characterizationofnovelhumanleukemiccelllinesselectedforre...
TRANSCRIPT

[CANCERRESEARCH56. 2573-2583. June 1. 1996]
ABSTRACT
Merbarone is a catalytic inhibitor of DNA topoisomerase (topo) II thatdoes not stabifize DNA-topo II cleavable complexes. Although the cytotoxicityof and resistanceto complex-stabilizingtopo II inhibitors, such asetoposide, is thought to be mediated through stabilization of these com
plexes, the mechanisms of cytotoxicity and resistance to catalytic inhibitors are not well known. To investigate this issue, we established 12merbarone-resistant cell lines from human leukemia CEM cells, designated CEM/M70-B1 through -B12. Assessed by either growth inhibitionor clonogenic assay, these cell lines are 3.5- to 6.6-fold resistant to merbarone, compared to the CEM parent cells. Karyotype analysis of three ofthe cell lines revealed that while CEM and drug-resistant cell lines hadchromosome abnormalities in common, indicating a common origin, twoof the merbarone-resistantlines(B! and B8) eachhad uniquestructuralmarkers. These novel cell lines are cress-resistant to complex-stabilizingtopo II inhibitors, etoposide, teniposide, amsacrine, and doxorubicin, butnotto othercatalyticinhibitors,aclarubicinor SN-22995.Ofconsiderableinterest, these cell llnes are cross-resistant to SN-38, a putative topo Iinhibitor, but cross-resistance to other topo I inhibitors (camptothecin andtopotecan) was lower and not seen in every cell line. In all 12 cell lines,there was a high correlation among drug resistance ratios between etoposide and teniposide and between merbarone and SN-38.By contrast,there was a low correlation between merbarone and etoposide and be
tween SN-38 and other topo I inhibitors. These results suggest thatresistance to merbarone and cross-resistance to etoposide might bethrough different mechanisms, whereas cross-resistance to SN-38 mightbe through a merbarone-related mechanism. Etoposide and SN-38 stabilized fewer DNA-topoisomerase complexes in CEM/M70-B cells than inCEM cells, but camptothecin stabilized more. Merbarone inhibited complex formation induced by etoposide in drug-sensitive and -resistant cells,but the degreeof inhibitionwas lowerin CEMIM7O-Bcells than in theparental cells. Moreover, merbarone did not affect complex formationstabilized by SN-38 or camptothecin. Immunoblot analysis of the CEM/M70-B cells showed decreased topo 1kw,increased topo II@3,and no changeof topoI protein,comparedto CEMcells.Weproposethehypothesisthatdecreased topo 1hz may play role in the resistance to merbarone that isdifferent from that to complex-stabffizing drugs. Cross-resistance to cat
alytic inhibitors may be due to reduced complex formation as a consequence ofdecreased topo 1kv.We also found that DNA-protein complexesstabilized by SN-38 might be different from those stabilized by topo LIinhibitors and blocked by merbarone. Judging from both the high correlation of drug sensitivities and complex-formation assays, we postulatethat mechanismsof cytotoxicityand cross-resistanceof SN-38in CEM/M70-B cells might be similar to those of merbarone. We believe that theCEM/M70-B cells are the first to be selected and characterized for resist
Received 2/12/96; accepted 3/26/96.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
I This work was supported in part by Research Grants CA-40570 (to W. T. B.)
and CA-4972l (to S. C. R.), Program Grants CA-23099 (to W. T. B.) and CA-20l80(to S. C. R.), and Cancer Center Support CORE Grant CA-21765, all from the NationalCancer Institute, Bethesda, MD; in part by Children's Cancer Research Fund (to S. C. R.);and in partby AmericanLebaneseSyrianAssociatedCharities.
2 Present address: Department of Surgery, Medical Institute of Bioregulation, Kyushu
University, Beppu, Japan.3 To whom requests for reprints should be addressed, at present address: Cancer Center
(M/C 569). University of Illinois at Chicago, 900 S. Ashland Avenue, Chicago, IL,60607-7173. Phone: (312) 355-0827; Fax: (312) 355-0194.
ance to a catalytic inhibitor of topo II. This study provides novel cell lineswith characteristics of resistance to topo II and topo I inhibitors.
INTRODUCTION
topo4 II is an essential enzyme that relaxes supercoiled DNA anddecatenates intertwined DNA by breakage and reunion of DNA double strands (1). This enzyme regulates DNA topology involved inDNA replication (2, 3), transcription (4), and repair (5). topo II is alsoan important target of anticancer agents; the cytocidal mechanism ofsome topo II inhibitors, VM-26 (6), m-AMSA (7), and doxorubicin(8), stabilize covalent complexes between topo II and DNA are relatedto the generation of DNA double-strand damage (9).
Recently, the relationship between cytotoxicities of topo II inhibitors and their effects on the cell cycle has been developed. VP-16 andVM-26 are known to inhibit cells entering mitosis and to inhibit cdc2kinase activity (10, 1 1). If the drug-induced DNA-topo II complexes
interfere with these functions of topo II in DNA synthesis, this G2arrest might be due to induction of G2 “checkpoint―machinery thatallows damaged DNA to be repaired before cells move to the next cellcycle stage (12, 13).
At high concentrations of drug in vitro, complex-stabilizing topo IIinhibitors have also been reported to prevent chromatid separation(14, 15). In temperature-sensitive top2 mutants of fission yeast, topoII is an essential enzyme for chromosome condensation and segregation (16, 17), and disturbance of mitosis by inhibitors of this enzymeis readily seen in human cancer cells. However, it is not knownwhether the inhibitory effects of topo II inhibitors on the cell cycle aredirect results from DNA damage after stabilization of DNA-topo IIcomplex formation.
In contrast to the complex-stabilizing topo II inhibitors, some drugshave inhibitory effects on catalytic activity of the enzyme but do notstabilize complexes. These are termed “catalytic―inhibitors of topo IIto distinguish them from the complex-stabilizing inhibitors and indude agents such as merbarone (18), aclarubicin ( 19), ICRF-193 (20),and SN-22995 (21).
Merbarone is a barbiturate derivative (22) presently in Phase IIstudy (23, 24). Drake et a!. (18) and Chen and Beck (21) reported thatthis drug inhibits cleavable complex formation induced by the complex-stabilizing inhibitor, VM-26. Similar inhibition of VM-26, VP16, or daunorubicin-induced complex-stabilization by catalytic inhibitors of topo II has been reported for aclarubicin (19, 25), ICRF-l93(26), and SN 22998 (21). Detailed mechanisms of merbarone actionafter the inhibition of topo II activity are unknown.
Work from this laboratory revealed that merbarone inhibits chromosome condensation and sister chromatid segregation in nonsynchronized human leukemia CEM cells (21) and chromosome division
in HeLa cells synchronized at metaphase (27). This agent also inducesG2 block with an inhibition of cdc2 kinase activity (27). However,
4 The abbreviations used are: topo, topoisomerase; VP-16, etoposide; VM-26. tenipo
side; m-AMSA, 4'(9-acridinylamino) methanesulfon-m-aniside, amsacrine; DOX, doxorubicin; VCR, vincristine; SN-38, 7-ethyl-lO-hydroxycamptothecin; CPT, camptothecin;CPT- I I, 7-ethyl-lO-[4-(l-piperidino)-1-piperidinoj carbonyloxy camptothecin; Pgp,P-glycoprotein; ACR, aclarubicin.
2573
Characterization of Novel Human Leukemic Cell Lines Selected for Resistance toMerbarone, a Catalytic Inhibitor of DNA Topoisomerase 111
Hiroki Kusumoto,2 Queen E. Rodgers, Friedrich Boege, Susana C. Raimondi, and William T. Beck3
Departments of Molecular Pharmacology [H. K.. Q. E. R.. W. T. B.] and Pathology and Laboratory Medicine (S. C. RI, St. Jude Children ‘sResearch Hospital. Memphis.Tennessee 38105; Departments of Pharmacology (W. T. B.] and Pathology (S. C. R.], College of Medicine, University of Tennessee. Memphis, Tennessee 38168; and Hauptlabor.Medizinische Poliklinik der Universitdt Warzburg. Wllrzburg. Germany (F. B.]
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

CHARACTERIZATIONOF MERBARONE.RESISTANTCELL LINES
merbarone produces the arrest in convergent doses that are within oneto five times the concentration that inhibits chromosome separation,whereas the concentrations of VM-26 to cause quantitatively similareffects are divergent; namely, VM-26 is 50- to 100-fold more efficientin causing G2 arrest than in inhibiting chromosome separation (27).Downes et a!. (28) found that there is a catenation-sensitive checkpoint mechanism for ICRF-193-induced G2 block that is sensitive toDNA damage induced by VM-26. ICRF-193 is also a catalytic inhibitor of topo II that inhibits chromosome condensation and sisterchromatid segregation (29). These data suggest that there might bedifferent cytocidal mechanisms of catalytic inhibitors compared tothose of complex-stabilizing inhibitors mediated by different topo IIfunctions.
To address this hypothesis, the study of resistance to catalyticinhibitors of topo II is likely to be informative. However, there are fewresistant cell lines to show the cross-resistance between two kinds of
topo II inhibitors (30). In fact, Chen and Beck (21) have found thatthere is no cross-resistance to merbarone or other catalytic topo IIinhibitors in CEM/VM-l and CEM/VM-1—5 cells that are 40- and140-fold resistant to VM-26 and cross-resistant to other complexstabilizing topo II inhibitors. This suggests that certain characteristicsof these resistant cell lines might not be sufficient to cause resistanceto merbarone.
To investigate the common and different cytocidal mechanisms ofdifferent types of topo II inhibitors, we selected and characterized 12merbarone-resistant clones from CEM cells. As far as we know, thisis the first report characterizing cell lines selected specifically forresistance to a catalytic inhibitor of topo II. These novel cell linesexpress phenotypes that differ from CEM/VM-1 and CEMIVM-1—5cells, and they are cross-resistant to SN-38, a putative topo I inhibitor,about which no resistant cell line has been reported. DNA-topocomplex-forming activities and enzyme levels in these cell lines werealso analyzed.
MATERIALS AND METHODS
Chemicals and Supplies. The following drugs were obtained: merbarone
from SmithKline Beecham (King of Prussia, PA), generously supplied by Dr.Randall Johnson; VM-26 from Bristol-Myers Squibb (Wallinglord, CT),through the courtesy of Dr. Byron Long; VP-16 and CPT from Sigma Chem
ical (St. Louis, MO); m-AMSA and SN-22995 from Dr. Bruce C. Baguley(University of Auckland, New Zealand); daunorubicin from Farmitalia-Carlo
Erba (Milan, Italy); aclarubicin from Dr. Ellen Friche (Rigshospitalet University, Copenhagen, Denmark); SN-22995 was also provided by Dr. TerryBeerman (Rosewell Park Memorial Institute, Buffalo, NY); topotecan andSN-38 from Dr. Peter J. Houghton (St. Jude Children's Research Hospital,Memphis TN); SN-38 was also provided by Upjohn Co. (Kalamazoo, MI),through the courtesy of Dr. Patrick McGovren; and VCR was from Eli LillyCo. (Indianapolis, IN), generously supplied by Dr. H. L. Pearce. MEM forsuspension culture, RPM! 1640, and 200 mM glutamine were from WhittakerBioproducts (Walkersville, MD), and fetal bovine serum was obtained fromHyclone Laboratories, Inc. (Logan, UT) or Sigma Chemical Co. MethoCult
M3100 was from Stem Cell Technologies, Inc. (Vancouver, BC), [3H]thymidine (specific activity, 20 Ci/mmol) was purchased from Amersham (Arlington
Heights, IL), and [‘4C]leucine(specific activity, 325 mCi/mmol) was fromNEN/DuPont (Boston, MA). Alkaline phosphatase-conjugated goat antirabbitIgG was purchased from Zymed (San Francisco, CA).
Cell Culture Conditions. Cells were cultured in MEM for suspensionculture with 10% fetal bovine serum at 37°C, 5% CO2. 95% air, in a humid
ified chamber. Merbarone-resist.ant cells were selected from the original parentCCRF-CEM cell line by continuous incubation with increasing concentrationsof merbarone, starting with I 3 @Mup to 70 @Mover 3 months. In the processof increasing drug, the cells were incubated with the same concentration of
drug for at least 1 week. When the damage to cells was severe, the drugconcentration was reduced for —1 week. After stabilizing cells at 70 p@M
merbarone, they were maintained at this concentration for an additional 3months. Then, 12CEMIM7O-Bcell lines were cloned by limiting dilution (31)and have now retained their resistance to merbarone for more than 2 years inthe absence of drug. All cells used for experiments were grown in drug-freemedia for at least 3 months.
Chromosome Analysis. Harvesting and processing of the cell lines and0-banding of chromosomes were performed using standard techniques.Twenty metaphases were evaluated per cell line, and the karyotype was writtenaccording to the International System for Human Cytogenetic Nomenclature
(ISCN 1991).
Doubling Time and Growth Inhibition Assay. Doubling times weredetermined by counting exponentially growing cells between 2 and 8 X 10@cells/rn] with a Coulter counter. For a growth inhibition assay, the cells were
incubated with drugs for 72 h. The percentage of counts of the drug-treatedcells to those of control cells was determined, and the concentration of drugrequired to inhibit growth of cells by 50% (IC50)for each drug was calculated.Fold-resistance was calculated by dividing the IC50 of the CEMIM7O-B celllines by the IC50 of that drug in the CEM parent line. Experiments were doneat least three different times, and averages of IC50 in resistant cell lines werecompared with that of the parent line by Student's t test.
Clonogenic Assay. CEM and drug-resistant sublines in mid-log phase werediluted to 10,000/ml in RPM! 1640 supplemented with 10% FBS and 2 mMglutamine. (Four-mi cell suspensions were sufficient to test one drug at sixconcentrations.) MethoCult complete medium was made as follows. One40-mI bottle of MethoCult M3100 was thawed, and the following were added,
with shaking: FBS (19.5 ml), glutamine (0.55 ml of 200 mMstock), and 39.6ml RPM! 1640. Drug solutions at 100X final concentrations were added toeach of six tubes (14 ml), followed by the addition of 4 ml MethoCuit“complete.―The tubes were vortexed, 0.4 ml of the diluted cells were added toeach tube, and the tubes were vortexed again three times to mix. Using 5-mlsyringes fitted with 16-guage blunt tip needles, the MethoCult cell suspension(1.1 ml) was transferred to 6-well (35-mm) cluster dishes (Costar #3506). Thedishes were then placed inside a humidified plastic box and incubated at 37°C;
then colonies were counted 7 days later under a microscope, using a grid.DNA-Protein Complex Formation Assay in Intact Cells. The methods
have been described previously (32). In brief, cellular DNA and protein werelabeled by incubating 3 x i0@ cells/mI at 37°Covernight with [‘4Clleucine(0.2 MCi/mi)and [3H]thymidine(0.6 MCi/mi).The indicated concentrations oftopo I or II inhibitors were added to cell suspensions, and the cells wereincubated for an additional 15 mm (for VP-l6) or 10 mm (for SN-38 and CPT).To study the effect of merbarone on complex-formation induced by othertopoisomerase inhibitors, cells were treated with merbarone for 30 mm beforethe addition of the other drugs at the indicated concentrations (21). Afterincubation with drug, reactions were stopped by adding 0.5 ml of a solutioncontaining 2.5% SDS, 10 mM EDTA (pH 8.0), and 0.8 mg/mi salmon spermDNA. Cell lysates were passed 20 times through a 22-gauge needle and thenheated to 65°Cfor 15 mm. KC1 was added to each preparation to a finalconcentration of 100 mM.The sample tubes were vortexed vigorously, put onice for 5 mm, and then centrifuged at 10,000 X g for 10 mm at 4°C.Each pelletwas washed three times with 1ml ofa solution containing 10msi Tris-HCI (pH8.0), 100 mMKC1,1 mtsiEDTA (pH 8.0), and 0.1 mg salmon sperm DNA/mI.The pellets were dissolved in 0.5 ml water and incubated at 65°Cfor 15 mm.The tubes were centrifuged for 15 s at 10,000 X g, and the supernatants weretransferredto scintillationvials.The sampleswere then countedin a scintillation counter (Beckman 7000). Results are expressed as the ratio of [3H]DNA:
[‘4C]protein,with the cpm of protein precipitated as the internal control for allsamples.Experimentsweredonein quadruplicate,and the averageof numberof complexes in each resistant cell line was compared with that of the parentcell line by Student's t test.
Immunodetection of Topoisomerases. To protect topo II@3protein in thesamplesfromdegradation,wepreparedthe samplesby a hot SDSmethod(30,33). Exponentially growing cells were harvested and rinsed once with PBSbuffer. After centrifugation at 400 X g for 5 mm, the pellets were immediatelylysed in 1x Laemmli buffer (34), prewarmed to 65°Cand sonicated for 10 s.Samples (50 i@g protein) were loaded onto a 7.5% SDS-PAGE gel andelectrophoresed at 25 mA for 6 h. Proteins were transferred to nitrocellulosepaper using a PolyBlot apparatus (American Bionetics, Emeryville, CA). Themembranes were incubated at 37°C for 2 h in “blocking―solution containing
3% (wlv) BSA, 5% (w/v) powdered milk, 0.2% (vlv) Tween 20, and 0.02%
2574
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

CHARACTERIZATIONOF MERBARONE-RESISTANTCELLLINES
(wlv) sodium azide in 10 mM Tris-HCI (pH 7.5) and 140 mM NaCI to blocknonspecific proteins. For detection of topo II a and (3, we used MAC antiserum
that was produced in rabbits immunized with a Mr 72,000 COOH-terminalfragment of the sequence of human topo Ha (35). To confirm the identity oftopo 11f3,we used a rabbit antiserum, 282, directed against a COOH-terminalpeptide consisting of residues 1586—1596of human topo IIf3(33). For identification of topo I, we used a rabbit antiserum Tl-l, directed against aminoacids 219—239 of human topo I, developed in one of our laboratories (36). The
MAC, 282, and 11-1 antisera were diluted 1:600, 1:5000, and 1:2000, respeclively, with the blocking solution, described above. For a control protein, weincubated each membrane with a 1:500 dilution of an antitubulin antiserum(Sigma) concomitantly with one of these antisera. The blots were incubatedwith the antisera solutions overnight, then washed, incubated with alkaline
phosphatase-linked goat antirabbit IgG, and developed with 5-bromo-4-chloro3-indolyl phosphate and nitro blue tetrazolium (33, 35). The blotted proteins onthe membrane were analyzed by densitometry (Scan Jet lic; Hewlett Packard),using NIH Imagel—55 software. The relative levels of a topoisomerase totubulin in each lane were calculated. Each experiment was done at least fivetimes with different preparations. The relative levels of topoisomerases inCEM/M70-B cells compared to those in CEM cells were calculated, and themeans ±SD between the parent cell line and CEMIM7Ocells were comparedby Student's t test.
RESULTS
Doubling Time. We did experiments five different times, and themeans and SD of doubling times between CEM and each CEM/M70-B cell line were compared by Student's t test. The doubling timeof CEM cells was 16. 1 h. In all CEMIM7O-B cell lines, the doublingtimes were longer than that in CEM cells, ranging between 17.7 h(B!) and 23.3 h (B7 and B! 1; Table 1). Ten of the 12 CEMIM7O-Bcell lines grew significantly more slowly than CEM cells.
Chromosome Analysis. We performed karyotype analysis onCEM and three merbarone-resistant cell lines. All had many numerical and structural abnormalities. Each cell line had metaphases withvariations, and each of the karyotypes were inferred from the 20metaphases evaluated. The CEM cell line was near-tetraploid, asreported earlier (37, 38): 94,XXXX,dup(l)(p32p36),+del(6)(q13),—8,—8,add(9)(p22)x2,+14,+20,+20 (Fig. 1A). The Bi cell linekaryotype was: 88-92,XXX,dup(l)(p32p36),add(3)(pl4),inv(3)(p13q27),del(4)(q12q25), +del(6)(q13),der(7)t(7;?12)(q36;ql2),—8-9,add(9)(p22)x2,— 12,—15,+20,+20,+mar (Fig. 1B).
The B8 cell line karyotype was: 90—93,XXXX,dup(1)(p32p36),add(3)(p14),inv(3)(p13q27),+7, —8,-9,add(9)(p22)x2,add(10)(p12),— 15,+20,+20+3mar (Fig.@ The B12 cell line karyotype was:
80—90,XXX,dup(1)(p32p36),add(3)(p 14),inv(3)(p 13q27), +del(6)(q13),—8,—9,add(9)(p22)x2,—12,—15,+20,+20 (Fig. 1D). In sum
Table 1 Doubling times of CEM and CEM/M70-Bcells
DoublingCell line time'@(h)
CEM 16.1±2.4CEM/M70
-BI 17.7 ±2.0-B2 23.1±32b-B3 22.5 ±31b-B4 19.7 ±1.7―-B5 20.7 ±1.8―-B6 18.9 ±1.8-B7 23.3±3.8―-B8 20.6 ±1.3―-B9 21.8 ±2.0―-BlO 22.2 ±25b-BlI 23.3 ±3.3―-Bl2 21.6±3.5―
‘IDoubling times were determined as described in “Materials and Methods.― Each
value represents the mean ±SD of five separate experiments.bp < o.os.
mary, all of the lines had chromosome abnormalities in common,indicating a common origin, most of which were observed in earlierstudies of the drug-resistant CEM cell lines (37, 38). The karyotypesof the Bi and B8 cell lines each had a unique structural marker:der(7)t(7;?12)(q36;q12) and add(l0)(pl 2), respectively.
Cross-Resistance of Merbarone-resistant Cell Lines. We exammed the growth inhibition induced by topo I and H inhibitors andVCR. In Table 2, we show the means ±SD of IC50s of differentagents in CEM cells, representing at least three different experiments
for each drug. Among the merbarone-resistant cell lines, the numbersin Table 2 represent relative resistance ratios of these cell lines,compared to the IC 505 of CEM cells. CEMIM7O-B cell lines were3.5- to 6.6-fold resistant to merbarone, and they were cross-resistantto VP-16 and VM-26 by 8.4- to 15.2-fold and 15.4- to 33.9-fold,respectively. All CEMIM7O-B cell lines were significantly resistant to
these three topo H inhibitors, as determined by Student's t test. For theother complex-stabilizing topo II inhibitors, m-AMSA and DOX, theresistance ratios were lower than for the epipodophyllotoxins, rangingfrom 3.0- and 7.9-fold, and between 1.8- and 3.9-fold for m-AMSAand DOX, respectively. Resistance ratios of CEMIM7O-B cells toother catalytic inhibitors, aclarubicin and SN-22995, were less than4-fold, and only four of the resistant cell lines showed statisticallysignificant cross-resistance ratios to SN-22995. These data suggest
that there might be different mechanisms of cytotoxicity or resistanceamong catalytic topo II inhibitors. In other words, we cannot explainthese results simply from catalytic inhibition of topo II by merbaroneandtheothertwodrugs.
Several of these cell lines tended to be slightly resistant to the topoI inhibitors, CPT and topotecan, but the resistance ratios were lessthan 3-fold. However, of considerable interest, 9 of 12 CEMIM7O-B
cell lines were resistant to SN-38, a putative topo I inhibitor, and anactive metabolite of CPT-l 1 (39), which is a water-soluble derivativeof CPT (40). The range of SN-38 resistance ratios was almost thesame as that for merbarone. Finally, CEMIM7O-B cells were assensitive as their parent cells to VCR.
Because the merbarone-resistant cell lines grew somewhat moreslowly than the parental CEM line, we wanted to assure ourselves thatthe resistance and cross-resistance profiles seen represented true resistance and not differences due to effects of the drugs on cellulargrowth delay (although by definition and the selection process thecells are resistant to merbarone). Accordingly, we examined theeffects of several drugs in a clonogenic survival assay. It can be seenin Table 3 that the B! and Bl2 sublines are 4.6- and 4.3-fold resistant,respectively, to the cytotoxic actions of merbarone, confirming thegrowth inhibition assays. That these are cytotoxic effects is seen in theproduction of apoptotic ladders within 48—72 h after merbaronetreatment (data not shown). In addition to the resistance to merbarone,
the B 1 and B 12 cells are cross-resistant to VP-l6 (25. 1- and I 2.5-fold,respectively) and SN-38 (6.8-fold) but are not cross-resistant to VCR.
Thus, by both growth inhibition and clonogenic survival, the B celllines are resistant to merbarone and cross-resistant to VP-l6 and
SN-38.Cross-Resistance Relationships. We found significant cross-re
sistance in the CEM/M70-B cells to drugs with different mechanisms
of action. However, we cannot determine whether this cross-resistance is due to a common mechanism among 12 clones, because of thevariations among the cell lines. That is, one hypothesized mechanism
of resistance to a particular drug in one cell line might not explain theresistance to other drugs in other cell lines. For example, CEMIM7O-Bl2 cells have the highest resistance to VP-16 and VM-26 in 12 celllines, but the resistance to merbarone is not as high as in the other 11cell lines. The following analysis provides clues to resolve this. Toinvestigate the cross-resistance relationships in 12 CEM/M70-B cell
2575
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

CHARACTERIZATIONOF MERBARONE-RESISTANTCELLLINES
Ay (12(1
ftI@*R S@$@@'!!
19 20
ft
‘ft15
Imar
16
22@,.@
21
1'
ft t H1xx
•@‘I,19@ISI1@ 20)1 mar0.•' 21$@l@i@@* 22
Fig. I. Representative karyotypes of CEM. CEM/M70-Bl. -B8, and -B12 cell lines. Arrows indicate consistent structural or numerical chromosomal abnormalities identified in eachline, and random occurrences were noted for each figure part. A, CEM cell line: 94, XXX,dup(lXp32p36), +del(6Xql3),—8,—8,add(9Xp22)x2,+14,—15,20,20,+22,+mar. The—x,—15+22 and+marwereobservedinthismetaphaseonly.B,Bl cellline:91,XXX,dup(lXp32p36),add(3Xp14),inv(3@pl3q27),del(4@ql2q25),+deI(6@ql3),der(7)t(7;?l2@q36;q12),—8,—9,—9,add(9Xp22)x2,—12,+ 14,—15,+20,+20,+mar. The —9 and + 14 were identified in this sole metaphase. C. B8 cell line: 9l,XXX.dup(lXp32p36),add(3Xpl4),inv(3)(pl3q27)—8,—9.add(9)(p22)x2,add(lOXpl2).—l5.+20.+20,+2mar. The —Xand —21were observed in this metaphase only. Most metaphases in this cell line hadthree additional copies of chromosome 7. D. B12 cell line: 88, XX,dup(lXp32p36),add(3Xpl4),inv(3Xpl3q27),+del(6)(ql3),—8,—9, add(9)(p22)x3,— 12,—l3,+ 14,—15,—16,—22.This is the only metaphase having an extra copy of chromosome 8,add(9p),+ 14 and markers and had random loss of chromosomes X, 13, 16, 20, and 22.
lines, we compared the resistance ratios of the 12 CEMIM7O-B celllines among pairs of drugs (Fig. 2). Comparison of the resistanceratios between VM-26 and VP-l6 revealed a high correlation coefficient (r = 0.81; Fig. 2A). We also found high correlations among
other complex-stabilizing inhibitors, e.g., for VM-26 and doxorubicin(r 0.78) and for VP-16 and m-AMSA (r 0.73). By contrast, therewas a low correlation between merbarone and VP-16 (r = 0.21; Fig.2B). Thus, CEMIM7O-B12 cells were the most resistant of the cell line
2576
)141( 1)511
“i@*_@iP$i@I@
‘IIxx
v$(@@L4@1@I(‘!
t@II 4@s'ii
13 14 17“so
18
PCi EtIl.::::115(1
@6•[email protected]@4@@U$7f 8I 9UI, 10@!@2@i 11ill 12
$1.1soa@,,@fill*1*1@$1131415161718
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

CHARACTERIZATIONOF MERBARONE-RESISTANTCELL LINES
C@ itic @a1L
liii 1%LfIp' 1!@'
I@*Id dbI‘14@ @15
ft.1mar 21@
Fig. 1. Continued
panel to VP-l6 and VM-26 but were not as resistant to merbarone aswere the others (Table 2). The correlation coefficients between merbarone and the other complex-stabilizing drugs were lower than 0.4:VM-26 (r = 0.12), m-AMSA (r 0.39), and DOX (r 0.08). Theseresults suggest that the mechanism of cross-resistance of CEMIM7O-Bcells to complex-stabilizing inhibitors might be different from that ofa catalytic inhibitor such as merbarone.
I$ft13
i9
D
ft •1@1PUI22@ X x
‘iie@2O
2577
11411 051(
Finally, the correlation of drug resistance between two topo Iinhibitors, CI―!'and SN-38, was not very strong (r 0.58), althoughit was statistically significant (Fig. 2C), and the correlation coefficientbetween SN-38 and topotecan was 0.50. However, the correlation
between merbarone and SN-38 was significantly high (r = 0.87;
Fig. 2D). There was also a relatively high correlation between SN-38and m-AMSA (r = 0.73), but there were low correlations to other
iI@@;
16$3.1 1710.1 18
)‘)J5*@(((Un19)6I
)Hi!,$in7I
$11\@amitt@I@IHI@10“I; 11I
@;12•lti@1t•t@a1p―
•01'.@@f19tail @2O@;.; mar4'.4.@ ‘@2'ISI' @221 X
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

Table 2 Resistanceandcross-resistance ratiosof CEM/M70-Bcell lines tomerbarone, othertopo 1 and topoII inhibitors,andVCRCell
lineMerbaroneVP-16VM-26m-AMSADOXSN22995ACRCFTSN-38TopotecanVCRCEM14.4±390±3OnM
4.l±l.8nM69±I9nMl8±l.4nM0.4±[email protected]±4.3nsi3.8±0.2nM2.9±1mMl4±3.2nM62±5.6nM
Table 3 Effectsof drugs on clonogenic surs'ival of merbarone-resistant celllinesCell
lineDrug
IC5@s―Merbarone
(SM)VP-16 (g.@M) SN-38 (nM)VCR(nsi)CEM
CEMIM7O-BlCEM/M70-Bl215b
69 (4.6x)'165 (4.3x)0.065
0.31'1.63 (25.1X) 2.1 (6.8X)0.81 (l2.5X) 2.1 (6.8X)25b
2.8 (1.1X)2.7 (1.1X)
3 4
CHARACrERIZATION OF MERBARONE-RESISTANTCELL LINES
BI 59b.cB2 39CB3 6.6'B4 6.6cB5 4ØCB6 6.4'B7 4.9'B8 6.5'B9 4.5'BlO 3.6'BIl 45CB12 3.5'
13.6'11.8'8.4'
13.3'14.6'13.6'9.8'
14.2'9.5'8.8'
13.6'15.2'
33.2'27.1'20.1'26.8'27.6'28.8c21.4'25.9'16.8'15.4c25.6'339C
complex-stabilizing inhibitors: VP-l6 (r = 0.01), VM-26 (r = 0.15),and DOX (r = 0.25). Judging from those results, we postulate that themechanism of cross-resistance to SN-38 might be related to that ofmerbarone resistance.
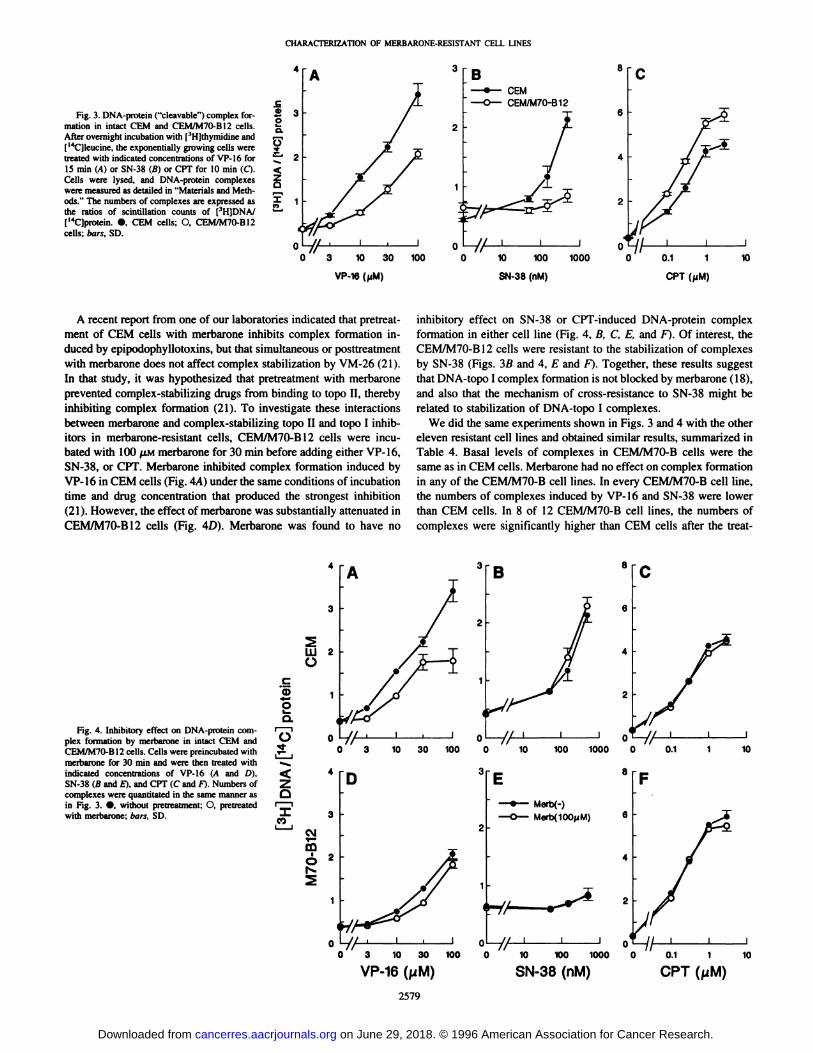
DNA-Protein Complex Formation Assay in Intact Cells. Toinvestigate the effect of topo II and topo I drugs on the merbaroneresistant cells, we measured DNA-protein complex formation in intactcells. Up to 300 @LMmerbarone-produced no DNA-protein complexesin either parental or drug-resistant cell lines. By contrast, at concentrations of VP-16 between 3 and 100 p.M for 15 mm, there was aproportional increase in the number of DNA-protein complexes in
7.9' 3.9' 2.8 2.4 2.5' 79C 1.6c 0.96.7' 3.2c 2.5 1.8 1.9' 2.4' 1.2 0.95.2c 1.8' 2.2 1.0 1.0 4.9' 1.3 0.87.5' 3.3' 2.6 1.8 1.7' 8.4c 1.8 0.85.6' 3.2 2.7 2.1 1.3 1.7 1.3 1.16.3c 2.8c 2.3 1.5 1.9' 6.1c 1.6' 0.95.9' 3.lc 3.0@ 3.3 2.1 2.6c 1.5 0.77.4' 3.4' 3.0' 1.9 2.2' 6.6' 1.4 0.76.6c 3.8 3.0' 2.5 1.9 45C 1.5 0.83.0 2.lc 2.4 1.5 1.0 1.5 1.3 1.24.8c 2.7' 2.8 1.5 1.5 2.4' 1.4 0.859C 3,5C 2.8c 1.7 l.6c 1.9 1.5 1.3
a Mean±SD of IC50.b Relative resistance ratio of M70-B cell lines (IC50 of CEM = 1).
C p < 0.05 by Student's t test.
CEM and CEMIM7O-B cells, but the maximum stabilization waslower in the drug-resistant cells (Fig. 3A). When we incubated CEMand CEMIM7O-B12 cells with the indicated concentrations of eitherCPT or SN-38 for 10 rain, the basal level of complex formation inCEM/M70-B12 cells without drug was the same as that in CEM cells.When we compared DNA-topoisomerase complex formation betweenCEM and CEMIM7O-B12 cells after drug treatment, we found that thenumbers of DNA-protein complexes induced by VP-16 in CEMIM7O-B12 cells were lower than in CEM cells. We obtained similar resultswith m-AMSA and VM-26 (data not shown). The reduced complexforming ability of CEMIM7O-B cells seems to be related to their
cross-resistance to VP-16. Interestingly, the number of complexesstabilized by SN-38 in CEMIM7O-B cells was lower in CEM cells, butthose stabilized by CPT were higher. From the perspective of therelationship between DNA-topo I complexes and cytotoxicity, thisdiscrepancy of results between two topo I inhibitors could partiallyexplain the selective resistance of CEMIM7O-B cells to SN-38. However, the increased numbers of stabilized complexes by CPT did notcontribute to the hypersensitivity of these cells to this drug; CEM/M70-B12 cells were, in fact, 1.6-fold resistant to it. These resultssuggest that there might be other mechanisms of cross-resistance to
topo I drugs in addition to or instead of DNA-protein complexstabilization ability.
. . •.
. r=0.207. p-0.52
5
Merbarone
D.
4>@(
.‘—@. • r-O.867
2 )@-@ [email protected]@@@@ I I I I
3 4 5 6 7
a IC5@,sweredeterminedby extrapolationfrom a graphofcolony survival(drugtreated/control) versus drug concentration.
b Mean of two separate experiments.
C Single experiment.
d Numbers in parentheses represent fold-resistance. See “Materials and Methods― for
details.
16
14
12
10
A B•
16
14
Co
cL 12>
10
8S
.5
20 30 40 6 7Fig. 2. Correlations of resistance ratios between pairs of topoisomerase inhibitors. The resistance ratios of 12 CEMIM7O-B celllines to two topoisomerase inhibitors were plotted in the graphs, andcorrelation coefficients were calculated. A, VM-26 versus VP-l6; B,merbarone versus VP-16; C, CPT versus SN-38; D, merbarone versusSN-38. See text and “Materialsand Methods―for details.
VM-26
10
8
6
4
2
r-O.580 Cp-O.048 •
S
I@ •@•
10
8
6
CO
cL>
COC')
zC')
COC')
zCl)
0 1 2 3CPT Merbarone
2578
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
o@ 0.1 1 10
0.1 10
CHARACI'ERIZATION OF MERBARONE-RESISTANTCELL LINES
3
2
B—.--. CEM
@ —0— CEM/M70-B12
0
C
C000.
z0
IC')
Fig. 3. DNA-protein (“cleavable―)complex formation in intact CEM and CEM/M70-Bl2 cells.Afterovernight incubation with [3H]thymidine and[‘@C]leucine,the exponentially growing cells weretreated with indicated concentrations of VP-l6 for15 mm (A) or SN-38 (B) or CPT for 10 mmn(C).Cells were lysed, and DNA-protein complexeswere measured as detailed in “Materialsand Methods.―The numbers of complexes are expressed asthe ratios of scintillation counts of [3HIDNA/[@CJprotein. •,CEM cells; 0, CEMIM7O-B12cells; bars, SD.
VP-16 (tiM) SN-38 (nM) CPT (SM)
A recent report from one of our laboratories indicated that pretreatment of CEM cells with merbarone inhibits complex formation induced by epipodophyllotoxins, but that simultaneous or posttreatmentwith merba.rone does not affect complex stabilization by VM-26 (21).In that study, it was hypothesized that pretreatment with merbaroneprevented complex-stabilizing drugs from binding to topo II, therebyinhibiting complex formation (21). To investigate these interactionsbetween merbarone and complex-stabilizing topo II and topo I inhibitors in merbarone-resistant cells, CEMIM7O-B12 cells were incubated with 100 p.i@imerbarone for 30 rain before adding either VP-l6,SN-38, or CPT. Merbarone inhibited complex formation induced byVP-16 in CEM cells (Fig. 4A) under the same conditions of incubationtime and drug concentration that produced the strongest inhibition(21). However, the effect of merbarone was substantially attenuated inCEMIM7O-B12 cells (Fig. 4D). Merbarone was found to have no
C000@
II
C-)I@I
zU'I
C,)
inhibitory effect on SN-38 or CPT-induced DNA-protein complexformation in either cell line (Fig. 4, B, C, E, and F). Of interest, theCEMIM7O-B12 cells were resistant to the stabilization of complexesby SN-38 (Figs. 3B and 4, E and F). Together, these results suggestthat DNA-topo I complex formation is not blocked by merbarone (18),and also that the mechanism of cross-resistance to SN-38 might berelated to stabilization of DNA-topo I complexes.
We did the same experiments shown in Figs. 3 and 4 with the othereleven resistant cell lines and obtained similar results, summarized inTable 4. Basal levels of complexes in CEMIM7O-B cells were thesame as in CEM cells. Merbarone had no effect on complex formationin any of the CEMIM7O-B cell lines. In every CEMIM7O-B cell line,the numbers of complexes induced by VP-16 and SN-38 were lowerthan CEM cells. In 8 of 12 CEM/M70-B cell lines, the numbers ofcomplexes were significantly higher than CEM cells after the treat
3 8A
2
4
3
W2C-)
0
4
3
Fig. 4. Inhibitory effect on DNA-protein complex formation by merbarone in intact CEM andCEM/M70-Bl2 cells. Cells were preincubated withmerbarone for 30 rain and were then treated withindicated concentrations of VP-16 (A and D),SN-38 (B and A), and CFF (C and F). Numbers ofcomplexes were quantitated in the same manner asin Fig. 3. •,without pretreatment; 0, pretreatedwith merbarone; bars, SD.
0@@@//l I I
0 10 100 1000
3
100
7//U I@ I
0 3 10 30
D
. —.—- Merb(-)
—0-- Merb(I00@iM)2
1
0_71/ I I
0 10 100 1000
SN-38 (nM)
0.//I I I
0 3 10 30 100
VP-16(,@iM) CPT(NM)10
2579
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

Merbarone VP-l6Cell line Control (100 @sM) (40 saM) Merbarone —sVP-16―SN-38 (0.3 @sM)CPT (0.6@M)CEM
0.32 ±0.01 0.34 ±0.00 2.20 ±0.02 1.45 ±0.021.62 ±0.041.06 ±0.05CEM/M70-Bl
0.38 ±0.01 0.36 ±0.01 0.9l'@±0.00 0.78c ±0.02l.20c ±0.04l.21c ±0.03-820.35 ±0.01 0.33 ±0.00 l.Otf ±0.01 [email protected]'@ ±0.091.13 ±0.07-B30.33 ±0.00 0.33 ±0.01 l.23c ±0.07 0.90c ±0.010.42c ±0.10l.68' ±0.08-B40.33 ±0.02 0.35 ±0.01 l.02c ±0.00 0.68c ±0.010.46c ±0.02l.52'@' ±o.io-B50.35 ±0.01 0.34 ±0.00 0.96c ±0.02 Ø,75C@ 0.030.67c ±0.061.20 ±0.09-B60.37 ±0.63 0.37 ±0.02 0.99c ±0.00 0.96 ±0.050.42c ±0.02152c,d@ [email protected] ±0.01 0.38 ±0.01 l.08c ±0.07 0.71c ±0.020.82c ±0.10l.77'@― ±o.os-880.36 ±0.00 0.38 ±0.00 0.94c ., 0.01 091b@ 0.03O.43c ±[email protected]@ 0.14-B9
0.33 ±0.01 0.35 ±0.00 l.1Oc ±0.07 0.74c ±0.010.48c ±0.04133c,d@ o.oi-BlO0.33 ±0.01 0.33 ±0.01 l.03c ±0.04 0.92c ±0.020.53c ±0.01l.46― ±o.o@-Bl
1 0.34 ±0.02 0.31 ±0.02 0.99c ±0.01 0.89c ±0.030.60― ±0.071.13 ±0.08-Bl20.33 ±0.00 0.32 ±0.00 l.04c ±0.04 0.72c ±0.040.66' ±0.021.18 ±0.06a
Results represent the means ± SD for four separate experiments. See “Materials and Methods― for details.
b Treatment with 40 @LMVP-l6 after pretreatment with 100 @Mmerbarone.C
CEM > M70-B cells, P < 0.05, compared to control.
d CEM < M70-B cells, P < 0.05, compared to control.
One way to attempt to understand the cytocidal mechanism ofcatalytic topo H inhibitors is to establish novel resistant cell lines tothese agents and compare their features to those of the parental cells.We selected novel merbarone-resistant CEMIM7O-B cells that arecross-resistant to both merbarone and complex-stabilizing topo Hinhibitors. Merbarone-resistant CEM/M70-B cells may provide insights into the following: (a) mechanisms of merbarone resistance; (b)the function of topo ilk; (c) differences in mechanisms of cytotoxicityamong catalytic topo H inhibitors; (d) mechanisms of action andresistance of SN-38; and most importantly, (e) mechanisms by whichtopoisomerase inhibition leads to cell death. These merbarone-resistant cells differ from the “atypical―multidrug resistant cell linesCEM/VM-! and CEM/VM-!—5 that express altered topo H (at-MDR;Ref. 41); those cell lines are resistant to complex-stabilizing topo IIinhibitors but not to merbarone or other catalytic inhibitors of topo II(2!, 41, 42). Single-strand conformational polymorphism analysisrevealed no mutations in the sequence of the topo II gene in themembrane-resistant cells, where two point mutations in CEMIVM-!and VM-l—5cells were found (Refs. 43 and 44; data not shown). Ina search of the literature, we found only two reports of cell lines
Table 5 Densitometric analysis of immunoblots of topo ila, topo 11f3,and topo 1 inmerbarone-resistant CEM cell line?
Cell lineRelative
proteinlevels―topo
Hatopo [email protected]
±0.l5c0.83 ±0.481.09 ±0.28-B20.30±0.09'@l44@ ±0.490.87 ±0.20-B30.32±0.lOc1.77 ±0.63'@1.45 ±0.42-840.35±0.06c1.82 ±0.69c0.74 ±0.20-B50.46±0.l3c2.02 ±0.97c0.82 ±0.16-860.59±0.l3'@2.10 ±O.8O'@1.39 ±0.66-B70.60±0.25c1.38 ±0341.02 ±0.42-B80.31±0.05c1.29 ±0.311.28 ±0.64-B90.42±0.l0'@1.40 ±0.611.33 ±0.45-BlO0.51±0.l5c1.53 ±0.761.09 ±0.19-Bl
10.43 ±0.101.33 ±0.511.21 ±0.28-Bl20.46±O.l3c1.36 ±0.561.74 ±0.77
CHARACTERIZATIONOF MERBARONE-RESISTANTCELL LINES
Table 4 DNA-topo complexes (3H1DNAJI'4Cjprotein in merbarone-resistant CEM-M7OB cellf'
OEM 01 82 B3 B4 B5 B6 B7 B8 89 BlO Bli B12
lbpollc@ —@
bpolfl L
bPoIr
-- - @--
-OOkd
,.-.-
Fig. 5. Immunoblot analysis of topo Ha, topo ll@,and topo I in CEM and merbaroneresistant cell lines. Total cell proteins from CEM and CEMIM7O-Bl--B12 cells wereelectrophoresed in 7.5% SDS-PAGE. topo Ha, topo ll@, and topo I proteins were blottedwith antisera described in “Materialsand Methods.―Shown are representative blots fromone experimenL Immunoblots were done with at least five separate preparations, andprotein levels were analyzed densitometrically. Relative protein amounts (mean ±SD)compared to those in CEM cells are given in Table 5. See “Materialsand Methods―andTable 5 for details.
ment with CPT. In the other four cell lines (-B2, -B5, -Bll, and -B 12),the numbers tended to be higher than in CEM cells but were notsignificantly different from CEM cells (Table 4). Compared to VP-16alone, pretreatment with merbarone reduced the number of complexesin both CEM and all CEMIM7O-B cell lines. However, as before, theeffect of merbarone on VP-16-stabilized DNA-protein complexes inCEMIM7O-B cells was less than in CEM cells (Table 4).
Immunodetection of topo II and topo I in Merbarone-resistantCEM Cells. A representativeresultof immunoblotsof topo Ha, topoHP,andtopoI in CEM andCEMIM7O-Bcellsis shownin Fig. 5.Densitometric analysis and comparison of the protein levels betweenCEM and CEMJM7O-B cells in five separate experiments revealedthat in every resistant cell line, the expression of topo Ha wassignificantly decreased to less than one-half of that in CEM cells(Table 5). The mean topo Ha level in 12 CEMIM7O-B cell lines wasreduced to 42% of that of CEM cells. With the exception of the B!cell line, topo ll@3was increased by 52% in 11 merbarone-resistant celllines. Four of the B cell lines expressed statistically higher levels oftopo H@than in CEM cells. In contrast to topo H levels, the expressionof topo I in all resistant cell lines was about the same as or a littlehigher than that in CEM cells (average topo I level, 117%, compared
to CEM), but no CEMIM7O-B cell line had a statistically differentlevel of topo I from that of CEM cells.
a Topo Ha was detected with the MAC antiserum. Topo 11(3 was detected with the
#282 antiserum, and topo I was detected with the T!-l antiserum. See “MaterialsandMethods―for details.
b Band intensities were determined as described in “Materials and Methods.― Each
value was made relative to that from the parental CEM cells and represents themean ±SD of at least five different preparations and experiments.
C p@ 0.05, compared to CEM cells.
2580
DISCUSSION
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

CHARACFERIZATION OF MERBARONE-RESISTANTCELL LINES
resistant to complex-stabilizing drugs that have distinctive cross
resistance to merbarone (30, 45).Our data suggest that the merbarone resistance of CEMIM7O-B
cells might be a consequence of decreased levels of topo 1hz. Thefinding is counter-intuitive; given the dominant-negative action ofcatalytic topo II inhibitors, we had, in fact, expected to see increasedtopo II levels. There are several reports showing decreased topo IIlevels or activity in cell lines selected for resistance to complexstabilizing inhibitors of topo II (Ref. 46 and references therein). Thisphenomenon is easily explained by the fact that these drugs capturethe topo H covalently bound to DNA and block the religation of theDNA (Ref. 46 and references therein). Under these circumstances,reduced topo II levels permit fewer DNA-topo II complexes to beformed and stabilized, leading to drug resistance. However, if there isa decreased topo II amount, as seen in CEMIM7O-B cells, or activity,lower concentrations of the catalytic inhibitors would be needed, andthe sensitivity of the cells would be expected to be increased (47, 48).The decreased topo 1hz suggests that there might be other factorscontributing to the resistance that do not correlate with the catalyticeffect of this drug. That CEMIM7O-B cells are not cross-resistant toother catalytic inhibitors might also support this idea. For instance,aclarubicin is a catalytic inhibitor that prevents topo H from performing its noncovalent DNA-binding reaction (48); however, there wasneither a correlation between drug sensitivity and topo II levels (49)nor between drug sensitivity and cell cycle-dependent activity of topoII in G2 or M phase (50). Since topo Ha is a structural component ofthe nuclear scaffold and mitotic chromosomes (5 1, 52), and becausemerbarone interferes with chromosome condensation and cell divisionmore strongly than it causes DNA damage, compared to VM-26 (27),it is possible that reduced structural topo Ha in the nuclei of CEM/M70-B cells buffers some unknown effects of merbarone on thisenzyme in the mitotic phase of the cell cycle.
For the cross-resistance of the merbarone-resistant cells to complex-stabilizing topo II inhibitors, reduced complex formation, perhaps due to reduced topo Ha levels, might be the main cause ofresistance. However, the cross-resistance profile of CEM/M70-B cellssuggests that the bases of resistance to merbarone differ from those ofcross-resistance to complex-stabilizing topo II inhibitors (Fig. 2).
Since merbarone inhibits topo H binding to DNA, thus preventingVM-26 from stabilizing the complexes between topo II and DNA (18,21), decreased inhibition of VP-l6-induced complex formation bymerbarone in CEMIM7O-B cells can be explained by decreased topoHa levels.
The physiological role of topo H@ or drug effects on this enzymehave not been studied in detail, and in CEMIM7O-B cells, althoughtopo II@3tends to be increased, this might not have a direct role inmerbarone resistance, since increased topo II@3is not seen in all of our12 CEMIM7O-B cell lines. Indeed, a correlation between intracellulartopo H@levels and sensitivity to topo II inhibitors is not a consistentfinding; the level of topo 11(3 is increased (53) in some drug-resistant
cell lines but decreased in others (30, 54). In a statistical analysis ofseveral leukemia cell lines, there was an inverse correlation between
the level of topo 11(3and resistance to doxorubicin (55). If increasedtopo 11f3plays a role in merbarone resistance, its decrease shouldresult in an increase in drug sensitivity. However, in CEM/VM-l—5cells, which express little or no topo ll@3(56), the sensitivity tomerbarone is about the same as in CEM cells (21), and topo 11(3is alsodecreased in mitoxantrone-resistant HL-60 cells, which are in factcross-resistant to merbarone (30).
Drake et a!. (57) speculated that topo II@3may play a primary rolein transcription, especially of rRNA that takes place in the nucleolus,rather than in replication or chromosome segregation. This idea is
supported by the facts that topo 11(3appears to be localized mainly in
the nucleolus (33, 58, 59), whereas topo ha is diffusely localized inboth nucleoli and nucleoplasm as a nuclear scaffold protein (34, 51,52, 58, 59). The expression of topo 11/3 protein appears to be constant
in interphase (59, 60) and present in quiescent cells (57), whereasexpression of topo ha is increased in S or G2 (60, 61) and decreasedin quiescent cells (57). These findings suggest that, compared to topolies, topo 1113might have a diminished role in cell cycle regulation andin the inhibition of chromosome condensation induced by merbarone(21, 27).
Decreased drug transport is a factor in some forms of drug resistance (41, 46), but we have not found any evidence to suggest that itis important in merbarone resistance. These cells display no crossresistance to VCR, a substrate for Pgp, and in preliminary studies, we
found no difference in [3H]VP-l6 accumulation between CEM andCEMIM7O-B cells.5 Moreover, the merbarone-resistant cells do notoverexpress Pgp or multidrug resistance-associated protein.6 It will beof interest to examine the transport of merbarone itself in these celllines. A mitoxantrone-resistant cell line has been reported to becross-resistant to topo H inhibitors, as well as the topo I inhibitorstopotecan and SN-38, but not to CPT (62). In that study, the crossresistance to topo I inhibitors did not appear to be due to alterations inthe enzyme, and decreased topotecan accumulation appeared to be dueto a novel, non-Pgp, non-multidrug resistance-associated proteinmechanism. Although the pattern of cross-resistance of that cell lineis different from CEMIM7O-B, merbarone resistance might involve, inpart, unknown mechanisms of decreased transport.
SN-38 is the active metabolite of CPT-ll (39), a derivative of CPT(40), and is converted by a carboxylesterase in the serum (39) or incancer cells (63). These drugs inhibit topo I activity (63—65).CVFinhibits topo I by stabilizing DNA-topo I cleavable complexes (66),and the cytocidal effects of CPT-11 and SN-38 are believed to berelated to the complex-stabilizing activity of SN-38. Although thereare few CPT-ll-resistant and no SN-38-resistant cell lines, reductionof topo I amount (64, 67—69)and activity (64, 68—70)and resistanceto the inhibitory effect of CPT-ll on enzyme activity (69, 70) havebeen reported to be related to the resistance, as in CPT-resistant celllines (71). Disturbance of conversion of CPT-ll to SN-38 has beenreported in one cell line (70). The resistance of these cell lines toSN-38 is less than that to CPT-l 1 (68—70).
We were intrigued to find that the CEMIM7O-B cells were crossresistant to SN-38, a putative topo I inhibitor. There have been but few
cell lines reported to be resistant to both topo H inhibitors and CPT-l1or SN-38 (62, 72—74).The mechanism of cross-resistance in each cellline is related to the combination of change in enzymatic levels oractivities of one enzyme and change of membrane transport of drugs
for the other topoisomerase. There are, however, many reports ofcollateral sensitivity of CPT-resistant cell lines to topo II inhibitors(75, 76) and of cell lines resistant to topo II inhibitors that are
hypersensitive to topo I inhibitors (77, 78); these may be due tocompensatory increases in topo I or topo II (16, 17). However, it isknown that topo II effects on chromosome condensation and segregation cannot be subserved by topo I (16, 17), and this may be why thecell lines resistant to topo II inhibitors are generally not cross-resistanttotopoIinhibitors.
Reduced DNA-topoisomerase complex formation may be a factorin the resistance of CEMIM7O-B cells to SN-38. However, it isdifficult to explain the discrepancy in the results of cross-resistanceand complex formation by CPT and SN-38. One may hypothesize thatSN-38 inhibits topo II as well as topo I activity, but this is unlikely,in part because of the lack of effect of merbarone on SN-38-induced
5 Q. E. Rodgers and W. T. Beck, unpublished observation.
6 H. Kusumoto and W. T. Beck, unpublished results.
2581
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

CHARACI'ERIZATION OF MERBARONE-RESISTANTCELL LINES
21. Chen, M., and Beck, W. T. Teniposide-resistant CEM cells, which express mutanttopoisomerase Ha, when treated with non-complex-stabilizing inhibitors of the enzyme, display no cross-resistance and reveal aberrant functions of the mutant enzyme.Cancer Res., 53: 5946—5953, 1993.
22. Cooney, D. A., Covey, J. M., Kay, 0. J., Da1a1,M., McMahon, J. B., and Johns, D. 0.Initial mechanistic studies with merbarone. Biochem. Pharmacol., 34: 3395—3398,1985.
23. Chang, A. Y., Kim, K., Glick, J., Anderson, T., Karp, D., and Johnson, D. Phase IIstudy of taxol, merbarone, and piroxantrone in stage IV non-small-cell lung cancer:The Eastern Cooperative Oncology Group results. J. Nail. Cancer Inst., 85: 388—394,1993.
24. Jones, D. V., Jr., Ajani, J. A., Winn, R. J., Daugherty, R. N., Levin, B., and Krakoff,I. H. A phase II study of merbarone in patients with adenocarcinoma of the pancreas.Cancer Invest., II: 667—669,1993.
25. Jensen, P. B., Jensen, P. S., Demant, E. J. F., Friche, E., S4rensen, B. S., Sehested,M., Wassermann, K., Vindel4w, L., Westergaard, 0., and Hansen, H. H. Antagonisticeffect of aclarubicin on daunorubicin-induced cytotoxicity in human small cell lungcancer cells: relationship to DNA integrity and topoisomerase H. Cancer Res., 51:5093—5099, 1991.
26. Ishida, R., Mild, T., Narita, T., Yui, R., Sato, M., Utsumi, K. R., Tanabe, K., andAndoh, T. Inhibition of intracellular topoisomerase II by antitumor bis(2, 6-dioxopiperazine) derivatives: mode of cell growth inhibition distinct from that of cleavable complex-forming type inhibitors. Cancer Rca., 51: 4909—4916, 1991.
27. Chen, M., and Beck, W. T. Differences in inhibition of chromosome separation and02 arrest by DNA topoisomerase II inhibitors merbarone and VM-26. Cancer Res.,55: 1509—1516,1995.
28. Downes, C. C., Clarke, D. J., Mullinger, A. M., Glme'nez-Abla'n, J. F., Creigliton,A. M., and Johnson, R. T. A topoisomerase 11-dependent 02 cycle checkpoint inmammalian cells. Nature (Lond.), 372: 467—470,1994.
29. Ishida, R., Sato, M., Narita, T., Utsumi, K. R., Nishimoto, T., Monte, T., Nagata, H.,and Andoh, T. Inhibition of DNA topoisomerase II by ICRF-193 inducespolyploidization by uncoupling chromosome dynamics from other cell cycle events.J. Cell Biol., 126: 1340—1351,1994.
30. Harker, W. 0., Slade, D. L., Drake, F. H., and Parr, R. L. Mitoxantrone resistance inHL-60 leukemia cells: reduced nuclear topoisomerase II catalytic activity and druginduced DNA cleavage in association with reduced expression of the topoisomeraselip isoform. Biochemistry, 30: 9953—9961,1991.
31. Norman, M. R., and Thompson, E. B. Characterization of a glucocorticoid-sensitivehuman lymphoid cell line. Cancer Res., 37: 3785—3791,1977.
32. Wolverton, J. S., Danks, M. K., Granzen, B., and Beck, W. T. DNA topoisomeraseII immunostaining in human leukemia and rhabdomyosarcoma cell lines and theirresponses to topoisomerase II inhibitors. Cancer Res., 52: 4248—4253, 1992.
33. Boege, F., Andersen A., Jensen, S., Zeidler, R., Kreipe, H. Proliferation-associatednuclear antigen 10-51 is identical with topoisomerase Ha. Delineation of a carboxy.terminal epitope with peptide antibodies. Am. J. Pathol., 146: 1302—1308,1995.
34. Laemmli, U. K. Cleavage of structural proteins during the assembly of the head ofbacteriophage T4. Nature (Lond.), 227: 680—685,1970.
35. Friche, E., Danks, M. K., Schmidt, C. A., and Beck, W. T. Decreased DNAtopoisomerase II in daunorubicin-resistant Ehrlich ascites tumor cells. Cancer Rca.,51: 4213—4218, 1991.
36. Baker, S. D., Wadkins, R. M., Stewart, C. F., Beck, W. T., and Danks, M. K. Cell cycleanalysis of amount and distributionof nuclear DNA topoisomeraseI as determinedbyfluorescencedigital imaging microscopy.Cytometry, 19: 134-145, 1995.
37. Beck, W. T., Cirtain, M. C., Danks, M. K., Felsted, R. L., Safa, A. R., Wolverton,J. S., Sunle, D. P., and Trent, J. M. Pharmacological, molecular, and cytogeneticanalysis of “atypical―multidrug-resistant human leukemic cells. Cancer Rca., 47:5455—5460, 1987.
38. Hill, A. B., Beck, W. T., and Trent, J. M. Cytogentic and molecular characterizationof tumors in nude mmccderived from a multidrug-resistant human leukemia cell line.Cancer Res., 48: 393—398,1988.
39. Kaneda, N., Nagata, H., and Yokokura, T. Metabolism and pharmacokinetics ofcamptothecin analog CPT-lI in the mouse. Cancer Res., 50: 1715—1720,1990.
40. Kunimoto, T., Nifta, K., Tanaka, T., Uehara, N., Babe, H., Takeuchi, M., Yokokura,T., Sawada, S., Miyasaka, T., and Mutai, M. Antitumor activity of 7-ethyl-10-[4-(l-piperidino)-1-piperidino]carbonyloxy-camptothecin, a novel water-soluble derivativeof camptothecm, against murine tumors. Cancer Res., 47: 5944—5947,1987.
41. Beck, W. T., Danks, M. K., Wolverton, J. S., Kim, R., and Chen, M. Drug resistanceassociated with altered DNA topoisomerase II. Adv. Enzyme Regul., 33: 113-127,1993.
42. Danks, M. K., Schmidt, C. A., Cirtain, M. C., StatIc, P., and Beck, W. T. Alteredcatalytic activity and DNA cleavage by DNA topoisomerase II from human leukemiccells selected for resistance to VM-26. Biochemistry, 27: 8861—8869,1988.
43. Bugg, B. Y., Danks, M. K., Beck, W. T., and Suttle, D. P. Expression ofmutant DNAtopoisomerase II in CCRF-CEM human leukemia cells selected for resistance toteniposide. Proc. Nail. Aced. Sci. USA, 88: 7634—7638, 1991.
44. Danks, M. K., Warmoth, M. R., Friche, E., Granzen, B., Bugg, B. Y., Barker, W. 0.,Zwelling, L., Futscher, B. W., Suttle, P., and Beck, W. T. Single-strand conformstional polymorphism analysis ofthe Mr 170,000 isoZymeofDNA topoisomerase II inhuman tumor cells. Cancer Res., 53: 1373—1379,1993.
45. Fauman, C., Allan, W. P., Hasinoff, B. B., and Yalowich, J. C. Collateral sensitivityto the bisdioxopiperazine, ICRF-187, in etoposide (VP-16)-resistant human leukemia1(562 cells. Proc. Am. Assoc. Cancer Res., 36: 317, 1995.
46. Beck, W. T., Danks, M. K., Wolverton, J. S., Chen, M., Granzen, B., Kim, R., andSuule, D. P. Resistance of mammalian tumor cells to inhibitors of DNA topoisomerase II. Adv. Pharmacol., 29B: 145—169,1994.
47. de Jong, S., Zijlstra, J. 0., Mulder, N. H., and de Vries, E. 0. E. Lack of cross
2582
complex formation (Fig. 4). Studies are in progress to understand thebasis of the SN-38 cross-resistance of our cells. The correlation ofIC5@sand stabilization of DNA-protein complexes between merbarone and SN-38 suggests that these agents have a mechanism incommon. In conclusion, our merbarone-resistant cell lines are noveltools to study resistance to topo II and topo I inhibitors. We arepresently examining the unresolved issues of resistance mentionedabove.
ACKNOWLEDGMENTS
We thank T. Huddleston, P. Mardis, E. Entrekin, and K. Wallace of the St.Jude Pathology Department for excellent karyotype analysis, Suzette Wingoand Dr. Raymond L. Blakley of the St. Jude Molecular Pharmacology Department for advice with clonogenic assays, and Linda Rawlinson of the Biomedical Communications Department for excellent preparation of the artwork. Weare most grateful to Fabrienne Holloway, Dolores Anderson, and Imella
Herrington for their skilled typing of the manuscript.
REFERENCES
1. Liu, L. F., Liu, C., and Alberta. B. M. Type II DNA topoisomerases: enzymes that canunknot a topologically knotted DNA molecule via a reversible double-strand break.Cell, 19: 697—707,1980.
2. Yang, L., Wold, M. S., Li, J. J., Kelly, T. J., and Liu, L. F. Roles of DNAtopoisomerases in simian virus 40 DNA replication in vitro. Proc. Natl. Acad. Sci.USA, 84: 950—954, 1987.
3. Brill, S. J., DiNArdo, S., Voelkel-Meiman, K., and Sternglanz, R. Need for DNAtopoisomerase activity as a swivel for DNA replication for transcription of ribosomalRNA. Nature (Lond.), 326: 414—416,1987.
4. Nelson, W. 0., Liu, L. F., and Coffey, D. S. Newly replicated DNA is associated withDNA topoisomerase II in cultured rat prostatic adenocarcinoma cells. Nature (Lond.),322: 187—189,1986.
5. Musk, S. R., and Steel, 0. G. The inhibition of cellular recovery in human tumourcells by inhibitors of topoisomerase. Br. J. Cancer, 62: 364—367,1990.
6. Ross, W., Rowe, T. C., Glisson, B., Yalowich, J., and Liu, L. F. Role of topoisomerase II in mediating epipodophyllotoxin-induced DNA cleavage. Cancer Rca., 44:5857—5860,1984.
7. Nelson, E. M., Tewey, K. M., and Liu, L. F. Mechanism of antitumor drug action:poisoning of mammalian DNA topoisomerase II on DNA by 4'-(9-acridinylamino)-methanesulfon-m-aniside. Proc. NatI. Acad. Sci. USA, 81: 1361—1365,1984.
8. Tewey, K. M., Rowe, T. C., Yang, L., Halligan, B. D., and Liu, L. F. Adriamycininduced DNA damage mediated by mammalian DNA topoisomerase II. Science(Washington DC), 226: 466—468,1984.
9. Liu, L. F. DNA topoisomerase poisons as anti-tumor drugs. Annu. Rev. Biochem., 58:351—375,1989.
10. Roberge, M. J., Th'ng, J., Hamaguchi, J., and Bradbury, E. M. The topoisomerase IIinhibitor VM-26 induces marked changes in histone Hl kinase activity, histone HIand H3 phosphorylation and chromosome condensation in G2 phase and mitotic BHKcells. J. Cell Biol., III: 1753—1763,1990.
11. Lock, R. B. Inhibition of p34@k2kinase activation, p34@2 tyrosine dephosphorylation, and mitotic progression in Chinese hamster ovary cells exposed to etoposide.CancerRes., 52: 1817—1822,1992.
12. Hartwell, L. H., and Weinert, T. A. Checkpoints: controls that ensure the order of cellcycle events. Science (Washington DC), 246: 629—633, 1989.
13. Murray, A. W. Creative blocks: cell-cycle checkpoints and feedback controls. Nature(Lond.), 359: 599—604,1992.
14. Downes, C. S., Mullinger, A. M., and Johnson, R. T. Inhibitors of DNA topoisomersac II prevent chromatid separation in mammalian cells but do not prevent exit frommitosis. Proc. Nail. Acad. Sci. USA, 88: 8895—8899,1991.
15. Summer, A. T. Inhibitors of topoisomerases do not block the passage of humanlymphocyte chromosomes through mitosis. J. Cell Sci., 103: 105—115, 1992.
16. Uemura, T., and Yanagida, M. Isolation of type I and II DNA topoisomerase mutantsfrom fission yeast: single and double mutants show different pbenotypes in cellgrowth and chromatin organization. EMBO J., 3: 1737—1744,1984.
17. Uemura, T., Ohkura, H., Adachi, Y., Morino, K., Shiozaki, K., and Yanagida, M.DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S. pombe. Cell, 50: 917—925,1987.
18. Drake, F. H., Hofmann, G. A., Mong, S., Bartus, J. 0., Hertzberg, R. P., Johnson,R. K., Mattern, M. R., and Mirabelli, C. K. In vitro and intracellular inhibition oftopoisomerase II by the antitumor agent merbarone. Cancer Res., 49: 2578—2583,1989.
19. Jensen, P. B., S4reosen, B. S., Demant, E. J. F., Sehested, M., Jensen, P. S.,Vindel4v, L., and Hansen, H. H. Antagonistic effect of aclarubicin on the cytotoxicityof etoposide and 4'-(9-acridinylamino) methanesulfon-m-aniside in human small celllung cancer cell lines and on topoisomerase 11-mediatedDNA cleavage. Cancer Res.,50:3311—3316,1990.
20. Tanabe, K., Ikegami, Y., Ishida, R., and Andoh, T. Inhibition of topoisomerase II byantitumor agents bis(2,6-dioxopiperazine) derivatives. Cancer Res., 51: 4903-4908,1991.
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

CHARACTERIZA11ONOF MERBARONE-RE5ISTANTCELL LINES
resistance to fostriecin in a human small-cell lung carcinoma cell line showingtopoisomerase n-related drug resistance. Cancer Chemother. Pharmacol., 28: 461—464, 1991.
48. Sørensen, B. S., Sinding, J., Andersen, A. H., Alsner, J., Jensen, P. B., andWestergaard, 0. Mode of action of topoisomerase 11-targetingagents at a specificDNA sequence. J. Mol. Biol., 228: 778—786,1992.
49. Jensen, P. B., Sørensen,B. S., Sehested, M., Demant, E. J. F., Kjeldsen, E, Friche,E., and Hansen, H. H. Different modes of anthracycline interaction with topoisomerase H. Biochem. Pharmacol., 45: 2025—2035, 1993.
50. Perez, C., Campayo, L., Navarro, P., Garcia-Bermejo, L, and Aller, P. The action ofthe DNA intercalating agents 4'-(9-acridinylamino) methanesulphon-m-arnside andhuman promonocytic cells: relationship between cell cycle and differentiation. Biochem. Phannacol., 48: 75—82,1994.
51. Earnshaw, W. C., Haffigan, B., Coke, C. A., Heck, M. M. S., and Liu, L. F.Topoisomerase II is a structural component of mitotic chromosome scaffolds. J. CellBiol.,109: 1706—1715,1985.
52. Taagepera, S., Ran, P. N., Drake, F. H., and Gorbsky, 0. J. DNA topoisomerase Hais the major chromosome protein recognized by the mitotic phosphoprotein antibodyMPM-2. Proc. Ned. Aced. Sci. USA, 90: 8407-8411, 1993.
53. Per, S. R., Mattern, M. R., Mirabelli, C. K., Drake, F. H., Johnson, R. K., and Crooke,S. T. Characterization of a sublime of P388 leukemia resistant to amsacrine: evidenceof altered topoisomerase II function. Mol. Pharmacol., 32: 17—25,1987.
54. Evans, C. D., Mirski, S. E. L., Danks, M. K., and Cole, S. P. C. Reduced levels oftopoisomerase Ha and H@in a multidrug-resistant lung-cancer cell line. CancerChemother. Pharmacol., 34: 242—248,1994.
55. Brown, 0. A., McPherson, P., Gu, L., Hedley, D. W., Toso, R., Deuchars, K. L.,Freedman, M. H., and Goldenberg, 0. J. Relationship ofDNA topoisomerase Ha and13expressionto cytotoxicityof antineoplasticagentsin humanacutelymphoblasticleukemia cell lines. Cancer Res., 55: 78—82,1995.
56. Chen. M., and Beck, W. T. DNA topoisomerase H expression, stability, and phosphorylation in two VM-26-resistant human leukemic CEM sublines. Oncol. Res., 7:109—117,1995.
57. Drake, F. H., Hofmann, 0. A., Bartus, H. F., Mattern, M. R., Crooke, S. T., andMirabelli, C. K. Biochemical and pharmacological properties ofpl7O and p180 formsof topoisomerase H. Biochemistry, 28: 8154—8160,1989.
58. Petrov, P., Drake, F. H., Loranger, A., Huang, W., and Hancock, R. Localization ofDNA topoisomerase H in Chinese hamster fibroblasts by confocal and electronmicroscopy. Exp. Cell Res., 204: 73—81,1993.
59. Zini, N., Santi, S., Ognibene, A., Ned, L. N., Valmori, A., Mariani, E., Negri, C.,Astaldi-Ricotti, B., and Maraldi, N. M. Discrete localization of different topoisomerases in HeLa and K562 cell nuclei and subnuclear fractions. Exp. Cell Res., 210:336—348,1994.
60. Woessner, R. D., Mattem, M. R., Mirabelli, C. K., Johnson, R. K., and Drake, F. H.Proliferation- and cell cycle-dependent differences in expression ofthe 170 kilodaltonand 180 kilodalton forms of topoisomerase H in NIH-3T3 cells. Cell Growth &Differ., 2: 209—214,1991.
61. Kimura, K., Saijo, M., Ui, M., and Enomoto, T. Growth state- and cell cycledependent fluctuation in the expression of two forms of DNA topoisomerase H andpossible specific modification of the higher molecular weight form in the M phase. J.Biol.Chem.,269: 1173—1176,1994.
62. Yang, C. J., Horton, J. K., Cowan, K. H., and Schneider, E. Cross-resistance tocamptothecin analogues in a mitoxantrone-resistant human breast carcinoma cellline is notdue to DNAtopoisomeraseI alterations.CancerRes.,55: 4004—4009,1995.
63. Kawato, Y., Aonuma, M., Hirota, Y., Kuga, H., and Sato, K. Intracellular roles ofSN-38, a metabolite of the camptothecin derivative CPT-1 1, in the antitumor effect ofCPT-ll.CancerRes.,51:4l87—4l91, 1991.
64. Andoh, T., Ishii, K., Suzuki, Y., Ikegami, Y., Kusunoki, Y., Takemoto, Y., andOkada, K. Characterization of a mammalian mutant with a camptothecin-resistantDNA topoisomerase I. Proc. Nail. Acad. Sci. USA, 84: 5565—5569,1987.
65. Niimi, S., Nakagawa, K., Sugimoto, T., Nishio, K., Fujiwara, Y., Yokoyama, S.,Terashima, Y., and Saijo, N. Mechanism of cross-resistance to a camptothecinanalogue (CPT-l 1) in a human ovarian cancer cell line selected by cisplatin. CancerRes., 52: 328—333,1992.
66. Hsiang, Y., Hertzberg, R., Hecht, S., and Liu, L. F. Camptothecin induces proteinlinked DNA breaks mediated via mammalian DNA topoisomerase I. J. Biol. Chem.,260: 14873—14878,1985.
67. Takeda, S., Shimazoe, T., Sato, K., Sugimoto, Y., Tsuruo, T., and Kano, A. Differential expression of DNA topo I gene between CPT-l 1-acquired and native-resistanthuman pancreatic tumor cell lines detected by RNA/PCR-based quantitation assay.Biochem. Biophys. Res. Common., 184: 618—625,1992.
68. Kijima, T., Kubota, N., and Nishio, K. Establishment of a CPT-l 1-resistant humanovarian cancer cell line. Anti-Cancer Res., 14: 799—803, 1994.
69. Saijo, N., Nishio, K., Kubota, N., Kanzawa, F., Shinkai, T., Karato, A., Sasaki, Y.,Eguchi, K., Tamura, T., Ohe, Y., Oshita, F., and Nishio, M. 7-Ethyl-lO-[4-(l-piperidino)-1-piperidino] carbonyloxy camptothecin: mechanism of resistanceand clinical trials. Cancer Chemother. Pharmacol., 34 (Suppl.): 5112—5117, 1994.
70. Kanzawa, F., Sugimoto, Y., Minato, K., Kasahara, K., Bungo, M., Nakagawa, K.,Fujiwara, Y., Liu, L. F., and Saijo, N. Establishment of a camptothecin analogue(CPT-ll)-resistant cell line of human non-small cell lung cancer: characterization andmechanism of resistance. Cancer Res., 50: 5919—5924,1990.
71. Andoh, T., and Okada, K. Drug resistance mechanisms of topoisomerase I drugs.Adv. Pharmacol., 29B: 93—103,1994.
72. Minato, K., Kanzawa, F., Nishio, K., Nakagawa, K., Fujiwara, Y., and Saijo, N.Characterization of an etoposide-resistant human small-cell lung cancer cell line.Cancer Chemother. Pharmacol., 26: 313—317,1990.
73. Takigawa, N., Ohnishi, T., Ueoka, H., Kiura, K., and Kimura, I. Establishment andcharacterization of an etoposide-resistant human small cell lung cancer cell line. ActaMedics Okayama, 46: 203-212, 1992.
74. Kubota, N., Nishio, K., Takeda, Y., Ohmori, T., Funayama, Y., Ogasawara, H., Ohira,T., Kunikane, H., Terashima, Y., and Saijo, N. Characterization of an etoposideresistant human ovarian cancer cell line. Cancer Chemother. Pharmacol., 34: 183—190, 1994.
75. Sugisnoto, Y., Tsukahara, S., Oh-ham, T., Isoe, T., and Tsuruo, T. Decreasedexpression of DNA topoisomerase I in camptothecin-resistant tumor cell lines asdetermined by a monoclonal antibody. Cancer Rca., 50: 6925—6930,1990.
76. Gupta, R. S., Gupta, R., Eng, B., Lock, R. B., Ross, W. E., Hertzberg, R. P., Caranfa,M. J., and Johnson, R. K. Camptothecin-resistant mutants of Chinese hamster ovarycells containing a resistant form of topoisomerase I. Cancer Res., 48: 6404—6410,1988.
77. Tan, K. B., Mattern, M. R., Eng, W. K., McCabe, F. L., and Johnson, R. K.Nonproductive rearrangement of DNA topoisomerase I and II genes: correlationwith resistance to topoisomerase inhibitors. J. Natl. Cancer Inst., 81: 1732—1735,
1989.78. Leferre, D., Riou, J. F., Ahomadadybe, J. C., Thou, D., Benard, J., and Riou, 0. Study
of molecular markers of resistance to mAMSA in a human breast cancer cell line.Biochem. Pharmacol., 41: 1967—1979,1991.
2583
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

1996;56:2573-2583. Cancer Res Hiroki Kusumoto, Queen E. Rodgers, Friedrich Boege, et al. Topoisomerase IIfor Resistance to Merbarone, a Catalytic Inhibitor of DNA Characterization of Novel Human Leukemic Cell Lines Selected
Updated version
http://cancerres.aacrjournals.org/content/56/11/2573
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/56/11/2573To request permission to re-use all or part of this article, use this link
on June 29, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from