calcium sensing receptor absence delays … sensing...calcium sensing receptor absence delays...
TRANSCRIPT

Calcium Sensing Receptor Absence Delays Postnatal BrainDevelopment via Direct and Indirect Mechanisms
Xiu-Ling Liu & Yu-Shan Lu & Jun-Ying Gao &
Charles Marshall & Ming Xiao & Deng-Shun Miao &
Andrew Karaplis & David Goltzman & Jiong Ding
Received: 16 January 2013 /Accepted: 18 March 2013 /Published online: 7 April 2013# Springer Science+Business Media New York 2013
Abstract Calcium sensing receptor (CaSR) is implicated inthe establishment of neural connections and myelin forma-tion. However, its contribution to brain development re-mains unclear. We addressed this issue by analyzing brainphenotype in postnatal CaSR null mice, a model of humanneonatal severe hyperparathyroidism. One- and 2-week-old CaSR null mice exhibited decreased brain weightand size with a developmental delay in expression ofproliferating cell nuclear antigen. Neuronal and glialdifferentiation markers, neuronal specific nuclear pro-tein, glial fibrillary acidic protein, and myelin basicprotein, were also decreased compared with age-matched wild-type littermates. Moreover, deletion ofthe parathyroid hormone gene that corrects hyperpara-thyroidism, hypercalcemia, hypophosphatemia, andwhole-body growth retardation normalized brain cell pro-liferation, but not differentiation, in CaSR null mice. Culturedneural stem cells (NSCs) derived from the subventricular zonesof CaSR null neonatal mice exhibited normal proliferationcapacity but decreased differentiation capacity, compared withwild-type controls. These results demonstrate that direct effectsof CaSR absence impair NSC differentiation, while secondary
effects of parathyroid hormone-related endocrine abnormalitiesimpair NSC proliferation, both of which contribute to delayedbrain development in CaSR null newborn mice.
Keywords Calcium sensing receptor . Parathyroidhormone . Gene knockout . Neural stem cells . Braindevelopment
Introduction
The extracellular calcium-sensing receptor (CaSR), amembrane spanning G protein-coupled receptor, playsa central role in maintaining systemic calcium homeo-stasis, predominately through negative-feedback regula-tion of parathyroid hormone (PTH) secretion [1–3].Furthermore, CaSR is implicated in cellular prolifera-tion, differentiation, or apoptosis in a variety of celltypes such as fibroblasts [4], vascular smooth musclecells [5], colon cells [6], and osteoblasts [7, 8].
There are increasing evidences indicating that CaSR isalso involved in various functions of neurons and glial cells(for a review, see [9]). Immunohistochemical analysis re-veals that CaSR is localized to nerve terminals through-out the adult rat brain [10]. In situ hybridizationdemonstrates that transcripts of CaSR and myelin basicprotein (MBP) share similar patterns of developmentalexpression in the postnatal rat brain [11]. An in vitrostudy indicates that CaSR participates in the differenti-ation of neural stem cells (NSCs) into oligodendrocytes[12]. Moreover, CaSR has been shown to regulate axo-nal growth of sympathetic neurons and dendritic growthof hippocampal pyramidal neurons isolated from fetalmouse brain [13]. Combined, these findings suggest acrucial role of CaSR in brain development, but direct invivo evidence is, however, lacking.
Xiu-Ling Liu and Yu-Shan Lu contributed equally to this work.
X.-L. Liu :Y.-S. Lu : J.-Y. Gao :M. Xiao (*) :D.-S. Miao :J. Ding (*)Department of Anatomy, Nanjing Medical University, Nanjing,Jiangsu 210029, People’s Republic of Chinae-mail: [email protected]: [email protected]
C. MarshallDepartment of Rehabilitation Sciences, University of KentuckyCenter for Excellence in Rural Health, Hazard, KY 41701, USA
A. Karaplis :D. GoltzmanDepartment of Medicine, McGill University, Montreal H3A 1A1QC, Canada
Mol Neurobiol (2013) 48:590–600DOI 10.1007/s12035-013-8448-0

In the present study, we employed newborn CaSR null(CaSR−/−) mice, a mouse model of human neonatal severehyperparathyrodism [14, 15] to address the contribution ofCaSR to mouse brain development. Moreover, the previousstudies suggest that secondary hyperparathyroidism mainlycauses bone abnormalities, delayed growth, and immaturedeath in CaSR−/− mice, which can be rescued by geneticablation of PTH [16, 17]. Therefore, to discern direct effectsof CaSR deficiency from secondary effects of hyperparathy-roidism on brain development, we compared brain pheno-types of CaSR−/− mice, CaSR−/−/PTH−/− mice, and wild-type (WT) littermates at 7 and 14 days after birth. We alsocultured NSCs derived from the subventricular zones (SVZ)of CaSR−/− and WT newborn mice and compared theirproliferation and differentiation capabilities. Our findingsreveal that both direct and indirect mechanisms contributeto brain developmental abnormalities in CaSR−/− mice.
Materials and Methods
Generation and Genotyping of Mice
The generation of the two parental strains of CaSR−/− andPTH−/− mice by homologous recombination in embryonicstem cells has been previously described [15, 18]. Briefly,fertile CaSR+/− and PTH+/− mice were mated to produceCaSR+/−/PTH+/− mice, which were then mated to generateCaSR−/−/PTH−/−, CaSR−/− mice, and WT pups. The geno-type of offspring mice was identified using polymerasechain reaction, as previously described [8, 17]. All animalexperimental protocols were conducted in accordance withinternational standards on animal welfare and the guidelinesof the Institute for Laboratory Animal Research of NanjingMedical University (approval ID 2008-00518).
Biochemical and Hormone Analyses
Serum calcium and phosphorus levels were measured using anautoanalyzer (Synchron 67, Beckman Instruments), and intactserum PTH was detected by a two-site immunoradiometricassay (Immutopics, San Clemente, CA, USA) [8, 17].
Brain Tissue Preparation
CaSR−/−, CaSR−/−/PTH−/−, and WT littermates weresacrificed under anesthesia by quick decapitation, at postna-tal days 7 and 14 (P7 and P14). Heads were immersed into4 % paraformaldehyde for 2 days, and then brains weredissected out and post-fixed in 4 % paraformaldehyde foranother 2 days. After dehydration by gradient alcohol,brains were embedded in paraffin and cut into series sagittalor coronal sections at 5 μm.
Nissl’s Staining
Paraffin-embedded brain sections were stained in 0.5 %cresyl violet for 10 min following dewaxing and rehydra-tion. After thorough washing with distilled water, brainsections were placed in acetic acid buffer for 2 min.Tissues were then dehydrated in a graded series of alcoholand vitrification in xylene. The Nissl stained sagittal brainsections were photographed using a digital microscope(Leica Microsystems, Wetzlar, Germany). The number ofbrain cells in the cerebral cortex per section and the crossareas of the cerebellum were quantitated using NorthernEclipse image analysis software (Empix Imaging, Inc.,New York, NY, USA).
Immunohistochemical Staining
Immunohistochemical protocols for paraffin tissue sectionshave been described previously [19]. Briefly, brain sectionswere incubated with the following primary antibodies:mouse anti-proliferating cell nuclear antigen (PCNA;1:500, Abcam), mouse anti-neuron-specific nuclear protein(NeuN; 1:1,000, Millipore), mouse anti-glial fibrillary acidicprotein (GFAP; 1:1,000, Millipore), mouse anti-MBP(1:1,000, Sigma), or mouse anti-parvalbumin (PV; 1:2,500,Millipore), overnight at 4 °C. After PBS rinsing, the sectionswere incubated with biotinylated goat anti-mouse or rabbit(1:400) for 1 h at room temperature and visualized using thediaminobenzidine visualization method (Elite ABC Kit,Vector, Burlingame, CA, USA).
Western Blotting
Proteins extracted from brains or cultured cells were loadedone per lane of sodium dodecyl sulfate–polyacrylamide gelelectrophoresis and separated in an electric field. After elec-trophoretic transfer onto polyvinylidene fluoride member(Millipore), immunoblotting was carried out as previouslydescribed [7, 20] using antibodies against NeuN (1:1,000),GFAP (1:1,000), MBP (1:5,000), PV (1:2,500), and β-tubulin (1:500; Santa Cruz). Bands were visualized usingECL chemiluminescence (Amersham) and quantitated byScion Image Beta 4.02 (Scion Corporation).
NSC Culture and Neurosphere Assay
Three, 4-day-old newborn CaSR−/− and WT pups were usedas cell donors. The SVZ was dissected and plated at 1×104
cells per well in an uncoated 24-well culture plate. NSCswere cultured in serum-free growth medium consisting ofDMEM/F12 (1:1) (Gibco), supplemented with 2 % B27(Gibco), 20 ng/ml EGF, and 20 ng/ml bFGF (Peprotech) toform neurospheres [20]. The number and diameter of the
Mol Neurobiol (2013) 48:590–600 591

neurospheres in each well were counted after 7 days in vitro(IV). Self-renewal was also assayed by dissociatingneurospheres in bulk and reculturing the cells at a constantdensity of 1×104 cells per well. The number and size ofsecondary or tertiary spheres were determined after 7 daysIV. Neurospheres were counted under an optical micro-scope, with a minimum cutoff of 40 μm in diameter [20].The diameter of randomly chosen neurospheres was deter-mined using Image-Pro Plus 6.0 Analysis System (MediaCybernetics Inc.)
Cell Proliferation Assays
Proliferation was determined using 5-bromodeoxyuridine(BrdU) incorporation [20]. The secondary neurosphereswere collected and gently mechanically dissociated.Dissociated cells were plated on 24-well culture platescoated with poly-L-ornithine (10 mg/ml) and laminin(5 μg/ml) and cultured for 48 h. Cells were incubatedwith 10 μM BrdU and different concentrations of CaCl2(baseline, 3 and 5 mM) for 24 h, then fixed andwashed. DNA was denatured by treating the cells for30 min with 2 N HCl at 37 °C. After another washing andblocking in 5 % bovine serum albumin for 30 min, thecoverslips were incubated with the primary mouse anti-BrdU antibody (1:15,000, Chemicon) for 1 h at 37 °Cfollowed by goat anti-mouse FITC antibody (1:200,Chemicon). The cells were then counterstained with Hoechst33342 (Sigma) and mounted with anti-fade medium.
NSC Differentiation Assays
The secondary neurospheres were triturated chemically andmechanically into cell suspension and plated on coatedcoverslips at a density of 2×105 cells/ml in DMEM/F12medium supplemented with 1 % FBS, but without EGFand bFGF. After 6 or 10 days IV, cells were probed withmouse anti-neuron-specific beta-3 tubulin (TUJ1; 1:2,000,Sigma), mouse anti-GFAP (1:800, Millipore), or rabbit anti-galactocerebroside (GalC; 1:1,000, Millipore) antibody. Thepercentages of TUJ1, GFAP, and GalC-positive cells to thetotal number of cell nuclei labeled with Hoechst 33342 wereanalyzed. In addition, neurite length per TUJ1-positive neu-ron and cross area of GalC-positive oligodendrocytes weremeasured using Image-Pro Plus 6.0 Analysis System.
TUNEL Assays
Apoptosis of NSCs was detected by TUNEL assay using the InSitu Cell Death Detection Kit (Roche Diagnostics GmbH,Mannheim, Germany). After washed with PBS and fixed by4 % paraformaldehyde, NSCs on coverslips were incubatedwith 20 μg/ml proteinase K for 10 min at room temperature,
followed by incubation in a TUNEL reaction mixture for60 min at 37 °C. After rinsing in PBS, randomly selected areasof the coverslips were examined by with a DIC-fluorescencemicroscope (Carl Zeiss Microimaging Inc., Germany). Thepercentage of TUNEL-positive cells was counted.
Statistics
The data are expressed as mean ± SEM. Comparisons amongCaSR−/−, CaSR−/− /PTH−/−, and WT groups were performedby using one-way ANOVA, followed by the Student–Newman–Keuls tests. Significance was accepted at P<0.05.
Results
Deletion of PTH Significantly Rescues Abnormal BrainMorphogenesis of the Postnatal CaSR−/− Mice
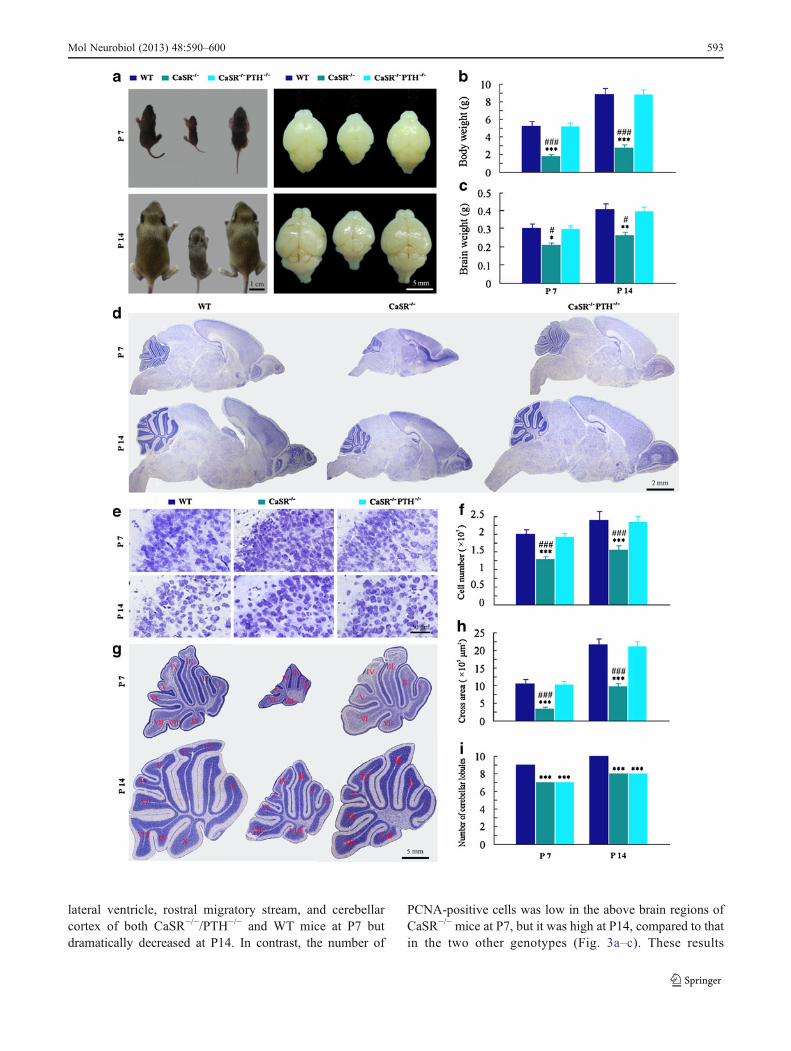
Initially, we addressed the interaction between CaSR and PTHon brain morphogenesis during postnatal development. P7 andP14CaSR−/−mice exhibited lower body and brain weights thantheir WT littermates, and deletion of PTH in CaSR−/− miceresulted in normalization of both body and brain weights(Fig. 1a–c). Nissl stained sagittal sections of P7 and P14CaSR−/− brain revealed small brain size with micropolygyriaand undifferentiated cytoarchitecture of the cerebral and cere-bellar cortices compared with WT controls (Fig. 1d, e, g). Cellcounting showed that the mean cell number per section of thecerebral cortex was low in CaSR−/− mice, compared withCaSR−/−/PTH−/− mice and their WT mice (Fig. 1f). Deletionof PTH in CaSR−/− mice significantly rescued the abnormalshape and structure of brain; however, the mild hypoplasia ofthe cerebellar lobules was still observed (Fig. 1h, i).
Deletion of PTH Completely Corrects Hyperparathyroidismand Accompanying Hypercalcemia and Hypophosphatemia
Serum biochemical analysis revealed that P14 CaSR−/− micedisplayed hypercalcemia, hypophosphatemia, and increased se-rum PTH compared to WT controls, whereas CaSR−/−/PTH−/−
mice exhibited normocalcemia, hyperphosphatemia, andundetectable serum PTH (Fig. 2a–c). These results are consistentwith previous reports [8, 16, 17].
Deletion of PTH Normalizes Neural Stem/PrecursorProliferation in the Postnatal CaSR−/− Mice
To determine whether decreased brain weight and size inCaSR−/− mice was associated with impaired proliferation ofneural stem/progenitor cells, the developmental expressionof PCNA in the forebrain and cerebellum was investigated.PCNA-positive cells were densely present in the SVZ of the
592 Mol Neurobiol (2013) 48:590–600
lateral ventricle, rostral migratory stream, and cerebellarcortex of both CaSR−/−/PTH−/− and WT mice at P7 butdramatically decreased at P14. In contrast, the number of
PCNA-positive cells was low in the above brain regions ofCaSR−/− mice at P7, but it was high at P14, compared to thatin the two other genotypes (Fig. 3a–c). These results
Mol Neurobiol (2013) 48:590–600 593

revealed a prolonged time course of neural stem/precursorproliferation in the postnatal CaSR−/− mice, which wasnormalized by PTH gene knockout.
Deletion of PTH Does Not Normalize Neural Stem/Precursor Differentiation in the Postnatal CaSR−/− Mice
To determine whether abnormal brain shape and structure inCaSR−/− mice was also related to altered neural stem/precursordifferentiation, postnatal developmental expression of a neuro-nal marker NeuN, an astrocyte marker GFAP, and an oligoden-drocyte marker MBP was examined in the forebrain andcerebellum by immunohistochemistry and immunoblotting.NeuN, GFAP, and MBP immunostainings were clearly ob-served in the telencephalon and cerebellum of WT mice at P7and further increased at P14. The immunostaining density ofthe above markers was significantly reduced in the CaSR−/−
brain. Deletion of PTH in CaSR−/−mice resulted in increases inthe immunoreactivities of NeuN, GFAP, and MBP, which,however, were generally weak relative to WT mice (Figs. 4aand 5a). Consistently, western blotting revealed that NeuN,GFAP, and MBP levels increased in the cerebellum and fore-brain of P7 and P14 CaSR−/−/PTH−/− mice relative to CaSR−/−
mice, but they were still lower thanWTmice, except for GFAPexpression in the forebrain at P14 (Figs. 4b, c and 5b, c). Inaddition, immunohistochemical and western blotting analysisof PV, a marker for GABAergic neurons, showed that Purkinjecell dendritic differentiationwas higher in P14CaSR−/−/PTH−/−
mice than CaSR−/− mice, but it was still lower than WTlittermates (Fig. 6a–c).
Deletion of CaSR Decreases DifferentiationBut Not Proliferation of Cultured NSCs In Vitro
In order to explore the direct influence of CaSR absence onneural cell proliferation and differentiation, NSCs derived formthe SVZ of newborn CaSR−/− mice were isolated and cultured.Neurosphere-forming assays revealed that the primary, second-ary and tertiary neurospheres from CaSR−/− mice were similarin number and diameter compared with those from WT mice(Fig. 7a–c). BrdU incorporation assay also revealed there wasno significant difference in the percentage of BrdU-positivecells between CaSR−/− and WT NSCs (Fig. 7d, e).
We further addressed the effects of CaSR deficiency ondifferentiation capability of NSCs. After 6 days cultured inthe differentiated medium, all WT NSCs had differentiatedinto glia or neurons, while about 30 % of CaSR−/− NSCsremained undifferentiated as revealed by analysis of thepercentages of TUJ1, GFAP, and GalC-positive cells(Fig. 8a, b). After 10 days, the percentages of differentiatedneuronal and glial cells were no different between the twogenotype groups (Fig. 8b). However, neurite length perTUJ1-positive neuron and cross area of GalC-positive oli-godendrocytes were lower in CaSR−/− group than WT groupat 6 and 10 days after inducing differentiation (Fig. 8c, d).Collectively, these in vitro results demonstrated that CaSRdeficiency had no effect on NSC proliferation and self-renew, but did impair NSC differentiation.
High Calcium Inhibits Proliferation and Increases Apoptosisof Cultured CaSR−/− NSCs
The above results demonstrated that CaSR deficiencyresulted in a prolonged time course of neural stem/precursorproliferation in vivo (Fig. 3), but had no effect on NSCproliferation in vitro (Fig. 8a, b). These data highly suggestthat abnormal neural stem/precursor proliferation inCaSR−/− mice is not due to direct effects of the CaR alter-ations, but due to secondary effects of hyperparathyroidism-related endocrine alterations including hypercalcemia. Inorder to prove this presumption, we observed the effects ofdifferent concentrations of CaCl2 on the proliferation ofCaSR−/− NSCs. As expected, CaSR−/− NSCs cultured with
Fig. 2 Serum biochemicalanalysis of 14 days old WT,CaSR−/−, and CaSR−/−/PTH−/−
littermates. a Serum calcium.b Serum phosphorus. c SerumPTH. Each value is the mean ±SEM of five mice per group.**P<0.01; ***P<0.001 vs.WT mice. ###P<0.001 vs.CaSR−/−/PTH−/− mice
�Fig. 1 Brain morphogenesis of WT, CaSR−/−, and CaSR−/−/PTH−/−
littermates at postnatal days 7 and 14 (P7 and P14). a–c Overallphenotypes of the whole body and brain (a), measurements on bodyweight (b), and brain weight (c). d Representative micrographs of Nisslstained sagittal brain sections. e High magnification showing thecytoarchitecture of the cerebral cortex. f Counts of brain cells in thecerebral cortex per sagittal sections. g High magnification showing thesize and shape of cerebellum. h The cross areas of the cerebellum. iThe number of cerebellar lobules. I-X represents ordinal number ofcerebellar lobules. Each value is the mean ± SEM of five mice pergroup. *P<0.05; **P<0.01; ***P<0.001 vs. WT mice. #P<0.05;###P<0.001 vs. CaSR−/−/PTH−/− mice
594 Mol Neurobiol (2013) 48:590–600
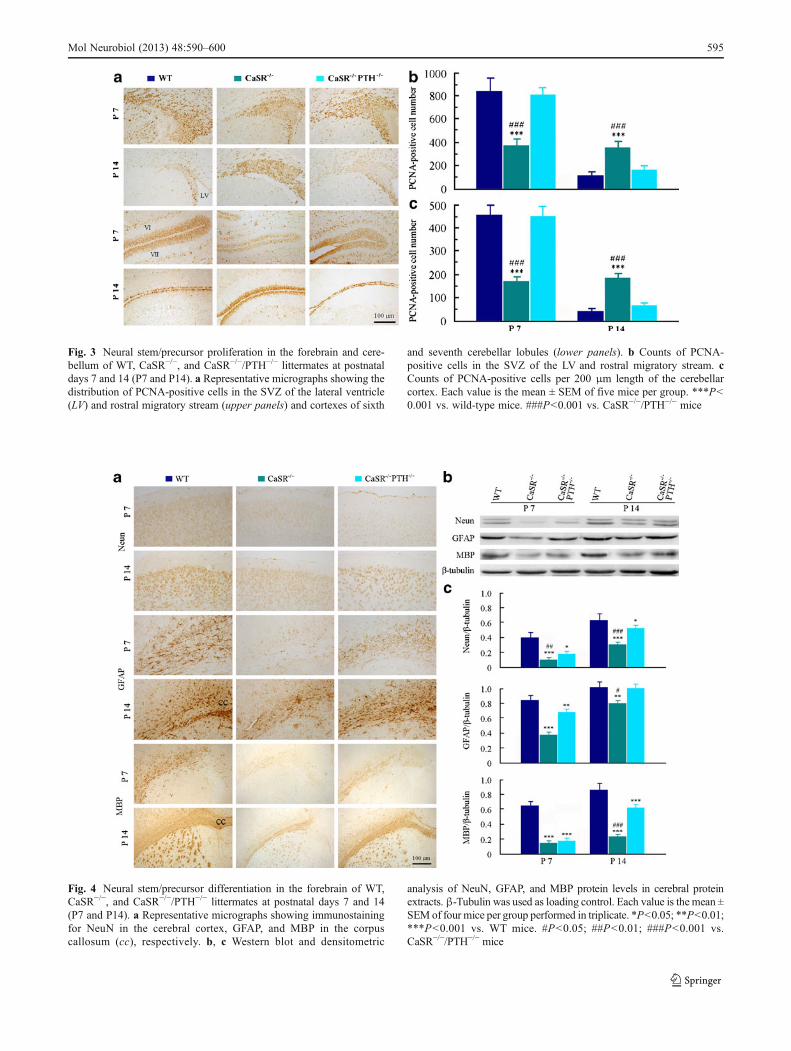
Fig. 3 Neural stem/precursor proliferation in the forebrain and cere-bellum of WT, CaSR−/−, and CaSR−/−/PTH−/− littermates at postnataldays 7 and 14 (P7 and P14). a Representative micrographs showing thedistribution of PCNA-positive cells in the SVZ of the lateral ventricle(LV) and rostral migratory stream (upper panels) and cortexes of sixth
and seventh cerebellar lobules (lower panels). b Counts of PCNA-positive cells in the SVZ of the LV and rostral migratory stream. cCounts of PCNA-positive cells per 200 μm length of the cerebellarcortex. Each value is the mean ± SEM of five mice per group. ***P<0.001 vs. wild-type mice. ###P<0.001 vs. CaSR−/−/PTH−/− mice
Fig. 4 Neural stem/precursor differentiation in the forebrain of WT,CaSR−/−, and CaSR−/−/PTH−/− littermates at postnatal days 7 and 14(P7 and P14). a Representative micrographs showing immunostainingfor NeuN in the cerebral cortex, GFAP, and MBP in the corpuscallosum (cc), respectively. b, c Western blot and densitometric
analysis of NeuN, GFAP, and MBP protein levels in cerebral proteinextracts. β-Tubulin was used as loading control. Each value is the mean ±SEM of four mice per group performed in triplicate. *P<0.05; **P<0.01;***P<0.001 vs. WT mice. #P<0.05; ##P<0.01; ###P<0.001 vs.CaSR−/−/PTH−/− mice
Mol Neurobiol (2013) 48:590–600 595

3 or 5 mM CaCl2 for 24 h showed a decrease in thepercentage of BrdU-positive cells compared with those inbaseline controls (1 mM CaCl2) (Fig. 9a, b). Moreover,CaSR−/− NSCs treated with high concentrations of CaCl2showed a high percentage of apoptosis (Fig. 9a, c). Thesedata suggest that calcium regulates NSC proliferation andapoptosis independent of CaSR.
Discussions
Since the identification of CaSR in 1993, its role in regulat-ing systemic calcium homeostasis has been extensivelystudied [1–3]. The contribution of this receptor to reproduc-tion, development, and birth has been acknowledged grad-ually in recent years [21]. The involvement of CaSR in
Fig. 6 GABAergicdifferentiation in the cerebellumof 14 days oldWT, CaSR−/−, andCaSR−/−/PTH−/− littermates.a Representative micrographsshowing parvalbumin (PV)-positive cerebellar Purkinjecells in the cerebellum. b, cWestern blot anddensitometric analysis of PVprotein levels in cerebellarprotein extracts. β-Tubulinwas used as loading control.Each value is the mean ± SEM offour mice per group performed intriplicate. **P<0.01; ***P<0.001 vs. WT mice. #P<0.05 vs.CaSR−/−/PTH−/− mice
Fig. 5 Neural stem/precursor differentiation in the cerebellum of WT,CaSR−/−, and CaSR−/−/PTH−/− littermates at postnatal days 7 and 14(P7 and P14). a Representative micrographs showing immunostainingfor NeuN, GFAP, and MBP in the cerebellum. b, c Western blot anddensitometric analysis of NeuN, GFAP, and MBP protein levels in
cerebellar protein extracts. β-Tubulin was used as loading control.Each value is the mean ± SEM of four mice per group performed intriplicate. *P<0.05; **P<0.01; ***P<0.001 vs. WT mice. #P<0.05;##P<0.01; ###P<0.001 vs. CaSR−/−/PTH−/− mice
596 Mol Neurobiol (2013) 48:590–600

establishment of neural connections and myelin formationhas also been addressed by a few studies [12, 13]. In thisstudy, for the first time, we provide direct in vivo evidencefor a necessary role of CaSR in postnatal brain developmentby analyzing brain phenotype of newborn CaSR−/− mice.Moreover, by generation of CaSR−/−/PTH−/− mice, we areable to separate direct effects of CaSR deficiency fromsecondary effects caused by hyperparathyroidism on braindevelopment. Our data suggest that the direct effect of CaSRdeficiency impairs brain cell maturation, rather than prolif-eration, during postnatal development. Consistently, in vitrodata further reveal that CaSR regulates differentiation, butnot generation and proliferation, of NSCs.
It is known that rodent brain development is not fullycompleted until approximately 4 weeks after birth [22, 23].Particularly, during postnatal weeks 1–2, a large numbers ofNSCs undergo robust proliferation and differentiation tomature neurons and glial cells, resulting in a dramatic in-crease in brain volume and size [24]. The postnatal CaSR−/−
mice show a prolonged time course of neural stem/precursorproliferation and small brain weight and size compared with
WT littermates from the same brood. However, decreasedbrain weight and delayed brain cell proliferation in CaSR−/−
mice can be totally normalized by backcrossing onto thePTH-deficient background. This finding indicates that CaSRis not involved in the proliferation of NSCs during postnataldevelopment. Further supporting this, in vitro data show thatthere are no significant differences in the proliferation andself-renew capabilities of the SVZ-derived NSCs from new-born CaSR−/− and WT mice. In agreement with the presentfindings, previous studies confirm that there is a very lowexpression of CaSR mRNA in cultured NSCs and in rat ormouse brain within a few days of postnatal life [11, 12, 25].All these evidences suggest that CaSR does not play acritical role in the proliferation of NSCs.
In agreement with our previous studies [8, 17], deletionof PTH corrects hyperparathyroidism, hypercalcemia,hypophosphatemia, and whole-body growth retardation inCaSR−/− mice. Moreover, P7 and 14 CaSR−/−/PTH−/− miceshow normal brain size and weight, although accompaniedwith the mild hypoplasia of the cerebellar lobules, comparedwith their WT littermates. Thus, we believe that retardation
Fig. 7 Self-renewal and proliferation capabilities of SVZ-derivedNSCs from newborn WT and CaSR−/− mice. a–c Neurosphere assay.Representative micrographs (a) and measurements on the number (b)and diameter (c) of the primary, secondary, and tertiary neurospheres
after 7 days in culture. d, e BrdU incorporation assay. Representativemicrographs (d) and measurements on the percentage (e) of BrdU-positive NSCs. Each value is the mean ± SEM of three independentexperiments
Mol Neurobiol (2013) 48:590–600 597
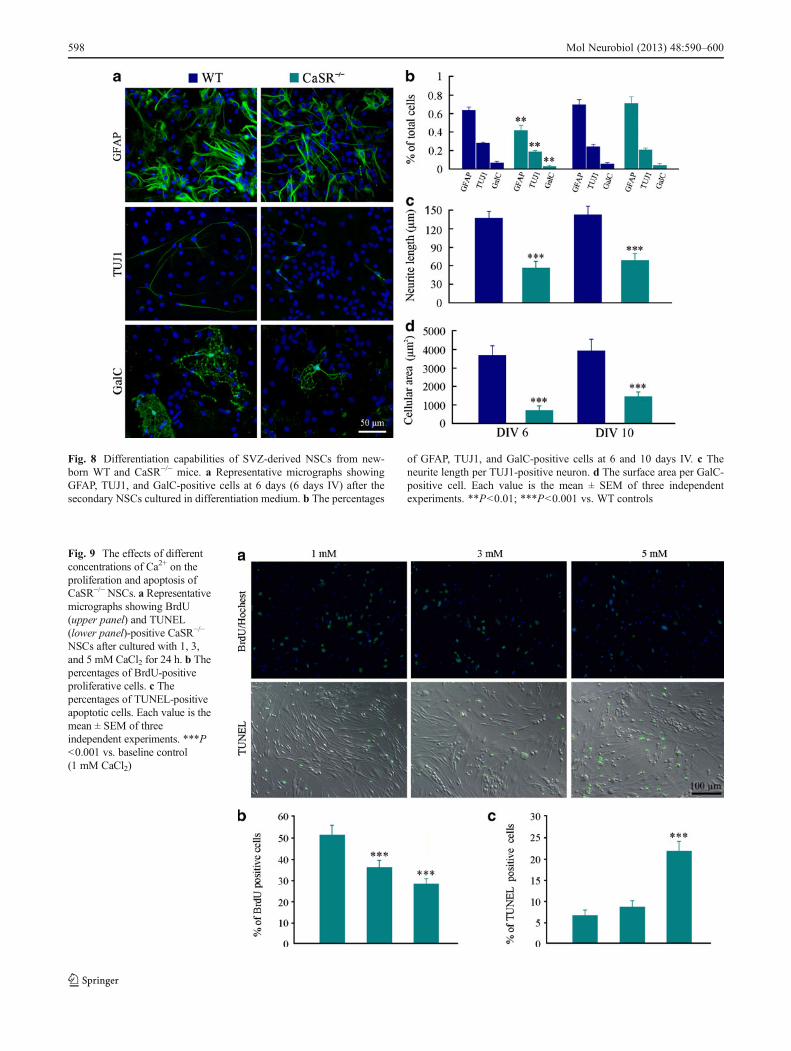
Fig. 9 The effects of differentconcentrations of Ca2+ on theproliferation and apoptosis ofCaSR−/−NSCs. a Representativemicrographs showing BrdU(upper panel) and TUNEL(lower panel)-positive CaSR−/−
NSCs after cultured with 1, 3,and 5 mM CaCl2 for 24 h. b Thepercentages of BrdU-positiveproliferative cells. c Thepercentages of TUNEL-positiveapoptotic cells. Each value is themean ± SEM of threeindependent experiments. ***P<0.001 vs. baseline control(1 mM CaCl2)
Fig. 8 Differentiation capabilities of SVZ-derived NSCs from new-born WT and CaSR−/− mice. a Representative micrographs showingGFAP, TUJ1, and GalC-positive cells at 6 days (6 days IV) after thesecondary NSCs cultured in differentiation medium. b The percentages
of GFAP, TUJ1, and GalC-positive cells at 6 and 10 days IV. c Theneurite length per TUJ1-positive neuron. d The surface area per GalC-positive cell. Each value is the mean ± SEM of three independentexperiments. **P<0.01; ***P<0.001 vs. WT controls
598 Mol Neurobiol (2013) 48:590–600

in brain cell proliferation in CaSR−/− mice is likely due tothe above-mentioned secondary effects of CaSR deficiency.This presumption is supported by our in vitro results show-ing that high calcium inhibits proliferation and increasesapoptosis of NSCs independent of CaSR. Indeed, calciumhomeostasis is crucial for development and survival of alltypes of cells in the central nervous system [26]. Ca2+
overload resulting from dysregulation of channels andpumps leads to death of neurons as well as glial cells[27–29]. In addition, PTH receptors are widely distributedin rodent brain, and PTH is able to cross the blood–brainbarrier, suggesting that serum PTH may affect brain devel-opment and function [30–34]. Clinical studies indicate thathigh serum PTH level is closely related to cognitive mal-function in elderly patients with primary hyperparathyroid-ism, and the associated dementia is often alleviated afterparathyroidectomy [35–38]. Moreover, whole-body growthretardation, especially feeding inability, affects nutrient ab-sorption, protein, and DNA synthesis and subsequentlyhampers brain development. Taken together, the present invivo and in vitro evidences support the idea that impairmentof brain cell proliferation in CaSR−/− mice is due to thesecondary effects, rather than a direct consequence ofCaSR deficiency.
The present results have demonstrated that CaSR is in-volved in the differentiation and maturation course of NSCs.Previous studies have revealed that expression of CaSRmRNA coincides with the developmental pattern of MBPexpression in the rat brain [11]. Reduced MBP levels in thecerebellum of 2-week-old CaSR−/−mice suggest an importantrole of CaSR in oligodendrocyte myelination [12]. In addition,it has been shown that CaSR mRNA expression rises in thehippocampus at a time when long-term potentiation can firstbe induced, indicating a possible role for CaSR in synapticplasticity [39]. A further study by Vizard et al. [13] revealedthat CaSR is necessary for axonal and dendritic growth ofcultured neurons. In agreement with these findings, the pres-ent study has shown that CaSR deficiency prolongs the timecourse of cultured NSCs differentiating into neurons, astro-cytes, and oligodendrocytes. Consistently, the in vivo datahave shown that, even after correction of hyperparathyroidismby knockout of PTH, decreases in NeuN, GFAP, and MBPexpression still occurs in the forebrain and cerebellum of P7and P14 CaSR−/− mice.
It should be mentioned that Kos and colleagues haveshowed that CaSR−/−/PTH−/− mice survive to adulthood[16]. These adult mice are indistinguishable from their con-trol littermates with respect to size, body weight, and be-havior, despite a much wider range of values for serumcalcium and renal excretion of calcium. These data indicatethat the abnormal NSC differentiation in CaSR−/−/PTH−/−
mice might ameliorate or even restore the normal levels inthe late stages of postnatal development. In agreement with
this view, our in vitro data have shown that about 30 % ofCaSR−/− NSCs remain undifferentiated at 6 days after in-ducing differentiation, but these cells can differentiate intoneuronal or glial cells at 10 days. Further studies are neces-sary to define the long-term interaction of CaSR and PTHon brain phenotype using older newborn or adultCaSR−/−/PTH−/− mice.
In summary, the present results reveal that delayed braindevelopment in CaSR−/− mice results from the direct effectsof CaSR absence and the secondary effects of parathyroidhormone-related endocrine abnormalities that impair NSCdifferentiation and proliferation, respectively. These find-ings also suggest that additional in vitro and in vivo studiesare necessary to remove secondary or compensatory effects,so direct effects of the gene deletion on animal phenotypescan be identified.
Acknowledgments This work was supported by grants from theNational Nature and Scientific Foundation of China (no. 81271210and no. 81230009), PAPD Foundation of Jiangsu Higher EducationInstitutions, and Qing Lan Project. We would like to thank ProfessorEdward Brown in the Brigham and Women’s Hospital, Harvard Med-ical School for providing CaSR+/− mice. We thank Dr. Hui Kong forexcellent work on culturing neural stem cells.
References
1. Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, KiforO, Sun A, Hediger MA, Lytton J, Hebert SC (1993) Cloning andcharacterization of an extracellular Ca(2+)-sensing receptor frombovine parathyroid. Nature 366:575–580
2. Brown EM, MacLeod RJ (2001) Extracellular calcium sensing andextracellular calcium signaling. Physiol Rev 81:239–297
3. Caudarella R, Vescini F, Buffa A, Rizzoli E, Ceccoli L, FrancucciCM (2011) Role of calcium-sensing receptor in bone biology. JEndocrinol Invest 34:13–17
4. McNeil SE, Hobson SA, Nipper V, Rodland KD (1998) Functionalcalcium-sensing receptors in rat fibroblasts are required for activa-tion of SRC kinase and mitogen-activated protein kinase in re-sponse to extracellular calcium. J Biol Chem 273:1114–1120
5. Molostvov G, Fletcher S, Bland R, Zehnder D (2008) Extracellularcalcium-sensing receptor mediated signalling is involved in humanvascular smooth muscle cell proliferation and apoptosis. CellPhysiol Biochem 22:413–422
6. Whitfield JF (2009) The calcium-sensing receptor–a driver ofcolon cell differentiation. Curr Pharm Biotechnol 10:311–316
7. Shu L, Ji J, Zhu Q, Cao G, Karaplis A, Pollak MR, Brown E,Goltzman D, Miao D (2011) The calcium sensing receptor medi-ates bone turnover induced by dietary calcium and parathyroidhormone in neonates. J Bone Miner Res 26:1057–1071
8. Sun W, Sun W, Liu J, Zhou X, Xiao Y, Karaplis A, Pollak MR,Brown E, Goltzman D, Miao D (2010) Alterations in phosphorus,calcium and PTHrP contribute to defects in dental and dentalalveolar bone formation in calcium-sensing receptor-deficientmice. Development 137:985–992
9. Bandyopadhyay S, Tfelt-Hansen J, Chattopadhyay N (2010)Diverse roles of extracellular calcium-sensing receptor in the cen-tral nervous system. J Neurosci Res 88:2073–2082
Mol Neurobiol (2013) 48:590–600 599

10. Ruat M, Molliver ME, Snowman AM, Snyder SH (1995) Calciumsensing receptor: molecular cloning in rat and localization to nerveterminals. Proc Natl Acad Sci U S A 92:3161–3165
11. Ferry S, Traiffort E, Stinnakre J, Ruat M (2000) Developmentaland adult expression of rat calcium-sensing receptor transcripts inneurons and oligodendrocytes. Eur J Neurosci 12:872–884
12. Chattopadhyay N, Espinosa-Jeffrey A, Tfelt-Hansen J, Yano S,Bandyopadhyay S, Brown EM, de Vellis J (2008) Calcium recep-tor expression and function in oligodendrocyte commitment andlineage progression: potential impact on reduced myelin basicprotein in CaR-null mice. J Neurosci Res 10:2159–2167
13. Vizard TN, O'Keeffe GW, Gutierrez H, Kos CH, Riccardi D, DaviesAM (2008) Regulation of axonal and dendritic growth by the extra-cellular calcium-sensing receptor. Nat Neurosci 11:285–291
14. Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ,Steinmann B, Levi T, Seidman CE, Seidman JG (1993)Mutations in the human Ca(2+)-sensing receptor gene cause fa-milial hypocalciuric hypercalcemia and neonatal severe hyper-parathyroidism. Cell 75:1297–1303
15. Ho C, Conner DA, Pollak MR, Ladd DJ, Kifor O, Warren HB,Brown EM, Seidman JG, Seidman CE (1995) A mouse model ofhuman familial hypocalciuric hypercalcemia and neonatal severehyperparathyroidism. Nat Genet 11:389–394
16. Kos CH, Karaplis AC, Peng JB, Hediger MA, Goltzman D,Mohammad KS, Guise TA, Pollak MR (2003) The calcium-sensing receptor is required for normal calcium homeostasis inde-pendent of parathyroid hormone. J Clin Invest 111:1021–1028
17. Liu J, Lv F, Sun W, Tao C, Ding G, Karaplis A, Brown E,Goltzman D, Miao D (2011) The abnormal phenotypes of cartilageand bone in calcium-sensing receptor deficient mice are dependenton the actions of calcium, phosphorus, and PTH. PLoS Genet 7:e1002294
18. Miao D, He B, Karaplis AC, Goltzman D (2002) Parathyroidhormone is essential for normal fetal bone formation. J ClinInvest 109:1173–1182
19. Hua X, Lei M, Zhang Y, Ding J, Han Q, Hu G, Xiao M (2007)Long-term D-galactose injection combined with ovariectomyserves as a new rodent model for Alzheimer's disease. Life Sci80:1897–1905
20. Kong H, Fan Y, Xie J, Ding J, Sha L, Shi X, Sun X, Hu G(2008) AQP4 knockout impairs proliferation, migration andneuronal differentiation of adult neural stem cells. J Cell Sci121:4029–4036
21. Riccardi D, Kemp PJ (2012) The calcium-sensing receptor beyondextracellular calcium homeostasis: conception, development, adultphysiology, and disease. Annu Rev Physiol 74:271–297
22. Tramontin AD, García-Verdugo JM, Lim DA, Alvarez-Buylla A(2003) Postnatal development of radial glia and the ventricularzone (VZ): a continuum of the neural stem cell compartment.Cereb Cortex 13:580–587
23. Hu F, Strittmatter SM (2004) Regulating axon growth within thepostnatal central nervous system. Semin Perinatol 28:371–378
24. Peretto P, Merighi A, Fasolo A, Bonfanti L (1999) Thesubependymal layer in rodents: a site of structural plasticity and
cell migration in the adult mammalian brain. Brain Res Bull49:221–243
25. Rogers KV, Dunn CK, Hebert SC, Brown EM (1997) Localizationof calcium receptor mRNA in the adult rat central nervous systemby in situ hybridization. Brain Res 744:47–56
26. Leclerc C, Néant I, Moreau M (2012) The calcium: an early signalthat initiates the formation of the nervous system during embryo-genesis. Front Mol Neurosci 5:3
27. Alberdi E, Sánchez-Gómez MV, Matute C (2005) Calcium andglial cell death. Cell Calcium 38:417–425
28. Beck A, Nieden RZ, Schneider HP, Deitmer JW (2004) Calciumrelease from intracellular stores in rodent astrocytes and neurons insitu. Cell Calcium 35:47–58
29. Bondarenko A, Svichar N, Chesler M (2005) Role of Na+-H+ andNa+-Ca2+ exchange in hypoxia-related acute astrocyte death. Glia49:143–152
30. Hashimoto H, Aino H, Ogawa N, Nagata S, Baba A (1994)Identification and characterization of parathyroid hormone/para-thyroid hormone-related peptide receptor in cultured astrocytes.Biochem Biophys Res Commun 200:1042–1048
31. Usdin TB, Gruber C, Bonner TI (1995) Identification and func-tional expression of a receptor selectively recognizing parathyroidhormone, the PTH2 receptor. J Biol Chem 270:15455–15458
32. Eggenberger M, Flühmann B, Muff R, Lauber M, LichtensteigerW, Hunziker W, Fischer JA, Born W (1996) Structure of a para-thyroid hormone/parathyroid hormone-related peptide receptor ofthe human cerebellum and functional expression in human neuro-blastoma SK-N-MC cells. Brain Res Mol Brain Res 36:127–136
33. Pang PK, Kaneko T, Harvey S (1988) Immunocytochemical dis-tribution of PTH immunoreactivity in vertebrate brains. Am JPhysiol 255:R643–R647
34. Wang T, Palkovits M, Rusnak M, Mezey E, Usdin TB (2000)Distribution of parathyroid hormone-2 receptor-like immunoreac-tivity and messenger RNA in the rat nervous system. Neuroscience100:629–649
35. Ohrvall U, Akerström G, Ljunghall S, Lundgren E, Juhlin C,Rastad J (1994) Surgery for sporadic primary hyperparathyroidismin the elderly. World J Surg 18:612–618
36. Tilvis RS, Kähönen-Väre MH, Jolkkonen J, Valvanne J, PitkalaKH, Strandberg TE (2004) Predictors of cognitive decline andmortality of aged people over a 10-year period. J Gerontol ABiol Sci Med Sci 59:268–274
37. Papageorgiou SG, Christou Y, Kontaxis T, Bonakis A,Anagnostouli M, Potagas C, Kalfakis N (2008) Dementia aspresenting symptom of primary hyperparathyroidism: favourableoutcome after surgery. Clin Neurol Neurosurg 110:1038–1040
38. Jorde R, Waterloo K, Saleh F, Haug E, Svartberg J (2006)Neuropsychological function in relation to serum parathyroid hor-mone and serum 25-hydroxyvitamin D levels. The Tromsø study. JNeurol 253:464–470
39. Chattopadhyay N, Legradi G, Bai M, Kifor O, Ye C, Vassilev PM,Brown EM, Lechan RM (1997) Calcium-sensing receptor in the rathippocampus: a developmental study. Brain Res Dev Brain Res100:13–21
600 Mol Neurobiol (2013) 48:590–600