developing protocols & procedures for ct data integrity
TRANSCRIPT

DEVELOPING PROTOCOLS & PROCEDURES FOR CLINICAL TRIAL DATA INTEGRITY
Dr. Bhaswat S. ChakrabortySenior Vice President, R&D
Cadila Pharmaceuticals Ltd.
1
Presented at the International Conference on CRO/Sponsor Summit on “Data Integrity in
Clinical Research” at Hyderabad, India, 21-22 July, 2016

CONTENTS Clinical trial data Approaches to build and maintain data integrity CT Data management system On-site monitoring Centralized monitoring Alternate monitoring Risk based monitoring (RBM) Data & safety monitoring plan (DSMP)
Data & safety monitoring board (DSMB) Training & inspection-readiness Concluding remarks
2
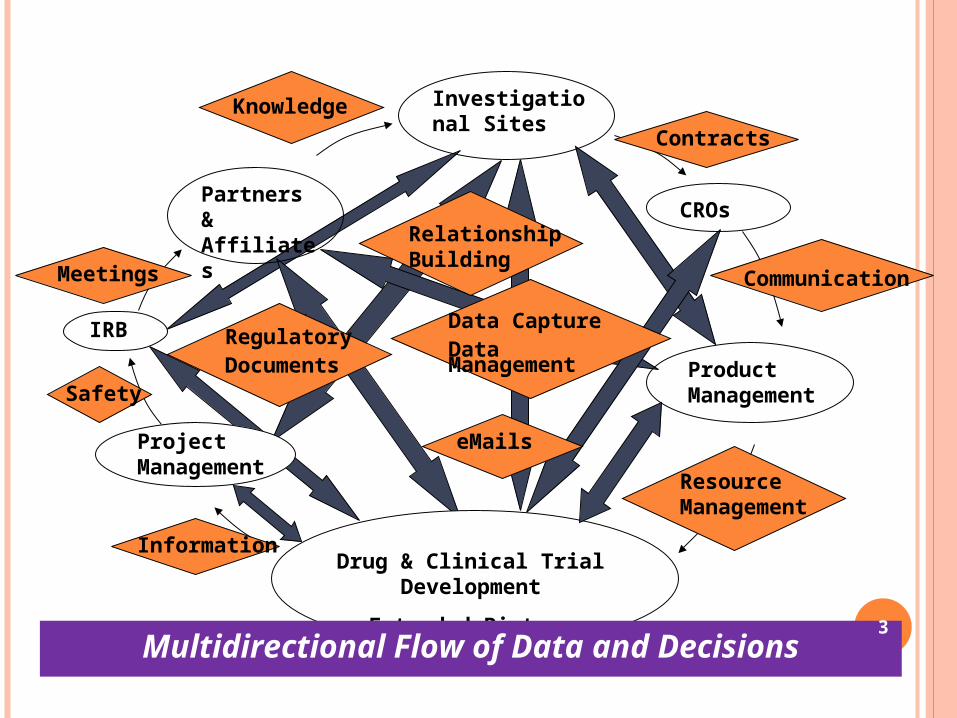
Investigational Sites
Product Management
Project Management
Drug & Clinical Trial Development
Extended Picture
IRB Regulatory Documents
Relationship Building
eMails
Partners & Affiliates
Meetings
CROs
ContractsKnowledge
Information
Safety
Communication
Resource Management
Data Capture Data Management
Multidirectional Flow of Data and Decisions 3

CLINICAL TRIAL DATA AND DOCUMENTS Study and site feasibility documents Protocol
Inclusion/Exclusion criteria Informed Consent Investigators brochure Training documents and data Data
Randomisation, Blinding CRF/ECRF (Demographics and site visit data) Primary and secondary outcome variables (end points) Clinical procedures and study conduct data Investigational products: Supply, Inventory, Handling & Usage, Retention Safety monitoring and signal detection Subject withdrawal and retention data Data and safety monitoring committee (activities, data, reports) Data management and data monitoring including SDV by Sponsor/CRO Data recording and reporting
Statistical analysis Study reporting ..
4

WHY DO WE NEED A DATA MANAGEMENT & DATA INTEGRITY SYSTEM? Enormous volumes of data
Example, a Phase-III trial in 10 centres with 100 patients each
60 pages of CRF for each recruited patient 20 fields each page
40 pages of screening form for each candidate patient 20 fields each page
[1000 (60 x 20)] + [1500 (40 x 20)] = 12, 00000 + 12, 00000= 24,00000 specific data points 5

CLINICAL TRIAL DATA Useful only if it is clean & accurate Data processing must be
real-time subject randomization management of clinical trials materials laboratory uploads patient diary data
Integrated Consistent Accurate
Data structures must be Standard Validated
Data transfer method must be Standard Validated 6

DATA INTEGRITY IN RESEARCH Research integrity depends on data integrity
Includes all aspects of collection, use, storage and sharing of data.
Data integrity is a shared responsibility Everyone involved in the research is responsible. The ultimate responsibility belongs to the PI. However, there is a broader role and responsibility for the
institute and scientific community. Transparency of the research data is required
7Free and accurate information
exchange is fundamental to scientific progress
Van Eyk J., JHU NHLBI Innovative Proteomics Center on Heart Failure

SOURCES OF DATA INTEGRITY & ITS LACK Data integrity is based on accurate and traceable:
Collection Organization & Storage Analysis Reporting
Data integrity can be compromised numerous ways: Malicious or unethical intentions Human mistakes and naivety Technical error Misinterpretation
8
Accurate & complete data = High quality data = QbD & RBM data = IRB+DSMP+DSMB data

CONSEQUENCES OF NON-INTEGRITY
Personal loss Blocked scientific
progression Impaired technology
development Damage to the
institution, sponsors or CRO
Tarnished public perception of science
Damage to or loss of patent protection 9
Van Eyk J., JHU NHLBI Innovative Proteomics Center on Heart Failure

APPROACHES TO BUILD AND MAINTAIN DATA INTEGRITY Monitoring, monitoring ,,,, monitoring
CTDM Systems On site monitoring Centralized monitoring
Clinical trial quality assurance units (QAUs) Sponsors often use internal or external QAUs Not required by regulation
QbD and Risk based monitoring Building QbD Risk identification & assessment Critical attributes and riskcategorization thereof Plans and processes Targeted monitoring 10
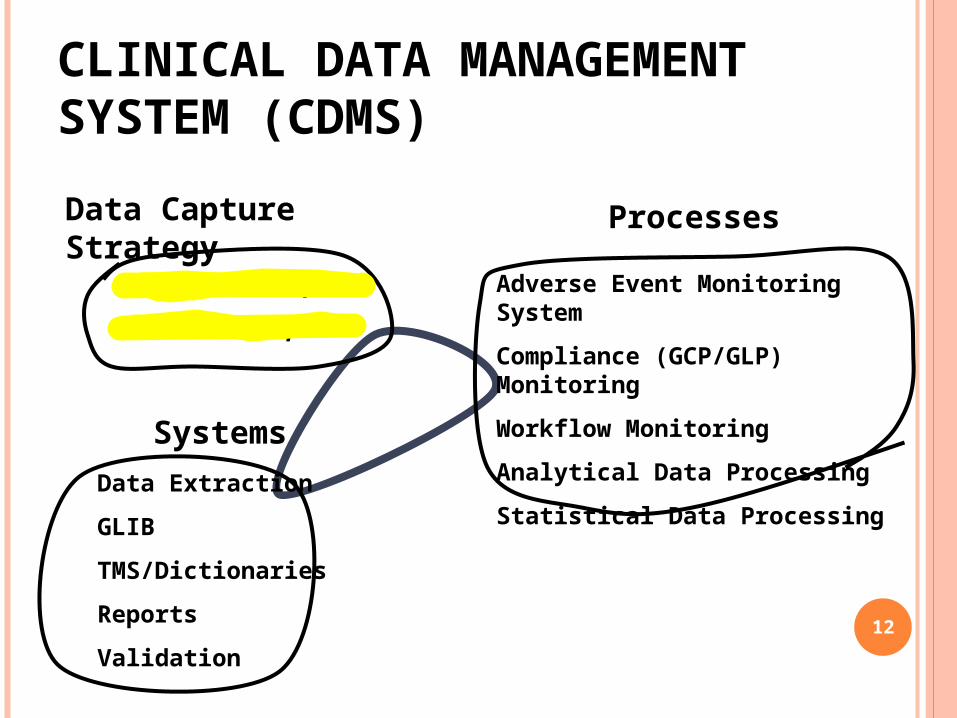
CLINICAL DATA MANAGEMENT SYSTEM (CDMS)Data Capture Strategy
Remote Data Capture
Portal Data Capture
Processes
Adverse Event Monitoring System
Compliance (GCP/GLP) Monitoring
Workflow Monitoring
Analytical Data Processing
Statistical Data ProcessingSystems
Data Extraction
GLIB
TMS/Dictionaries
Reports
Validation12

CRF Database
Data Entry Layout
CRF Maker CRF
(Form) Editor
Edited
Hard Copy
Electronic Case Report Forms13

FIRST & SECOND ENTRY DATABASES
First entry (first database) For accurately capturing the data to be cleaned &
analyzed by SAS datasets, data entry personnel entered data from paper or eCRFs
Second entry (second database) Errors can due to data transcription Thus all data are entered a second time into a second,
but identical, database by a second personnel Entered values are compared between the two databases If the two entries match, the entered value was accepted If not matched, the personnel makes a decision whether
the initial or second data entry value is correct.
14
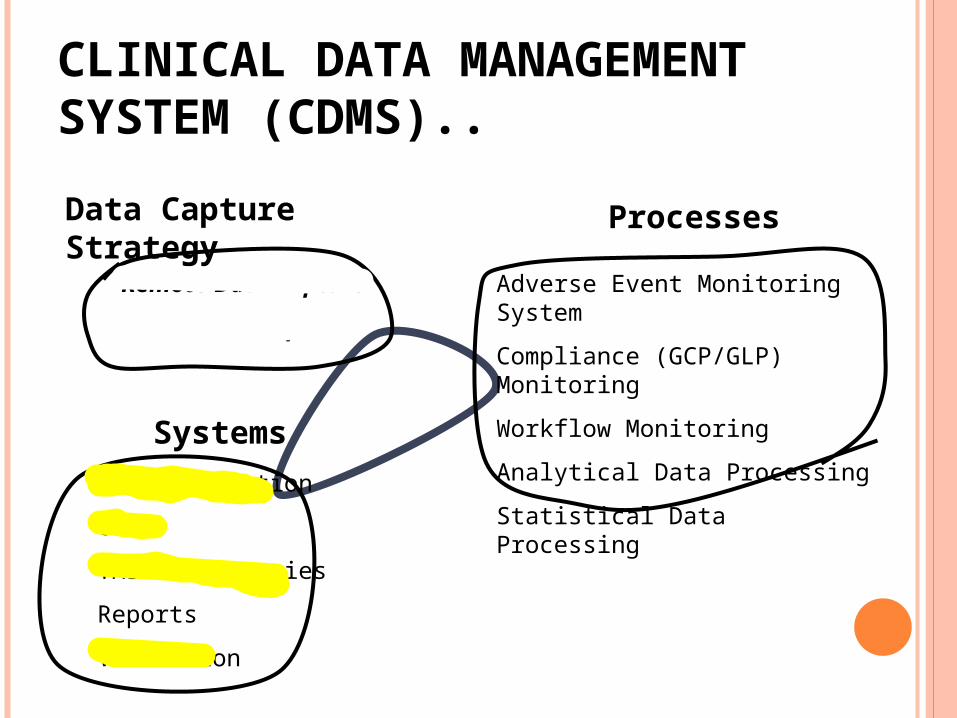
CLINICAL DATA MANAGEMENT SYSTEM (CDMS)..Data Capture Strategy
Remote Data Capture
Portal Data Capture
Processes
Adverse Event Monitoring System
Compliance (GCP/GLP) Monitoring
Workflow Monitoring
Analytical Data Processing
Statistical Data ProcessingSystems
Data Extraction
GLIB
TMS/Dictionaries
Reports
Validation
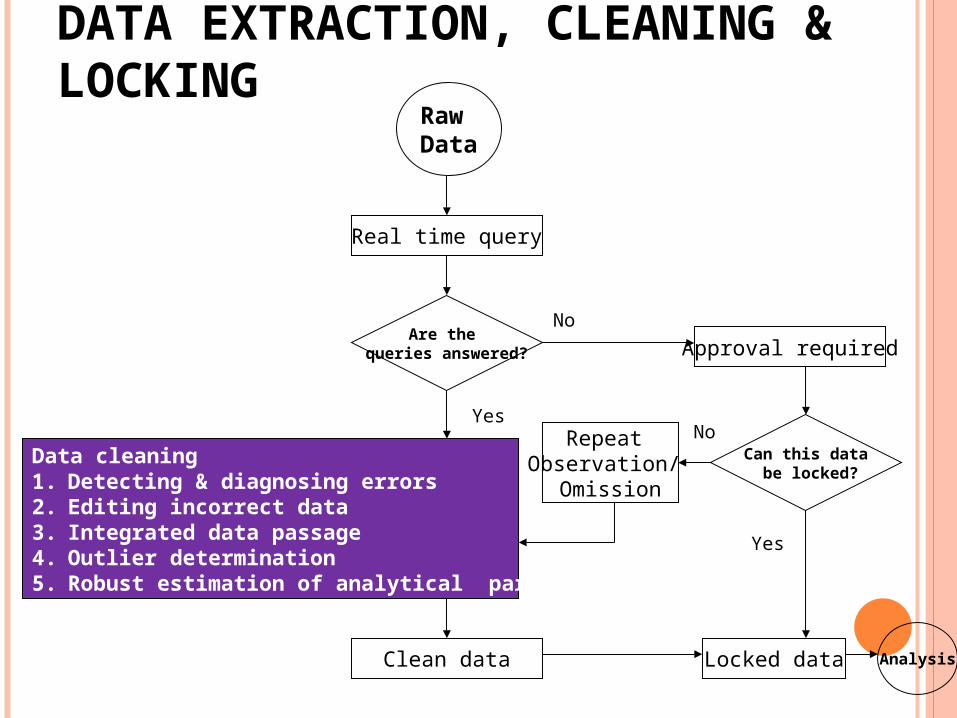
Raw Data
DATA EXTRACTION, CLEANING & LOCKING
Real time query
Are the queries answered?
Yes
No
Data cleaning1. Detecting & diagnosing errors2. Editing incorrect data3. Integrated data passage4. Outlier determination5. Robust estimation of analytical parameters
Clean data
Approval required
Can this data be locked?
Yes
NoRepeat Observation/
Omission
Locked data Analysis

ON-SITE MONITORING FOR INTEGRITY Effective monitoring is critical to:
Human subject protection Conduct of high‐quality studies Data Integrity
On-site monitoring An in-person evaluation at the investigational sites Can identify
data entry errors missing data in source records or CRFs provide assurance that study documentation exists assess compliance with the protocol and investigational product quality of the overall conduct of the trialat that site particularly helpful early in a study, especially if protocol is complex
and includes novel procedures lead to meaningful training efforts 17

CENTRALIZED MONITORING A remote evaluation carried out by sponsor personnel or CRO
By clinical monitors, data management personnel, or statisticians At a location other than the sites Can provide many of the capabilities of on-site monitoring as well as
value additions Success of centralized monitoring depend on various factors
Use of electronic systems Access to subjects’ electronic records Timeliness of data entry from paper CRFs Must ensure that record keeping, data entry, and reporting and
supporting source data are well-defined & accessible Must also identify in their monitoring plan when one or more on-site
monitoring visits are required
18Alternative monitoring techniques can exist

ALTERNATE MONITORING Monitor/review data quality
missing data, inconsistent data, data outliers, and potential protocol deviations
Conduct statistical analyses to identify data trends not easily detected by onsite monitoring, such as checks of range, consistency, completeness, unusual distribution of data
within and between sites Analyze site characteristics, performance metrics
high screen failure, withdrawal rates, high eligibility violations, delays in reporting ..
Verify critical source data remotely where accessible; CRF data are according to the protocol?
Complete administrative and regulatory tasks IRB approvals, IP accountability, randomization and CRF data, previously
requested CRF corrections are made Communication with Study Site Staff
Teleconferences, videoconferencing, email Review site’s processes, procedures, and records technique
19

RISK-BASED MONITORING Basis: Monitoring activities prevent or mitigate important
and likely sources of error in conduct, collection, and reporting of critical data and processes necessary for human subject protection and trial integrity
Importance of Critical Quality Factors: Procedures critical to collecting reliable data for study endpoints Consistency across sites or in a highly specific manner in some sites Procedures that won’t significantly impact data analysis or subject
safety
1. Identify Critical Data and Processes to be Monitored: IC verification, adherence to protocol eligibility criteria,
accountability and administration of IP, conduct, documentation & assessments related to study endpoints & red safety assessments
Procedures essential to trial integrity, e.g., blinding is maintained, both at the site level and at the sponsor level 20
Some types of errors in CT is more important than others (error in age v/s error in endpoint)

RISK-BASED MONITORING..2. Risk Assessment:
Risk identification based on trial design or investigational product Risks assessed and prioritized by considering the following:
the likelihood of errors occurring the impact of such errors on human subject protection and trial integrity the extent to which such errors would be detectable
3. Factors to consider while developing a monitoring plan: Complexity of the study design may require increased frequency and extent
of review (adaptive designs, stratified designs, complex dose titrations..)4. Monitoring Plan:
Description of each monitoring method & how it will be used to address important risks and ensure the validity of critical data
Criteria for determining the timing, frequency, and extent of planned monitoring activities
5. Documentation of monitoring: Date of the activity and the individual(s) conducting and participating in it Summary of the data or activities reviewed Description of noncompliances, potential noncompliance, data irregularities.. A description of any actions taken
21
Use the results of risk assessment in developing monitoring plan and type and intensity of monitoring to
address this risks
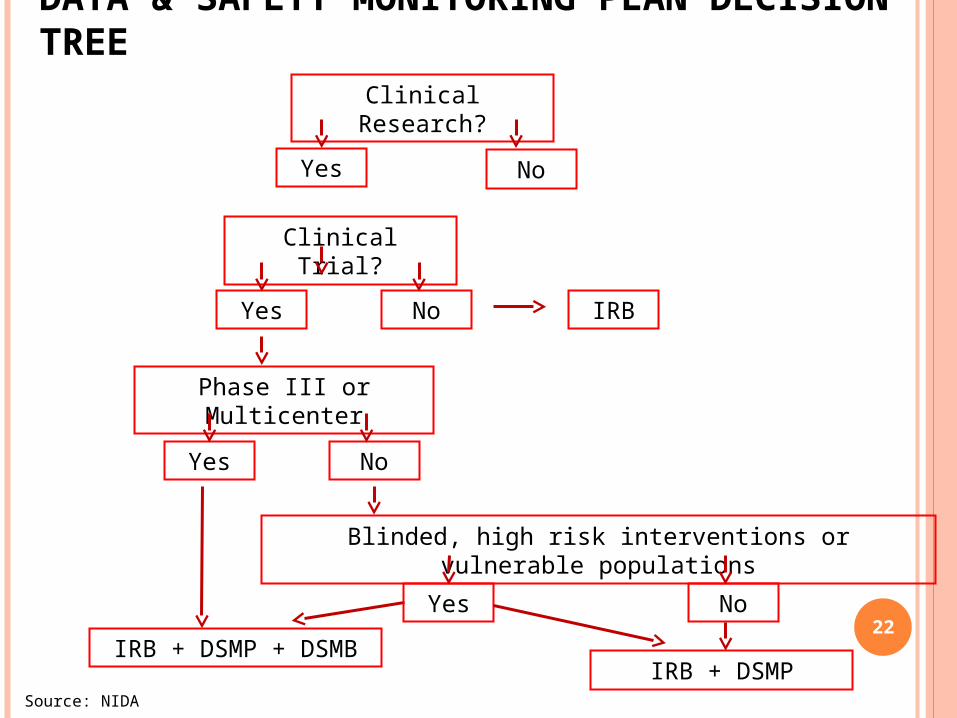
DATA & SAFETY MONITORING PLAN DECISION TREE
22
Clinical Research?
Yes No
Clinical Trial?
Phase III or Multicenter
Blinded, high risk interventions or vulnerable populations
Yes No IRB
Yes No
IRB + DSMPIRB + DSMP + DSMB
Yes No
Source: NIDA

MONITORING REQUIRED IN DIFFERENT PHASES OF STUDY OF CLINICAL TRIAL Phase I trial involves relatively high risk to a small N
Usually the study investigator performs continuous monitoring of safety
Phase II trial follows phase I with more N Toxicity and outcomes are confounded by disease process Monitoring similar to that of a phase I trial or additional monitoring
by experts or DSMB Phase III trials frequently compares a new treatment
to a standard treatment or to no treatment Large N Short-term risk is low, but long term effects of IP to achieve
significant safety or efficacy difference from the control May require a DSMB to perform monitoring functions
23

DATA AND SAFETY MONITORING PLAN (DSMP) & BOARD (DSMB) DSMPs promote data integrity as well as safety and
protection of trial subjects Many common activities with other trials and some unique
activities for a particular trial depending on the risk, size and complexity
Subject safety, data integrity and validity are the foremost consideration for a DSMP
For low risk studies an annual safety monitoring (by PI) may be enough
For higher risks studies, safety monitoring will have to be done more frequently as required by an established DSMB/DMC
Usually DSMBs are not required for Phase I studies but only for Phase II and III, but exceptions exists 24

WHEN DO YOU NEED A DSMB? When do you need a DSMB?
To ensure that participants are not exposed to undue risk To ensure that the study will yield usable results To do Interim Analyses and/or change protocol study design based on
IA To maintain trial integrity To ensure unbiased, timely publication of results
Composition: Researcher(s)/Clinician knowledgeable of the science of the study Biostatistician and/or Epidemiologist (study design/analysis) Research subject advocate and/or ethicist (research ethics)
SOP The DSMB chairman and members (quorum) participate Frequency at which the study will be evaluated, e.g. annually,
quarterly etc. Adverse event reporting requirements A description of interim efficacy analyses, if appropriate Study stopping rules, if appropriate The distribution of DSMB reports
25

DATA AND SAFETY MONITORING PLAN (DSMP) The specifics of a DSMP depend upon the nature, size,
complexity, and risk level To reduce harm or injury to study individuals The minimum required content for a DSMP includes:
Assessment of levels of risk A plan for safety review (by whom and how often)
Anticipated adverse events AE grading and attributions A plan for unanticipated and/or SAE reporting A plan for periodic or annual reporting of AEs
Ensure compliance with the principles and process of IC Assessment of protocol compliance including violations and/or
deviations A plan for compliance with privacy related regulations (e.g., HIPAA)26

TRAINING OF CLINICAL INVESTIGATORS & DATA PROCESSORS/ANALYSTS Clinical trial monitors train the CI and site staff during a study
Training on the conduct of a study and appropriate instruction during the study (e.g., when changes are made to the protocol)
Teleconferences, webcasts, online training modules and communication methods for training and feedback & significant change notification
Sponsors who plan less frequent monitoring should consider Monitoring plus discussion of CI’s and site staff’s responsibilities, feedback, and
additional training Train the trainers, data managers, analysts & report writers Delegating monitoring to a CRO
Need written transfer of any obligations from a sponsor to a CRO & CRO requires to comply with the regulations
Sponsors should evaluate CRO compliance with regulatory requirements & contractual obligations
e.g., periodic review of monitoring reports and vendor performance or quality metrics, communication between the sponsor & CRO regarding monitoring progress and findings
Sponsors are ultimately responsible for all data & monitoring27

PREPARING AS A SPONSOR OR A CRO FOR FDA AUDIT Registration of studies on http://www.clinicaltrials.gov/
For CTs of drugs and biologics other than Phase-I Selection and monitoring of clinical investigators
TMF & SMFs Sponsor/CRO provide the investigators with all necessary information prior to
initiation of CT Serious deviations from the approved investigational plan (which includes the
study protocol) or FDA regulations Terminated sites and the circumstances Non-compliant investigator still continuing in the study
Selection of Monitors Criteria for selecting monitors and how monitors meet those criteria How more than one person handles monitoring
Monitoring procedures and activities Procedures, frequency, scope, and process to monitor the progress of the clinical
investigation Relevant SOPs are followed Activities Review of Site Records
28

PREPARING AS A SPONSOR OR A CRO FOR FDA AUDIT.. QAUs (CT QAUs are not required by regulation)
However, if the sponsor/CRO has a QAU, its organization & operation should be described in SOPs; separation of functions between QA auditing and monitoring of clinical trials
List of audited studies Safety/AE reporting
Safety reports of potential serious risks within 15 calendar days; within 7 calendar days if unexpected fatal or life-threatening suspected AE
AE reported to participating investigators (and to reviewing IRBs for device studies) as required by the regulations.
Procedures (e.g., frequency, scope) the sponsor/CRO uses for the receipt, evaluation, and monitoring of safety information/unanticipated AEs
All Study Tabulations All Investigator Tabulations
All pertinent studies (form 1572) included in the above two categories Reasons for non-inclusion
Data tabulations on each subject in each clinical trial in an NDA 29

PREPARING AS A SPONSOR OR A CRO FOR FDA AUDIT ...
Electronic records and electronic signatures Regulatory requirements for the clinical data are the same regardless of
the type of computer system used by the clinical site Data must be reliable and usable for evaluating the safety and/or
effectiveness of FDA-regulated products Electronic Records (CFR Part 11) and electronic signatures are maintained in
electronic format (in some cases in addition to paper format); SOPs for procedures Data collection:
Procedures for collection, retention, and transmission of data at each clinical site; File transfer protocols to and from processing center to and from site
Original data entries and changes cannot be made by anyone other than the clinical investigators
How electronic data are reviewed during monitoring visits; SOPs for such reviews Data Security;
Clear, justified authority to access the system How the paper & computerized systems are accessed (e.g., password protected,
access privileges, user identification How information is captured related to the creation, modification, or deletion of
electronic records (e.g., audit trails, date/time stamps); backup, disaster recovery, and/or contingency plans; error messages or system failures handled/corrected
How the system and data are handled during site closure30

CONCLUDING REMARKS A CT is as good as the quality of its data In an effort to ensure the integrity of clinical trial data, the
FDA has released requirements Monitoring of data collection, review and analysis is
essential to ensure data integrity Traditionally monitoring required an in-depth and comprehensive
examination of all collected data. The risk-based monitoring (RBM) system is fundamentally
different as to how data managers review clinical data Does not mandate a specific methodology but requireds an ideal
strategy that allows for faster time to market, reduces site monitoring costs and frees up time and resources for value-added tasks
For complex Phase III (sometimes Phase II) trials require a DSMP as well as a DSMB for overall data integrity or to stop the trial
Training and audit (FDA/Client) readiness for data integrity ensures high success rates
31

THANK YOU VERY MUCH