axin2 regulates chondrocyte maturation and axial skeletal development
TRANSCRIPT

Axin2 Regulates Chondrocyte Maturation and Axial SkeletalDevelopment
Debbie Y. Dao,1,2 Xue Yang,1 Lisa M. Flick,1 Di Chen,1 Matthew J. Hilton,1 Regis J. O’Keefe1,3
1Department of Orthopaedics, Center for Musculoskeletal Research, University of Rochester School of Medicine, Rochester, New York 14642,2Department of Pathology, Center for Musculoskeletal Research, University of Rochester School of Medicine, Rochester, New York 14642,3Department of Orthopaedics and Rehabilitation, University of Rochester Medical Center, 601 Elmwood Avenue, Box 665, Rochester, New York14642
Received 19 February 2009; accepted 11 June 2009
Published online 27 July 2009 in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/jor.20954
ABSTRACT: Axis inhibition proteins 1 and 2 (Axin1 and Axin2) are scaffolding proteins that modulate at least two signaling pathways thatare crucial in skeletogenesis: the Wnt/b-catenin and TGF-b signaling pathways. To determine whether Axin2 is important in skeletogenesis,we examined the skeletal phenotype of Axin2-null mice in a wild-type or Axin1þ/� background. Animals with disrupted Axin2 expressiondisplayed a runt phenotype when compared to heterozygous littermates. Whole-mount and tissue b-galactosidase staining of Axin2LacZ/LacZ
mice revealed that Axin2 is expressed in cartilage tissue, and histological sections from knockout animals showed shorter hypertrophic zonesin the growth plate. Primary chondrocytes were isolated from Axin2-null and wild-type mice, cultured, and assayed for type X collagen geneexpression. While type II collagen levels were depressed in cells from Axin2-deficient animals, type X collagen gene expression was enhanced.There was no difference in BrdU incorporation between null and heterozygous mice, suggesting that loss of Axin2 does not alter chondrocyteproliferation. Taken together, these findings reveal that disruption of Axin2 expression results in accelerated chondrocyte maturation. Inthe presence of a heterozygous deficiency of Axin1, Axin2 was also shown to play a critical role in craniofacial and axial skeletondevelopment. � 2009 Orthopaedic Research Society. Published by Wiley Periodicals, Inc. J Orthop Res 28:89–95, 2010
Keywords: Axin; endochondral; chondrocyte; b-catenin; TGF-b
During embryonic development, skeletal elements areformed via two distinct processes: intramembranousand endochondral ossification. Several molecularsignals coordinating endochondral bone formation havebeen discovered, including Wnt/b-catenin signaling andTGF-b signaling. Recent studies have demonstratedthat Wnt/b-catenin signaling is required to driveproper osteoblast and chondrocyte differentiation dur-ing endochondral ossification.1–3 Additionally, TGF-bhas been shown to stimulate early stages of chondro-genesis and chondrocyte proliferation while inhibitingthe terminal differentiation of chondrocytes.4–6 Thus,Wnt and TGF-b signaling appear to oppose oneanother during select periods of endochondral bonedevelopment.
Axins regulate both the Wnt/b-catenin and TGF-bsignaling pathways. In the absence of Wnt signal, Axinsbind GSK-3b and b-catenin, facilitating GSK-3b-medi-ated phosphorylation of b-catenin, marking the proteinfor ubiquitination and proteasomal degradation.7 Axinsare stabilized when phosphorylated by GSK-3b.8 Inthe presence of Wnt signal, Axins are recruited to theWnt co-receptor LRP5 or 6, and are dephosphory-lated.9,10 The protein complex formed by Axin is there-fore destabilized, allowing b-catenin to accumulate andtranslocate to the nucleus where it regulates genetranscription through TCF and LEF transcriptionfactors.11,12 In a negative feedback loop, Wnt/b-catenin/TCF signaling induces Axin2 expression, which in turninhibits canonical Wnt signaling.13,14 While Axinsinhibit Wnt signaling, they enhance TGF-b signaling intwo ways. First, Axins facilitate the phosphorylation and
activation of Smad3.15 Activated Smad3 associates withSmad4, translocates to the nucleus, and initiates genetranscription. Second, Axins facilitate phosphorylationof the inhibitory Smad7, which competitively inhibitsSmad3 activity. Phosphorylation of Smad7 marks it forubiquitination and proteasomal degradation.16
Axin1 and Axin2 are master scaffolding proteinsoriginally identified as negative regulators of canonicalWnt/b-catenin signaling (see review17). Althoughthese proteins are similar in function, Axin1 appearsto be ubiquitously expressed, while Axin2 has amore restricted expression pattern.18 Further, Axin2expression is directly induced by canonical Wnt signal-ing and therefore acts in a negative feedback loop.8,13 Inaddition to interactions with several canonical Wntsignaling proteins,9,19 the Axin proteins have beenshown to interact with MEKK1 and Smad3 establishingtheir importance in other key signaling pathways.15,20
Given the role of Axins in at least two pathwaysrelevant to endochondral ossification and embryogenesisin general, it is likely that Axins specifically contribute tonormal skeletogenesis. Therefore, we examined Axin2expression in the cartilage of Axin2LacZ/LacZ mice andexamined the skeletal phenotype of Axin2-deficientmice to ascertain the effects of disrupted Wnt/b-cateninsignaling on endochondral ossification. Intramem-branous bone formation has already been shown toinvolve Axin2 as Axin2-deficient mice are characterizedby craniofacial defects.21 Interestingly, mice deficient inthe functional homolog Axin1 die in utero and arecharacterized by the presence of axis determinationdefects22–24; however, mice heterozygous for a mutationin Axin1 survive with no abnormalities. Hence, wealso explored the skeletal phenotype of Axin2-deficientmice on a genetic background of Axin1 heterozygo-sity to determine if the presence of Axin2 in Axin1
JOURNAL OF ORTHOPAEDIC RESEARCH JANUARY 2010 89
Correspondence to: Regis J. O’Keefe (T: 585-275-1261; F: 585-756-4727; E-mail: [email protected])
� 2009 Orthopaedic Research Society. Published by Wiley Periodicals, Inc.

heterozygotes is compensates for the absence of Axin1.We hypothesized that Axin2 would be expressed incartilage cells, that Axin2-deficient mice would havesignificant changes in endochondral skeletal develop-ment when compared to heterozygous or wild-typelittermate controls, and that these alterations would beeven more profound in the Axin1þ/�; Axin2�/� animals.
METHODSAxin2-Deficient MiceAxin2LacZ/LacZ mice were a generous gift from Dr. Wei Hsu andhave been described by his group.21 In this article, the term‘‘Axin2�/�’’ connotes ‘‘Axin2LacZ/LacZ.’’ Care and use of exper-imental animals complied with the guidelines and policies ofthe University Committee on Animal Resources at theUniversity of Rochester.
Histology and b-Galactosidase StainingWhole-mount embryo or frozen tissue section b-galactosidasestaining was performed as previously described.21,25 Stainedtissue sections were then washed in PBS, counterstained withnuclear fast red, dehydrated, and coverslipped using standardmounting media.
Bromodeoxyuridine (BrdU) labeling was achieved in1-week-old mice by administering the labeling reagent(1 mL/100g body weight; Zymed, San Francisco, CA) 3 h beforesacrifice via i.p. injection. BrdU incorporation was examinedusing immunohistochemistry on paraffin-embedded sectionswith a primary mouse monoclonal antibody against BrdU (LabVision, Fremont, CA).
RNA Extraction and Quantitative Reverse-Transcriptase PCRfrom ChondrocytesChondrocytes were isolated from the sterna and ribs of 3-day-old mice as previously described.26 Cells were plated in 12-wellplates at 5� 104 cells per well for RNA isolation. Total RNAwas extracted from primary chondrocytes using the Trizol(Invitrogen, Carlsbad, CA) protocol following the manufac-turer’s recommendations. One microgram of total RNA wasreverse-transcribed using the i-Script cDNA synthesis kit(Biorad, Hercules, CA) following the manufacturer’s recom-mended protocol. Two microliters of reverse-transcribed cDNAwas used for quantitative PCR. cDNA levels were measured inreal-time using the fluorescent dye SYBR Green I (SYBR GreenPCR Master Mix, Applied Biosystems, Foster City, CA) andspecific primers designed for mouse type X collagen (Forward:50-ACC CCA AGG ACC TAA AGG AA-30; Reverse: 50-CCC CAGGAT ACC CTG TTT TT-30), type II collagen (Forward: 50-ACTGGT AAG TGG GGC AAG AC-30; Reverse: 50-CCA CAC CAAATT CCT GTT CA-30), and b-actin (Forward: 50-TGT TAC CAACTG GGA CGA CA-30; Reverse: 50-CTG GGT CAT CTT TTCACG GT-30). The PCR reaction used the RotorGene real-timeDNA amplification system (Corbett Research, Sydney, Aus-tralia) and the following protocol: 958C denaturation step for10 min followed by 45 cycles with denaturation for 30 s at 958C,annealing for 30 s at 558C, and extension for 30 s at 728C.Detection of the fluorescent product occurred after eachextension period. PCR products were subjected to melting curveanalysis, and the data were analyzed and quantified with theRotorGene analysis software. Gene expression was normalizedto b-actin expression levels.
Micro-CT and Skeletal StainingEmbryos were fixed overnight in 10% neutral bufferedformalin following evisceration and skinning, then dehydrated
in a graded series of ethanol. Fixed embryos were scanned at aresolution of 12.5 mm using a ScanCo Medical VivaCT40(Basserdorf, Switzerland) with x-ray settings of 55 kVp and145 mA, and an integration time of 300 ms. Three-dimensionalcomposite images were created with a threshold value of150. Skeletal staining of whole embryos using alcian blueand alizarin red was performed as previously described,27
immediately following micro-CT analysis.
RESULTSAxin2 Knockout Mice Display a Runt PhenotypeYu and colleagues have demonstrated that Axin2plays a critical role in intramembranous bone formationsuch that disruption of Axin2 in mice results inskeletal abnormalities, particularly a craniosynostosis-like phenotype.21 Measurement of Axin2�/� andAxin2þ/� littermates reveals an overall runt phenotypein the null mice (Fig. 1A). One-week-old Axin2�/� mice(n¼ 11) had an approximate 12.5% decrease inshoulder-to-rump length when compared to hetero-zygous littermates (n¼ 12) (Fig. 1B). Accordingly, theAxin2�/� mice weighed less, averaging 3.8 g at 1 week,compared to Axin2þ/� littermates, which averaged 4.5 gat the same time point (Fig. 1C). This decrease in bodysize suggests that Axin2 plays a critical role not only inintramembranous bone formation of the skull, but alsoin endochondral bone formation, which is critical todevelopment of the axial and appendicular skeleton. Nodifference in body size or weight was observed betweenheterozygous and homozygous wild-type animals.
Axin2 Is Expressed in CartilageIt has previously been established that Axin2 isspecifically expressed in neural crest-derived skeletalelements during postnatal development.21 Whole-mount b-galactosidase staining of E13.5 Axin2LacZ/LacZ
embryos reveals Axin2 expression in cartilaginousareas of the axial and appendicular skeleton duringembryonic development (Fig. 2A). Thus, positivelystained regions at this stage reveal that Axin2 isexpressed in tissues derived from paraxial and lateral-plate mesoderm, as well as in neural crest derivatives.
Axin2 continues to be expressed in cartilaginouselements postnatally. At 1 week of age, b-galactosidasestaining of frozen tissue sections from Axin2�/� micereveals Axin2 expression in chondrocytes of the ribs,vertebra, and long bone growth regions, specifically inperipheral epiphyseal chondrocytes and prehypertrophic/hypertrophic chondrocytes (Fig. 2). These findings areconsistent with the idea that Axin2 functions duringendochondral bone formation, and likely accounts for therunt phenotype observed in Axin2-null mice.
Axin2 Regulates Chondrocyte MaturationWhile defects in intramembranous bone formationleading to craniosynostosis in Axin2�/� mice have beenattributed to abnormal osteoblast proliferation anddifferentiation,28 the defects observed during endochon-dral bone formation appear to result exclusively fromabnormal chondrocyte maturation. Histological sections
90 DAO ET AL.
JOURNAL OF ORTHOPAEDIC RESEARCH JANUARY 2010

of distal femurs from 1-week-old Axin2�/� mice (n¼7)reveal thinner hypertrophic and columnar zones whencompared to Axin2þ/� littermates (n¼13) (Fig. 3A–D).This finding is consistent with an overall acceleration inboth the initiation of hypertrophy and terminal differ-entiation processes resulting in shorter limb length,reduced rib cage size, and a shorter axial skeleton. Toexamine whether loss of Axin2 disrupts chondrocyteproliferation, BrdU staining was performed on growthregion chondrocytes of 1-week-old Axin2þ/� (n¼13) andAxin2�/� (n¼7) hindlimb sections. No difference wasobserved in BrdU labeling between these two groups(Fig. 3E), suggesting that Axin2 does not regulate
Figure 1. One-week-old Axin2-null mice (n¼11) display a runtphenotype compared to heterozygous littermates (n¼12). (A) Plainx-ray; (B) body length in centimeters; (C) mass in grams. (B) and(C) are shown as the mean�SD. *p< 0.05 using unpaired t-test.
Figure 2. Axin2 is expressed in cartilage during development.(A) b-galactosidase staining of whole wild-type or Axin2-null embryos at day E13.5. Arrowhead indicates b-galactosidaseactivity. Representative photomicrographs (original magnification,�100) of b-galactosidase stained frozen sections: tibia (B) andribs (C).
ROLE OF AXIN2 IN SKELETOGENESIS 91
JOURNAL OF ORTHOPAEDIC RESEARCH JANUARY 2010
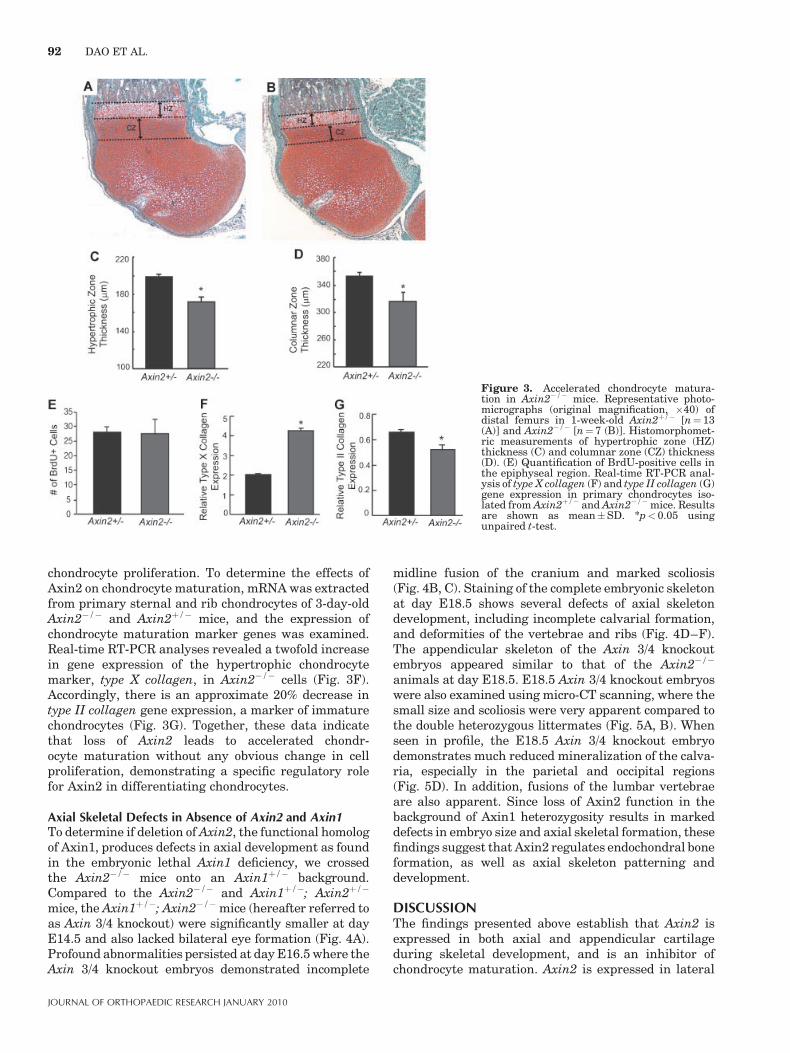
chondrocyte proliferation. To determine the effects ofAxin2 on chondrocyte maturation, mRNA was extractedfrom primary sternal and rib chondrocytes of 3-day-oldAxin2�/� and Axin2þ/� mice, and the expression ofchondrocyte maturation marker genes was examined.Real-time RT-PCR analyses revealed a twofold increasein gene expression of the hypertrophic chondrocytemarker, type X collagen, in Axin2�/� cells (Fig. 3F).Accordingly, there is an approximate 20% decrease intype II collagen gene expression, a marker of immaturechondrocytes (Fig. 3G). Together, these data indicatethat loss of Axin2 leads to accelerated chondr-ocyte maturation without any obvious change in cellproliferation, demonstrating a specific regulatory rolefor Axin2 in differentiating chondrocytes.
Axial Skeletal Defects in Absence of Axin2 and Axin1To determine if deletion of Axin2, the functional homologof Axin1, produces defects in axial development as foundin the embryonic lethal Axin1 deficiency, we crossedthe Axin2�/� mice onto an Axin1þ/� background.Compared to the Axin2�/� and Axin1þ/�; Axin2þ/�
mice, the Axin1þ/�; Axin2�/� mice (hereafter referred toas Axin 3/4 knockout) were significantly smaller at dayE14.5 and also lacked bilateral eye formation (Fig. 4A).Profound abnormalities persisted at day E16.5 where theAxin 3/4 knockout embryos demonstrated incomplete
midline fusion of the cranium and marked scoliosis(Fig. 4B, C). Staining of the complete embryonic skeletonat day E18.5 shows several defects of axial skeletondevelopment, including incomplete calvarial formation,and deformities of the vertebrae and ribs (Fig. 4D–F).The appendicular skeleton of the Axin 3/4 knockoutembryos appeared similar to that of the Axin2�/�
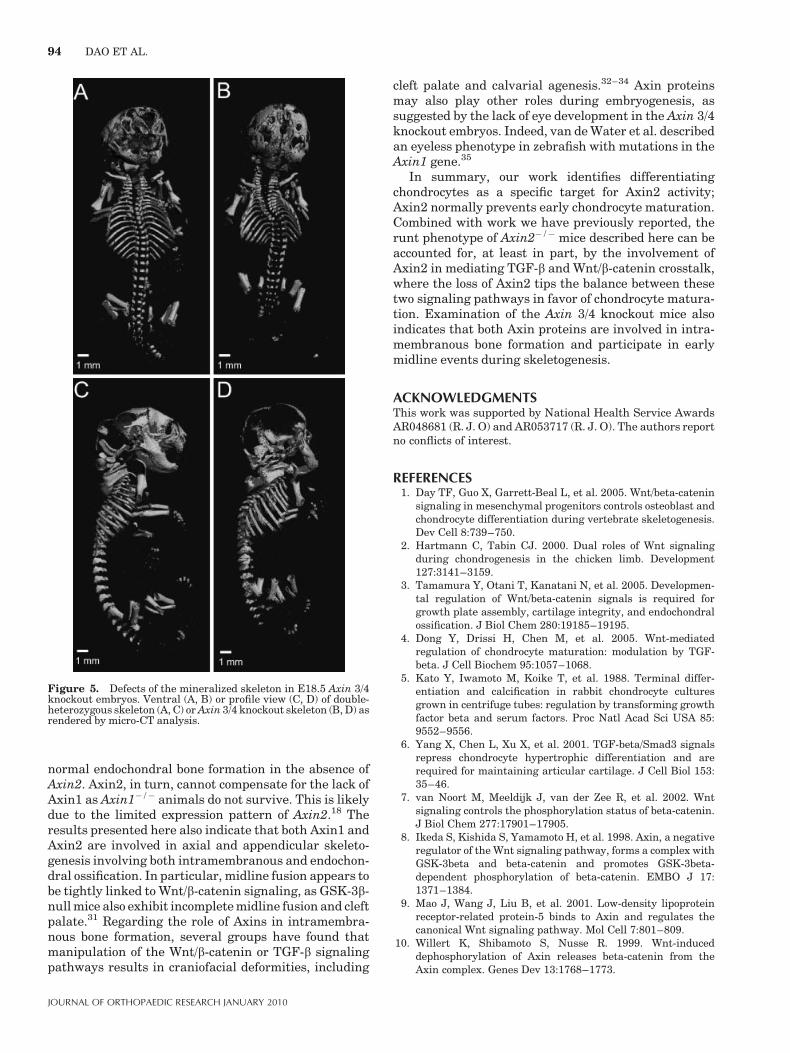
animals at day E18.5. E18.5 Axin 3/4 knockout embryoswere also examined using micro-CT scanning, where thesmall size and scoliosis were very apparent compared tothe double heterozygous littermates (Fig. 5A, B). Whenseen in profile, the E18.5 Axin 3/4 knockout embryodemonstrates much reduced mineralization of the calva-ria, especially in the parietal and occipital regions(Fig. 5D). In addition, fusions of the lumbar vertebraeare also apparent. Since loss of Axin2 function in thebackground of Axin1 heterozygosity results in markeddefects in embryo size and axial skeletal formation, thesefindings suggest that Axin2 regulates endochondral boneformation, as well as axial skeleton patterning anddevelopment.
DISCUSSIONThe findings presented above establish that Axin2 isexpressed in both axial and appendicular cartilageduring skeletal development, and is an inhibitor ofchondrocyte maturation. Axin2 is expressed in lateral
Figure 3. Accelerated chondrocyte matura-tion in Axin2�/� mice. Representative photo-micrographs (original magnification, �40) ofdistal femurs in 1-week-old Axin2þ/� [n¼ 13(A)] and Axin2�/� [n¼ 7 (B)]. Histomorphomet-ric measurements of hypertrophic zone (HZ)thickness (C) and columnar zone (CZ) thickness(D). (E) Quantification of BrdU-positive cells inthe epiphyseal region. Real-time RT-PCR anal-ysis of type X collagen (F) and type II collagen (G)gene expression in primary chondrocytes iso-lated from Axin2þ/� and Axin2�/� mice. Resultsare shown as mean�SD. *p< 0.05 usingunpaired t-test.
92 DAO ET AL.
JOURNAL OF ORTHOPAEDIC RESEARCH JANUARY 2010

plate and paraxial mesoderm-derived tissue, namely inthe cartilage of limbs, spine, and ribs. Interestingly, weshow that Axin2 expression is primarily restrictedto hypertrophic chondrocytes and a subset of themost peripheral epiphyseal chondrocytes. Loss of Axin2function accelerates hypertrophic differentiation,resulting in reduced endochondral bone growth and arunt phenotype in mutant mice. Therefore, these dataemphasize the important role Axin2 plays duringendochondral bone formation.
Previous work has shown that Axin2 regulatesboth proliferation and differentiation in osteoblasts,primarily through its actions in Wnt/b-catenin signal-ing.28 In this article, we show that Axin2 also affectschondrocytes. It is probable that the lack of a chondrocyteproliferation phenotype in Axin2�/� mice is due to thefact that Axin2 expression is primarily restricted todifferentiating chondrocytes in the prehypertrophic andhypertrophic zones, but not proliferating chondrocytes.The accelerated cartilage differentiation phenotype islikely caused by an increase in localized Wnt signaling inthose differentiating cells, which is similar to cartilagephenotypes of animal models with Wnt gain-of-functionmodifications.
Although the role of Axin2 in inhibiting Wnt signalinghas been well established, recently Axins have beenshown to play active roles in the TGF-b and JNK
signaling pathways as well. Axins enhance TGF-bsignaling by facilitating Smad3 phosphorylation andactivation, as well as by enhancing Smad7 phos-phorylation and degradation.15,16 In mitogen-activatedprotein kinase (MAPK) signaling, Axins facilitate theactivation of c-Jun N-terminal Kinase (JNK), a MAPKregulator, through interactions with the protein kinasesMEKK1, 4, and 7.20 Recently, we have shown that Axinsare negatively regulated by TGF-band mediate crosstalkbetween the TGF-b and Wnt signaling pathways inchondrocytes.29 The overall effect of this crosstalk is anenhancement of b-catenin signaling and an inhibition ofSmad3 signaling that results in chondrocyte maturation.This is consistent with the in vivo findings presentedhere, such that disruption of Axin2 signaling results inaccelerated chondrocyte maturation and shortening ofendochondral bones. b-Catenin gain-of-function in vivoproduces a similar phenotype (shorter limbs and cranio-facial abnormalities), which provides further evidencefor the role of Axin proteins in the regulation of thissignaling cascade.30 A mechanism through which thisoccurs may lie in the removal of Axin2 as a mediator thatbalances TGF-b and Wnt/b-catenin signaling, which arethought to oppose one another during chondrocyteproliferation and maturation.
Interestingly, Axin1 protein is ubiquitously ex-pressed, but does not apparently function to preserve
Figure 4. Profound abnormalities inAxin 3/4 knockout embryos. (A) Wholemounts of representative E14.5 embryoslacking one or more copies of the Axin1or Axin2 genes. Clockwise from theupper left, the genotypes are: Axin2þ/�,Axin2�/�, Axin1þ/�; Axin2þ/�, and Axin3/4 knockout. Ventral (B) and dorsal (C)views of E16.5 Axin 3/4 knockout embryos.Skeletal staining of E18.5 Axin1þ/�
Axin2þ/� (D), Axin2�/� (E), and Axin 3/4knockout (F) embryos.
ROLE OF AXIN2 IN SKELETOGENESIS 93
JOURNAL OF ORTHOPAEDIC RESEARCH JANUARY 2010
normal endochondral bone formation in the absence ofAxin2. Axin2, in turn, cannot compensate for the lack ofAxin1 as Axin1�/� animals do not survive. This is likelydue to the limited expression pattern of Axin2.18 Theresults presented here also indicate that both Axin1 andAxin2 are involved in axial and appendicular skeleto-genesis involving both intramembranous and endochon-dral ossification. In particular, midline fusion appears tobe tightly linked to Wnt/b-catenin signaling, as GSK-3b-null mice also exhibit incomplete midline fusion and cleftpalate.31 Regarding the role of Axins in intramembra-nous bone formation, several groups have found thatmanipulation of the Wnt/b-catenin or TGF-b signalingpathways results in craniofacial deformities, including
cleft palate and calvarial agenesis.32–34 Axin proteinsmay also play other roles during embryogenesis, assuggested by the lack of eye development in the Axin 3/4knockout embryos. Indeed, van de Water et al. describedan eyeless phenotype in zebrafish with mutations in theAxin1 gene.35
In summary, our work identifies differentiatingchondrocytes as a specific target for Axin2 activity;Axin2 normally prevents early chondrocyte maturation.Combined with work we have previously reported, therunt phenotype of Axin2�/� mice described here can beaccounted for, at least in part, by the involvement ofAxin2 in mediating TGF-b and Wnt/b-catenin crosstalk,where the loss of Axin2 tips the balance between thesetwo signaling pathways in favor of chondrocyte matura-tion. Examination of the Axin 3/4 knockout mice alsoindicates that both Axin proteins are involved in intra-membranous bone formation and participate in earlymidline events during skeletogenesis.
ACKNOWLEDGMENTSThis work was supported by National Health Service AwardsAR048681 (R. J. O) and AR053717 (R. J. O). The authors reportno conflicts of interest.
REFERENCES1. Day TF, Guo X, Garrett-Beal L, et al. 2005. Wnt/beta-catenin
signaling in mesenchymal progenitors controls osteoblast andchondrocyte differentiation during vertebrate skeletogenesis.Dev Cell 8:739–750.
2. Hartmann C, Tabin CJ. 2000. Dual roles of Wnt signalingduring chondrogenesis in the chicken limb. Development127:3141–3159.
3. Tamamura Y, Otani T, Kanatani N, et al. 2005. Developmen-tal regulation of Wnt/beta-catenin signals is required forgrowth plate assembly, cartilage integrity, and endochondralossification. J Biol Chem 280:19185–19195.
4. Dong Y, Drissi H, Chen M, et al. 2005. Wnt-mediatedregulation of chondrocyte maturation: modulation by TGF-beta. J Cell Biochem 95:1057–1068.
5. Kato Y, Iwamoto M, Koike T, et al. 1988. Terminal differ-entiation and calcification in rabbit chondrocyte culturesgrown in centrifuge tubes: regulation by transforming growthfactor beta and serum factors. Proc Natl Acad Sci USA 85:9552–9556.
6. Yang X, Chen L, Xu X, et al. 2001. TGF-beta/Smad3 signalsrepress chondrocyte hypertrophic differentiation and arerequired for maintaining articular cartilage. J Cell Biol 153:35–46.
7. van Noort M, Meeldijk J, van der Zee R, et al. 2002. Wntsignaling controls the phosphorylation status of beta-catenin.J Biol Chem 277:17901–17905.
8. Ikeda S, Kishida S, Yamamoto H, et al. 1998. Axin, a negativeregulator of the Wnt signaling pathway, forms a complex withGSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J 17:1371–1384.
9. Mao J, Wang J, Liu B, et al. 2001. Low-density lipoproteinreceptor-related protein-5 binds to Axin and regulates thecanonical Wnt signaling pathway. Mol Cell 7:801–809.
10. Willert K, Shibamoto S, Nusse R. 1999. Wnt-induceddephosphorylation of Axin releases beta-catenin from theAxin complex. Genes Dev 13:1768–1773.
Figure 5. Defects of the mineralized skeleton in E18.5 Axin 3/4knockout embryos. Ventral (A, B) or profile view (C, D) of double-heterozygous skeleton (A, C) or Axin 3/4 knockout skeleton (B, D) asrendered by micro-CT analysis.
94 DAO ET AL.
JOURNAL OF ORTHOPAEDIC RESEARCH JANUARY 2010

11. Behrens J, von Kries JP, Kuhl M, et al. 1996. Functionalinteraction of beta-catenin with the transcription factor LEF-1. Nature 382:638–642.
12. Molenaar M, van de Wetering M, Oosterwegel M, et al. 1996.XTcf-3 transcription factor mediates beta-catenin-inducedaxis formation in Xenopus embryos. Cell 86:391–399.
13. Jho EH, Zhang T, Domon C, et al. 2002. Wnt/beta-catenin/Tcfsignaling induces the transcription of Axin2, a negativeregulator of the signaling pathway. Mol Cell Biol 22:1172–1183.
14. Leung JY, Kolligs FT, Wu R, et al. 2002. Activation of AXIN2expression by beta-catenin-T cell factor. A feedback repressorpathway regulating Wnt signaling. J Biol Chem 277:21657–21665.
15. Furuhashi M, Yagi K, Yamamoto H, et al. 2001. Axinfacilitates Smad3 activation in the transforming growth factorbeta signaling pathway. Mol Cell Biol 21:5132–5141.
16. Liu W, Rui H, Wang J, et al. 2006. Axin is a scaffold protein inTGF-beta signaling that promotes degradation of Smad7 byArkadia. EMBO J 25:1646–1658.
17. Kikuchi A. 1999. Roles of Axin in the Wnt signalling pathway.Cell Signal 11:777–788.
18. Chia IV, Costantini F. 2005. Mouse Axin and Axin2/conduct-ing proteins are functionally equivalent in vivo. Mol Cell Biol25:4371–4376.
19. Luo W, Lin SC. 2004. Axin: a master scaffold for multiplesignaling pathways. Neurosignals 13:99–113.
20. Zou H, Li Q, Lin SC, et al. 2007. Differential requirement ofMKK4 and MKK7 in JNK activation by distinct scaffoldproteins. FEBS Lett 581:196–202.
21. Yu HM, Jerchow B, Sheu TJ, et al. 2005. The role of Axin2 incalvarial morphogenesis and craniosynostosis. Development132:1995–2005.
22. Gluecksohn-Schoenheimer S. 1949. The effects of a lethalmutation responsible for duplications and twinning in mouseembryos. J Exp Zool 110:47–76.
23. Greenspan RJ, O’Brien MC. 1986. Genetic analysis ofmutations at the fused locus in the mouse. Proc Natl AcadSci USA 83:4413–4417.
24. Zeng L, Fagotto F, Zhang T, et al. 1997. The mouse fused locusencodes Axin, an inhibitor of the Wnt signaling pathway thatregulates embryonic axis formation. Cell 90:181–192.
25. Whiting J, Marshall H, Cook M, et al. 1991. Multiple spatiallyspecific enhancers are required to reconstruct the pattern ofHox-2.6 gene expression. Genes Dev 5:2048–2059.
26. Li TF, Darowish M, Zuscik MJ, et al. 2006. Smad3-deficientchondrocytes have enhanced BMP signaling and accelerateddifferentiation. J Bone Miner Res 21:4–16.
27. McLeod MJ. 1980. Differential staining of cartilage and bonein whole mouse fetuses by alcian blue and alizarin red S.Teratology 22:299–301.
28. Liu B, Yu HM, Hsu W. 2007. Craniosynostosis caused byAxin2 deficiency is mediated through distinct functions ofbeta-catenin in proliferation and differentiation. Dev Biol 301:298–308.
29. Dao DY, Yang X, Chen D, et al. 2007. Axin1 and Axin2 areregulated by TGF- and mediate cross-talk between TGF- andWnt signaling pathways. Ann NY Acad Sci 1116:82–99.
30. Akiyama H, Lyons JP, Mori-Akiyama Y, et al. 2004.Interactions between Sox9 and beta-catenin control chondro-cyte differentiation. Genes Dev 18:1072–1087.
31. Liu KJ, Arron JR, Stankunas K, et al. 2007. Chemical rescueof cleft palate and midline defects in conditional GSK-3betamice. Nature 446:79–82.
32. Brault V, Moore R, Kutsch S, et al. 2001. Inactivation of thebeta-catenin gene by Wnt1-Cre-mediated deletion results indramatic brain malformation and failure of craniofacialdevelopment. Development 128:1253–1264.
33. Chang J, Sonoyama W, Wang Z, et al. 2007. NoncanonicalWnt-4 signaling enhances bone regeneration of mesenchymalstem cells in craniofacial defects through activation of p38MAPK. J Biol Chem 282:30938–30948.
34. Ito Y, Yeo JY, Chytil A, et al. 2003. Conditional inactivation ofTgfbr2 in cranial neural crest causes cleft palate and calvariadefects. Development 130:5269–5280.
35. van de Water S, van de Wetering M, Joore J, et al. 2001.Ectopic Wnt signal determines the eyeless phenotype ofzebrafish masterblind mutant. Development 128:3877–3888.
ROLE OF AXIN2 IN SKELETOGENESIS 95
JOURNAL OF ORTHOPAEDIC RESEARCH JANUARY 2010