asl to2 codice: procedura tecnica pr-grc-001 dm · - dm 20 febbraio 2007 - dm 20 marzo 2007 - d.lgs...
TRANSCRIPT


ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 2 di 34
2
PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDALE SUI DISPOSITIVI MEDICI”
INDICE 1. Introduzione pag. 3 2. Riferimenti pag. 3 3. Destinatari pag. 4 4. Scopo pag. 4 5. Valore pag. 4 6. Campo d’applicazione pag. 4 7. Definizioni pag. 4 8. Responsabilità pag. 5 9. Compiti del Responsabile della Vigilanza sui Dispositivi Medici pag. 5 10. Compiti dei Referenti degli Ospedali Amedeo di Savoia/Birago di Vische – Maria Vittoria - San Giovanni Bosco e dei Distretti
pag. 6
11. Compiti del Referenti de i Presidi Sanitari - Strutture Accreditate – Strutture Private
pag. 7
12. Compiti degli Operatori Sanitari pag. 7 13. Modalità di Segnalazione al Ministero di Incidente o Mancato Incidente pag. 8
13.1) Tipologia di eventi da segnalare pag. 8 13.2) Fase della notifica pag. 12 13.3) Fase di gestione dei Dispositivi oggetto della segnalazione pag. 14 13.4) Fase dell’indagine pag. 15 13.5) Fase dell’azione valutativa e correttiva pag. 15 13.6) Raccolta ed archiviazione pag. 15
14. Tracciabilità pag. 15 15. Formazione pag. 16 16. Monitoraggio della procedura pag. 16 17. P arametri di controllo pag. 16 18. Revisione pag. 16 Allegato 1: Glossario pag. 17 Allegato 2: Tabella delle responsabilità pag. 20 Allegato 3: Fac simile verbale di consegna del Dispositivo Medico- pag. 22 Allegato 4:Flow Chart della segnalazione Strutture Private – Accreditate e Presidi Sanitari
pag.
23
Allegato 5: Flow Chart della segnalazione Ospedali Amedeo di Savoia/Birago di Vische - Maria Vittoria San Giovanni Bosco e Distretti
pag.
24
Allegato 6: Scheda Ministeriale per incidente con DISPOSITIVO MEDICO
pag.
25
Allegato 7: Scheda Ministeriale per incidente con DISPOSITIVO MEDICO-DIAGNOSTICO IN VITRO
pag.
30

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 3 di 34
3
1.
INTRODUZIONE
Lo scopo principale del sistema di vigilanza sui Dispositivi Medici è di migliorare il livello di sicurezza di Pazienti, Operatori, Utilizzatori ed altri in un sistema più ampio di Risk Management, ponendo così i presupposti per una riduzione della possibilità del ripetersi di un incidente o di altri eventi indesiderati.
Questo può essere attuato attraverso la valutazione degli incidenti segnalati e la diffusione delle informazioni utili a questo scopo.
La vigente normativa, linee guida, raccomandazioni regionali, nazionali ed internazionali sulla tematica sono state recentemente aggiornate. Esse forniscono sufficienti e dettagliate indicazioni sulle modalità procedurali e sui compiti dei vari attori coinvolti nel sistema di vigilanza.
La presente procedura rappresenta e descrive in particolare il sistema aziendale per la notifica e la valutazione di eventi avversi, conosciuta come Vigilanza dei Dispositivi Medici.
L’ASL TO2 con deliberazione n. 804/001A/2007 del 28 dicembre 2007 ha individuato, in ottemperanza ai disposti Ministeriali, il Responsabile Aziendale della Vigilanza sui Dispositivi Medici nella:
Dr.ssa Anna BRUNETTI–Direttore SoSD Vigilanza sui Dispositivi Medici e Referente Risk Management Ospedale Maria Vittoria/Amedeo di Savoia/Birago di Vische Sede lavorativa: Direzione Sanitaria Ospedale Maria Vittoria - Via Luigi Cibrario, 72 – 10144 Torino Tel.: 0114393202/302 Cell.: 3484303219 e-mail: [email protected]
- D.Lgs 507/92 2. RIFERIMENTI
- D.Lgs 46/97 - DM 318/98 - DM 23 luglio 98 - D.Lgs 332/2000 - DM 3 febbraio 2003 - DM 24 maggio 2004 - DM 14 luglio 2004 - D.Lgs 304/04 - DM 26 gennaio 2005 - D.Lgs 67/05 - DM 2 agosto 2005 - DM 22 settembre 2005 - DM 15 novembre 2005 - DM 23 gennaio 2007

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 4 di 34
4
- DM 20 febbraio 2007 - DM 20 marzo 2007 - D.Lgs 37/2010 - D.G.R. n. 14-8500 del 31 marzo 2008 “indicazione alle aziende sanitarie regionali per
la gestione del rischio clinico e l’attivazione dell’unità di gestione del rischio clinico e prime linee di indirizzo su tematiche di particolare interesse” – linea di indirizzo 5/2007
- Circolare Ministeriale n. 59849 26 luglio 2004 - Circolare Ministeriale n. 9693 04.08.04 - Circolare Regione Piemonte n. 13638 21.09.04 - “Guidelines on a medical devices vigilance system” – European Commission – aprile
2001 - “Guidelines on a medical devices vigilance system” – European Commission – rev. 5
aprile 2007 - “Guidelines on a medical devices vigilance system” – European Commission – rev. 6
dicembre 2009 - Atti del “Corso per i referenti delle aziende sanitarie e dei servizi regionali” anno 2006 –
ISS/Ministero della Salute - atti del corso “Rischio Clinico connesso all’uso dei Dispositivi Medici” anno 2011 –
Regione Piemonte - .ministerosalute.
Questa procedura è diretta a coloro i quali in azienda espletano attività di vigilanza sui dispositivi medici ed agli operatori sanitari, relativamente alle azioni a cui sono tenuti per legge in merito agli adempimenti sulla materia.
3. DESTINATARI
Questa procedura intende essere ausilio, guida e standardizzazione delle azioni da eseguire per gli operatori che, nell’ambito di attività di vigilanza, interpretano e leggono l’applicazione delle indicazioni normative nazionali e regionali in materia di dispositivi medici, messi a disposizione a titolo gratuito o oneroso, anche nell’ottica di omogeneizzare le azioni.
4. SCOPO
Questa procedura ha carattere operativo interno. Sono naturalmente vincolanti i disposti di legge nazionali e di recepimento delle direttive CEE concernenti i dispositivi medici.
5. VALORE
Ambito territoriale dell’ASL TO2 6. CAMPO D’APPLICAZIONE
Vengono adottate le definizioni contenute nel glossario riportato sul sito internet del Ministero della Salute – sezione Dispositivi Medici – (Allegato1), con alcune integrazioni aggiornate in base al D.Lgs 37/2010 ed alla MEDDEV 2.12.-1 rev. 6.
7. DEFINIZIONI

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 5 di 34
5
Il rispetto di quanto riportato in questa procedura, da parte di chi esercita la vigilanza, non assolve gli operatori sanitari dall’adempimento degli obblighi posti dalla vigente normativa in merito alla segnalazione d’incidenti aventi per oggetto i dispositivi medici e alle sanzioni che derivano dalle violazioni delle prescrizioni relative alla materia (arresto sino a 6 mesi ed ammenda da 7.200 a 43.200 €)
8. RESPONSABILITÀ
I dati relativi agli incidenti devono essere infatti segnalati, per obbligo di legge, dagli operatori sanitari mediante le specifiche schede di rapporto per i dispositivi medici predisposte dal Ministero della Salute.
Le schede redatte dagli operatori sanitari devono essere inviate al Referente di Area o di Struttura e da questi al Responsabile Aziendale della Vigilanza sui Dispositivi Medici nel più breve tempo possibile, onde consentire il rispetto delle tempistiche per i vari adempimenti previsti e comunque deve essere rispettato quanto indicato nello specifico paragrafo (paragrafo 13 pag. 8).
Le responsabilità associate alla gestione della presente procedura sono riportate nella tabella 1 (Allegato2).
1) Ricevere le schede ministeriali di segnalazione d’incidente con dispositivi medici, redatte dagli operatori delle strutture sanitarie attive nell’ambito territoriale dell’ASL TO2.
9. COMPITI DEL RESPONSABILE DELLA VIGILANZA SUI DISPOSITIVI MEDICI
2) Ricevere le segnalazioni, da operatori e/o utenti dell’ASL TO2, di ogni altro inconveniente con dispositivi medici che, pur non integrando le caratteristiche dell’incidente, possa consentire l’adozione delle misure atte a garantire la protezione e la salute di pazienti ed utilizzatori.
3) Verificare la completezza e congruità dei dati contenuti nelle segnalazioni degli operatori effettuando le eventuali necessarie precisazioni, valutando inoltre la necessità di trasmissione delle stesse al Ministero della Salute oltre che al fabbricante e/o mandatario anche per il tramite del fornitore.
4) Trasmettere al Ministero della Salute, nel caso degli Ospedali Amedeo di Savoia/Birago di Vische, Maria Vittoria, San Giovanni Bosco e dei Distretti, le schede di segnalazione d’incidente.
5) Assicurare che le informazioni su tutti gli incidenti e mancati incidenti comunicati al Ministero della Salute siano raccolte, ordinate ed accessibili in un unico luogo.
6) Provvedere affinché tutte le informazioni relative agli incidenti e ogni altro inconveniente di cui ai punti 1 e 2, che coinvolgono un dispositivo medico in azienda, siano rapidamente e ufficialmente portate a conoscenza degli utilizzatori e di eventuali servizi interessati (ingegneria clinica, servizio tecnico, servizio provveditorato, servizio farmaceutico…), di norma per posta elettronica. Questa diffusione dovrà avvenire per il tramite dei referenti di area o struttura. Dovrà inoltre provvedere affinché le stesse informazioni siano inviate anche al fabbricante o mandatario o distributore (anche per il tramite dei referenti di area) nonché alla Direzione Generale e al Funzionario Regionale preposto.

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 6 di 34
6
7) Esaudire in maniera rapida e per quanto possibile completa ogni richiesta d’informazione supplementare, proveniente dal Ministero della Salute, finalizzate alla valutazione dei rischi di un dispositivo medico.
8) Provvedere alla consultazione, almeno settimanale, del sito internet relativo a dispositivi medici del Ministero della Salute nella specifica sezione dedicata ( ://www.ministerosalute.it/dispositivi/dispomed. ) per gli aggiornamenti relativi alla materia.
9) Trasmettere le informative e le comunicazioni sui dispositivi medici provenienti dal Ministero della Salute, dall’Assessorato Regionale, dai fabbricanti o autonomamente reperite, nell’ambito territoriale di riferimento secondo quanto valutato rilevante e di pertinenza dei vari settori. La trasmissione dovrà avvenire, di norma, utilizzando gli indirizzi di posta elettronica appositamente individuati dai responsabili di settore.
10) Fornire a tutti gli operatori sanitari le informazioni ed i chiarimenti di competenza richiesti e dare supporto per l’adempimento degli obblighi normativi.
11) Provvedere all’archiviazione, ove possibile informatizzata, della documentazione relativa ai dispositivi medici sia per quanto riguarda i rapporti d’incidente sia per ciò che concerne segnalazioni circolari, informative, comunicazioni, avvisi.
12) Segnalare al Referente di area o struttura le anomalie procedurali rilevate nell’applicazione della presente procedura.
Il Responsabile di Vigilanza dell’ASL TO2 si avvale della collaborazione di Referenti di area, individuati nei Direttori o loro incaricati: Sanitari Ospedalieri, di Distretto, Dipartimento Farmaceutico, Servizio Provveditorato, Servizio Economato, Servizio Tecnico, Ingegneria Clinica, Medicina Legale.
10. COMPITI DEI REFERENTI DEGLI OSPEDALI AMEDEO DI SAVOIA/BIRAGO DI VISCHE – MARIA VITTORIA - SAN GIOVANNI BOSCO E DEI DISTRETTI
Tra i Referenti il Responsabile Aziendale della vigilanza può individuare eventuali sostituti per i periodi di assenza dal servizio contrattualmente previsti.
Ai Referenti compete la diffusione capillare “a cascata” presso il proprio settore di pertinenza, ivi compresi Medici di Medicina Generale, Pediatri di Libera Scelta, Farmacie Comunali, delle segnalazioni ed informazioni trasmesse dal Responsabile Aziendale della Vigilanza sui Dispositivi Medici.
I Referenti provvedono alla verifica dei contenuti ed alla trasmissione al Responsabile Aziendale della Vigilanza, delle schede di segnalazione di incidente nel più breve tempo possibile onde consentire il rigoroso rispetto delle tempistiche previste dalla vigente normativa (2 giorni di calendario in caso di serio pericolo per la salute pubblica; 10 giorni di calendario per gli incidenti; 30 giorni di calendario per gli altri casi).

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 7 di 34
7
Essi forniscono inoltre l’adeguato supporto agli operatori per la compilazione delle suddette schede e possono a loro volta avvalersi del Responsabile Aziendale della Vigilanza a cui comunque devono fare riferimento.
Come previsto nelle linee d’indirizzo regionali sulla materia, nelle strutture sanitarie private insistenti sul territorio dell’ASL TO2
11. COMPITI DEI REFERENTI DEI PRESIDI SANITARI - STRUTTURE ACCREDITATE – STRUTTURE PRIVATE
deve
I nominativi dei responsabili così individuati, i loro recapiti telefonici ed e-mail nonché ogni relativa variazione, dovranno essere tempestivamente comunicati al Responsabile di Vigilanza dell’ASL TO2. In assenza di altre indicazioni verrà comunque considerato quale referente il Direttore o Responsabile Sanitario della struttura stessa.
essere formalmente individuata la figura di un Referente per la vigilanza che farà capo al Responsabile di Vigilanza sui Dispositivi Medici dell’ASL TO2.
Ai Referenti dei Presidi, Strutture Accreditate, Strutture Private insistenti sul territorio dell’ASL TO2 compete:
1. La diffusione capillare agli operatori della propria struttura delle segnalazioni ed informazioni trasmesse dal Responsabile Aziendale della Vigilanza sui Dispositivi Medici.
2. La ricezione delle segnalazioni di incidente provenienti dagli operatori della propria struttura e la verifica per ciò che attiene la loro correttezza, completezza e congruità.
3. La trasmissione al Ministero della Salute, al Fabbricante e/o Mandatario anche per il tramite del Fornitore ed al Responsabile Aziendale della Vigilanza sui Dispositivi Medici delle schede di segnalazione di cui al punto precedente nel rispetto dei termini di legge (2 giorni di calendario in caso di serio pericolo per la salute pubblica; 10 giorni di calendario per gli incidenti; 30 giorni di calendario per gli altri casi)
Si ricorda che, per le strutture private, il corretto funzionamento del sistema di vigilanza sui dispositivi medici è previsto venga a costituire oggetto di verifica in occasione dei periodici sopralluoghi da parte delle Commissioni di Vigilanza dell’ASL.
12. COMPITI DEGLI OPERATORI SANITARI Ogni operatore sanitario ha l’obbligo normativo
Sempre per obbligo normativo gli operatori sanitari sono tenuti a comunicare al fabbricante e/o mandatario, anche per il tramite del fornitore, ogni incidente che coinvolge un dispositivo medico ed ogni altro inconveniente che, pur non integrando le caratteristiche dell’incidente può consentire l’adozione delle misure atte a garantire la protezione e la salute di utilizzatori e pazienti.
di effettuare la segnalazione al Ministero della Salute di incidenti (come da definizione in allegato 1) che hanno coinvolto i dispositivi medici.
Le modalità con cui assolvere detti obblighi nell’ASL TO2 sono specificate nell’apposito paragrafo della presente procedura.

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 8 di 34
8
Si precisa che l’accezione di “operatore sanitario” è da intendersi riferita a coloro che abbiano in concreto utilizzato o impiantato il Dispositivo interessato dall’evento ovvero abbiano avuto diretta conoscenza dell’incidente/altro inconveniente in momento successivo (visite di controllo, follow up o altro.)
13. MODALITÀ DI SEGNALAZIONE AL MINISTERO DI INCIDENTE
Al Ministero della Salute è obbligatorio segnalare (per il tramite delle apposite schede ministeriali - Allegati 6 e 7) ogni
1 3.1 Tipologia Di Eventi Da Segnalare
- incidenteÈ inoltre necessario per obbligo di legge che, da parte degli operatori sanitari,
venga data al Responsabile Aziendale della Vigilanza, al fabbricante, al mandatario o al distributore del prodotto, comunicazione di ogni altro inconveniente coinvolgente Dispositivi Medici o concernente il loro uso e le procedure pre-uso, anche riferito a qualsiasi non conformità e non ricompreso nei casi definiti di incidente. Questo è stato definito dalla normativa perché possa essere consentita l’adozione delle misure atte a garantire la protezione e la salute di pazienti ed utilizzatori.
(cfr anche definizione nel glossario allegato 1).
In caso di dubbio sull’opportunità di effettuare una segnalazione o meno si deve propendere per la segnalazione dell’incidente.
Le MEDDEV rev. 6 suggeriscono come linea guida sui tipi d’incidenti da segnalare quanto segue:
Viene considerato incidente un qualsiasi malfunzionamento o deterioramento nelle caratteristiche e/o nelle prestazioni di un dispositivo, così come una qualsiasi inadeguatezza dell’etichettatura e delle istruzioni per l’uso, che hanno causato o potrebbero aver causato, sia in modo diretto che indiretto:
1. la morte di un paziente, utilizzatore o altra persona 2. un grave peggioramento del loro stato di salute, vale a dire:
- malattia grave o lesione con rischio di morte - compromissione permanente di una funzione del corpo o danno permanente ad una struttura corporea - situazioni che hanno richiesto un intervento medico o chirurgico per impedire una menomazione permanente di una funzione del corpo o danno permanente ad una struttura corporea (es.: un aumento clinicamente rilevante della durata di una procedura chirurgica, oppure una condizione che richiede il ricovero o un prolungamento significativo del ricovero in atto) - ogni danno indiretto quale conseguenza di risultati incorretti di strumentazioni diagnostiche o di test IVD, quando siano stati utilizzati rispettando le istruzioni d’uso del fabbricante - sofferenza fetale, morte del feto o ogni altra anomalia congenita o difetti della nascita.
I criteri per la segnalazione dell’incidente sono quindi: A. l’evento è accaduto:

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 9 di 34
9
malfunzionamento/ peggioramento delle caratteristiche o delle prestazioni del dispositivo in caso di test diagnostici in vitro, risultati falsi negativi o falsi positivi che non rientrano nei limiti delle prestazioni di chiarate nei test reazione avversa inattesa effetti d’interazione con altre sostanze o prodotti danno/distruzione del dispositivo (es.: incendio) terapia inappropriata inaccuratezza dell’etichettatura o delle istruzioni per l’uso (incluso anche il materiale promozionale)
B. opinione di professionisti del settore sanitario
si sospetta che un dato dispositivo possa aver contribuito all’incidente
risultato di una verifica del fabbricante evidenza derivata da incidenti simili avvenute precedentemente qualsiasi altra evidenza venuta a conoscenza dell’operatore
C.
Incidenti, in genere, da non segnalare al Ministero della Salute, ma, nel caso, solo al Fabbricante:
situazioni che abbiano causato, o che possano aver causato la morte o un grave peggioramento dello stato di salute di un paziente, utilizzatore o altra persona
• Difetto di un dispositivo nuovo riscontrato dall’utilizzatore prima del suo impiego
Esempi:
: indipendentemente dalla presenza di raccomandazioni nelle istruzioni per l’uso fornite dal fabbricante, difetti del dispositivo che possono essere normalmente individuati dall’utilizzatore e che non hanno causato alcun danno non richiedono di essere segnalati al Ministero (al fabbricante sì).
- la confezione di un dispositivo sterile monouso è etichettata con l’avviso “non utilizzare se la confezione è aperta o danneggiata”. Avendo riscontrato, prima dell’utilizzo, un evidente danno alla confezione, il dispositivo non viene utilizzato.
- Un set per la somministrazione endovenosa perde la protezione all’estremità rendendo il percorso del fluido non sterile. Il dispositivo non viene utilizzato.
- Uno speculum vaginale presenta diverse fratture e al momento di azionare l’impugnatura il dispositivo si rompe. Il dispositivo non viene utilizzato.
- Il contenuto di un flacone di un kit IVD, etichettato come liofilizzato viene, prima dell’utilizzo, rinvenuto allo stato fluido. Il dispositivo non viene utilizzato.
• Incidenti causati dalle condizioni del paziente:
Esempi:
quando si dispone di informazioni per cui la causa prima dell’incidente può essere ricondotta alle condizioni del paziente, l’incidente non richiede di essere segnalato al Ministero. Per giustificare l’assenza di segnalazione si deve però disporre di informazioni atte a poter concludere che il dispositivo ha funzionato come previsto e che non ha provocato né contribuito a provocare l’incidente. Queste informazioni devono essere condivise da parte di persone qualificate in grado di esprimere un giudizio anche dal punto di vista medico.
- La revisione anticipata di un impianto ortopedico che si mobilizza in quanto il paziente sta sviluppando un’osteolisi che non è conseguenza diretta dell’impianto (conclusione suffragata dall’opinione di un medico competente).

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 10 d i 34
10
- Un paziente muore dopo un trattamento dialisi. Il paziente aveva una patologia renale all’ultimo stadio ed è morto per insufficienza renale. Il dispositivo ha funzionato correttamente e l’incidente non è ad esso imputabile.
• Durata prevista per l’impiego o per la validità del Dispositivo Medico:
Esempi:
quando l’incidente è dovuto al fatto che il dispositivo è stato usato oltre la data limite di validità specificata dal fabbricante ed il tipo di evento non era inusuale, non è richiesta la segnalazione al Ministero. La valutazione della necessità o meno della segnalazione deve essere basata sulle informazioni riportate nella documentazione tecnica di prodotto o nelle istruzioni per l’uso.
- perdita del segnale al termine della durata dell’esercizio di un pacemaker. Gli indicatori hanno segnalato, nei tempi debiti e secondo le specifiche del dispositivo, la necessità della sostituzione. Il pacemaker deve essere espiantato chirurgicamente.
- Si osserva un contatto insufficiente degli elettrodi di un defibrillatore sul paziente. La defibrillazione non è stata possi bile a causa del loro scarso contatto con il torace. La data di scadenza degli elettrodi era indicata sull’etichetta, ma era stata superata.
- Un paziente è stato ricoverato per ipoglicemia causata dalla somministrazione di un errato dosaggio di insulina a seguito del risultato di un test sulla glicemia. Le indagini hanno evidenziato che le strisce reattive del test erano state usate dopo la data di scadenza indicata.
• Funzionamento corretto del sistema di protezione contro eventuali difetti:
Esempi:
si tratta di incidenti che non hanno provocato danni, grazie ad una caratteristica specifica di progetto che protegge il dispositivo contro il rischio che un difetto si tramuti in danno. Tali incidenti non necessitano di segnalazione.
- Una pompa per infusione si ferma a causa di un malfunzionamento segnalato da un adeguato allarme e non causa nessuna lesione al paziente
- Un’incubatrice controllata da microprocessore ha un guasto che viene segnalato da un adeguato allarme acustico e non c’è alcun peggioramento dello stato del paziente
- Un analizzatore di laboratorio si blocca durante un’analisi a causa di un malfunzionamento del sistema di pipettaggio dei campioni, ma l’operatore riceve un appropriato messaggio d’errore. Nessun risultato viene riferito.
• Effetti collaterali attesi e prevedibili:
Esempi:
quando il dispositivo viene utilizzato secondo l’uso previsto, non è necessaria la segnalazione al Ministero nel caso si verifichino effetti collaterali prevedibili e clinicamente accettabili in vista del beneficio per il paziente, o che sono stati specificamente indicati nelle informazioni fornite dal fabbricante oppure che presentano una predittività certa, funzionale o numerica. La documentazione relativa ad un particolare effetto collaterale deve essere disponibile nella documentazione tecnica del dispositivo.
- Un paziente con noti disturbi di claustrofobia, colto da seria crisi d’ansia nello spazio ristretto di una macchina per RM si procura lesioni. I rischi connessi alla claustrofobia sono conosciuti e descritti nella documentazione informativa sul dispositivo.
- Un paziente, a seguito dell’utilizzo di un defibrillatore in situazione d’emergenza, riporta ustioni di secondo grado. La valutazione del rischio documenta che tali ustioni sono accettabili alla luce del potenziale beneficio per il paziente e tale eventualità è segnalata nelle istruzioni per l’uso. La frequenza delle ustioni è compresa nel range specificato nel master record relativo al dispositivo.
- Un paziente presenta reazioni tissutali (allergia al nichel) già note in precedenza e descritte nella documentazione informativa del dispositivo.

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 11 d i 34
11
• Errori d’impiego ed impiego anomalo:
Alcuni esempi, sicuramente non esaustivi, di Incidenti da segnalare:
l’utilizzo del dispositivo in modo diverso da quello indicato dal fabbricante, anche con conseguenze di ordine medico, non comporta la segnalazione al Ministero.
Un paziente muore dopo l’uso di un defibrillatore ed è segnalato un problema con l’apparecchio.
Un paziente subisce un’ustione durante l’uso di un bisturi elettrico utilizzato in conformità alle istruzioni del fabbricante.
Una pompa d’infusione a causa di un malfunzionamento si arresta, ma non si aziona l’allarme previsto. Non si verifica alcun danno al paziente. Ciò deve essere segnalato anche se non si è verificato danno in quanto in una situazione differente avrebbe potuto causare un grave peggioramento dello stato di salute.
Una pompa d’infusione somministra una dose errata a causa d’incompatibilità tra pompa e set d’infusione utilizzato. Se la combinazione tra la pompa ed il set usato è conforme alle istruzioni per l’uso, sia della pompa sia del set, l’incidente deve essere segnalato anche se il paziente non ha riportato danni.
Un catetere aortico con palloncino presenta una perdita dovuta ad una manovra inappropriata con il dispositivo utilizzato, causando una situazione potenzialmente pericolosa per il paziente. Si ritiene che la manovra inappropriata sia dovuta ad un’inadeguatezza delle istruzioni per l’uso.
Un catetere si è rotto durante l’inserimento, anche se non vi è sospetto di manovra inappropriata. La rottura è avvenuta in un punto in cui la parte distaccata poteva essere facilmente estratta. Tuttavia questa è stata chiaramente una circostanza fortunata, dal momento che, se il catetere si fosse rotto in un punto leggermente diverso, sarebbe stato necessario un intervento chirurgico per recuperare l’estremità danneggiata.
Particelle di vetro vengono trovate in un contenitore per lenti a contatto. Perdita di segnale dopo che un pacemaker è giunto ad esaurimento. L’indicatore per la
sostituzione non si è attivato in tempo utile, nonostante quanto previsto dalle specifiche del dispositivo.
In un sistema a raggi x, durante l’esame di un paziente, una componente compie un movimento incontrollato ed il paziente viene colpito dall’intensificatore d’immagine riportando frattura nasale. Il sistema è stato istallato, manutenuto ed usato conformemente alle istruzioni del fabbricante
Viene richiesta la revisione prematura di un impianto ortopedico dovuta ad un cedimento. Sebbene al momento dell’evento non sia stata determinata la causa, l’incidente deve essere segnalato
Viene rilevato un difetto del software di un pacemaker Il test di fatica compiuto su una valvola cardiaca biologica in commercio ha rilevato un
deterioramento prematuro. Questo costituisce un pericolo per la salute pubblica. Vengono utilizzate strisce per il test di glucosio nel sangue secondo le istruzioni del fabbricante,
ma la lettura fornisce valori errati portando alla somministrazione di una quantità scorretta di insulina con conseguente shock ipoglicemico.
L’operatore rileva l’errore di abbinamento dei risultati analitici al codice paziente da parte di un analizzatore automatico.
Durante la manutenzione di un analizzatore per autodiagnosi è stato rilevato che una vite atta a posizionare l’unità riscaldante dell’analizzatore nella posizione corretta si è allentata. A causa di questo fatto potrebbe accadere che l’unità riscaldante si sposti dalla propria posizione e che la misurazione avvenga a temperatura non corretta, producendo risultati errati.

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 12 d i 34
12
1 3.2 Fase Della Notifica
In caso d’incidente, l’operatore sanitario pubblico o privato che ne è stato spettatore, attore o ne è venuto a conoscenza, è tenuto a far pervenire al referente di struttura o di area per la vigilanza sui dispositivi medici, nel modo più veloce possibile (comunque entro 2 giorni di calendario in caso di serio pericolo per la salute pubblica, 10 giorni di calendario per gli incidenti ed entro 30 giorni di calendarioN.B.: i termini sopra indicati sono quelli tassativi entro i quali il Ministero della Salute deve ricevere la documentazione, pertanto è utile che i documenti ai settori aziendali competenti vengano fatti
per gli altri casi.)
pervenire prima.
Esistono due tipologie di schede: una per i dispositivi medici ed un’altra per i dispositivi medico-diagnostici in vitro (rispettivamente allegati 1 e 4 del DM 15 nov. 2005, reperibili on line agli indirizzi ://www.ministerosalute.it/imgs/C_17_pagineAree_26_listaFile_itemName_3_file. e ://www.ministerosalute.it/imgs/C_17_pagineAree_29_listaFile_itemName_2_file. ).
Pur non essendoci campi obbligatori nella compilazione della scheda è comunque importante che siano riportati più dati possibile riguardanti l’identificazione del dispositivo medico nonché fabbricante/produttore/distributore (i servizi di settore che provvedono agli acquisti possono e devono fornire tutto l’adeguato supporto necessario alla compilazione del campo), il codice di classificazione unica nazionale dispositivi medici (CND) e, non appena sarà disponibile, il numero di repertorio del Dispositivo Medico (anche questo potrà essere richiesto ai servizi che provvedono agli acquisti). L’importanza di questi codici risiede nel fatto che permettono d’identificare univocamente il dispositivo medico.
I rapporti dovrebbero contenere tutti i dettagli del caso, tuttavia la segnalazione non deve essere ritardata oltre il lecito, correndo il rischio di superare le tempistiche previste di legge, nel tentativo di ottenere ed inserire informazioni addizionali: in tal caso può essere più utile trasmettere la scheda incompleta con l’indicazione in calce che i dati mancanti verranno trasmessi a strettissimo giro di posta. Tale affermazione deve poi essere soddisfatta dal dichiarante.
I tempi brevi di trasmissione definiti dalla normativa sottolineano come sia necessario che gli operatori sanitari provvedano alla segnalazione senza ritardo affinché non incorrano nelle sanzioni previste di legge (arresto sino a 6 mesi ed ammenda da 7.200 a 43.200 € salvo che il fatto non costituisca reato).
In caso di Struttura Privata, Accreditata o Presidio Sanitario, il Referente di struttura per la Vigilanza sui Dispositivi Medici, effettuata una

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 13 d i 34
13
valutazione di completezza e congruità dei dati riportati nella scheda di segnalazione ed apportate assieme al segnalante le eventuali correzioni e/o puntualizzazioni (in caso d’urgenza di trasmissione inserirà direttamente le integrazioni datando e firmando quanto aggiunto), provvederà direttamente alla trasmissione
1. al Ministero della Salute (Dipartimento dell’innovazione-Direzione Generale dei Farmaci e Dispositivi Medici-Ufficio V – Via Giorgio Ribotta, 5 - 00144 ROMA – telefax 0659943812);
via posta o fax avendo cura di rispettare i tempi previsti di legge:
2. al fabbricante o mandatario o distributore; 3. al Responsabile Aziendale per la Vigilanza sui Dispositivi Medici.
I Referenti di Struttura o di Area degli Ospedali Amedeo di Savoia/Birago di Vische, Maria Vittoria, San Giovanni Bosco e dei Distretti dovranno trasmettere le schede, anticipando il documento via telefax (n. 0114393273) e quindi per posta ordinaria, esclusivamente al Responsabile Aziendale per la Vigilanza sui Dispositivi Medici in tempi utili affinché possa essere rispettata la tempistica prevista per l’inoltro al Ministero della Salute.
Il Responsabile Aziendale per la Vigilanza sui Dispositivi Medici, eseguita a sua volta sui documenti ricevuti una verifica di congruità, completezza, correttezza e rispetto dei termini, effettuerà eventuali rilievi al responsabile di struttura provvedendo quindi alla trasmissione per conoscenza dell’accaduto alla Direzione Sanitaria d’Azienda; ai Direttori dei Servizi Dipartimentali coinvolti negli acquisti (quali provveditorato, farmaceutico, tecnico) per quanto di loro eventuale competenza, ivi comprese le possibili restituzioni dei lotti di prodotti alle Ditte e sospensione dei pagamenti; ai servizi/strutture che potrebbero essere eventualmente interessati; alla Regione Piemonte (Assessorato Tutela della Salute e Sanità, Settore Ispettivo e Controllo di Qualità in Materia Sanitaria – C.so Regina Margherita, 153 bis – 10122 Torino) e, solo nel caso degli Ospedali Aziendali e dei Distretti, anche al Ministero della Salute.
Le richieste di ulteriori notizie e delucidazioni da parte del Ministero o dell’Assessorato Regionale dovranno essere soddisfatte nel più breve tempo possibile da parte del Responsabile della Vigilanza sui Dispositivi Medici con la collaborazione degli operatori segnalanti ed ogni altra professionalità necessaria. Nel caso a ciò provveda direttamente il Referente di Area o di Struttura dovrà comunque darne informazione anche al Responsabile Aziendale della Vigilanza.
Solo nelle strutture private, convenzionate e nei Presidi insistenti sul territorio dell’ASLTO2, conformemente a quanto indicato nelle linee d’indirizzo regionali, il Responsabile/Referente individuato all’interno delle struttura dovrà pertanto farsi carico autonomamente della raccolta, verifica di completezza, correttezza, congruità e dell’inoltro al Ministero della Salute nei succitati termini definiti di legge, delle notifiche d’incidente identificate nel proprio ambito di competenza. Egli provvederà inoltre ad inviare contestualmente, per conoscenza, copia della notifica anche al Responsabile di Vigilanza dell’ASL TO2 e ad avvisare il fabbricante/mandatario/distributore.

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 14 d i 34
14
Il Responsabile Aziendale sulla vigilanza sarà comunque disponibile a fornire tutto l’eventuale supporto ritenuto necessario.
Si ricorda che, per ciò che concerne le strutture private, il corretto funzionamento del sistema di vigilanza sui dispositivi medici è previsto che venga a costituire oggetto di verifica in occasione dei periodici sopralluoghi da parte delle Commissioni di Vigilanza dell’ASL.
Se disponibile il dispositivo medico oggetto della segnalazione deve essere conservato presso il servizio segnalante, adottando i seguenti accorgimenti previsti dal Ministero della Salute:
1 3.3 Fase Di Gestione Dei Dispositivi Oggetto Delle Segnalazioni
- Segregazione in apposito spazio. - Se non utilizzato
-
il dispositivo deve, per quanto possibile, essere conservato nella confezione originale. Se utilizzato il dispositivo non dovrà essere pulito, manipolato o disinfettato e dovrà essere conservato negli appositi contenitori utilizzati per i rifiuti speciali oppure in contenitori rigidi con tappo a pressione o a vite tipo quelli utilizzati per la conservazione dei prelievi bioptici o anatomici, salvo diversa modalità più idonea di gestione comunicata dal Ministero della Salute o dall’Autorità Giudiziaria. Il contenitore dovrà
- In mancanza di differente indicazione da parte del Ministero della Salute, o da parte dell’Autorità Giudiziaria (per i casi che ne abbiano previsto il coinvolgimento), il Dispositivo Medico deve essere restituito al Fabbricante secondo le sue istruzioni.
essere opportunamente etichettato (riportare la data dell’incidente, la data di compilazione della notifica, il nominativo dell’operatore che ha rilevato l’incidente e le iniziali del paziente/operatore che è stato coinvolto nell’incidente). Nel caso il fabbricante richieda una conservazione differente del materiale rispetto a quella sopra indicata dovrà darne precisazione scritta.
- Il fabbricante/mandatario/distributore deve ottenere il dispositivo medico oggetto di segnalazione per poter adempiere ai propri obblighi normativi d’indagine finalizzata ad intraprendere eventuali azioni correttive. Nella trasmissione del rapporto iniziale d’incidente dovrà essere specificato che il fabbricante/mandatario/distributore è stato reso consapevole che eventuali analisi anche distruttive del Dispositivo Medico potranno iniziare solo dopo 10 giorni dalla ricezione del rapporto iniziale redatto dall’operatore, a meno che le autorità competenti non si oppongano a dette analisi.
- L’affidamento alla Ditta del dispositivo medico dovrà avvenire utilizzando modulistica appositamente predisposta
- Nel caso il fabbricante non richieda la restituzione del Dispositivo Medico coinvolto nell’evento, il Responsabile della Vigilanza ne darà comunicazione ai
riportante l’impegno sottoscritto da parte del fabbricante/mandatario/distributore a non iniziare le analisi sul materiale prima che siano trascorsi 10 giorni dalla ricezione del rapporto iniziale redatto dall’operatore (fac simile Allegato3). La sottoscrizione dell’impegno potrà essere effettuata dalla persona delegata dalla Ditta al ritiro del Dispositivo Medico.

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 15 d i 34
15
competenti uffici ministeriali, precisando che, qualora non pervengano indicazioni entro 90 giorni la struttura sanitaria si riterrà autorizzata a disporne nel modo più opportuno, ivi compresa l’alienazione.
- Previ accordi con il fabbricante/fornitore, dei quali dovranno essere informati sia il Responsabile della Vigilanza per i Dispositivi Medici sia il settore responsabile dell’acquisto, sarà possibile, salvo diversa indicazione, procedere alla restituzione anche del lotto di prodotti di cui fa parte il dispositivo oggetto di segnalazione.
L’indagine sull’evento segnalato è di competenza del fabbricante e gli operatori sanitari dovranno collaborare con lui fornendo le notizie necessarie per la valutazione dell’evento pur senza rivelare l’identità del paziente.
1 3.4 Fase Dell’indagine
Le note informative, gli aggiornamenti sulla sicurezza dei dispositivi medici e le risultanze delle indagini attuate a seguito di notifiche di incidenti pervenute dai fabbricanti e dal Ministero della Salute, saranno trasmesse, a cura del Responsabile Aziendale per la Vigilanza ai Responsabili/Referenti dei settori interessati (di norma tramite e-mail). Questi ultimi provvederanno quindi alla diffusione capillare ed all’applicazione dei provvedimenti individuati.
1 3.5 Fase Dell’azione Valutativa E Correttiva
Le risultanze di indagini su dispositivi medici coinvolti in incidenti/inconvenienti, qualora pervengano direttamente agli operatori o ai referenti di area o struttura, dovranno essere da questi trasmesse in copia anche al Responsabile Aziendale della Vigilanza.
Le segnalazioni pervenute in materia di Dispositivi Medici e le notifiche d’incidente saranno conservate in originale presso gli uffici che hanno provveduto direttamente alla trasmissione al Ministero della Salute in appositi Dossier separati ed archiviati per data di ricezione e numero di protocollo.
1 3.6 Raccolta Ed Archiviazione
Come previsto dalle linee d’indirizzo 5/2007 della Regione Piemonte,
14. TRACCIABILITÀ
i servizi deputati agli acquisti (provveditorato, economato, farmaceutico, tecnico…) dovranno adottare al più presto una procedura di monitoraggio
- nome del fornitore
sui prodotti acquistati e distribuiti, che consenta di agevolare qualsiasi azione correttiva o cautelativa predisposta dal Ministero o dai Fabbricanti e che preveda la registrazione almeno di:
- nome commerciale del prodotto - numeri di serie e lotto - struttura/reparto a cui il dispositivo medico viene consegnato I reparti/servizi/ambulatori/uffici, ogni volta che ciò sia possibile, analogamente a
quanto avviene per i dispositivi medici impiantati in sala operatoria ove l’etichetta

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 16 d i 34
16
viene allegata all’atto operatorio del paziente, dovranno accludere le etichette dei dispositivi medici utilizzati sui pazienti alla documentazione sanitaria degli stessi onde consentirne la tracciabilità.
Per ciò che riguarda la necessità di conoscere il numero d’iscrizione al Repertorio dei Dispositivi Medici, nonché l’indicazione contenuta nelle linee regionali d’indirizzo di classificare i dispositivi medici acquistati dalle strutture sanitarie in classi e sottoclassi omogenee adottando la classificazione CND, si affida la responsabilità di provvedere al più presto
Ai servizi aziendali deputati agli acquisti potranno rivolgersi gli operatori per conoscere il numero d’iscrizione a repertorio dei Dispositivi Medici, da inserire nel rapporto d’incidente.
a quanto necessario ai servizi aziendali deputati agli acquisti.
Si prevede che la tracciabilità dei dispositivi medici sarà oggetto di valutazione da parte degli organismi regionali
Sarà cura del Responsabile della Vigilanza dei Dispositivi Medici, in collaborazione con l’Area di Formazione prevedere appositi corsi sulla materia rivolti a operatori sanitari nonché a Direttori/Referenti delle strutture sanitarie affinché diffondano le conoscenze al personale del settore di pertinenza e prevedano specifici richiami sull’argomento nell’ambito di riunioni di settore.
15. FORMAZIONE
Il monitoraggio dell’applicazione della presente procedura è affidato al Responsabile della Vigilanza sui Dispositivi Medici ed ai Referenti/Responsabili delle strutture coinvolte.
16. MONITORAGGIO DELLA PROCEDURA
- Invio al Ministero della Salute entro i termini di 2, 10 e 30 giorni delle segnalazioni previste di legge
17. PARAMETRI DI CONTROLLO
- Diffusione, in ambito aziendale (di norma tramite e-mail), delle segnalazioni ed informative ministeriali, regionali e dei fabbricanti, inerenti i Dispositivi Medici
- Adozione di procedure di monitoraggio sui prodotti acquistati, in grado di permetterne la tracciabilità.
Questa procedura è sottoposta a revisione, in caso di necessità e di adeguamenti alla normativa, da parte del Responsabile della Vigilanza sui Dispositivi Medici
18. REVISIONE

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 17 d i 34
17
Allegato 1
GLOSSARIO Accessorio Prodotto che, pur non essendo un dispositivo, sia destinato in modo specifico dal fabbricante ad essere utilizzato con un dispositivo per consentirne l'utilizzazione prevista dal fabbricante stesso.
Comunicazione relativa ad un’azione correttiva in campo da parte del fabbricante/mandatario ai clienti/utilizzatori.
Avviso di Sicurezza (FSN)
Azione mirata all’eliminazione delle cause di una potenziale non conformità o di altre criticità. Azione Correttiva
Misura intrapresa dal fabbricante per ridurre il rischio di morte o grave peggioramento dello stato di salute legato all’utilizzo di un Dispositivo Medico già commercializzato. Queste misure possono essere segnalate con un avviso di sicurezza e possono prevedere: la riconsegna del DM al fornitore, la modifica del DM, la sostituzione del DM, la distruzione del DM, l’adozione da parte dell’acquirente di modifiche o variazioni progettuali del fabbricante, raccomandazioni da parte del fabbricante sull’utilizzo del DM, la modifica dell’etichettatura o delle istruzioni per l’uso, la modifica della gestione clinica del paziente, avvisi relativi a cambiamento delle modalità di utilizzo del DM.
Azione Correttiva in Campo (FSCA/ recall)
Dispositivi diagnostici e IVD non agiscono direttamente sull’individuo. Il danno può quindi verificarsi come conseguenza di una decisione medica o di un provvedimento intrapreso o no, sulla base delle informazioni/risultati forniti dal dispositivo. Il danno può quindi derivare da diagnosi errata o ritardata, trattamento ritardato o inappropriato ecc…
Danno Indiretto
Destinazione L'utilizzazione alla quale è destinato il dispositivo secondo le indicazioni fornite dal fabbricante sull'etichetta, nelle istruzioni per l’uso e/o nei materiali pubblicitari.
Qualsiasi strumento, apparecchio, impianto, software, sostanza o altro prodotto, utilizzato da solo o in combinazione, compresi gli accessori tra cui il software destinato dal fabbricante ad essere impiegato specificamente con finalità diagnostiche e/o terapeutiche e necessario al corretto funzionamento del dispositivo stesso, destinato dal fabbricante ad essere impiegato sull’uomo a fini di:
Dispositivo medico
- diagnosi, prevenzione, controllo, trattamento o attenuazione di malattie; - diagnosi, controllo, trattamento, attenuazione o compensazione di una ferita o di un handicap; - studio, sostituzione o modifica dell'anatomia oppure di un processo fisiologico; - controllo del concepimento, che non eserciti nel o sul corpo umano l’azione principale cui è destinato con
mezzi farmacologici, immunologici o mediante processi metabolici, ma la cui funzione possa essere coadiuvata da tali mezzi.
Qualsiasi dispositivo medico collegato per il suo funzionamento ad una fonte di energia elettrica o a qualsiasi altra fonte di energia diversa da quella prodotta direttamente dal corpo umano o dalla gravità.
Dispositivo medico attivo
Qualsiasi dispositivo medico attivo destinato ad essere impiantato interamente o parzialmente mediante intervento chirurgico o medico nel corpo umano o mediante intervento medico in un orifizio naturale e destinato a restarvi dopo l’intervento.
Dispositivo medico impiantabile attivo
Qualsiasi dispositivo medico composto da un reagente, da un prodotto reattivo, da un calibratore, da un materiale di controllo, da un kit, da uno strumento, da un apparecchio, un'attrezzatura o un sistema, utilizzato da solo o in combinazione, destinato dal fabbricante ad essere impiegato in vitro per l'esame di campioni provenienti dal corpo umano, inclusi sangue e tessuti donati, unicamente o principalmente allo scopo di fornire informazioni su uno stato fisiologico o patologico, o su una anomalia congenita, o informazioni che consentono la determinazione della sicurezza e della compatibilità con potenziali soggetti riceventi, o che consentono il controllo delle misure terapeutiche. I contenitori dei campioni sono considerati dispositivi medico-diagnostici in vitro. Si intendono per contenitori di campioni i dispositivi, del tipo sottovuoto o no, specificamente destinati dai fabbricanti a ricevere direttamente il campione proveniente dal corpo umano e a conservarlo ai fini di un esame diagnostico in vitro. I prodotti destinati ad usi generici di laboratorio non sono dispositivi medico-diagnostici in vitro a meno che, date le loro caratteristiche, siano specificamente destinati dal fabbricante ad esami diagnostici in vitro.
Dispositivo medico-diagnostico in vitro
Dispositivo destinato ad essere utilizzato una sola volta per un solo paziente. Dispositivo monouso

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 18 d i 34
18
Qualsiasi dispositivo destinato ad essere utilizzato da un medico debitamente qualificato per lo svolgimento di indagini cliniche di cui all'allegato 7 , punto 2.1 (D.Lgs 507/92 e s.m.i.), in un ambiente clinico umano adeguato; per l'esecuzione delle indagini cliniche, al medico debitamente qualificato è assimilata ogni altra persona, la quale, in base alle qualifiche professionali, sia autorizzata a svolgere tali indagini.
Dispositivo per indagini cliniche
Qualsiasi dispositivo fabbricato appositamente, sulla base della prescrizione scritta di un medico debitamente qualificato che precisi, sotto la propria responsabilità, le caratteristiche specifiche di progettazione e destinato ad essere utilizzato solo per un determinato paziente; i dispositivi fabbricati con metodi di produzione in serie che devono essere adattati per soddisfare un'esigenza specifica del medico o di un altro utilizzatore professionale non sono considerati dispositivi su misura.
Dispositivo su misura
Atto o omissione che produce risultati diversi da quelli definiti al fabbricante o attesi dall’operatore Errore di Utilizzo
Fabbricante La persona fisica o giuridica responsabile della progettazione, della fabbricazione, dell'imballaggio e dell'etichettatura di un dispositivo in vista dell'immissione in commercio a proprio nome, indipendentemente dal fatto che queste operazioni siano eseguite da questa stessa persona o da un terzo per suo conto. Gli obblighi del presente decreto che si impongono al fabbricante valgono anche per la persona fisica o giuridica che compone, provvede all'imballaggio, tratta, rimette a nuovo, etichetta uno o più prodotti prefabbricati o assegna loro la destinazione di dispositivo in vista dell'immissione in commercio a proprio nome. I predetti obblighi non si applicano alla persona la quale, senza essere il fabbricante compone o adatta dispositivi già immessi in commercio in funzione della loro destinazione ad un singolo paziente.
Ogni evento che possa costituire un imminente rischio di morte, di grave peggioramento dello stato di salute o di malattia seria, e che richieda un tempestivo intervento correttivo (es.: eventi significativi e di natura imprevedibile tali da generare allarme e rappresentare un pericolo potenziale per la salute pubblica tipo HIV e Creutzfeldt-Jacob o possibilità di decessi multipli in brevi intervalli di tempo). In questi casi la segnalazione dell’incidente deve pervenire al Ministero della Salute entro 2 giorni di calendario.
Grave pericolo per la salute pubblica
La prima messa a disposizione a titolo oneroso o gratuito di dispositivi, esclusi quelli destinati alle indagini cliniche, in vista della distribuzione o utilizzazione sul mercato comunitario, indipendentemente dal fatto che si tratti di dispositivi nuovi o rimessi a nuovo.
Immissione in commercio
Senza alcun ritardo Immediatamente
Incidente La condizione in cui qualsiasi disfunzione o deterioramento delle caratteristiche o delle prestazioni, nonché qualsiasi carenza nell'etichettatura o nelle istruzioni per l'uso di un dispositivo medico abbiano causato, direttamente o indirettamente, un grave peggioramento dello stato di salute o la morte del paziente o di un utilizzatore. Per grave peggioramento dello stato di salute si deve intendere: una malattia o lesione con pericolo per la vita; una menomazione di una funzione del corpo o una lesione di una struttura corporea; una condizione che rende necessario un intervento medico o chirurgico per impedire una menomazione di una funzione del corpo o una lesione di una struttura corporea; una condizione che causa l'ospedalizzazione o il prolungamento dell'ospedalizzazione.
Mandatario La persona fisica o giuridica stabilita nel territorio dell'Unione Europea che, dopo essere stata espressamente designata dal fabbricante, agisce e può essere interpellata dalle autorità nazionali competenti e dagli organismi comunitari in vece del fabbricante per quanto riguarda gli obblighi che il presente decreto impone a quest'ultimo.
Fase in cui il dispositivo è stato reso disponibile all'utilizzatore finale in quanto pronto per la prima utilizzazione sul mercato comunitario secondo la sua destinazione d'uso.
Messa in servizio
Colui che ha in concreto utilizzato o impiantato il Dispositivo Medico interessato dall’evento ovvero che abbia avuto diretta conoscenza dell’incidente/inconveniente avvenuto in un momento successivo (in occasione di visite di controllo o follow up o altro)
Operatore Sanitario
Gli Organismi Notificati (anche indicati come Organismi Designati nelle direttive comunitarie e nei decreti legislativi di recepimento), sono Enti pubblici o privati, autorizzati dalle Autorità competenti dei singoli Stati membri ad espletare, su richiesta delle ditte fabbricanti, le procedure di valutazione conformità e di certificazione dei dispositivi medici previste dalla normativa vigente in materia di dispositivi medici.
Organismo notificato

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 19 d i 34
19
L’elenco degli Organismi autorizzati dai diversi Stati viene comunicato alla Commissione Europea ed agli altri Stati membri dell’Unione Europea e pubblicato in un apposito Registro Comunitario.
Segnalazione di qualsiasi inconveniente, evento o non conformità concernenti l’uso o le procedure per l’uso correlate ad un Dispositivo Medico e che non siano ricomprese nelle caratteristiche dell’incidente.
Reclamo
Atto, od omissione di un atto, da parte di un operatore o utilizzatore , di un dispositivo medico come risultato di una condotta che va oltre ogni possibilità di controllo del rischio da parte del fabbricante
Utilizzo Anomalo

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 20 d i 34
20
Tabella 1 Allegato 2
TABELLA DELLE RESPONSABILITÀ
Attori Responsabilità attribuite
Operatore Sanitario
(pubblico o privato)
- segnalazione mediante compilazione della scheda min isteriale degli incidenti, nonché di ogni altro inconveniente coinvolgente dispostivi medici, di cui viene a diretta conoscenza
- trasmissione della scheda compilata al referente di area o di struttura per la vigilanza nel più breve tempo possibile e comunque entro 2 giorni di calendario in caso di serio pericolo per la salute pubblica, 10 giorn i di calendario per g li incidenti, 30 giorn i di calendario per gli altri casi
- segnalazione dei reclami - conservazione corretta dei d ispositivi medici oggetto di segnalazione - consentire, per quanto possibile, la tracciabilità dei dispositivi medici
impiegati sui pazienti - collaborare con il Min istero della Salute e con il fornitore fornendo le notizie
eventualmente rich ieste - prendere atto ed adottare le indicazioni contenute nelle informat ive trasmesse
e riferite ai dispositivi medici
- rispetto delle tempistiche previste di legge e nella procedura aziendale
Referente di Struttura delle Strutture Private – Accreditate e Presidi Sanitari
- aggiornare, se necessario, l’indirizzo e-mail, con controllo giornaliero, al quale ricevere dal Responsabile Aziendale per la Vig ilanza sui Dispositivi Medici le segnalazioni sulla materia, ivi compresi gli avvisi di sicurezza
- diffusione capillare presso gli operatori della struttura di pertinenza delle segnalazioni ed informazioni tras messe dal Responsabile Aziendale per la Vig ilanza sui Dispositivi Medici
- ricezione delle segnalazioni di incidente e di inconvenienti coinvolgenti dispositivi medici da parte degli operatori della struttura di pertinenza
- supporto agli operatori per la compilazione delle schede ministeriali di segnalazione
- verifica per ciò che attiene correttezza, completezza e congruità delle schede di segnalazione compilate
- trasmissione al Ministero della Salute, al Responsabile Aziendale della Vig ilanza ed al fabbricante o mandatario o distributore delle schede nel rispetto delle tempistiche previste dalla normat iva e dalla procedura aziendale
- corretta archiviazione delle notifiche d’incidente e delle segnalazioni di inconvenienti con dispositivi medici
- controllo della corretta applicazione della procedura aziendale nella struttura di pertinenza
- collaborazione con il Responsabile Aziendale della Vig ilanza
Referenti di Area o Struttura Ospedaliera (Direttori Sanitari degli Ospedali Amedeo di Savoia/Birago di Vische – Maria Vittoria – San Giovanni Bosco; Direttori di Distretto, Direttori dei
- aggiornare, se necessario, l’indirizzo e-mail, con controllo giornaliero, al quale ricevere dal Responsabile Aziendale per la Vig ilanza sui Dispositivi Medici le segnalazioni sulla materia, ivi compresi gli avvisi di sicurezza
- diffusione capillare presso gli operatori della struttura di pertinenza delle segnalazioni ed informazioni tras messe dal Responsabile Aziendale per la Vig ilanza sui Dispositivi Medici
- ricezione delle segnalazioni di incidente e di inconvenienti coinvolgenti dispositivi medici da parte degli operatori della struttura di pertinenza

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 21 d i 34
21
Dipartimenti/Servizi Farmaceutico, Provveditorato, Economato, Tecnico, Ingegneria Clinica)
- supporto agli operatori per la compilazione delle schede ministeriali di segnalazione
- verifica per ciò che attiene correttezza, completezza e congruità delle schede di segnalazione compilate
- trasmissione al Responsabile Aziendale della Vigilanza delle schede nel rispetto delle tempistiche previste dalla normat iva e dalla procedura aziendale
- controllo della corretta applicazione della procedura aziendale nella struttura di pertinenza
- collaborazione con il Responsabile Aziendale della Vig ilanza
Servizi deputati agli acquisti (Provveditorato/Economato; Farmaceutico; Tecnico)
In particolare: - fornire supporto urgente agli operatori nella compilazione della scheda di
segnalazione ministeriale per ciò che attiene i dati relat ivi al fabbricante/produttore/distributore ed il numero di repertorio dei Dispositivi Medici
- adottare un’adeguata procedura di monitoraggio sui prodotti acquistati e distribuiti tale da agevolare qualsiasi azione correttiva/cautelativa predisposta dal Ministero o dai Fabbricanti e d ’identificazione rapida dei repart i/settori aziendali a cui i p rodotti sono stati consegnati
- provvedere a quanto previsto dalla vigente normativa in merito alla conoscenza del numero d’iscrizione al repertorio dei Dispositivi Medici
- provvedere ad applicare le linee d’indirizzo regionali in merito alla classificazione dei dispositivi medici acquistati in classi e sottoclassi omogenee adottando la classificazione CND
Responsabile Aziendale della Vigilanza sui Dispositivi Medici
- ricezione delle segnalazioni di incidente e di inconvenienti coinvolgenti dispositivi medici redatti dagli operatori, verificarne completezza, correttezza e congruità ed effettuare le eventuali necessarie precisazioni
- trasmettere le schede, quando dovuto, al Ministero della Salute, alla Regione Piemonte ed al fabbricante/produttore/distributore
- fornire ad operatori e referenti di area e struttura informazioni, chiarimenti e supporto tecnico
- consultazione almeno settimanale del sito ministeriale sui dispositivi medici - trasmissione tempestiva ai referenti di area e struttura delle info rmative e
comunicazion i riferite ai dispositivi medici secondo quanto valutato rilevante e di pertinenza dei vari settori
- corretta archiviazione delle schede di notifica di incidente, di inconvenienti coinvolgenti dispositivi medici e delle info rmative e comunicazioni riferite a dispositivi medici
- controllo della corretta applicazione della procedura aziendale - revisione della procedura tecnica aziendale per l’attuazione della vig ilanza
sui dispositivi medici

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 22 d i 34
22
FAC SIMILE Allegato 3
Verbale di consegna di Dispositivo Medico oggetto di
incidente segnalato al Ministero della Salute
In data odierna il dispositivo medico:
(nome commerciale)………………………………………………………della Ditta……………………….………………..
che è stato oggetto, in data…………………..di □ Rapporto di incidente al Ministero della Salute
□ Segnalazione di inconveniente
viene consegnato al Sig.…………………………………………………………………………………………………….incaricato
al ritiro dalla Ditta interessata.
Quanto sopra avviene in conformità a quanto previsto in materia dal Ministero della Salute
(Direzione Generale dei Farmaci e dei Dispositivi Medici), non essendo pervenute diverse indicazioni
dagli organismi competenti.
Una volta effettuate le indagini del caso, si ricorda alla Ditta, che prende in consegna il materiale,
l’obbligo di dare le dovute informazioni al Ministero della Salute in merito al tipo d’indagini eseguite
ed all’esito delle stesse.
L’incaricato della Ditta, che prende in consegna il materiale, attesta
per conto del Fabbricante/Mandatario/Distributore l’impegno a non iniziare
le indagini su quanto consegnato prima che siano trascorsi 10 giorni dalla
data di ricezione del rapporto prodotto dall’operatore sanitario dell’ASL TO2.
Firma leggibile di chi ritira il materiale per conto della Ditta
……………………………………..
Firma leggibile di chi consegna il materiale
per conto della Struttura Sanitaria
………………………………… Data …………………………………………… Consegnare una copia a chi ritira il Dispositivo Medico Consegnare una copia al Responsabile della Vigilanza Dispositivi Medici Trattenere l’originale presso la struttura che consegna il Dispositivo Medico.

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 23 d i 34
23
Allegato 4
FLOW CHART DELLA SEGNALAZIONE STRUTTURE PRIVATE – ACCREDITATE E PRESIDI SANITARI
Si attivano procedure di
reparto affinché il
fatto non si ripeta e si informa il
Responsabile
Evento coinvolgente Dispositivo Medico
L’operatore è in grado di gestire la notifica in prima
persona?
Cercare il Capo Sala o il Responsabile
Medico per la necessaria assistenza
È realmente un incidente?
Si compila una lettera di segnalazione indicando l’accaduto al Responsabile di Struttura che lo invierà in copia al Resp. della vigilanza
Si compila la scheda di segnalazione per il Ministero
Ci sono i dati più importanti?
(dati della struttura e del segnalante, dati sul disp. medico e sul fabbricante o distributore, dati
sull’accaduto)
Si cerca telefonicamente chi può di fornirli (es.: provveditorato, farmacia, servizio tecnico. La classificazione CND si trova on-line sul sito ministeriale)
Persistono dubbi?
Contattare il Referente di Struttura sui dispositivi medici per le necessarie indicazioni
Si inv ia la scheda al Referente di Struttura per i Dispositivi Medici
Contattare il Responsabile Aziendale per la Vigilanza per eventuale supporto
Termine della procedura
NO
NO
NO
NO
NO
SI
SI
SI
SI
SI
è davvero un Disp. Medico ed è stato usato secondo le
indicazioni d’uso?
La scheda è compilata correttamente?
SI
Contattare il segnalante ed apportare le correzioni
Trasmissione della Scheda al Ministero e Copia al
Responsabile Aziendale per la Vigilanza. Comunicare l’accaduto anche al
fabbricante/mandatario/distributore
Persistono dubbi? NO
SI
NO
► N.B.: il rapporto deve essere trasmesso NEL PIÙ BREVE TEMPO POSSIBILE per poter rispettare le tempistiche previste di legge

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 24 d i 34
24
Allegato 5
FLOW CHART DELLA SEGNALAZIONE
OSPEDALI AMEDEO DI SAVOIA/BIRAGO DI VISCHE – MARIA VITTORIA – SAN GIOVANNI BOSCO E DISTRETTI
Evento coinvolgente Dispositivo Medico
L’operatore è in grado di gestire la notifica in prima
persona?
Cerca il Capo Sala o il Responsabile Medico per la
necessaria assistenza
È realmente un incidente?
Si compila una lettera di segnalazione indicando l’accaduto al Responsabile di Struttura che lo invierà in copia al Resp. della vigilanza
Si compila la scheda di segnalazione per il
Ministero
Ci sono i dati più importanti?
(dati della struttura e del segnalante, dati sul disp. medico e sul fabbricante o distributore, dati
sull’accaduto)
Si cerca telefonicamente chi può di fornirli (es.: provveditorato, farmacia, servizio tecnico. La classificazione CND si trova on-line sul sito ministeriale)
Persistono dubbi?
Contattare il Referente di area/struttura sui disp. medici
Persistono dubbi?
Contattare il responsabile aziendale per la vigilanza
Si invia la scheda al Referente
di area/struttura per i Dispositivi Medici
La scheda è compilata
correttamente?
Contattare il segnalante ed apportare le correzioni
Persistono dubbi?
Trasmissione della scheda al Responsabile Aziendale per la
vigilanza per l’inoltro agli organismi preposti
Si attivano procedure di
reparto affinché il fatto non si ripeta e si informa il
Responsabile di Struttura
Elaborazione congiunta della scheda di segnalazione e trasmissione agli organismi di competenza da parte del resp. Az.le per la vigilanza
Termine della procedura
NO
NO
NO
NO
NO
NO
NO
SI
SI
SI
SI
NO
SI
SI
SI
SI
è davvero un Disp. Medico ed è stato usato secondo le
indicazioni d’uso?
► N.B.: il rapporto deve essere trasmesso NEL PIÙ BREVE TEMPO POSSIBILE per poter rispettare le tempistiche previste di legge

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 25 d i 34
25
Allegato 6
(on line all’indirizzo: http://www.ministerosalute.it/imgs/C_17_pagineAree_39_listaFile_itemName_2_file.doc)
Modulo Ministeriale Da Utilizzare Per Incident Con DISPOSITIVO MEDICO
Allegati n. 1 e n. 7 al DM 15 novembre 2005 (le definizioni sono state modificate in base a quanto riportato nel D.Lgs
37/2010 e MEDDEV 2.12-1 rev. 6)
Rapporto di incidente o di mancato incidente da parte di operatori sanitari al Ministero della Salute
(artt. 9 e 10, D.Lgs. n. 46 del 1997; art. 11, D.Lgs. n. 507 del 1992)
Rapporto interno n. ……….
Rapporto relativo a:
Incidente Mancato incidente
A) Dati relativi al luogo dove si è verificato l’episodio 1. Denominazione della struttura (utilizzare la denominazione ufficiale della struttura)
2. Reparto
3. Dati dell’operatore sanitario che ha rilevato l’episodio (nome, cognome, qualifica)
Telefono
Fax
4. Data dell'episodio
5. Azienda Ospedaliera o Azienda Sanitaria Locale competente per territorio (utilizzare la denominazione ufficiale della struttura). La ASL va indicata in caso di segnalazione di operatore sanitario operante in struttura sanitaria privata o pubblica non aziendale, di medico di medicina generale o pediatra di libera scelta, di farmacista
6. Dati del responsabile della vigilanza (nome, cognome, servizio di appartenenza)
Dr.ssa Anna BRUNETTI – Direzione Sanitaria Ospedale Maria Vittoria – ASL TO2
B) Dati relativi al dispositivo medico
Fabbricante (nome, ragione sociale e indirizzo)

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 26 d i 34
26
Fornitore (nome, ragione sociale e indirizzo)
Nome commerciale ed eventuale modello del dispositivo
Descrizione del dispositivo medico
N. codice del dispositivo assegnato dal fabbricante
Numero di lotto o di serie
Data di scadenza
Codice Classificazione unica nazionale dispositivi medici (CND)
Codice numerico che contraddistingue il dispositivo nella banca dati del Ministero della Salute (1)
Dispositivo su misura Se Sì, specificare il campo di applicazione, la tipologia e l’origine del materiale (vedi tabella costituente l’allegato 7) (2)
Sistemi o kit
Prodotto sterile
Non sterile
Dispositivo monouso pluriuso
Dispositivo in commercio in sperimentazione clinica
In caso di dispositivo in sperimentazione clinica, indicare il n°. di codice della sperimentazione
(1) Questo campo dovrà essere compilato a partire dalla data che sarà stabilita con apposito decreto ministeriale in relazione allo stato di avanzamento delle procedure di informatizzazione dei dati sui dispositivi medici.
(2) Benché il decreto legislativo 46/97 non preveda espressamente la segnalazione di incidenti e mancati incidenti concernenti dispositivi medici su misura, la loro comunicazione all’Autorità Competente da parte dell’operatore sanitario è richiesta sul piano della deontologia professionale.
C) DATI RELATIVI ALL’EVENTO L'episodio ha coinvolto: il paziente l'operatore
Se sì, età ……… ………
Se sì, iniziali ……… ……… (nome-cognome)
Nel caso di dispositivo impiantato
Data dell’impianto (se conosciuta) ………………… …….

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 27 d i 34
27
Dati sull’utilizzo del dispositivo
Il dispositivo è stato utilizzato Sì No
Motivo per il quale è stato utilizzato (o si intendeva utilizzare) il dispositivo; per i dispositivi impiantabili, indicare anche la specifica diagnosi: _______________________________________________________________
_______________________________________________________________
_______________________________________________________________
_______________________________________________________________
Nel caso di effettivo utilizzo del dispositivo: procedura diagnostica, clinica, chirurgica, contatto con il paziente, tempo di permanenza, durata della procedura, etc.: _______________________________________________________________
_______________________________________________________________
_______________________________________________________________
_______________________________________________________________
_______________________________________________________________
Descrizione dell'incidente o del mancato incidente
_______________________________________________________________
_______________________________________________________________
_______________________________________________________________
_______________________________________________________________
_______________________________________________________________
_______________________________________________________________
Conseguenza dell'incidente (vedi definizioni pag. 5)
- decesso
- intervento chirurgico
- intervento medico specifico
- ospedalizzazione o prolungamento ospedalizzazione
- altro*
*(specif icare, ad es. prolungamento dello stato di malattia dopo dimissione ospedaliera, menomazione di una funzione corporea, ecc.) ……………………………………………………………………………………………………………………………………
………………………………………………………………………………………
Numero di pezzi coinvolti ……………………
Il dispositivo (“specifico pezzo”) coinvolto nell’incidente o mancato incidente è disponibile: Sì
No
Se sì, dove: ………………………………………………………...

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 28 d i 34
28
Azioni intraprese dall’operatore o dalla struttura in cui opera per la gestione del dispositivo medico oggetto di segnalazione e del lotto di provenienza
Informativa al fabbricante/distributore
Informazione alla Direzione sanitaria/Direzione generale
Comunicazione al responsabile della vigilanza
Altro…………………………………………………………………………………………………………
………………………………………………………………………………………………………………
………………………………………………………
Altre eventuali informazioni che il segnalatore intende fornire al Ministero della Salute
_______________________________________________________________
_______________________________________________________________
_______________________________________________________________
_______________________________________________________________
_______________________________________________________________
_______________________________________________________________
Data di compilazione del presente rapporto: _____________________
D) Dati del compilatore
Legale rappresentante della struttura
Operatore sanitario
Responsabile della vigilanza
Nome e cognome:________________________________________________
*Qualifica:_______________________________________________________
*Struttura sanitaria di appartenenza __________________________________
*Telefono_______________________________________________________
*Fax___________________________________________________________
*E-mail_________________________________________________________
Firma:_________________________________________________________ *= l’indicazione può essere omessa se è stata barrata la casella operatore sanitario ed il compilatore corrisponde alla f igura del campo 3 della parte A
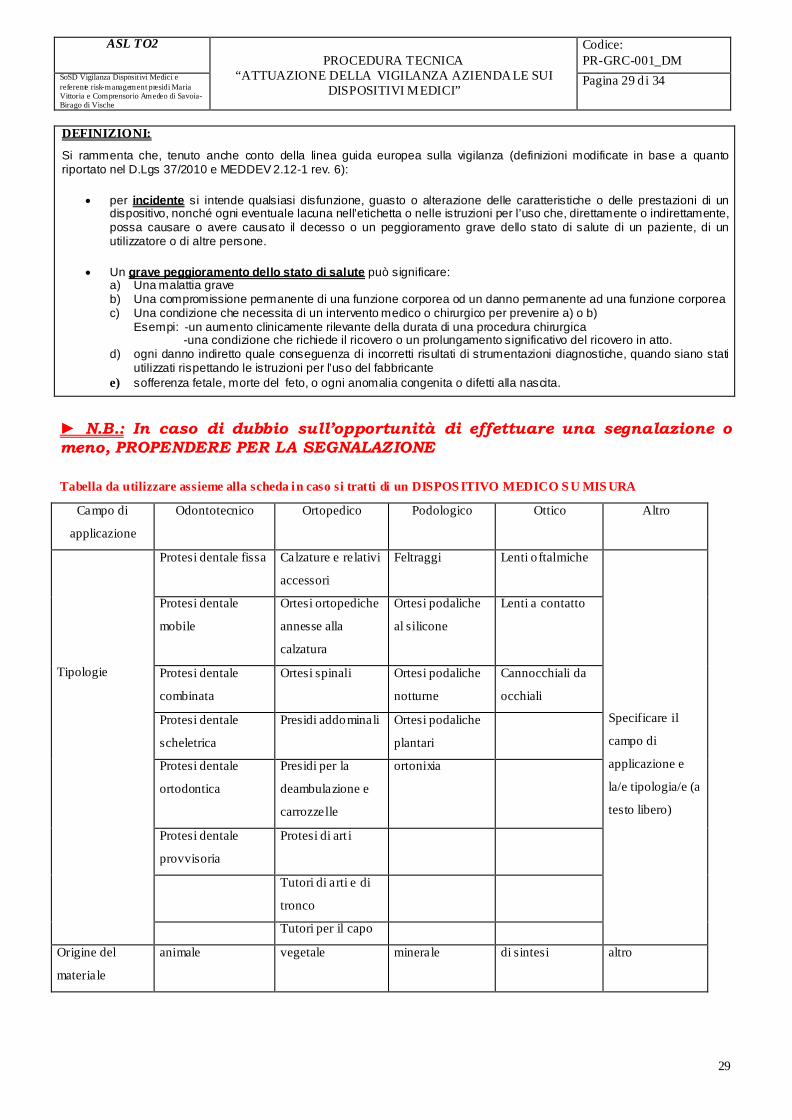
ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 29 d i 34
29
DEFINIZIONI:
Si rammenta che, tenuto anche conto della linea guida europea sulla vigilanza (definizioni modificate in base a quanto riportato nel D.Lgs 37/2010 e MEDDEV 2.12-1 rev. 6):
• per incidente si intende qualsiasi disfunzione, guasto o alterazione delle caratteristiche o delle prestazioni di un dispositivo, nonché ogni eventuale lacuna nell’etichetta o nelle istruzioni per l’uso che, direttamente o indirettamente, possa causare o avere causato il decesso o un peggioramento grave dello stato di salute di un paziente, di un utilizzatore o di altre persone.
• Un grave peggioramento dello stato di salute può significare: a) Una malattia grave b) Una compromissione permanente di una funzione corporea od un danno permanente ad una funzione corporea c) Una condizione che necessita di un intervento medico o chirurgico per prevenire a) o b)
Esempi: -un aumento clinicamente rilevante della durata di una procedura chirurgica -una condizione che richiede il ricovero o un prolungamento significativo del ricovero in atto.
d) ogni danno indiretto quale conseguenza di incorretti risultati di strumentazioni diagnostiche, quando siano stati utilizzati rispettando le istruzioni per l’uso del fabbricante
e) sofferenza fetale, morte del feto, o ogni anomalia congenita o difetti alla nascita.
► N.B.: In caso di dubbio sull’opportunità di effettuare una segnalazione o meno, PROPENDERE PER LA SEGNALAZIONE
Tabella da utilizzare assieme alla scheda in caso si tratti di un DISPOS ITIVO MEDICO S U MIS URA
Campo di
applicazione
Odontotecnico Ortopedico Podologico Ottico Altro
Tipologie
Protesi dentale fissa Calzature e relativi
accessori
Feltraggi Lenti o ftalmiche
Specificare il
campo di
applicazione e
la/e tipologia/e (a
testo libero)
Protesi dentale
mobile
Ortesi ortopediche
annesse alla
calzatura
Ortesi podaliche
al silicone
Lenti a contatto
Protesi dentale
combinata
Ortesi spinali Ortesi podaliche
notturne
Cannocchiali da
occhiali
Protesi dentale
scheletrica
Presidi addominali Ortesi podaliche
plantari
Protesi dentale
ortodontica
Presidi per la
deambulazione e
carrozzelle
ortonixia
Protesi dentale
provvisoria
Protesi di art i
Tutori di arti e di
tronco
Tutori per il capo
Origine del
materiale
animale vegetale minerale di sintesi altro

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 30 d i 34
30
Allegato 7
Modulo Ministeriale Da Utilizzare Per Incidente Con
(on line all’indirizzo: http://www.ministerosalute.it/imgs/C_17_pagineAree_29_listaFile_itemName_3_file.doc)
DISPOSITIVI MEDICO-DIAGNOSTICI IN VITRO
Allegato n. 4 al DM 15 novembre 2005 (definizioni modificate in base a quanto riportato nel D.Lgs 37/2010 e MEDDEV
2.12-1 rev. 6)
Rapporto di incidente o di mancato incidente da parte di operatori sanitari al Ministero della Salute
(art. 11, D.Lgs. n. 332 del 2000)
Rapporto interno n. ……….
Rapporto relativo a:
Incidente Mancato incidente
A) Dati relativi al luogo dove si è verificato l’episodio
1. Denominazione della struttura
(utilizzare la denominazione
ufficiale della struttura )
2. Reparto
3. Dati dell’operatore sanitario che ha rilevato l’episodio (nome, cognome, qualifica)
Telefono
Fax
4. Data dell’episodio
5. Azienda Ospedaliera o Azienda Sanitaria Locale competente per territorio (utilizzare la denominazione ufficiale della struttura). La ASL va indicata in caso di segnalazione di operatore sanitario operante in struttura sanitaria privata o pubblica non aziendale, di medico di medicina generale o pediatra di libera scelta, di farmacista
6. Dati del responsabile della vigilanza (nome, cognome, servizio di appartenenza)
Dr.ssa Anna BRUNETTI – Direzione Sanitaria Ospedale Maria Vittoria – ASL TO2

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 31 d i 34
31
B) Dati relativi al dispositivo medico-diagnostico in vitro
Fabbricante (nome, ragione sociale e indirizzo)
Fornitore (nome, ragione sociale e indirizzo)
Nome commerciale ed eventuale modello del dispositivo
Descrizione del dispositivo medico-diagnostico in vitro
N. codice del dispositivo assegnato dal fabbricante
Numero di lotto o di serie
Data di scadenza
Codice Classificazione nazionale (1)
Codice numerico che contraddistingue il dispositivo nella banca dati del Ministero della Salute (2)
Identificazione del tipo del dispositivo: - Allegato II elenco A - Allegato II elenco B - Test autodiagnostico - Altro tipo di dispositivo
Sistemi o kit
Dispositivo in commercio
Dispositivo destinato alla valutazione delle prestazioni
Prodotto sterile Altro stato microbiologico
Versione del software (Se presente)
(1) Questo campo dovrà essere compilato quando sarà disponibile la classif icazione nazionale per IVD. (2) Questo campo dovrà essere compilato a partire dalla data che sarà stabilita con apposito decreto ministeriale in relazione allo stato di avanzamento delle procedure di informatizzazione dei dati sui dispositivi medico-diagnostici in vitro.
C) DATI RELATIVI ALL’EVENTO
L'episodio ha coinvolto: il paziente l'operatore
Se sì, età ……… ………
Se sì, iniziali ……… ……… (nome-cognome)
Dati sull’utilizzo del dispositivo medico-diagnostico in vitro
Il dispositivo è stato utilizzato Sì No
Motivo per il quale è stato utilizzato ( o si intendeva utilizzare) il dispositivo:

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 32 d i 34
32
Descrizione dell'incidente o del mancato incidente
_________________________________________________________________________________________
_________________________________________________________________________________________
_________________________________________________________________________________________
_________________________________________________________________________________________
_________________________________________________________________________________________
_________________________________________________________________________________________
Conseguenza dell'incidente (vedi definizione pag. 5):
- decesso
- intervento chirurgico
- intervento medico specifico
- ospedalizzazione o prolungamento ospedalizzazione
- altro* ………………………………………………………...
*(specificare, ad es. prolungamento dello stato di malattia dopo dimissione ospedaliera, menomazione di una
funzione corporea, ecc.)
……………………………………………………………………………………………………………………………………
………………………………………………………………………………
Numero di pezzi coinvolti:………….
Il dispositivo coinvolto nell’incidente o mancato incidente è disponibile: Sì
No
Se sì, dove:……………………………………………...
Azioni intraprese dall’ operatore o dalla struttura in cui opera per la gestione del dispositivo medico-diagnostico in vitro oggetto di segnalazione e del lotto di provenienza
Informativa al fabbricante/distributore
Informazione alla Direzione sanitaria /Direzione generale
Comunicazione al responsabile della vigilanza
Altro…………………………………………………………………………………………………………………….……….
………………………………………………………………………………………………………………………………….

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 33 d i 34
33
...................................................................................................................................................................................
Altre eventuali informazioni che il segnalatore intende fornire al Ministero della Salute
__________________________________________________________________________________________
__________________________________________________________________________________________
__________________________________________________________________________________________
__________________________________________________________________________________________
__________________________________________________________________________________________
Data di compilazione del presente rapporto: _____________________
D) Dati del compilatore
Legale rappresentante della struttura
Operatore sanitario
Responsabile della vigilanza
Nome e cognome: _________________________________________________________________
*Qualifica: _________________________________________________________________________________
*Struttura sanitaria di appartenenza _____________________________________________________________
*Telefono__________________________________________________________________________________
*Fax______________________________________________________________________________________
*E-mail____________________________________________________________________________________
Firma: ___________________________________________________________________________________ *= l’indicazione può essere omessa se è stata barrata la caselle operatore sanitario ed il compilatore corrisponde alla f igura del campo 3 della parte A

ASL TO2 PROCEDURA TECNICA
“ATTUAZIONE DELLA VIGILANZA AZIENDA LE SUI DISPOSITIVI MEDICI”
Codice: PR-GRC-001_DM
SoSD Vigilanza Disposit ivi Medici e referente risk-management presidi Maria Vittoria e Comprensorio Amedeo di Savoia-Birago di Vische
Pagina 34 d i 34
34
DEFINIZIO NI: Si rammenta che, tenuto anche conto della linea guida europea sulla vigilanza (definizioni modif icate in base a quanto riportato nel D.Lgs 37/2010 e MEDDEV 2.12-1 rev. 6):
• per incidente si intende qualsiasi disfunzione, guasto o alterazione delle caratteristiche o delle prestazioni di un dispositivo, nonché ogni eventuale lacuna nell’etichetta o nelle istruzioni per l’uso che, direttamente o indirettamente, possa causare o avere causato il decesso o un peggioramento grave dello stato di salute di un paziente, di un utilizzatore o di altre persone.
• Un grave peggioramento dello stato di salute può signif icare: a) Una malattia grave b) Una compromissione permanente di una funzione corporea od un danno permanente ad una funzione corporea c) Una condizione che necessita di un intervento medico o chirurgico per prevenire a) o b)
Esempi: -un aumento clinicamente rilevante della durata di una procedura chirurgica -una condizione che richiede il ricovero o un prolungamento signif icativo del ricovero in atto.
d) ogni danno indiretto quale conseguenza di incorretti risultati di strumentazioni diagnostiche o di test IVD, quando siano stati utilizzati rispettando le istruzioni per l’uso del fabbricante
e) sofferenza fetale, morte del feto, o ogni anomalia congenita o difetti alla nascita.
► N.B.: può essere difficile determinare se un grave peggioramento dello stato di salute a un paziente possa essere imputabile ai risultati errati ottenuti con IVD o se il danno possa essere invece imputabile ad un errore dell’utilizzatore o a terzi. In queste circostanze è auspicabile inviare comunque la segnalazione. Devono inol tre essere anche segnalate le carenze nelle informazioni fornite dal fabbricante che hanno causato o avrebbero potuto causare danno all’utilizzatore.