chapter i - information and library network...
TRANSCRIPT

, ...................................................... ,... .... . ..............,... , .,...,.,.,.,.,,. ............................... ,
CHAPTER I

CHAPTER - I
I. Introduction
Copper 1s an essential trace element and the third most abundant transition
metal in the human body following iron and zlnc. It IS second only to iron in its
prevalence in redox active metalloproteins. Copper Ions, as centers of the active slte
of a large number of biologically important metalloproteins play an essential role In
b~ological processes [ l ] i.e. electron transfer, oxldat~on and dioxygen transport.
Based on the function of the protein, the structure of the active site and/or the
spectroscopic behaviour of the copper ions, the copper protelns are classified Into
four types. They are:
Blue copper proteins
Normal copper prote~na
Dinuclear copper proteins
Multlcopper oxidase
From a structural and spectroscopic polnt of view, the three main types of
biologically active copper centers found in the copper proteins may be distinguished
according to a generally accepted convention deriving mainly from their electron
paramagnetic resonance spectra.
Type 1 (TI), have 'blue' copper centers, consist of a large family of electron
transferring proteins w ~ t h a trigonal planar N2S' chromophore, together with one or
two weakly coordinating axial ligands (S and/or 0). The typical spectroscopic
features (e.g. small 41 value and intense blue colour due to a LMCT Cu(II) -t S') are

dominated by the unique, short copper-thiolate bond (2.13 A) [2]. This Cu-S bond
has unusually high 'covalent' character, interpreted as substantial delocalisation of
the S @) rr - orbital with the copper orbitals: the half - filled redox active orbital of
the blue copper site has only 40% d.2.; character [3].
Type 2 (T2), have 'non-blue' copper centers, and similar to common Cu(I1)
coordination complexes containing an N, 0 chromophore with tetragonal symmetry.
T h ~ s class of copper proteins consists of several mononuclear copper proteins, albeit
with very different biochemical functions. Examples include copper-zlnc
superoxide dismutase (Cu-Zn SOD), dopamine-phydroxylase (Dm), phenylalanine
hydroxylase (PhH) and galactose oxidase (GaOx).
Type 3 /T3), have copper dimers. The nitrogens come from histidine groups, the
sulphurs from methionine and cysteine, and the oxygens from a carboxylic acid In
the proteln. Water, hydroxide and alkoxide oxygens are also used.
Both Type I and Type 2 copper proteins are not focus of our interest since
they contain monocopper at their active sites. Hence it is worth to discuss in detail
on Type 3 copper proteins that contain coupled bicopper center at their actwe site.
1.1. Dinuclear Copper Proteins containing the Type-3 site
Three well known coupled copper proteins are hernocyanin, tyrosinase and
catechol oxidase. Due to strong anti-femomagnetic coupling between both Cu(1I)
centers, the oxy-form of a type-3 is EPR silent. (-23 > 600 cm.').

Hemocyanin (Greek for 'blue bloods') function 1s the dioxygen carners for
the mollusks and arthropods [4] The crystal structures of both the oxy and
deoxyfonn of the L~mulus subunlt II hemocyamn have been determined [5, 61 In
the deoxygenated form, the two Cu(1) Ions are ca 4 6 A apart and are both
coordlnated by three ~m~dazole nltrogens from h~stidine res~dues In a tr~gonal
envlronment
F I ~ I 1 Schemat~c draw~ng of the actlve site of hemocyanln and its reactlon w ~ t h 02
In the oxygenated fonn (Fig I 11, the d~oxygen is bound as peroxo (02 ' ) 111 a
4-4 geometry between the two copper Ions, the Cu(I1)-Cu(I1) d~stance 1s reported
to be about 3 6 A The copper Ions are both coordlnated 1n a square pyram~dal
envlronment In the equatorial plane each copper ion is bound to two h~s t~dlne
tutrogens and two-peroxo oxygen atoms At the axlal posltlon each copper Ion is
weakly cwrdlnated by hlstidlne nitrogen Oxyhemocyanin exhlblts two Intense
absorption In the visible reglon, at ca 350 (E = ca 20000 dm3 moll cm ') and at 580

nrn (E = ca. 1000 dm3 m01.'cxn.~), both attributable to a peroxo to Cu(II), ligand to
metal charge-transfer (LMCT) band. The extremely low v(o.0, frequency (ca.750
cm") observed in the resonance Raman spectrum revealed that the 0-0 band is
significantly we&ened by the coordination of the oxygen to the copper ions [7].
The oxy form of the protein is EPR silent, due to a strong antiferromagnetic
interaction between the two copper(I1) ions (-2J > 600 cm").
Tyrosinase
Tyroslnase is a mono-oxygenase found in microorganisms, plants and
animals and catalyses the o-hydroxylation of monophenols to o-diphenols and
further oxidat~on to o-quinones [g]. It is responsible for the chemistry involved in
skin tanning and for the browning reaction observed when mushrooms, potatoes and
fru~ts are injured and exposed to dioxygen. The actlve site of tyrosinase is believed
to be similar to that of hemocyanin, but tyrosinase has the additional feature that the
slte is h~ghly accessible to substrates, which bind directly to the copper center [g]. A
proposed mechanism of tyrosinase catalysis is depicted in Fig. 1.2.
Fig. 1.2. Proposed mechanism of tyrosinase catalysis

Catch01 oxidases are ub~quitous plant enzymes containing a dlnuclea
copper center Without act~ng on tyrosine, they only catalyze the oxidation of a
broad range of o-diphenols to the corresponding o-qumones [lo] coupled w ~ t h the
reduct~on of oxygen to water T h ~ s reactlon is of great importance In rned~cal
diagnosis for the determlnat~on of the hornonally active catecholamines adrenaline,
nonadrenallne and dopa [ l l ] Secondary reactions (melanin formation) follow after
oxidation of the substrate in the presence of polyphenol oxidases, which cause the
brown color of Injured plant [12] The copper In the lsolated catechol oxidases is
found to be EPR s~lent and has been asslgned to an ant~ferrornagnetically spln
coupled Cu(L1)-Cu(I1) pair [13] The proteln part of the W - V i s spectrum of the oxy
catechol ox~dase from Ipomoea batatas exhib~ts an intense absorption band at 343
nm and a weaker band at 580 nrn, corresponding to the peroxo complexes of
hernocyanin and tyrosinase They are asslgned to peroxo + Cu(I1) charge transfer
transitions [14] w ~ t h an 0-0 stretching vtbrat~on band at 749 crn lndicatlng a
posslble p - 4 - $ - bndging mode of the peroxo group XAS lnvestlgatlons on the
nat~ve met forms of catechol oxidases from Lycopus europueus and I Batatus have
revealed that the actlve site conslsts of a dlcopper(I1) center, In which the metal
ztorns are coordinated by four N/O donor l~gands Multiple scanenng EXAFS
calculations have shown hlgh significance for one or two coordlnat~ng hlstldine
residues [15] The short metal-metal dlstance of 2 9 A and the results of EPR
~nvestigations indlcate a p - hydroxo bndged dicopper(I1) active site in the met
forms of the proteins [16]

1.2. Need for Synthetic Models
Over the past decade, much progress has been made in the structural
characterization of copper proteins by spectroscopic techniques, X-ray analysis and
site-directed mutagenesis [17]. However, specific reaction pathways and
mechanisms of reactlon often remain unclear. In order to understand their catalytic
mechanisms and their unusual spectroscopic characteristics, the development of
relatively simple, synthetic models of the active site structures of copper proteins
therefore remains of considerable interest [18].
1.3. Significance of Synthetic Models
Low molecular we~ght model compounds can be examined easler than the
metalloenzyme itself, active site analogues can help to elucidate details of the
active site of many metalloenzymes.
Once a structural model is obtained, this may lead to the development of a
coordination complex with reactivity properties similar to that of the native
metalloprotelns: a functional model.
Low molecular weight synthetic model compounds can be obtained relatively
easy in large amounts, which is often not the case for native metalloproteins.
Moreover, synthetic models are relatively easy to modify, allowing optlmizat~on
of ligand-design in terms of reactivity and selectivity, in contrast to the
metalloenzyme itself.

1.4. Literature Overview
Model complexes for dinuclear copper proteins can be prepared using
dinucleating ligands, designed to accommodate two copper ions in close proximity.
The term "dinucleating ligand" is introduced in 1970 by Robson [I91 to describe the
class of polydentate chelating ligands, able to bind simultaneously two metal ions.
Since then, a very large number of such ligands are designed and their coordination
compounds are thoroughly investigated. The possible applications of the complexes
with this type of ligands vary from modeling the active sites of many metallo
enzymes [20-221 to hosting and canying small molecules [23-251 or catalysis [26,
271.
Among many different types of dinucleating ligands, the phenol based
compartmental ligands attracted particularly w ~ d e attention of scientists. The term
"compartmental" is introduced to indicate a ligand containing two adjacent, similar
or dissimilar coordination sites [20]. Particular interest in the ligands having distinct
donor sets has resulted from the recent recognition of the asymmetric nature of a
number of dimetallic hiosites [28, 291.
Understanding the ability of individual metal ions to play possibly different
functions in dinuclear sites In metalloenzymes lead to the design of a great number
of symmetric and asymmetric dinucleating ligands, where two compartments would
provide a same or different coordination sumounding for the two metal ions. An
extraordinary number of multidentate dinucleating phenoxide ligands have been
studied, and a representative collection of these types of ligands is listed in Table.
1.1-3.

Table I. 1 . Syrnmctncal Saturated Dinucleahng L~gands
L~gands Reference Ligands Reference
(:I ""0 [31, 341
I I R- CN Br
v
)$, [361 h f ~
R R
VI1
R = CI, R'=CH, R - CH,. R'= CI
IW a:. ".:D

Table 1.1 Symmetrical Saturated Dinucleating Ligands (Continued)
Ligands Reference Ligands Reference
XI11 XIV
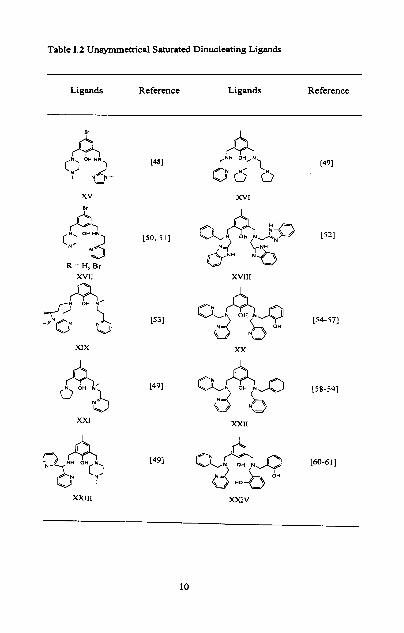
Table 1.2 Unsymmetrical Saturated Dinucleating Ligands
Ligands Reference Ligands Reference
XIX XX
A 0 OH Q 1 aNAa [1&591
XXI
(=j N b
XXIl
Xxlll XXIV

Table 1.3 Unsymmemcal Unsaturated Dinucleating Ligands
L~gands Reference Ligands Reference
xxv XXVI
A, "2
XXIX
Y
Y - OK NH,
XXVIIl
XXX
1651 N, OH d
I

1.5. Dicopper(II) Complexes as Structural and Functional Models for Type3
Copper Proteins
T.N. Sorrel1 et al. [67], one of the pioneers in the study of copper proteins,
have attempted to make a model for the active site of oxidized hemocyanin
derivatives. They synthesized and X-ray crystallographically characterized both p-
o-acetato and p-1,3-azido copper(I1) complexes (A) with a Cu-Cu separation >3.5 A.
X= OAc, N,
(A)
Variable temperature magnetic susceptibility measurements demonstrate strong
antiferromagnetic coupling (2J = -1800 cm.') for the azido derivative but negligible
magnetic interaction between the copper ions in the acetato bridged complex.
K.D. Karlin et al. [68] reported on the synthesis, structural and spectroscopic
comparisons of analogous phenolate and X (where X = OH, C1', Br', OBz', OAc-)
loubly bridged dinuclear copper (11) complexes (B), which act as a suitable model
For met-hernocyanin derivatives. Stmctural comparison reveal the presence of a
larger X atom results in distortion away from pure square pyramidal geometry with
an opening of the Cu-0-Cu bridging angle resulting in a greater Cu-Cu separation.
Temperature dependent magnetic measurements of the structurally characterized
complexes reveal that the halide bridged complexes are the least (25 = -335 cm")

strongly coupled and the OK bridged complex is the most strongly coupled (21
-600 em.'), while the azide complex falls in between (25 = -440 cm-').
X = N;, CI., Br, OBz, OAc-
(B)
On the basis of the C U - O ~ ~ ~ ~ ~ ~ ~ ~ - C U angles, the coupling should increase in the
d~rection OH. C: p-l,l-N; < Bi but the opposite trend is actually observed and is
attnbuted to the modulating effect of the exogenous bridges.
Rajendiran et al. [69] have studied the synthesis, spectral and
electrochemical behaviour of dicopper(I1) complex ( C ) derived from pentadentate
dinucleating ligand. Two acetates bridge the two coppers through basal and apical
pos~tions to provide the square pyramidal geometry around the distorted square base.
The variable temperature magnetic studies reveal weak antiferromagnetic interaction
(2J = -93 cm") between the two metal centers. The cyclic voltammetric behaviour
of the complexes shows the quasireversible nature of the process.

Belle et al. [70] have studied the PH induced changes of redox, specboscopic,
structural properties and catecholase activity of the dicopper(I1) complexes (D) and
(E). They also correlated the PH dependent catalytic abilities of the complexes with
changes in the coordination sphere of the metal centers. p-hydroxo bridged
complexes ffom HLC~], HLF and bH3 exhibit a catecholase activity.
R- F, CF,, OCH,
Modification of R-substituent induces a drastic effect on the catecholase activity; the
presence of an electron-donating group on the ligand increases this activity, whereas
the reverse effect is observed with an electron withdrawing group.
J.D. Crane et al. [71] have reported the systematic design, synthesis and
characterization of symmetrical and unsymmetrical dinuclear copper(I1) complexes
(F, G and H) that can be viewed as first generation models for the dinuclear copper
centers in metalloproteins and enzymes.

J. Reim and B. Krcbs [72] have studied the synthesis of a series of
symmetrical and unsymmetrical dinuclear copper(II) complexes (I and J) as a
ptential structural and functional models for the active site of catechol oxidase.
The cyclic voltammogram reveal irreversible nature of the redox process and are
attributed to changes in the coordination geometry or coordination number upon
change of the oxidation state even to the expulsion of metal ions from the
coordination sphere. Investigation of the catecholase activity of compounds shows
that these complexes have significant catalytic activity with respect to the aerial
oxidation of 3, 5-DTBC to its corresponding o-quinone. But no clear relationship
between the electrochemical properties of the complexes and the catecholase activ~ty
exists.
The symmetrical dicopper(I1) complexes (K) with exogenous bridging motifs
like OAc, OH and Br have been synthesized and studied by Kandaswamy et al. [73].
They reported that the complexes undergo quasireversible reduction steps at
negative potential. The reduction potential is exogenous donor dependent and
follows the order O M e OH> Br. The -2J value ofthe complexes calculated !?om

variable temperature magnetic moment also follows the same trend as the
:lactnx.hernical data.
R = CH,, R' = CI, CH, R = CI, CH, R = H, CH,
R"= CH,, C,Hl, R' = CH,, C,Hll X = OMe, OH, Br X = OH, Br, OAc
(K) (L) (M)
Eventhough, the study on unsymmetrical dicopper(l1) complexes is sparse,
Kandaswamy et al. also succeeded in synthesizing the complexes (L) of the same
with OH, Br and OAc as the bridging units [74]. The exogenous dependent
reduction potential is observed and it follows the order OH > Br > OAc. The lower
-21 value observed in these complexes reveal the reduction in electron density and
the distorted geometry around the copper center. The interesting feature noticed in
the complex (M) [9] is that the CufJI) ion present in the oxime compartment reduces
at a lesser potential compared to the other Cu(I1) ion present in the piperazinc
compartment because of the oxime compartment has three imino nitrogen
coordinating sites.
Fenton and coworkers [76] have studied the crystal structure of the
complexes (N and 0) . The coordination environment at each Cu atom is a square
pyramidal. The magnetic moment of complex (N) is 1.85 PB at room temperature

and the moment is practically independent of temperature down to liquid N2
temperature.
On the other hand, the magnetic moment of ( 0 ) is subnormal at room temperature
(1.19 p8 per Cu) and decreases with decreasing temperature to 0.63 peat 82 K. The
results suggest the operation of an antiferromagnetic interaction between the two
Cu(1I) ions. The complexes are also differenttated by cyclic voltammetic studies.
The facile reduction of (N) relative to ( 0 ) certainly relates to the square pyramidal
geometry about the copper atoms in (N) and the involvement of soft Br- anion. The
reduction of the Cu atoms of (0) is more difficult because the square planar
environment about the metals in an unfavorable geometric environment for Cu(1).

1.6. Scope of thia Thesis
In o rda to understand the structure and functions of the actlve site m proteln
units, it is necessary to synthesize discrete molecular systems whose metal
environmmt is similar to that of the active sites of proteins. Binuclear copper
complexes arc found to mimic the active site of the type-3 copper proteins. Besides
m~m~cking the active site, these binuclear copper(I1) complexes are found interesting
due to its significance and application in the fields of magneto chemistry, catalysis,
matenal science, superconductivity and redox chemistry.
One way to mimic the dinuclear slte of type-3 copper proteins with model
compound is to make use of ligand systems, which can bind two metal Ions and
bring them together. Our aim is to understand the influence of ligand on the
structure, spectral, electrochemical, magnetic and catalytic propemes. Hence we
have focused on the building block for the ligand and their corresponding
complexes. End-off compartmental hgands arc generally used slnce they provlde
d~stlnct coordination environments. Many of this subgroup binucleating ligands are
derived from a 2, 6-disubstituted phenol. Ligands of this type strongly favour the
formation of dimetallic species because of the enforced ideal distance between the
donor sets and the presence of the endogenous bridging phenolate group. Moreover,
they also provide space for the coordination of one or two exogenous bndgiilg
llgand to the metal center.
The goal of research descnbed in this thesis is the synthesis and
characterization of symmehical and unsymmetrical binucleating ligands and thelr
dicopper(I1) complexes with exogenous bridging motifs.

The research approach described in this thesis can be divided into 6 chapters.
A literature overview of a number of multidentate symmemcal and unsymmetrical
monophmoxide dinucleating tigands and their dicopper(I1) complexes which act as
a potential structural and functional models for the active site of type-3 copper
proteins are discussed in chapterl. In chapter 2, various instmentation techniques,
which are employed to characterize the ligands and to study the properties of the
complexes, are discussed. Chapter 3 describes the synthesis and studies of
unsymmetrical saturated binucleating ligands and their dicopper (II) complexes.
Synthesis and studies of unsymmetrical saturated binucleating ligands and their
dicopper(I1) complexes arc discussed in chapter 4. In chapter 5, synthesis, spectral,
electrochemical, magnetic and catecholase activity of dicopper(I1) complexes
derived from unsymmetrical unsaturated ligands are discussed. Chapter 6 contains
general conclusions and suggestions for further research and ends with the list of
publications that are resulted from this thesis.

1. E.I. Solomon, B.L. Hemming and D.E. Root, in Bioinorganic Chemistry of
copper, K.D. Karlii and Z. Tyeklar (Eds.), Chapman & Hall, London, 1993,
Metal ions in Biological systems, H . Sigel (Ed.), Marcel Dekker Inc., New
York, 1981, 13.
2. (a) E.I. Solomon and M.D. Lowery, Sclence, 1993,259, 1575.
(b) J.A. Guckert, M.D. Lowery and E.I. Solomon, ' Am. Chem. Soc., 1995,
117,2817.
3. S. Larson, A. Broo and L.J. Sjolin, Phys. Chem., 1995, 99, 4860.
4. E.I. Solomon, M. J. Baldwin and M.D. Lowery, Chem. Rev , 1992,92,521.
5. K.A. Magnus, H. Ton-that and J. Carpenter, Chem. Rev., 1994, 94, 727.
6. N. Kitajima and Y. Moro-oka, Chem. Rev., 1994, 94, 737.
7. E.I. Solomon, M.J. Baldwin and M.D. Lowery, Chem. Rev., 1992,92,521.
8. N . Kitajima and Y. Moro-oka, Chem. Rev., 1994, 94, 737.
9 N. Kitajima and Y. Moro-oka, Chem. Rev., 1994,94, 737.
10. M. Tremolicres and J.B. Bieth, Phylochem~st~ . 1984, 23, 501.
11. D. Meiwes, B. Ross, M. Kiesshauer, K. Cammann, H. Witzel, M. Knoll, M.
Borchardt and C. Sandermaier, Lab. Med., 1992, 15, 24
12. K. Lerch, Enrym. Brown. Prev., 1995, 600,64.
13. A. Rompel, H. Fischer, K. Buldt-Karentzopoulos, D. Meiwes, F. Zippel,
H.H. Nolting, C. Hermes, B. Krebs and H. Witzel, J. Inorg. Biochem., 1995,
59, 715.
14. (a) N.C. Eickman, R.S. Himrncrwright and E.I. Solomon, Proc. Acad. Sci.
USA. 1979,76,2094.

@) E.I. Solomon, M.J. Baldwin and M.D. Lowny, Chem. Rev., 1992, 92,
521.
15. F. Zippel, F. Ahlcrs, B. Krebs, S. Behning, K. Buldt-Karcnt~~poulos, H.
Witzel and M. Oversluizen, Daresbury Annual Report, D a r e s b q
Laboratory, Daresbury, UK, 1994,95, 102.
16 B. Krebs, K. Buldt-Karentzopoulos, C E~cken, A. Rompel, H. Wltzel, A.
Feldrnann, R. Kruth, J. Reim, W. Stemforth, S. Te~pel, F. Z~ppel, S.
Schindler and F. Wiesemann, in: DFG Deutsche Forschungsgemeinschaft
(Ed.), Bioinorganzc Chemistry: Transztzon Metals m Biology and thezr
Coordination Chemistry, D.14, VCH, Welnheim, 1997, p. 616.
17. W. Kaim and J. Rall, Angew. Chem. Int, Ed Engl., 1996, 35,43.
18. N. Kitaj~ma and Y. Moro-oka, Chem. Rev., 1994, 94,737.
19. R. Robson, Inorg. Nucl. Chem. Lett, 1970, 6 , 125.
20 D.E. Fenton, Inorg. Chem. Commun, 2002,5,537.
21. K.D. Karlin, J.C. Hayes, Y. Gultnch, R.W. Cruse, J.W Mckown, J.P
Hutchinson and J. Zubieta, J. Am. Chem. Soc., 1984,106,2121.
22. E. Lambea, B. Chabut, S. Chardon-Noblat, A. Deronz~er, G. Chpttard, A
Bousseksou, J.P. Tuchagues, J. Laug~er, M. Bardet and J.M. Latour, J. Am
Chem. Soc., 1997,119,9424.
23. N.N. Murthy, M. Mahroof-Tahir and K.D. Karlin, Inorg. Chem., 2001, 40,
628.
24. F. Meyer and P. Rutsch, Chem. Commun., 1998, 1037.
25. M. Suzuki, H. Knatomi and I. Murase, Chem. let!., 1981, 1745.
26. P. Gamez, J. Von Harras, 0 . Roubeau, W.L. driesscn and 1. Reedjik, Inorg.
Chtm. Acta., 2001, 324, 27.

27. S. Torelli, C. Belle, I. Gautier-Luneau, J.L. Piem, E. Saint-Aman, J.M.
Latour, L. Lc paper and D. Luneau, Inorg. Chem., 2000.39.3526.
28. T. Klabunde, C. Eicken, J.C. Sacchcttini and B. b b s , Nut. struct. Biol. 5
(dec), 1998, 1084.
29. E.I. Solomon, U.M. Sundaram and T.E. Machonkin, Chem. Rev., 1996, 96,
2563.
30. R, Gupta, S. Mukherjee and R. Mukherjee, J. Chem. Soc. Dalton Trans.,
1999,4025.
31. J. Reim and B. Krebs, J. Chem. Soc. Dalton Trans., 1997, 3793.
32. (a) C.K. Williams, N.R. Brooks, M.A. Hillmycr and W.B Tolman, J. Chem.
Soc Chem. Commun., 2002,2132.
(b) N.V. Kaminskaia, B. Spingler, and S.J. Lippard, J Am. Chem. Soc. ,
2000, 122, 6411.
33 (a) B.P. Murch, P.D. Boyle and L. JT. Que, J Am Chem Soc., 1985, 107,
6728.
(b) B.P. Murch, F.C. Bradley, P.D. Boyle, V. Papaefthymiou and L. Jr. Que,
J Am Chem. Soc., 1987,109,7993.
34 T.M. Rajendiran, R. Venkatesan, P.S. Rao and M. Kandaswamy,
Polyhedron, 1998,17,3427.
35. M.S. Mashuta, R.J. Webb, J.K. Mccushker, E.A. Schmit, K.J. Oberhausen,
J.F. Richardson, R.M. Buchanan and D.N. Hendrickson, J. Am Chem So=.,
1992,114,3815.
36. P. Amudha, M. Kandaswamy, L. Govindaswamy and D. Velmumgan, Inorg.
Chem., 1998,37,4486.

37. (a) M.A. de Brito, A.J. Bortoluzzi, A. Gnatti, A.S. Ceccato, A.C. Joussef
and S.M. Drechsel, Acta C?ystallogr., Sect. C , 2000, C56, 1188.
@) V.D. Campbell, E.J. Parson and W.T. P d g t o n , Inorg. Chem., 1993,
32, 1773.
38. T.N. Sorrell, C.0' Conner, O.P. Anderson and J.H. Reibenspies, J. Am.
Chem. Soc., 1985,107,4199.
39. (a) S. Uhlenbrock and B. Krcbs, Angew Chem. Int. Ed. Engl., 1992, 31,
1647.
@) H.P. Berends and D.W. Stephen, Inorg Chem., 1987.26, 749.
40. A.J. Bortoluzzi, A. Neves, I. Vencato, C. Zucco and M. Homer, Acta
Crystallogr. Sect. C, 1999, C55, 1634.
(b) B. Krebs, K. Schepers, B. Bremer, C Henkel, E. Althaus, W. Muller-
Warmuth, K. Griesar and W. Haase, Inorg Chem., 1994,33, 1907.
41. T. Ookubo, H. Sug~moto, T. Nagayama, H. Masuda, T. Sato, K. Tanka,
Maeda, H. Okawa, Y . Hayashi, A. Uehara and M.J. Suzuki, J . Am. Chem.
Soc.. 1996, 118, 701.
42. K.D. Karlin, R.W. Cruse, Y Gultneh, A. Farooq, J.C. Hayes and J.J.
Zubieta,
J. Am. Chem. Soc., 1987,109,2668.
43 K. Schepers, B. Bremer, B. Krebs, G. Henkel, E. Althaus, B. Mosel and W.
Muller-Wannuth, Angew.Chem Inr. Ed. Engl., 1990,29, 531.
44. A.S. Borovik, M.P. Hendrich, T.R. Holman, E. Munch, V. Papaefiymiou
and L. Jr. Que, J. Am. Chem. Soc., 1990,112,603 1.
45. M. Suzuki, H. Kanatomi and I. Murase, Bull. Chem Soc. Jpn., 1984, 57, 36.

46. S. Torclli, C. Belle, I. Gauticr-Luneau, S. Hamman and J.L. Pierre, Inorg.
Chrm. Acta. 2002,333, 144.
47. H.-R.Chang, H. Diril, M.J. Nilges, X. Zhang, J.A. Potenza, H.J. Schugar,
D.N. Hmdrikson, S.S. Isied, J, Am. Chem. Soc., 1988, 110, 625.
48. J. Reim and B. Krebs J. Chem. Soc.. Dalton Trans., 1997,3793.
49. M. Lubben and B.L. Feringa, J. Org. Chem., 1994,59,2227.
50. J.D. Crane, D.E. Fenton, J.M. Latour, A.J. Smith, J. Chem. Soc. Dalton
Trans., 1991,2979.
51. M. Lubben, R. Hage, A. Meetsma, K. Byma and B.L. Feringa, Inorg. Chem.,
1995,34,2217.
52. P. Kamaras, M.C. Cajulis, M. Rapta, G.A. Brewer and G.B. Jameson, J Am.
Chem Soc., 1994,116, 10334.
53. N.N. Murthy, M.M. Tahir and K.D. Karl~n, Inorg Chem., 2001, 40.
54. P. Karsten, A. Neves, A.J. Bortoluzzi, J. Strahle and C. Maichle-Mossmer,
Inorg. Chem. Commun., 2002, 5 , 434.
55. P. Karstcn, A. Neves, A.J. Bonoluzzi, M. Lanznaster and V. Drago, Inorg
Chem., 2002,41,4624.
6 . M. Lamaster. A. Neves, A.J. Bonoluwi, B. Szpoganicz and E. Schwingel,
Inorg. Chem., 2002,41,5641.
7 . E . Lambert, B. Chabut, S. Chardon-Noblat, A. Deronzler, G. Chottard,
A.Bousseksou and J.M. Tuchagues, J. Am. Chem. SOC., 1997,119, 9424.
8. W . Kanda, W. Moneta, M. Bardet, E. Bernard, N. Debaecker, J. Laugier,
A. Bousseksou, S. Chardon-Noblat and J. -M. Latour, Angau. Chem. Int.Ed,
Engl., 1995, 34, 588.

59. L. Dubois, D. -F. Xiang, X-S. Tan, J. Pccaut, P. Jones. S. Baudron, L. Le
Pape, J-M. Latour, C. Baffert, S. Chardson-Noblat, M. -N. Collomb and A.
Dcronzier, Inorg. Chem., 2003,42, 750.
60. C. Belle, I. Gautier-Luneau, J. -L. Pierre and C. Scheer, Inorg. Chem., 1996,
35,3706.
61. C. Belle, I. Gautier-Luneau, L. Kannazin, J. -L. Pierre, S. Albedyhl, B. Krebs
and M. Bonin, Eur, J. horg . Chem., 2002, 3087.
62. H. Adams, S. Clunas and D.E. Fenton, Inorg. Chem. Commun., 2001,4,667.
63. H. Adams, D.E. Fenton, S.R. Haque, S.L. Heath, M. Ohba, H. Okawa and
S.E. Spey, J. Chem. Soc. Dalton Trans, 2000, 1849.
64. H. Adams, D.E. Fenton, P.E. McHugh and T. Potter, J. Inorg. Chim. Acta,
2002,331,117.
65. H. Adams, S. Clunas, D.E. Fenton and D. N. Towers, J. Chem. Soc. Dalton
Trans., 2002, 3933.
66. J.D. Crane, D.E. Fenton, J.M. Latour and A.J. Smith, J. Chem. Soc. Dalton
Trans., 1991,2979.
67. T.N. Sorrell, C.J. 0' Comer, 0 . P Anderson and J.H. Reibenspies, J Am.
Chem. Soc., 1985,107,4199.
68. K.D. Karlin, A. Farooq, J.C. Hayes, B.I. Cohen, T.M. Rowe, E. Sinn and J .
Zubieta, Inorg. Chem., 1987,26, 1271.
69. T. M. Rajmdiran, R. Kannappan, R. Venkatesan, P. Sarnbasiva Rao and M.
Kandaswamy, Polyhedron, 1999,18, 3085.
70. C. Belle, C. Beguin, I. G. Luneau, S. Hamman, C. Philouze, J.L. Pierre, F.
Thomas and S. Torelli, Inorg. Chem., 2002, 41,479.
71. J.D. Crane, D.E. Fenton, J.M. Latour and A.J. Smith, J. Chem. Soc. Dalton

Trans., 1991,2979.
72. J. Reim and B. Krebs, J. Chem. Soc. Dalton Trans., 1997,3793.
73. P . Amudha, M. Kandaswamy, L. Govindaswamy and D. Velmumgan, Inorg.
Chem., 1998,37,4486.
74. P. Amudha, M. Thinunavalavan and M. Kandaswamy, Polyhedron, 1999,18,
1363.
75. D. Saravanakumar, N. Sengottuvelan, G. Priyadarshini, M. Kandaswamy and
H. Okawa, Polyhedron, 2003, article in press.
76. H. Adams, D.E. Fenton, S.R. Haque, S.L. Heath, M. Ohba, H. Okawa and E.
Spey, J. Chem. Soc. Dalton Trans., 2000, 1849.