deletion analysis of the chinese hamster … · indirect assay for cellular dhfr activity that...
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY @ 1988 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 263, No. 31, Issue of November 5, pp. 16274-16282,1988 Printed in U.S.A.
Deletion Analysis of the Chinese Hamster Dihydrofolate Reductase Gene Promoter*
(Received for publication, April 11, 1988)
Carlos J. Ciudad, Gail Urlaub, and Lawrence A. ChasinS From the Department of Biological Sciences, Columbia University, New York, New York 10027
Deletion analysis of the 5’ flank of the Chinese ham- ster dihydrofolate reductase (dhfr) gene reveals a pro- moter region starting 48 base pairs upstream of the major transcriptional start site. A dhfr minigene con- taining approximately 900 base pairs of 6’ flank and one small intron was used as a wild-type standard. Seven deletions were created with BAL-3 1. Promoter activity was measured in three ways: 1) transient expression of the dhfr gene; 2) frequence of transfec- tion of dhfr- Chinese hamster cells to a dhfr+ pheno- type; and 3) RNase protection analysis of dhfr tran- scripts in pooled populations of permanently trans- fected cells. The transient expression assay was developed in this work for the rapid analysis of dhfr promoter mutants; this assay could be of general use for analyzing constructs carrying dhfr as a reporter gene. Two of the deletions define a requirement for part or all of the sequence GGGCGT located 48 base pairs upstream of the major transcriptional start site. This site has been shown to bind transcription factor Spl in the mouse dhfr gene. The function of the major promoter is independent of the function of the minor promoter. These minigene constructs also contain cryptic promoters located upstream of the natural start sites, probably in the plasmid vector. Transcripts orig- inating from these upstream sites are inefficiently spliced, but do result in messenger RNA molecules that are translated into active dihydrofolate reductase.
The dihydrofolate reductase (dhfr) gene belongs to the housekeeping category, being expressed to some extent in all cells. Because of its essential role in de nouo DNA synthesis, DHFR’ has been a common target for cancer chemotherapy (Bertino, 1979). Although it is widely expressed, the dhfr gene is a highly controlled gene, subject to cell cycle regulation at the transcriptional and posttranscriptional levels (for review see Chasin, 1986).
The dhfr genes in mouse and Chinese hamster use multiple transcription initiation sites; in the hamster the two predom- inant sites are a minor (15%) one at -107 bp and a major (85%) one at -63 bp relative to the dhfr translational start (Mitchell et al., 1986). The dhfr promoter region, like other housekeeping genes such as hprt, ada, pgk, and hmgCoA reductase, lacks the TATA box which is necessary for normal transcription initiation in many eukaryotic genes (Dynan,
* This work was supported by National Institutes of Health Grant GM 22629. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ To whom correspondence should be addressed. The abbreviations used are: DHFR, dihydrofolate reductase; bp,
base pair(s); CHO, Chinese hamster ovary.
1986). One the other hand, it includes several boxes thought to be involved in controlling eukaryotic transcription initia- tion: three distal C-rich GC boxes, four proximal G-rich GC boxes, and two CAA boxes. The presence of these boxes in the hamster dhfr 5’ flank makes this gene a possible candidate to be regulated by the transcription factors Spl and CTF, which bind to GGGCGG and CAAT boxes, respectively. In order to determine which DNA sequences are important for the correct transcription initiation and expression of the dhfr gene and preliminary to the exploration of the cellular factors involved, we have performed a deletion analysis of the dhfr promoter, measuring the gene activity both transiently and in pooled permanent transformants. We also describe an indirect assay for cellular DHFR activity that could prove useful for gene transfer analysis where dhfr is a reporter gene.
MATERIALS AND METHODS
Cell Culture-Conditions for the monolayer culture of CHO cells have been described previously (Urlaub et al., 1985b). Complete medium was a modification (Coon and Weiss, 1969) of Ham’s nutrient mixture F-12 (Ham, 1965) supplemented with 10% (v/v) fetal calf serum. The CHO cell lines used in this study were UA21 (Urlaub et al., 1983), DG44 (Urlaub et al., 1986) and MK42 (Nunberg et al., 1978), containing 1, 0, and 200-300 copies of the dhfr gene, respec- tively.
DNA Construction-Deletion mutants were derived from the Chinese hamster dhfr construct pDCHlP, which is referred to as the “full minigene.” pDCHlP was constructed by cloning the EcoRI-PstI fragment of a cDNA clone, pA3-A35 (Melera et al., 1984) into the polycloning region of pUC119 (Vieira and Messing, 1987). The PstI site at the 3’ end of this insert was destroyed by digesting with the enzyme, blunt-ending with T4 polymerase, and religating. A 1.3- kilobase pair EcoRI fragment spanning the 5’ end of the gene, isolated from pDCHl (Venolia et al., 1987) was cloned into the EcoRI site of this plasmid. The insert of this construct, pDCHlP, contains the six exons of the gene, intron 1, approximately 900 bp of the 5’ flank and the first of the two major polyadenylation sites in exon 6 (Fig. 1).
The exonuclease BAL-31 was used to create deletions (Maniatis et al., 1982) from the unique BssHII site a t position -240 (all numbering is relative to the translational start site as +l). The DNA was then treated with NdeI to cleave all of the deleted molecules at this unique upstream site (position -839). Upon filling in and religation using Klenow polymerase and T, DNA ligase, respectively, these constructs were transformed into Escherichia coli strain 7118 (Messing et al., 1977).
DNA Sequencing-CHO dhfr promoter deletion mutants were sequenced directly (Hattori and Sakaki, 1986) from the recombinant pUC119 plasmids.
Transfections-Plasmid DNA was prepared by alkaline lysis, phenol extraction, and Bio-Gel A15 chromatography and used to transfect dhfr- DG44 cells. Two methods of transfections were used (i) the calcium phosphate method (Wigler et al., 1979) in experiments performed to study transformation frequency and RNA analysis and ii) the Polybrene method (Chaney et al., 1986) in those experiments where the transient gene activity was measured. For the transfor- mation frequency experiments, transfectants were selected in F-12 medium lacking hypoxanthine following a 6-h exposure to DNA and a 24-h expression period in complete medium. For the generation of populations of dhfr cotransfectants for RNA analysis, 2 pg of dhfr
16274

Dhfr Promoter Deletions 16275
deletion mutant plasmid DNA, 0.4 pg of plasmid pNEO-BPV100 (Luskey and Botchan, 1984) carrying the bacterial Tn5 ne0 gene, and 17.6 pg of carrier calf thymus DNA were used to transfect DG44 cells. In this case, transfectants were selected for resistance to G418 (Ge- neticin) at a final concentration of active material in the media of 400 pg/ml. After 9 days, viable colonies were harvested, pooled, and expanded.
RNA Analysis-dhfr transcripts were characterized by RNase pro- tection using the two riboprobes shown in Fig. 1. The recombinant plasmid pMGA, containing no introns, has been described previously (Carothers et al., 1988). pMGS17 contains a single intron, the 304-bp intron 1. A 1010-bp SrnaI-SstI fragment containing exon 1, intron 1, exon 2, and part of exon 3 was isolated from pDCHlP and subjected to limited digestion with BAL-31, for other purposes. The resulting molecules were cloned into the HincII site of pGEMl (Promega Biotec). One clone was isolated that contained an 813-bp insert, having lost 93 bp from its 3' end and approximately 104 bp from its 5' end. This fragment was oriented such that its 3' end was adjacent to the SP6 promoter. Probes were prepared after cleavage of pMGA at the AuaI site in the 5' flank or pMGS17 at the BarnHI site in the vector (Fig. 1).
RNA probes (Melton et al., 1984) were hybridized overnight to 60 pg of total RNA (Chirgwin et al., 1979) at 48 "C in 60 p1 of the hybridization buffer. After the addition of 0.3 ml of RNase digestion buffer (10 mM Tris-HC1, pH 7.5,5 mM EDTA, 300 mM NaCl, 40 pg/ ml RNase A, 1000 units/ml RNase Tl), the samples were incubated at room temperature for 30 min. The nucleic acid was isolated, and samples corresponding to 3, 10, or 30 pg of the original RNA were electrophoresed in 4% polyacrylamide gels containing 8 M urea. The amounts of protected fragments were quantified using a Bio-Rad densitometer, correcting for film nonreciprocity as described previ- ously (Orlofsky and Chasin, 1985).
Enzyme Assay-Cell extracts were prepared and assayed for DHFR-specific catalytic activity as described previously (Urlaub and Chasin, 1980).
dhfr Transient Expression Assay-The method is based on the incorporation of radioactive deoxyuridine into cellular DNA. This incorporation depends on the generation of tetrahydrofolate by DHFR from folate supplied in the medium. The tetrahydrofolate is used in the reductive methylation of deoxyuridylate to thymidylate. The latter is subsequently incorporated into DNA, which is then isolated by a simple trichloroacetic acid precipitation procedure.
Dishes (35 mm) were seeded with 3 X lo6 dhfr- DG44 cells the night before transfection. The monolayers were then treated with 7 pg of Polybrene (Sigma) and 5 pg of supercoiled or linearized dhfr plasmid in 0.7 ml of complete medium for 1 h at 37 'C in a 5% CO, incubator. No carrier DNA was used. The cells were then subjected to a dimethyl sulfoxide shock (10% dimethyl sulfoxide in medium for 4 min at room temperature, followed by a rinse in complete medium). After 24 h in complete medium, the monolayers were rinsed with medium lacking glycine, hypoxanthine, and thymidine and then incubated with 1 ml of this medium containing 2 pCi 6-[3H]deoxyu- ridine (20 mCi/mmol, Du Pont-New England Nuclear). After 24 h, the cells were rinsed twice with phosphate-buffered saline and lysed in 1 ml of 0.1% sodium dodecyl sulfate. The lysate plus a 1-ml water rinse of the dish was combined in a test tube in ice. An equal volume of 20% trichloroacetic acid containing 2 X M deoxyuridine was added and the mixture incubated on ice for 30 min. The trichloroa- cetic acid precipitate was collected by filtration on glass microfiber filters (Whatman GF/A) which were subsequently washed five times with 2 ml of 10% trichloroacetic acid containing lo" M deoxyuridine, dried, and counted in a scintillation counter. The development as well as the characteristics of this assay are described in Fig. 3 of the present paper.
RESULTS
Deletion Mutants of a Chinese Hamster dhfr Minigem- The starting point for deletion mutagenesis of the dhfr 5'- flanking region was a minigene (carried by recombinant plas- mid pDCHlP, Fig. 1, top) containing only the first small (304 bp) intron of the 5 found in the dhfr genomic gene. Approxi- mately 900 bp of 5' genomic flank are present at the 5' end of the minigene. Although at the 3' end the construct is truncated within exon 6, it includes a major polyadenylation site found within that exon. The elimination of the large
introns (introns 2-5) reduces the size of the dhfr gene from 25 kilobase pairs to about 2 kilobase pairs. We have shown previously that this minigene sequence efficiently transforms DHFR-negative CHO cells to a DHFR-positive phenotype (Mitchell et al., 1986) and that its transcripts are correctly polyadenylated in transfected cells (Venolia et al., 1987).
Seven 5' deletion mutants were created by cleavage of pDCHlP DNA at a unique BssHII site at position -240 (all numbering is relative to the translational start site being +l ; the major transcriptional start site is at position -63) followed by limited digestion with the exonuclease BAL-31. Before ligation, the BAL-31 digests were cleaved at a unique up- stream NdeI site, such that all of the deletions had a common 5' end at position -839 and a 3' end that ranged from positions -240 to -79 (Table I and Fig. 2).
Development of a Transient Expression Assay for the dhfr Gene-The phenotypic analysis of in vitro mutagenesis prod- ucts can be facilitated by the relatively rapid measurement of gene expression following transient transfection. Most often, mutated promoter regions are joined to the bacterial chlor- amphenicol acetyltransferase gene as a reporter for such experiments. We reasoned that the construction of such chi- meras should not be necessary in the case of dhfr, since the enzyme activity specified by this gene lends itself to simple metabolic analysis. The conversion of [3H]deoxyuridine to [3H]TMP and its subsequent incorporation into DNA is de- pendent on DHFR activity, and the extent of this incorpora- tion can be easily measured in the acid-precipitable fraction of a cell monolayer. We have developed such an assay for the measurement of dhfr gene activity following transfection of DHFR-deficient CHO cells. Some of the parameters of this assay using pDCHlP DNA as a wild-type minigene are shown in Fig. 3. With the idea of applying this assay to many mutants, we adopted the Polybrene method (Chaney et aL, 1986) for gene transfer, since this procedure requires the least manipulation (Polybrene and soluble DNA are simply added to a cell culture for a short time). As can be seen in Fig. 3A, dhfr gene expression was maximal after exposure to DNA for only 1 h; activity gradually decreased with longer exposure times. The amount of incorporation was dependent on in- creasing amounts of plasmid DNA up to 10 pg/dish, but activity fell off sharply when this amount was doubled (Fig. 323). It is likely that this decrease is due to the toxic effect of the higher amount of DNA. An expression period was found to be required between the application of DNA and the [3H] deoxyuridine labeling. As can be seen in Fig. 3C, after a lag of 3-6 h, incorporation increased with increasing time for expression. Expression periods longer than 24 h resulted in even greater labeling (data not shown); much of this increase would be expected if the cell number was increasing with time. A similar situation is seen when labeling time is varied (Fig. 3 0 ) : the amount of incorporation increased linearly with labeling time for at least 24 h. The relatively low background incorporation displayed by the DHFR-deficient recipient cells and the specific requirement for a dhfr gene (see below) attest to the fact that the measured incorporation is dependent on DHFR activity. It should be noted that during the 24-h labeling period, those recipient cells that are not expressing dhfr are starving for glycine, a purine, and thymidine. On the basis of these experiments, the following standard assay con- ditions were adopted exposure of cells to 5 pg of DNA for 1 h, an expression period in complete medium for 24 h, and a 24-h labeling period. In most experiments the pDCHlP min- igene yielded about 5,000-10,000 cpm under these conditions. For comparison, equal numbers of exponentially growing cells of the CHO cell line UA21, carrying one copy of the endoge-

16276 Dhfr Promoter Deletions A2 B pAl I I I
pDCHl P I I I I ,
.... . .. . . . . .. . . . . . . .. . . .. . . .. . . . . . . . . . . . . ".... .. 872
627
563
- 193
- 148
~ 414
pMGSl7
__._ 452. 56 ._.__.- 812
553
508
- %____ ~ ____. 1 9 3 56
- .._____ /... .- '49, 56
FIG. 1. Structure of the dhfr recombinant plasmids used. Open boxes represent exons, with shaded areas indicating 5'- and 3"untranslated regions. The narrower box at the 5' end of exon 1 represents a minor transcriptional start site. Wavy lines denote plasmid sequences; a thick line represents intron 1. Restrictive sites are AI, A d ; A 5 AvaII; B, BamHI; E, EcoRI; and P, PstI. The position of the proximal major polyadenylation site in exon 6 is indicated. Top, pDCHlP, used as the starting point for making deletion mutants. The dhfr sequences have been inserted into the polycloning region of pUC119. Middle, pMGA, used to synthesize an mRNA antisense riboprobe. Bottom, pMGS17, used to produce a riboprobe containing intron 1. The riboprobes produced from the two plasmids are indicated by the dotted lines beneath each map. The expected protected fragments from the RNA analysis shown in Figs. 5 and 6 are depicted by the solid lines below the probe. The sizes of the protected fragments are also indicated.
TABLE I Location and effects of deletion mutations
Mutant number
DDCHlP ___
12 22 26 24
410 414 413
5' flank remaining
-900 -240 - 189 -166 - 145 -111 -105 -79
expressionb Transient
100 105 ND 101 55 53 26 21
Transfection frequency'
100 90 60 50 25 20 3 1
' Transcriptional startsd
Copy no.' Minol' Majol' Upstread
9 100 100 100 17 48 92 125
ND ND ND ND 25 0 42 147 25 0 29 148 25 0 31 184 21 0 0 180 25 0 0 130
Relative to the translational start site. All mutants retain the 5' genomic sequence from approximately -900
Relative to pDCHlP, after subtraction of the signal obtained with the pUC119 vector alone. Gross values were typically 7000 cpm for pDCHlP and 300 cpm for pUC119.
e Measured as the slope of the linear part of a curve relating transfection frequency to amount of DNA applied and expressed relative to the value for pDCHlP in the same experiment (typically transfectants/pg of DNA/ recipient cell).
From densitometric analysis of RNase protection experiments. RNA was prepared from populations of pooled permanent transfectants selected for receipt of the cotransferred ne0 gene.
e The average number of dhfr minigene copies per cell was calculated from densitometric measurements of slot blots.
'The major transcriptional start site is at position -63, and the minor start is a t position -107. Values are expressed relative to pDCHlP and have been normalized for gene copy number. A value of zero indicates that no band was apparent; ND, not done.
Total upstream starts, corrected for gene copy number and expressed relative to pDCH1P. The transcription starts for pDCHlP were: major promoter, 45%; minor promoter, 18%; and upstream promoter(s), 37%.
to -842.
nous dhfr gene, incorporated [3H]deoxyuridine at more than 100 times this rate.
Mutant Analysis by Transient Expression-Supercoiled DNA from each of the mutants was assayed for transient expression of the dhfr gene using the standard conditions described above. As can be seen in Table I, all of the plasmids that contained the dhfr coding sequence gave rise to detectable DHFR activity in this assay. Mutant 413, in which all but 16 base pairs upstream of the major transcriptional start site were deleted, yielded reduced (20%) activity relative to the full minigene construct (pDCHlP, with 5' flank to approxi-
mate position -900); its activity was clearly and reproducibly higher than the vector control. The effect of more extensive deletion of the 5' flank was tested by using linearized frag- ments of the full minigene that had been cut at a unique AurII site 41 base pairs downstream of the major transcriptional start site (and 23 base pairs upstream of the translational initiation site). This DNA produced a transient expression signal that was 10% of that yielded by the positive control, the full minigene linearized outside of the dhfr sequence (by cleavage with PuuII at sites 88 bp upstream and 190 bp downstream of the 5' and 3' dhfr vector joints, respectively).
Dhfr Promoter Deletions 16277
Thus, even truncation within the 5“untranslated leader re- gion does not eliminate a background of DHFR activity in this assay. This residual activity could be the result of some promoter activity downstream of the Awl1 site or it could be caused by cryptic promotion in host sequences recruited dur- ing the 48-h expression and assay period. The latter is more likely, since no downstream transcription start sites were found in the RNA analysis to be described below.
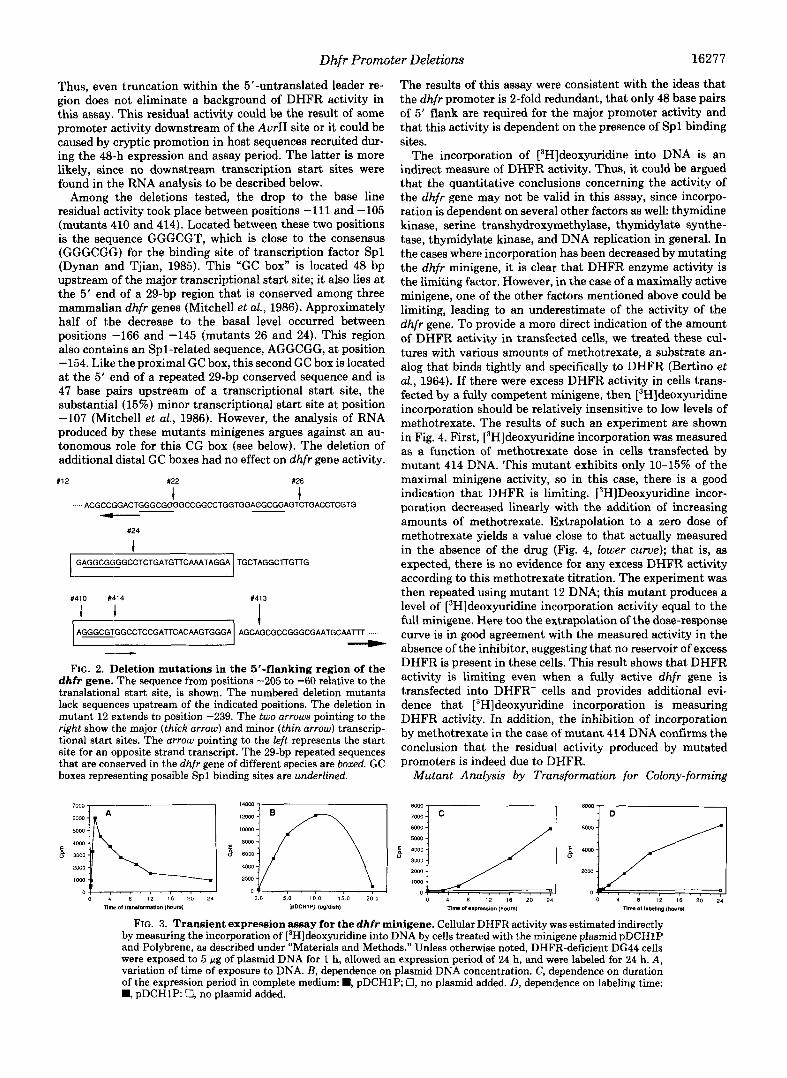
Among the deletions tested, the drop to the base line residual activity took place between positions -111 and -105 (mutants 410 and 414). Located between these two positions is the sequence GGGCGT, which is close to the consensus (GGGCGG) for the binding site of transcription factor Spl (Dynan and Tjian, 1985). This “GC box” is located 48 bp upstream of the major transcriptional start site; it also lies at the 5’ end of a 29-bp region that is conserved among three mammalian dhfr genes (Mitchell et al., 1986). Approximately half of the decrease to the basal level occurred between positions -166 and -145 (mutants 26 and 24). This region also contains an Spl-related sequence, AGGCGG, at position -154. Like the proximal GC box, this second GC box is located at the 5’ end of a repeated 29-bp conserved sequence and is 47 base pairs upstream of a transcriptional start site, the substantial (15%) minor transcriptional start site at position -107 (Mitchell et al., 1986). However, the analysis of RNA produced by these mutants minigenes argues against an au- tonomous role for this CG box (see below). The deletion of additional distal GC boxes had no effect on dhfr gene activity.
#I 2 #22
I #26
1 ..... ACGCCGGACTGGGCGGGGCCGGCCTGGTGGAGGCGGAGTCTGACCTCGTG ”
#24
1 GAGGCGGGGCCTCTGATGTTCAAATAGGA TGCTAGGClTGlTG
#410 #414
I I #413
1 AGGGCGTGGCCTCCGATTCACAAGTGGGA AGCAGCGCCGGGCGAATGCAAAm .....
FIG. 2. Deletion mutations in the 5”flanking region of the dhfr gene. The sequence from positions -205 to -60 relative to the translational start site, is shown. The numbered deletion mutants lack sequences upstream of the indicated positions. The deletion in mutant 12 extends to position -239. The two arrows pointing to the right show the major (thick arrow) and minor (thin arrow) transcrip- tional start sites. The urrow pointing to the left represents the start site for an opposite strand transcript. The 29-bp repeated sequences that are conserved in the dhfr gene of different species are boxed. GC boxes representing possible Spl binding sites are underlined.
E 8
:Kl 3ow
2040
io00
0 0 4 8 12 16 20 24
10000
4000
2000
0 0 5 0 1 0 0 1 5 0 2 0 0
The results of this assay were consistent with the ideas that the dhfr promoter is 2-fold redundant, that only 48 base pairs of 5’ flank are required for the major promoter activity and that this activity is dependent on the presence of Spl binding sites.
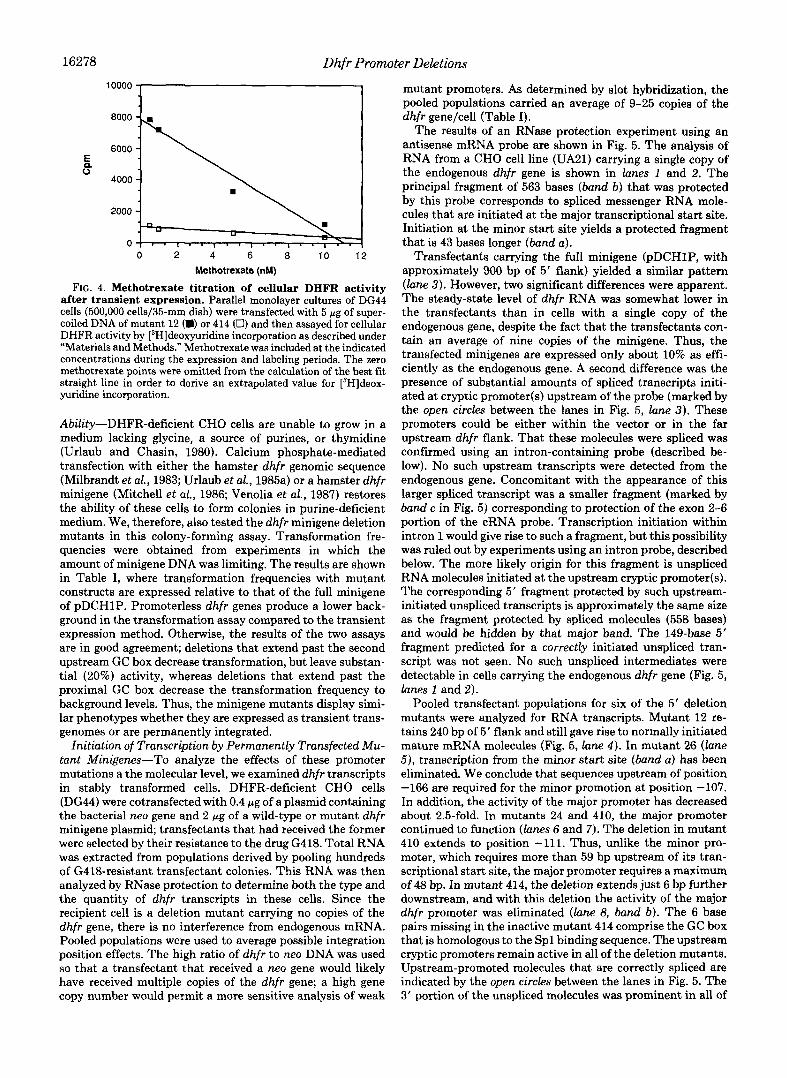
The incorporation of [3H]deoxyuridine into DNA is an indirect measure of DHFR activity. Thus, it could be argued that the quantitative conclusions concerning the activity of the dhfr gene may not be valid in this assay, since incorpo- ration is dependent on several other factors as well: thymidine kinase, serine transhydroxymethylase, thymidylate synthe- tase, thymidylate kinase, and DNA replication in general. In the cases where incorporation has been decreased by mutating the dhfr minigene, it is clear that DHFR enzyme activity is the limiting factor. However, in the case of a maximally active minigene, one of the other factors mentioned above could be limiting, leading to an underestimate of the activity of the dhfr gene. To provide a more direct indication of the amount of DHFR activity in transfected cells, we treated these cul- tures with various amounts of methotrexate, a substrate an- alog that binds tightly and specifically to DHFR (Bertino et al., 1964). If there were excess DHFR activity in cells trans- fected by a fully competent minigene, then [3H]deoxyuridine incorporation should be relatively insensitive to low levels of methotrexate. The results of such an experiment are shown in Fig. 4. First, [3H]deoxyuridine incorporation was measured as a function of methotrexate dose in cells transfected by mutant 414 DNA. This mutant exhibits only 10-15% of the maximal minigene activity, so in this case, there is a good indication that DHFR is limiting. 13H]Deoxyuridine incor- poration decreased linearly with the addition of increasing amounts of methotrexate. Extrapolation to a zero dose of methotrexate yields a value close to that actually measured in the absence of the drug (Fig. 4, lower curue); that is, as expected, there is no evidence for any excess DHFR activity according to this methotrexate titration. The experiment was then repeated using mutant 12 DNA; this mutant produces a level of [3H]deoxyuridine incorporation activity equal to the full minigene. Here too the extrapolation of the dose-response curve is in good agreement with the measured activity in the absence of the inhibitor, suggesting that no reservoir of excess DHFR is present in these cells. This result shows that DHFR activity is limiting even when a fully active dhfr gene is transfected into DHFR- cells and provides additional evi- dence that [3H]deoxyuridine incorporation is measuring DHFR activity. In addition, the inhibition of incorporation by methotrexate in the case of mutant 414 DNA confirms the conclusion that the residual activity produced by mutated promoters is indeed due to DHFR.
Mutant Analysis by Transformation for Colony-forming
FIG. 3. Transient expression assay for the dhfr minigene. Cellular DHFR activity was estimated indirectly by measuring the incorporation of [3H]deoxyuridine into DNA by cells treated with the minigene plasmid pDCHlP and Polybrene, as described under “Materials and Methods.” Unless otherwise noted, DHFR-deficient DG44 cells were exposed to 5 pg of plasmid DNA for 1 h, allowed an expression period of 24 h, and were labeled for 24 h. A, variation of time of exposure to DNA. B, dependence on plasmid DNA concentration. C, dependence on duration of the expression period in complete medium: B, pDCH1P 0, no plasmid added. D, dependence on labeling time: B, pDCH1P: 0, no plasmid added.
16278 Dhfr Promc
2000 1 0 2 4 6 8 10 12
Methotrexate (nM)
FIG. 4. Methotrexate titration of cellular DHFR activity after transient expression. Parallel monolayer cultures of DG44 cells (500,000 cells/35-mm dish) were transfected with 5 p g of super- coiled DNA of mutant 12 (D) or 414 (0) and then assayed for cellular DHFR activity by [3H]deoxyuridine incorporation as described under “Materials and Methods.” Methotrexate was included at the indicated concentrations during the expression and labeling periods. The zero methotrexate points were omitted from the calculation of the best fit straight line in order to derive an extrapolated value for [3H]deox- yuridine incorporation.
Ability-DHFR-deficient CHO cells are unable to grow in a medium lacking glycine, a source of purines, or thymidine (Urlaub and Chasin, 1980). Calcium phosphate-mediated transfection with either the hamster dhfr genomic sequence (Milbrandt et al., 1983; Urlaub et al., 1985a) or a hamster dhfr minigene (Mitchell et al., 1986; Venolia et al., 1987) restores the ability of these cells to form colonies in purine-deficient medium. We, therefore, also tested the dhfr minigene deletion mutants in this colony-forming assay. Transformation fre- quencies were obtained from experiments in which the amount of minigene DNA was limiting. The results are shown in Table I, where transformation frequencies with mutant constructs are expressed relative to that of the full minigene of pDCH1P. Promoterless dhfr genes produce a lower back- ground in the transformation assay compared to the transient expression method. Otherwise, the results of the two assays are in good agreement; deletions that extend past the second upstream GC box decrease transformation, but leave substan- tial (20%) activity, whereas deletions that extend past the proximal GC box decrease the transformation frequency to background levels. Thus, the minigene mutants display simi- lar phenotypes whether they are expressed as transient trans- genomes or are permanently integrated.
Initiation of Transcription by Permanently Transfected Mu- tant Minigenes-To analyze the effects of these promoter mutations a the molecular level, we examined dhfr transcripts in stably transformed cells. DHFR-deficient CHO cells (DG44) were cotransfected with 0.4 wg of a plasmid containing the bacterial neo gene and 2 pg of a wild-type or mutant dhfr minigene plasmid; transfectants that had received the former were selected by their resistance to the drug G418. Total RNA was extracted from populations derived by pooling hundreds of G418-resistant transfectant colonies. This RNA was then analyzed by RNase protection to determine both the type and the quantity of dhfr transcripts in these cells. Since the recipient cell is a deletion mutant carrying no copies of the dhfr gene, there is no interference from endogenous mRNA. Pooled populations were used to average possible integration position effects. The high ratio of dhfr to neo DNA was used so that a transfectant that received a neo gene would likely have received multiple copies of the dhfr gene; a high gene copy number would permit a more sensitive analysis of weak
Iter Deletions
mutant promoters. As determined by slot hybridization, the pooled populations carried an average of 9-25 copies of the dhfr gene/cell (Table I).
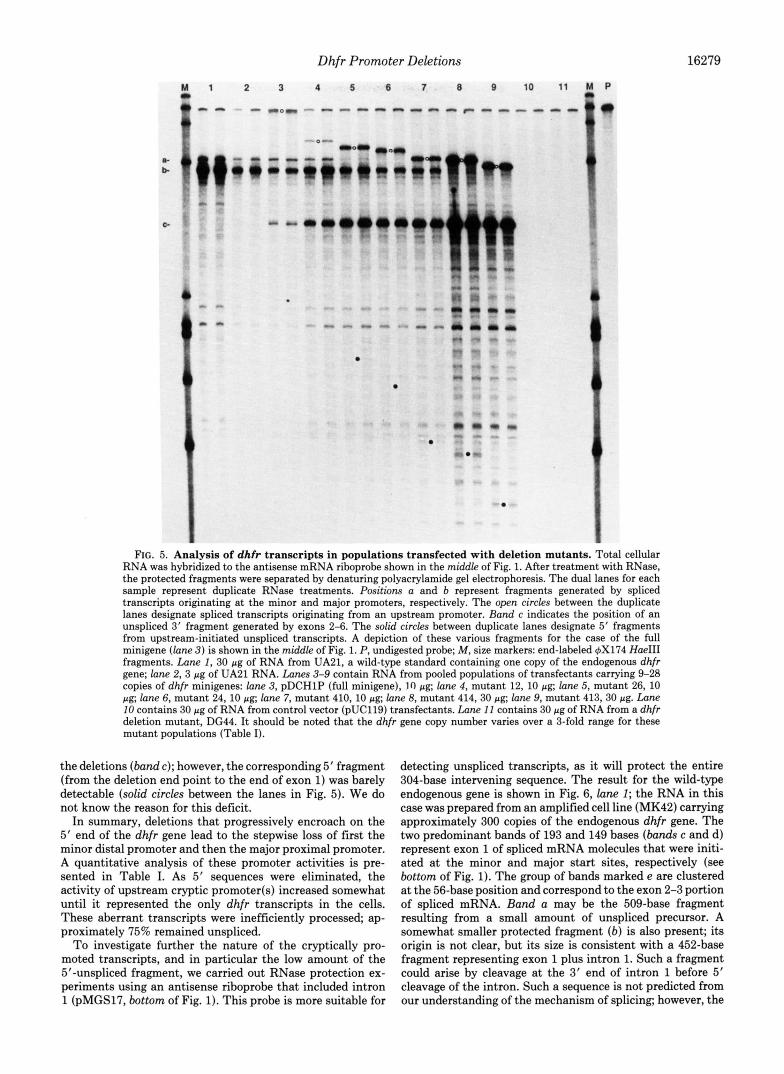
The results of an RNase protection experiment using an antisense mRNA probe are shown in Fig. 5. The analysis of RNA from a CHO cell line (UA21) carrying a single copy of the endogenous dhfr gene is shown in lanes 1 and 2. The principal fragment of 563 bases ( b a d b) that was protected by this probe corresponds to spliced messenger RNA mole- cules that are initiated at the major transcriptional start site. Initiation at the minor start site yields a protected fragment that is 43 bases longer (band a).
Transfectants carrying the full minigene (pDCHlP, with approximately 900 bp of 5’ flank) yielded a similar pattern (lane 3) . However, two significant differences were apparent. The steady-state level of dhfr RNA was somewhat lower in the transfectants than in cells with a single copy of the endogenous gene, despite the fact that the transfectants con- tain an average of nine copies of the minigene. Thus, the transfected minigenes are expressed only about 10% as effi- ciently as the endogenous gene. A second difference was the presence of substantial amounts of spliced transcripts initi- ated at cryptic promoter(s) upstream of the probe (marked by the open circles between the lanes in Fig. 5, lane 3 ) . These promoters could be either within the vector or in the far upstream dhfr flank. That these molecules were spliced was confirmed using an intron-containing probe (described be- low). No such upstream transcripts were detected from the endogenous gene. Concomitant with the appearance of this larger spliced transcript was a smaller fragment (marked by band c in Fig. 5) corresponding to protection of the exon 2-6 portion of the cRNA probe. Transcription initiation within intron 1 would give rise to such a fragment, but this possibility was ruled out by experiments using an intron probe, described below. The more likely origin for this fragment is unspliced RNA molecules initiated at the upstream cryptic promoter(s). The corresponding 5’ fragment protected by such upstream- initiated unspliced transcripts is approximately the same size as the fragment protected by spliced molecules (558 bases) and would be hidden by that major band. The 149-base 5’ fragment predicted for a correctly initiated unspliced tran- script was not seen. No such unspliced intermediates were detectable in cells carrying the endogenous dhfr gene (Fig. 5, lanes I and 2).
Pooled transfectant populations for six of the 5‘ deletion mutants were analyzed for RNA transcripts. Mutant 12 re- tains 240 bp of 5‘ flank and still gave rise to normally initiated mature mRNA molecules (Fig. 5, lane 4) . In mutant 26 (lane 5) , transcription from the minor start site (band a) has been eliminated. We conclude that sequences upstream of position -166 are required for the minor promotion at position -107. In addition, the activity of the major promoter has decreased about 2.5-fold. In mutants 24 and 410, the major promoter continued to function (lanes 6 and 7). The deletion in mutant 410 extends to position -111. Thus, unlike the minor pro- moter, which requires more than 59 bp upstream of its tran- scriptional start site, the major promoter requires a maximum of 48 bp. In mutant 414, the deletion extends just 6 bp further downstream, and with this deletion the activity of the major dhfr promoter was eliminated (lane 8, band b). The 6 base pairs missing in the inactive mutant 414 comprise the GC box that is homologous to the Spl binding sequence. The upstream cryptic promoters remain active in all of the deletion mutants. Upstream-promoted molecules that are correctly spliced are indicated by the open circles between the lanes in Fig. 5. The 3’ portion of the unspliced molecules was prominent in all of
Dhfr Promoter Deletions 16279
M l 2 3 4 5 6 7 8 9 10 1 1 M P 0 m
"
t
i r - -. 1 . c. , .
-- o a - 0 "." .""" ." .
. . a . .
I .- a
0
FIG. 5. Analysis of dhfr transcripts in populations transfected with deletion mutants. Total cellular RNA was hybridized to the antisense mRNA riboprobe shown in the middle of Fig. 1. After treatment with RNase, the protected fragments were separated by denaturing polyacrylamide gel electrophoresis. The dual lanes for each sample represent duplicate RNase treatments. Positions a and b represent fragments generated by spliced transcripts originating at the minor and major promoters, respectively. The open circles between the duplicate lanes designate spliced transcripts originating from an upstream promoter. Band c indicates the position of an unspliced 3' fragment generated by exons 2-6. The solid circles between duplicate lanes designate 5' fragments from upstream-initiated unspliced transcripts. A depiction of these various fragments for the case of the full minigene ( l a n e 3 ) is shown in the middle of Fig. 1. P, undigested probe; M , size markers: end-labeled 4x174 Hue111 fragments. Lane 1, 30 pg of RNA from UA21, a wild-type standard containing one copy of the endogenous dhfr gene; lane 2 , 3 pg of UA21 RNA. h n e s 3-9 contain RNA from pooled populations of transfectants carrying 9-28 copies of dhfr minigenes: lane 3, pDCHlP (full minigene), 10 pg; lane 4, mutant 12, 10 pg; lane 5, mutant 26, 10 pg; lane 6, mutant 24,lO pg; lane 7, mutant 410,lO pg; lane 8, mutant 414,30 pg, lane 9, mutant 413,30 pg. Lane 10 contains 30 pg of RNA from control vector (pUC119) transfectants. Lane 11 contains 30 pg of RNA from a dhfr deletion mutant, DG44. It should be noted that the dhfr gene copy number varies over a 3-fold range for these mutant populations (Table I).
the deletions (band c); however, the corresponding 5' fragment (from the deletion end point to the end of exon 1) was barely detectable (solid circles between the lanes in Fig. 5). We do not know the reason for this deficit.
In summary, deletions that progressively encroach on the 5' end of the dhfr gene lead to the stepwise loss of first the minor distal promoter and then the major proximal promoter. A quantitative analysis of these promoter activities is pre- sented in Table I. As 5' sequences were eliminated, the activity of upstream cryptic promoter(s) increased somewhat until it represented the only dhfr transcripts in the cells. These aberrant transcripts were inefficiently processed; ap- proximately 75% remained unspliced.
To investigate further the nature of the cryptically pro- moted transcripts, and in particular the low amount of the 5"unspliced fragment, we carried out RNase protection ex- periments using an antisense riboprobe that included intron 1 (pMGS17, bottom of Fig. 1). This probe is more suitable for
detecting unspliced transcripts, as it will protect the entire 304-base intervening sequence. The result for the wild-type endogenous gene is shown in Fig. 6, lane 1; the RNA in this case was prepared from an amplified cell line (MK42) carrying approximately 300 copies of the endogenous dhfr gene. The two predominant bands of 193 and 149 bases (bands c and d) represent exon 1 of spliced mRNA molecules that were initi- ated at the minor and major start sites, respectively (see bottom of Fig. 1). The group of bands marked e are clustered at the 56-base position and correspond to the exon 2-3 portion of spliced mRNA. Band a may be the 509-base fragment resulting from a small amount of unspliced precursor. A somewhat smaller protected fragment (b ) is also present; its origin is not clear, but its size is consistent with a 452-base fragment representing exon 1 plus intron 1. Such a fragment could arise by cleavage at the 3' end of intron 1 before 5' cleavage of the intron. Such a sequence is not predicted from our understanding of the mechanism of splicing; however, the

16280 Dhfr Promoter Deletions
F
8- b-
C-
d-
e-
FIG. 6. Analysis of dhfr transcript splicing in pooled popu- lations of transfected cells. RNase protection was carried out as described for Fig. 5. The riboprobe used contained intron 1 and is shown at the bottom of Fig. 1. Positions c and d represent fragments generated by spliced transcripts originating at the minor and major promoters, respectively. Band e is the result of protection of the exon 2-3 portion of the probe by spliced mRNA. Band a represents un- spliced correctly initiated transcripts. Band b may represent aber- rantly processed transcripts (see text). The solid circles denote up- stream-initiated unspliced transcripts and the open circles upstream- initiated spliced transcripts. Lanes MI and M2 contain size markers, end-labeled 6x174 Hue111 and pBR322 MspI fragments, respectively. Lane I contains 0.6 pg of RNA from amplified cell line MK42, carrying approximately 300 copies of the endogenous dhfr gene. Lanes 2-4 contain RNA from pooled populations of cells transfected by the following minigenes: lane 2, pDCHlP, the full minigene, 30 pg; lane 3, mutant p414,15 pg; lane 4, mutant p413,15 pg.
Occurrence of small amounts of such aberrant in vivo splicing products cannot be ruled out.
The results with cells transfected by the full minigene are presented in lane 2 of Fig. 6. The major protected species were the same as those produced by the wild-type gene. The analy-
TABLE I1 Expression of dhjr transcripts in transfected populations
Mutant 5' flank dhfr Enzyme number remaininf mRNAb activitf :$:$
pDCHlP -900 100 100 100 12 -240 152 101 95 26 -166 124 155 98 24 -145 97 52 57
410 -111 122 56 54 414 -105 66 17 26 41 3 -79 47 5 20
Relative to the translational start site. 'Densitometric measurements from the experiment shown in Fig.
1 were corrected for length and amount of total RNA analyzed. The values for all spliced species were then added and expressed relative to pDCH1P.
e DHFR activity relative to pDCHlP (0.94 mIU/mg). For compar- ison, DHFR activity in CHO cells carryingone copy of the endogenous dhjr gene is 3 mIU/mg (Urlaub et al., 1983).
dPlating efficiency in DHFR selective medium lacking glycine, purines, and thymidine, relative to pDCH1P.
sis of the extreme deletion mutants 414 (lane 3) and 413 (lane 4 ) again showed evidence of initiation a t upstream promoters. The solid circles point out bands corresponding to 5' frag- ments from unspliced upstream-initiated transcripts, whereas the open circles indicate their spliced counterparts. Both types of protected fragment exhibit a size reduction corresponding to the size of the BAL-31 deletions. In agreement with the experiment shown in Fig. 5, no correctly initiated transcripts (positions c and d ) were produced by these mutants, both spliced and unspliced upstream-initiated transcripts were pro- duced, and the amounts of these transcripts were comparable to correctly initiated transcripts produced by the full mini- gene. This intron-containing probe detected no substantial promoter activity in the 304-bp intron 1, since no unaccounted for major band of constant size in the region between 56 bases (exons 2-3) and 361 bases (exons 2-3 plus intron 1) was present. The idea of transcription initiation within intron 1 had been considered as an explanation for the excess of 3' fragments seen in the transfected cells in Fig. 5.
The presence of substantial amounts of spliced transcripts initiating upstream of the normal transcriptional start sites in the promoterless mutants 413 and 414 raised the possibility that these molecules could function as messenger RNA. En- zymatic assay of cell extracts from the corresponding trans- fected populations revealed significant levels of DHFR activ- ity (Table 11). Indeed, the corresponding transfected popula- tions are able to grow in DHFR selective medium, albeit at a somewhat reduced plating efficiency (Table 11). The question then arises as to why few if any dhfr" transfectants were produced in the direct selection experiments using plasmid DNA of mutants 413 and 414 (Table I, column 4). One possibility is that a long expression period is necessary when using these "promoterless" mutants. In the direct selection, 24 h elapses between DNA exposure and plating in selective medium; when G418 transfectants are selected first, the dhfr genes are afforded an effective expression period of over 2 weeks. To test this idea, the direct selection experiment was repeated, comparing mutant 413 plasmid DNA and the full minigene pDCHlP and allowing the transfected recipient cells 1, 3,5, 7, and 10 days of growth in nonselective medium before plating in a medium lacking purines. Whereas few transfectants (1 x lO-'/cell) were observed with 1-3 days expression, this number increased after 5 days and by 10 days had achieved a value of 2 X 10-4/cell, 15% of the level produced by the wild-type minigene.

Dhfr Promoter Deletions 16281
DISCUSSION
Previous studies of the Chinese hamster, mouse, and human dhfr promoters have elucidated the following features: 1) multiple transcriptional start sites, with the proximal site predominating (Mitchell et al., 1986; Sazer and Schimke, 1986; Chen et al., 1984); 2) bidirectional transcription, with the opposite strand transcription initiating within 200 base pairs of the major dhfr start site (Crouse et al., 1985; Farnham et al., 1985; Mitchell et al., 1986); and 3) the conservation among the three species of a 30-bp sequence that starts 48-57 bp upstream of the transcriptional start site (Mitchell et al., 1986), with this sequence being present in multiple copies in the rodents (McGrogan et al., 1985; Mitchell et al., 1986). The conserved sequence includes no TATA box, but contains a GC box at the 5’ end that can act as a binding site for the transcription factor Spl (Dynan et al., 1986); at the 3‘ end of the region there is a conserved CAA box that represents a small AT-rich island in the GC-rich flank.
Our analysis of seven deletion mutations shows that the major promoter of the Chinese hamster dhfr gene functions with as little as 48 bp upstream of the major transcriptional start (in mutant 410) and that it requires the presence of the putative Spl binding sequence at the 5‘ end of this 48-bp region. The only difference between mutants 410 and 414 is the absence of this box in the latter; this absence correlates with the failure to initiate transcription at the major site. Notably lacking in mutant 410 is a CAAAT box located 72 bp upstream of the major transcriptional start (at position -135); this sequence is probably functioning in the minor promoter, rather than acting as a classical upstream CAAAT box. Whether the proximal CAA box is necessary for transcription awaits site-directed mutagenesis experiments. It is also clear that the activity of the major promoter is independent of the minor promoter. Our in uiuo results with the hamster dhfr gene confirm and extend the experiments of Farnham and Schimke (1986), who showed that a deletion extending past the proximal GC box inactivated the major promoter of the mouse dhfr gene in a cell-free transcription system.
The RNase protection analysis allowed us to examine the separate effects of deletions on the minor upstream promoter. More than 59 base pairs upstream of the transcriptional start are needed for minor promoter function in stable transform- ants, since this promoter was inactive in mutant 26. Notably, the presence of a GC box located 47 bp upstream of the minor start site (at position -153) was not sufficient. It should also be noted that the proximal GC box that functions in the major promoter and overlaps the minor start site cannot substitute for the upstream sequences. This result stands in contrast not only to that found for the major promoter, but also to the results of Farnham and Schimke (1986) using the mouse cell- free transcription system. It also is in contrast with the quantitative analysis of the transient expression assay, where inclusion of a the second GC box did yield increased DHFR activity. However, in view of the upstream starts revealed by the RNA analysis, measurements based on the amounts of gene product alone must be considered less reliable.
There are several reasons why the minor promoter might require a longer upstream region than that analogous to the 48-bp major promoter sequence. The sequence of the GC box upstream of the minor start site, AGGCGG, may not be close enough to the consensus sequence for Spl binding (GGCGG). The next perfect match to the consensus is not found until position -195, which is 89 bp upstream of the minor start site. In the mouse gene, the homologous GC box does match all six bases of the consensus sequence. Second, the minor promoter lies closer to the start site for opposite strand
transcription; interference by the opposite strand promoter may place additional requirements on this upstream dhfr promoter. Third, the minor promoter, being an intrinsically weaker promoter, may be more sensitive to interference from far upstream cryptic promoters that appear as an apparent artifact in this test system (see below). In any case, it is not yet clear that the minor promoters found in the rodent dhfr genes serve any physiological purpose, since minigenes lacking these promoters are able to efficiently transform DHFR- deficient cells and since the human gene lacks these redun- dant structures (Chen et aZ., 1984).
Deletion of 5”flanking sequences from this minigene re- veals the presence of cryptic promoter(s) lying upstream of the two dhfr promoters. The exact transcription initiation sites of these promoters was not determined here; all we know is that they are located further upstream than position -472, the extent of the probes used here. Only about 50 bp of dhfr genomic flank remain beyond the 5’ limit of probe protection. Since we see no promoter activity from this region in the endogenous dhfr gene, we think it likely that these cryptic promoters lie in the pUCl19 vector. Although these cryptic promoters are evidently artifacts of genetic engineering, they do raise several points of interest. First, the ability of RNA polymerase to read through the dhfr promoter region may be somewhat inhibited by dhfr flanking sequences, as the up- stream transcripts increase about 2-fold with progressive dele- tion of these sequences (Table I, last column). This inhibition may be related to competition from the major dhfr promoter, the activity of which decreases modestly as these sequences are deleted, or to the activity of the minor promoter, which is located in this region. A promoter on the opposite strand is also located here, and its activity may also impede the up- stream transcription. A second point of interest is that the correct splicing of intron 1 is inhibited (but not eliminated) in the transcripts that are initiated upstream. Thus 5’ RNA sequences can interfere with splicing at bona fide splice sites at least 150 bases downstream. Third, the upstream-initiated transcripts that are spliced reach substantial levels in trans- fected populations (approximately 50% of that found in cells carrying a single copy of the endogenous dhfr gene). These messenger RNA molecules are translated, since transfected cell populations containing only these RNA species contain substantial levels of DHFR enzyme activity. The translational efficiency of these mRNA molecules is apparently less than that of wild-type mRNA, as can be seen in the ratio of DHFR activity in spliced RNA levels in Table 11. Since the upstream- initiated mRNA molecules are likely to contain additional upstream translational start signals, this lower efficiency is not surprising (e.g. see Liu et al., 1984). The ability of trans- fectants with low enzyme levels to grow in DHFR-selective medium is in agreement with several earlier studies (Urlaub and Chasin, 1980; Crouse et al., 1983; Venolia et al., 1987).
The transient expression assay developed here provides a rapid and convenient means of testing the relative activity of mutated dhfr genes after transfection into DHFR-deficient recipient cells. The incorporation of [3H]deoxyuridine into DNA has also been used for assaying the thymidylate synthe- tase gene after transfection of mouse cell mutants lacking that enzyme activity (Kaneda et al., 1987), and an analogous assay for the hypoxanthine phosphoribosyltransferase gene has been described (Melton et al., 1986). Although the pro- moterless dhfr gene exhibits measurable incorporation, the relative activities calculated after subtraction of this back- ground are in reasonable agreement with the alternative meas- urements of gene activity (transfection frequencies and tran- script quantitation in permanently transformed cells). The

16282 Dhfr Promoter Deletions
use of Polybrene for gene transfer also facilitates the assay of Altman, S. (1987) Nucleic Acids Res. 16,1259-1270
rate calcium phosphate-DNA coprecipitates is avoided. In 228-232 Lee, F., Mulligan, R., Berg, P., and Ringold, G. (1981) Nature 294 ,
addition to assaying the activity of the dhfr promoters this Liu, C., Simonsen, C. C., and Levinson, A. D. (1984) Nature 3 0 9 , method may find general utility in systems where the dhfr 82-85 gene is used as a reporter gene joined to other control se- Luskey, M., and Botchan, M. R. (1984) Cell 36,391-401 quences (Lee et al., 1981; Kaufman and Sharp, 1982). Maniatis, T., Fritsch, E. F., and Sambrook, J. (1982) Molecular
Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory,
a large number of samples, as the preparation of many sepa- Kaufman, R. J, and Sharp, P. A. (1982) J. Mol. B i d 169,601-621
REFERENCES Bertino, J. R. (1979) Cancer Res. 39,293-304 Bertino, J. R., Booth, B. A., Bieber, A. L., Cashmore, A., and Sarto-
relli, A. C. (1964) J. Biol. C k m . 2 3 9 , 479-485 Carothers, A. M., Urlaub, G., Grunberger, D., and Casin, L. A. (1988)
Somatic Cell Mol. Genet. 14,169-183 Chaney, W. G., Howard, D. R., Pollard, J. W., Sallustio, S., and
Stanley, P. (1986) Somatic Cell Mol. Genet. 12,237-244 Chasin, L. A. (1986) in Molecular Cell Genetics (Gottesman, M. M.,
ed) pp. 449-488, Wiley & Sons, New York Chen, M.-J., Shimada, T., Moulton, A. D., Cline, A., Humphries, R.
K., Maizel, J., and Nienhuis, A. W. (1984) J. Biol. Chem. 2 5 9 , 3933-3943
Chirgwin, J. M., Przybyla, A. E., MacDonald, R. J., and Rutter, W. J. (1979) Biochemistry 18,5294-5299
Coon, H. G., and Weiss, M. (1969) Proc. Natl. Acad. Sci. U. S. A. 6 2 ,
Crouse, G. F., McEwan, R. N., and Pearson, M. L. (1983) Mol. Cell.
Crouse, G. F., Leys, E. J., McEwan, R. N., Frayne, E. G., and Kellems, R. E. (1985) Mol. Cell. Biol. 6, 1847-1858
Dynan, W. S. (1986) Trends Genet. 2 , 196-197 Dynan, W. S., and Tjian, R. (1985) Nature 3 1 6 , 774-778 Dynan, W. S., Sazer, S., Tjian, R., and Schimke, R. T. (1986) Nature
Farnham, P. J., Abrams, J. M., and Schimke, R. T. (1985) Proc. Natl.
Farnham, P. J., and Schimke, R. T. (1986) Mol. Cell. Biol. 6 , 365-
Ham, R. G. (1965) Proc. Natl. Acad. Sci. U. S. A. 63,288-293 Hattori, M., and Sakaki, Y. (1986) Anal. Biockm. 152,232-238 Kaneda, S., Takeishi, K., Ayusawa, D., Shimizu, K., Seno, T., and
852-859
BWl. 3,257-266
319,246-248
Acad. Sci. U. S. A. 82,3978-3982
371
~~
Cold Spring Harbor, New York McGrogan, M., Simonson, C. C., Smouse, D. T., Farnham, P. J., and
Schimke, R. T. (1985) J. Biol. Chem. 2 6 0 , 2307-2314 Melera, P. W., Davide, J. P., Hession, C. A., and Scotta, K. W. (1984)
Mol. Cell. BioL 4, 38-48 Melton, D. A., Krieg, P. A., Rebagliatti, M. R., Maniatis, T., Zinn,
K., and Green, M. R. (1984) Nucleic Acids Res. 12, 7035-7056 Melton, D. W., McEwan, C., McKie, A., and Reid, M. (1986) Cell 44,
Messing, J., Gronenborn, B., Muller-Hill, B., and Hofschneider, P.
Milbrandt, J. D., Azizkhan, J. C., and Hamlin, J. L. (1983) Mol. Cell.
Mitchell, P. J., Carothers, A. M., Han, J. H., Harding, J. D., Kas, E., Venolia, L., and Chasin, L. A. (1986) Mol. Cell. Bwl. 6 , 425-440
Nunberg, J. H., Kaufman, R. J., Schimke, R. T., Urlaub, G., and Chasin, L. A. (1978) Proc. Natl. Acad. Sci. U. S. A. 7 6 , 5553-5556
Orlofsky, A., and Chasin, L. A. (1985) Mol. Cell. BWl. 6,214-225 Sazer, S., and Schimke, R. T. (1986) J. BioL C h m . 261,4685-4690 Urlaub, G., and Chasin, L. A. (1980) Proc. Natl. Acad. Sci. U. S. A.
Urlaub, G., Kas, E., Carothers, A. M., and Chasin, L. A. (1983) Cell
Urlaub, G., Carothers, A. M., and Chasin, L. A. (1985a) Proc. Natl.
Urlaub. G.. McDowell. J.. and Chasin. L. A. (1985b) Somatic Cell
319-328
(1977) Proc. Natl. Acad. Sci. U. S. A. 74,3642-3646
BWl. 3,1274-1282
77,4216-4220
33,405-412
Acad. SC~. U. S. A. 82,1189-1193
MOL &At . 11,71-j7 Urlaub. G.. Mitchell. P. J.. Kas. E.. Funanape. V. L., Myoda, T. T.,
and Hamlin, J. L. (1986) Somatic' Cell M o r Genet. 12,555-566 Venolia, L., Urlaub, G., and Chasin, L. A. (1987) Somatic Cell Mol.
Genet. 13,491-504 Vieira, J., and Messing, J. (1987) Methods Enzymol. 163, 6-11 Wigler, M., Pellicer, A., Silverstein, S., Axel, R., Urlaub, G., and
Chasin, L. A. (1979) Proc. Natl. Acad. Sci. U. S. A. 76,1373-1376