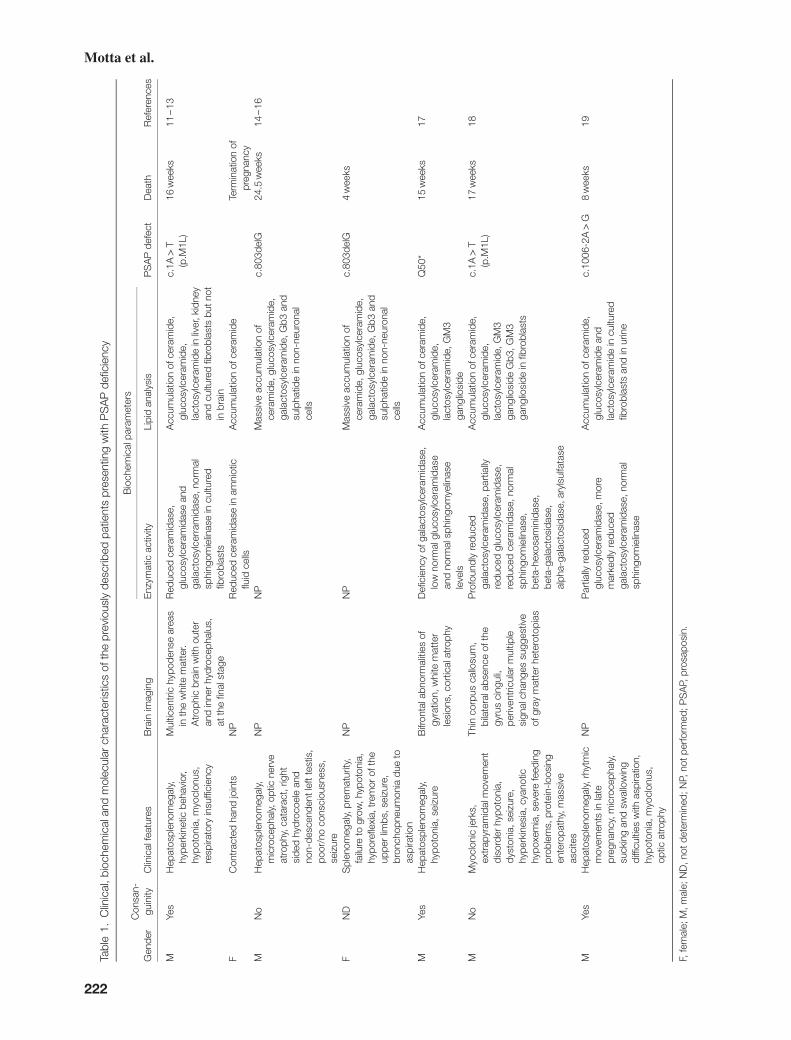
clinical, biochemical and molecular characterization of ... · f nd splenomegaly, prematurity,...
TRANSCRIPT

Clin Genet 2016: 90: 220–229Printed in Singapore. All rights reserved
© 2016 John Wiley & Sons A/S.Published by John Wiley & Sons Ltd
CLINICAL GENETICSdoi: 10.1111/cge.12753
Original Article
Clinical, biochemical and molecularcharacterization of prosaposin deficiency
Motta M., Tatti M., Furlan F., Celato A., Di Fruscio G., Polo G., Manara R.,Nigro V., Tartaglia M., Burlina A., Salvioli R. Clinical, biochemical andmolecular characterization of prosaposin deficiency.Clin Genet 2016: 90: 220–229. © John Wiley & Sons A/S. Published byJohn Wiley & Sons Ltd, 2016
Prosaposin (PSAP) deficiency is an ultra-rare, fatal infantile lysosomalstorage disorder (LSD) caused by variants in the PSAP gene, with sevensubjects reported so far. Here, we provide the clinical, biochemical andmolecular characterization of two additional PSAP deficiency cases.Lysoplex, a targeted resequencing approach was utilized to identify thevariant in the first patient, while quantification of plasma lysosphingolipids(lysoSLs), assessed by liquid chromatography mass spectrometry(LC-MS/MS) and brain magnetic resonance imaging (MRI), followed bySanger sequencing allowed to attain diagnosis in the second case. Functionalstudies were carried out on patients’ fibroblast lines to explore the functionalimpact of variants. The two patients were homozygous for two differenttruncating PSAP mutations (c.895G>T, p.Glu299*; c.834_835delGA,p.Glu278Aspfs*27). Both variants led to a complete lack of processedtranscript. LC-MS/MS and brain MRI analyses consistently provided adistinctive profile in the two children. Quantification of specific plasmalysoSLs revealed elevated levels of globotriaosylsphingosine (LysoGb3) andglucosylsphingosine (GlSph), and accumulation of autophagosomes, due toa decreased autophagic flux, was observed. This report documents thesuccessful use of plasma lysoSLs profiling in the PSAP deficiency diagnosis,as a reliable and informative tool to obtain a preliminary information ininfantile cases with complex traits displaying severe neurological signs andvisceral involvement.
Conflict of interest
The authors declare no conflict of interest.
M. Mottaa, M. Tattib, F. Furlanc,A. Celatoc, G. Di Frusciod,e,G. Poloc, R. Manarac,V. Nigrod,e, M. Tartagliaa,A. Burlinac and R. Salviolib
aGenetics and Rare Diseases ResearchDivision, Ospedale Pediatrico BambinoGesù, Rome, Italy, bDepartment ofHaematology, Oncology and MolecularMedicine, Istituto Superiore di Sanità,Rome, Italy, cDivision of InheritedMetabolic Diseases, University Hospital,Padua, Italy, dDepartment ofBiochemistry, Biophysics and GeneralPathology, Second University of Naples,Naples, Italy, and eTelethon Institute ofGenetics and Medicine (TIGEM), Naples,Italy
Key words: autophagy – brainmagnetic resonance imaging –lysosphingolipids – prosaposindeficiency – saposins
Corresponding author:Dr Rosa Salvioli, Department ofHaematology, Oncology and MolecularMedicine, Istituto Superiore di Sanità,Viale Regina Elena, 299, 00161, Rome,Italy.Tel.: +390649902887;fax: +390649387086;e-mail: [email protected]
Received 18 December 2015, revisedand accepted for publication 29January 2016
Prosaposin (PSAP) deficiency (MIM #611721) is a fatalinfantile lysosomal storage disease (LSD) inherited as anautosomal recessive trait. The disease, which is rarelydiagnosed, is caused by variants in the PSAP gene (MIM#176801), and is characterized by whole lack of PSAPprotein (1). PSAP encodes a 65/70 kDa multifunctionalhighly conserved glycoprotein. The fully glycosylated(70 kDa) form is secreted by cells in cerebrospinal fluid,maternal milk, seminal plasma, pancreatic juice, bileand brain, and is involved in many biological func-tions, including the development of the nervous and
reproductive systems (2–4). Differently, the partiallyglycosylated (65 kDa) PSAP form is targeted to lyso-somes, where its proteolytic cleavage releases four smallactive peptides, known as saposins (Saps) A, B, C and D(1). Saps are involved in sphingolipids (SLs) breakdownacting as enzymatic activators (5–7), and are requiredfor the hydrolysis of galactosylceramide (GalCer) (SapA), catabolism of cerebroside sulfate (CS), globotriao-sylceramide (Gb3Cer) and lactosylceramide (LacCer)(Sap B), degradation of glucosylceramide (GlCer) (SapC), and hydrolysis of ceramide (Cer) (Sap D). Inherited
220

Two new cases of prosaposin deficiency
deficiency of Sap A, Sap B or Sap C lead to sphingolipi-doses, which are described as variant forms of late-onsetKrabbe disease (8), metachromatic leukodystrophy (9),and Gaucher disease (10), respectively. Mutation in SapD domain of PSAP, causing Farber disease, has beenreported only in a mouse model (11). PSAP deficiencycombines the features of all the above mentioned disor-ders. Affected individuals show a severe neurovisceraldystrophy associated with the storage of complex SLs(i.e. GlCer, LacCer, Gb3Cer, CS, and Cer in multiple tis-sues), and some related N-deacylated forms in plasmaand brain. Neurologic symptoms may be caused eitherby the toxic build-up of different lipids and/or by loss ofPSAP neurotrophic function.
To date, seven patients with PSAP deficiency, carryingfour different variants at the homozygous state havebeen described (12–20). Two recurrent variants havebeen reported; c.1A>T (p.Met1Leu) was documentedin three individuals from eastern Europe (12–14, 19),while the single nucleotide deletion, c.803delG, wasidentified in two apparently unrelated subjects from thesame geographical district of eastern Slovakia (15, 16).The two remaining variants were also loss-of-functionmutations (18, 20). Genotypes and phenotypes of thesepatients are summarized in Table 1.
In this study, we provide the clinical, biochemi-cal and molecular characterization of two additionalPSAP-deficient subjects, and show that autophagyflux is dysregulated in primary fibroblasts of thetwo affected individuals. Finally, we demonstratethat plasma lysosphingolipids (lysoSLs) profile andbrain MRI are reliable diagnostic tools for this raredisease.
Materials and methods
Cell culture and treatments
Three primary fibroblast lines obtained fromPSAP-deficient patients (P1, PSAPp.E299*/E299*; P2,PSAPp.E278Dfs*27/E278Dfs*27; PFW, PSAPp.M1L/M1L) wereutilized (19). These lines and control cells were grown inDulbecco’s Modified Eagle Medium (DMEM) supple-mented with 10% fetal bovine serum, 2 mM glutamine,100 units/ml of penicillin and 100 μg/ml streptomycin.Fibroblasts were used between passages 4 and 12. Toinduce starvation, cells were cultured under amino aciddeprivation in Earle’s balanced salt solution (EBSS)for 2 h. To inhibit autophagic flux, fibroblasts wereincubated with the lysosomotropic agent, chloroquine(20 μM), for 2 h.
Molecular analysis
Variant screening was performed by Lysoplex, a tar-geted resequencing panel including 891 genes involvedin lysosomal, endocytic and autophagic pathways (21)(patient 1), or Sanger sequencing of the entire PSAP cod-ing sequence (patient 2) (RefSeq: NM_001042466). Forthe latter, primer pairs and PCR conditions are availableupon request.
Total RNA was extracted from fibroblasts follow-ing the RNeasy Mini protocol (Quiagen, Valencia,CA), and cDNA was prepared using SuperScript™ IIIReverse Transcriptase (Invitrogen, Carlsbad, CA) witholigo(dT)20 according to the manufacturer’s instruc-tions. PSAP cDNA was amplified utilizing the primerpair PSAP_Fw 5′-CT ATG TAC GCC CTC TTCCTC CTG-3′ and PSAP_Rv 5′-CTA GTT CCA CACATG GCG TTT GC-3′. GAPDH cDNA amplification(GAPDH_Fw 5′-CTA CAC TGA GCA CCA GGT G-3′
and GAPDH_Rv 5′-CCT CTT GTG CTC TTG CTGG-3′) was performed as loading control.
Biochemical assessment
Concentration of globotriaosylsphingosine (LysoGb3),sphingosylphosphorylcholine (SPC), glucosylsph-ingosine (GlSph) and psychosine (Psy) in plasmawere measured by liquid chromatography massspectrometry (LC-MS/MS). Briefly, a mix of ace-tone:methanol:water 45:45:10 (v/v) with internalstandard (1-β-d-glucosylsphingosine from plant) wasadopted to extract lysoSLs from EDTA-plasma sam-ples. The clear supernatant was dried under a streamof nitrogen and residues were reconstituted in acetoni-trile:water 50:50 (v/v) containing 0.1% formic acid. Thechromatographic separation was performed on a BEHC18 column 2.1× 50 mm 1.7 μm (Waters, Milford, MA),using a gradient of water and acetonitrile containing0.1% formic acid. The mass spectrometer Waters XevoTQ MS was used as detector and quantification (22, 23).
GCase activity was measured utilizing4-methylumbelliferyl-β-d-glucopyranoside (4-MU-Glc)(Sigma-Aldrich, St. Louis, MO) as substrate. Briefly,the assay mixture contained, in the final volume of0.2 ml, 2.5 mM 4-MU-Glc, 0.1% Triton X-100, 0.25%(w/v) sodium taurocholate, 0.1–0.2 mol/l sodiumcitrate-phosphate buffer, pH 5.6 and an appropriateamount of enzyme source. Assay mixtures were incu-bated at 37∘C for 30 min. The extent of reaction wasestimated fluorimetrically (10).
Western blot analysis
Cell samples were lysed in cold lysis buffer (50 mMTris-base, 1.0 mM Ethylenediaminetetraacetic acid(EDTA), 150 mM NaCl, 0.1% Sodium Dodecyl Sul-fate (SDS), 1% NP-40, 0.5% sodium deoxycholate,pH 7.4), containing protease inhibitors (Complete™,Roche Diagnostics, Risch-Rotkreuz, Switzerland),centrifuged at 15,000 g for 15 min and the super-natants were used as fibroblast homogenates. Proteins(5–20 μg) were resuspended in Laemmli buffer, loadedon SDS-PAGE, and then transferred on polyvinylidenedifluoride membranes (Bio-Rad Laboratories, Hercules,CA), as previously described (10). Rabbit polyclonalanti-human PSAP (kind gift of Mia Horowitz, Tel AvivUniversity, Israel), rabbit polyclonal anti-Sap C (10),mouse monoclonal anti-GCase 8E4 (kindly providedby Hans Aerts, University of Amsterdam, the Nether-lands), rabbit polyclonal anti-LC3 (MBL International,
221
Motta et al.
Tabl
e1.
Clin
ical
,bio
chem
ical
and
mol
ecul
arch
arac
teris
tics
ofth
epr
evio
usly
desc
ribed
patie
nts
pres
entin
gw
ithP
SA
Pde
ficie
ncy
Bio
chem
ical
para
met
ers
Gen
der
Con
san-
guin
ityC
linic
alfe
atur
esB
rain
imag
ing
Enz
ymat
icac
tivity
Lipi
dan
alys
isP
SA
Pde
fect
Dea
thR
efer
ence
s
MYe
sH
epat
ospl
enom
egal
y,hy
perk
inet
icbe
havi
or,
hypo
toni
a,m
yocl
onus
,re
spira
tory
insu
ffici
ency
Mul
ticen
tric
hypo
dens
ear
eas
inth
ew
hite
mat
ter.
Atr
ophi
cbr
ain
with
oute
ran
din
ner
hydr
ocep
halu
s,at
the
final
stag
e
Red
uced
cera
mid
ase,
gluc
osyl
cera
mid
ase
and
gala
ctos
ylce
rram
idas
e,no
rmal
sphi
ngom
ielin
ase
incu
lture
dfib
robl
asts
Acc
umul
atio
nof
cera
mid
e,gl
ucos
ylce
ram
ide,
lact
osyl
cera
mid
ein
liver
,kid
ney
and
cultu
red
fibro
blas
tsbu
tnot
inbr
ain
c.1A
>T
(p.M
1L)
16w
eeks
11–1
3
FC
ontr
acte
dha
ndjo
ints
NP
Red
uced
cera
mid
ase
inam
niot
icflu
idce
llsA
ccum
ulat
ion
ofce
ram
ide
Term
inat
ion
ofpr
egna
ncy
MN
oH
epat
ospl
enom
egal
y,m
icro
ceph
aly,
optic
nerv
eat
roph
y,ca
tara
ct,r
ight
side
dhy
droc
oele
and
non-
desc
ende
ntle
ftte
stis
,po
or/n
oco
nsci
ousn
ess,
seiz
ure
NP
NP
Mas
sive
accu
mul
atio
nof
cera
mid
e,gl
ucos
ylce
ram
ide,
gala
ctos
ylce
ram
ide,
Gb3
and
sulp
hatid
ein
non-
neur
onal
cells
c.80
3del
G24
.5w
eeks
14–1
6
FN
DS
plen
omeg
aly,
prem
atur
ity,
failu
reto
grow
,hyp
oton
ia,
hypo
refle
xia,
trem
orof
the
uppe
rlim
bs,s
eizu
re,
bron
chop
neum
onia
due
toas
pira
tion
NP
NP
Mas
sive
accu
mul
atio
nof
cera
mid
e,gl
ucos
ylce
ram
ide,
gala
ctos
ylce
ram
ide,
Gb3
and
sulp
hatid
ein
non-
neur
onal
cells
c.80
3del
G4
wee
ks
MYe
sH
epat
ospl
enom
egal
y,hy
poto
nia,
seiz
ure
Bifr
onta
labn
orm
aliti
esof
gyra
tion,
whi
tem
atte
rle
sion
s,co
rtic
alat
roph
y
Defi
cien
cyof
gala
ctos
ylce
ram
idas
e,lo
wno
rmal
gluc
osyl
cera
mid
ase
and
norm
alsp
hing
omye
linas
ele
vels
Acc
umul
atio
nof
cera
mid
e,gl
ucos
ylce
ram
ide,
lact
osyl
cera
mid
e,G
M3
gang
liosi
de
Q50
*15
wee
ks17
MN
oM
yocl
onic
jerk
s,ex
trap
yram
idal
mov
emen
tdi
sord
erhy
poto
nia,
dyst
onia
,sei
zure
,hy
perk
ines
ia,c
yano
tichy
poxe
mia
,sev
ere
feed
ing
prob
lem
s,pr
otei
n-lo
osin
gen
tero
path
y,m
assi
veas
cite
s
Thin
corp
usca
llosu
m,
bila
tera
labs
ence
ofth
egy
rus
cing
uli,
periv
entr
icul
arm
ultip
lesi
gnal
chan
ges
sugg
estiv
eof
gray
mat
ter
hete
roto
pias
Pro
foun
dly
redu
ced
gala
ctos
ylce
ram
idas
e,pa
rtia
llyre
duce
dgl
ucos
ylce
ram
idas
e,re
duce
dce
ram
idas
e,no
rmal
sphi
ngom
ielin
ase,
beta
-hex
osam
inid
ase,
beta
-gal
acto
sida
se,
alph
a-ga
lact
osid
ase,
aryl
sulfa
tase
Acc
umul
atio
nof
cera
mid
e,gl
ucos
ylce
ram
ide,
lact
osyl
cera
mid
e,G
M3
gang
liosi
deG
b3,G
M3
gang
liosi
dein
fibro
blas
ts
c.1A
>T
(p.M
1L)
17w
eeks
18
MYe
sH
epat
ospl
enom
egal
y,rh
ytm
icm
ovem
ents
inla
tepr
egna
ncy,
mic
roce
phal
y,su
ckin
gan
dsw
allo
win
gdi
fficu
lties
with
aspi
ratio
n,hy
poto
nia,
myo
clon
us,
optic
atro
phy
NP
Par
tially
redu
ced
gluc
osyl
cera
mid
ase,
mor
em
arke
dly
redu
ced
gala
ctos
ylce
ram
idas
e,no
rmal
sphi
ngom
ielin
ase
Acc
umul
atio
nof
cera
mid
e,gl
ucos
ylce
ram
ide
and
lact
osyl
cera
mid
ein
cultu
red
fibro
blas
tsan
din
urin
e
c.10
06-2
A>
G8
wee
ks19
F,fe
mal
e;M
,mal
e;N
D,n
otde
term
ined
;NP,
notp
erfo
rmed
;PS
AP,
pros
apos
in.
222

Two new cases of prosaposin deficiency
Woburn, MA), mouse monoclonal anti-p62 (Santa CruzBiotechnology, Dallas, Inc., TX), and mouse mono-clonal anti-actin (Sigma-Aldrich) were used as primaryantibodies. Horseradish Peroxidase (HRP)-conjugatedanti-rabbit IgG and anti-mouse IgG (GE Healthcare,Little Chalfont, UK) were used as secondary antibodies.Immunoreactive proteins were detected by an ECLAdvance™ Western Blotting Detection kit, accordingto the manufacturer’s instructions (GE Healthcare).Band intensity was determined by densitometry usingalphaview sa software (Protein Simple, San Jose, CA).
Immunofluorescence analysis
Fibroblasts (20× 103) were seeded on glass coverslips,fixed with 3% paraformaldehyde for 30 min at 4∘Cand permeabilized with 0.5% Triton X-100 for 10 minat room temperature. For single immunofluorescence,cells were incubated with anti-GCase 8E4 primary anti-body for 1 h at room temperature, rinsed three timeswith PBS, and then incubated with anti-mouse IgGsecondary antibody conjugated with Alexa Fluor 488(Molecular Probes, Eugene, OR) for 1 h at room tem-perature. For double immunostaining, cells were incu-bated 1 h with anti-Lamp 1, mouse monoclonal pri-mary antibody, rinsed twice with Phosphate BufferedSaline (PBS), and incubated 1 h with anti-mouse IgGsecondary antibody conjugated with Alexa Fluor 488.Cells were then rinsed twice with PBS, incubated 1 hwith anti-human PSAP, rinsed twice with PBS andincubated 1 h with anti-rabbit IgG secondary antibodyconjugated with Alexa Fluor 555 (Molecular Probes).Finally, glass coverslips were mounted on microscopeslides using the Vectashield antifade medium containing4′,6-Diamidine-2′-phenylindole (DAPI) (Vector Labora-tories, Burlingame, CA) and analyzed by a Leica TCSSP2 AOBS apparatus (Leica Lasertechnik, Heidelberg,Germany), utilizing excitation spectral laser lines at 405,488 and 555 nm, tuned with an acousto-optical tunablefilter. Image acquisition and processing were conductedusing the leica confocal software 2.3 (Leica) andadobe photoshop 7.0 (Adobe Systems Incorporated,Mountain View, CA). Signals from different fluorescentprobes were taken in sequential scanning mode.
Lipid analysis
For each line, cells from two 75 cm2 flasks werecultured for 18 days, harvested using trypsin, and pel-leted by centrifugation. Cell pellets were suspendedin 500 μl 0.9% NaCl and lipids were extracted withchloroform/methanol 1:1 (v/v) for 24 h at 40∘C. Phos-pholipids were degraded by mild alkaline hydrolysiswith methanolic sodium hydroxide (50 mM) for 2 h at37∘C and neutralized with acetic acid. Lipid extracts,after desalting, were applied on high-performancethin-layer chromatography (HPTLC) silica-gel plates(Merck, Darmstadt, Germany) and developed withchloroform/methanol/CaCl2 0.22% (60:35:8). SLs werestained with 3% cupric acetate in 8.5% phosphoric acidin water, and identified by their Rf values.
Results
Clinical description
Patient 1P1 (Fig.1a, left) is the first born of non-consanguineousparents with a history of a previous late miscarriage. Shewas born at term (38th weeks) of a pregnancy presentingmaternal diabetes and, during the last month, intrauter-ine growth retardation (IUGR). Birth weight, length andhead circumference were all at the third percentile (W2.410 kg, L 45 cm, HC 32 cm) with normal Apgar score.She presented neonatal apnoea and desaturation, hypoto-nia, and generalized myoclonic jerks. Blood tests showednormal blood count, coagulation and liver function. Aninfective disease was ruled out. Repolarization abnor-mality was noted by electrocardiogram (ECG). Abdom-inal ultrasounds pointed out a transient building up offluids surrounding spleen and liver. Electroencephalo-gram (EEG) recording revealed right temporal spikes andwaves. Brain magnetic resonance imaging (MRI), per-formed at the age of 10 days, displayed T2-hypointensesmall foci of deep white matter, marked thinning of thecorpus callosum and a reduced frontal-polar gyrification.The basal ganglia were also impaired (Fig.1a, right).
The patient was referred to the Division of InheritedMetabolic Diseases of the Padua University Hospital atthe age of 2 months, after an episode of severe apnoeaand cyanosis due to aspiration and bronchopneumonia,followed by myoclonic jerks. The patient was micro-cephalic (HC 35.5 cm) and showed coronal and lamb-doid synostosis. She presented hepatosplenomegaly withliver and spleen at 4 cm below the costal edge. Glob-ally, clinical picture was poor with severe sucking dif-ficulties, which needed nasogastric tube feeding. Froma neurological point of view, she had poor eye contactand fixation, severe generalized hypotonia, contractedjoints (arthrogrypotic-like hands), and opisthotonus. Shealso presented increased startle responses, cloniformmovements and hiccups. General blood testing revealedsevere anemia and thrombocytopenia, requiring trans-fusion. Ophtalmologic examination was unremarkable.Levels of angiotensin converting enzyme (ACE) wereperformed and found increased (130 U/l and 190 U/l,with normal value ranging between 8 and 52 U/l). A bonemarrow aspirate smear revealed the presence of foamycell. Afterwards, the patient started presenting daily mul-tiple seizures, which were treated pharmacologically(levetiracetam and vigabatrin) with poor responsiveness.At the age of 3 months, a second brain MRI was car-ried out showing the disappearance of deep white mat-ter small lesions with persistence of the thinning of thecorpus callosum, the immature frontal-polar gyrificationand basal ganglia involvement progressing in signs ofatrophy (Fig. S1a, Supporting information). The visualevoked potentials (VEPs) showed high voltage record-ings with increased latencies. The brainstem auditoryevoked response (BAER) showed impaired central con-duction velocities. At 4 months of age, she presented acoarse facies with visceral enlargement and severe respi-ratory difficulties needing oxygen ventilation. She died
223

Motta et al.
(a)
(b)
Fig. 1. Clinical features of the subjects included in the study. (a) P1 at 5 months of age (left) and her brain magnetic resonance imaging (MRI, right).A diffuse and largely symmetric T2-hypointense small foci in the deep white matter are more evident in the parietal–occipital regions (black arrows).These lesions have signal characteristics similar but the same as the cortex (in particular, on T1-weighted imaging, some lesions appeared slightlymore intense) and are consistent, according to size, site and signal characteristics, with small foci of coagulative necrosis. Severe thinning of the corpuscallosum is also present, together with a reduced gyrification, especially at the level of the frontal-polar regions, and basal ganglia ischemic anoxic insult,that appear hyperintense on T1-weighted imaging. (b) P2 at 2 months of age (left) and her brain MRI (right) showing decreased gyrification, blurringand thickening of the prefrontal cortex consistent with cortical displasia (arrowheads) and numerous punctate T2-hypointense foci in the supratentorialdeep white matter (black arrows).
at the age of 7 months due to severe respiratory and neu-rological impairment.
Patient 2P2, a Macedonian female (Fig. 1b, left), was con-ceived from non-consanguineous parents. Family his-tory was unremarkable. She was born at term (40thweeks+ 4 days) of a pregnancy presenting maternal dia-betes. Birth weight and length were at the 25th–50thpercentile, whereas head circumference was below third
percentile (W 3.140 kg, L 52 cm, HC 31 cm). Amnioticfluid at birth was meconium stained (membrane rupture<12 h before birth), and the patient needed a respiratorysupport. Apgar scores were 5, 8, and 8. Intravenousantibiotic treatment was started. Despite this and a neg-ative chest X-ray findings, in the following days shecontinued to require oxygen. She had severe hypoto-nia with poor sucking needing nasogastric tube feeding.She also presented contracted joints (arthrogrypotic-likehands and foot). EEG performed at 3 days of life
224

Two new cases of prosaposin deficiency
(a)
(b)
Fig. 2. Molecular analysis. (a) Chromatograms showing the homozygous state for truncating mutations affecting the PSAP gene in the two subjectsincluded in the study (P1 and P2). Reference DNA sequence and the predicted respective amino acid sequence are given. (b) Analysis of the PSAPcDNA (1581 bp) levels in control (C), P1, P2, and PFW cDNA samples obtained from primary skin fibroblasts lines. Lane M, 100 bp ladder molecularweight marker. GAPDH cDNA (222 bp) amplification was performed as loading control.
showed high amplitude bouffées especially at the pos-terior regions, and antiepileptic drug treatment wasstarted (phenobarbital). A brain MRI was performed at1 week of age showing mild hypomyelination, decreasedgyrification with blurring and thickening of the pre-frontal cortex, and numerous punctate T2-hypointensefoci in the supratentorial deep white matter togetherwith globus pallidum involvement (Fig. 1b, right andFig. S1b).
The patient was referred to the Division of InheritedMetabolic Diseases of the Padua University Hospital
at the age of 2 months. She was microcephalic (HC33.5 cm) and presented hepatosplenomegaly withliver at 3.5 cm and spleen at 5 cm below the costaledge. Though nasogastric tube feeding, the patientunderwent three episodes of aspiration pneumoniatreated with antibiotics. Globally, clinical picturewas very poor. Neurological examination showeda poorly responsive patient with eyes opening anddecerebrate posturing as response to pain stimula-tion. She also presented severe generalized hypotonia,contracted joints (arthrogrypotic-like hands), tongue
225

Motta et al.
Table 2. Plasma lysoSLs profile in PSAP deficiency and other sphingolipidoses
LysoGb3a SPCa GlSpha Psya
Controls (n=100) 0.0–0.6 3.7–15.7 1.1–3.0 0.0–1.0PSAP deficiency P1 8.8 15.1 53.3 1.7PSAP deficiency P2 5.1 22.5 61.1 0.9Fabry disease (n= 16) 2.8–48.0 6.4–23.0 2.1–4.1 NDGaucher disease (n=9) 0.2–1.1 11.6–48.5 41.0–500.0 NDNiemann-Pick type A/B disease (n=1) 0.4 4273.0 2.8 NDNiemann-Pick type C disease (n=11) 0.2–0.4 18.4–75.9 2.4–7.7 ND
LysoGb3, globotriaosylsphingosine; nd, not determined; PSAP, prosaposin; SPC, sphingosylphosphorylcholine; GlSph, glucosylsph-ingosine; Psy, psychosine.alysoSLs values expressed as nmol/l.
fasciculations and thrusting and chaotic movements ofthe legs. General blood testing revealed severe ane-mia and thrombocytopenia, which required blood celltransfusion. Metabolic investigations including aminoacids, organic acids, sialo transferrin, and Very LongChain Fatty Acids (VLCFA) were normal. On the basisof the previous patient, ACE was measured and foundto be increased (300 U/l, normal values 8–52 U/l). Thelast EEG recording showed multifocal epileptiformdischarges and worsening background activity. She diedat the age of 3 months.
Molecular findings
Scanning of the entire coding sequence of the PSAPgene revealed the homozygous c.895G>T nucleotidechange in exon 8 in P1 (Fig. 2a, left). This pointmutation was predicted to introduce a stop codon atposition 299 (p.Glu299*). Both parents were heterozy-gous for the change. P2 was found to be homozygousfor a dinucleotide deletion c.834_835delGA, affect-ing the same exon (Fig. 2a, right), and resulting ina frameshift and premature stop 81 bases downstream(p.Glu278Aspfs*27). Both parents were heterozygousfor the same variation. The content of PSAP cDNA infibroblasts from both patients was analyzed, and com-pared with that of control cells and a PSAP-deficientcase (PFW), carrying a missense mutation (c.1A>T,p.Met1Leu). In both cases, the analysis pointed out anextremely reduced PSAP cDNA level, indicating decayof the transcript (Fig. 2b).
Plasma lysoSLs analysis by LC-MS/MS
Quantitative analysis of plasma lysoSLs in PSAP defi-ciency and other sphingolipidoses by LC-MS/MS isshown in Table 2. These data indicate that individ-ual LSD presents a specific lysoSLs accumulation pat-tern. LysoGb3 is particularly high in Fabry disease,while a markedly increased level of SPC is observed inNiemann-Pick type A/B, and a high GlSph content is spe-cific of Gaucher disease. Remarkably, both LysoGb3 andGlSph were found to be increased in the two patients withPSAP deficiency. The level of these two SLs was elevatedcompared with the control values, but in the low range of
patients with Fabry disease or Gaucher disease. SPC con-centration was comparable to the high control values inP1 and more elevated in P2. Psy value was slight higherin P1 than control.
Biochemical findings in PSAP-deficient fibroblasts
Consistent with the dramatically reduced PSAP tran-script levels, Western blot analysis highlighted theabsence of PSAP protein in patient-derived fibroblastlines. Sap C, was not detected in cells, suggestingimpaired synthesis of the precursor protein (Fig. 3a).Similar results were obtained by immunofluorescenceanalysis (Fig. 3b). In control cells, PSAP was observedto colocalize with Lamp 1, a marker of lysosomes.In contrast, no staining of PSAP was detectable incells from both patients. In order to check whether theabsence of Sap C might affect the GCase stability (24),the amount of the enzyme was analyzed by Western blotand found to be reduced to about 26% in P1 and 15% inP2 (Fig. 3c). This result was confirmed by immunoflu-orescence analysis showing a reduced punctate enzymepattern in affected cells compared with control fibrob-lasts (Fig. 3d). Consistent with these data, a decreasedenzymatic activity of GCase (32% and 26% in P1 andP2, respectively) was observed (Fig. 3e).
To verify the storage of lipids, the total lipid extractsfrom fibroblasts of both patients were compared withthose obtained from control fibroblasts. The amount ofGlCer, LacCer, and Gb3Cer was dramatically increasedin P1 and P2, whereas these lipids were below the levelof visual detection in control cells (Fig. 3f). The amountof sphingomyelin (SM) was also sharply increased inPSAP-deficient fibroblasts, while the plasma level ofSPC was only slightly enhanced (Table 2).
Impaired autophagy in PSAP-deficient fibroblasts
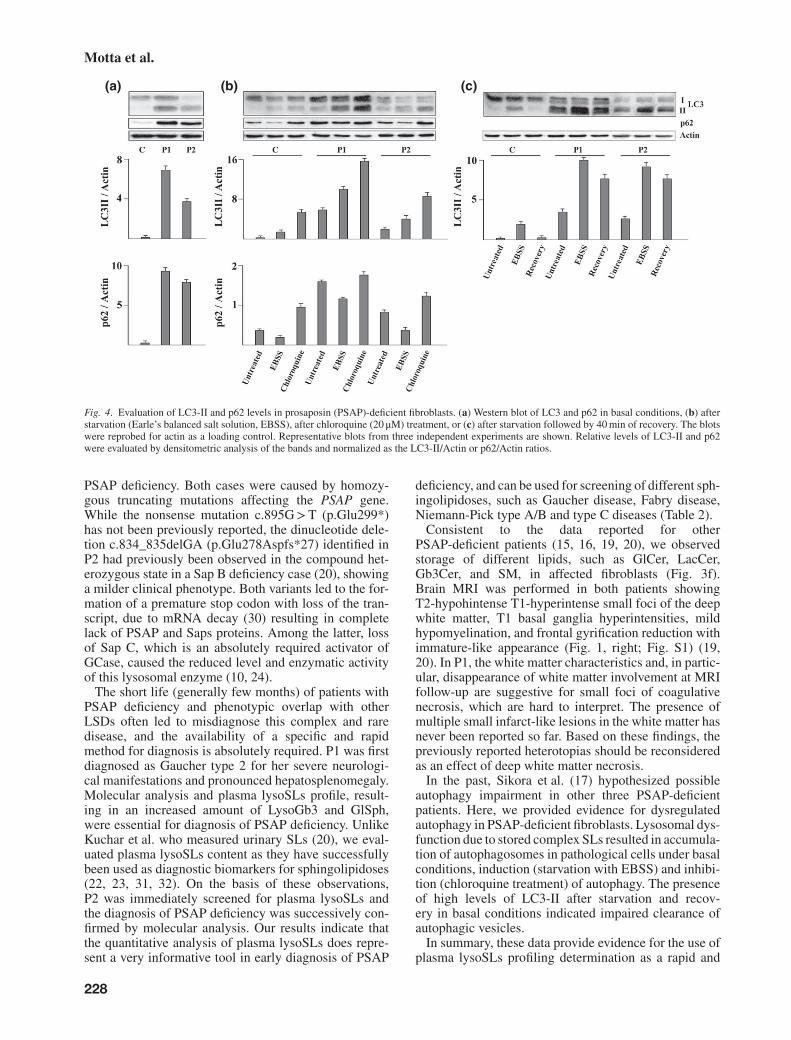
Lysosomal dysfunction due to complex lipid storagemay lead to impairment of autophagy as reported inseveral LSDs (10, 25–28). Based to these observa-tions, we examined endogenous levels of two widelyused autophagy markers, microtubule-associated pro-tein1light chain 3 (LC3) and p62/SQSTM1 (p62), incontrol and PSAP-deficient cells. LC3 is present in
226

Two new cases of prosaposin deficiency
(a) (b)
(c)
(d)
(e)
(f)
Fig. 3. Prosaposin (PSAP), saposin C (Sap C) and GCase levels, enzyme activity, and lipid analysis in PSAP-deficient fibroblasts. (a, c) Western blotsof PSAP, Sap C (a) and GCase (c) in control and PSAP-deficient cells (P1 and P2). Normalization of protein loading was performed using anti-actinantibody. Representative blots from three independent experiments are shown. (b, d) Immunofluorescence of Lamp 1, PSAP (b), and GCase (d) in controland pathological fibroblasts. Nuclei were counterstained with DAPI. Scale bar, 10 μm. (e) Enzymatic activity of GCase in control and PSAP-deficientfibroblasts was evaluated in three independent experiments. (f) Analysis of sphingolipids (SLs) in control, P1 and P2 cells by high-performancethin-layer chromatography (HPTLC). As standards (St), glucosylceramide (GlCer), lactosylceramide (LacCer), globotriaosylceramide (Gb3Cer) andsphingomyelin (SM) were utilized.
two forms, a cytosolic one (LC3-I) which is con-verted into lipidated form (LC3-II), that is associ-ated with autophagosomal membranes and thereforestrongly correlated with the number of autophago-somes. p62 is incorporated into autophagosome throughdirect binding to LC3 and is degraded by autophagy(29). Western blot and densitometric analyses clearlyshowed increased basal levels of LC3-II and p62 inPSAP-deficient fibroblasts compared with control cells(Fig. 4a), indicating that autophagosome number wasincreased either for enhanced autophagy or defectiveclearance of autolysosomes. To distinguish betweenthese two possibilities, cells were cultured in pres-ence of EBSS or chloroquine to induce autophagyand inhibit lysosomal degradation, respectively. TheLC3-II amount was documented to further increaseafter EBSS or chloroquine. The same behavior, butwith lower values of LC3-II was observed in control
fibroblasts (Fig. 4b). As expected, in control cellsp62 levels decreased following induction of autophagyand increased following its inhibition. A similar pat-tern was documented in P1 and P2 cells (Fig. 4b).These findings document that the fusion of autophago-somes with lysosomes is not blocked in PSAP-deficientfibroblasts. Remarkably, when cells were allowed torecover in complete medium for 40 min after star-vation, LC3-II totally disappeared in control fibrob-lasts, whereas only a partial recovery was documentedin P1 and P2 cells, indicating that the clearance ofautolysosomes is delayed in PSAP-deficient fibroblasts(Fig. 4c).
Discussion
In this study, we reported on the clinical, biochemi-cal and molecular characterization of two subjects with
227
Motta et al.
(a) (b) (c)
Fig. 4. Evaluation of LC3-II and p62 levels in prosaposin (PSAP)-deficient fibroblasts. (a) Western blot of LC3 and p62 in basal conditions, (b) afterstarvation (Earle’s balanced salt solution, EBSS), after chloroquine (20 μM) treatment, or (c) after starvation followed by 40 min of recovery. The blotswere reprobed for actin as a loading control. Representative blots from three independent experiments are shown. Relative levels of LC3-II and p62were evaluated by densitometric analysis of the bands and normalized as the LC3-II/Actin or p62/Actin ratios.
PSAP deficiency. Both cases were caused by homozy-gous truncating mutations affecting the PSAP gene.While the nonsense mutation c.895G>T (p.Glu299*)has not been previously reported, the dinucleotide dele-tion c.834_835delGA (p.Glu278Aspfs*27) identified inP2 had previously been observed in the compound het-erozygous state in a Sap B deficiency case (20), showinga milder clinical phenotype. Both variants led to the for-mation of a premature stop codon with loss of the tran-script, due to mRNA decay (30) resulting in completelack of PSAP and Saps proteins. Among the latter, lossof Sap C, which is an absolutely required activator ofGCase, caused the reduced level and enzymatic activityof this lysosomal enzyme (10, 24).
The short life (generally few months) of patients withPSAP deficiency and phenotypic overlap with otherLSDs often led to misdiagnose this complex and raredisease, and the availability of a specific and rapidmethod for diagnosis is absolutely required. P1 was firstdiagnosed as Gaucher type 2 for her severe neurologi-cal manifestations and pronounced hepatosplenomegaly.Molecular analysis and plasma lysoSLs profile, result-ing in an increased amount of LysoGb3 and GlSph,were essential for diagnosis of PSAP deficiency. UnlikeKuchar et al. who measured urinary SLs (20), we eval-uated plasma lysoSLs content as they have successfullybeen used as diagnostic biomarkers for sphingolipidoses(22, 23, 31, 32). On the basis of these observations,P2 was immediately screened for plasma lysoSLs andthe diagnosis of PSAP deficiency was successively con-firmed by molecular analysis. Our results indicate thatthe quantitative analysis of plasma lysoSLs does repre-sent a very informative tool in early diagnosis of PSAP
deficiency, and can be used for screening of different sph-ingolipidoses, such as Gaucher disease, Fabry disease,Niemann-Pick type A/B and type C diseases (Table 2).
Consistent to the data reported for otherPSAP-deficient patients (15, 16, 19, 20), we observedstorage of different lipids, such as GlCer, LacCer,Gb3Cer, and SM, in affected fibroblasts (Fig. 3f).Brain MRI was performed in both patients showingT2-hypohintense T1-hyperintense small foci of the deepwhite matter, T1 basal ganglia hyperintensities, mildhypomyelination, and frontal gyrification reduction withimmature-like appearance (Fig. 1, right; Fig. S1) (19,20). In P1, the white matter characteristics and, in partic-ular, disappearance of white matter involvement at MRIfollow-up are suggestive for small foci of coagulativenecrosis, which are hard to interpret. The presence ofmultiple small infarct-like lesions in the white matter hasnever been reported so far. Based on these findings, thepreviously reported heterotopias should be reconsideredas an effect of deep white matter necrosis.
In the past, Sikora et al. (17) hypothesized possibleautophagy impairment in other three PSAP-deficientpatients. Here, we provided evidence for dysregulatedautophagy in PSAP-deficient fibroblasts. Lysosomal dys-function due to stored complex SLs resulted in accumula-tion of autophagosomes in pathological cells under basalconditions, induction (starvation with EBSS) and inhibi-tion (chloroquine treatment) of autophagy. The presenceof high levels of LC3-II after starvation and recov-ery in basal conditions indicated impaired clearance ofautophagic vesicles.
In summary, these data provide evidence for the use ofplasma lysoSLs profiling determination as a rapid and
228

Two new cases of prosaposin deficiency
informative preliminary test that can be used to directdiagnostic genetic analysis in metabolic disorders withcomplex phenotypes lacking pathognomonic features.This report also documents that brain MRI can be usefulto ascertain if neurological involvement present in someinfants correlate with PSAP deficiency.
Supporting Information
Additional supporting information may be found in the onlineversion of this article at the publisher’s web-site.
Acknowledgements
This work was partially supported by Associazione COMETA–ASMME (Associazione Studio Malattie Metaboliche Ereditarie).
References1. Sandhoff K, Kolter T, Harzer K. Sphingolipid activator proteins.
In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, KinzlerKW, Volgstein B, eds. The metabolic and molecular bases of inher-ited disease, 8th edn, Vol. 3. New York, NY: MC-Graw-Hill, 2001:3371–3388.
2. Kondoh K, Hineno T, Sano A, Kakimoto Y. Secretion of sphingolipidhydrolase activator precursor, prosaposin. Biochem Biophys Res Com-mun 1991: 181: 286–292.
3. Sylvester SR, Skinner MK, Griswold MD. A sulfated glycoproteinsynthesized by Sertoli cells and by epididymal cells is a component ofthe sperm membrane. Biol Reprod 1984: 31: 1087–1101.
4. Hineno T, Sano A, Kondoh K, Ueno S, Kakimoto Y, Yoshida K. Secre-tion of sphingolipid hydrolase activator precursor, posaposin. BiochemBiophys Res Commun 1991: 176: 668–674.
5. O’Brien JS, Kishimoto Y. Saposin proteins: structure, function, and rolein human lysosomal storage disorders. FASEB J 1991: 5: 301–308.
6. Vaccaro AM, Salvioli R, Tatti M, Ciaffoni F. Saposins and theirinteraction with lipids. Neurochem Res 1999: 24: 307–314.
7. Schulze H, Sandhoff K. Sphingolipids and lysosomal pathologies.Biochim Biophys Acta 2014: 1841 (5): 799–810.
8. Spiegel R, Bach G, Sury V et al. A mutation in the saposin A codingregion of the prosaposin gene in an infant presenting as Krabbe disease:first report of saposin A deficiency in humans. Mol Genet Metab 2005:84: 160–166.
9. Stevens RL, Fluharty AL, Kihara H et al. Cerebroside sulfatase activatordeficiency induced metachromatic leukodistrophy. Am J Hum Genet1981: 33: 900–906.
10. Vaccaro AM, Motta M, Tatti M et al. Saposin C mutations in Gaucherdisease patients resulting in lysosomal lipid accumulation, saposin Cdeficiency, but normal prosaposin processing and sorting. Hum MolGenet 2010: 19: 2987–2997.
11. Matsuda J, Kido M, Tadano-Aritomi K et al. Mutation in saposin Ddomain of sphingolipid activator protein gene causes urinary systemdefects and cerebellar Purkinje cell degeneration with accumulation ofhydroxy fatty acid-containing ceramide in mouse. Hum Mol Genet 2004:13: 2709–2723.
12. Harzer K, Paton BC, Poulos A et al. Sphingolipid activator proteindeficiency in a 16-week-old atypical Gaucher disease patient and his fetalsibling: biochemical signs of combined sphingolipidoses. Eur J Pediatr1989: 149: 31–39.
13. Schnabel D, Schröder M, Furst W et al. Simultaneous deficiency ofsphingolipid activator proteins 1 and 2 is caused by a mutation in
the initiation codon of their common gene. J Biol Chem 1992: 267:3312–3315.
14. Bradòva V, Šmíd F, Ulrich-Bott B, Roggendorf W, Paton BC, HarzerK. Prosaposin deficiency: further characterization of the sphingolipidactivator protein-deficient sibs. Hum Genet 1993: 92: 143–152.
15. Hulkova H, Cervenkova M, Ledvinova J et al. A novel mutation in thecoding region of the prosaposin gene leads to a complete deficiency ofprosaposin and saposins, and is associated with a complex sphingolipido-sis dominated by lactosylceramide accumulation. Hum Mol Genet 2001:10: 927–940.
16. Elleder M, Jirasek A, Smid F, Ledvinova J, Besley GT, Stopekova M.Niemann-Pick disease type C with enhanced glycolipid storage. Reporton further case of so-called lactosylceramidosis. Virchows Arch 1984:402: 307–317.
17. Sikora J, Harzer K, Elleder M. Neurolysosomal pathology in human pros-aposin deficiency suggests essential neurotrophic function of prosaposin.Acta Neuropathol 2007: 113: 163–175.
18. Millat G, Verot L, Rodriguez-Lafrasse C et al. Fourth reported fam-ily with prosaposin deficiency. 14th ESGLD Workshop (September18th–21st, 2003). Podebrady/Prague, Czech Republic.
19. Elleder M, Jerabkova M, Befekadu A et al. Prosaposin deficiency -a rarely diagnosed, rapidly progressing, neonatal neurovisceral lipidstorage disease. Report of a further patient. Neuropediatrics 2005: 36:171–180.
20. Kuchar L, Ledvinova J, Hrebiceck M et al. Prosaposin deficiency andsaposin B deficiency (activator-deficient metachromatic leukodistrophy):report on two patients detected by analysis of urinary sphingolipids andcarrying novel PSAP gene mutations. Am J Hum Genet 2009: 149A:613–621.
21. Di Fruscio G, Schulz A, De Cegli R et al. Lysoplex: an efficient toolkitto detect DNA sequence variations in the autophagy-lysosomal pathway.Autophagy 2015: 11: 928–938.
22. Boutin M, Gagnon R, Lavoie P, Auray-Blais C. LC-MS/MS analysisof plasma lyso-Gb3 in Fabry disease. Clin Chim Acta 2012: 414:273–280.
23. Welford RW, Garzotti M, Marques Lourenço C et al. Plasma lysosph-ingomyelin demonstrates great potential as a diagnostic biomarker forNiemann-Pick disease type C in a retrospective study. PLoS One 2014:9 (12): e114669.
24. Sun Y, Qi X, Grabowski GA. Saposin C is required for normal resistanceof acid β-glucosidase to proteolytic degradation. J Biol Chem 2003: 278:31918–31923.
25. Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann-Pick Cdisease is dependent upon Beclin-1 and responsive to lipid traffickingdefects. Hum Mol Genet 2007: 16: 1495–1503.
26. Settembre C, Fraldi A, Jahreiss L et al. A block of autophagy inlysosomal storage disorders. Hum Mol Genet 2008: 17: 119–129.
27. Fukuda T, Ewan L, Bauer M et al. Dysfunction of endocytic andautophagic pathways in a lysosomal storage disease. Ann Neurol 2006:59: 700–708.
28. Tessitore A, Pirozzi M, Auricchio A. Abnormal autophagy, ubiquitina-tion, inflammation and apoptosis are dependent upon lysosomal storageand are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics2009: 2: 1–12.
29. Jiang P, Mizushima N. LC3- and p62-based biochemical methods for theanalysis of autophagy progression in mammalian cells. Methods 2015:75: 13–18.
30. Brogna S, Wen J. Nonsense-mediated mRNA decay (NMD) mecha-nisms. Nat Struct Mol Biol 2009: 16: 107–113.
31. Aerts JM, Groener JE, Kuiper S et al. Elevated globotriaosylsphingosineis a hallmark of Fabry disease. Proc Natl Acad Sci U S A 2008: 105:2812–2817.
32. Dekker N, van Dussen L, Hollak CE et al. Elevated plasma glucosylsph-ingosine in Gaucher disease: relation to phenotype, storage cell markers,and therapeutic response. Blood 2011: 118: e118–e127.
229