bisulfite sequencing - haowulab.org · varleyk e et al. genome res. 2013;23:555-567 in human...
TRANSCRIPT

Bisulfitesequencing
1
AdvancesinGeneticsVolume70201027- 56
http://dx.doi.org/10.1016/B978-0-12-380866-0.60002-2
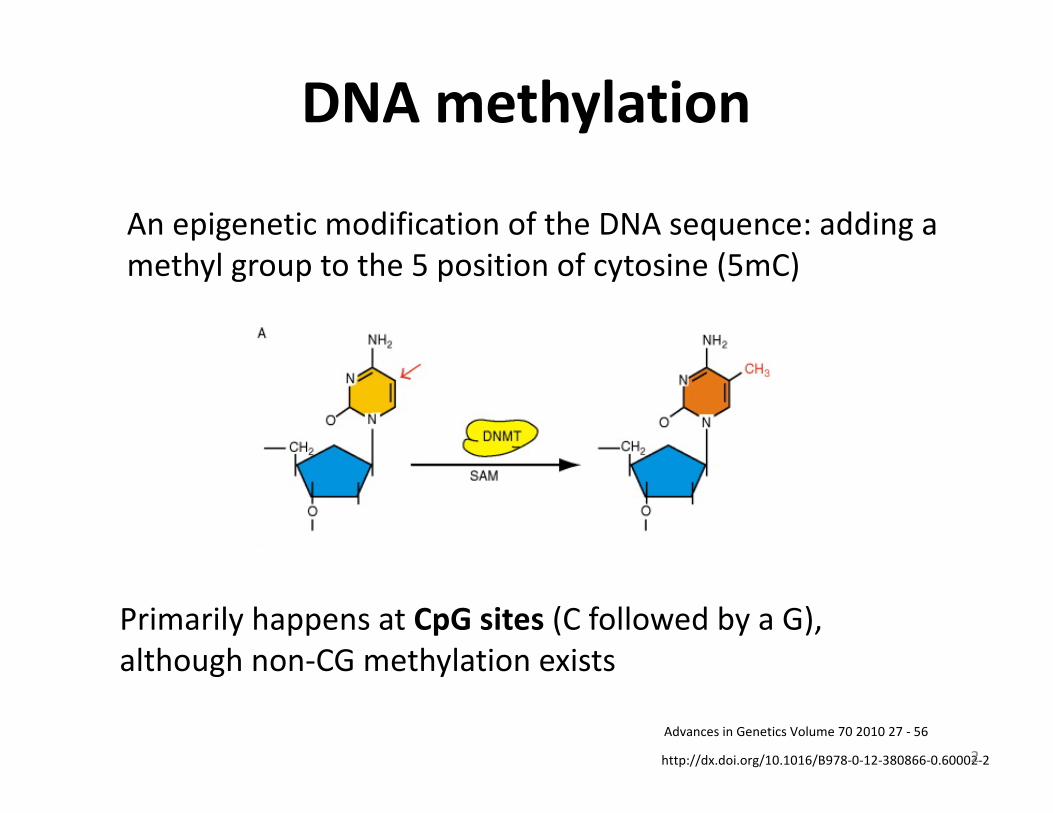
AnepigeneticmodificationoftheDNAsequence:addingamethylgrouptothe5positionofcytosine(5mC)
PrimarilyhappensatCpGsites(CfollowedbyaG),althoughnon-CGmethylationexists
DNAmethylation
2
Varley K E et al. Genome Res. 2013;23:555-567
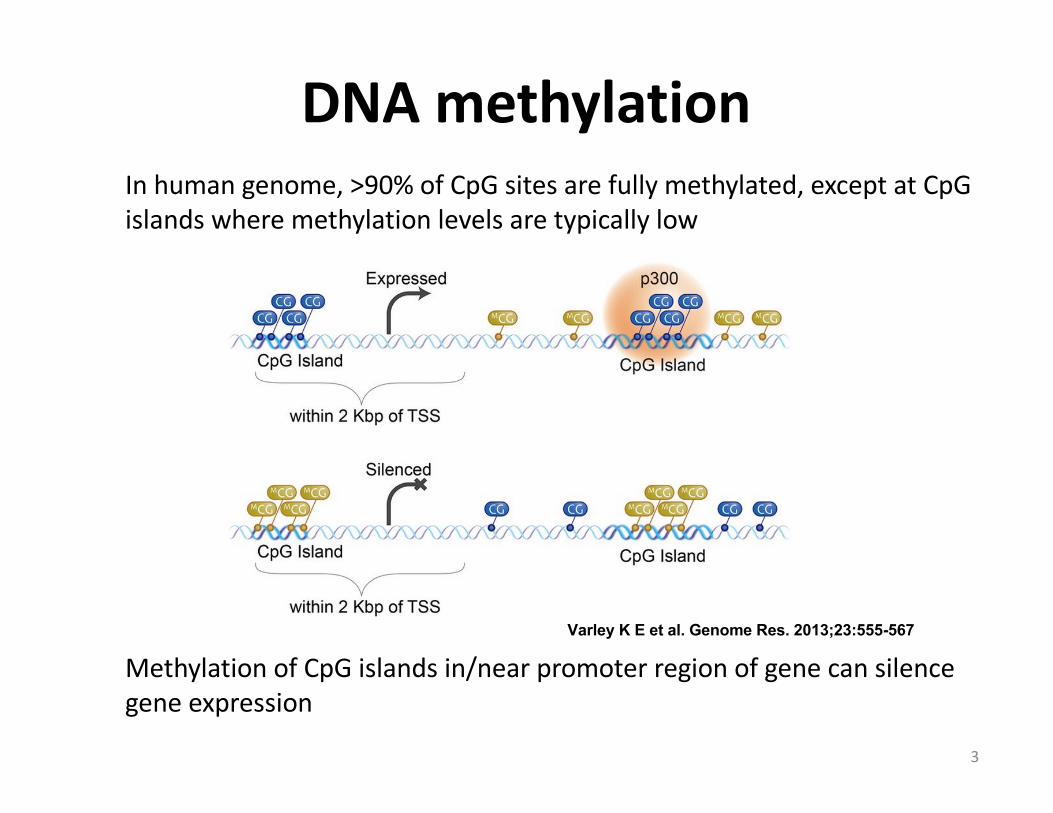
Inhumangenome,>90%ofCpGsitesarefullymethylated,exceptatCpGislandswheremethylationlevelsaretypicallylow
MethylationofCpGislandsin/nearpromoterregionofgenecansilencegeneexpression
DNAmethylation
3

• Importantingeneregulation– Methylationofpromoterregionscansuppressgeneexpression
• Playscrucialroleindevelopment– Heritableduringcelldivision
– Helpscellsestablishidentityduringcell/tissuedifferentiation
• Canbeinfluencedbyenvironment– GoodcandidatetomediateGxE interactions
FunctionofDNAmethylation
4

SequencingapproachesforDNAmethylation
• Canbedividedintotwocategories– Capture-basedorenrichment-basedsequencing
• Usemethyl-bindingproteinsorantibodiestocapturemethylatedDNAfragments,thensequencefragments
• Resolutionislow:cantypicallyquantifytheamountofDNAmethylationin100-200bp regions
– Bisulfite-conversion-basedsequencing• Bisulfitetreatmentconvertsunmethylated C’stoT’s• Sequencingconverteddatagivessingle-bp resolution• CanmeasuremethylationstatusofeachCpG site• Untilrecently,notpossibletodistinguish5mCfrom5hmC
• Focusofthislecture:bisulfitesequencing
5

Capture-basedsequencingapproaches• AllinvolvecaptureofmethylatedDNAfollowedbysequencing
• MeDIP-seq (MethylatedDNAImmunoPrecipitation)1
– LikeChIP-seq,butusesantibodyagainstmethylatedDNA
– Assessesrelativeratherthanabsolutemethylationlevels• Problem:don’tobserveunmethylated DNAfragments,onlymethylatedones
• Anotherproblem:immunoprecipitation maybeaffectedbyCpG density
– MEDIPS2 isapopulartoolforanalysis
• Captureviamethyl-bindingdomainproteins:MBD-seq3/MIRA-seq4,methylCap-seq5
• Captureviamethyl-sensitiverestrictionenzymes(MRE-seq)6
6
1Weberetal.(2005)NatGenet;2Chavezetal.(2010)GenRes;3Serreetal.(2010)NAR4Rauchetal.(2010)Methods; 5Brinkmanetal.(2010)Methods; 6Maunakeaetal.(2010)Nature

Bisulfitesequencing(BS-seq)• Technologyinanutshell:– TreatfragmentedDNAwithbisulfite
• Unmethylated CwillbeconvertedtoU,amplifiedasT• MethylatedCwillbeprotectedandremainC• Nochangeforotherbases
– AmplifythetreatedDNA– SequencetheDNAsegments– Alignsequencereadstogenome
7

Reducedrepresentationbisulfitesequencing(RRBS)1,2
• Goal:affordablealternativetogenome-widesequencing
– BynarrowingfocustoCpG-richareas,reduce#ofreadsnecessaryto
obtaindeepcoverageofpromoterregions
– Interrogates~1%ofthegenomebut5-10%ofCpG sites
• Approach:enrichforCpG-richsegmentsofgenome
– MspI restrictionenzyme cutsatCpG sites,leavingfragmentswithCpGs at
eitherend:
– Sizeselectionforfragmentsof40-220bpmaximizescoverageofpromoter
regionsandCpG islands
– Bisulfitetreat,amplify,end-sequence,andalignfragmentstogenome
81Meissner(2005)NAR;2Guetal.(2011)NatProtoc
CCGGCCGG
IllustrationofbisulfiteconversionBMC Bioinformatics 2009, 10:232 http://www.biomedcentral.com/1471-2105/10/232
Page 2 of 9(page number not for citation purposes)
detect the methylation pattern of every C in the genome.Nevertheless, the mapping of millions of bisulfite reads tothe reference genome remains a computational challenge.
ProblemsFirst, the searching space is significantly increased relativeto the original reference sequence. Unlike normalsequencing, the Watson and Crick strands of bisulfite-treated sequences are not complementary to each otherbecause the bisulfite conversion only occurs on Cs. As aresult, there will be four distinct strands after PCR ampli-fication: BSW (bisulfite Watson), BSWR (reverse comple-ment of BSW), BSC (bisulfite Crick), and BSCR (reversecomplement of BSC) (Figure 1). During shotgun sequenc-ing, a bisulfite read is almost equally likely to be derivedfrom any of the four strands.
Second, sequence complexity is reduced. In the mamma-lian genome, although ~19% of the bases are Cs and
another 19% are Gs, only ~1.8% of dinucleotides are CpGdinucleotides. Because C methylation occurs almostexclusively at CpG dinucleotide, the vast majority of Cs inBSW and BSC strands will be converted to Ts. Therefore,most reads from the above two strands will be C-poor.However, PCR amplification will transcribe all Gs as Cs inBSWR and BSCR strands, so reads from those two strandsare typically G-poor and have a normal C content. As aresult, we expect the overall C content of bisulfite reads tobe reduced by ~50%.
Third, C to T mapping is asymmetric. The T in the bisulfitereads could be mapped to either C or T in the referencebut not vice versa. This phenomenon not only increasesthe search space for mapping but also complicates thematching process (Figure 2). Efficient implementation ofsuch asymmetric C/T matching is critical for mappinghigh-throughput bisulfite reads to the reference genome
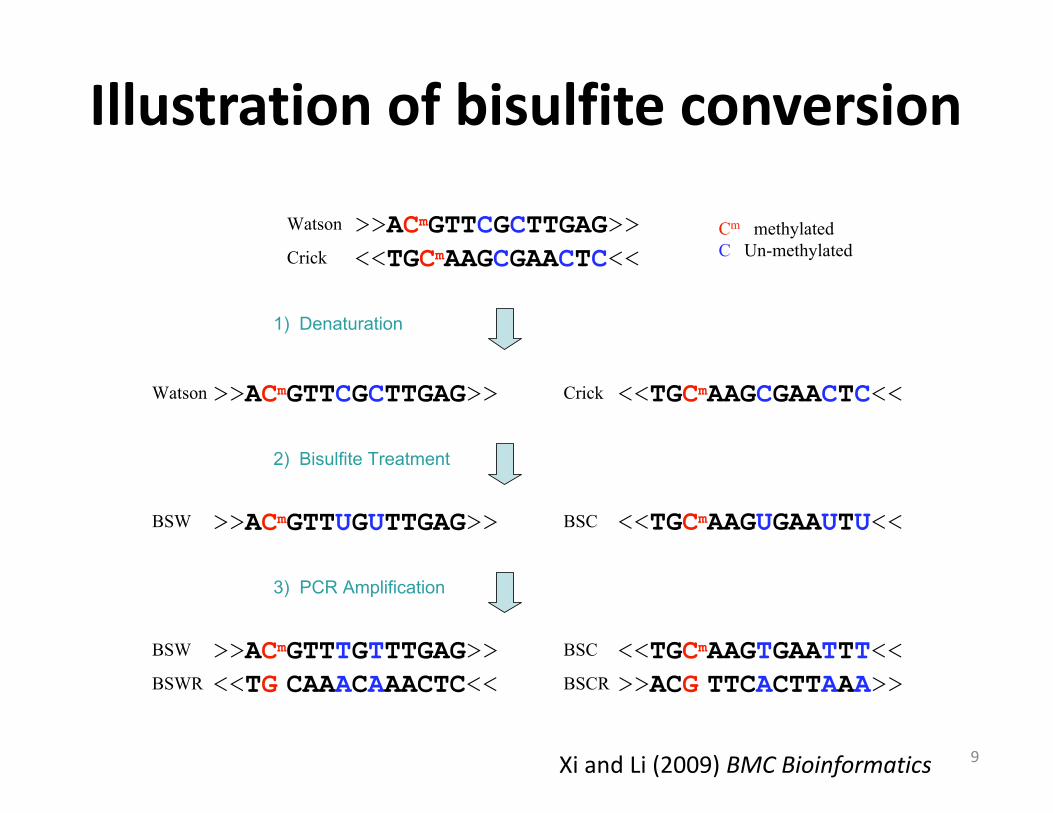
Pipeline of bisulfite sequencingFigure 1Pipeline of bisulfite sequencing. 1) Denaturation: separating Watson and Crick strands; 2) Bisulfite treatment: converting un-methylated cytosines (blue) to uracils; methylated cytosines (red) remain unchanged; 3) PCR amplification of bisulfite-treated sequences resulting in four distinct strands: Bisulfite Watson (BSW), bisulfite Crick (BSC), reverse complement of BSW (BSWR), and reverse complement of BSC (BSCR).
>>ACmGTTCGCTTGAG>> <<TGCmAAGCGAACTC<<
Watson
Crick
Watson Crick
>>ACmGTTUGUTTGAG>> <<TGCmAAGUGAAUTU<<
<<TGCmAAGTGAATTT<<
>>ACG TTCACTTAAA>><<TG CAAACAAACTC<<
>>ACmGTTTGTTTGAG>>BSW
BSWR
BSW
BSC
BSCR
BSC
Cm methylatedC Un-methylated
1) Denaturation
2) Bisulfite Treatment
3) PCR Amplification
>>ACmGTTCGCTTGAG>>
<<TGCmAAGCGAACTC<<
XiandLi(2009)BMCBioinformatics 9

AlignmentofBS-seq• Problem:readscannotbedirectlyalignedtothereferencegenome.– FourdifferentstrandsafterbisulfitetreatmentandPCR– C-Tmismatcheswillmeanunmethylated readscan’tbealignedtothecorrectposition• Unmethylated CpGs willalignwithTpGs orlikelynotatall• Willleadtoastrongbiasinfavorofmethylatedreads
• Onepossiblesolutioninsilico bisulfiteconversion– SwitchallC’stoT’sinbothreadsandreferencesample– Usethisforalignment,thenchangebacktooriginal
10
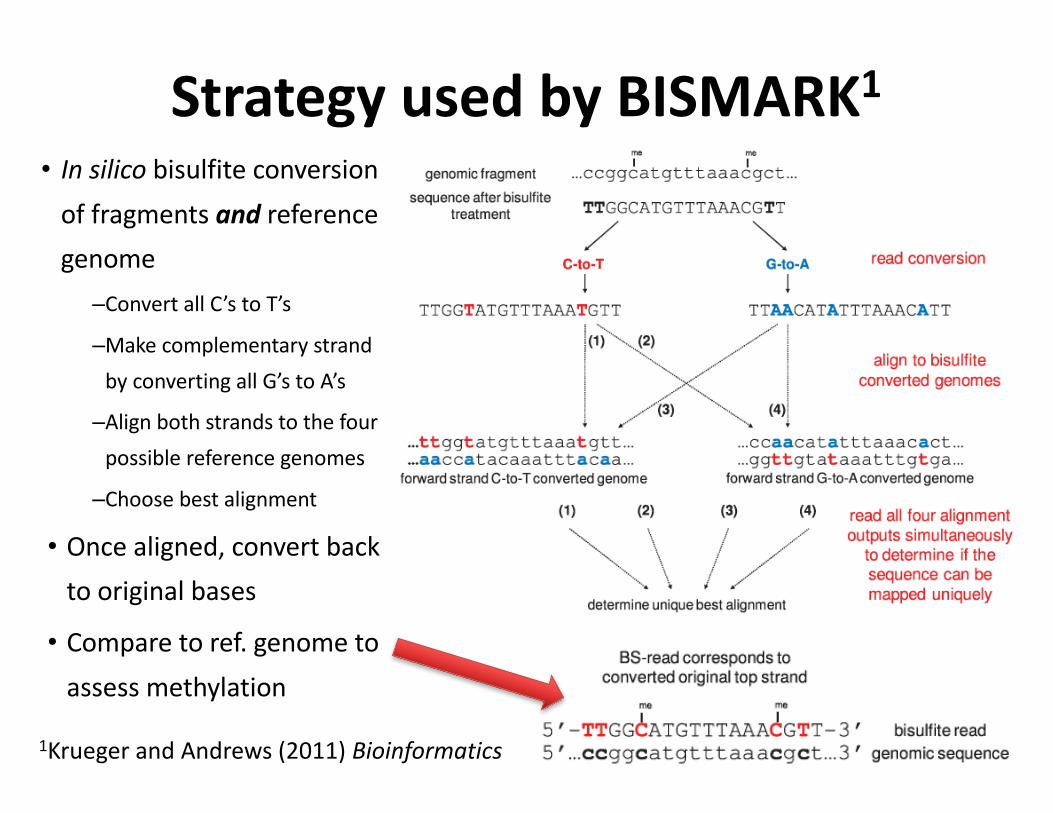
• Insilico bisulfiteconversionoffragmentsand referencegenome–ConvertallC’stoT’s
–MakecomplementarystrandbyconvertingallG’stoA’s
–Alignbothstrandstothefourpossiblereferencegenomes
–Choosebestalignment
• Oncealigned,convertbacktooriginalbases
• Comparetoref.genometoassessmethylation
StrategyusedbyBISMARK1
111KruegerandAndrews(2011)Bioinformatics

• Possibleproblemswithinsilico approach
– ByconvertingallC’stoT’s,reducesequencecomplexityto3bases
– Largersearchspaceforpossiblealignments
– Couldleadtomismatchesornon-uniquemapping
Alignmentissues
12
BMC Bioinformatics 2009, 10:232 http://www.biomedcentral.com/1471-2105/10/232
Page 3 of 9(page number not for citation purposes)
and is still lacking in current short read alignment soft-ware.
A common approach to overcome these issues is to con-vert all Cs to Ts and map the converted reads to the con-verted reference; then, the alignment results are post-processed to count false-positive bisulfite C/T alignmentsas mismatches, where a C in the BS-read is aligned to a Tin the reference [2]. Although this all-inclusive C/T con-version is effective for reads derived from the C-poorstrands, it is not appropriate for reads derived from the G-poor strands, where all the Cs are actually transcribedfrom Gs by PCR amplification and thus could not be con-verted to Ts during bisulfite treatment. During shotgunsequencing, however, a bisulfite read is almost equallylikely to be derived from either the C-poor or the G-poorstrands. There is no precise way to determine the original
strand a bisulfite read is derived from. Furthermore, byignoring the C/T mapping asymmetry, this strategy gener-ates a large number of false-positive bisulfite mappingsand greatly increases the computational load in a quad-ratic manner with an increase in the size of the referencesequence. In order to accurately extract the true bisulfitemappings in the post-processing stage, all mapping loca-tions have to be recorded, even the non-unique map-pings. Therefore, this approach is only practical for smallreference sequences, where only the C-poor strands aresequenced. For example, Meissner et al. used this map-ping strategy for reduced representation bisulfite sequenc-ing (RRBS) [2], where the genomic DNA was digested bythe Mspl restriction enzyme and 40–220 bp segmentswere selected for sequencing. The reference sequence (~27M nt) is only about 1% of the whole mouse genome, cov-ering 4.8% of the total CpG dinucleotides.
Mapping of bisulfite readsFigure 2Mapping of bisulfite reads. 1) Increased search space due to the cytosine-thymine conversion in the bisulfite treatment. 2) Mapping asymmetry: thymines in bisulfite reads can be aligned with cytosines in the reference (illustrated in blue) but not the reverse.
>>ATTTCG>>
>>ATACTTCGATGATCTCGCAAGACTCCGGC>>
ATTTCG ATTTCGATTTCG
Bisulfite Read
Reference
Bisulfite Read Reference
C
T
C
T
1) Multiple Mapping
2) Mapping AsymmetryXiandLi(2009)BMCBioinformatics
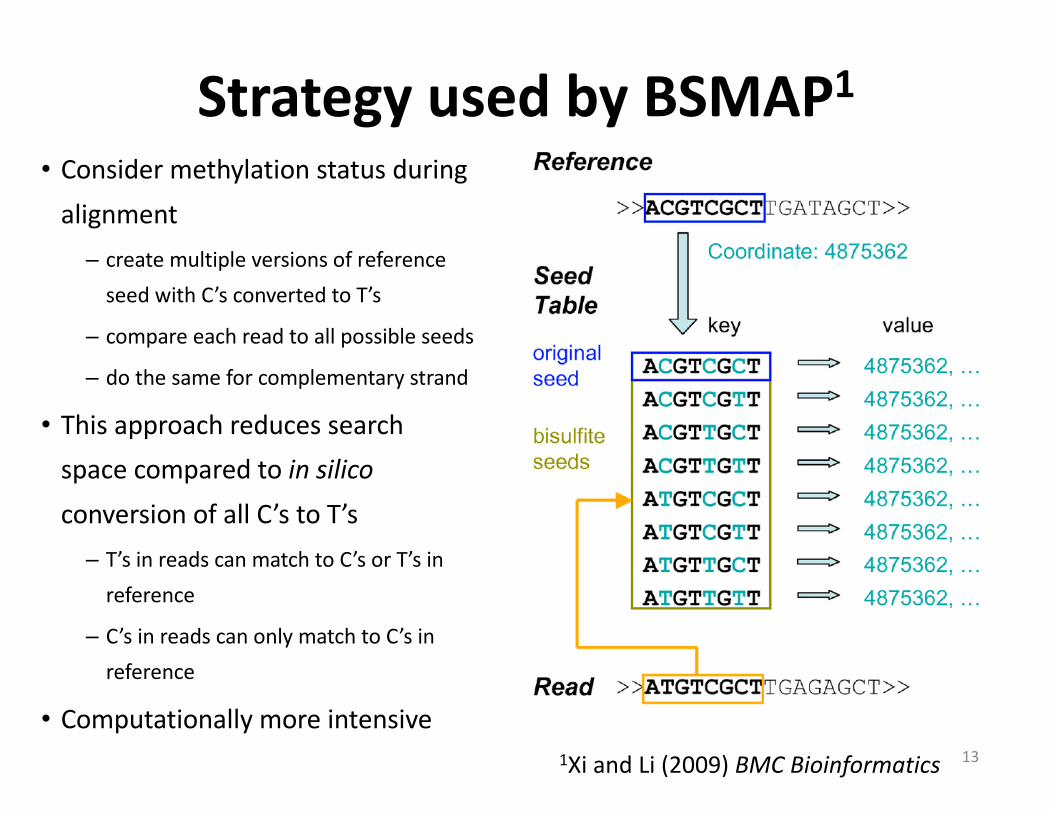
• Considermethylationstatusduringalignment– createmultipleversionsofreferenceseedwithC’sconvertedtoT’s
– compareeachreadtoallpossibleseeds
– dothesameforcomplementarystrand
• ThisapproachreducessearchspacecomparedtoinsilicoconversionofallC’stoT’s– T’sinreadscanmatchtoC’sorT’sinreference
– C’sinreadscanonlymatchtoC’sinreference
• Computationallymoreintensive
StrategyusedbyBSMAP1
131XiandLi(2009)BMCBioinformatics

Whichalignmentsoftwareisbest?
14
• AdvantagesofBSMAP:– reducessearchspacebyeliminatingmappingofC’stoT’s
– greaterproportionofuniquelymappingreads1
• AdvantagesofBISMARK:– muchfasterthanBSMAPandotherprograms1
– uniquenessofmappingindependentofmethylationstatus1
– moreuser-friendlyintermsofextractingdata,interfacingwithothersoftware1
• Ingeneral,BISMARKseemstobethepopularchoice
1Chatterjeeetal.(2012)NAR

Otheraligners
15
• AlignmentofRRBSdata
– Chatterjee etal.notesitismuchfasterifweuseinformationonMspI cutpoints to“reduce”referencegenomeinsilico1
– RRBSMAP:aversionofBSMAPthatdoesexactlythat2
– Hasoptiontoworkwithdifferentrestrictionenzymes
• Manyotheralignersforbisulfitesequencingdata
– OneusefulreviewoftheseisHackenberg etal.3
1Chatterjeeetal.(2012)NAR;2Xietal.(2012)Bioinformatics;3Hackenbergetal.(2012):Chapter2in“DNAMethylation– FromGenomicstoTechnology”Tatarinova (Ed.)http://www.intechopen.com/books
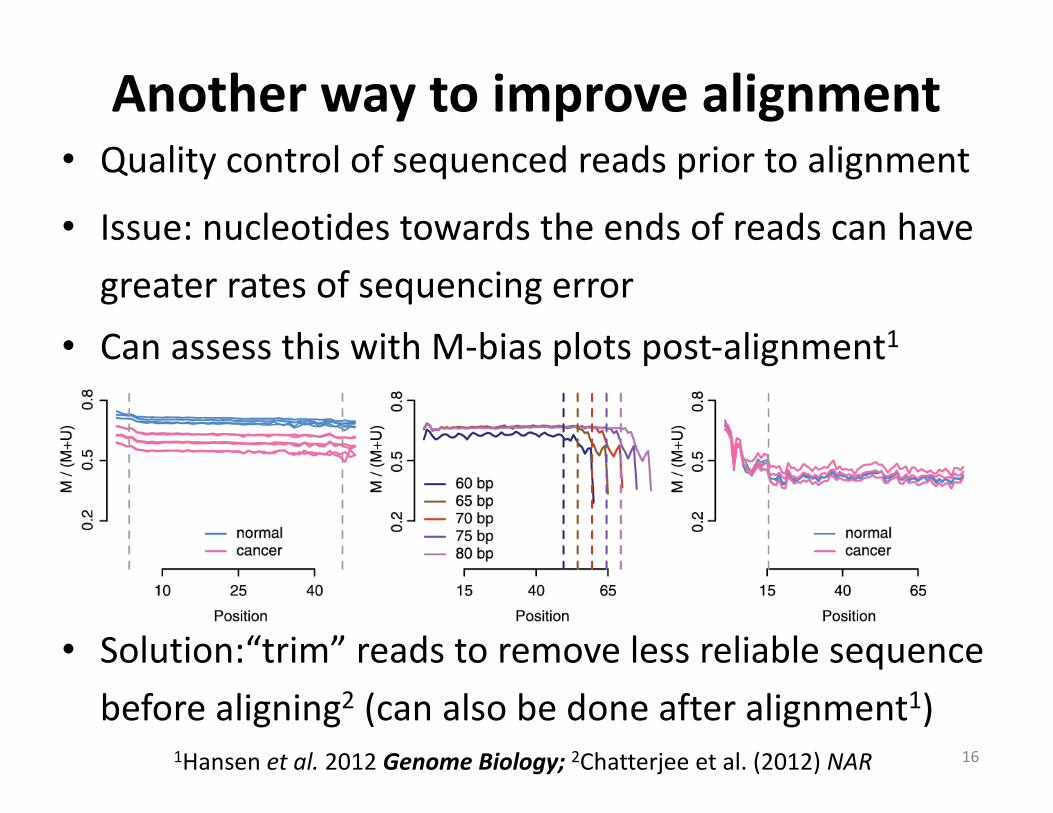
Anotherwaytoimprovealignment
16
• Qualitycontrolofsequencedreadspriortoalignment
• Issue:nucleotidestowardstheendsofreadscanhavegreaterratesofsequencingerror
• CanassessthiswithM-biasplotspost-alignment1
• Solution:“trim”readstoremovelessreliablesequencebeforealigning2 (canalsobedoneafteralignment1)
1Hansenetal.2012GenomeBiology;2Chatterjeeetal.(2012)NAR
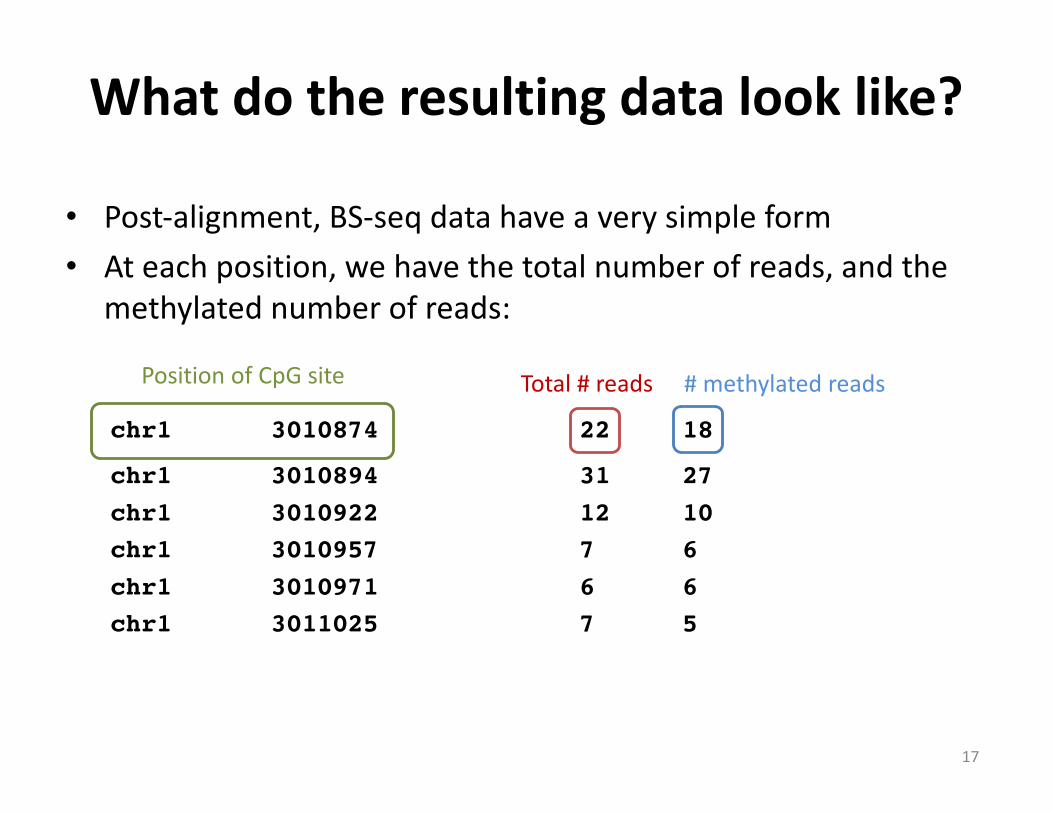
• Post-alignment,BS-seq datahaveaverysimpleform• Ateachposition,wehavethetotalnumberofreads,andthe
methylatednumberofreads:
chr1 3010874 22 18
chr1 3010894 31 27chr1 3010922 12 10chr1 3010957 7 6chr1 3010971 6 6chr1 3011025 7 5
Total#reads #methylatedreadsPositionofCpG site
Whatdotheresultingdatalooklike?
17

StudydesignforBS-seq studies• Highcostsà fewsamplestypicallyanalyzed• Twocommonstudydesigns– Analysisofasinglesample:
• Goal:observemethylationpatternsacrossgenome• Commonlydonetocharacterizemethylome foraparticularcelltypeorspecies
– Comparisonofseveralsamples:• Typicalgoal:comparemethylationlevelsbetweengroups• Differentialmethylationanalysis• ComparedwithChIP-seq andRNA-seq,methodsarestillinearlystage,andareoftenadhoc
18

StudydesignforBS-seq studies• Becausesofewsamplesareinvolvedinmoststudies,itiscrucialtoavoidallformsofheterogeneity– Inlargestudieswecanadjustfordifferencesviacovariates– WithsmallNmodelsoftencannotaccommodatecovariates
• Heterogeneity=differencesbetweensamplesotherthanvariableofinterest– Inadvertentdifferencesintissuesampled– Differencesincelltypemixingproportions– Geneticdifferencesbetweenindividuals– Agedifferencesbetweensamples– Different#ofpassagesforcelllines
19

Avoidingheterogeneity• Canavoidheterogeneitywithcarefulstudydesign
– Stringentcontroloftissuedissectionfortissuesampling
– Analysisofhomogeneouscelltypeswheneverpossible
– Useofwithin-individualcomparisonstoavoidgeneticand
demographicdifferences
• Example:pairedtumorandnormalsamplesfromsamepatients
• Ifnotpossible,matchcarefullyforethnicity,age,gender
– Carefulcontrolofcelllineexperiments
20

QualitycontrolofalignedBS-seq data
21
• Goal:removesiteslikelytobelow-qualityornon-informative– Bestfilteringstrategywilldependonstudydesignandgoals
• Filteringbasedonnon-uniquealignment– Willmostlyhappennaturallyduringalignmentprocess– Post-alignment,CpG siteswithunusuallyhighreadcountaresuspect
• Removalofsiteswithlowcoverage(often<5or10totalreads)– Appropriatecutoffwillvarydependingonanalysismethodused– Formethodsthatmodelreadcount,cansetcutofflower
• Filteringbasedonlackofvariability– Ifthegoalisdifferentialmethylationanalysis,removesiteswith0%of
readsmethylatedinallsamples,or100%methylatedinallsamples– Incontrast,ifgoalistocharacterizemethylationpatternsinaparticular
genome,keepthesesites!

Differentialmethylationanalysis• Typicalgoal:comparemethylationlevelsbetweentwogroups– Example:tumorvs.normaltissuesamples
– Important:dogroupscontainbiologicalreplicates?
– Somestudiesmaycompare1tumorto1normalsample
– Otherstudieswillinclude2ormorereplicatesofeach
• PopularadhocapproachesforthiscomparisonareFisher’sexacttestandtwo-groupt-test
• Wewillshowwhythesecanbeproblematic
22

Fisher’sexacttestwith2samples• Ifwehaveonlyonesamplepergroup(nobiologicalreplicates),Fisher’sexacttestisanaturalchoice
• Example:singleCpG sitesequencedfor2samples– Fortumorsample,32/44methylatedreads
– Fornormalsample,8/12methylatedreads
• CanthenperformFisher’sexacttestonthefollowingtable:
• OR=1.33
• p=.73
Methylated Unmeth. Totalreads
Tumor 32 12 44
Normal 8 4 12
Total 40 16 56
23

Fisher’sexacttestinmethylKit• Forcomparisonsbetweentwosamples,Fisher’sexacttestisareasonablechoice– EasytocarryoutinRusingfisher.test()function
– Alternatively,methylKit1 isasuiteofRfunctionsthatfacilitatesanalysisofgenome-widemethylationdata
– Differentialmethylationanalysisviaeither• Fisher’sexacttest(forcomparisonsbetweentwosamples)
• Logisticregressionbasedonmethylationproportions– Analogoustotwo-groupt-test,butwithcovariates
• Canperformanalysisinuser-definedtilingwindows– However,basedonsimplecollapsingofinformationacrosssitesratherthan
smoothing241Akalinetal.2012GenomeBiology

Fisher’sexacttestwith>2samples• ForFisher’sexacttestwithbiologicalreplicates,needtocollapsereadinformationwithingroups
• Example:singleCpG sitesequencedfor4samples– For2tumorsamples,32/44and4/10methylatedreads
– For2normalsamples,8/12and12/34methylatedreads
• CouldthenperformFisher’sexacttestonthefollowingtable:
• OR=2.6
• p=.0264
Methylated Unmeth. Totalreads
Tumor 36=32+4 18 54 =44+10
Normal 20= 8+12 26 46 =12+34
Total 56 44 100
25

ProblemwithFisher’sexacttest• ToperformFisher’sexacttestfor>2samples,wehaveto
collapsereadinformationacrosssampleswithineachgroup
• Bydoingthis,weareignoringinformationonbiologicalvariationbetweensamples– Biologicalvariation:naturalvariationinunderlyingfractionofDNA
methylatedbetweensamplesinthesamecondition
– Technicalvariation:variationinestimationofmethylationlevelsduetorandomsamplingofDNAduringsequencing1
• Bycollapsing,weareassumingthat:– sampleswithinagroupinherentlyhavethesameunderlying
fractionofDNAmethylated
– anyvariationbetweensamplesisduetotechnicalvariation
261Hansenetal.2012GenomeBiology

Naïvet-test• Example:singleCpGsitesequencedfor4samples
– For2tumorsamples,32/44and4/10methylatedreads
– For2normalsamples,8/12and12/34methylatedreads
• Fort-test,computeaproportionforeachsample– .727and.400fortumorsamples
– .667and.353fornormalsamples
• Differenceinmeanproportions=.563- .510=.053
• T-statistic=0.2375
• p=.834
27

Problemwitht-test• Toperformt-test,computedaproportionforeachsample
– Testinherentlygivesequalweighttoeachsample
– Doesnotaccountfortechnicalvariationinproportionestimates
– Recall:Technicalvariation=variationinestimationofmethylationlevelsduetorandomsamplingofDNA
– Canexpectthisvariationtobelowerforsampleswithmorereads
• Onepossiblesolutionwouldbetoincorporateweightsbasedonreadcount
• However,anotherissuewiththisapproachisthesmallnumberofsamples– WithN=4,thet-testhasverylittlepowerduetolowdf
28

Fisher’sexactvs.t-test• Thetwotestsyieldedverydifferentresults
– Fisher’sexactp=.0264
– T-testp=.834
• Maindifference:unitofobservation(readsvs.samples)
• Fisher’stestwasbasedon100“independent”reads– Readsareactuallynotindependentifthereisbiologicalvariation
– Correlatedwithineachsample,sincesampleshavedifferentmethylationfractions
• T-testwasbasedon4samples– Treatedsamplesasequallyinformative,whenreallytheyarenot
– For2tumorsamples,32/44and4/10methylatedreads
– For2normalsamples,8/12and12/34methylatedreads29

Needbetterapproaches• Problem:wanttotestmanysiteswithfewsamples
– Limitedinformationavailableateachsiteduetolow#ofsamples
• Solution:approachesthatborrowinformationacrosssites
– Smoothingapproachesthatshareinformationacrossnearbysites
• Usefulinsinglesampleanalysesthataimtocharacterizethegenome
• Usefulfordetectingdifferentialmethylatedregions(DMRs)ofthegenome
– Bayesianhierarchicalmodelthatborrowsinformationacrossthe
genome
• Usefulfordetectingdifferentiallymethylatedloci(DMLs)
30

Smoothingapproaches
• Firstconsideranalysisofasinglesample
• Goalhereistoidentifymethylatedregionsorloci:– CanestimateproportionofreadsthataremethylatedateachCposition,but:
• Variabilityinestimationneedstobeconsidered
• SpatialcorrelationamongnearbyCpG sitescanbeutilizedtoimproveestimation
– Methylatedregions(orstates)canbedeterminedbysmoothingbasedmethodsusingtheestimatedmethylationproportionasinput
31
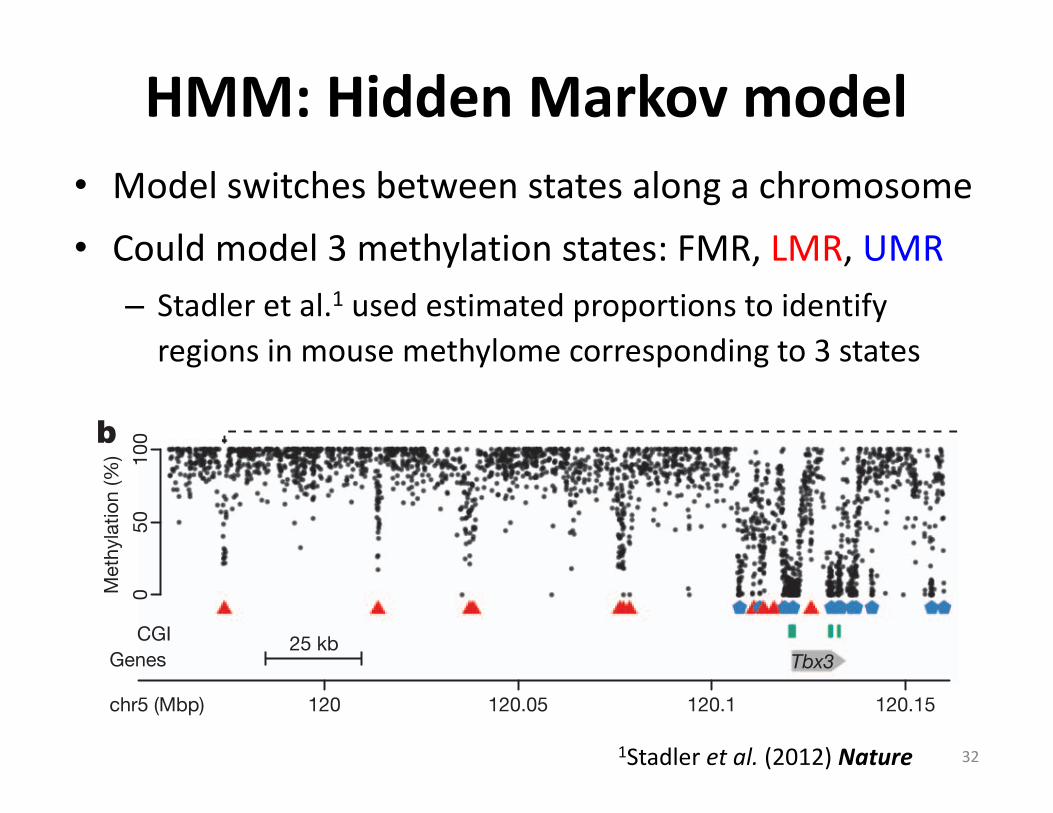
HMM:HiddenMarkovmodel• Modelswitchesbetweenstatesalongachromosome• Couldmodel3methylationstates:FMR,LMR,UMR
– Stadleretal.1 usedestimatedproportionstoidentifyregionsinmousemethylomecorrespondingto3states
DNase-I-hypersensitive sites (DHS), a unique chromatin state thatdepends on DNA-binding factors10–12. In fact, at least 80% of LMRsand 90% of UMRs overlap with DHS (Fig. 2 and SupplementaryFig. 2). LMRs are unlikely novel promoters as we find only weak signalfor RNA polymerase II (Fig. 2 and Supplementary Fig. 3) and no RNAsignal abovewhat we observe atmethylated regions evenwhen using astrand-specific protocol that does not require polyadenylation (Sup-plementary Fig. 3). Next, we explored if LMRs could represent distalregulatory regions, such as enhancers. Indeed, LMRs are stronglyenriched for chromatin features such as highH3K4monomethylation(H3K4me1) signal relative to H3K4 trimethylation (H3K4me3) andthe presence of p300 histone acetyltransferase, which are predictivefeatures of enhancers13 (Fig. 2). This indicates that a subset of LMRsare enhancers that, in light of the absence of H3K27me3 and thepresence of H3K27ac, are presumably active14 (Fig. 2b). Transgenicassays further show that individual LMRs increase the activity of alinked promoter and experimentally function as enhancers (Sup-plementary Fig. 4). We thus conclude that many LMRs, identifiedsolely by their DNA methylation pattern, represent active regulatoryregions.To investigate LMR features further, we combined newly generated
and published data sets for several DNA-binding factors and addi-tional histone modifications (Supplementary Table 1, Fig. 2b andSupplementary Figs 5 and 6). LMRs and UMRs are depleted for theheterochromatic histone modification H3K9me2 in agreement withthe absence of this mark at active chromatin6. Most DNA-bindingfactors show enrichment not only at UMRs, which are mostly pro-moters, but also at LMRs. Factors enriched at LMRs in stem cellsinclude pluripotency transcription factors such as Nanog, Oct4 andKlf4, but also structural DNA-binding factors such as the insulator
protein CTCF15 and members of the cohesin complex (Fig. 2b andSupplementary Fig. 5), both of which bind promoters and distalregulatory regions16. Notably, not all factors occupy distal andproximal regulatory regions with equal preferences. Smad1 binds toneither LMRs nor UMRs, whereas some bind primarily at UMRs, suchas KDM2A and Zfx, and others such as Nanog and Esrrb show higherenrichment at LMRs (Fig. 2b and Supplementary Fig. 5). In summary,several lines of evidence including genomic position, conservation,chromatin state, regulatory activity and transcription factor occupancysupport the hypothesis that LMRs are indeed active distal regulatoryregions.InterestinglyLMRsshowastrongpresenceof5-hydroxymethylcytosine
(5hmC), consistent with recent reports of 5hmC presence at enhancerregions17–19. One candidate protein responsible for catalysing 5hmC,Tet1 (refs 20, 21), is enriched at both UMRs and LMRs (Fig. 2b).To ask if LMRs are also present in other mammals we performed
HMM segmentation of a human stem cell methylome3, which alsoidentifies LMRswith similar features, indicating that these are a generalcharacteristic of mammalian methylomes (Supplementary Fig. 7).
Transcription factor binding creates LMRsTodetermine howLMRs are formed,we investigated theDNA-bindingprotein CTCF, which binds to regulatory regions including promoters,enhancers and insulators22,23.Wedetermined the genome-wide bindingof CTCF by chromatin immunoprecipitation followed by sequencing(ChIP-seq) (Supplementary Fig. 8), revealing high occupancy at bothUMRs and LMRs (Fig. 2b and Supplementary Fig. 5). A composite viewof DNA methylation shows an average methylation of 20% at CTCFbinding sites with increasing methylation adjacent to it (Supplemen-tary Fig. 9), in line with a previous report in primates24. If reducedmethylation is a general feature of CTCF-occupied sites, inclusion ofDNA methylation data should improve prediction of CTCF binding.
020
4060
8010
0M
ethy
latio
n (%
)
01
23
Enric
hmen
t
FMR UMR LMR
Tet15hmC.GLIB5hmC.CMSSmad1STAT3n-MycZfxKDM2AE2f1EsrrbKlf4NanogOct4Smc3Smc1NipblCTCFH3K27acH3K27me3H3K9me2p300Pol IIH3K4me3H3K4me2H3K4me1DNase IMethylation
a
b
UMRLMR
FMR
Mea
n co
nser
vatio
n Conservation
0 3–3 3–3 3–3
3–3 3–3 3–3
0.1
0.2
0.3
Enric
hmen
t (lo
g 2)
0
DNase I
0.0
0.5
1.0
1.5
Position around segment middle (kb)
Enric
hmen
t (lo
g 2)
00
00
H3K4me3 Pol II
H3K4me1 p300
0.0
0.5
1.0
0.0
1.0
2.0 1.
50.
00.
51.
01.
5
0.0
0.3
0.6
0.9
Figure 2 | General features of LMRs. Composite profiles 3 kb aroundsegment midpoints. a, Evolutionary conservation based on multi-speciesalignments (upper left). Enrichment of DNase I tags (lower left). Chromatinfeatures that predict enhancer function are enriched at LMRs (middle andright). b, Heat map of methylation levels, histone modifications and proteinbinding (H3K4me1 signal rescaled for visibility).
a c d
e f
b
025
5075
100
Met
hyla
tion
(%)
FMRLMRUMR
−3 0 3
Position around middle (kb)
0 5 10 15 20
0.00
0.10
Distance to TSS (log2 nt)
Den
sity
FMRLMRUMR
12
22
44
32
FMR(2,485.0 Mbp)
57
3
13 7
20
UMR(27.9 Mbp)
34
25
34
33
LMR(12.0 Mbp)
Promoter Exon Intron Repeat Intergenic
89 (1)
2 (1)9 (98)
CpG islands
FMR UMR LMR
(n = 15,974)
Methylation (%)
Frac
tion
of C
pGs
0.0
0.25
0.5
0−10
10−2
0
20−3
0
30−4
0
40−5
0
50−6
0
60−7
0
70−8
0
80−9
0
90−1
00
6.5% 4.1% 89.4%0
5010
0M
ethy
latio
n (%
)
CGITbx3
120 120.05 120.1 120.15chr5 (Mbp)
Genes
LMR
25 kb
Figure 1 | Features of the mouse ES cell methylome. a, Distribution of CpGmethylation frequency for all CpGs with at least tenfold coverage. Of allcytosines, 4.1% show intermediate methylation levels. b, Representativegenomic region. Computational segmentation identifies UMRs (bluepentagons), LMRs (red triangles) and FMRs (unmarked). Each dot representsone CpG (CpG islandsmarked in green). Included is an independently verifiedLMR upstream of Tbx3. Mbp, million base pairs. c, Composite profile of CpGmethylation for all three groups. kb, kilobases. d, Distances to TSS.e, f, Distribution of all three classes among genome features. e, A smallpercentage of LMRs overlap with CpG islands. Numbers indicate observedpercentage of overlaps per group (expected percentage in parentheses).f, Distribution of the regions throughout the genome.
ARTICLE RESEARCH
2 2 / 2 9 D E C E M B E R 2 0 1 1 | V O L 4 8 0 | N A T U R E | 4 9 1
Macmillan Publishers Limited. All rights reserved©2012
321Stadleretal.(2012)Nature

Smoothingsequencingdata• Problemwithdirectlysmoothingtheproportions:
– Doesn’tconsidertheuncertaintyinproportionestimates– EstimatesmorevariableforCpG siteswithlowreadcounts– Maywanttoputlessweightontheseestimates
• Abetterapproach:BSmooth model1
– Alocal-likelihoodsmoothingapproach– Keyassumptions:
• Truemethylationlevelπjisasmoothcurveofgenomiccoordinates.• TheobservedcountsMj followabinomial(Nj,πj)distribution.• BinomialassumptionaccountsfordifferencesinvariationforsampleswithdifferenttotalreadcountsNj
331Hansenetal.2012GenomeBiology

BSmoothsmoothing• NotationforCpG sitej:
– Nj,Mj:#totaland#methylatedreads– πj:underlyingtruemethylationlevel– lj:location
• Model:
whereβ0,β1,andβ2 varysmoothlyalongthegenome.
• Fitthisasaweightedgeneralizedlinearmodel(glm)• Obtainasmoothedmethylationestimateforeachpositionalongthegenomeusingslidingwindowapproach
M j ~ Bin(N j,π j )
log(π j / (1−π j )) = β0 +β1l j +β2l j2
34Hansenetal.2012GenomeBiology

Slidingwindowapproach• Choosewindowsize(eitherdistanceor#CpGsites)• Foreverygenomiclocationlj,usedatainwindowsurroundinglj
• Fitweightedglmforalldatainwindow,whereweightfordatapointk dependsinverselyon:– thevarianceofestimatedπk, estimatedasπk(1-πk)/Nk
– distanceofCpGsitefromwindowcenter|lk – lj |
• Estimationofβ0,β1,andβ2 inwindowsurroundingljprovidesestimateofπj
M j ~ Bin(N j,π j )
log(π j / (1−π j )) = β0 +β1l j +β2l j2
35Hansenetal.2012GenomeBiology
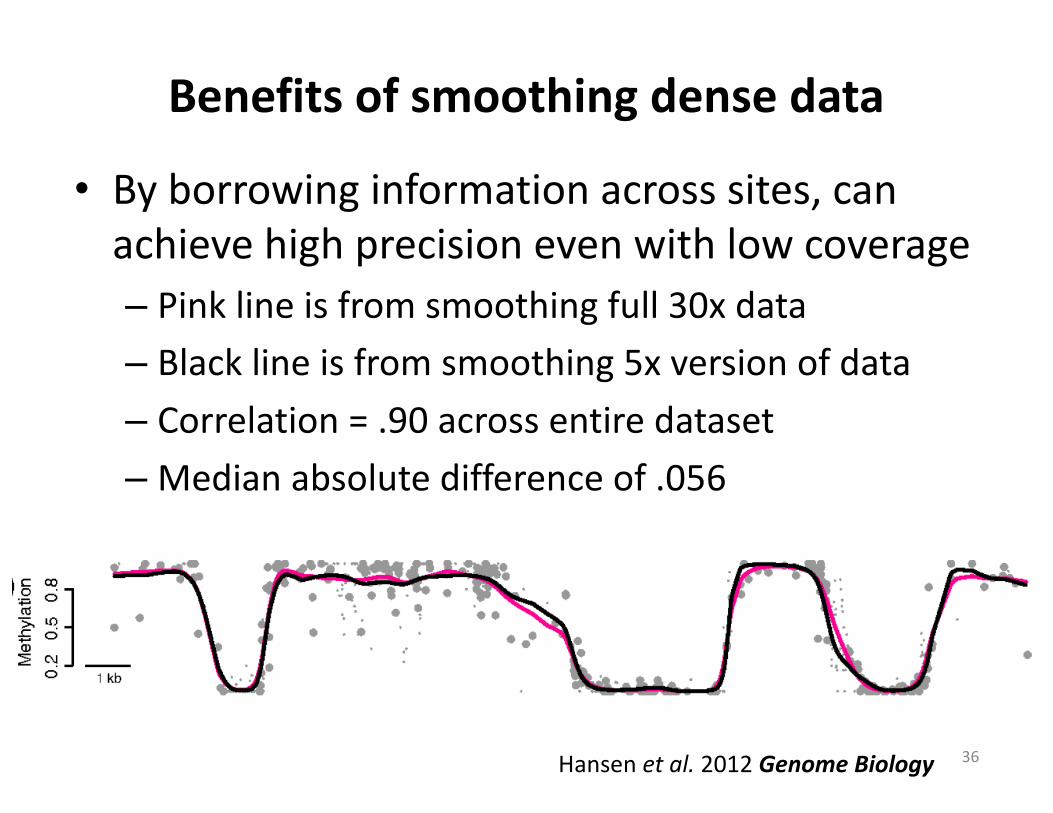
Benefitsofsmoothingdensedata
• Byborrowinginformationacrosssites,canachievehighprecisionevenwithlowcoverage– Pinklineisfromsmoothingfull30xdata– Blacklineisfromsmoothing5xversionofdata– Correlation=.90acrossentiredataset–Medianabsolutedifferenceof.056
36Hansenetal.2012GenomeBiology

Smootheddifferentialmethylationanalysis• Goal:identifyregionsdifferentiallymethylated(DMRs)betweengroups
• BSmooth computesat-test-likestatistic– Signal-to-noiseratiobasedonsmootheddataformultiplesamples
– Essentiallytheaveragedifferencebetweensmoothedprofilesfrom2groups,dividedbyestimatedstandarderror
– Whenbiologicalreplicatesareincluded,thisstatisticcorrectlyaccountsforbiologicalvariation
• IdentifyDMRsasregionswherethisstatisticexceedssomecutoff 37Hansenetal.2012GenomeBiology

BsmoothfunctionsimplementedinBioconductorpackagebsseq1
• Functionsfor– Smoothing– Smoothedt-tests– DMRidentification– Visualizationofresults– Fisher’sexacttest(notsmoothed)
• Canbeimplementedinparallelcomputingenvironmenttospeedupcalculation
381Hansenetal.2012GenomeBiology

Usebsseq
• FirstcreateBSseq objects• UseBSmooth functiontosmooth.• fisherTests performsFisher’sexacttest,ifthere’sno
replicate.• BSmooth.tstat performst-testwithreplicates.• dmrFinder callsDMRsbasedonBSmooth.tstat results.

library(bsseq)library(bsseqData)
## take chr21 on BS.cancer.ex to speed up calculationdata(BS.cancer.ex)ix = which(seqnames(BS.cancer.ex)=="chr21")BS.chr21 = BS.cancer.ex[ix,]
## use BSmooth to smooth and call DMRBS.chr21 = BSmooth(BS.chr21) ## this takes 1-2 minutes
## perform t-testBS.chr21.tstat = BSmooth.tstat(BS.chr21,
c("C1","C2","C3"),c("N1","N2","N3"))
## call DMRdmr.BSmooth <- dmrFinder(BS.chr21.tstat, cutoff = c(-4.6, 4.6))
40

Anotherapproach:Bayesianhierarchicalmodel1
• Hierarchicalmodeltoseparatelymodelbiologicalandtechnicalvariation– Biologicalvariation:naturalvariationinunderlyingfractionof
DNAmethylatedbetweensamplesinthesamecondition– Technicalvariation:variationinestimationofmethylationlevels
duetorandomsamplingofDNAduringsequencing1
– Manymethodsonlycaptureoneortheother– Fisher’sexacttest:technicalvariationonly– Naïvet-test:biologicalvariationonly
• Shrinkageapproachallowsustoborrowinformationaboutvariationacrossgenome– EspeciallyusefulwheninformationperCpG siteislimitedbylow
numberofsamples411Fengetal.2014NucleicAcidsResearch

Beta-binomialhierarchicalmodel• “ThemostnaturalstatisticalmodelforreplicatedBS-seq DNA
methylationmeasurements”1
• SamplingofreadsforeachCpG sitewillfollowabinomialdistribution– OutofNreadscoveringaparticularsite,howmanyaremethylated?
– Thisnumberwillfollowabinomial(N,π)distribution
– However,πmayvaryacrossreplicates
• Tomodelthebiologicalvariationofπacrossreplicates,thebetadistributionisanaturalchoice
• Beta-binomialdistributionusedtomodelmethylatedreadsinDSS2,BiSeq3,MOABS4,RADMeth5,MethylSig6
421Robinsonetal.2014;2Fengetal.2014;3Hebestreitetal.2013;4Sunetal.2014;5Dolzhenko&Smith2014;6Parketal.2014
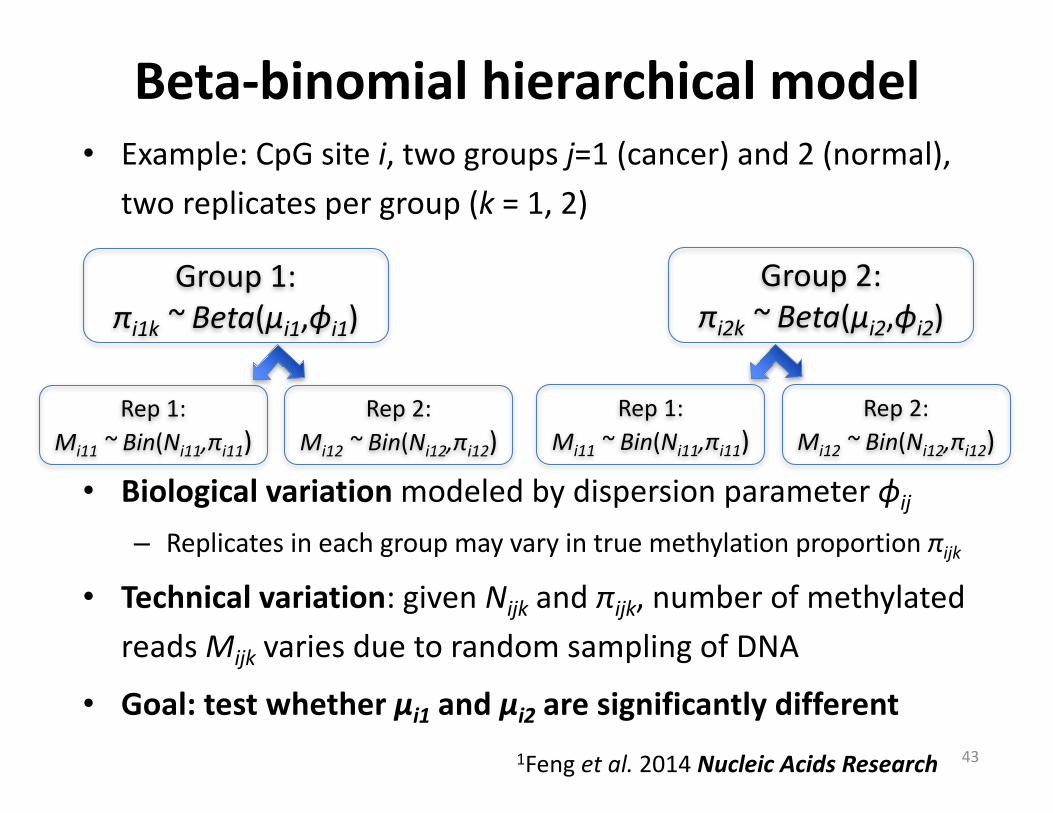
Beta-binomialhierarchicalmodel• Example:CpG sitei,twogroupsj=1(cancer)and2(normal),
tworeplicatespergroup(k =1,2)
• Biologicalvariationmodeledbydispersionparameterϕij
– Replicatesineachgroupmayvaryintruemethylationproportionπijk
• Technicalvariation:givenNijk andπijk,numberofmethylatedreadsMijk variesduetorandomsamplingofDNA
• Goal:testwhetherμi1 andμi2 aresignificantlydifferent43
Group1:πi1k ~Beta(μi1,ϕi1)
Group2:πi2k ~Beta(μi2,ϕi2)
Rep1:Mi11 ~Bin(Ni11,πi11)
Rep2:Mi12 ~Bin(Ni12,πi12)
Rep1:Mi11 ~Bin(Ni11,πi11)
Rep2:Mi12 ~Bin(Ni12,πi12)
1Fengetal.2014NucleicAcidsResearch

Motivationforshrinkageapproach• Hierarchicalmodel:
• Goal:aftercorrectlymodelingdifferentsourcesofvariation,testwhetherμi1 andμi2 aresignificantlydifferentatCpG i
• Possiblelimitationofmodel:withsmallnumberofsamples,estimationofparametersmaybepoor– Inparticular,difficulttoaccuratelyestimatedispersionϕij withonly2-
3replicatespergroup
– Estimatesmayvarywildlyduetosmallnumbers
• Solution:borrowinformationfromCpG sitesacrossthegenometoobtainreasonableestimatesofϕij
44
Mijk ~ Binomial Nijk ,π ijk( )π ijk ~ Beta µij ,φij( )
1Fengetal.2014NucleicAcidsResearch

• Toobtainstableestimatesofdispersionwithfewsamples,we:– imposealog-normalprioronϕ:– useinformationfromallCpGs inthegenometoestimatethe
parametersmj andrj2
• Choiceoflog-normalpriorwasmotivatedbydistributionofdispersioninbisulfitesequencingdata– RRBSdatafrommouseembryogenesisstudy
(Smithetal.2012Nature)– Estimationrobusttodeparture
fromlog-normality– Priorprovidesagood“referee”– Encouragesdispersionestimates
tostaywithinbounds45
Smith et al. data
log(estimated dispersion)−7 −6 −5 −4 −3 −2 −1
Estimatingdispersionparameter
φij ~ lognormal mj ,rj2( )
1Fengetal.2014NucleicAcidsResearch

WaldtestforDML,basedonhierarchicalmodel1
• DML:DifferentiallyMethylatedLoci– TestfordifferentialmethylationateachCpG site
• Atsitei,test:
• Basicalgorithm:– Usenaïveestimatesofϕ acrossgenometoestimateprior
– Foreachsitei,estimateμi1 andμi2 asproportionofmethylatedreadsforeachgroup
– Bayesianestimationofϕij basedondataandprior
– Pluginestimatesofμij andϕij tocreateWaldstatisticofform
Xijk|Nijk, pijk ⇠ Bin(Nijk, pijk)
pijk ⇠ Beta(µik,�i)
H0 : µi1 = µi2
In beta distribution
E(X) =↵
↵+ �⌘ µ; V (X) =
↵�
(↵+ �)2(↵+ � + 1)⌘ �
2
�2 = µ ⇤ (1� µ) ⇤ 1
↵+�+1
� ⌘ 1↵+�+1
In beta-binomial
E(X) =N↵
↵�⌘ Nµ
V (X) =N↵�(↵+ � +N)
(↵+ �)2(↵+ � + 1)⌘ �
2
�2 = Nµ ⇤ (1� µ)⇤
1
46
ti =µi1 − µi2
Var µi1 − µi2( )1Fengetal.2014NucleicAcidsResearch

UsingDSS tocallDMLandDMRs• DSScanidentifydifferentiallymethylatedloci (DML)andregions (DMRs)– DMLidentifiedviaWaldtest,basedonp-valuethreshold– DMRscalledfromDMLbasedonuser-specifiedcriteria(regionlength,p-valueandeffectsizethresholds)
• NewfeaturesinDSS– Accommodatessingle-replicatestudiesbysmoothingdatafromnearbyCpG sitestoform“pseudo-replicates”1
– Inclusionofdesignmatrixtoallowcovariatesandamoregeneralexperimentaldesign2
47
1Wuetal.NucleicAcidsResearch2015.2Parketal.Bioinformatics 2016.

BS-seqexperimentundergeneraldesign
• Generalexperimentaldesign:– Multiplegroups.– Multiplefactors,crossed/nested.– Continuouscovariates.
• Limiteddataanalysismethodswithnotsogoodproperties:– BiSeq andRADMeth,bothbasedongeneralizedlinearmodel(GLM).
– Computationallydemanding.– Numericallyunstable.

DSS-general
• SupposetheinputdataincludeN CpGsitesandD samples.• Notations:
– Yid,mid:methylatedandtotalcountsforith CpGanddthdataset.
– πid,Φi:meananddispersion.– X:fullrankeddesignmatrixofdimensionDbyp.
• Countsaremodeledbyabeta-binomialregression:
• DMLdetectionisachievedbyageneralhypothesistesting:whereCisap-vector.
This is for slides
Yid ⇠ beta-bin(mid,⇡id,�i)
g(⇡id) = xd�i
1
This is for slides
Yid ⇠ beta-bin(mid,⇡id,�i)
g(⇡id) = xd�i
H0 : CT�i = 0, where C is a p-vector.
1

Ourapproach- approximation
• Beta-binomialregression.• Transformation:– g(Y/m)asresponseordata–Whatisg(·)?
• Applyinggeneralized(weighted)leastsquaretoestimateparameters,butwithcaution!

Choiceofthelinkfunction
• arcsinelink:• “Variancestabilizationtransformation”forbinomialproportion:– Varianceofthetransformeddatadoesnotdependonmean(butondispersion),soleastsquareapproachispossible.
– logitorprobit transformeddataneedsiterativeproceduresincevariancedependsonmean.
– Morelinearthanlogit orprobit,especiallyattheboundaries.
This is for slides
Yid ⇠ beta-bin(mid,⇡id,�i)
g(⇡id) = xd�i
H0 : CT�i = 0, where C is a p-vector.
g(x) = arcsin(2x� 1).
1

Parameterestimation
Considering a transformation Zid = arcsin(2Yid/mid � 1). We have:
E[Zid] ⇡ arcsin(2E[Yid]/mid � 1) = arcsin(2⇡id � 1) = xd�i.
The variance of Zid can be obtained as follows (refer to Supplementary Materials for more details)
var(Zid) ⇡1 + (mid � 1)�i
mid. (2)
Given dispersion parameter �i, a GLS method can be applied to estimate the regression
coe�cients �i. To be specific, define the following covariance matrix:
Vi = diag
✓1 + (mid � 1)�i
mid
◆,
then
�i = (XTV
�1i X)�1XT
V�1i Z.
For beta-binomial model, there are several ways to estimate dispersion parameter such as
maximizing likelihood, and using Pearson �2 or deviance statistics. Here we propose to use
Pearson �2 statistics based on transformed linear model to estimate �i because it is less
computationally demanding and has relatively good property. We first let �i = 0 and the initial
covariance matrix is Vi0 = diag(1/mid). Then the parameter estimator from GLS with covariance
matrix V0 is: �(0)i = (XT
V�1i0 X)�1XT
V�1i0 Z.
Consider Pearson chi-square statistics �2i =
Pdmid(Zid � xd�0
i )2. Let �2
i = �2i /(D � p), an
estimator for �i is obtained as below (detailed derivations provided in Supplementary Materials):
�i =D(�2
i � 1)Pd(mid � 1)
. (3)
Note that our model is based on beta-binomial distribution and hence 0 < �i < 1, which requires
1 < �2i <
Pd(mid�1)
D + 1. However, because of random variation and approximation bias, it is
possible that �2i does not satisfy the constraints. To avoid this, we take an ad hoc procedure to
force �i to be bounded by 0.001 and 0.999. This procedure achieves some “shrinkage” e↵ects,
which helps stabilize the result.
Given estimated dispersion, the estimate of variance structure is
Vi = diag
1 + (mid � 1)�i
mid
!.
18
Considering a transformation Zid = arcsin(2Yid/mid � 1). We have:
E[Zid] ⇡ arcsin(2E[Yid]/mid � 1) = arcsin(2⇡id � 1) = xd�i.
The variance of Zid can be obtained as follows (refer to Supplementary Materials for more details)
var(Zid) ⇡1 + (mid � 1)�i
mid. (2)
Given dispersion parameter �i, a GLS method can be applied to estimate the regression
coe�cients �i. To be specific, define the following covariance matrix:
Vi = diag
✓1 + (mid � 1)�i
mid
◆,
then
�i = (XTV
�1i X)�1XT
V�1i Z.
For beta-binomial model, there are several ways to estimate dispersion parameter such as
maximizing likelihood, and using Pearson �2 or deviance statistics. Here we propose to use
Pearson �2 statistics based on transformed linear model to estimate �i because it is less
computationally demanding and has relatively good property. We first let �i = 0 and the initial
covariance matrix is Vi0 = diag(1/mid). Then the parameter estimator from GLS with covariance
matrix V0 is: �(0)i = (XT
V�1i0 X)�1XT
V�1i0 Z.
Consider Pearson chi-square statistics �2i =
Pdmid(Zid � xd�0
i )2. Let �2
i = �2i /(D � p), an
estimator for �i is obtained as below (detailed derivations provided in Supplementary Materials):
�i =D(�2
i � 1)Pd(mid � 1)
. (3)
Note that our model is based on beta-binomial distribution and hence 0 < �i < 1, which requires
1 < �2i <
Pd(mid�1)
D + 1. However, because of random variation and approximation bias, it is
possible that �2i does not satisfy the constraints. To avoid this, we take an ad hoc procedure to
force �i to be bounded by 0.001 and 0.999. This procedure achieves some “shrinkage” e↵ects,
which helps stabilize the result.
Given estimated dispersion, the estimate of variance structure is
Vi = diag
1 + (mid � 1)�i
mid
!.
18
Considering a transformation Zid = arcsin(2Yid/mid � 1). We have:
E[Zid] ⇡ arcsin(2E[Yid]/mid � 1) = arcsin(2⇡id � 1) = xd�i.
The variance of Zid can be obtained as follows (refer to Supplementary Materials for more details)
var(Zid) ⇡1 + (mid � 1)�i
mid. (2)
Given dispersion parameter �i, a GLS method can be applied to estimate the regression
coe�cients �i. To be specific, define the following covariance matrix:
Vi = diag
✓1 + (mid � 1)�i
mid
◆,
then
�i = (XTV
�1i X)�1XT
V�1i Z.
For beta-binomial model, there are several ways to estimate dispersion parameter such as
maximizing likelihood, and using Pearson �2 or deviance statistics. Here we propose to use
Pearson �2 statistics based on transformed linear model to estimate �i because it is less
computationally demanding and has relatively good property. We first let �i = 0 and the initial
covariance matrix is Vi0 = diag(1/mid). Then the parameter estimator from GLS with covariance
matrix V0 is: �(0)i = (XT
V�1i0 X)�1XT
V�1i0 Z.
Consider Pearson chi-square statistics �2i =
Pdmid(Zid � xd�0
i )2. Let �2
i = �2i /(D � p), an
estimator for �i is obtained as below (detailed derivations provided in Supplementary Materials):
�i =D(�2
i � 1)Pd(mid � 1)
. (3)
Note that our model is based on beta-binomial distribution and hence 0 < �i < 1, which requires
1 < �2i <
Pd(mid�1)
D + 1. However, because of random variation and approximation bias, it is
possible that �2i does not satisfy the constraints. To avoid this, we take an ad hoc procedure to
force �i to be bounded by 0.001 and 0.999. This procedure achieves some “shrinkage” e↵ects,
which helps stabilize the result.
Given estimated dispersion, the estimate of variance structure is
Vi = diag
1 + (mid � 1)�i
mid
!.
18
Considering a transformation Zid = arcsin(2Yid/mid � 1). We have:
E[Zid] ⇡ arcsin(2E[Yid]/mid � 1) = arcsin(2⇡id � 1) = xd�i.
The variance of Zid can be obtained as follows (refer to Supplementary Materials for more details)
var(Zid) ⇡1 + (mid � 1)�i
mid. (2)
Given dispersion parameter �i, a GLS method can be applied to estimate the regression
coe�cients �i. To be specific, define the following covariance matrix:
Vi = diag
✓1 + (mid � 1)�i
mid
◆,
then
�i = (XTV
�1i X)�1XT
V�1i Z.
For beta-binomial model, there are several ways to estimate dispersion parameter such as
maximizing likelihood, and using Pearson �2 or deviance statistics. Here we propose to use
Pearson �2 statistics based on transformed linear model to estimate �i because it is less
computationally demanding and has relatively good property. We first let �i = 0 and the initial
covariance matrix is Vi0 = diag(1/mid). Then the parameter estimator from GLS with covariance
matrix V0 is: �(0)i = (XT
V�1i0 X)�1XT
V�1i0 Z.
Consider Pearson chi-square statistics �2i =
Pdmid(Zid � xd�0
i )2. Let �2
i = �2i /(D � p), an
estimator for �i is obtained as below (detailed derivations provided in Supplementary Materials):
�i =D(�2
i � 1)Pd(mid � 1)
. (3)
Note that our model is based on beta-binomial distribution and hence 0 < �i < 1, which requires
1 < �2i <
Pd(mid�1)
D + 1. However, because of random variation and approximation bias, it is
possible that �2i does not satisfy the constraints. To avoid this, we take an ad hoc procedure to
force �i to be bounded by 0.001 and 0.999. This procedure achieves some “shrinkage” e↵ects,
which helps stabilize the result.
Given estimated dispersion, the estimate of variance structure is
Vi = diag
1 + (mid � 1)�i
mid
!.
18
This is for slides
Yid ⇠ beta-bin(mid,⇡id,�i)
g(⇡id) = xd�i
1
• Model:
• Transformation:
• Leastsquareestimator:

Two-stepestimation• Dispersionestimation
– Estimatebysettingdispersionto0.– EstimatevariancebasedonPearson’schi-squarestatistics:
,– Dispersioncanbederivedas:
– Restriction:
• ParameterestimationusingGLSbasedon
Considering a transformation Zid = arcsin(2Yid/mid � 1). We have:
E[Zid] ⇡ arcsin(2E[Yid]/mid � 1) = arcsin(2⇡id � 1) = xd�i.
The variance of Zid can be obtained as follows (refer to Supplementary Materials for more details)
var(Zid) ⇡1 + (mid � 1)�i
mid. (2)
Given dispersion parameter �i, a GLS method can be applied to estimate the regression
coe�cients �i. To be specific, define the following covariance matrix:
Vi = diag
✓1 + (mid � 1)�i
mid
◆,
then
�i = (XTV
�1i X)�1XT
V�1i Z.
For beta-binomial model, there are several ways to estimate dispersion parameter such as
maximizing likelihood, and using Pearson �2 or deviance statistics. Here we propose to use
Pearson �2 statistics based on transformed linear model to estimate �i because it is less
computationally demanding and has relatively good property. We first let �i = 0 and the initial
covariance matrix is Vi0 = diag(1/mid). Then the parameter estimator from GLS with covariance
matrix V0 is: �(0)i = (XT
V�1i0 X)�1XT
V�1i0 Z.
Consider Pearson chi-square statistics �2i =
Pdmid(Zid � xd�0
i )2. Let �2
i = �2i /(D � p), an
estimator for �i is obtained as below (detailed derivations provided in Supplementary Materials):
�i =D(�2
i � 1)Pd(mid � 1)
. (3)
Note that our model is based on beta-binomial distribution and hence 0 < �i < 1, which requires
1 < �2i <
Pd(mid�1)
D + 1. However, because of random variation and approximation bias, it is
possible that �2i does not satisfy the constraints. To avoid this, we take an ad hoc procedure to
force �i to be bounded by 0.001 and 0.999. This procedure achieves some “shrinkage” e↵ects,
which helps stabilize the result.
Given estimated dispersion, the estimate of variance structure is
Vi = diag
1 + (mid � 1)�i
mid
!.
18
Considering a transformation Zid = arcsin(2Yid/mid � 1). We have:
E[Zid] ⇡ arcsin(2E[Yid]/mid � 1) = arcsin(2⇡id � 1) = xd�i.
The variance of Zid can be obtained as follows (refer to Supplementary Materials for more details)
var(Zid) ⇡1 + (mid � 1)�i
mid. (2)
Given dispersion parameter �i, a GLS method can be applied to estimate the regression
coe�cients �i. To be specific, define the following covariance matrix:
Vi = diag
✓1 + (mid � 1)�i
mid
◆,
then
�i = (XTV
�1i X)�1XT
V�1i Z.
For beta-binomial model, there are several ways to estimate dispersion parameter such as
maximizing likelihood, and using Pearson �2 or deviance statistics. Here we propose to use
Pearson �2 statistics based on transformed linear model to estimate �i because it is less
computationally demanding and has relatively good property. We first let �i = 0 and the initial
covariance matrix is Vi0 = diag(1/mid). Then the parameter estimator from GLS with covariance
matrix V0 is: �(0)i = (XT
V�1i0 X)�1XT
V�1i0 Z.
Consider Pearson chi-square statistics �2i =
Pdmid(Zid � xd�0
i )2. Let �2
i = �2i /(D � p), an
estimator for �i is obtained as below (detailed derivations provided in Supplementary Materials):
�i =D(�2
i � 1)Pd(mid � 1)
. (3)
Note that our model is based on beta-binomial distribution and hence 0 < �i < 1, which requires
1 < �2i <
Pd(mid�1)
D + 1. However, because of random variation and approximation bias, it is
possible that �2i does not satisfy the constraints. To avoid this, we take an ad hoc procedure to
force �i to be bounded by 0.001 and 0.999. This procedure achieves some “shrinkage” e↵ects,
which helps stabilize the result.
Given estimated dispersion, the estimate of variance structure is
Vi = diag
1 + (mid � 1)�i
mid
!.
18
Considering a transformation Zid = arcsin(2Yid/mid � 1). We have:
E[Zid] ⇡ arcsin(2E[Yid]/mid � 1) = arcsin(2⇡id � 1) = xd�i.
The variance of Zid can be obtained as follows (refer to Supplementary Materials for more details)
var(Zid) ⇡1 + (mid � 1)�i
mid. (2)
Given dispersion parameter �i, a GLS method can be applied to estimate the regression
coe�cients �i. To be specific, define the following covariance matrix:
Vi = diag
✓1 + (mid � 1)�i
mid
◆,
then
�i = (XTV
�1i X)�1XT
V�1i Z.
For beta-binomial model, there are several ways to estimate dispersion parameter such as
maximizing likelihood, and using Pearson �2 or deviance statistics. Here we propose to use
Pearson �2 statistics based on transformed linear model to estimate �i because it is less
computationally demanding and has relatively good property. We first let �i = 0 and the initial
covariance matrix is Vi0 = diag(1/mid). Then the parameter estimator from GLS with covariance
matrix V0 is: �(0)i = (XT
V�1i0 X)�1XT
V�1i0 Z.
Consider Pearson chi-square statistics �2i =
Pdmid(Zid � xd�0
i )2. Let �2
i = �2i /(D � p), an
estimator for �i is obtained as below (detailed derivations provided in Supplementary Materials):
�i =D(�2
i � 1)Pd(mid � 1)
. (3)
Note that our model is based on beta-binomial distribution and hence 0 < �i < 1, which requires
1 < �2i <
Pd(mid�1)
D + 1. However, because of random variation and approximation bias, it is
possible that �2i does not satisfy the constraints. To avoid this, we take an ad hoc procedure to
force �i to be bounded by 0.001 and 0.999. This procedure achieves some “shrinkage” e↵ects,
which helps stabilize the result.
Given estimated dispersion, the estimate of variance structure is
Vi = diag
1 + (mid � 1)�i
mid
!.
18
DSS-general
then�i = (XTV �1
i X)�1XTV �1i Z.
For beta-binomial model, there are several ways to estimate dispersionparameter such as maximizing likelihood, and using Pearson �
2 or deviancestatistics. Here we propose to use Pearson �
2 statistics based on transformedlinear model to estimate �i because it is less computationally demanding andhas relatively good property. We first let �i = 0 and the initial covariancematrix is Vi0 = diag(1/mid). The parameter estimator from GLS withcovariance matrix Vi0 is: �(0)
i = (XTV �1i0 X)�1XTV �1
i0 Z.
Consider Pearson chi-square statistics �2i =
Pd mid(Zid � xd�0
i )2.
Let �2i = �
2i /(D � p), an estimator for �i is obtained as below (detailed
derivations provided in Supplementary Materials):
�i =D(�2
i � 1)P
d(mid � 1). (3)
Our model is based on beta-binomial distribution and hence 0 < �i < 1,which requires 1 < �
2i <
Pd(mid�1)
D + 1. However, because of randomvariation and approximation bias, it is possible that �2
i does not satisfy theconstraints. To avoid this, we restrict �i to be bounded by 0.001 and 0.999.
Given estimated dispersion, the estimate of variance structure is now
Vi = diag
1 + (mid � 1)�i
mid
!.
GLS procedure is applied once more based on Vi, and the updated estimatesfor regression coefficients and covariance matrix are obtained as
�i = (XT V �1i X)�1XT V �1
i Z,
and⌃i ⌘ \var(�i) = (XT V �1
i X)�1.
The estimation procedure utilizes two GLS for each CpG site withoutrelying on intensive iterative algorithm. It has profound connection witha beta-binomial GLM, in which the initial regression coefficients areestimated from logistic regression and using Pearson �
2 statistics to estimatedispersion parameter similar to equation (3) (Hinde and Demetrio, 1998).The covariance structure Vi is diagonal matrix. Thus our GLS procedure isalso a wighted least square, where weight for sample d is V �1/2
id .
2.4 Hypothesis testing
Hypothesis testing for differential methylation at CpG site i can beformulated as: H0 : CT�i = 0. Here C is a p-vector. The procedure isvery general and can test any linear combination of the effects. For example,to test the effect of factor k, C will be a vector having 1 at the k
th elementand 0’s in all others. With point estimation and estimated covariance matrix,the null hypothesis is tested through a standard Wald test procedure. TheWald test statistics is calculated as:
ti =CT �iqCT ⌃iC
.
The Wald test statistics approximately follow normal distribution, andthe p-values can be obtained accordingly. False discovery rate (FDR) willbe estimated using established procedures such as Benjamini-Hochberg’smethod (Benjamini and Hochberg, 1995).
2.5 Simulation settings
In all simulations, data are generated semi-parametrically. The countsare generated from the data model described in Equation (1) with modelparameters estimated from the human lung adenocarcinoma dataset. Themodel is 2 ⇥ 2 factorial design with 20,000 CpG sites for 3 replicatesin each condition group (12 datasets in total). For each factor, 5% of theCpG sites are DML, and the DM status for two factors are independentlygenerated. The regression coefficients �g (g = 0, 1, 2) are simulated in
the following way: (1) �0 (the intercept) is randomly sampled from theestimated intercepts from real data; (2) �1 and �2 are set to be 0 if the CpGsite is not DML, and sampled from N(0, 1) if the CpG site is DML; (3)The dispersion parameter �i’s are independently generated from log-normaldistribution with mean -3 and standard deviation 0.7, which are similar tothe real data estimates.
We also perform simulations when data are generated from a GLM with“logit” link to assess the robustness of our method. In this case, since thescales of the coefficients under “logit” link are greater than those from“arcsine” link for the same data (by a ratio of approximately 2.3), wemultiply 2.3 for �g’s for all simulations using “logit” link.
3 RESULTS3.1 Simulation
Comprehensive simulation studies are conducted to evaluate theperformance of DSS-general from several different aspects.
3.1.1 Dispersion estimation Dispersion parameter is an importantcomponent in various types of differential analysis for sequencingdata. Improved dispersion estimation from RNA-seq and BS-seqin two-group comparison has been shown to lead to better resultsin differential expression and differential methylation analyses(Robinson and Smyth, 2007; Wu et al., 2013; Feng et al., 2014;Love et al., 2014).
The estimated dispersions from DSS-general are compared tothe true ones in this simulation. Overall, the Pearson correlationbetween estimated and true dispersions is moderate at around 0.4.Data exploration indicates that large differences of estimated andtrue dispersions are mostly comes from the following two types ofCpG sites: (1) those with average methylation levels very close to0 or 1 and (2) those with low sequencing depth. For those CpGsites, it is not surprising to see estimates with large variation dueto low in-data information. The correlation indeed improves to 0.55when restricted to those with average methylation levels between0.3 and 0.7, and to 0.74 when further restricted to those with averagesequencing depth of at least 30. Figure ?? shows a plot for estimatedvs true dispersions for CpG sites with methylation levels between0.3 and 0.7. Sites with different levels of sequencing depth arerepresented by different colors. It shows reasonably good dispersionestimation.
3.1.2 DML detection accuracy We next compare the DMLdetection accuracies from several methods, including DSS-general,RADMeth, BiSeq and a binomial GLM with “logit” link incomparison. Here, the proportion of true positives among a givennumber of top-ranked CpGs is used as criterion. This refers to truediscovery rate (TDR) hereafter. Higher TDR is expected from bettermethod. This criterion is also referred to precision–recall analysis.Because the proportion of true positives is usually very low in DManalysis (5% in simulation setting), TDR is a better measurementof the accuracy for genome-wide differential analysis than receiveroperating characteristic (ROC) (Davis and Goadrich, 2006).
Each simulation is repeated 50 times to obtain the averageTDR estimates. Figure 1(A) shows the TDR curves up to top1,000 (5% of total) CpG sites when data are simulated using“arcsine” link function. It can be seen that DSS-general outperformsother methods for all top ranked CpG sites. For example, amongtop 200 ranked CpGs from DSS-general, 99.8% are true DML,whereas the percentages are 93.6%, 82.4%, and 38.4% from
3
DSS-general
then�i = (XTV �1
i X)�1XTV �1i Z.
For beta-binomial model, there are several ways to estimate dispersionparameter such as maximizing likelihood, and using Pearson �
2 or deviancestatistics. Here we propose to use Pearson �
2 statistics based on transformedlinear model to estimate �i because it is less computationally demanding andhas relatively good property. We first let �i = 0 and the initial covariancematrix is Vi0 = diag(1/mid). The parameter estimator from GLS withcovariance matrix Vi0 is: �(0)
i = (XTV �1i0 X)�1XTV �1
i0 Z.
Consider Pearson chi-square statistics �2i =
Pd mid(Zid � xd�0
i )2.
Let �2i = �
2i /(D � p), an estimator for �i is obtained as below (detailed
derivations provided in Supplementary Materials):
�i =D(�2
i � 1)P
d(mid � 1). (3)
Our model is based on beta-binomial distribution and hence 0 < �i < 1,which requires 1 < �
2i <
Pd(mid�1)
D + 1. However, because of randomvariation and approximation bias, it is possible that �2
i does not satisfy theconstraints. To avoid this, we restrict �i to be bounded by 0.001 and 0.999.
Given estimated dispersion, the estimate of variance structure is now
Vi = diag
1 + (mid � 1)�i
mid
!.
GLS procedure is applied once more based on Vi, and the updated estimatesfor regression coefficients and covariance matrix are obtained as
�i = (XT V �1i X)�1XT V �1
i Z,
and⌃i ⌘ \var(�i) = (XT V �1
i X)�1.
The estimation procedure utilizes two GLS for each CpG site withoutrelying on intensive iterative algorithm. It has profound connection witha beta-binomial GLM, in which the initial regression coefficients areestimated from logistic regression and using Pearson �
2 statistics to estimatedispersion parameter similar to equation (3) (Hinde and Demetrio, 1998).The covariance structure Vi is diagonal matrix. Thus our GLS procedure isalso a wighted least square, where weight for sample d is V �1/2
id .
2.4 Hypothesis testing
Hypothesis testing for differential methylation at CpG site i can beformulated as: H0 : CT�i = 0. Here C is a p-vector. The procedure isvery general and can test any linear combination of the effects. For example,to test the effect of factor k, C will be a vector having 1 at the k
th elementand 0’s in all others. With point estimation and estimated covariance matrix,the null hypothesis is tested through a standard Wald test procedure. TheWald test statistics is calculated as:
ti =CT �iqCT ⌃iC
.
The Wald test statistics approximately follow normal distribution, andthe p-values can be obtained accordingly. False discovery rate (FDR) willbe estimated using established procedures such as Benjamini-Hochberg’smethod (Benjamini and Hochberg, 1995).
2.5 Simulation settings
In all simulations, data are generated semi-parametrically. The countsare generated from the data model described in Equation (1) with modelparameters estimated from the human lung adenocarcinoma dataset. Themodel is 2 ⇥ 2 factorial design with 20,000 CpG sites for 3 replicatesin each condition group (12 datasets in total). For each factor, 5% of theCpG sites are DML, and the DM status for two factors are independentlygenerated. The regression coefficients �g (g = 0, 1, 2) are simulated in
the following way: (1) �0 (the intercept) is randomly sampled from theestimated intercepts from real data; (2) �1 and �2 are set to be 0 if the CpGsite is not DML, and sampled from N(0, 1) if the CpG site is DML; (3)The dispersion parameter �i’s are independently generated from log-normaldistribution with mean -3 and standard deviation 0.7, which are similar tothe real data estimates.
We also perform simulations when data are generated from a GLM with“logit” link to assess the robustness of our method. In this case, since thescales of the coefficients under “logit” link are greater than those from“arcsine” link for the same data (by a ratio of approximately 2.3), wemultiply 2.3 for �g’s for all simulations using “logit” link.
3 RESULTS3.1 Simulation
Comprehensive simulation studies are conducted to evaluate theperformance of DSS-general from several different aspects.
3.1.1 Dispersion estimation Dispersion parameter is an importantcomponent in various types of differential analysis for sequencingdata. Improved dispersion estimation from RNA-seq and BS-seqin two-group comparison has been shown to lead to better resultsin differential expression and differential methylation analyses(Robinson and Smyth, 2007; Wu et al., 2013; Feng et al., 2014;Love et al., 2014).
The estimated dispersions from DSS-general are compared tothe true ones in this simulation. Overall, the Pearson correlationbetween estimated and true dispersions is moderate at around 0.4.Data exploration indicates that large differences of estimated andtrue dispersions are mostly comes from the following two types ofCpG sites: (1) those with average methylation levels very close to0 or 1 and (2) those with low sequencing depth. For those CpGsites, it is not surprising to see estimates with large variation dueto low in-data information. The correlation indeed improves to 0.55when restricted to those with average methylation levels between0.3 and 0.7, and to 0.74 when further restricted to those with averagesequencing depth of at least 30. Figure ?? shows a plot for estimatedvs true dispersions for CpG sites with methylation levels between0.3 and 0.7. Sites with different levels of sequencing depth arerepresented by different colors. It shows reasonably good dispersionestimation.
3.1.2 DML detection accuracy We next compare the DMLdetection accuracies from several methods, including DSS-general,RADMeth, BiSeq and a binomial GLM with “logit” link incomparison. Here, the proportion of true positives among a givennumber of top-ranked CpGs is used as criterion. This refers to truediscovery rate (TDR) hereafter. Higher TDR is expected from bettermethod. This criterion is also referred to precision–recall analysis.Because the proportion of true positives is usually very low in DManalysis (5% in simulation setting), TDR is a better measurementof the accuracy for genome-wide differential analysis than receiveroperating characteristic (ROC) (Davis and Goadrich, 2006).
Each simulation is repeated 50 times to obtain the averageTDR estimates. Figure 1(A) shows the TDR curves up to top1,000 (5% of total) CpG sites when data are simulated using“arcsine” link function. It can be seen that DSS-general outperformsother methods for all top ranked CpG sites. For example, amongtop 200 ranked CpGs from DSS-general, 99.8% are true DML,whereas the percentages are 93.6%, 82.4%, and 38.4% from
3

Hypothesistesting
• Fortesting– Variance/covariancematrixestimates:
–Waldteststatisticsfor
This is for slides
Yid ⇠ beta-bin(mid,⇡id,�i)
g(⇡id) = xd�i
H0 : CT�i = 0, where C is a p-vector.
1
Then the coe�cient estimate is: �i = (XTV
�1i X)�1XT
V�1i Z, and the estimated covariance
matrix of the parameter estimates is ⌃i ⌘ \var(�i) = (XTV
�1i X)�1.
This estimation procedure utilizes two GLS estimations for each CpG site, which is
computationally very e�cient without relying on any iterative algorithm. The above procedure
has profound connection with a beta-binomial GLM, in which the initial regression coe�cients are
estimated from logistic regression and using Pearson �2 statistics to estimate dispersion
parameter in the similar way as equation (3) [44].
4.3.1 Hypothesis testing
Hypothesis testing for di↵erential methylation at CpG site i can be formulated as: H0 : CT�i = 0.
Here C is a p-vector. The procedure is very general and can test any linear combination of the
e↵ects. For example, to test the e↵ect of factor k, C will be a vector having 1 at the kth element
and 0’s in all others. With point estimates and estimated covariance matrix, the null hypothesis
is tested through a standard Wald test procedure. The Wald test statistics is calculated as:
ti =C
T �ipCT ⌃iC
The Wald test statistics approximately follow normal distribution, so that the p-values can be
obtained accordingly. False discovery rate (FDR) will be estimated using established procedures
such as Benjamini-Hochberg’s method [45].
4.4 Simulation settings
For all simulations, the data are generated semi-parametrically, i.e., the counts are generated from
the data model described in Equation (1) with model parameters estimated from the human lung
adenocarcinoma dataset. In all simulations, data are generated for a 2⇥ 2 factorial design, with
20,000 CpG sites and 3 replicates in each condition (12 datasets in total). For each factor, we
assume 5% of the CpG sites are DML, and the DM status for two factors are independent. The
regression coe�cients �g (g = 0, 1, 2) are simulated in the following way. �0 (the intercept) is
randomly sampled from the estimated intercept of real data. �1 and �2 are set to be 0 if the CpG
19
Then the coe�cient estimate is: �i = (XTV
�1i X)�1XT
V�1i Z, and the estimated covariance
matrix of the parameter estimates is ⌃i ⌘ \var(�i) = (XTV
�1i X)�1.
This estimation procedure utilizes two GLS estimations for each CpG site, which is
computationally very e�cient without relying on any iterative algorithm. The above procedure
has profound connection with a beta-binomial GLM, in which the initial regression coe�cients are
estimated from logistic regression and using Pearson �2 statistics to estimate dispersion
parameter in the similar way as equation (3) [44].
4.3.1 Hypothesis testing
Hypothesis testing for di↵erential methylation at CpG site i can be formulated as: H0 : CT�i = 0.
Here C is a p-vector. The procedure is very general and can test any linear combination of the
e↵ects. For example, to test the e↵ect of factor k, C will be a vector having 1 at the kth element
and 0’s in all others. With point estimates and estimated covariance matrix, the null hypothesis
is tested through a standard Wald test procedure. The Wald test statistics is calculated as:
ti =C
T �ipCT ⌃iC
The Wald test statistics approximately follow normal distribution, so that the p-values can be
obtained accordingly. False discovery rate (FDR) will be estimated using established procedures
such as Benjamini-Hochberg’s method [45].
4.4 Simulation settings
For all simulations, the data are generated semi-parametrically, i.e., the counts are generated from
the data model described in Equation (1) with model parameters estimated from the human lung
adenocarcinoma dataset. In all simulations, data are generated for a 2⇥ 2 factorial design, with
20,000 CpG sites and 3 replicates in each condition (12 datasets in total). For each factor, we
assume 5% of the CpG sites are DML, and the DM status for two factors are independent. The
regression coe�cients �g (g = 0, 1, 2) are simulated in the following way. �0 (the intercept) is
randomly sampled from the estimated intercept of real data. �1 and �2 are set to be 0 if the CpG
19

UseDSS
• Inputdataobjecthasthesameformatasbsseq.• DMLtest performsWaldtestateachCpG.• callDML/callDMR callsDMLorDMR.
## two group comparisondmlTest <- DMLtest(BSobj, group1=c("C1", "C2", "C3"),
group2=c("N1","N2","N3"),smoothing=TRUE, smoothing.span=500)
dmrs <- callDMR(dmlTest)## A 2x2 designDMLfit = DMLfit.multiFactor(RRBS, design, ~case+cell) DMLtest = DMLtest.multiFactor(DMLfit, term="case")

Conclusions• Analysisofgenome-widebisulfitesequencingdatapresentssomeuniquechallenges– Alignmentofreadscanbecomplicated– Manyteststobeperformed,butnumberofsamplessequencedislimitedbycostsinmostexperiments
– Beta-binomialmodeliswidelyused.
56

Forsoftware/analysis• Akalin etal.2012GenomeBiology13:R87.MethylKit paper.• Chatterjee etal.(2012)NucleicAcidsResearch.40(10):e79.Comparesaligners.• Chavezetal.(2010)GenomeResearch20:1441-50.MEDIPSsoftware.• Dolzhenko andSmith(2014)BMCBioinformatics 15:215.RADMeth.• Feng,Conneely,andWu(2014)NucleicAcidsResearch42(8):e69,DSSfortwo-group.• Hansenetal.(2012)GenomeBiology 13:R83. Bsmooth paper.• Hebestreit,Dugas,andKlein(2013)Bioinformatics 29:1647-53.BiSeq.• KruegerandAndrews(2011)Bioinformatics27(11):1571-2.BISMARKaligner.• Parketal.(2014)Bioinformatics 30:2414-22.MethylSig.• Robinsonetal.(2014)FrontiersinGenetics5:324.ReviewofmethodsforDMLandDMR• Stadler etal.(2012)Nature480:490-6.Mousemethylome paperthatusedHMM.• Sunetal.(2014)GenomeBiology 15:R38.MOABS.• Wuetal.(2015)NucleicAcidsResearch.43(21):e141.DSS-singleforsinglereplicates.• ParkandWu(2016)Bioinformatics32(10),1446-1453. DSS-generalforgeneraldesign.• XiandLi(2009)BMCBioinformatics 10:232.BSMAPaligner.• Xietal.(2012)Bioinformatics 28(3):430-2.RRBSMAPaligner.
57
References

Fordifferentsequencingtechnologies• Bocketal.(2010)NatBiotech 28(10):1106-16.ComparesRRBS,MeDIP-seq,others• Brinkmanetal.(2010)Methods52:232-236.MethylCap-seq.• Gu etal.(2011)NatProtoc 6(4):468-81.Genome-wideRRBSprotocol.• Maunakea etal.(2010)Nature466:253-7.MRE-seq.• Meissner (2005)NucleicAcidsResearch.33:5868-77.OriginalRRBSpaper.• Rauchetal.(2010)Methods52:213-7.MIRA-seq.• Serre etal.(2010)NucleicAcidsResearch.38:391-9.MBD-seq.• Weberetal.(2005)NatGenet 37:853-62.OriginalMeDIP paper.
58
References