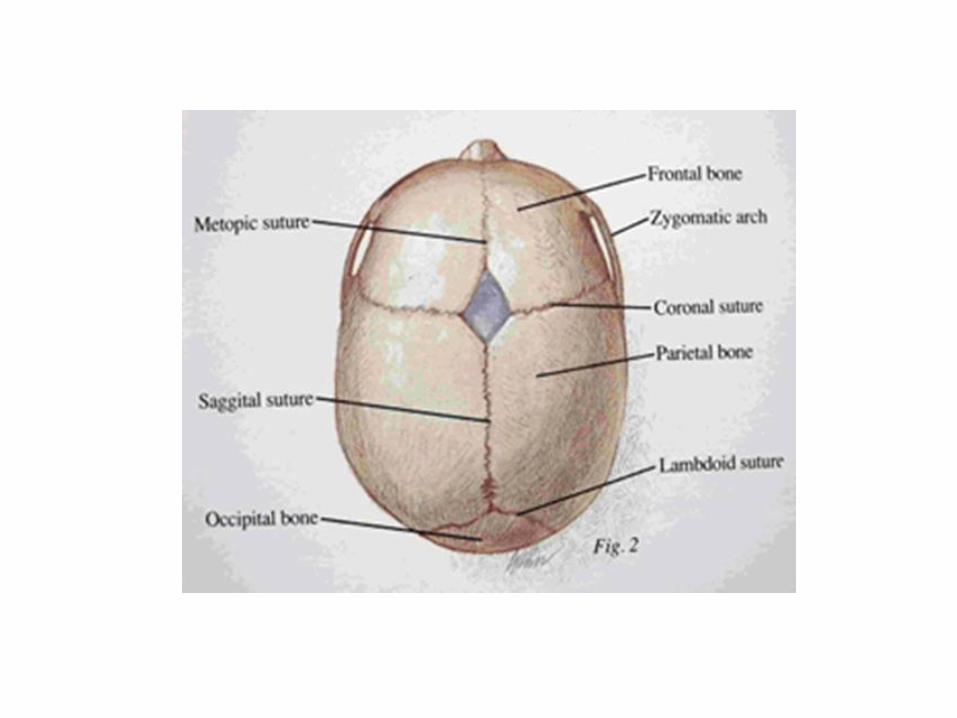

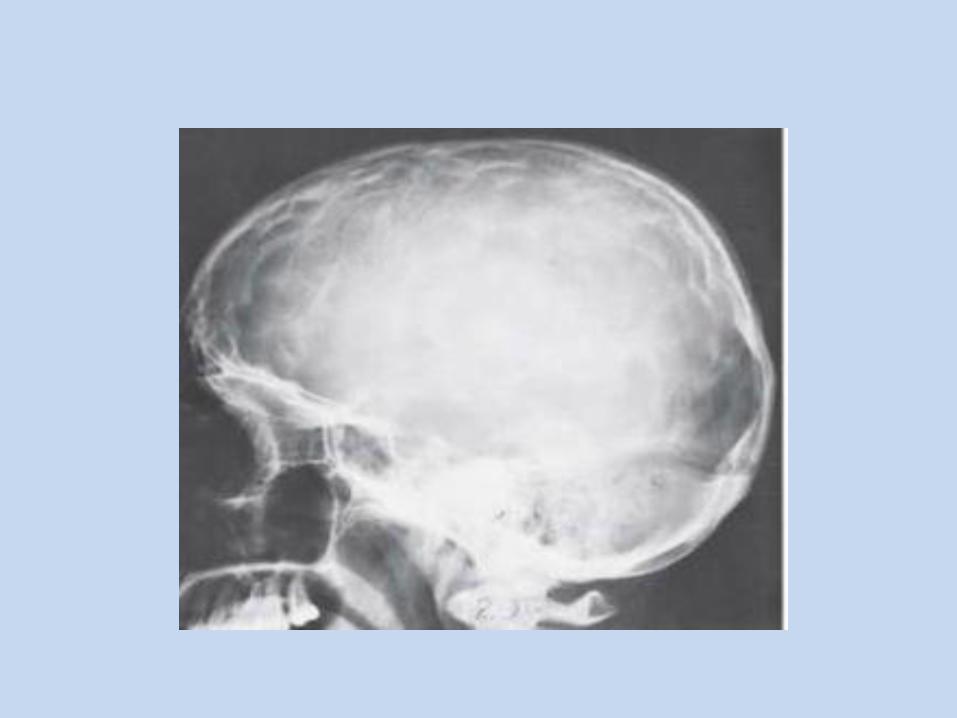
Etiology: Inherited as autosomal dominant pattern. It caused by mutation of FGFR-2 gene. Clinically: - Coronal and sagittal sutures show early ossification. - Anteroposterior diameter of the skull is smaller than transverse diameter ( broad face and skull). - Patients with Crouzon's syndrome have a characteristic face that is often described as froglike.

- Hypoplastic maxilla. Malocclusion. - Progressing optic nerve atrophy leads to vision impairment. - Impairment of hearing. - Short upper lip with cleft lip. - Hypertelorism, shallow orbits and protruding eyeballs. - Unilateral or bilateral posterior crossbite. - Relative mandibular macrognathia.


X-Ray: Digital markings of skull, maxillary hypoplasia and small paranasal sinuses.

4- Apert Syndrome Diffinition A developmental malformation characterized by malformations of the skull, face, hands and feet. It is characterized by: - Craniosynostosis. - Cone shaped calvarium (skull cap) - midface hypoplasia ocular manifestations - syndactyly( two or more digits are fused together) of the hands and feet.

Oral manifestations - Maxillary hypoplasia - Dental crowding, impacted teeth, delayed eruption, ectopic eruption, supernumerary teeth, and thick gingiva - Skeletal and dental anterior open bite.


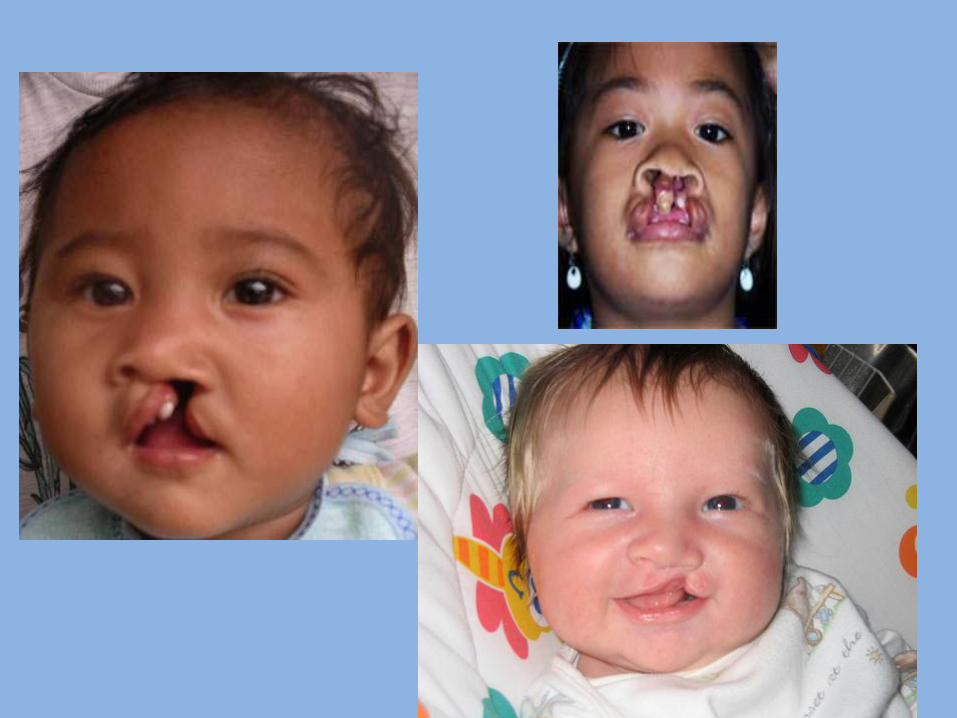
5- Cleft lip and Palate There are congenital clefts which may affect lip, alveolus or palate.
Etiology: clefts are said to be due to multifactorial inheritance ( there is genetic predisposition coupled by environmental factor).
Pathogenesis:
- Clefts of the lip result from failure of fusion of maxillary process with the median nasal process.
- Median clefts of upper lip result from defective formation of median nasal process( globulomaxillary process).
- Alveolar clefts result from failure of fusion of maxillary process with the median nasal process.
- Clefts of the palate result from failure of fusion of the two palatine processes.

- Clefts of the lower lip result from failure of fusion of
the two mandibular processes. Classification according to Association with other syndromes 1. Syndromic A. Treacher collins syndrome B. Pierre robin syndrome C. Apert’s syndrome 2. Non-syndromic Anatomical classification A. Prealveolar clefts (lip clefts). B. Alveolar clefts. C. Postalveolar clefts (palatal clefts). (Unilateral, bilateral, complete and incomplete.)

Veau classification for cleft lip Class I: unilateral notching of the vermilion border that does not extend into the lip. Class II: unilateral notching of the vermilion extends into the lip but does not involve the floor of the nose. Class III: unilateral clefts of the vermilion border extending through the lip into the floor of the nose. Class IV: Any bilateral cleft of the lip, exhibiting incomplete notching or a complete cleft.

Veau classification for cleft palate Class I: A cleft limited to the soft palate Class II: defect of the hard and soft palate; extend not farther than the incisive foramen and therefore are limited to the secondary palate only. They may be complete or incomplete. A complete cleft includes the soft and hard palate, extending to the incisive foramen. An incomplete cleft involves the uvula, soft palate and a portion of the hard palate, not extending to the incisive foramen. Class lll: Complete unilateral clefts extending from the uvula to the incisive foramen in the midline and the alveolar process unilaterally. Class IV: complete bilateral clefts involving the soft and hard palate and the alveolar process on both sides of the premaxilla, leaving it free
and often mobile.


Submucosal clefts are not included in this system of classification, but they can be identified clinically by the presence of a bifid uvula, palpable notching of the posterior portion of the hard and soft palate, and the presence of a zonapellucida (a thin, translucent membrane) covering the defect. Nasopharyngial complications 1. Eustachian tube dysfunction. 2. Recurrent otitis media 3. Hearing deficits. 4. Palatal pharyngeal incompetence results from failure of the soft palate and pharyngeal wall to make contact during swallowing and speech, thus preventing the necessary muscular seal between the nasopharynx and the oropharynx.

Dental complications - Abnormalities of tooth number, size, morphology, calcification, and eruption. - Both deciduous and permanent dentitions may be affected. - The lateral incisor in the vicinity of the cleft is often involved and usually missing. - Teeth outside the cleft area exhibit developmental defects to a greater degree than is seen in unaffected patients. - The prevalence of hypodontia increases directly with the severity of the cleft. - Complete unilateral and bilateral alveolar clefts are often associated with supernumerary teeth. - Tooth formation is often delayed, and enamel hypoplasia, microdontia or macrodontia, and fused teeth are often seen.



6- Oblique Facial Cleft Definition: Developmental congenital cleft, which runs from the inner canthus of the eye to ala of the nose along the path of nasolacrimal duct, and often extends into the lip and primary palate.
Etiology: Due to failure of fusion of lateral nasal process and maxillary process.

7- Transverse Facial Cleft
Definition: This is a cleft running from the angle of the mouth to the ear.
Etiology: Due to failure of fusion of maxillary and mandibular processes.

8- Macrostomia: Means large mouth and is due to early arrest of fusion between maxillary and mandibular processes. 9- Microstomia: Means small mouth and is due to excessive closure of the maxillary and mandibular processes.

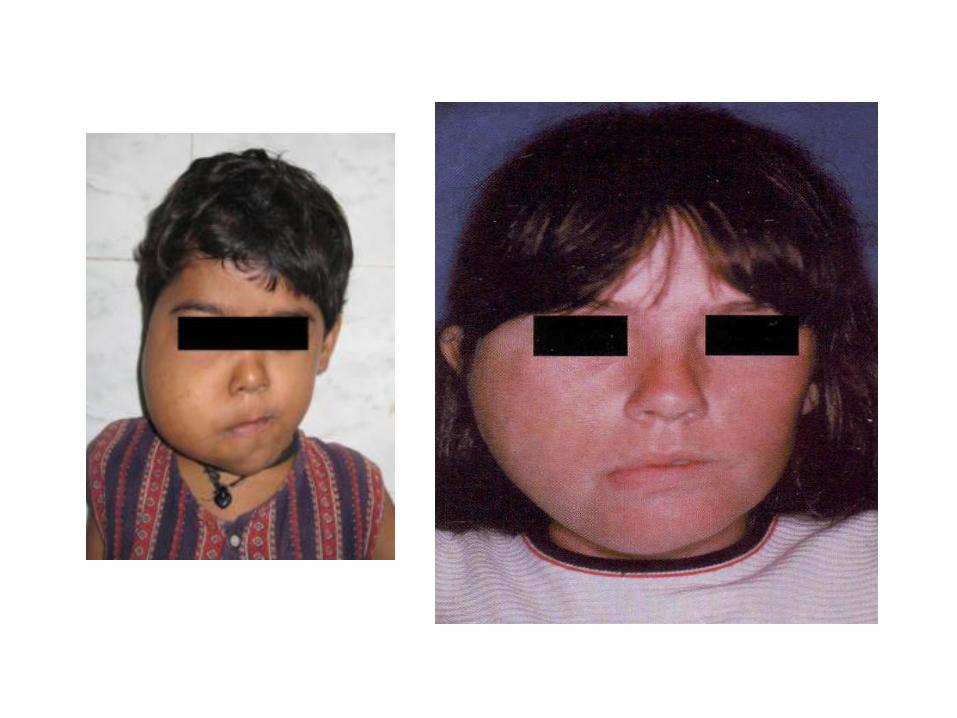
10-Facial Hemihypoplasia (Hemifacial atrophy) Definition: A characteristic loss in the soft tissues below the skin, usually on one side of the face, in some severe cases, the underlying bone is also affected. Etiology: - Peripheral nerve dysfunction. - Trauma or infection of the growth centers. - Genetic causes have been proposed. Clinically: - Starts in 1st or 2nd decade of life. - Left side more commonly affected. - Affected side may be hyperpigmented. - Hollowing of cheek and the orbit. - Jaw bones and roots of teeth on affected side may exhibit delayed development and retarded tooth eruption.

Radiographically: Deficient root development or root resorption.
Prognosis: The condition progresses slowly for a few years and remains stable.

11. Hemifacial hyperplasia (hemifacial hypertrophy) Definition Abnormal enlargement of one side of the face. Etiology Multiple etiologic factors have been suggested including vascular or lymphatic abnormalities, endocrine dysfunction, and chromosome abnormalities (heredity), but the cause remain obscure. Clinically - Asymmetry often is noted at birth (congenital) and continues until the patient's overall growth ceases. - The changes involve all the tissues on the affected side. - Unilateral macroglossia. -Enlargement of oral soft tissues and jaws on the affected side. - The crowns and roots of the teeth on the affected side are larger.

Developmental disturbances of the jaws 1- Agnathia : Congenital absence of jaw, classified into:
Complete agnathia: absence of the maxilla or the mandible.
Partial agnathia : absence of a part of one jaw as follows:
Maxilla:
- Absence of one maxillary process.
- Absence of the premaxilla.
Mandible:
- Unilateral or bilateral absence of the condyle.
- Unilateral or bilateral absence of the ramus.
- Absence of one mandibular process.
Unilateral absence of the ramus usually accompanied by deformity of the pinna of the ear on the affected side.

2- Micrognathia: Definition: Abnormal small jaw, classified into: Relative (False) micrognathia : due to disproportion between size of the jaw and size of skull or teeth, the jaw is still within normal size. True microagnathia : the jaw is smaller than normal & it is divided into: 1. Congenital true micrognathia : usually hereditary and associated with malformations such as Treacher Collins syndrome and Pierre Robin syndrome. 2. Acquired true micrognathia : it is due to environmental factors as infection or trauma or irradiation , which disturb the growth centers of the jaw.

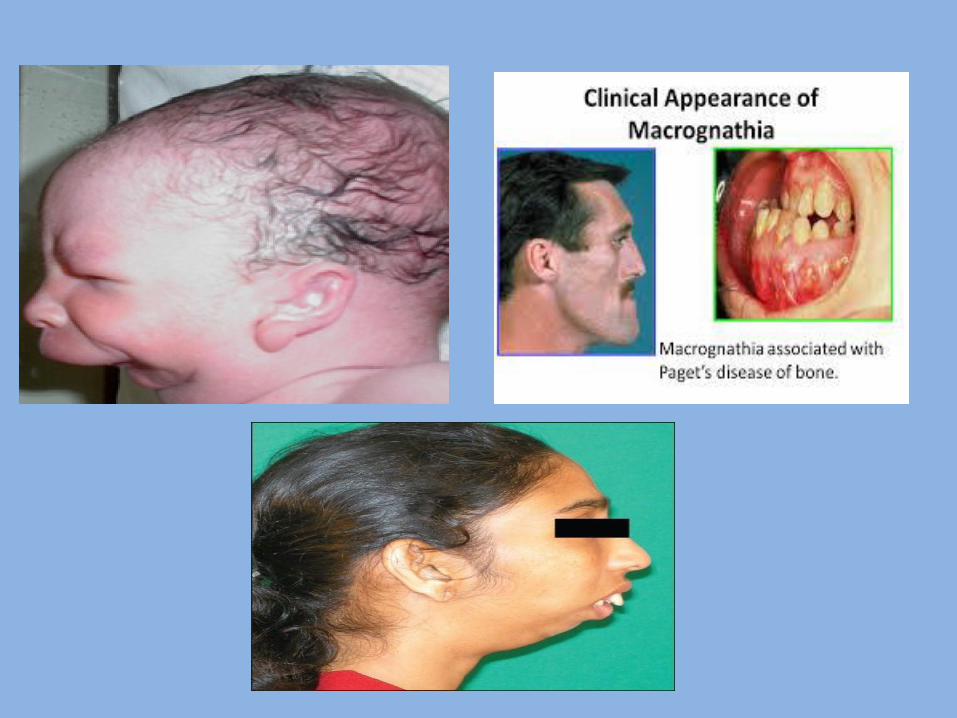
3- Macrognathia It means abnormal large jaw. It is classified into: Relative macrognathia: Due to disproportion between size of the jaw and size of skull or teeth as in Crouzon syndrome. True macrognathia :it is divided into: 1. Congenital true macrognathia : it is due to hereditary factors. 2. Acquired true macrognathia : it is due to bone diseases as Paget,s disease of bone , or hormonal imbalance as acromegaly , or other factors.

Developmental Disturbances Of Palate 1- Cleft palate
2- Exostoses:
Definition: They are localized bony Protuberances that arise from the cortical plate.
- Torus Palatinus: Hereditary condition
It is a common exostosis that occurs in the midline of the hard palate.
It presents as flat, spindle, nodular or lobular bony hard mass.
Most palatal tori are small and asymptomatic but in some cases, the thin overlying mucosa may become ulcerated.

-Torus mandibularis: It is a common exostosis that develops along the lingual aspect of the mandible above the mylohyoid line in the region of the premolars. Mostly it occurs bilateral, asymptomatic single nodules although multiple lobules are not uncommon. Tori, if large can interfere with stability of dentures and may need surgical removal or denture relief. - Tori considered to be hamartomatous condition.

Developmental disturbance of the lips:
1- Cleft lip
2- Commissural Lip Pits:
They are small mucosal invaginations that occur at the corners of the mouth on the vermilion border.
They may be unilateral or bilateral.
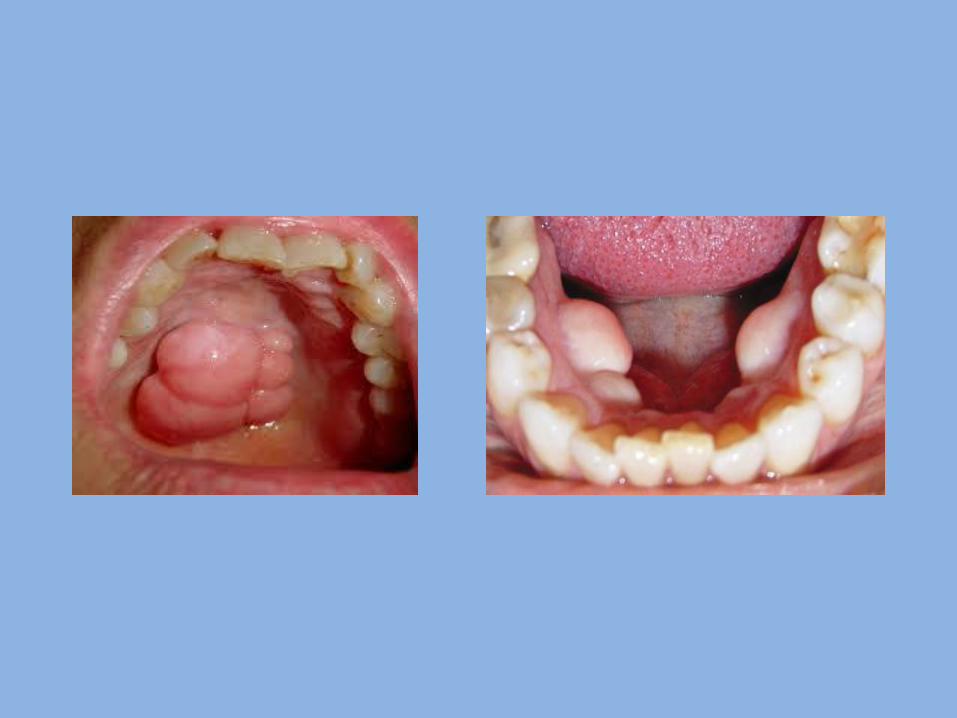
3- Paramedian lip pits and fistulae:
They are rare congenital invaginations of the lower lip they are believed to arise from persistence of the lateral sulci on the embryonic mandibular arch.
They are usually inherited as an autosomal dominant trait in combination with cleft lip and/ or cleft palate.
They present as bilateral and symmetric fistulas on either side of the midline of the vermilion of the lower lip.
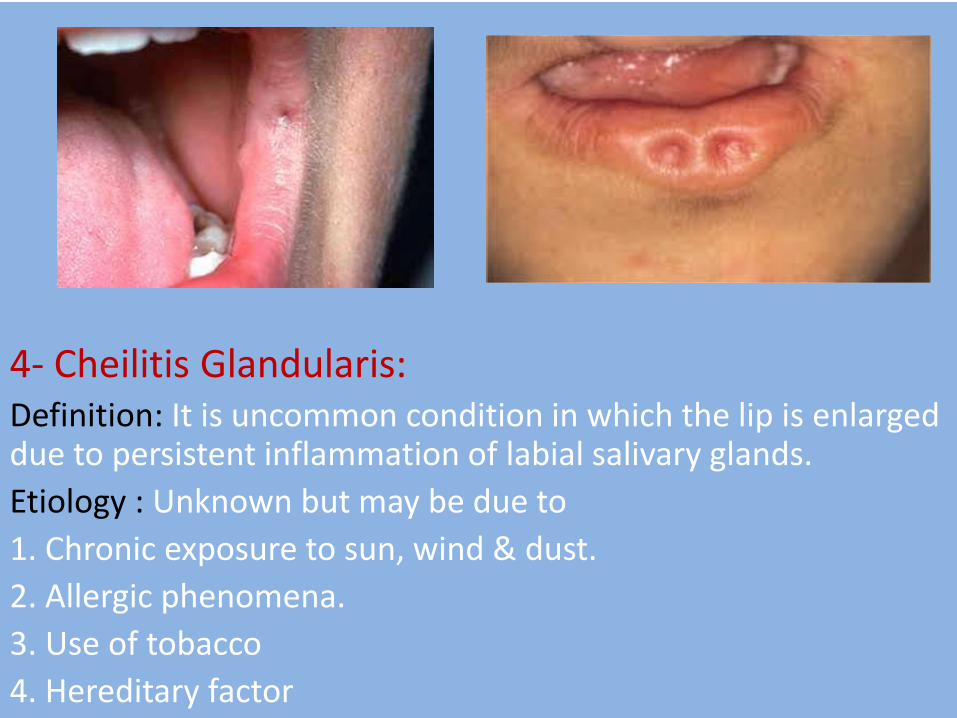
4- Cheilitis Glandularis: Definition: It is uncommon condition in which the lip is enlarged due to persistent inflammation of labial salivary glands.
Etiology : Unknown but may be due to
1. Chronic exposure to sun, wind & dust.
2. Allergic phenomena.
3. Use of tobacco
4. Hereditary factor

Clinical features: - Age : young adults - Sex : males more than female - site: lower lip - The lip is enlarged , firm , everted exposing it is mucosal surface to the sun so the minor labial mucous glands become nodular & the orifices of their ducts become inflamed & dilated giving the labial mucosa a red macular appearance. - The condition is considered to be precancerous.

5- Cheilitis Granulomatosa Definition: It is a condition in the lip; characterized by presence of chronic soft swelling in the upper lip. Etiology: It may occur due to allergy to some factors as: Cosmetics, Food, Oral hygiene products (e.g. tooth paste, mouth rinses) and Dental restorative materials. Clinical features : age: young adults sex : equal site: upper lip Characters: soft enlarged, firm and everted lip, tongue may be involved.
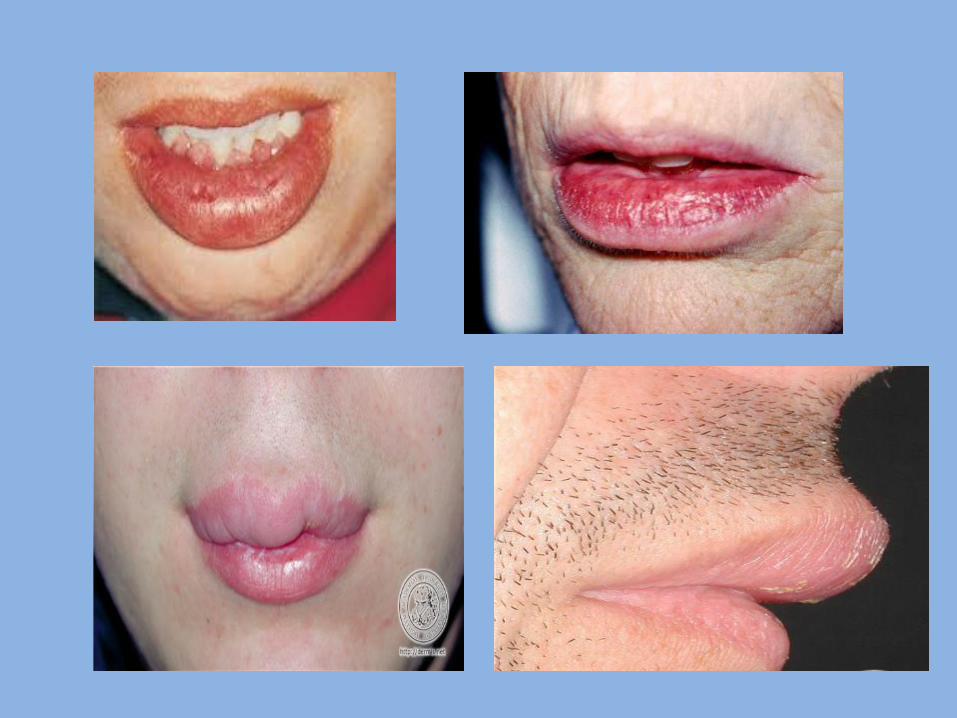

Melkerson Rosenthal Syndrome: 1. Cheilitis granulomatosa. 2. Facial paralysis. 3. Fissured tongue.
6- Double lip: - It is rare anomaly characterized by presence of fold of tissue on the mucosal side of the lip. - The upper lip is affected much more often than the lower lip. - The condition may be congenital or acquired later on life. - Congenital cases may result from malformation of the labial sulcus.
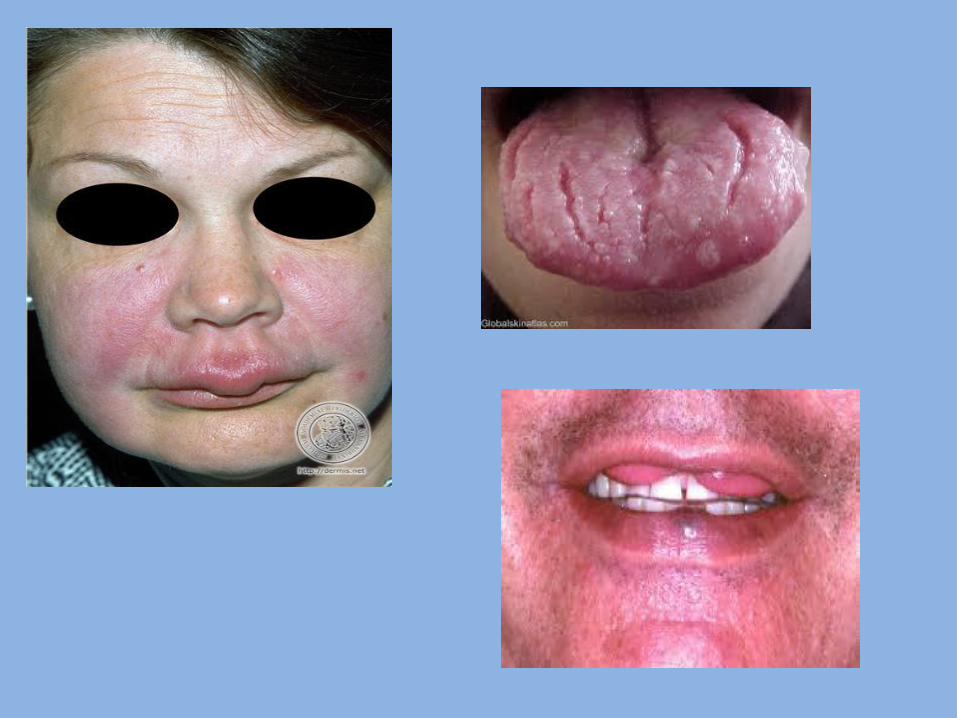

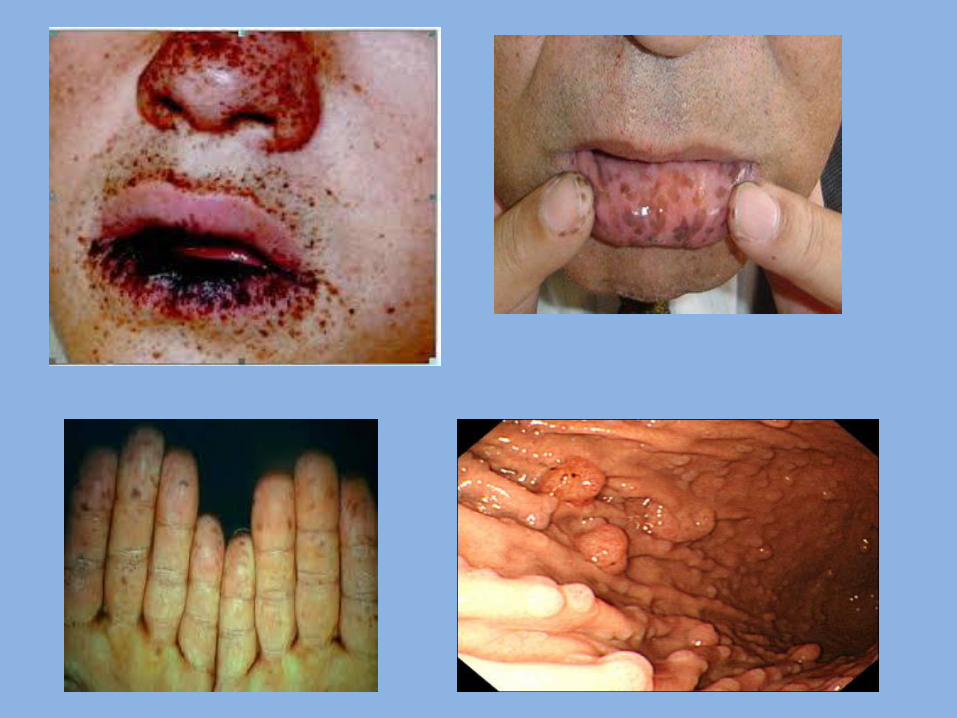
7- Peutz-Jegher’s syndrome (hereditary intestinal polyposis): Definition: It is hereditary malformation characterized by presence of multiple polyps in small intestine. - Transmitted as autosomal dominant trait. Clinical features: - Generalized intestinal benign polyps (in Small intestine). These polyps cause mechanical obstruction, resulting in attacks of abdominal pain. These polyps are hamartomas(polyps are projections of mucous membrane). - Spots of melanin pigmentation: 1. Around the mouth, nostrils & eyes. 2. On the oral mucosa ( lip , gingiva& palate). 3. On the skin of the dorsal surface of hands and feet.

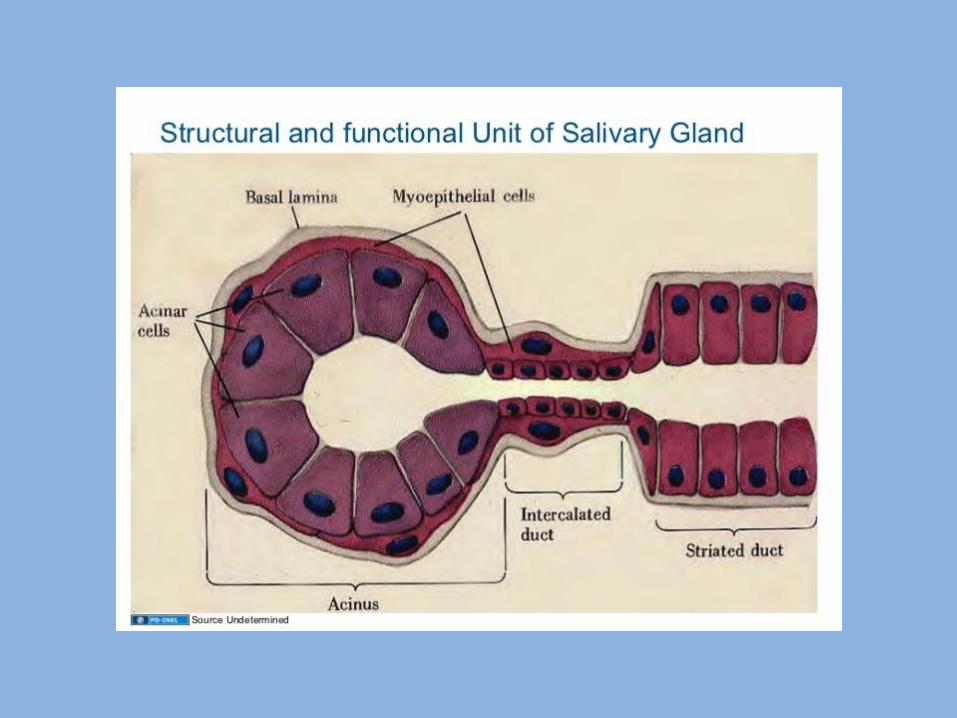
Developmental disturbances of Salivary Glands
1- Aplasia:
- It means a complete absence of one or more of the salivary glands.
- It results from failure of the terminal portion of the gland bud to differentiate into salivary tissue.
- The condition of no clinical significance unless the remaining salivary glands fail to compensate for the decrease in saliva production, xerostomia will result.
2- Atresia:
This is a condition in which there is congenital absence or occlusion of one or more of the ducts of major salivary glands.

- It result from degeneration or failure of canalization of the proximal part of the salivary gland after the distal part differentiated into salivary gland tissue. 3- Aberrancy: - It is an ectopic lesion in which normal secreting Salivary gland tissues develop at an abnormal position. - It has no clinical or pathological significance apart from the fact that the aberrant tissue may be the site of development of a cyst or neoplasms. - Most famous example is Staphne defect. Latent Bone Cyst (static bone cyst, Stafne defect, Lingual mandibular salivary gland depression): - It represent developmental ectopic defect containing a portion of the sub mandibular gland or rarely sublingual gland develops in bony cavity in the lingual surface of body of the mandible. - It mostly presents as a unilateral asymptomatic radiolucency below the mandibular canal in the posterior mandible between the molar teeth and the angle of the mandible.

- The condition is static and never increase in size. - Injecting radio-opaque material through the parent gland will confirm the nature of the lesion.

Developmental disturbances of the buccal mucosa
1- Fordyce’s granules: They are ectopic collections of sebaceous glands present in the oral mucosa. Pathogenesis: May result from inclusion of ectoderm possessing the potentialities to form skin appendages at the line of fusion of the maxillary and mandibular processes during embryonic life. Clinical features: - Common site: Buccal mucosa opposite to molars, lips & anterior tonsillar pillar. - Small yellow spots, single or multiple separated or forming large plaque, may be raised above the surface. - Scattered along linea alba buccalis, may grow in size with age. - Present in 80% of population so considered to be normal variant of oral mucosa.

Histological features: - They are consisting of sebaceous follicles. Sometimes lying free in the CT. (dermis). - Connected to the surface epithelium, directly or by a duct which may be blocked by keratin. They are not related to root of hair follicles.

2- White spongy nevus: It is a hereditary, autosomal dominant developmental malformation of the oral mucosa. Nevus: developmental malformation of skin or mucous membrane arising from cells native to skin or mucous membrane and resembling neoplasm. Nevi are of 3 types: - Keratotic ( white spongy nevus) - Melanotic ( pigmented nevi) - Vascular ( hemangioma and lymphangioma) Common site: Entire oral mucosa, buccal mucosa, floor of mouth & ventral surface of the tongue. Clinical features: - It appears as thick, soft, spongy, fissured, folded, greyish - white & diffuse areas bilaterally. - May appear at birth, in infancy, or in children and It reaches full severity at puberty and then remains static.
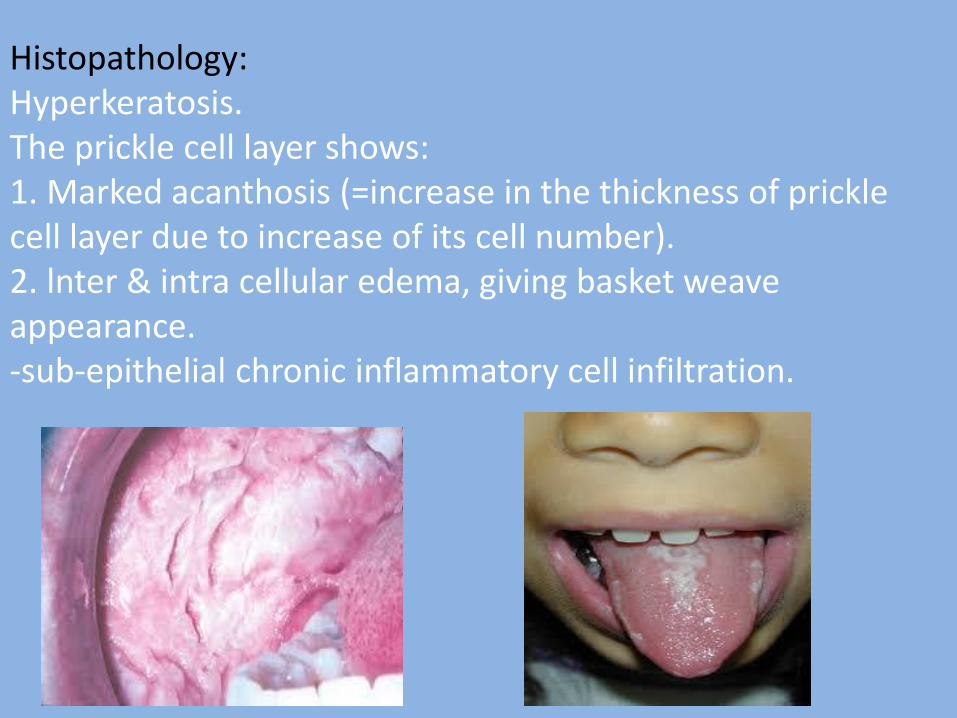
Histopathology: Hyperkeratosis. The prickle cell layer shows: 1. Marked acanthosis (=increase in the thickness of prickle cell layer due to increase of its cell number). 2. lnter & intra cellular edema, giving basket weave appearance. -sub-epithelial chronic inflammatory cell infiltration.


3- Hereditary benign intraepithelial dyskeratosis (red eye): - It is a hereditary disease affecting the oral mucosa, conjunctiva & sometimes the cornea - Dyskeratosis: abnormal keratinization of epithelial cells. - Common site: Buccal, labial mucosa, floor of mouth & lateral borders of the tongue, conjunctiva & cornea. - Clinical features: It resembles the white spongy nevus. - Eye lesion: Corneal opacities (white plaque), temporary blindness, and conjunctiva is congested so the term red eye. Histopathology: it consists of: Hyper-parakeratosis. The lesion similar to white spongy nevus except the presence of eosinophilic dyskeratotic cells in prickle cell layer, most of the cells show perinuclear condensation of keratin. These cells described as “cell within cell dyskeratosis”


4- Leukodema: Definition - Generalized opacification of the buccal mucosa that is regarded as a variation of normal. - There is an alteration of oral epithelium characterized by the intracellular accumulation of fluid (edema) within the spinous cell layer. - Etiology of this condition is unknown. Clinically -The affected mucosa shows an asymptomatic, diffuse, translucent, grayish-white appearance. It affects the buccal mucosa bilaterally. -When stretched, the appearance is greatly decreased. -No treatment is necessary.


Developmental disturbances of the tongue
1- Aglossia
- It means congenital absence of the tongue.
- It is due to failure of the development of the anterior two thirds of the tongue from the ventro - medial surface of the first arch.
- lt disturb the speech, mastication & swallowing.
The tuberculum impar may develop in the place of the normal tongue and improves the speech.
2- Microglossia:
- It means abnormal small tongue.
- The condition is of little clinical significance.

3- Macroglossia
It means abnormal large tongue. Types: 1. Congenital macroglossia: Etiology: - Cretinism (congenital malformation). - Monogolism. - Lymphangioma or hemangioma. - Fibrous and muscular hypertrophy. 2. Acquired Macroglossia Etiology: - Myxedema(adult hypothyroidism). - Acromegaly(adult hyperpituitrism). - Blockage of the lingual lymphatic vessels as in malignant neoplasm or infection.

- Tumors as papilloma , fibroma, carcinoma , or sarcoma. - Chronic granulomas as actinomycosis, syphilis, or T.B. Clinical features: - Enlarged tongue. - displacement & spacing of teeth & malocclusion. - Scalloping of borders of the tongue, because they are compressed against the teeth & interdental spaces. - in acquired macroglossia the edges of the tongue are scalloped as they fit against the teeth and interdental spaces. - In congenitaI macroglossia the edges are smooth.


4- Ankyloglossia(Tongue Tie) It is a developmental malformation, in which the tongue movements are restrained. Types: 1. True (inferior): may be complete or partial: - (complete) ankyloglossia: it is due to failure of separation of the tongue from the floor of the mouth. - Partial ankyloglossia ( commonest type): due to abnormal short lingual frenum , or the lingual frenum attached to the tip of the tongue. 2. Superior ankyloglossia: Always, it is associated with cleft palate. It is due to the dorsal surface of the tongue is connected by a membrane to one side of the palate.
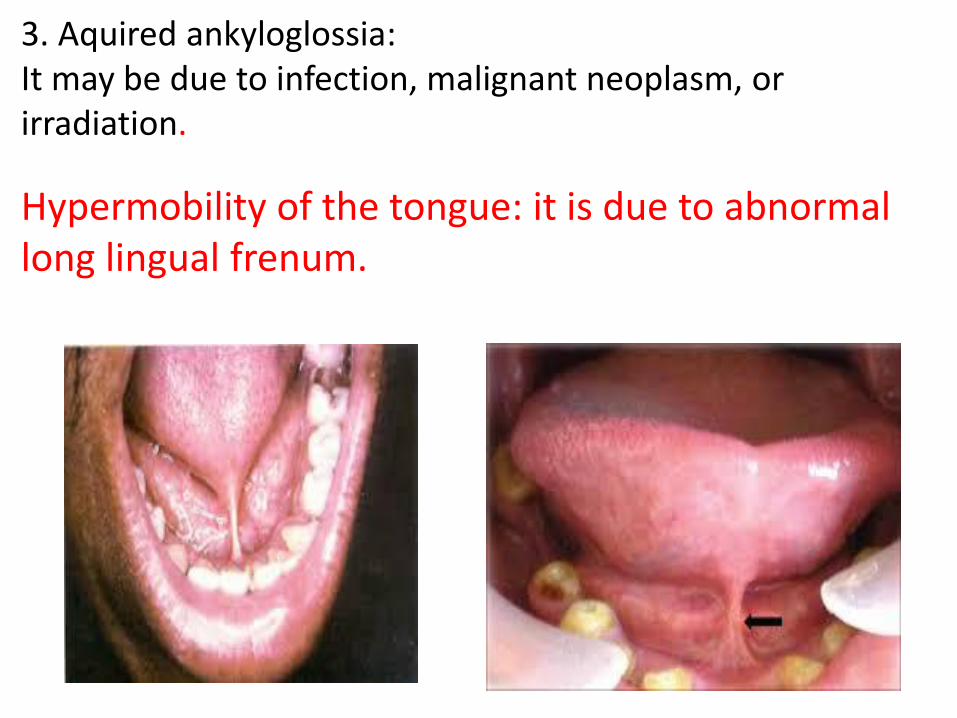
3. Aquired ankyloglossia: It may be due to infection, malignant neoplasm, or irradiation. It may be due to infection, malignant neoplasm, or irradiation.
Hypermobility of the tongue: it is due to abnormal long lingual frenum.

5- Fissured tongue (scrotal tongue): The tongue having on it is dorsal surface (of anterior, 2/3) in addition to the normal median raphe, symmetrical or semi-symmetrical grooves, or fissures. Etiology: Frequently, it is congenital, but because it increase with age , it may be acquired due to: 1. Chronic trauma 2. Vitamin deficiency 3. Tobacco smoking 4. Psychological stress 5. Infection as syphilis Clinical features: The fissures are depapillated. The fissures are classified according to their arrangement into: a. Transverse b. Cerebriform c. Foliaceous d. Irregular
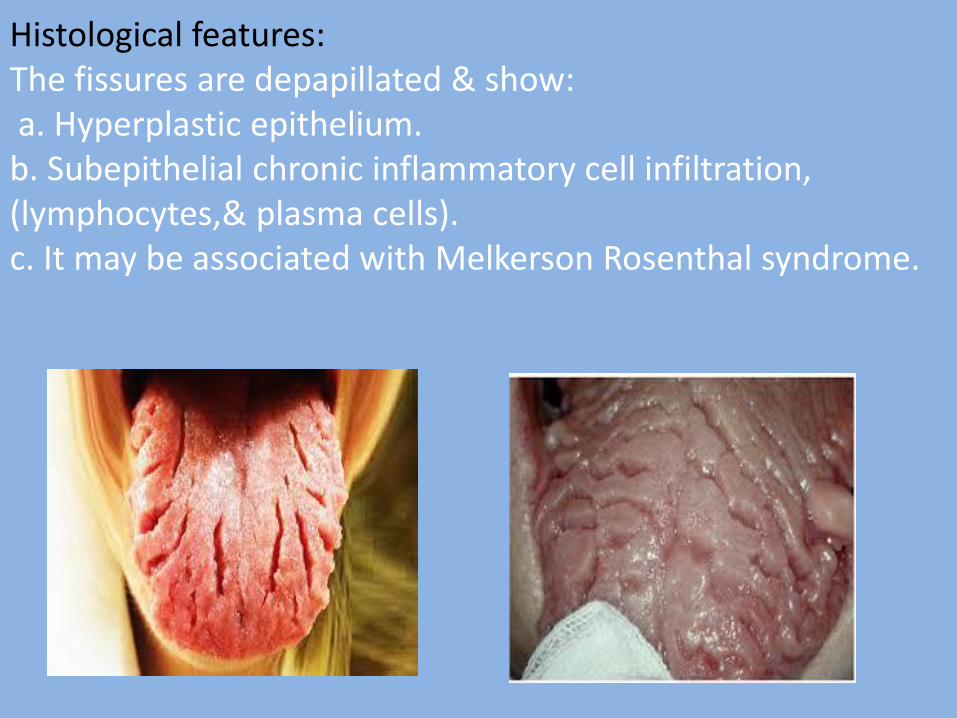
Histological features: The fissures are depapillated & show: a. Hyperplastic epithelium. b. Subepithelial chronic inflammatory cell infiltration, (lymphocytes,& plasma cells). c. It may be associated with Melkerson Rosenthal syndrome.

6- Median rhomboid glossitis Definition: Congenital malformation characterized by an area of redness and loss of lingual papillae, situated on the dorsum of the tongue in the midline immediately in front of the circumvallate papillae.
Etiology: Median rhomboid glossitis is thought first to be created by failure of the tuberculum impar to retract or withdraw before fusion of the two halves of the anterior 2/3 of the tongue .
Now it is well known as a chronic fungal infection, and usually is a type of oral candidiasis.
Clinically:
- The typical appearance of the lesion is an oval or rhomboid shaped area located in the midline of the dorsal surface of the tongue, just anterior (in front) of the sulcus terminalis.
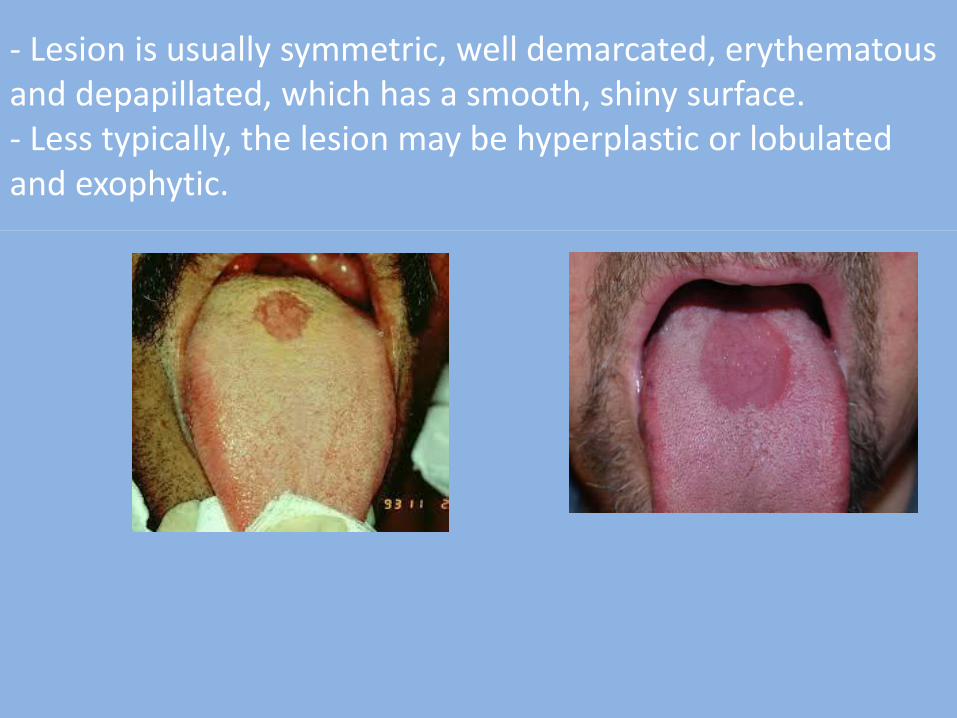
- Lesion is usually symmetric, well demarcated, erythematous and depapillated, which has a smooth, shiny surface. - Less typically, the lesion may be hyperplastic or lobulated and exophytic.

7- Geographic tongue (glossitis areata exfoliativa, erthema migrans , benign migratory glossitis) It is of unknown etiology or due to emotional stress.
Clinical features:
Age: unhealthy children, sometimes older individuals especially women.
Characters: The anterior 2/3 of the tongue show many small red patches
characterized by:
1. Desquamation of the filiform papillae.
2. The fungiform are retained & appear red dots.
3. Each patch appears as a small red area round or oval in shape with a slightly elevated yellowish irregular border.
4. Grow slowly & coalesce, forming larger patches.
5. Each patch persists for about one week and then disappears only to
reappear at another area.
6. The whole condition regresses spontaneously &may recur later.

Histopathology - At the periphery, elongation of the rete ridges is noted with associated hyperparakeratosis and acanthosis. - Toward the center of the lesion, corresponding to the erythematous area, loss of filiform papillae with migration and clustering neutrophils within the epithelium is seen. - The predominant inflammatory infiltrate in the lamina propria is neutrophils with admixture of chronic inflammatory cells.

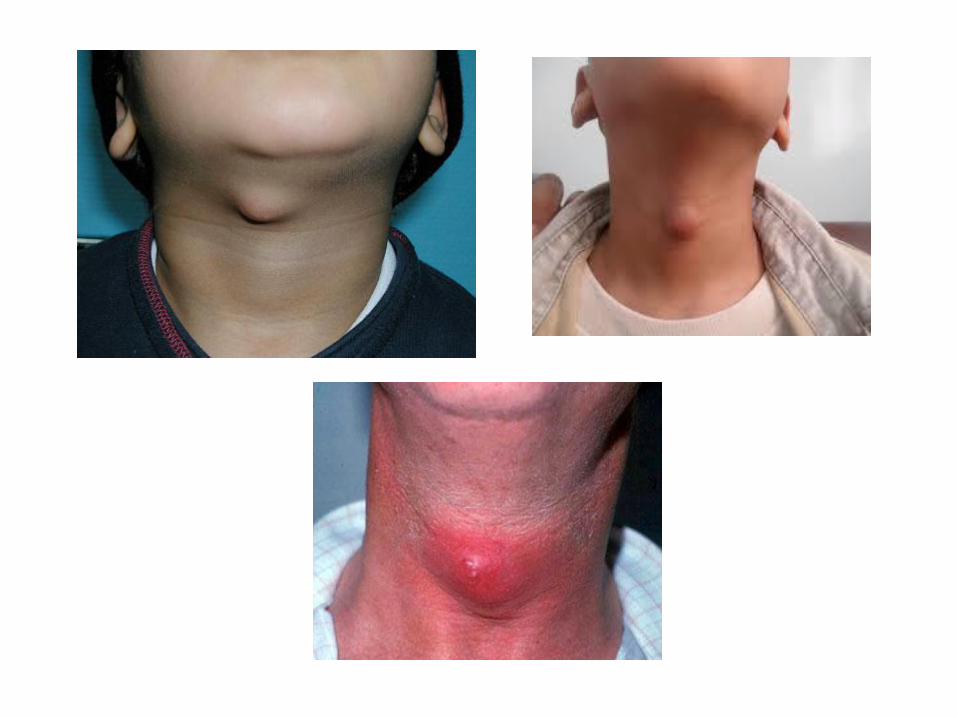
8- Thyroglossal tract cyst: It is a developmental true cyst which is a pathological cavity lined by epitheliam contain fluid or semi - fluid. Pathogenesis: It arises from proliferation & cystic degeneration of the epithelial remnant of the thyroglossal tract. Clinical features: It appears as a slowly growing swelling occurs in the midline , & classified into: 1. Supra-hyoid cyst which occurs in the floor of mouth, or the tongue. 2. Infra hyoid cyst which occurs in the midline of the neck & moves upwards downwards during swallowing. Sometimes, it ruptures forming a fistula with overlying skin or mucosa.
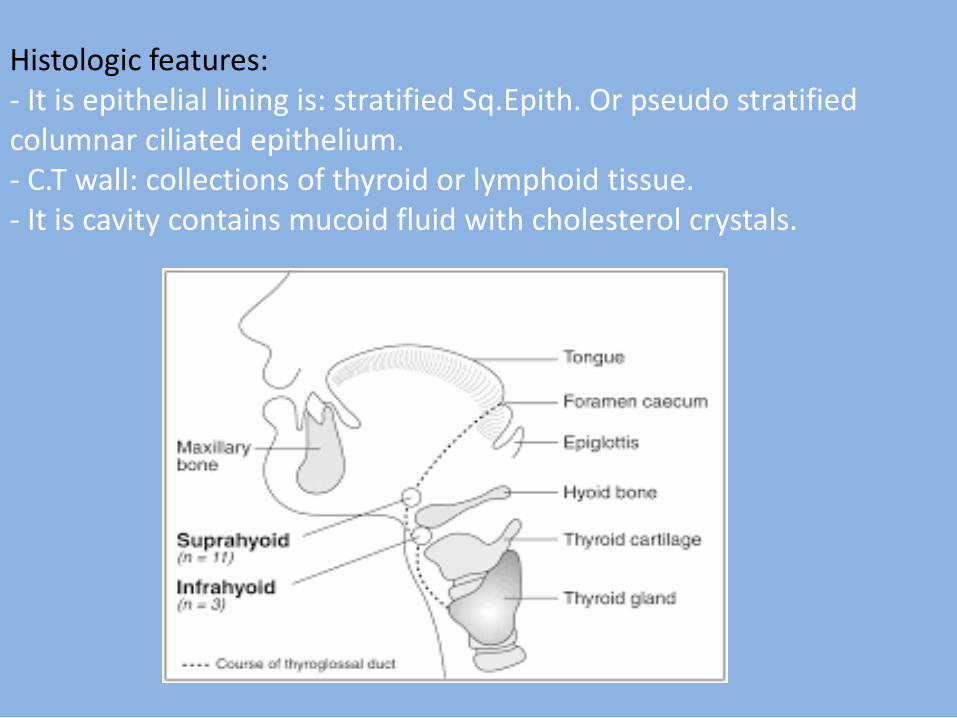
Histologic features: - It is epithelial lining is: stratified Sq.Epith. Or pseudo stratified columnar ciliated epithelium. - C.T wall: collections of thyroid or lymphoid tissue. - It is cavity contains mucoid fluid with cholesterol crystals.

9-Lingual thyroid nodule
It is an ectopic thyroid tissue occurs in the tongue
Pathogenesis:
Deficient migration may occur at the area of foramen caecum
and differentiation of the epithelial tract into thyroid tissue takes place while it still lies in the substance of the tongue.
Clinical features:
It appears as a nodular mass; occurs in the midline in the area of the foramen caecum, & covered by normal mucosa. Histological features:
- It consists of collection of thyroid tissue.
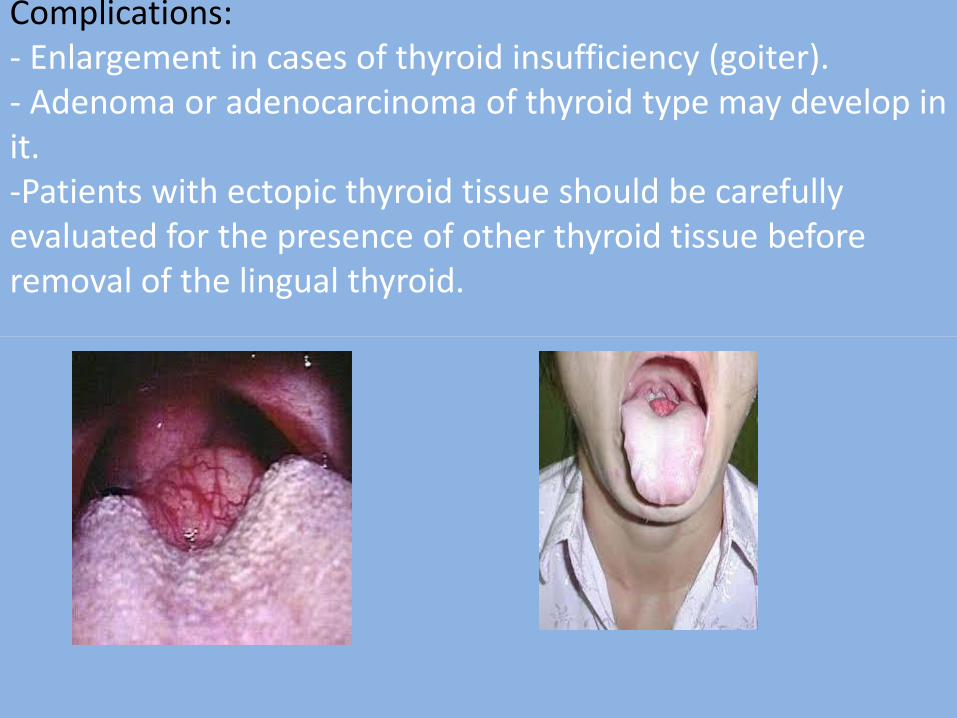
Complications: - Enlargement in cases of thyroid insufficiency (goiter). - Adenoma or adenocarcinoma of thyroid type may develop in it. -Patients with ectopic thyroid tissue should be carefully evaluated for the presence of other thyroid tissue before removal of the lingual thyroid.

10- Hairy tongue (Black hairy tongue): - It is characterized by marked accumulation of keratin on the filiform papillae of the dorsal tongue resulting in a hair- like appearance.
- This condition represents an increase in keratin production or a decrease in normal keratin desquamation. Other possible associated factors include:
1. Heavy smoking.
2. Antibiotic therapy.
3. Poor Oral Hygiene.
4. Radiation therapy.
5. Overgrowth of fungal or bacterial organisms
6. Use of oxidizing mouth washes or antacids.
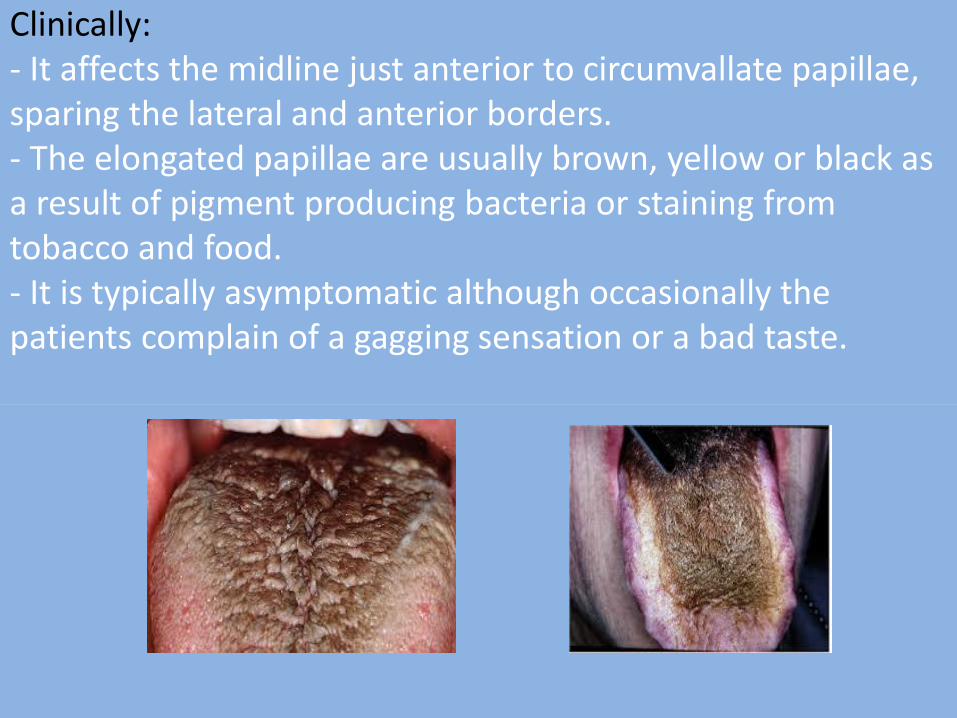
Clinically: - It affects the midline just anterior to circumvallate papillae, sparing the lateral and anterior borders. - The elongated papillae are usually brown, yellow or black as a result of pigment producing bacteria or staining from tobacco and food. - It is typically asymptomatic although occasionally the patients complain of a gagging sensation or a bad taste.
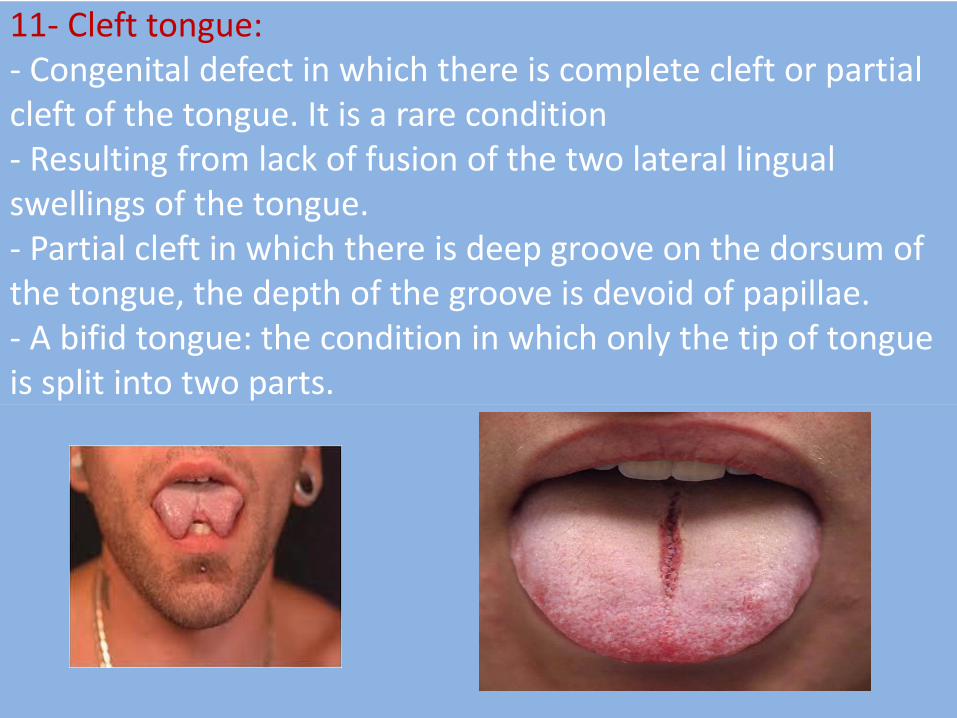
11- Cleft tongue: - Congenital defect in which there is complete cleft or partial cleft of the tongue. It is a rare condition - Resulting from lack of fusion of the two lateral lingual swellings of the tongue. - Partial cleft in which there is deep groove on the dorsum of the tongue, the depth of the groove is devoid of papillae. - A bifid tongue: the condition in which only the tip of tongue is split into two parts.

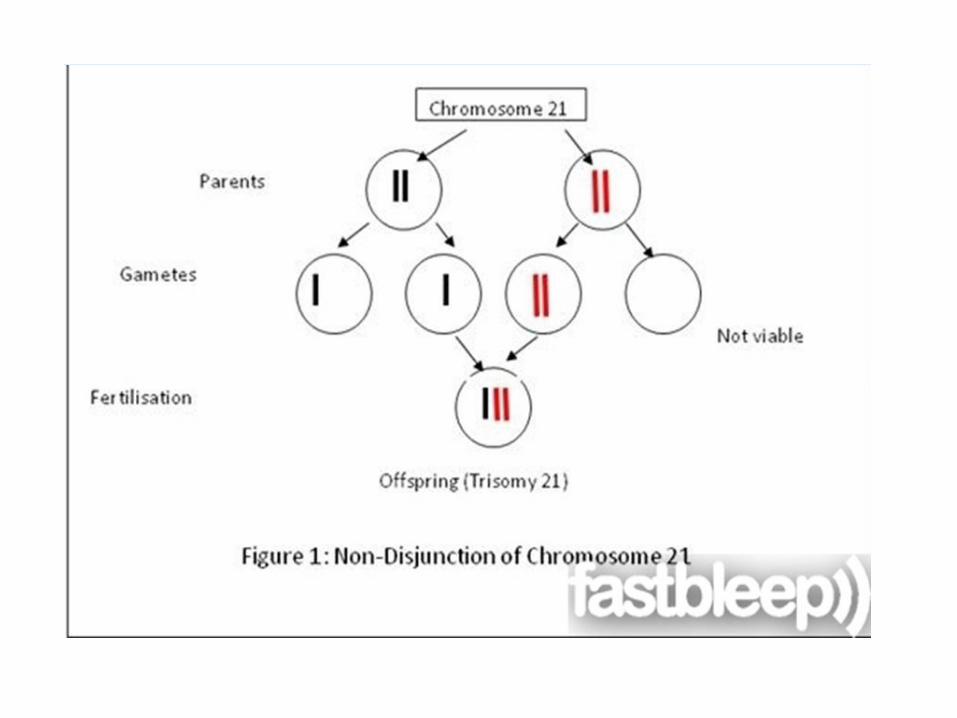
Common syndromes Mongolism (Down,s syndrome , trisomy 21 chromosome)
- It is a developmental disturbances, due to trisomy 21 chromosomes which results from failure of separation of the 21 chromosome pair at the reduction division (meiosis).
Clinical features:
1. Macroglossia ¯ognathia.
2. Mongloid slant of the eyes.
3. Multiple congenital malformations of the heart & joints.
4. Sexual underdevelopment.
5. Mental retardation.
6. High arched palate

7. Microdontia 8. enamel hypoplasia 9. malformed teeth. 10- Decreased incidence of caries, and increased incidence of periodontal disease.

Thank you