article 1 receptor autoantibody increases in serum of
TRANSCRIPT

Cellular & Molecular Immunology 209
Article
Volume 5 Number 3 June 2008
Agonistic AT1 Receptor Autoantibody Increases in Serum of Patients with Refractory Hypertension and Improves Ca2+ Mobilization in Cultured Rat Vascular Smooth Muscle Cells Feng Zhu1, 2, Yanxiang Sun1, 2, Yuhua Liao1, 3, Yumiao Wei1, Ming Chen1, Min Wang1 and Zihua Zhou1 Agonistic AT1 receptor autoantibodies (AT1-AAs) have been described in the patients with malignant hypertension or preeclampia. Furthermore, AT1-AAs were highly associated with refractory hypertension. Function of vascular smooth muscle cells (VSMCs) is important in the regulation of blood pressure. We investigated and compared the ability of angiotensin II (Ang II) and AT1-AAs to stimulate the intracellular calcium mobilization and cellular proliferation of rat VSMCs. Twenty-two patients with refractory hypertension, 24 patients with non-refractory hypertension and 37 normotensives were recruited. The serum of each patient was detected for the presence of AT1-AAs by ELISA. Ang II and the AT1-AAs from the sera of patients were used to stimulate rat VSMCs in vitro. AT1-AAs were detected in 10/22, 3/24 and 3/37 of patients with refractory hypertension, non-refractory hypertension and normotensives, respectively. AT1-AAs led the increase intracellular calcium mobilization in a dose-dependent manner and cellular proliferation of VSMCs just as Ang II. Both of these effects caused by AT1-AAs were blocked with losartan or a peptide corresponding to a part of the second extracellular loop of AT1 receptor. Since AT1-AAs exhibited pharmacological activity in rat VSMCs just as Ang II, they might play a role in the elevation of peripheral vascular resistance and in vascular remodeling. And AT1-AAs were suggested to involve in resistance to antihypertensive therapy. Cellular & Molecular Immunology. 2008;5(3):209-217. Key Words: autoimmunity; antibody; hypertension; angiotensin II receptor type 1 Introduction Only 2.9% hypertensive patients in China have blood
pressure (BP) levels that are well controlled (1). Hyper- tension in some of these patients persists despite multiple antihypertensive medications and excellent compliance. Refractory hypertension is conventionally defined as systolic or diastolic BP that remains uncontrolled despite sustained therapy with at least three different classes of anti- hypertensive agents. True refractory hypertension is estimated
to affect less than 5% of the general population with hypertension (2). However, these patients are at greater risk for stroke, renal insufficiency and morbid cardiovascular events than BP well-controlled patients (3). Many factors including obesity, excessive dietary salt ingestion, alcohol consumption and sleep apnea are found contributing to resistance to antihypertensive therapy (4, 5).
Agonistic AT1 receptor autoantibodies (AT1-AAs) were first described by Wallukat et al. (6). The patients with preeclampsia developed the autoantibodies capable of activating the angiotensin II receptor type 1 (AT1 receptor). AT1-AAs were also found in other severe hypertensive conditions including malignant hypertension and secondary hypertension (7). However, their potential contribution to refractory hypertension has received little attention. In our previous studies, we found that AT1-AAs were highly associated with refractory hypertension (8). And they were also found to be associated with stroke in hypertensive patients (9).
In the previous studies, AT1-AAs were suggested to play an important role in the pathogenesis of preeclampsia. AT1-AAs induced signaling in vascular cells and trophoblasts including activator protein-1 (AP-1) and nuclear factor- kappa B (NF-κB) (10, 11), and activated calcineurin-NFAT (nuclear factor of activated T cells) through increased
1Laboratory of Cardiovascular Immunology, Institute of Cardiology, Union Hospital, Tongji Medical College, Huazhong Science & Technology University, Wuhan 430022, China; 2These authors contributed equally to this work; 3Corresponding to: Dr. Yuhua Liao, Laboratory of Cardiovascular Immunology, Institute of Cardiology, Union Hospital, Tongji Medical College of Huazhong University of Science and Technology, 1277 Jiefang Road, Wuhan 430022, China. Fax: +86-27-8572-7140, E-mail: liaoyh27@ hotmail.com
Received Mar 13, 2008. Accepted May 19, 2008. Copyright © 2008 by The Chinese Society of Immunology

210 Agonistic AT1 Receptor Autoantibodies in Hypertension
Volume 5 Number 3 June 2008
intracellular Ca2+ mobilization in Chinese hamster ovary cells transferred with the rat AT1A receptor (12). However, there were limited data related to the role of AT1-AAs in pathogenesis of hypertension. Function of vascular smooth muscle cells (VSMCs) is important in the regulation of BP. Because intracellular Ca2+ ([Ca2+]i) is a major determinant of VSMCs tone and function (13), we investigated and compared the ability of angiotensin II (Ang II) and AT1-AAs to stimulate the intracellular Ca2+ mobilization and cellular proliferation of rat VSMCs. Materials and Methods Patients This study was approved by the Ethics Committee of Union Hospital. The patients with essential hypertension participated in the study after written permission. Exclusion criteria included renal disease, liver disease, chronic inflammatory disease, sleep apnea, malignant hypertension and secondary hypertension. And patients with auto- antibodies against α1-adrenergic receptor in circulation were also excluded. Prior to drug therapy, all the patients were advised to reduce dietary sodium intake and alcohol consumption. Hypertension was treated systemically in an open trial with a stepwise dosing regimen. All patients were treated with the long-acting calcium antagonist felodipine of 5 mg once a day for 2 weeks. Additional drugs and dose increments were prescribed in two further steps. Hydro- chlorothiazide 12.5 mg/d and metoprolol 50 mg/d or enalapril 10 mg/d were added if BP was greater than 140/90 mmHg after two weeks of felodipine administration. Then, after two additional weeks, if BP was not below 140/90 mmHg, the doses of felodipine and metoprolol or enalapril were doubled for an additional 4 weeks. Hypertension was considered refractory if BP cannot be reduced to target level after combination three drugs therapy. According to the decrease of BP, the patients were assigned to two groups, a refractory hypertension (RH) group and a non-refractory (NRH) group. Twenty-two patients with refractory hyper- tension were recruited randomly. Twenty-four gender-, age- and weight-matched patients were selected as the NRH group. And the control group included 37 normotensives. The clinical data of the subjects are presented in Table 1. Blood samples for the tests were obtained from each participant. Peptide synthesis Two different peptides were synthesized by the type PSSM-8 Peptide Synthesizer (Shimadzu Company, Japan). The peptides had the following amino acid sequences: P1, IHRNVFFIENTNITVCAFHYESQNSTL; and P2, AFHYESQ. P1 corresponding to the second extracelluar loop of the human AT1 receptor was used in the ELISA assay and preparation of the immunoglobulin (Ig) fractions. P2 corre- sponding to portion of the second extracelluar loop of the human AT1 receptor was an epitope recognized by AT1-AAs from preeclamptic individuals (6), and was used in antibody neutralization experiment.
Preparation of Ig fraction The serum of each patient was checked for the presence of AT1-AAs by ELISA, which was performed as described previously (8), the titer was shown by the value of P/N when the sera were diluted in 1:40 [(P/N = (the A value of test − the A value of blank) / (the A value of negative control − the A value of blank)]. Preparation of the Ig fractions was outlined in detail earlier (6). Briefly, the Ig fractions in the sera were isolated by ammonium sulfate precipitation at a saturation of 40%. The Ig fractions from positive AT1-AAs sera were loaded on a sepharose 4B CNBr-activated gel (Pharmacia, USA) to which the P2 peptide was covalently linked. The antibodies were eluted with 3 mol/L potassium thiocyanate (pH = 7.4) followed by immediate extensive dialysis against phosphate buffered saline; the purity was detected by SDS-PAGE. Cell culture Rat AT1A receptors, an important subset of AT1 receptors which are abundantly expressed in VSMCs, show a high degree of sequence identity to the human AT1 receptor (16, 17). We used VSMCs from Sprague Dawley rats (200-250 g) in this study. VSMCs were isolated after a well established procedure (18). Briefly, the vessels of the rat thoracic aortae were cleared of fat and incubated for 10 min at 37°C in Hanks’ balanced salt solution containing 0.2 mg/ml elastase type III (Sigma, USA), 2 mg/ml collagenase type II (Gibco, USA) and 0.1 mg/ml soybean trypsin inhibitor (Sigma, USA). This procedure ensured removal of the endothelial cells. The
cleaned vessels were cut into 1- to 2-mm pieces and digested
in the fresh Hanks' balanced salt solution with enzyme. The cell suspension was centrifuged at 200 × g for 5 minutes, and
the pellet was resuspended in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA) containing 10% heat- inactivated fetal calf serum (Gibco, USA), 2 mmol/L L- glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. For measurement of [Ca2+]i, the dispersed cells were aliquoted into 35 mm glass dishes (BD, USA) at 37°C in a humidified 5% CO2. And cells were studied between day 5 and day 7 of the primary culture. Cells in subculture passages
Table 1. Clinical data of patients with hypertension RHT (n = 22) NRH (n = 24) Gender (male/female) 10/12 12/12 Age (years) 54.1 ± 9.6 53.7 ± 9.2 BMI (kg/m2) 25.5 ± 1.47 25.2 ± 1.42 Before treatment SBP (mmHg) 180.0 ± 15.3* 168.9 ± 15.0 DBP (mmHg) 104.6 ± 11.0 99.1 ± 11.4 Heart rate (bpm) 85.7 ± 9.7 83.0 ± 9.9 After treatment SBP (mmHg) 153.5 ± 8.6** 130.4 ± 7.1 DBP (mmHg) 92.1 ± 8.6** 80.3 ± 6.2 Heart rate (bpm) 77.1 ± 7.2 78.0 ± 5.0
*p < 0.05, **p < 0.01, compared with the NRH group.

Cellular & Molecular Immunology 211
Volume 5 Number 3 June 2008
5-10 were used for the measurement of cellular proliferation. Cell culture purity was routinely tested with smooth muscle α-actin staining. Immunofluorescence Immunofluorescence was done as described previously (6). AT1-AAs at 1:40 dilution were used as first antibodies. FITC conjugated anti-human IgG (Zhongshan Biological Technology Ltd, China) was used as secondary antibody. And the Ig fractions from AT1-AAs negative sera were used as the negative control (control IgG). The preparations were observed in a fluorescent microscope (Nikon, Japan). Measurement of Intracellular Ca2+ fluorescence intensity [Ca2+]i was measured by using the fluorescent dye Fluo-3/AM (19, 20). Twenty-four hours before the experiment, VSMCs were incubated in serum-free medium. On the day of the experiment, cells were washed twice with fluorescence buffer (140 mmol/L NaCl, 1 mmol/L MgCl2, 1.5 mmol/L CaCl2, 5 mmol/L KCl, 10 mmol/L HEPES, 5 mmol/L glucose, pH 7.4). The cells were loaded with 10 μmol/L Fluo-3/AM and 0.02% pluronic F-127 (Sigma, USA) in darkness for 40 minutes at 37°C in fluorescence buffer. Excessive dye was removed by rinsing twice with fluorescence buffer. Experiments were performed at room temperature under laser scanning confocal microscope (FV-500, Olympus, Japan). Each experiment was repeated at least 3 times, and at least 20 cells were tested. Changes of fluorescence intensity (FI) of individual cells were detected in FLUO3 mode of the microscope, scanning one frame every second and scanning 320 frames every cell. And peak FI value of each cell was recorded. Cellular proliferation assessment VSMC proliferation was determined by a BrdU cell proliferation assay kit (Exalpha Biologicals, Inc, USA). Cells (2.5 × 103/well) were plated into 96-well plates. Prior to
experiments, cells were growth-arrested in serum-deprived DMEM for 24 h (time 0). Cells were then stimulated with test reagents or antibodies. Each reagent or antibody was tested in 5 wells. Remain procedures were performed as described in the kit protocol. Statistical Analysis Data were expressed as mean ± standard error of the mean (SEM). Student’s t test was used for comparison of variables between groups. Significance of incidence was checked by the χ2 test. Values of p < 0.05 were considered to denote statistical significance. Results Incidence of the AT1-AAs Ten patients (10/22) in the RH group and 3 patients (3/24) in the NRH group had detectable AT1-AAs (Figure 1). The difference between the prevalence of two groups was statistically significant (χ2 = 4.630, ν = 1, p < 0.05). There were 3 normotensives (3/37) also had positive AT1-AAs, and all of them were completely healthy. There were no significant difference in the prevalence between the NRH group and normotensives. Sera of 5 autoantibody-positive hypertensive patients (3 from the RH group and 2 from the NRH group) were investigated 6 months later. All of them remained positive, even if BP was well controlled. Recognition of the AT1 receptor by AT1-AAs We showed the fluorescent photomicrographs of VSMCs exposed to AT1-AAs and antibodies from negative serum (Figure 2). FITC-labeled anti-human antibody (secondary antibody) produced the green staining in VSMCs, which were incubated with AT1-AAs at 1:40 dilution previously (Figure 2A). But the same cells exposed to the antibodies from AT1-AAs negative serum produced the negative staining (Figure 2B). The results indicated AT1-AAs could bind the antigen of VSMCs, whereas the IgG from negative serum could not.
RH NRH Normotensive Figure 1. Prevalence of autoantibody against the AT1 receptor in the patients with primary hypertension control subjects. Top part of the bars represented number of autoantibody-positive individuals; bottom part, number of autoantibody-negative individuals. *p < 0.05.
A B
Figure 2. Immunofluorescence images of VSMCs exposed to AT1-AAs. (A) VSMCs were stained with anti-AT1-AAs, whichpurified from the sera of refractory patients, and FITC-anti-human IgG, then were observed by fluorescent microscope. (B) The Igfractions from AT1-AAs negative sera were used as the negative control.

212 Agonistic AT1 Receptor Autoantibodies in Hypertension
Volume 5 Number 3 June 2008
Intracellular [Ca2+]i stimulated by Ang II via AT1 receptor The changes of Ca2+ FI of individual cell were traced, and peak FI value of each cell was recorded. Ang II at 10-6 mol/L stimulated an acute increase [Ca2+]i (Figure 3A). Losartan
D
C
B
A
Figure 3. Increasing intracellular free Ca2+ is mediated by Ang II in VSMC. (A) Fluo-3/AM-loaded VSMC were stimulated by Ang II (10-6 mol/L) and change in intracelluar free Ca2+ was monitored by change of fluorescence intensity. (B) Losartan (10-6
mol/L) had no effect in stimulation of intracellular Ca2+. (C) Increasing Ca2+ response triggered by Ang II was inhibited by 10-6
mol/L losartan. (D) Fluo-3/AM-loaded VSMCs were treated with different concentrations of Ang II (10-9, 10-8, 10-7 and 10-6 mol/L).Ang II stimulated an increase in [Ca2+]i in a dose-dependent manner. Arrows indicate time when activator or inhibitor was added.
E
D
C
B
A
n = 21 n = 21 n = 21 n = 22 Figure 4. AT1-AAs stimulated Ca2+ transients in VSMCs. Fluo-3/AM-loaded VSMCs were treated with (A) 1:50 dilution AT1-AAspositive serum, (B) 1:50 dilution AT1-AAs negative serum, (C) 1:10 purified AT1-AAs and (D) 1:10 control IgG. (E) AT1-AAs positive serum stimulated a significant [Ca2+]i response in comparison withnegative serum. And purified AT1-AAs stimulated a significant [Ca2+]i response in comparison with control IgG. *p < 0.05.
Cellular & Molecular Immunology 213
Volume 5 Number 3 June 2008
(10-6 mol/L, Merck Co., Inc, USA), an AT1 receptor antagonist, had no effects on intracellular [Ca2+]i (Figure 3B). The increased intracellular [Ca2+]i stimulated by 10-7 mol/L Ang II could be completely inhibited by 10-6 mol/L losartan (Figure 3C). Then, Fluo-3/AM loaded VSMCs were stimulated by series doses of Ang II (10-9, 10-8, 10-7, and 10-6 mol/L). As shown in Figure 3D, Ang II stimulated an increase of [Ca2+]i in a dose-dependent manner. The data indicated that an increase in [Ca2+]i of VSMCs induced by Ang II was a result of the AT1 receptor activation. Dose-dependent stimulation of [Ca2+]i mediated by AT1-AAs The primary goal of our study was to examine the ability of AT1-AAs to stimulate an increase of [Ca2+]i through the AT1 receptor in VSMCs. Initially, we evaluated the serum from a refractory patient, who had positive AT1-AAs. AT1-AAs positive serum (1:50 dilution) stimulated a significant [Ca2+]i response in comparison with negative serum (Figures 4A and 4B). Then, AT1-AAs purified from the positive individuals were used to stimulate VSMCs. The [Ca2+]i response induced by AT1-AAs (1:10 dilution) was measured as a square- shaped increase FI (Figure 4C). In contrast, after addition of control IgG purified from negative serum, FI almost maintained the baseline level through all observation (Figure 4D). The average peak [Ca2+]i FI induced by positive serum was significantly higher than the value of the [Ca2+]i
response induced by negative serum [(2.60 ± 0.25) × 106 versus (1.47 ± 0.24) × 106, p < 0.05, Figure 4E]. Mean peak FI of the [Ca2+]i response induced by AT1-AAs was significantly higher than the value of the [Ca2+]i response induced by control IgG [(5.14 ± 0.47) × 106 versus (1.14 ± 0.14) × 106, p < 0.05, Figure 4E].
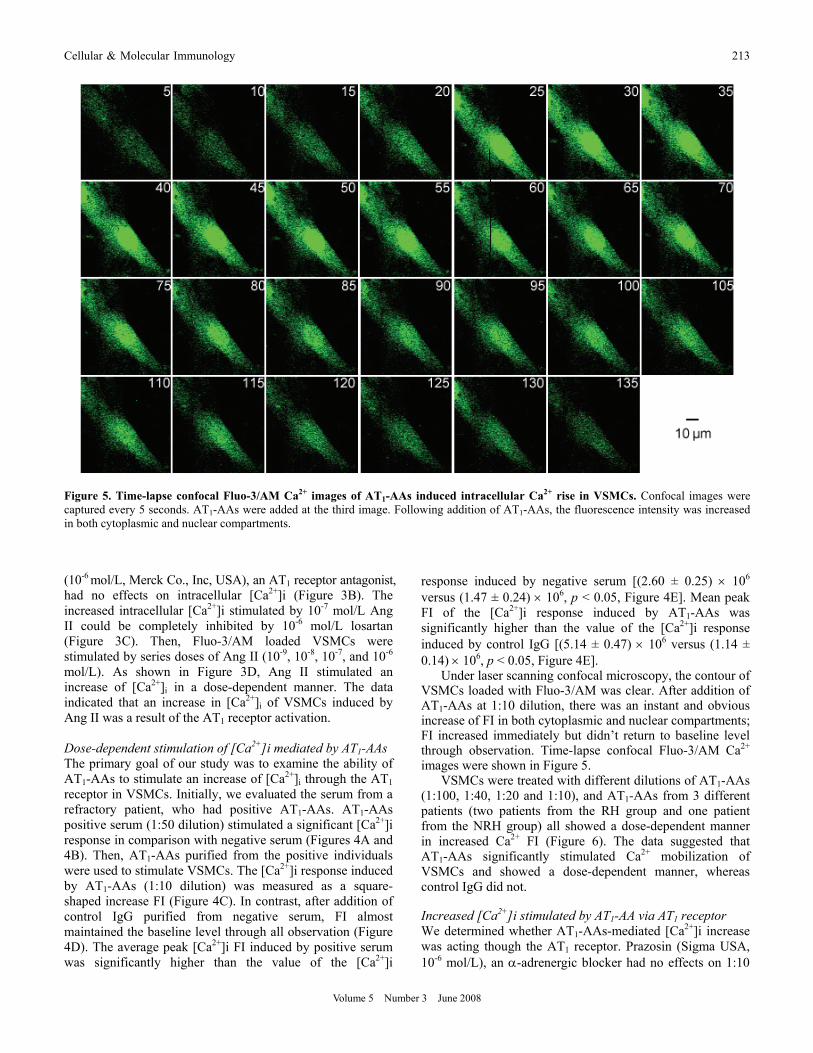
Under laser scanning confocal microscopy, the contour of VSMCs loaded with Fluo-3/AM was clear. After addition of AT1-AAs at 1:10 dilution, there was an instant and obvious increase of FI in both cytoplasmic and nuclear compartments; FI increased immediately but didn’t return to baseline level through observation. Time-lapse confocal Fluo-3/AM Ca2+ images were shown in Figure 5.
VSMCs were treated with different dilutions of AT1-AAs (1:100, 1:40, 1:20 and 1:10), and AT1-AAs from 3 different patients (two patients from the RH group and one patient from the NRH group) all showed a dose-dependent manner in increased Ca2+ FI (Figure 6). The data suggested that AT1-AAs significantly stimulated Ca2+ mobilization of VSMCs and showed a dose-dependent manner, whereas control IgG did not. Increased [Ca2+]i stimulated by AT1-AA via AT1 receptor We determined whether AT1-AAs-mediated [Ca2+]i increase was acting though the AT1 receptor. Prazosin (Sigma USA, 10-6 mol/L), an α-adrenergic blocker had no effects on 1:10
Figure 5. Time-lapse confocal Fluo-3/AM Ca2+ images of AT1-AAs induced intracellular Ca2+ rise in VSMCs. Confocal images were captured every 5 seconds. AT1-AAs were added at the third image. Following addition of AT1-AAs, the fluorescence intensity was increased in both cytoplasmic and nuclear compartments.

214 Agonistic AT1 Receptor Autoantibodies in Hypertension
Volume 5 Number 3 June 2008
dilution AT1-AAs-mediated [Ca2+]i increase (Figure 7A). In contrast, the functional response of [Ca2+]i of AT1-AAs at 1:10 dilution was inhibited by the AT1 receptor antagonist, losartan at 10-6 mol/L [(5.14 ± 0.47) × 106 versus (0.96 ± 0.13) × 106, p < 0.05, Figures 7B and 7C]. We also tested the specificity of AT1-AAs via the AT1 receptor with P2 peptide, which was corresponded to the portion of the second extracelluar loop of the AT1 receptor. When AT1-AAs were incubated with excessive amount of peptide P2 previously, mean peak Ca2+ FI decreased significantly in comparison with AT1-AAs at 1:10 dilution [(5.14 ± 0.47) × 106 versus (1.73 ± 0.25) × 106, p < 0.05, Figure 7C]. All the results above indicated that AT1-AAs stimulate increased intra- cellular Ca2+ mobilization by activation of the AT1 receptor.
E
D
C
B
A
Figure 6. AT1-AAs showed a dose-dependent manner in theincrease of [Ca2+]i in VSMCs. Fluo-3/AM-loaded VSMC were stimulated with different dilutions of AT1-AAs (A) 1:100, (B) 1:40, (C) 1:20 and (D) 1:10. (E) AT1-AAs stimulated an increase in [Ca2+]i in a dose-dependent manner. AT1-AAs from 3 differentpatients (two patients from the RH group and one patient from the NRH group) all showed a dose-dependent manner in [Ca2+]iincrease.
C
B
A
n = 21 n = 21 n = 22 n = 21 Figure 7. Increased [Ca2+]i stimulated by AT1-AAs was a result of activation of the AT1 receptor. (A) Previous incubation with prazosin (10-6 mol/L) did not block increased [Ca2+]i mediated by AT1-AAs. (B) Increasing Ca2+ response triggered by AT1-AAs wasinhibited by losartan (10-6 mol/L) completely. (C) Specificity of increased [Ca2+]i by AT1-AAs was determined by using the AT1receptor-specific antagonists and an AT1 receptor-specific epitope peptide P2.

Cellular & Molecular Immunology 215
Volume 5 Number 3 June 2008
VSMC proliferation induced by Ang II and AT1-AAs Cellular proliferation, determined by the Brdu chemilu- minescent assay, was measured in VSMCs after timed exposures to Ang II at 10-7 mol/L or AT1-AAs at 1:20. Optical density at 450 nm of ELISA assay represented level of cellular proliferation. The results were present in Figure 8. Compared with serum-deprived DMEM, Ang II stimulated cellular proliferation within 6 hours (0.162 ± 0.018 versus 0.094 ± 0.014, p < 0.05). On end of the test (24th hour), the cellar proliferation of VSMCs stimulated by Ang II was significant in comparison with only DMEM treated cells (0.310 ± 0.023 versus 0.119 ± 0.013, p < 0.05). When VSMCs were exposed to AT1-AAs, similar results were obtained. AT1-AAs also induced VSMC proliferation in comparison with control IgG within 6 hours (0.162 ± 0.018 versus 0.094 ± 0.014, p < 0.05). On end of the test, the cellular proliferation induced by AT1-AAs was significant in comparison with control IgG (1.244 ± 0.038 versus 1.033 ± 0.027, p < 0.05). Further test founded cellular proliferation induced by Ang II or AT1-AAs could be inhibited completely by the AT1 receptor antagonist, losartan (0.310 ± 0.023
versus 0.156 ± 0.031, p < 0.05; 1.244 ± 0.038 versus 1.015 ± 0.031, p < 0.05). And proliferation induced by AT1-AAs could also be blocked by peptide P2 (1.244 ± 0.038 versus 1.068 ± 0.036, p < 0.05). Discussion The important findings in this study were that AT1-AAs from hypertensives stimulated Ca2+ increasing in VSMCs and were related to therapy resistance in hypertension. We showed AT1-AAs were present in 10/22 patients with refractory hypertension. In comparison, in the subgroup of non- refractory hypertension, incidence of the antibodies was 3/24. In a larger hypertensive population we analyzed earlier, 42/98 patients with refractory hypertension got positive AT1-AAs, which were much higher than incidence in the NRH group and health control (8). The positive rate of AT1-AAs in refractory hypertension was significantly higher than the rate in well-controlled hypertension. All the clinical data indicated refractory hypertension was highly associated with the autoantibodies against the AT1 receptor.
Fu et al. first found AT1-AAs were present in the circulation of hypertensives and might be involved in the pathogenesis of malignant hypertension (7). The antibodies displayed a positive chronotropic effect on cultured neonatal rat cardiomyocytes and were proven to have an agonistic character as Ang II. The definitive role of AT1-AAs in the pathogenesis of primary hypertension is unclear, but Ang II is known to cause the contraction of VSMCs in blood vessels. And increased [Ca2+]i is the signal activating the contractile apparatus of VSMCs to cause a contraction (13). We studied the AT1 receptor agonistic response with the trace of intracellular free Ca2+ of VSMCs when exposed to AT1-AAs. The autoantibody exhibited cross-reactivity with rat AT1A receptor of VSMCs, and behaved as an agonist. AT1-AAs from all 3 different sera were capable of leading the increase intracellular Ca2+ mobilization and showed a dose-dependent manner. This effect was similar to that of Ang II. As an increase in [Ca2+]i stimulates VSMC contraction, all the experiments in vitro indicated AT1-AA was a vasoconstrictor to elevate the peripheral resistance. The specificity of the responses of VSMCs induced by AT1-AAs via the AT1 receptor was confirmed by using losartan as well as a peptide from the second extracellular loop of the AT1 receptor. In conclusion, AT1-AAs increase Ca2+ in VSMCs via activation of the AT1 receptor and may play a role in elevating peripheral vascular resistance.
Ang II is believed to play a pivotal role in the development of hypertension since it acts as a growth promoting factor in VSMCs and results in vascular remodeling (21, 22). According to present data, AT1-AAs were suggested to have similar effects. Dechend et al reported that AT1-AAs induced signaling in vascular cells including AP-1 and NF-κB activation (10, 11). Both of these nuclear factors participate in inflammation (23, 24). Wallukat et al. found monocytes, which were pivotal to vascular wall inflammatory processes, could be stimulated by AT1-AAs to adhere, produce tissue
B
A
Figure 8. Effects of AT1-AAs or Ang II on VSMC proliferation.(A) VSMCs were exposed to serum-free DMEM only, serum-free DMEM supplemented with Ang II (10-7 mol/L) and serum-free DMEM supplemented with losartan (10-6 mol/L) prior to Ang II. (B) VSMC were exposed to serum-free DMEM supplemented withcontrol IgG (1:20), serum-free DMEM supplemented with AT1-AAs (1:20), serum-free DMEM supplemented with losartan (10-6
mol/L) prior to AT1-AAs and serum-free DMEM supplemented with excessive peptide P2 prior to AT1-AAs. And the proliferation of VSMCs was determined by Brdu cell proliferation assay kit as decribed in material and methods

216 Agonistic AT1 Receptor Autoantibodies in Hypertension
Volume 5 Number 3 June 2008
factor, and probably reactive oxygen species (25). Both of their findings indicated that AT1-AAs involved in the inflammation of vascular walls, which is an important mechanism of vascular remodeling. In the animal model, the antibodies against the AT1 receptor produced by active immunization with the second extracellular loop of the human AT1 receptor were able to induce pathological structural changes of artery after one year (26). All the results in the laboratory suggest AT1-AAs may play a role in vascular remodeling. Whether or not AT1-AAs contribute to vascular remodeling in hypertension needs a long term follow-up with carotid intima-media thickening of the patients who have positive antibody.
Refractory hypertension is unusual in clinical practice, thanks to the widespread availability of antihypertensive drugs. However, its prevalence increases with increasing severity of BP and high prevalence of target organ damage (27). A maximum dose of drug or combinations of three or more drugs seems to be effective in this situation. But this therapy is not cost-effective and will bring many side effects of drug; both will lead to poor compliance of patients. So before treatments, there should be proper evaluation of such patients to determine the factor(s) responsible for resistance to therapy (28).
In present study, many factors including renal diseases, excessive sodium intake, alcohol, sleep apnea and obesity were all excluded before the therapy. AT1-AAs seemed to be an independent factor. Based on this recognition, AT1 receptor blockers (ARBs) are appropriate antihypertensive
drugs for the individuals with positive antibodies. Some refractory hypertensives with positive antibodies had already benefited from the use of ARBs in our earlier study (29). The antibody is also common in malignant hypertension. Fu and coworkers reported that AT1-AAs were present in 14% patients with malignant essential hypertension and 33% patients with malignant secondary hypertension (7). Very elevated BP is usually accompanied by acute and severe renal failure. Under the condition of highly increased serum creatinine and potassium, the use of ARBs is often reluctant. According to our practice, ARBs were beneficial and safe for the patients with the positive antibodies, when the serum creatinine and potassium levels were monitored. Since our practices were neither randomly assigned nor blinded and subjects were too small, we could not draw a firm conclusion that ARBs could benefit patients with positive antibody in refractory or malignant hypertension. As the effect of AT1-AAs can be neutralized by a peptide from the second extracellular loop of the AT1 receptor, immune adsorption (IA) is an alternative. IA had been used to remove auto- antibody against the α1-adrenoreceptor, which is another important autoantibody in refractory hypertension and had been proved effective (30). Because of its high cost, IA can not be used widely.
Even if the presence of AT1-AAs is a common event in patients with refractory and malignant hypertension, the occurrence of autoantibody can not only be explained as a consequence of vascular damages of hypertension and the release of immunogenic parts of the receptor. Because we
also observed 3 of 37 normotensives had positive response in ELISA and some BP well-controlled patients always maintained positive antibodies in circulation. And there was little evidence to support molecular mimicry theory to explain the AT1 receptor autoantibody phenomenon, which could well explain the antibodies directed against other G-protein receptors in Chagas’ disease or in myasthenia gravis (31, 32). There were several speculations on the origin of AT1-AAs in previous works. In preeclampia, diminished placental perfusion could alter the AT1 receptor expression in an aberrant fashion, thereby permitting autoantibody production (6). And in allograft renal-rejection subjects, post- transplantation reperfusion injury may alter the intragraft
expression of AT1 receptor, change its density, or cause conformational changes; a permissive environmental pheno- menon might enhance local intragraft immunoreactivity owing to an activated innate immune response (33). It seems that both of the speculations could not explain the AT1-AAs phenomenon in hypertension. We hypothesize that the interactions between underlying multiple genotypes and the environment factors (hypertension, pregnancy, post- transplantation reperfusion and others) may lead to an altered immune response to self antigens including the AT1 receptors (34). To prove the hypothesis, we intend to set up a blood- sample library of the individuals with positive antibodies to do further research.
In summary, we found 10/22 patients with refractory hypertension and 3/24 patients with non-refractory hypertension had autoantibodies directed against the AT1 receptor. We showed that the antibodies could lead an increase intracellular Ca2+ mobilization and induce proliferation of VSMCs through the AT1 receptor. The results indicated the autoantibody might play a role in the elevation of peripheral vascular resistance and in the development of vascular remodeling in patients with primary hypertension, especially in the patients with refractory hypertension. The detection of such autoantibody may assist in the selection of an appropriate antihypertensive drug for an individual. Acknowledgements This work was funded by National Natural Science Foundation of China (Project No. 30300133 and Project No. 30600235). References
1. Liu LS, Gong LS. Prevention and treatment of high blood pressure in China part I. Chin J Hyper. 2000;12:94-101.
2. Alper AB J, Calhoun DA. Contemporary management of refractory hypertension. Curr Hypertensn Rep. 1999;1:402-407.
3. Isaksson H, Ostergren J. Prognosis in therapy-resistant hypertension. J Int Med. 1994;236:643-649.
4. Goodfriend TL, Calhoun DA. Resistant hypertension, obesity, sleep apnea, and aldosterone: theory and therapy. Hypertension. 2004;43:518-524.
5. The Seventh Report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. JAMA. 2003;289:2560-2572.

Cellular & Molecular Immunology 217
Volume 5 Number 3 June 2008
6. Wallukat G, Homuth V, Fischer T, et al. Patients with preeclampsia develope agonistic antibodies against the angiotension AT1 receptor. J Clin Invest. 1999;103:945-952.
7. Fu ML, Herlitz H, Schulze W, et al. Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J Hypertens. 2000;18:945-953.
8. Liao YH, Wei YM, Wang M, et al. Autoantibodies against AT1-receptor and α1-adrenergic receptor in patients with hypertension. Hypertens Res. 2002;25:641-646.
9. Zhou ZH, Liao YH, Wang M, et al. Role of autoantibodies against angiotensin II type 1 receptor in hypertensive patients with cerebral sroke. Chin J Clin Rehabil. 2004;8:6944-6949.
10. Dechend R, Homuth V, Wallukat G, et al. AT1 receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation. 2000;101:2382-2387.
11. Dechend R, Viedt C, Müller DN, et al. AT1 Receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation. 2003;107:1632-1639.
12. Thway TM, Shlykov SG, Day MC, et al. Antibodies from preeclamptic patients stimulate increased intracellular Ca2+ mobilization through angiotensin receptor activation. Circulation. 2004;110:1612-1619.
13. Horowitz A, Menice CB, R Laporte, KG Morgan. Mechanisms of smooth muscle contraction. Physiol Rev. 1996;76:967-1003.
14. Schwartz, SM, Ross R. Cellular proliferation in atherosclerosis and hypertension. Prog Cardiovasc Dis. 1984;26:355-372.
15. Berk BC, Vekshtein V, Gordon HM, Tsuda T. Angiotensin II- stimulated protein synthesis in cultured vascular smooth muscle cells. Hypertension. 1989;13:305-314.
16. Mauzy CA, Hwang O, Egloff M, Wu LH, Chung FZ. Cloning, expression, and characterization of a gene encoding the human angiotensin II type 1A receptor. Biochem Biophys Res Commun. 1992;186:277-284.
17. Furuta H, Guo DF, Inagami T. Molecular cloning and sequencing of the gene encoding human angiotensin II type 1 receptor. Biochem Biophys Res Commun. 1992;183:8-13.
18. Gopalakrishnan V, Xu Y, Sulakhe PV, et al. Vasopressin (V1) receptor characteristics in rat aortic smooth muscle cells. Am J Physiol. 1991;261:H1927-H1936.
19. Minta A, Kao JP, Tsien RY. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J Biol Chem. 1989;264:8171-8178.
20. Cobbold PH, Rink TJ. Fluorescence and bioluminescence measurement of cytoplasmic free calcium. Biochem J. 1987; 248:313-328.
21. Berk BC, Corson MA. Angiotensin II signal transduction in
vascular smooth muscle: role of tyrosine kinases. Circ Res. 1997;80:607-616.
22. Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639-672.
23. Muller DN, Dechend R, Mervaala EMA, et al. NF-κB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension. 2000;35:193-201.
24. Kranzhöfer R, Schmidt J, Pfeiffer CAH, Hagl S, Libby P, Kübler W. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:1623-1629.
25. Wallukat G, Bochnig N, Homuth V, et al. Agonistic AT1 receptor autoantibodies and monocyte stimulation in hypertensive patients. Am J Hypertens. 2003;16:827-833.
26. Wang B, Liao YH, Zhou Z, et al. Arterial structural changes in rats immunized by AT1-receptor peptide. Heart Vessels. 2005;20: 153-158.
27. Cuspidi C, Macca G, Sampieri L, et al. High prevalence of cardiac and extracardiac target organ damage in refractory hypertension. J Hypertens. 2001;19:2063-2070.
28. Ram CV. Management of refractory hypertension. Am T Ther. 2003;10:122-126.
29. Liao YH, Wei YM, Wang M, et al. Pathogenesis and clinical intervention in refractory hypertensive patients: Focus on the serum autoantibodies against AT1 receptors. Zhongguo Ye Jin Gong Ye Yi Xue Zha Zhi. 2001;18:65-69.
30. Homuth V, Stabroth C, Wallukat G, et al. Agonistic antibodies against the α1-receptor in patients with refractory hypertension and their removal by immune adsorption. Hypertonie. 2003.
31. Masuda MO, Levin M, De Oliveira SF, et al. Functionally active cardiac antibodies in chronic Chagas’ disease are specifically blocked by Trypanosoma cruzi antigens. FASEB J. 1998;12: 1551-1558.
32. Schwimmbeck PL, Dyrberg T, Drachman DB, Oldstone MB. Molecular mimicry and myasthenia gravis. An autoantigenic site of the acetylcholine receptor α -subunit that has biologic activity and reacts immunochemically with herpes simplex virus. J Clin Invest. 1989;84:1174-1180.
33. Dragun D, Muller DN, Brasen JH, et al. Angiotensin II Type 1- receptor activating antibodies in renal-allograft rejection. N Engl J Med. 2005;352:558-569.
34. Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune disease. Nature. 2005;435:584-589.