computational structure-based redesign of enzyme activity cheng-yu chen, ivelin georgiev, amy...
Post on 22-Dec-2015
217 views
TRANSCRIPT

Computational Structure-Based Redesign of Enzyme Activity
Cheng-Yu Chen, Ivelin Georgiev, Amy C.Anderson, Bruce R.Donald
A Different computational redesign strategy
Yizhou Yin
Mar06, 2009

- Protein design:straightforward design vs. Directed mutation
De Novo vs. redesign
- Computational structure-based redesignGMEC (global minimum energy confirmation)
- ROSETTA (RosettaDesign, …)
1) Energy Function
2) Conformational Sampling
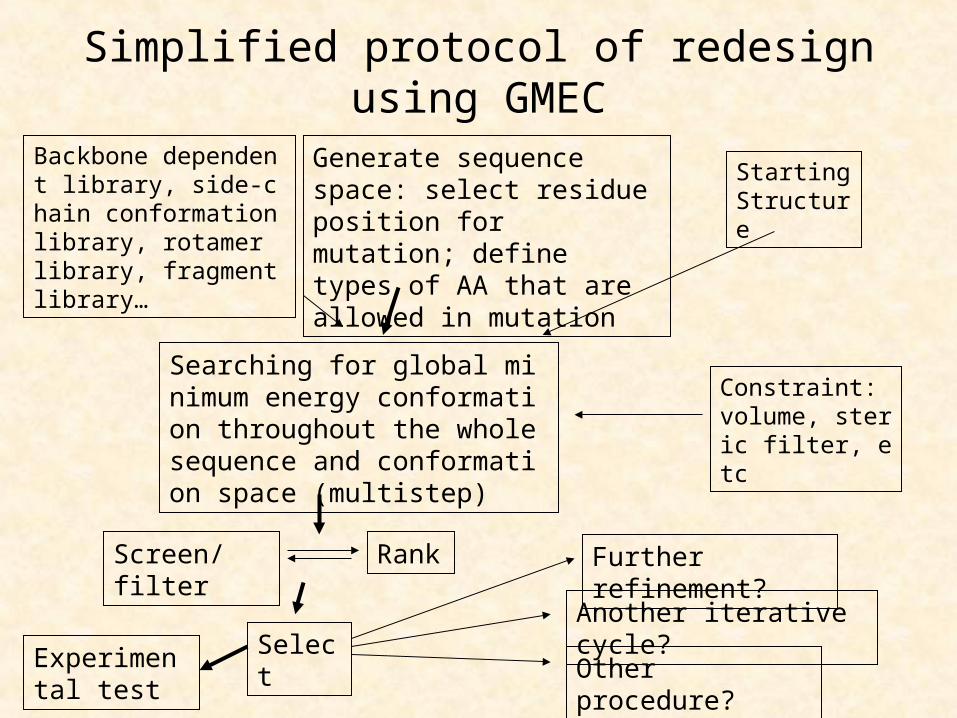
Simplified protocol of redesign using GMEC
Generate sequence space: select residue position for mutation; define types of AA that are allowed in mutation
Constraint: volume, steric filter, etc
Backbone dependent library, side-chain conformation library, rotamer library, fragment library…
Searching for global minimum energy conformation throughout the whole sequence and conformation space (multistep)
Starting Structure
Screen/filter Rank
Select
Further refinement?
Another iterative cycle?
Other procedure?Experimental test

Ensemble-based protein redesignBackbone dependent library, side-chain conformation library, rotamer library, fragment library…
Generate sequence space: select residue position for mutation (steric shell); define types of AA that are allowed in mutation
Starting Structure, targeted substrate, cofactor
Active site mutation
Filters: sequence-space filter, k-point, volume filter
K* algorithm: search and score
Rank + Select
Bolstering Mutation
Self-Consistent Mean Field entropy-based method
Experimental verification
MinDEE/A* algorithm: search and score
Experimental verification
Multiple pruning methods

K* algorithm- For a given protein-substrate complex, K* compute
s a provably-accurate ε-approximation to the binding constant KA
- K*= [Σexp(-Eb/RT)] / [Σexp(-El/RT)·Σexp(-Ef/RT)] b B∈ l L ∈ f F∈ B, L, F are rotamer-based ensembles; E is the conformation energy
- Several algorithms are used to prune the candidate sequences at different steps so that the searching in the sequence space will be more efficient.

For each allowable mutated sequence:
Step1 Molecular ensemble is generated, then pruned by steric, volume filters.
Step2 After constrained energy minimization, the conformation is enumerated by A*.
Step3 The scores from step2 are used to compute there separate partition functions, which is then combined to
compute K* score.

SCMF entropy-based method
Si = - ∑p(a︱ i) ln p(a︱ i) a A∈ i
p(a︱ i) = ∑ p(r︱ i) r R∈ a
- Ai is the set of AA types allowed at position i; p(a) is the probability of having AA type a at i. Ra is the set of rotamers for AA type a and p(r) is the probability of having rotamer r for AA type a at i.
- Higher entropy implies higher probability of multiple AA types, hence higher tolerance to mutation at position i.

Example of GrsA-PheA’sspecificity switched from Phe to Leu
- GrsA-PheA is the phenylalanine adenylation domain of the nonribosomal peptide synthetase (NRPS) enzyme gramicidin S synthetase A, whose cognate substrate is Phe.

-7 residues at the active site are allowed to mutate to (G, A, V, L, I, W, F, Y, M)
-only sequences with up to two mutations were considered, give the number candidates: 1450 (6.44 x 10<7>)
-After pruning, the number of sequences evaluated by K*: 505 (1.12 x 10<7>)
-Top ten sequences were experimentally verified.
-7 residues were selected by SCMF and were allowed to mutate to different subset of AA.
-Up to 3-point mutations were considered.
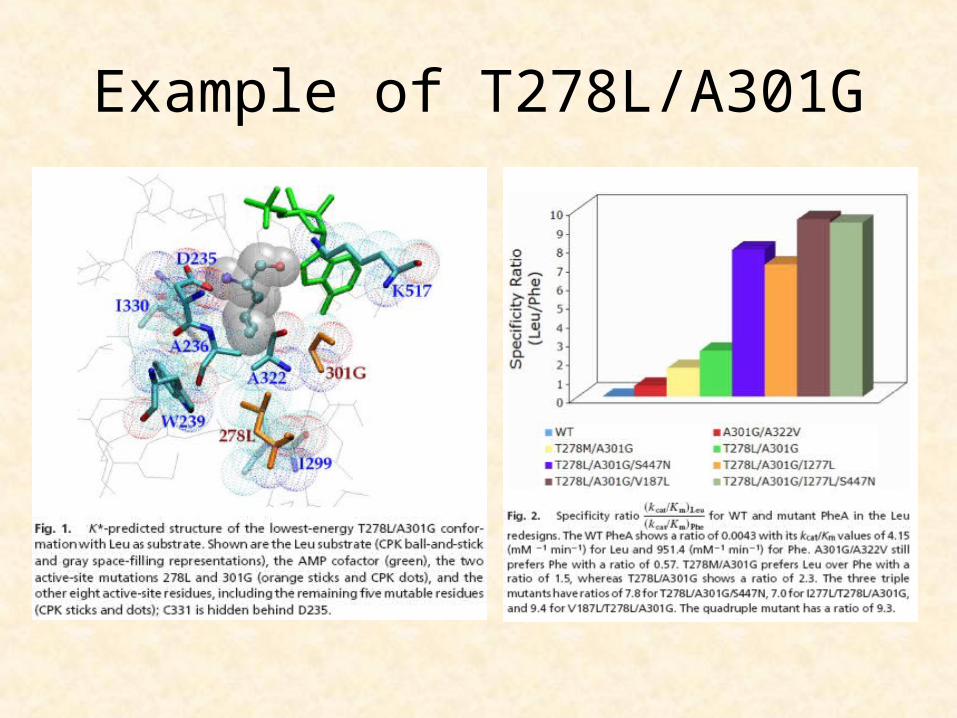
Example of T278L/A301G
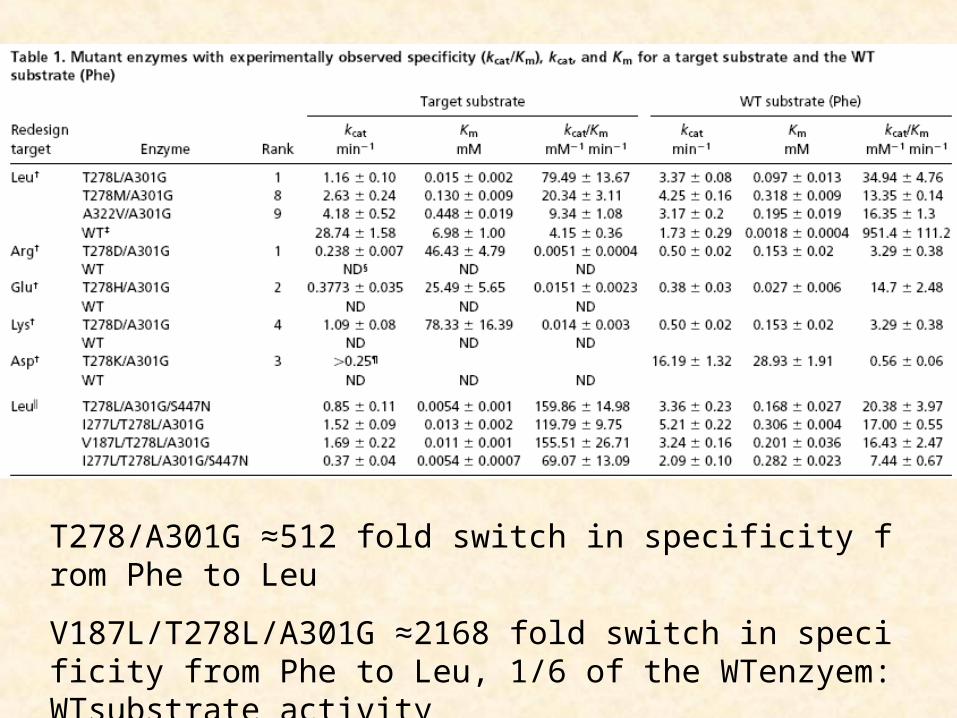
T278/A301G ≈512 fold switch in specificity from Phe to Leu
V187L/T278L/A301G ≈2168 fold switch in specificity from Phe to Leu, 1/6 of the WTenzyem:WTsubstrate activity

ensemble based vs. non-ensemble based
1) searching for best conformation
2) Searching for best mutation with best conformation
3) Other redesign
4) Other than redesign
structure-based design vs. other computational design/ evolution
Comparison in efficiency, accuracy

- Will there be any better “hybrid” methods?
- How to appropriately decide the sampling size based on the redesign methods?
- Any other new strategy?