business plan 2013-2014 - gov.uk€¦ · 2013-14 business plan the medicines and healthcare...
TRANSCRIPT

Business Plan 2013-2014

2 of 25

0 of 25
2013-14 Business Plan The Medicines and Healthcare Products Regulatory Agency is an Executive Agency of the
Department of Health and a government trading fund, with a mission to protect and improve
the health of millions of people every day through the effective regulation of medicines and
devices, underpinned by science and research.
April 2013
Contents 1. Introduction 2. Financial context 3. Core functions 4. Strategic activities for 2013-14
4.1 – NIBSC activities for 2013-14 4.2 – CPRD activities for 2013-14 Annex A – Table of core function targets for 2013-14 Annex B – Table of strategic activities for 2013-14

1. Introduction This is the first annual business plan for the new 2013-18 Corporate Plan1 of the Medicines and Healthcare Products Regulatory Agency. This plan has been prepared in the context of a particularly critical year in the development of the Agency as we enter our new Corporate Plan period. The Corporate Plan summarises the key risks and opportunities facing us as a whole over the next five years, but the next year’s business will be taken forward within the following key points of context: • From April 2013, the full enlargement of the Agency to cover both the Clinical Practice
Research Datalink (CPRD) and the National Institute for Biological Standards and Control (NIBSC). This enlargement significantly strengthens our capability and capacity – both in the regulation of the new generation of biological products, and in supporting clinical trial activity in the UK and building the use of real world data in support of the MHRA’s core regulatory functions.
• Real challenges in our operating environment, at a time when expectations and regulatory
activities are growing. In both medicines and devices work, we are facing reductions in income, increases in activity (particularly in relation to vigilance) and continued gaps in funding of critical EU activities, notably in relation to pharmacovigilance.
• A large amount of EU-driven activities, including major negotiations of new legislation
relating to clinical trials and devices regulation, and substantial efforts needed to implement new requirements in areas such as falsified medicines and pharmacovigilance;
• New top management. A new Chair, Sir Gordon Duff, took up post at the beginning of
2013, and a new Chief Executive is to be appointed in the first half of the business year as a result of the planned retirement of Sir Kent Woods.
The main focus of this annual business plan is to set out our key actions that will contribute to the strategic objectives established in the 2013-18 Corporate Plan. These are: • work to define the role and added value of the regulator – notably in building alliances
with healthcare professionals and patient groups to ensure that our work as a regulator, and the data and information that we generate and use in our work in relation to medicines and devices, informs and influences clinical practice
• work to contribute to the Government’s wider Life Sciences and Growth agendas, notably
through helping innovation in both the medicines and devices sector into use as quickly as safely possible
• strengthening the vigilance of medicines and devices once they are in use – including by
exploring how data and information about real-world use can be established and used in a more systematic manner, thereby supplementing traditional signals-based vigilance systems
• strengthening the security and oversight of the supply chains of the two highly globalised
industries that we regulate 1 The 2013-18 Corporate Plan is available at www.mhra.gov.uk.
1 of 25

• ensuring that we manage our business in a highly efficient and effective manner, and
continue to offer a high quality service that provides value for money to the industries that we regulate.
This plan focuses on the critical strategic and developmental work that we will be undertaking in these key areas in the coming year. It does not set out all of our work – much of our core work is properly the subject of specific, internal business plans for our divisions and centres. For completeness the next section summarises the key functions of our enlarged organisation, and other aspects of our core business are summarised in the specific targets at annex A. The remainder of this plan is structured as follows: • Section 2: summary of the financial and operating information for the organisation,
including the budget for 2013-14. • Section 3: summary of core functions of the enlarged organisation • Section 4: summary narrative of the Agency’s key strategic activities for 2013-14.
- 4.1: summary narrative with a list of NIBSC-specific activities. - 4.2: summary narrative with a list of CPRD-specific activities.
• Annex A: specific targets relating to the core functions of the enlarged MHRA’s business, particularly chosen to reflect aspects of our business where the industries we regulate have a choice about whether to use the UK regulator or another EU regulator.
• Annex B: table showing the key strategic activities, with outputs, referred to in narrative form in section 4.
2 of 25

2. Financial context We are a Government Trading Fund. We derive all of our income for medicines regulation from fees and most of our income for devices regulation from a service level agreement with the Department of Health. The operations of CPRD are funded by fees, with investments funded jointly by the MHRA and the Department of Health. Approximately 50% of NIBSC’s revenue is derived from fees it charges for its services including the sale of biological standards, and from research funding; the remainder is financed from the Department of Health. On 1 April 2013, we begin a new five year financial objective period. The objective, set out in a new HM Treasury minute, is expected to achieve, averaged over the five year period as a whole, a return of at least 3.5% in the form of a surplus on ordinary activities before interest payable and dividends expressed as a percentage of average capital employed. We will aim to ensure that annual plans contribute to the success of this financial objective and that there is no cross-subsidy between each of our parts. Our regulatory role within Europe is changing, and this will be accompanied by a more constrained level of regulatory income. The combined effect of reduced income from marketing authorisations and an increasing proportion of income being set by Europe is the most important strategic regulatory issue facing us. In a period of activity growth, the trading fund model has been very conducive to maintaining a funded, influential role, and we have had the discretion to shape it. Whilst it remains the most appropriate model in principle, lower future licensing income combined with the new pharmacovigilance system has the potential to challenge the sustainability of our more extended role. We will be flexible and seek efficiencies wherever possible in order to keep operating costs down. In the area of devices regulation, we will seek to influence the development of a more resilient funding model which operates within a wider EU network and shares the cost of safer products more broadly between industry and government. This business plan sets out a considerable amount of new work in relation to devices, and this needs to be reconciled with increasing funding restrictions. A more resilient funding model, allied to a risk-based approach to our regulatory activity, and supported by the appropriate IT tools, will be necessary to ensure that we have the resources to deliver this work.
3 of 25

4 of 25
3. Core functions This section sets out a number of core functions specific to each part of the Agency – the MHRA, NIBSC and CPRD. The two diagrams list the core functions and show synergies between each part. Targets related to these core functions are at Annex A.
MHRA
We approve clinical trial authorisation applications; assess marketing authorisation applications and variations; review applications for herbal and homeopathic medicine registrations/licences; assess the suitability of products approved under EU parallel trading provisions; contribute to EU and international guidelines towards common data requirements to facilitate development of new medicines; and provide scientific advice on proposals for development for new medicinal products. For devices, we instead register and audit the UK notified bodies that are responsible for approving medical devices, and undertake consultations on medical devices incorporating medicinal substances.
Through licensing and inspecting all UK manufacturers, wholesale dealers and importers of medicines and by inspecting clinical trials and toxicology laboratories, we seek to ensure compliance with the standards that apply to the manufacture and supply of medicines on the UK market. We will continue to gather intelligence, undertake investigations and initiate appropriate action in relation to suspected illegal advertising, manufacture, importation and sale or supply of medicines for human use.
We operate post-marketing surveillance and other systems for reporting, investigating and monitoring adverse reactions to medicines and adverse incidents involving devices; and for taking any necessary action to safeguard public health, for example through safety warnings or removing or restricting the availability of products. Through our laboratory testing services we determine whether medicines meet agreed quality standards and identify counterfeit medicines. We classify whether or not materials and products are medicines, monitor the importation of unlicensed medicines and ensure that appropriate action is taken following reports of defective medicines.
NIBSC
Through NIBSC, we are responsible for developing and producing over 90% of the international standards in use around the world to assure the quality of biological medicines. NIBSC is the UK's Official Medicines Control Laboratory (OMCL), responsible for testing biological medicines within the framework of the EU.
CPRD
CPRD is the new English NHS observational data and interventional research service, jointly funded by the NHS National Institute for Health Research (NIHR) and MHRA. CPRD will be working to increase the use of its anonymised data in research studies, and to help the way clinical trials of innovative medicines can be undertaken in order to maximise the health gain that can be achieved.

Core functions of MHRA, NIBSC and CPRD:
5 of 25
Medicines and Healthcare Products Regulatory Agency
MHRA •Operating a system of licensing, classification, monitoring (post-marketing surveillance) and enforcement for medicines. •Discharging statutory obligations for medical devices, including designating and monitoring the performance of notified bodies. •Ensuring statutory compliance in medicines clinical trials and assessing medical device clinical trials proposals. •Promulgating good practice in the safe use of medicines and medical devices. •Regulating the safety and quality of blood and blood components. •Discharging the functions of the UK Good Laboratory Practice Monitoring Authority (GLPMA). •Managing the activities of the British Pharmacopoeia (BP).
National Institute for Biological Standards Board (NIBSC)
•Devising and drawing up standards for the purity and potency of biological substances, and designing appropriate test procedures. •Preparing, approving, holding and distributing standard preparations of biological substances. •Providing, or arranging for the provision of, laboratory facilities for the testing of biological substances; carrying out such testing; examining records of manufacture and quality control and reporting on the results. •Carrying out, or arranging for the carrying out of, research in connection with biological standards and control functions.
Clinical Practice Research Datalink (CPRD)
•Maximising the way anonymised NHS and other health related data enable observational research to improve and safeguard public health. •Developing innovative IT solutions to improve the efficiency in interventional research. •Both undertaken in a manner to strongly contribute to the UK wealth agenda.
Corporate Divisions: Human Resources, Operations and Finance, Communications, Policy and Information Management
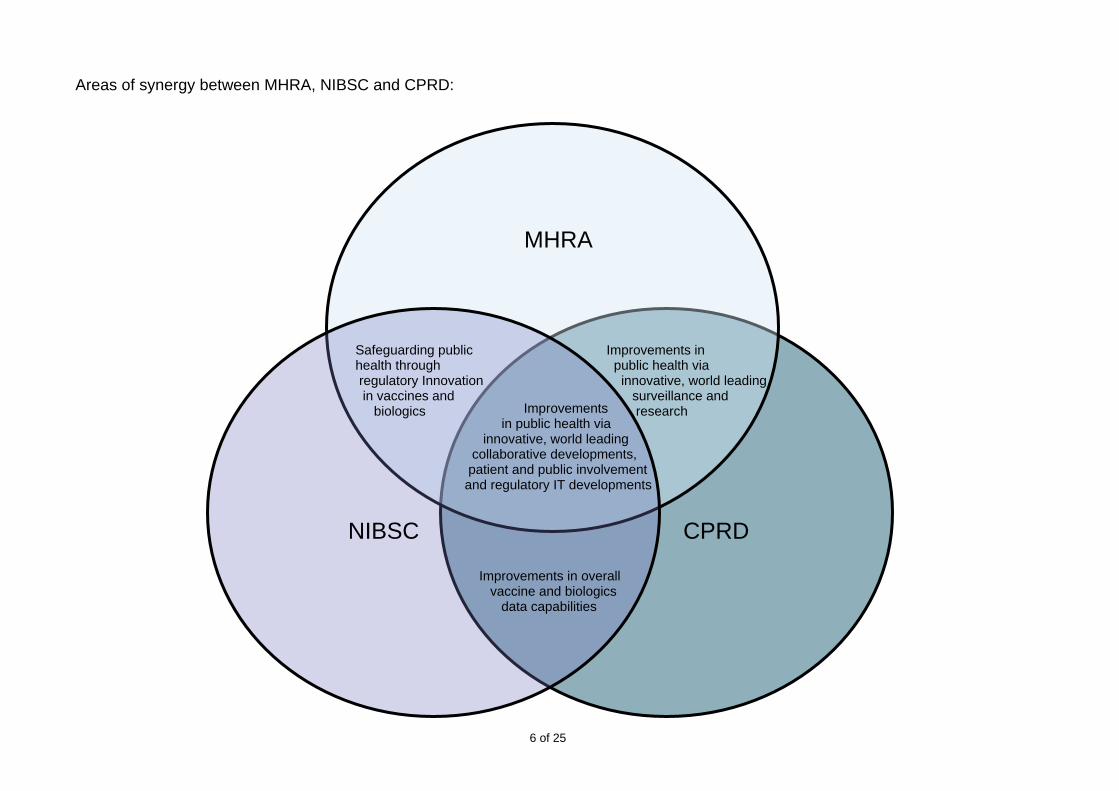
6 of 25
MHRA
NIBSC CPRD
Improvements in public health via innovative, world leading surveillance and research Improvements
in public health via innovative, world leading collaborative developments, patient and public involvement and regulatory IT developments
Improvements in overall vaccine and biologics data capabilities
Safeguarding public health through regulatory Innovation in vaccines and biologics
Areas of
synergy between MHRA, NIBSC and CPRD:

4. Strategic activities for 2013-14 This section sets out our key activities in 2013-14 that will contribute to delivery of the strategic aims, objectives and outcomes identified in our 2013-18 Corporate Plan. The key activities are arranged under the Corporate Plan’s five themes: • Theme 1: The role of regulation and the regulator • Theme 2: Bringing innovation safely to market • Theme 3: Strengthening surveillance • Theme 4: Safe products and secure supply in globalised industries • Theme 5: Achieving excellence – a well-run, efficient and effective organisation Broadly, these activities fall into 3 types: • Work to scope the major themes of the Corporate Plan, and to establish clearer outcomes for
the broad strategic objectives. Some of this work will need to bring together diverse activities, review them and set a reinforced strategic direction for them. This is particularly the case for work on engaging with healthcare professionals and patient groups, and for work to build more data systems that can support vigilance through more systematic evidence about the real-world use of products. We will set up an external reference group on our corporate strategy to support that strategic thinking and review the assumptions underlying our Corporate Plan.
• Activities that we have identified in-house, including a number of key activities arising from an
extensive exercise carried out in 2012-13 to ensure a successful integration of NIBSC and to develop the work of the CPRD.
• Activities to which we are committed as part of wider policy and regulatory developments in the
EU and through the wider UK Government. Work in this area includes our contribution to EU negotiations and the wider Government’s Life Sciences and Growth initiatives.
We envisage that over this first year of our new Corporate Plan, we will make solid progress on a range of key activities of central relevance to its objectives; and on work that will better define a set of milestones, indicators and outcomes for a range of strategic projects over the next five years. This will inform future annual business plans.
7 of 25

heme 1: The role of regulation and the regulator T
We are working towards the following key Corporate Plan objectives:
o To make best use of data available through CPRD and other sources, to provide more information about the performance of medicines and devices (benefit:risk) to influence clinical practice.
o To build partnerships to ensure that information about the performance of medicines and devices influences clinical practice in the interests of patients.
o To enhance understanding of the role our regulation should play as part of wider public health and health care systems.
This area of the Corporate Plan needs perhaps the greatest amount of further scoping and definition, and this will be the focus of our work in this area in 2013-14. As part of this, we will seek to make best use of the expertise of NIBSC and CPRD, and to further integrate our work on both medicines and devices (for example harnessing synergies in EU negotiation, vigilance and enforcement). We will concentrate on three key activities: 1A) Working with the Department of Health on a programme to rebalance the roles of product
and professional regulation in relation to sale and supply of medicines by pharmacies, with the aim of using professional regulation and standards where possible in preference to detailed legislation to regulate professionals’ behaviour. Developing proposals to address the issue of how most effectively to protect the public in relation to dispensing errors will be a priority. A new programme board with a recently appointed independent chair will agree a programme of activity, including the specific outputs to be achieved in 2013-14.
1B) Reviewing our current practices in working with healthcare professionals and patient groups to ensure that our work as a regulator is informed by real-world evidence from the use of medicines and devices once they are on the market – using data and research expertise from CPRD – and in turn effectively informs and influences clinical practice. We intend to set up a Corporate Strategy Reference Group to produce a detailed action plan.
1C) Reviewing our role in communicating with the public and patients about incidents leading to
public health concerns relating to specific products. Public and patient communication was one of the areas highlighted in the Howe review of the actions of the Department of Health and the MHRA on PIP breast implants. In the past, we have focused on informing healthcare professionals and the NHS to support the advice they give individual patients and the public; we need to consider whether this needs strengthening. We will therefore clarify our role and responsibilities in this area, and will actively contribute to ensuring an integrated approach across the new health and social care network when incidents or public health concerns arise, so that all stakeholders receive clear, consistent advice and messages.
8 of 25

T
heme 2: Bringing innovation safely to market
We are working towards the following key Corporate Plan objectives:
o To influence thinking and regulation in the EU and globally to achieve a convergence of standards – ensuring effective EU laws and progressing international thinking about global governance.
o To explore, in the context of Government interest in adaptive licensing, how we might redraw the boundary between pre- and post- market entry regulation to enable earlier patient access to beneficial innovative products, while managing associated risks.
o To realise the full benefits of NIBSC and CPRD to support innovation and contribute to the Government life sciences and growth agendas.
Key work areas in 2013-14 include the following: 2A) Completing the undertakings assigned to the MHRA in the Government’s Life Sciences and
Growth reviews, notably: • working with the wider Department of Health to take forward and complete work on an
Early Access scheme, responding to the public consultation. Much of the outstanding work now relates to the economic and financial underpinning of this proposed scheme, on which the Department is leading. However, we aim to complete this national scheme to allow unlicensed use of promising products at later stages of their development during the coming year
• progressing work on a pilot on adaptive licensing. We will follow up last year’s work to inform stakeholders of the existence of flexibilities in the licensing process which allow promising products in areas of high unmet clinical need into use on a controlled, conditional basis, subject to close monitoring and oversight. Since this pilot will need to be based in existing EU regulations, we will actively promote and participate in discussions and practical action within the EU (notably in the European Medicines Agency) and globally. We will explore how CPRD could contribute to setting up and running the controlled, monitored environment that will be necessary support to pilots to get products into earlier use than would normally have happened
• opening an Innovation Office to help companies, academics and individuals, who have developed a novel medicine or device, or a distinctly novel approach to developing or manufacturing a product. We will ensure systematic arrangements, linked to the Innovation Office, for utilising the intelligence gained through MHRA, including NIBSC and CPRD, for horizon scanning and planning purposes.
2B) Increasing the oversight of Notified Bodies, by undertaking joint inspections of them with
other Member States and the European Commission following agreement on joint methodologies.
9 of 25

2C) Improving clinical trials regulation, influencing the revision of EU rules and working with the
Health Research Agency on a single authorisation process for both clinical and ethical approvals.
2D) Maximising the availability of real world data from CPRD to all parties involved in the
research and development of innovative medicines. 2E) Establishing sixteen new or replacement biological standards and reference materials
needed for accurate measurement and dosing of biological medicines through either WHO endorsement or CE marking.
10 of 25

Theme 3: Strengthening surveillance
We are working towards the following key Corporate Plan objective:
o To strengthen systems that collect and use information about the performance of medicines and medical devices.
Key work areas in 2013-14 include the following: 3A&B) Implementing the new EU pharmacovigilance rules and communicating these changes
effectively to healthcare professionals and patients. Also ensuring innovative medicines are supported by risk management plans as a step towards more proactive, strategic vigilance to supplement signals-based systems.
3C) Exploring how best to link Yellow Card data, NIBSC’s laboratory-based research and
analysis and CPRD records into a routine model of vigilance and surveillance for vaccines and other biologicals, establishing cross-agency groups to take this work forward.
3D) Promoting the use of Sentinel, in particular the Pharmacovigilance module, to other EU
regulators. We believe that increasing the number of EU regulators that use Sentinel will help increase sharing of information, spread best practice in business processes and therefore enable better public health decision making.
3E) Helping shape the discussion at EU level on strengthening the vigilance of devices, through
work to further strengthen the links between the Heads of Medicines Agencies and the leads on medical devices regulation across the EU. Through this work, we will continue to improve the risk-based approach to device vigilance, including development of enhanced signal detection methodologies, undertaking a full review of adverse incident management processes and supporting IT.
3F) Contributing, including in the context of the Keogh Review into cosmetic surgery, to
planning how developments such as Unique Device Identifiers (UDIs) can contribute to effective tracking of clinical outcomes in real use. We will complete a pilot study to evaluate the feasibility of recording UDIs within patient electronic records and work with the European Commission and Member States to develop a strategy and timetable for delivery of an EU UDI database.
3G) Developing a strategy with the Department of Health for the Central Alerting System (CAS),
to ensure timely and clear information is cascaded effectively to healthcare professionals.
3H) Reviewing the benefits of bringing together medicines and devices adverse incident reporting under the Yellow Card brand, and exploring and developing a strategy for mobile-enabled technology for adverse incident reporting.
3I) Exploring options to establish a new Reference Laboratory function for quality assurance of in vitro diagnostic devices.
11 of 25

T
heme 4: Safe products and secure supply in globalised industries
We are working towards the following key Corporate Plan objective:
o To work with UK, EU and global partners to address the challenges posed by increasingly globalised medicines and devices industries – not least to combat counterfeiting and ensure a more secure supply chain.
Key work areas in 2013-14 include the following: 4A) Reducing the amount of inspections relating to medicines and devices that EU and US
inspectors carry out in each other’s countries, progressing work to share the outcomes and intelligence from inspections as part of risk identification. We will establish agreed methodologies, underpinned by confidentiality agreements.
4B&C) Implementing the Falsified Medicines Directive – taking forward work to implement the internet logo requirement, and promoting cost effective implementation of the safety features provisions. We will be work with the European Commission and Member States to ensure security of supply for the importation of Active Pharmaceutical Ingredients (APIs) after the API rules come into force in July 2013.
4D) Seeking to reduce the numbers of counterfeits purchased on websites – by continuing to
share information with other regulators on illegal activities; undertaking public awareness campaigns about the dangers of buying medicines and devices on the internet; and developing agreements and working practices with Domain Name Registrars and Registrees to close down offending websites.
4E) Developing international relationships to increase further the reach of NIBSC Standards,
and exploring new opportunities with the British Pharmacopoeia (BP) for BP monographs in the biologicals arena.
12 of 25

Theme 5: Achieving excellence – a well-run, efficient and effective organisation We are working towards the following key Corporate Plan objectives: o To regulate effectively and in a proportionate way. o To deliver organisational efficiency and value for money. o To manage our external relationships effectively. o To ensure a skilled and motivated workforce. o To effectively organise, prioritise and monitor our work.
We are undertaking the following key cross-Agency activities in this area, a number of which build on our merger project, to ensure that the new organisation is fit for purpose. In addition, below are some activities that NIBSC and CPRD are carrying out in support of their specific functions.
5A) Taking forward our Regulatory Excellence Programme, focusing on proportionate, risk-based approaches to regulation. The aims of the Programme are to:
• ensure that we deliver on regulatory expectations across Government, particularly as these relate to reducing regulatory burdens and regulating in an proportionate and risk-based manner
• facilitate and ensure timely delivery of our key regulatory undertakings, including those mentioned elsewhere in this plan, supporting the Life Sciences and Growth agendas, the revision of clinical trials legislation and implementing new EU falsified medicines rules
• manage other key EU legislative negotiations, including: - taking forward renegotiation of EU Devices legislation, developing European
Parliament and Council positions that reflect as far as possible the views of the UK Government
- developing an appropriate regime for regulating nicotine as part of changes to tobacco regulation
- work in relation to a proposal on EU pharmacovigilance fees expected this year • deliver the undertakings we have made on regulatory burden reduction and regulatory
simplification, including: - reducing the amount of guidance on our website by 40% - completing the undertakings we have made in the Red Tape Challenge, and
completing work with industry to measure the impact of these burden reductions - scoping options for further work to simplify regulation, especially through reducing
regulatory administrative burdens (e.g. in relation to variations).
5B) Running our Operational Excellence programme to manage and prioritise key efficiency activities, such as completing both the data assurance and Information Processing Unit (IPU) projects and improving device vigilance processes.
13 of 25

5C) Progressing work to strengthen our financial resilience, focusing on two key areas:
• Ensuring that, following the merger, our financial models, fee structure and
management reporting reflect and support the new organisation. Important to this work will be identifying a financial model for NIBSC within the Trading Fund to support investment decision making over a five year trading fund cycle.
• Continuing action to align our resources and activities to the external operating environment (see section 2 on the financial context).
5D) Developing an Information Systems Strategy to include how we will progress the
Government’s ‘Digital by Default’ agenda, and improve the IT infrastructure and keep it up to date.
5E) Developing the RamaXL service further, enabling added functionality. RamaXL provides all
company information in one secure package and it is the first port of call for companies to check their data. We will draw on stakeholder feedback to develop a business case to enhance the RamaXL service, such as enabling tracking of marketing authorisation applications for medicines and, possibly, devices. Subject to approval of the business case, we will look to complete development by the end of 2014.
5F) Integrating the NIBSC and MHRA infrastructure in two parts – telephony, instant
messaging, room booking and other capabilities integrated first, followed by all other infrastructure, such as corporate email branding.
5G&H) Establishing a people strategy for the new organisation:
5G – Developing a Workforce Planning Strategy to encompass the different employment pressures on MHRA, CPRD and NIBSC, including improvements to recruitment processes and retention. 5H – Developing a Learning and Development Strategy to encompass the different needs of MHRA, NIBSC and CPRD and to reflect the shared services programme within the Civil Service Reform plan.
5I&J) Delivering our Communications Strategy:
5I – For internal communications, launching our new intranet site and implementing our new Employee Communications and Engagement Strategy. 5J – Through external communications, building the brand and reputation of the new Agency family, positioning us as a leading regulator and a science and research supporting organisation. We will plan a strategy to achieve this.
5K) Completing a strategic analysis of the Agency’s long term facilities needs with the aim of
ensuring our facilities are modern and fit for purpose.
14 of 25

4.1 NIBSC activities for 2013-14 The NIBSC-MHRA merger presents the opportunity to create a global powerhouse for supporting safe and effective development, manufacture and use in patients of biologics, which are now beginning to dominate the pharmaceutical landscape. NIBSC is already the world leader in standardisation of biologics, and during the coming year we will maintain that position through establishing over 25 new or replacement biological standards and reference materials needed for consistent manufacture and accurate dosing of biological medicines. The standards will be validated either through the WHO’s Expert Committee on Biological Standards (ECBS) or through CE marking. We will also continue to extend the global reach of our standards and reference materials, working with existing partnerships in the USA, India and China, and taking advantage of synergies with the British Pharmacopeia. We expect that this will lead to further global harmonisation of quality standards, which is essential in the increasingly globalised world of medicines production. We will aim in particular to support the development of high quality biosimilar versions of ‘first generation’ biologics. This should lead to lower drug costs and increased medicines access, while mitigating the risk of adverse impact from poor quality products. It will also cement further NIBSC’s position as the global hub for biological standardisation and result in increased sales that will contribute to long term financial sustainability. NIBSC will also continue to provide the UK's Official Medicines Control Laboratory (OMCL) function, carrying out independent batch by batch testing of vaccines and blood-derived products to protect the health of UK and European citizens. This activity is also important to maintain specialist expertise that can be deployed in the event of a problem with these sensitive and high profile medicines. In recent years biologics manufacturing has been increasingly concentrated outside the UK, so it is increasingly challenging to maintain this in-depth knowledge of key products. We therefore aim to increase the breadth of products tested at NIBSC in the coming year, focusing especially on vaccines. Selection of an OMCL is the prerogative of manufacturers, so we aim to use the combined strength of the new Agency to promote the benefits of the UK as a route for licensing and testing of biologics. We will create a new Centre for Regulatory Science Research built around a small number of core research programmes. This will be supplemented by external research funding, and we will develop specific alliances with key academic centres. Regenerative medicine will be a key area of activity, capitalising on the work done to establish the UK Stem Cell Bank. We will take advantage of synergies across the enlarged MHRA to develop an integrated strategy that aims to support the emerging field through development of physical support materials, provision of expert advice and shaping sensible and practical regulation. The combined expertise of the organisation in biologics will be unparalleled. We will develop integrated cross-agency systems for sharing of information and expertise, and for providing expert evidence-based advice. Our aim is be to make the UK the country of choice, and the Agency the regulator of choice, for biologics manufacturers, and to establish the enlarged organisation as the clear international leader in biologics.
15 of 25

In addition to the activities outlined above, NIBSC will undertake the following activities in 2013-14: NA) Implementing a new electronic protocol submission system for Medicines Control Testing to
improve Batch Release efficiency, and seeking customer feedback on this. NB) Strengthening the MHRA’s research base by establishing a new Centre for Regulatory
Science Research for Biological Medicines, with outreach to the academic community.
16 of 25

4.2. CPRD activities for 2013-14 The CPRD operation within the Agency presents the opportunity to create new systems and methods for helping to bring treatments to the market at the earliest opportunity, and track use in a large population post launch to ensure the balance of benefit and risk serves patients appropriately. It will also enable the development of a surveillance system that combines the benefits of both the existing Yellow Card system and having large population cover Real World Data (RWD). In addition, it will enable the generation of specific datasets that will improve the understanding of medicine use in pregnancy and children. And by having access to linked data across a large population, researchers will be able to better understand the journey of care and the most appropriate way to use the right treatment at the appropriate point in a disease. CPRD has already been able to help with some device vigilance issues and is working to ensure that via appropriate coding of devices, in exiting datasets, more post marketing knowledge can be obtained. Similarly CPRD hopes that with the changes via NHS commissioning it will gain access to schools vaccination data, potentially leading to a national complete vaccination register that is not available in any other country. CPRD operates in the global market for health data and amongst big pharmaceutical companies and key regulators has a unique place, based upon the many aspects that stem from the NHS and added value from how CPRD enables access to high quality data. CPRD is also working with the MHRA’s funding partner, the National Institute for Health Research (NIHR), to improve the efficiency of clinical trials and ensure that the UK is considered a key place to undertake drug development research and trials at all stages in the process to market. How drugs and devices are used in the real world, and what their benefit:risk outcomes are, is a global question in relation to which the Agency, in particular through CPRD, punches above its weight and will continue to do so. In addition to the activities outlined above, CPRD will undertake the following activities in 2013-14:
CA) Exploring and defining how best CPRD will work and engage with health bodies in the new NHS Commissioning landscape to ensure that research, using both observational and interventional methodologies, plays a maximum role in ensuring the NHS provides the highest level of appropriate care.
CB) Implementing the CPRD Customer Relationship Management system that will enable easy outputs to be identified from all CPRD activities and improve the efficiency of the organisation.
CC) Enabling the potential for research on a greater number of drugs by providing (1) access to Primary Care data from at least three of the GP IT systems with a total population cover of ten million; and (2) access to Hospital Drug Administration data in conjunction with CPRD’s external partner, for a population cover of five million.
17 of 25

18 of 25
CD) Launching CPRD Trailviz, the CPRD clinical trial feasibility tool, as the first part of the
CPRD modular Clinical Trial Management System, with an offer to the research community of being able to link with up to 14 national NHS datasets.
CE) Launching a Pregnancy and Children's Database enabling a highly detailed understanding
of drugs used in these groups. CF) Establishing a launch plan for the CPRD electronic Case Report Form, and plans for
integration between this system and the primary care EHR systems.

Annex A Table of core function targets for 2013-14 In order to deliver the core responsibilities set out in chapter 2 in the most effective and efficient way, we will work to the following targets:
Activities Targets
Medicines Licensing – validation of applications
• 100% of Type IA variations validated within 30 days or receipt.
• 98% of Type IB/II variations validated within 14 days of receipt.
• For new Marketing Authorisation applications, 100% of validation reports produced within 14 days.
• 98% of Change of Ownership applications granted within 42 days of receipt
Medicines Licensing – assessment of applications
• The assessment of applications for new Marketing Authorisations for UK only: 97% assessed in 150 days
• The assessment of applications for new Marketing Authorisations in European (MR, DC & centralised) procedures: 97% assessed within the designated time
• The assessment of Type IB minor and Type II major variation applications in National and European (MR, centralised) procedures: 97% assessed within the designated time.
• The assessment of applications for clinical trials of medicines in the UK: 98% in 30 days (all trial phases) and an average time of 14 days (Phase I trials)
Assessment of Clinical trials and investigations
• Timescales for clinical investigation notifications for medical devices: maximum of 60 days with an overall
average of 54 days or less
Capturing and analysing adverse event reports –making reports available, issuing alerts and acting on
• Maximum timescales between receipt of reports and making them available for evaluation and analysis: For fatal and serious device adverse incidents: 95% within 2 working days and 100% within 3 working days
• Medical Device Alerts will be issued: 95% within 10 days, 100% within 15 days
19 of 25

signals
• For fatal UK adverse drug reactions: 90% within 24 hours, 100% within 72 hours
• For serious UK adverse drug reactions: 95% within 72 hours, 100% within 5 days
• Ensure all UK potential signals (relating to medicines) from whatever source are acted on promptly: 85% initially evaluated within 5 working days
Publication of UK assessment reports for new Marketing Authorisations
• The publication of UK assessment reports for new Marketing Authorisations and major non-safety variations of clinical importance:98% within 60 days of grant of new authorisations and 98% within 40 days of grant of the major variation
Standards and control • Biologics standards supply - 93% of all materials supplied within 6 working days
• Batch release activity - Completion of all requested OCABR and non-EU testing within agreed timelines; Time taken to issue Batch Release Certificate after last item received from the manufacturer should be no more than 10 days for Blood Products and 60 days for Vaccines.
NIBSC research activity • >80 papers and scientific review articles authored in calendar year 2013.
• Over £2.5 million in externally awarded research grant/contract funding utilised in the financial year 2013-2014.
Answering Freedom of Information requests, letters and Parliamentary Questions
• In working towards achieving 100% compliance, ensure that at least 92% of requests under the Freedom of Information Act are replied to within 20 working days.
• Return responses to Parliamentary Questions (PQs) to the Department of Health by noon on the date specified with less than 5% returned to MHRA by the Department for rewriting.
• Return Ministerial correspondence (POs) drafts to the Department of Health within 4 working days of receipt in at least 80% of cases with less than 5% returned to MHRA by the Department for rewriting.
Finance – income and expenditure position
• Achieve an income and expenditure surplus during 2013-14 and, as a minimum, exceed a 3.5% per annum return on capital employed, without cross-subsidising between MHRA, NIBSC and CPRD
20 of 25

Annex B Table of strategic activities for 2013-14
No Activity Outputs
Theme 1 – The role of regulation and the regulator
1A Rebalancing product and professional regulation
Work programme developed by Programme Board and agreement with the Department of Health by quarter one on outputs for 2013-14.
1B Real world evidence informing our work and influencing clinical practice
Corporate Strategy Reference Group set up by end of quarter two to produce a detailed action plan by the end of 2013-14.
1C Strengthening stakeholder communications and engagement
Current practice identified and integrated healthcare professional engagement strategy developed by quarter two. A risk communications framework developed and implemented by January 2014.
Theme 2 – Bringing innovation safely to market
2A Contributing to the Government’s Life Sciences and Growth reviews
Response to public consultation on early access scheme published in quarter one; Innovation Office opened in quarter one with arrangements introduced by quarter three for utilising the intelligence gained through MHRA, including NIBSC; a report and recommendations from the Expert Group on Innovation in Healthcare published in quarter two and an Adaptive Licensing pilot programme progressed in 2013-14.
2B Joint inspections of notified bodies Joint methodologies agreed and full programme of joint inspections taken place in 2013-14.
2C Clinical trials Regulation negotiation European Parliament and Council positions developed that reflect as far as possible the views of the UK Government by quarter four.
2D Real world data for clinical trials CPRD modular software developments, with beta launches during 2013-14.
2E New/replacement biological standards
At least 16 new or replacement standards through either WHO or CE marking route by quarter four.
21 of 25
Theme 3 – Strengthening surveillance
3A EU pharmacovigilance implementation and communication
Operation of the pharmacovigilance legislation audited by September 2013; Joint Action milestones delivered according to project plan; Commission proposal on fees influenced during quarter two. Changes communicated to pharmaceutical industry, healthcare professionals by quarter two and a plan for communicating to patients developed by end quarter three.
3B Risk-management plans for innovative medicines
Develop process for publishing summaries of Risk Management Plans (RMPs) by January 2014.
3C Pharmacovigilance monitoring of vaccines and biologicals
Cross-agency groups with clear goals, covering vaccines (Vision Network), Biosimilars, Blood related products, and regenerative medicines by quarter two.
3D Promotion of Sentinel Pharmacovigilance module
Memorandum of understanding signed with at least one other national competent authority and implementation in progress by quarter 4.
3E Improving device vigilance Full review of adverse incident management processes and supporting IT by quarter four.
3F Unique Device Identifiers (UDI) Pilot study to evaluate the feasibility of recording UDI within patient electronic records completed and strategy and timetable for delivery of EU UDI database developed by quarter four.
3G Central Alerting System (CAS) strategy
Review of CAS completed and recommendations put forward to Department of Health by end quarter one.
3H Promotion of adverse incident reporting
Decision taken by end quarter one on whether to bring medicines and devices incident reporting together. Yellow Card scheme promoted in 2013 and a strategy for mobile-enabled technology scoped by end quarter four.
3I Establishing an IVD Reference laboratory function
Opportunity analysed and business case developed for decision by quarter 3
22 of 25

Theme 4 – Safe products and secure supply in globalised industries
4A Coordinated international inspections on medicines and medical devices
Coordinated methodologies, underpinned by confidentiality agreements, established by quarter three.
4B Protecting the legitimate supply chain from counterfeits
Statutory Instrument transposing the Falsified Medicines Directive and assessment of options for the logo/distance selling by quarter two.
4C Access to imported Active Pharmaceutical Ingredients (APIs)
By quarter two, implementation of API requirements that ensures security of supply.
4D Internet sale of medical products Public awareness campaign launched in 2013, including Operation Pangea VI and agreements/working practices developed with Domain Name Registrars and Registrees by 2014.
4E Extending reach of standards to support global quality of biologics
Agree integrated strategy for partnering with international pharmacopoeias, and identify new opportunities for BP monographs in the biologicals arena; four revenue share projects completed by quarter four.
Theme 5 – Achieving excellence – a well-run, efficient and effective organisation
5A Regulate effectively and proportionately
Projects delivered to timescales to be agreed with the Regulatory Excellence programme. Key outputs include the Red Tape Challenge outcome published and an implementation plan developed by quarter one; 40% reduction in the amount of guidance on MHRA’s website by end year.
5B Organisational efficiency Projects delivered to timescales agreed with the Operational Excellence steering Group. Key outputs include phase II of the Information Processing Unit (IPU) Excellence project re-organisation completed by end July 2013.
5C Finance reporting Management reporting redesigned, incorporating NIBSC's and CPRD’s operational needs by quarter one. Financial model for NIBSC within the Trading Fund identified by quarter two. The fee and resource impact of the new pharmacovigilance regime identified and the fee setting framework for MHRA within the EU system agreed with HM Treasury by quarter two. Resource and cost information required to manage a sustainable role in Europe identified and collected by quarter three. MHRA’s financial and fee setting model updated to reflect the new organisation by quarter three.
23 of 25
5D IT strategy and procurement Information Systems strategy developed and published by quarter one. Transition of the services which move to the bespoke applications contract by 30 April 2013. Re-procurement of the Infrastructure Operations element of the current IT contract in time for transition to be completed by December 2014.
5E Services to external customers Produce a business case for internal agreement by quarter two to develop the RamaXL service further. Subject to the agreement of the business case, complete the development by end 2014.
5F Integrated NIBSC/MHRA infrastructure provision
Linkages between telephony, instant messaging and room booking etc in place by July 2013 and full merger by January 2014.
5G Workforce Planning Strategy Workforce Planning Strategy in place by January 2014.
5H Learning and Development Strategy Learning and Development Strategy in place by September 2013.
5I Employee communications New intranet site launched quarter one.
5J Communications about new organisation
Plan in place to build the brand and position the organisation by September 2013.
5K Analysis of long term facilities needs Completing a strategic analysis of Agency long term facilities needs by quarter two.
NIBSC
NA Improving Batch Release efficiency Improve Batch Release efficiency through implementing new electronic protocol submission system for Medicines Control Testing and gathering customer feedback by quarter four.
NB Regulatory Science Research New Centre for Regulatory Science Research for Biological Medicines established; agreed projects initiated and oversight mechanisms established by end quarter two. New alliances with two academic centres in place by quarter four.
CPRD
CA CPRD and the new NHS Commissioning landscape
The potential for CPRD to work and engage with health bodies in the new landscape scoped by quarter three. The original CPRD business case reviewed by quarter two.
CB Customer Relationship CPRD Customer Relationship Management system fully implemented by end quarter three.
24 of 25
25 of 25
Management
CC Access to Primary Care and Hospital Drug Administration (HDA) data
Access Primary Care data enabled from at least 3 of the GP IT systems with a total population cover of 10 million by the end of quarter two. Access to HDA data for a population cover of 5 million by the end of quarter two.
CD CPRD Trailviz clinical trial feasibility tool
Tool formally launched by mid quarter three. An offering to the research community of being able link with up to 14 national NHS datasets by quarter three.
CE Pregnancy and Children's database Database launched by end quarter three.
CF CPRD electronic Case Report Form Launch plan in place and plans in place for integration between this system and the primary care EHR systems by the end quarter three.

26 of 25

27 of 25
Business Plan 2013-2014 © Crown Copyright April 2013 151 Buckingham Palace Road London SW1W 9SZ United Kingdom T: 0203 080 6000 E: [email protected] mhra.gov.uk