clinical round - mondino€¦ · anamnesi familiare • madre di 55 anni, residente presso...
TRANSCRIPT

CLINICAL ROUND Dr. Gianpaolo M. D. Toscano
Scuola di Specializzazione in Neurologia
Moderatrice: Dr.ssa Roberta Zangaglia
UO Parkinson e Disturbi del Movimento IRCCS C. Mondino - Pavia

Caso Clinico
• M.M., ♂ 30 anni
• Ritardo mentale
• Sindrome distonica (spalla, collo) e discinetica
• Anomalie comportamentali (irritabilità, aggressività, acatisia)
• Residente presso RSD dall’età di 16 anni

Anamnesi Familiare • Madre di 55 anni, residente presso struttura
protetta psichiatrica • Padre biologico deceduto • Due fratelli (per linea materna):
- 29 anni, probabile ritardo mentale - 12 anni, adottato, non altre informazioni
• Probabili origini sarde (linea materna)
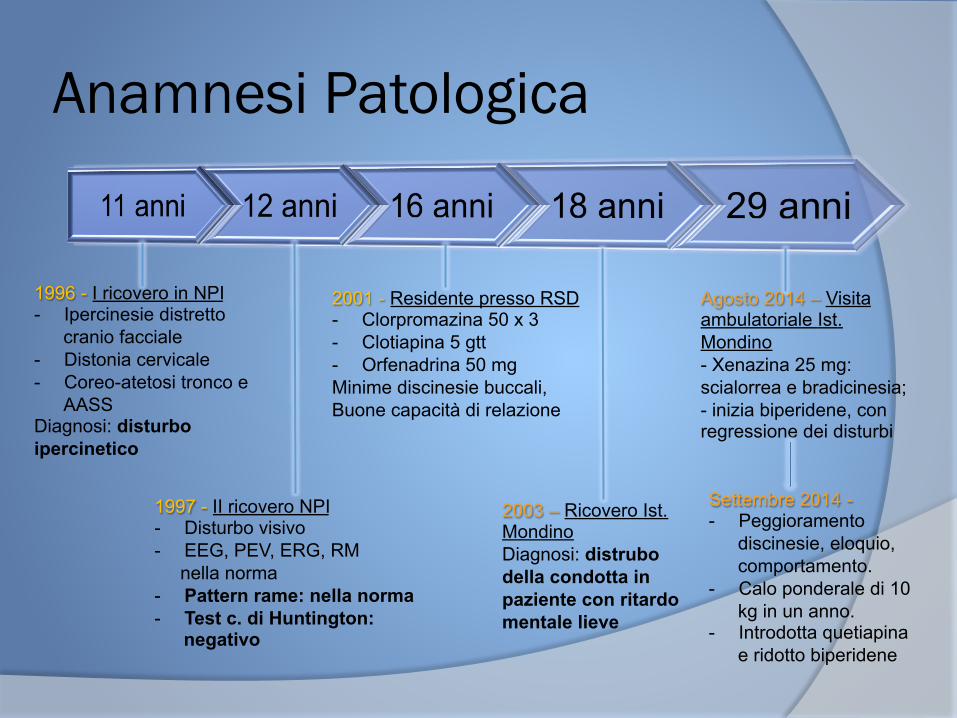
Anamnesi Patologica
1996 - I ricovero in NPI - Ipercinesie distretto
cranio facciale - Distonia cervicale - Coreo-atetosi tronco e
AASS Diagnosi: disturbo ipercinetico
1997 - II ricovero NPI - Disturbo visivo - EEG, PEV, ERG, RM nella norma - Pattern rame: nella norma - Test c. di Huntington:
negativo
2001 - Residente presso RSD - Clorpromazina 50 x 3 - Clotiapina 5 gtt - Orfenadrina 50 mg Minime discinesie buccali, Buone capacità di relazione
Agosto 2014 – Visita ambulatoriale Ist. Mondino - Xenazina 25 mg: scialorrea e bradicinesia; - inizia biperidene, con regressione dei disturbi
Settembre 2014 - - Peggioramento
discinesie, eloquio, comportamento.
- Calo ponderale di 10 kg in un anno.
- Introdotta quetiapina e ridotto biperidene
2003 – Ricovero Ist. Mondino Diagnosi: distrubo della condotta in paziente con ritardo mentale lieve

Quadro Clinico Esame Obiettivo: Soggetto normotipo, sottopeso. Turricefalia, palato ogivale, micrognazia. Distonia della spalla (> a dx) con rotazione del tronco e della spalla, cifosi dorsale e distonia cervicale compensatoria, antero-latero deviazione dx del capo, grave contrattura dei trapezi e dell’elevatore della scapola bilateralmente.
Esame Neurologico: Vigile, orientato. Disartria moderato-severa. Discinesie buccali. Tic e movimenti stereotipati. Acatisia, iperattività. Arti sup: Ipertono misto prossimale bilaterale. ROT scattanti per dx>sn. Movimenti atetosici delle dita della mano sn. Movimenti atetosici degli arti superiori con componente distonica. Arti inf: Rinforzo del tono prossimale. ROT scattanti per dx>sn. RCP: Babinski a dx, scorretto a sx. Sensibilità, coordinazione: nella norma. Stazione eretta: spinta + con recupero. Deambulazione: disarmonica, scarto di lateralità bilaterale.

Diagnosi Differenziale • Distonia primaria (ereditaria)
• Malattia di Huntington giovanile (forma rigida)
• Forme giovanili di gangliosidosi GM1 o GM2
• Malattia di Wilson

Approfondimenti diagnostici • Eco addome: fegato nei limiti morfovolumetrici,
contorni netti, ecostruttura omogenea, indenne da lesioni.
• Valutazione oculistica: ODV conta dita a 5 metri con lenti, OSV conta dita a 2 metri con lenti; cornea trasparente; papilla pallida; macula e albero vascolare nei limiti.
• PEV: o Pattern Reversal - Valutazione della morfologia, della
latenza e dell’ampiezza dell’onda P100: risposte assenti in OD, assenti in OS.
o PEV da stimolo flash con valutazione di ElettroRetinoGramma - Per stimolazione OD e OS ERG nella norma, risposte corticali nella norma.
• ENG: nella norma.

RM encefalo • Ispessimento diffuso delle ossa della teca
cranica. • Fossa cranica posteriore di piccole dimensioni. • Corpo calloso nastriforme, rostro ipertrofico e
tozzo. • Foci di suscettibilità magnetica in corrispondenza
dei nuclei pallidi bilateralmente, di significato distrofico aspecifico da accumulo di sostanze ferromagnetiche: ipointensità in T1 nel contesto di diffusa lieve iperintensità in T1 dei nuclei pallidi

T1
T2 T2
Flair

FFE SWI

Malattia di Wilson?
II dosaggio (in corso di ricovero): - Cupremia 121 µg/dl (60-160) - Ceruloplasminemia 35 mg/dl (20-60) - Cupruria 24 ore: 64 µg/24 h (15-70)
I dosaggio (pre-ricovero): - Ceruloplasminemia 13 mg/dl (20-60) - Cupruria 24 ore: 160 µg/24 h (15-70)

Score sviluppato all’VIII International Meeting on Wilson’s disease, Leipzig 2001
∗If no quantitative liver copper available, ∗∗or typical abnormalities at brain magnetic resonance imaging. KF, Kayser–Fleischer; ULN, upper limit of normal.

Malattia di Wilson?

NBIA (Neurodegeneration with Brain Iron Accumulation) • Gruppo di disordini ereditari caratterizzati
da disturbi extrapiramidali, disabilità intellettiva e accumulo di depositi di ferro nei gangli della base
• 8 AR, 1 AD, 1 X-linked dominante
• Patologia ‘ultra-rara’ meno di 1/1.000.000
• Spettro clinico vario

Distribution of NBIA subtypes in the North American database. NBIA: neurodegeneration with brain iron accumulation, PKAN: pantothenate kinase-associated neurodegeneration, PLAN: phospholipase A2-associated neurodegeneration, INAD: infantile neuroaxonal dystrophy, MPAN: mitochondrial membrane protein-associated neurodegeneration, BPAN: beta-propeller protein-associated neurodegeneration, FAHN: fatty acid hydroxylase-associated neurodegeneration, CoPAN: Coenzyme A synthase protein-associated neurodegeneration, NF: neuroferritinopathy, KRS: Kufor-Rakeb syndrome, ACP: aceruloplasminemia.
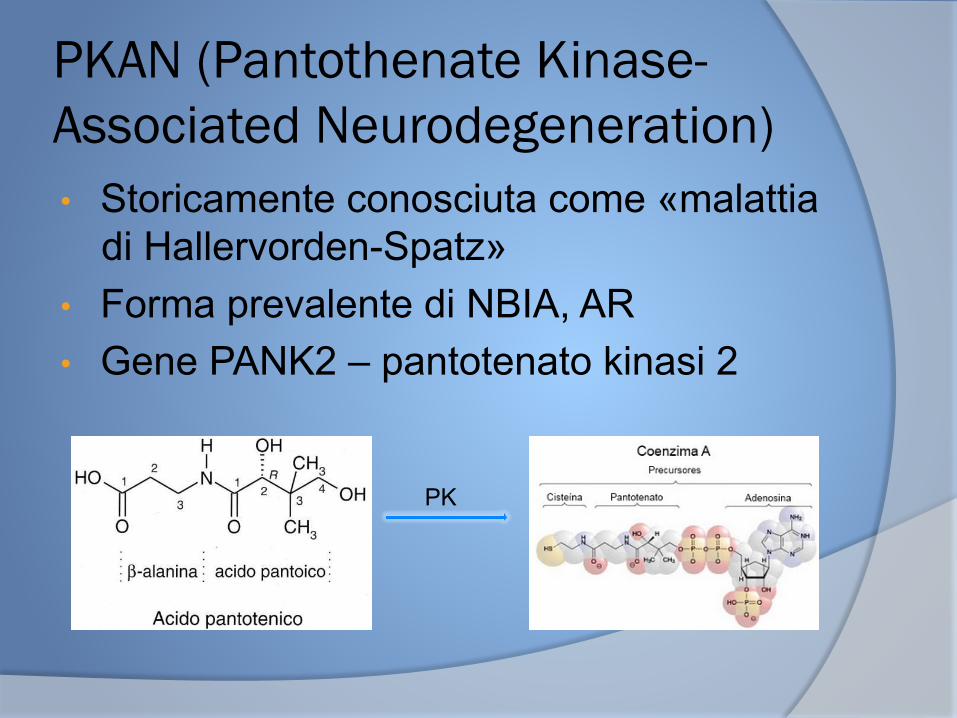
PKAN (Pantothenate Kinase-Associated Neurodegeneration) • Storicamente conosciuta come «malattia
di Hallervorden-Spatz» • Forma prevalente di NBIA, AR • Gene PANK2 – pantotenato kinasi 2
PK

PKAN: varianti FORMA CLASSICA FORMA ATIPICA
Presentazione Precoce (<6 aa) Tardiva, in età adolescenziale o giovanile
Decorso Rapidamente progressivo, sedia a rotelle entro l’adolescenza
Lento, aspettativa di vita normale
Caratteristiche - Distonia, prevalentemente AAII
- Segni piramidali - Emeralopia con ERG
anormale
- Palilalia simil-parkinson, Ipofonia, Disartria
- Distonia - Parkinsonismo - Spasticità di vario grado
Interessamento bulbare
Presente: compromissione di linguaggio e deglutizione con malnutrizione e frequenti polmoniti ab ingestis
Raro
Marker Clinico - Postura opistotonica - Distonie indotte dall’azione

PKAN: RM encefalo • Le imagini T2 pesate evidenziano pallidi ipointensi
con una regione di iperintensità localizzata anteromedialmente: “eye of the tiger sign”
• Sensibilità e specificità non assolute: ü alcuni casi senza segno ü segno riscontrato in MPAN,
intossicazione da CO, MSA, neuroferritinopatia..

PKAN: terapia
• Terapia sintomatica per la distonia e la spasticità: anticolinergici, bdz, baclofen, tossina botulinica
• DBS
• Terapia ferrochelante

PLAN (Phospholipase A2-associated Neurodegeneration) • Seconda NBIA più frequente • Gene PLA2G6 (PLA2 Ca-indipendente) • La forma infantile-onset è chiamata “distrofia
neuroassonale infantile” (INAD)
FORMA CLASSICA FORMA ATIPICA Presentazione Precoce (6 mesi-3 anni), con
rallentamento dello sviluppo e poi deterioramento in tutti i domini
Fase tardiva dell’infanzia
Decorso Aggressivo, ipotonia del tronco precoce (DD con SMA)
Lento
Caratteristiche - Atrofia ottica, strabismo, Ny à ipovisus, cecità
- Ipotonia e areflessia à tetraplegia spastica
- Ritardo mentale, epilessia
- Disfunzione cerebellare: atassia della marcia, disartria
- Ipotonia e areflessia - Spasticità di vario grado, distonia - Ritardo mentale, epilessia

PLAN: RM encefalo INAD: - Atrofia cerebellare (100% delle INAD) - Iperintensità sostanza bianca in T2 - Assottigliamento del corpo calloso e chiasma ottico
aNAD: - Atrofia cerebellare non costantemente presente
Gli accumuli di ferro possono non diventare mai evidenti!

PLAN: terapia • - Terapie per epilessia, spasticità,
parkinsonismo (precoci fluttuazioni motorie ed esacerbazioni sintomi neuropsichiatrici!)
• Nuove tecniche di imaging hanno mostrato nel cervello del topo mutante ridotto incorporamento di ac. docosaesaenoico e alterazione biosegnale del calcio

MPAN (Mithocondrial Membrane Protein Associated Neurodegeneration)
• Mutazione in c19orf12: associazione con membrana mitocondriale, ruolo nel metabolismo degli acidi grassi
• Entro i 10 anni ma anche nel giovane adulto, andatura spastica, atrofia ottica, disartria, disturbo comportamentali e psichiatrici
• Distonia limitata a piedi e mani, se presente • Lenta progressione, sopravvivenza anche oltre i
20 anni

BPAN (Beta-Propeller Protein-Associated Neurodegeneration)
• Ereditarietà X-linked dominante
• Epilessia, segni piramidali e disturbi del sonno (ipersonnia, insonnia, RBD) in infanzia à parkinsonismo, distonia, demenza
• Movimenti stereotipati delle mani: “S. di Rett atipica”


Non PKAN PKAN
RM encefalo: accumulo di ferro
Distonie orofacciali, tic, disartria, impulsività
Frequenza
RM: assottigliamento corpo calloso, assenza “eye of tiger sign”
Atrofia ottica MPAN
PLAN
PLAN

Wilson? Altro?
PKAN?
PLAN?
MPAN?

Conclusioni • NBIA comprende un gruppo eterogeneo di
patologie • I meccanismi fisiopatologici sono ancora poco
conosciuti
• Criteri diagnostici non esistenti, overlapping clinico frequente
• Diagnosi clinica complicata, terapia inadeguata

Grazie per l’attenzione