computational approaches to antibody design: … approaches to antibody design: improvements to the...
TRANSCRIPT

Computational Approaches to Antibody Design:
Improvements to the Predictions of Structure,
Stability and Affinity
David A. Pearlman
Schrödinger, Inc.
Cambridge, MA
Biologics Conference
February 2015

Who is Schrödinger ?
Computational solutions
to drug discovery:
Software and services
• Founded 23 years ago
– Richard Friesner (Columbia)
& Bill Goddard (CalTech)
• ~250 employees, 50%
Ph.D.
• 300+ commercial
customers (including all
top 30 Pharma
companies); 2000+
academic groups; 100+
government agencies

Why are you here?

• Too slow
• Too expensive
• Too hard
• Don’t provide answers to the
questions you have
Sometimes experiments are just not enough
Computational Chemistry
MIGHT BE

Obtain antibody model
Theoretical
Calculations
Improved antibodies
Structure-based antibody modeling
Xray crystallographic
or
Homology methods

Obtain antibody model
Theoretical
Calculations
Improved antibodies
What can we do with theory?
• ID mutations to improve affinity/stability
• Epitope ID
• in silico affinity maturation / library design
• Humanization
• Extension to new species and constructs
• ADC site ID
• Stabilization (cysteine disulphides)
• Aggregation avoidance
• Enzyme design
• And many more…
Identifying favorable
mutations to create
disulphide bonds using a new
approach (2014) (Salem,
Adzhigirey, Sherman &
Pearlman, Prot Eng Design
and Select 27 365-374)
o Residue scanning applied to enzyme design (2014) (Sirin,
Kumar, Martinez, Karmilowicz, Abramov, Martin & Sherman)
J. Chem. Inf. Mod. 54 2334-2346
o Residue scanning applied to enzyme design II (2014) (Sirin,
Pearlman, Sherman) Proteins 82 3397-3409
Obtain antibody model
Theoretical
Calculations
Improved antibodies
Structure-based antibody modeling
Predicting Fv from sequence

Predicting antibody CDR: The H3 loop is difficult
For antibody, L1→L3, H1, H2 usually “pretty good”
using homology models.
But H3 is a problem:
Framework L1 L2 L3 H1 H2 H3
Length
range
6-13 3-3 7-8 7-9 5-6 10-13
RMS (Å) 0.9 1.0 0.5 1.4 1.3 1.1 3.3
Based on blinded prediction of 9 antibody structures, using four
different “best practice” approaches.
Almagro et al. (2011) Proteins 79 3050-3066
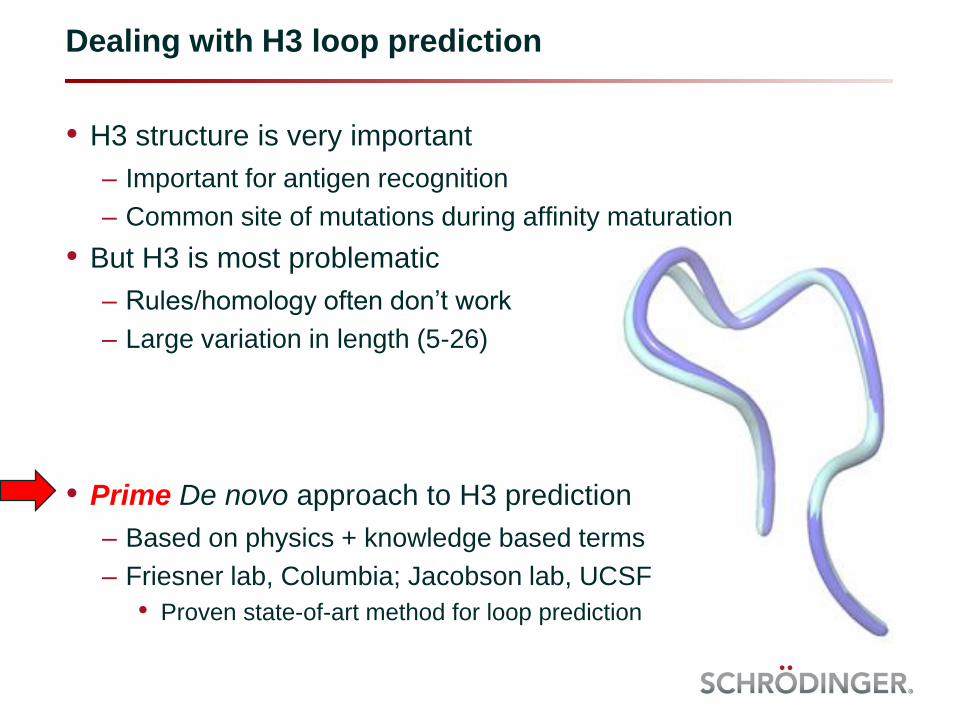
• H3 structure is very important
– Important for antigen recognition
– Common site of mutations during affinity maturation
• But H3 is most problematic
– Rules/homology often don’t work
– Large variation in length (5-26)
• Prime De novo approach to H3 prediction
– Based on physics + knowledge based terms
– Friesner lab, Columbia; Jacobson lab, UCSF
• Proven state-of-art method for loop prediction
Dealing with H3 loop prediction

• Organizers: J Almagro, A Teplyakov, J Luo, RW Sweet, S
Kondagantlil, F Hernandez-Guzman, R Stanfield, GL Gilliland
• 7 Participants:
– Schödinger; CCG; Accelrys; Rosetta (Jeff Grey @John Hopkins)),
Macromoltek; Astellas Pharma + Osaka U; PIGS server
• Predict 11 unpublished structures:
– 4 human Ab + 6 mouse Ab + 1 rabbit Ab
• Two stages:
– Stage 1: Predict full Fv from sequence
– Stage 2: Predict H3 given xray coords of remainder of structure
The 2nd Blinded Antibody Modeling Assessment (AMA-II)
2013

Antibody Assessment II publication
Proteins: Structure, Function, and
Bioinformatics
Special Issue: Antibody Modeling Assessment
II
Volume 82, Issue 8 August 2014
Our contribution:
K. Zhu, T. Day, D. Warshaviak, C. Murrett, R.
Friesner, D.A. Pearlman (2014) Antibody
structure determination using a
combination of homology modeling,
energy-based refinement, and loop
prediction Proteins: Struct, Funct and Bioinf
82 1646-1655.

Method Fv RMSD Framework
RMSD
All loops
RMSD –H3
H3 RMSD
Schrödinger 1.1 ± 0.2Å 0.8 ± 0.2Å 1.1 ± 0.4Å 2.7 ± 0.8Å
Accelrys 1.1 ± 0.3Å 0.9 ± 0.3Å 1.1 ± 0.5Å 3.0 ± 1.1Å
CCG 1.1 ± 0.2Å 0.9 ± 0.3Å 1.0 ± 0.3Å 3.3 ± 0.9Å
Rosetta (Jeff Grey) 1.1 ± 0.2Å 0.8 ± 0.2Å 1.1 ± 0.4Å 2.6 ± 0.9Å
Macromoltek 1.4 ± 0.2Å 1.2 ± 0.2Å 1.2 ± 0.3Å 3.0 ± 1.0Å
Astellas + Osaka U 1.1 ± 0.2Å 0.8 ± 0.2Å 1.0 ± 0.2Å 2.3 ± 0.6Å
PIGS server 1.2 ± 0.1Å 0.9 ± 0.2Å 0.9 ± 0.4Å 3.1 ± 1.1Å
Average 1.1 ± 0.2Å 0.9 ± 0.2A 1.1 + 0.4Å 2.8 ± 0.9Å
AMA-II : Overall results for Round 1:
Full Fv from sequence
• All methods are generally producing decent models
• H3 is the recurrent problem

Method H3 RMSD
(Round 1)
H3 RMSD
(Round 2)
Schrödinger 2.7 ± 0.8Å 1.4 ± 1.1Å
Accelrys 3.0 ± 1.1Å 2.3 ± 1.0Å
CCG 3.3 ± 0.9Å 2.5 ± 1.6Å
Rosetta (Jeff Grey) 2.6 ± 0.9Å 2.1 ± 1.1Å
Macromoltek 3.0 ± 1.0Å 3.3 ± 1.2Å
Astellas + Osaka U 2.3 ± 0.6Å 1.4 ± 1.9Å
PIGS server 3.1 ± 1.1Å
Average 2.8 ± 0.9A 2.2 ± 0.9Å
AMA-II : Overall results for Round 2:
Predict H3, given xray structure of remainder of Fv
• Impressive automated prediction using Prime

Obtain antibody model
Theoretical
Calculations
Improved antibodies
Structure-based antibody modeling
FEP
(Free Energy
Perturbation)

• How does this residue change affect:
– Stability
– Affinity (to other molecules)
Which way do we go? Residue mutation studies
ABC AXC
?
• Examples:
– Theoretical residue / alanine scanning
– In silico affinity maturation (multiple simultaneous mutations)
– Binary protein design decisions (“Make B or X?”)
– Evaluating possible non-natural amino acids
– Fundamental questions in all protein design

• Empirical scoring methods
– Approximate
– Fast
– Can only predict parameterized moieties
• MM-GBSA
– Approximate (implicit solvent)
– Fast (< 1 minute per calculation)
• FEP (Free Energy Perturbation)
– Precise (mean unsigned error ~1 kcal/mol)
– Computationally more expensive, explicit solvent
– ~1 calculation per GPU processor/day
– Requires huge amt conformational sampling
Predicting free energy changes: Affinity/Stability of
Residue A to Residue B
ABC AXC
?
Physics-based methods
(Can predict non-standard AA)

FEP: A 30 year ride to a robust approach
Hype Cycle for FEP
1985
1987
mid
90s
2015
Relative free energies of amino acid
sidechains (Science, Nature)
(Kollman, McCammon)
1989: D.A. Pearlman &
P.A. Kollman ``Free Energy
Perturbation Calculations:
Problems and Pitfalls Along the
Gilded Road.'' (In: Computer
Simulation of Biomolecular
Systems: Theoretical and
Experimental Applications pp. 101-
119, Escom)Relative free energies
of ethane and
methanol (Jorgensen)
Many publications;
Good ideas, but
tools not quite
there, especially
computer speeds
needed for
adequate sampling

FEP: Technologies Facilitate a Robust Solution
Improved force field…….
Enhanced sampling........
Hardware acceleration…
Automated setup………..
Error estimates………….
OPLS2
REST
GPU
FEP Mapper
Cycle Closure
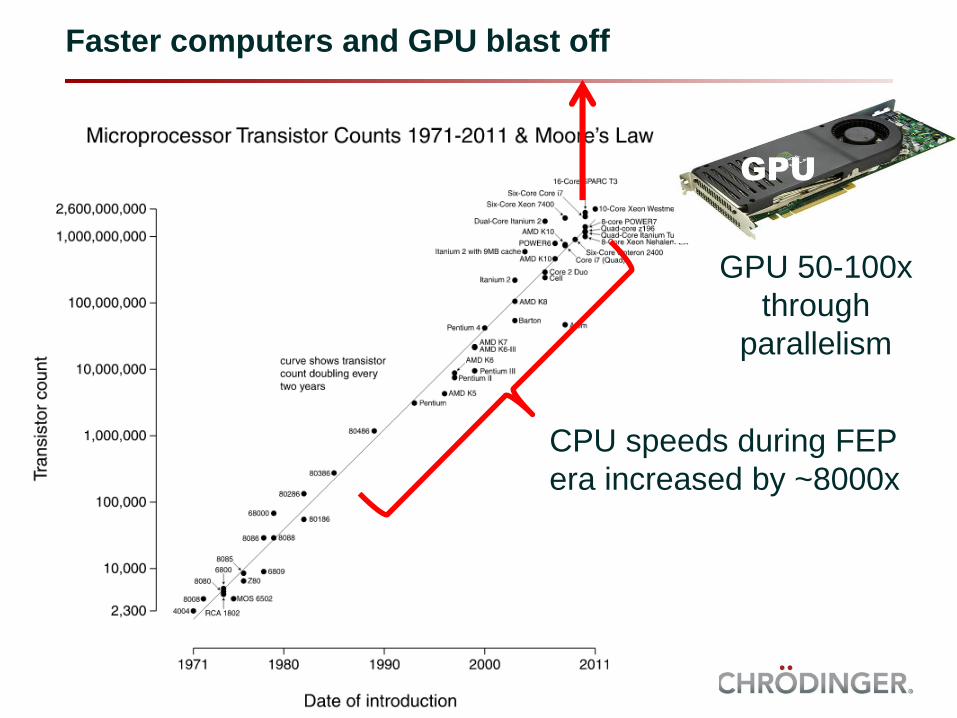
Faster computers and GPU blast off
CPU speeds during FEP
era increased by ~8000x
GPU
GPU 50-100x
through
parallelism

Schrodinger’s GPU Cluster
400 GPUs: Two 200 GPU racks
Roughly the same
processing power as
the total of every
home PC in the USA
in 1987
Year Relative amount
sampling
How long would
it take?
Sufficient for
accuracy?
1987 1x 1 month on
supercomputer
No
2014 3000x 1 day on GPU Yes

Predicting Protein Stability
Folding/unfolding
equilibrium

How we calculate relative stability using FEP
DDGstability = DG1 – DG2
= DGA – DGB
A
B
1 2
Vertical processes
are experimental
Horizontal
processes more
easily calculated
theoretically
Calculations use
MD sampling &
statistical
mechanics
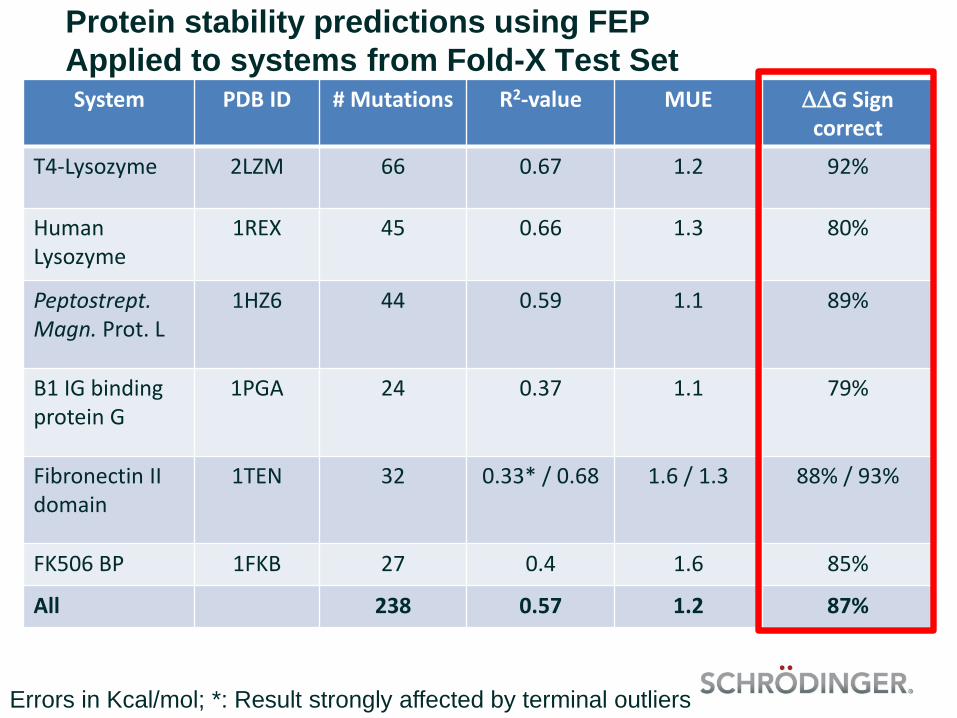
Protein stability predictions using FEP
Applied to systems from Fold-X Test SetSystem PDB ID # Mutations R2-value MUE DDG Sign
correct
T4-Lysozyme 2LZM 66 0.67 1.2 92%
Human Lysozyme
1REX 45 0.66 1.3 80%
Peptostrept. Magn. Prot. L
1HZ6 44 0.59 1.1 89%
B1 IG binding protein G
1PGA 24 0.37 1.1 79%
Fibronectin II domain
1TEN 32 0.33* / 0.68 1.6 / 1.3 88% / 93%
FK506 BP 1FKB 27 0.4 1.6 85%
All 238 0.57 1.2 87%
Errors in Kcal/mol; *: Result strongly affected by terminal outliers

Aggregate FEP stability predictions
Slope = 1.3
Offset = -0.2
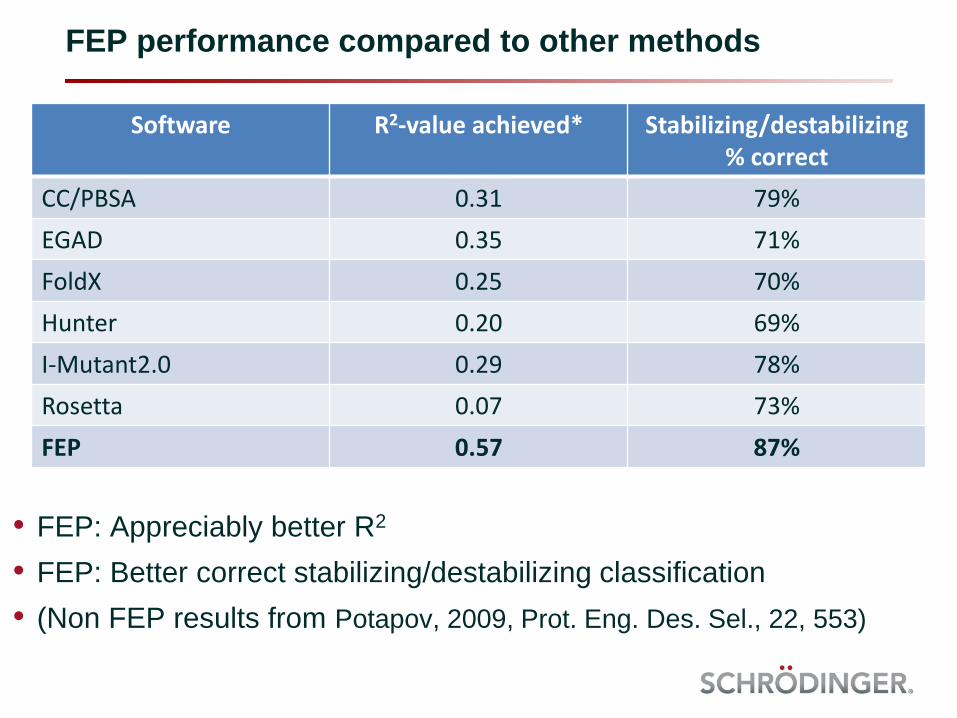
FEP performance compared to other methods
Software R2-value achieved* Stabilizing/destabilizing% correct
CC/PBSA 0.31 79%
EGAD 0.35 71%
FoldX 0.25 70%
Hunter 0.20 69%
I-Mutant2.0 0.29 78%
Rosetta 0.07 73%
FEP 0.57 87%
• FEP: Appreciably better R2
• FEP: Better correct stabilizing/destabilizing classification
• (Non FEP results from Potapov, 2009, Prot. Eng. Des. Sel., 22, 553)

FEP for affinity already validated for small mol ligands
-15
-14
-13
-12
-11
-10
-9
-8
-7
-6
-5
-4
-15 -14 -13 -12 -11 -10 -9 -8 -7 -6 -5 -4
BACE
CDK2
JNK1
MCL1
ΔG
FEP
(kc
al/m
ol)
ΔG Expt. (kcal/mol)|ΔΔGFEP – ΔΔGExpt.| (kcal/mol)
Pe
rce
nta
ge
46.2%
24.8%
15.4%
7.4% 6.2%
0%
10%
20%
30%
40%
50%
< 0.6 0.6-1.2 1.2-1.8 1.8-2.4 >2.4
• Over 500 perturbations tested for 17 systems w/ identical automated
protocol
– RMSE ≈ 1.2 kcal/mol

• We can predict antibody structure (CDR) from sequence
• H3 is problematic, but our Prime approach seems big step forward
• FEP calculations have come of age for stability / affinity
– More reliable than established prediction tools
– Physics based, applicable to predictions for non-standard amino acids
– Can now be run overnight on single GPU
– Suitable for incorporation in process of biologics design
The view from here…

• Kai Zhu
• Tyler Day
• Dora Warshaviak
• Colleen Murrett
• Richard Friesner
Acknowledgements
• Thomas Steinbrecher
• Woody Sherman
• Robert Abel
DRUG DISCOVERY COLLABORATIONS
Antibody predictions FEP Calculations
Visit us at
Booth # 3
