Área enfermedades hematológicas atención...
TRANSCRIPT

Área Enfermedades hematológicas
Atención farmacoterapéutica alpaciente con
Trombocitopenia Inmune Primaria (PTI)
Autores: Carolina Alarcón Payer
UGC de Farmacia de GranadaJose Manuel Puerta Puerta
UGC Hematología y HemoterapiaHospital Universitario Virgen de las Nieves
Complejo Hospitalario Universitario Granada
Basada en el de Dipiro JT et al. Pharmacotherapy: A Pathophysiologic Approach, ninth edition, 2014

• Enfermedad de carácter autoinmune mediada por autoAcs frente a lasplaquetas del tipo IgG, frente a Ags de la membrana de la plaqueta(GPIIb/IIIa), que provoca la reducción del número de plaquetas por dosmotivos:– Disminución en la producción medular.– Aumento de la destrucción extravascular, fundamentalmente a nivel esplénico.
• Incidencia en la población general de 1 caso por 10.000 habitantes.Constituye la primera causa de trombocitopenia en la práctica clínica.
• Puede afectar a pacientes de todas las edades. En niños, es más común entrelos 3-5 años, afectando a ambos sexos por igual. En los adultos, es mas comúnen mujeres, y en jóvenes, entre 2ª y 3ª década de la vida.
• Aunque se conocen a día de hoy la implicación de factores genéticos yadquiridos, la etiología no está del todo clara.
• Los términos “púrpura” e “idiopática” no se consideran actualmenteadecuados, pero por su significado histórico y uso cotidiano en el argotmédico, se mantiene el uso del acrónimo PTI para la trombopenia inmuneprimaria.

Enfermedad autoinmune que cursa con trombopenia aislada, con un recuento de
plaquetas inferior a 100x109/L, sin justificación por otras enfermedades o problemas clínicos.
Su diagnóstico se hace por exclusión y puede aumentarse el riesgo de sangrado, aunque la
clínica hemorrágica no es constante en todos los pacientes.

� La PTI es un trastorno autoinmune en el que la aparición de anticuerposantiplaquetarios sigue siendo el mecanismo patogénico central. Segeneran autoanticuerpos, generalmente de la clase IgG, contra ciertosantígenos plaquetarios, especialmente glicoproteinas IIb/ IIIa y Ib/IX.
� La sensibilización plaquetaria comienza con la fijación de autoanticuerposa glicoproteínas de las plaquetas. El complejo antígeno-anticuerpo escaptado y fagocitado por macrófagos u otra célula presentadora deantígeno (APC) fundamentalmente en el bazo. Las glicoproteínas sondegradadas a péptidos, expresándose de novo en la superficie de la célulaAPC a través de moléculas HLA de clase II. Estas glicoproteínas sonpresentadas de novo a células Th colaboradoras mediante la presencia demoléculas coestimulatorias para la activación de linfocito T, como son elantígeno 154 y su ligando CD 40.

Las células Th activadas producen citoquinas (interleukina-2 e interferón)que promueven la diferenciación de células B y producción deautoanticuerpos. Las células T reguladoras (Tregs) normalmente inhibenla actividad de Th y la proliferación celular, pero su función en la PTI estáafectada
Los autoanticuerpos también se unen a los megacariocitos de la médulaósea, afectando de esta manera la maduración de los megacariocitos y laproducción de plaquetas. Una vía alternativa de la destrucción de lasplaquetas es por células T citotóxicas autoreactivas. Las células Tc en lamédula ósea también podrían inhibir la megacariopoyesis ytrombopoyesis, aunque esto no ha sido aún demostrado.
� El papel de las infecciones, tales como H. pylori, no ha sidocompletamente aclarada, pero se ha demostrado reactividad cruzadaentre los antígenos bacterianos y las glicoproteínas de las plaquetas.

Fisiopatología de la PTI. Cines DB, Blanchette VS. Immune thrombocytopenicpurpura. N Engl J Med. 2002 Mar 28;346(13):995-1008.

• Mayoritariamente, aunque no en el 100% de los pacientes, seobservan lesiones purpúricas (petequias y equímosis).
• Existe posibilidad de hemorragias en mucosas (gingivorragia,epistaxis, hematuria, menorragia, melenas…). Los casos dehemorragias intracraneales son raros.
• Puede existir con relativa frecuencia, antes del diagnóstico, unantecedente de infección vírica en las semanas previas, a modo decatarro de vías respiratorias altas o cuadro gastrointestinal.
• El cuadro hemorrágico suele tener una evolución de pocos días.

• Fundamentalmente clínico y por exclusión.– Trombopenia aislada con normalidad de las otras series
hematopoyéticas, cualitativa y cuantitativamente.
• Obligatorio, y más en casos asintomáticos, descartarpseudotrombopenia mediante la realización del recuento en tubode citrato y estudio morfológico de sangre periférica, para ladetección de agregados plaquetarios.
• Estudio de médula ósea en pacientes mayores de 60 años paraexcluir displasias, y se recomienda previo a la realización deesplenectomía en los casos indicados.
• Las tasas de falsos positivos y negativos en los estudios de autoAcsespecíficos frente a glucoproteínas plaquetarias, hace de estaprueba no imprescindible para el diagnóstico.

Pruebas complementarias recomendadas para el diagnóstico de PTI
Estudios sistemáticos básicos Hemograma con reticulocitosEstudio morfológico de sangre periféricaEstudio de coagulaciónBioquímica básicaDosificación de inmunoglobulinas
Estudios de autoinmunidad Test de Coombs directoScreening de Acs antinucleares, antifosfolípidos, y antitiroideos y pruebas de función tiroidea
Estudios microbiológicos VIH, VHB, VHC, parvovirus, CMV, VEB, mycoplasma, H. pilory mediante prueba de aliento o Ag en heces
Estudio de médula ósea En > 60 años, refractarios a la 1ª línea de tratamiento, atipias en SP, antes de la esplenectomía
Estudios de imagen Ecografía abdominal para estudio de esplenomegaliaEstudio de cinética plaquetar
Otros estudios recomendados a considerar según evolución
Prueba de embarazo en mujeres en edad fértil, estudio de Acs antiplaquetas
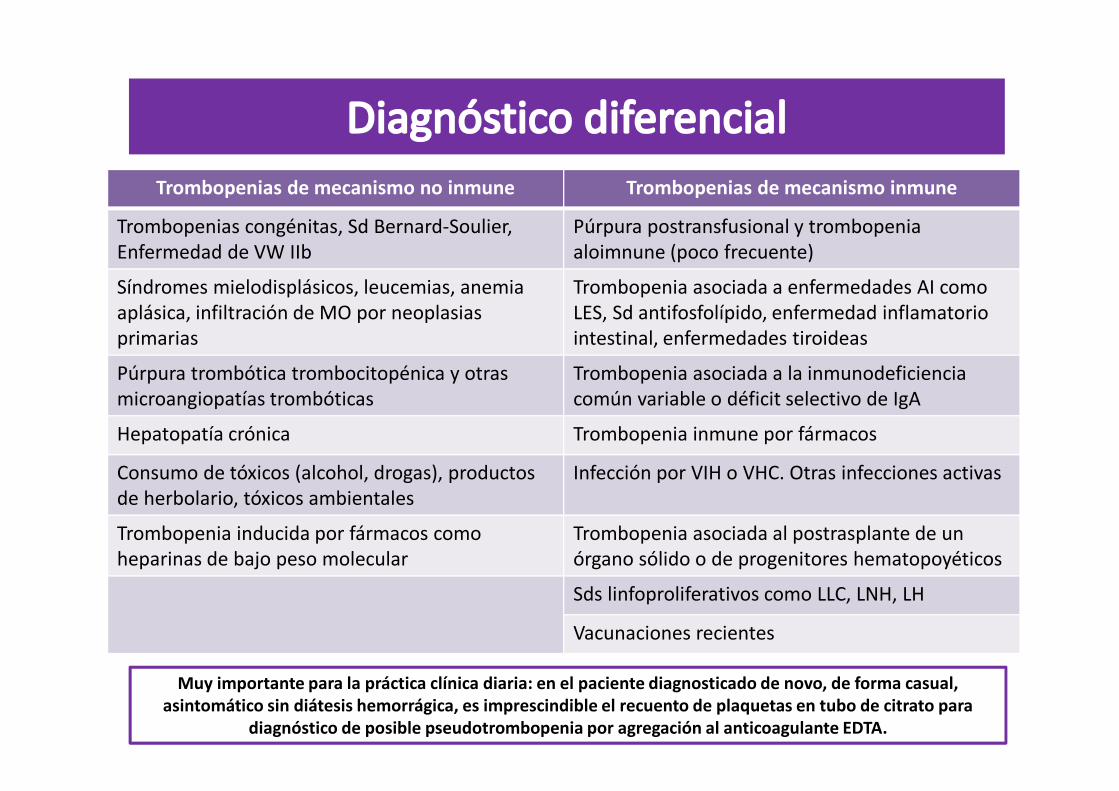
Trombopenias de mecanismo no inmune Trombopenias de mecanismo inmune
Trombopenias congénitas, Sd Bernard-Soulier, Enfermedad de VW IIb
Púrpura postransfusional y trombopeniaaloimnune (poco frecuente)
Síndromes mielodisplásicos, leucemias, anemia aplásica, infiltración de MO por neoplasias primarias
Trombopenia asociada a enfermedades AI como LES, Sd antifosfolípido, enfermedad inflamatorio intestinal, enfermedades tiroideas
Púrpura trombótica trombocitopénica y otras microangiopatías trombóticas
Trombopenia asociada a la inmunodeficiencia común variable o déficit selectivo de IgA
Hepatopatía crónica Trombopenia inmune por fármacos
Consumo de tóxicos (alcohol, drogas), productos de herbolario, tóxicos ambientales
Infección por VIH o VHC. Otras infecciones activas
Trombopenia inducida por fármacos como heparinas de bajo peso molecular
Trombopenia asociada al postrasplante de un órgano sólido o de progenitores hematopoyéticos
Sds linfoproliferativos como LLC, LNH, LH
Vacunaciones recientes
Muy importante para la práctica clínica diaria: en el paciente diagnosticado de novo, de forma casual, asintomático sin diátesis hemorrágica, es imprescindible el recuento de plaquetas en tubo de citrato para
diagnóstico de posible pseudotrombopenia por agregación al anticoagulante EDTA.

El diagnóstico de PTI se realiza por exclusión sistemática de otras causas de trombopenia y por el cuadro clínico del paciente, basándose en una buena anamnesis, exploración física, hemograma
y estudio morfológico de SP.
No se recomienda de forma sistemática el estudio de Ac antiplaquetas ni el estudio de MO en < 60
años. Sí el estudio de pseudotrombopenia por agregación

• Su incidencia es mayor entre los 2-8 años y es frecuente elantecedente de un episodio infeccioso desencadenante.
• Suele tender a la remisión espontánea, y en la decisión de inicio detratamiento hay que tener en cuenta que el niño, al disponer deuna mayor proyección de años y expectativa de vida, es muyimportante la valoración de efectos adversos y secuelas, de laenfermedad en si y de los tratamientos. La teórica menor tasa decomorbilidad en el niño facilita el manejo de la enfermedad.
• Para el inicio del tratamiento, se debe hacer una evaluación clínicade la gravedad en función de la clínica hemorrágica y de factores deriesgo que incrementen el riesgo de hemorragias graves como laintracraneal.
• Considerar hospitalización en un paciente diagnosticado de novocon factores de riesgo hemorrágico, hemorragia activa o si elrecuento plaquetario es < 20x109/L, evitando los inyectablesintramusculares y contraindicando el uso de AAS o sus derivados.

• Incidencia estimada de 1 caso por cada 1000-10.000 gestaciones yconstituye la causa más común de trombopenia aislada en el primertrimestre del embarazo. En mujeres ya diagnosticadas de PTI, el embarazopuede desencadenar una exacerbación o recaída.
• El diagnóstico está basado en la exclusión de otras causas detrombopenia, ya que hasta en un 10% de los embarazos puede haberrecuentos bajos de plaquetas debidos a múltiples causas, algunas de ellasespecíficas del embarazo.
• La morbi-mortalidad materna y la trombopenia neonatal suelen serbajas. Las complicaciones hemorrágicas en el niño también son bajas y sincorrelación con los recuentos plaquetarios de la madre.
• Se conoce un aumento de complicaciones hemorrágicas en mujeres concifra de plaquetas inferior a 50x109/L a las que se les practica cesárea,pero no cuando el parto es vía vaginal.
• La indicación para iniciar el tratamiento, decisión basada en lacolaboración entre hematólogos y obstetras, debe basarse en el riesgo dehemorragia de la mujer gestante con PTI. La cifra de plaquetas en partosvaginales debería ser > 50x109/L, y en la cesárea o para poner anestesiaepidural > 80x109/L.

• Objetivo: mantener una cifra de plaquetas en un nivel seguro querevierta y evite una hemorragia grave, en función del perfil delpaciente.
• No es recomendable tratar a pacientes asintomáticos, contrombopenias leves o moderadas, en los que los efectossecundarios del tratamiento podrían ser más importantes que lapropia enfermedad.
• El riesgo de hemorragia grave se asocia a recuentos inferiores a 10-30x109/L y en pacientes de edad avanzada. Recuentos ≥ 30x109/Lreducen de forma notable el riesgo de hemorragia.
• Aproximadamente un 10% de pacientes adultos puedenexperimentar una remisión espontánea sin tratamiento.
• La decisión de iniciar el tratamiento, se debe basar en fundamento,por la presencia de clínica hemorrágica, o con recuentosplaquetarios inferiores a 20x109/L .

Criterios de respuesta al tratamiento en la PTI
Remisión Completa (RC) Recuento de plaquetas > 100x109/L sin hemorragia.
Respuesta (R) Recuento de plaquetas ≥ 30x109/L, o incremento en más de 2 veces la cifra basal y sin hemorragia.
No Respuesta (NR) Recuento de plaquetas < 30x109/L, o incremento inferior a 2 veces la cifra basal y sin hemorragia.
Pérdida de Respuesta Recuento de plaquetas < 100x109/L o clínica hemorrágica (conseguida RC previa) o recuento < 30x109/L o menos de 2 veces el valor basal o hemorragia (conseguida al menos R).
Corticodependencia Necesidad de mantener dosis repetidas o mantenidas de corticoterapia con el que mantener una cifra de plaquetas ≥ 30x109/L para evitar la diátesis hemorrágica.
PTI Refractaria Situación en la que no se consigue respuesta o ésta se pierde tras realización de esplenectomía, con necesidad de tratamiento de forma continuada para disminuir el riesgo de hemorragia.
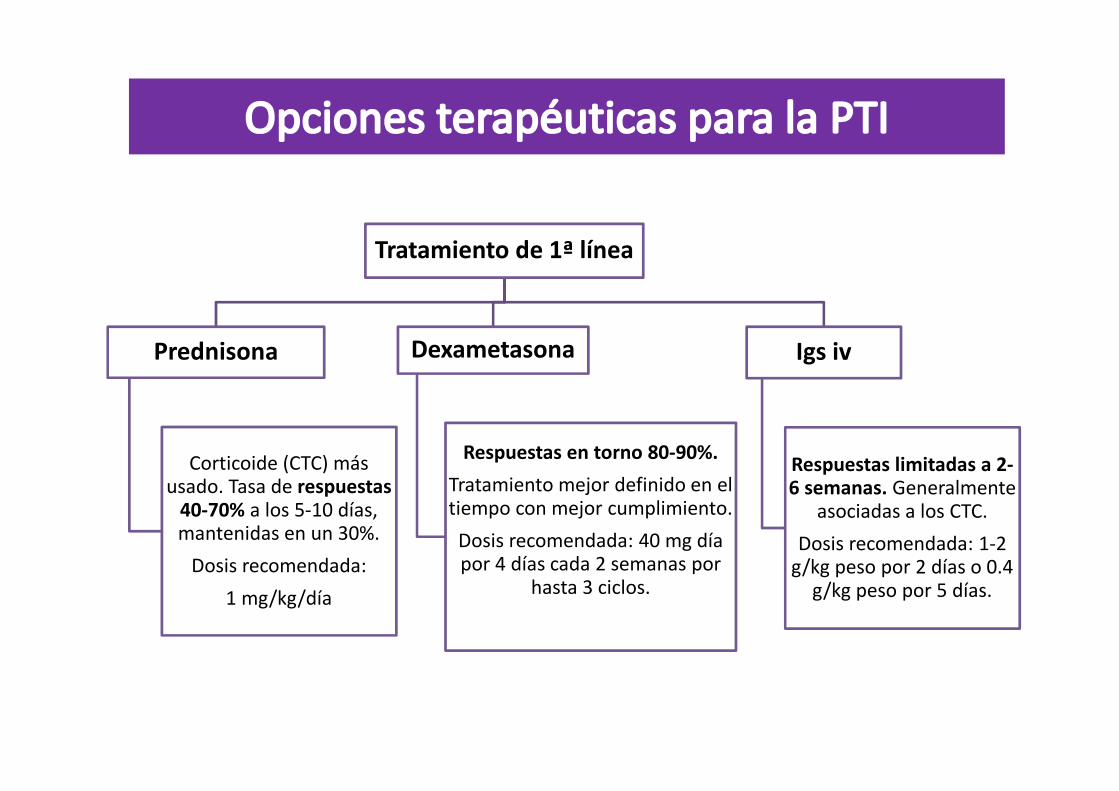
Tratamiento de 1ª línea
Prednisona
Corticoide (CTC) más usado. Tasa de respuestas
40-70% a los 5-10 días, mantenidas en un 30%.
Dosis recomendada:
1 mg/kg/día
Dexametasona
Respuestas en torno 80-90%.
Tratamiento mejor definido en el tiempo con mejor cumplimiento.
Dosis recomendada: 40 mg día por 4 días cada 2 semanas por
hasta 3 ciclos.
Igs iv
Respuestas limitadas a 2-6 semanas. Generalmente
asociadas a los CTC.
Dosis recomendada: 1-2 g/kg peso por 2 días o 0.4
g/kg peso por 5 días.

Manifestacioneshemorrágicas
Recuento de plaquetas
< 20x109/L 20-30x109/L 30-50x109/L
Asintomático Corticoidesa No tratarc No tratar
Púrpura menor Corticoides Corticoides No tratar
Hemorragias mucosas 1. Corticoides2. Hospitalización
Corticoides Corticoides
Hemorragia grave, con riesgo vital
1. Hospitalización2. Igs ivb
3. Corticoides4. Transfusión UTP5. Factor VII activado
1. Hospitalización2. Igs iv3. Corticoides
1. Hospitalización2. Corticoides3. Igs iv
a. Prednisona 1-2 mg/kg peso/día o su equivalente Dexametasona.b. Inmunoglobulina humana por vía iv 1-2 g/kg peso por 2 días o 0.4 g/kg peso por 5 días.c. Tratamiento indicado en pacientes que aunque estén asintomáticos, sean > 60 años o con factores de
riesgo hemorrágico como HTA, enfermedad ulcerosa, cirugía reciente…

Manifestacioneshemorrágicas
Recuento de plaquetas
< 20x109/L 20-30x109/L 30-50x109/L
Asintomático No tratara No tratarb No tratar
Púrpura menor 1. Igs iv2. Corticoides vo
No tratar No tratar
Hemorragias mucosas 1. Igs ivc
2. Hospitalización3. Corticoides vod
No tratar No tratar
Hemorragia grave, con riesgo vital
1. Hospitalización2. Igs iv3. Corticoides ive
4. Corticoides vo
1. Hospitalización2. Igs iv3. Corticoides iv4. Corticoides vo
1. Hospitalización2. Igs iv3. Corticoides iv4. Corticoides vo
a. Aunque sí indicado si hay factores de riesgo como edad < 3 años, HTA, enfermedad ulcerosa, estilo de vida.b. Sí indicado si hay factores de riesgo como edad < 3 años, HTA, enfermedad ulcerosa, estilo de vida…c. Inmunoglobulina humana iv a dosis de 1 g/kg peso/día por 1 día.d. Corticoides orales a dosis altas, como por ejemplo, prednisona 4-8 mg/kg peso/día por 7 días, con
disminución de dosis paulatina y retirada en 21 días.e. Corticoides iv a dosis altas, como o por ejemplo, metilprednisolona 30 mg/kg de peso/día por 3-7 días.
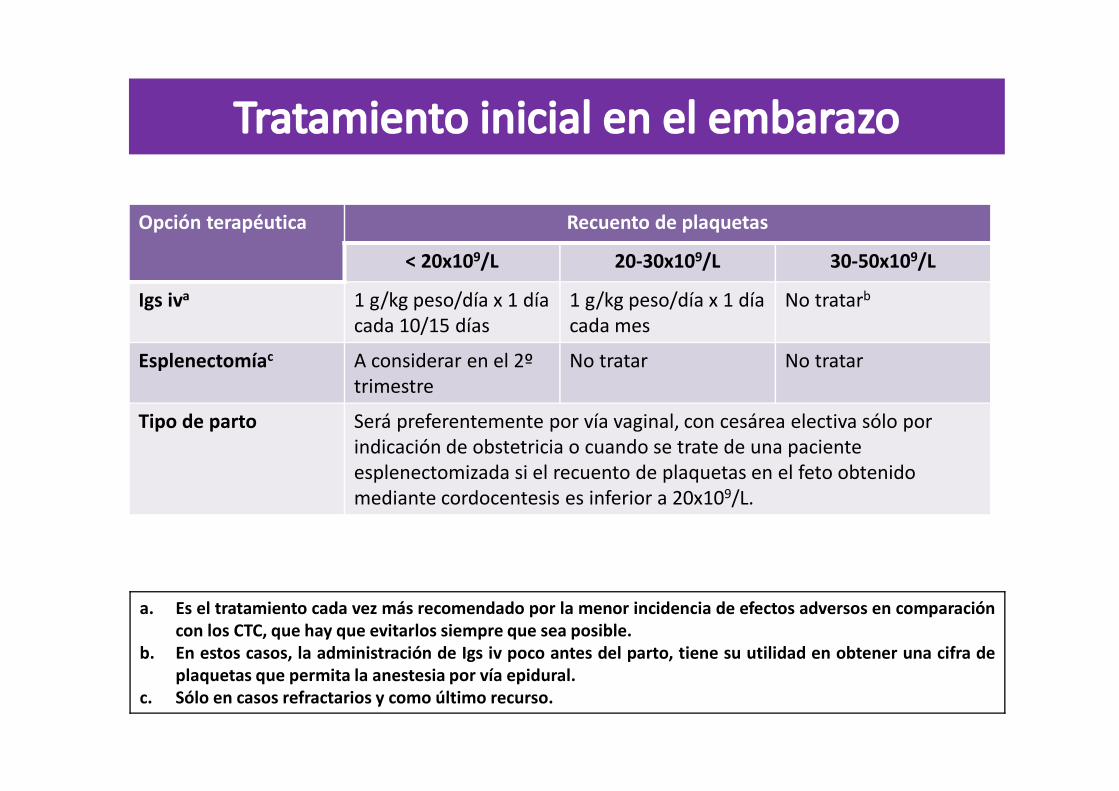
Opción terapéutica Recuento de plaquetas
< 20x109/L 20-30x109/L 30-50x109/L
Igs iva 1 g/kg peso/día x 1 día cada 10/15 días
1 g/kg peso/día x 1 día cada mes
No tratarb
Esplenectomíac A considerar en el 2º trimestre
No tratar No tratar
Tipo de parto Será preferentemente por vía vaginal, con cesárea electiva sólo por indicación de obstetricia o cuando se trate de una paciente esplenectomizada si el recuento de plaquetas en el feto obtenido mediante cordocentesis es inferior a 20x109/L.
a. Es el tratamiento cada vez más recomendado por la menor incidencia de efectos adversos en comparacióncon los CTC, que hay que evitarlos siempre que sea posible.
b. En estos casos, la administración de Igs iv poco antes del parto, tiene su utilidad en obtener una cifra deplaquetas que permita la anestesia por vía epidural.
c. Sólo en casos refractarios y como último recurso.

Tratamiento de 2ª línea
Esplenectomía
De 1ª elección en pacientes corticodependientes-
resistentes. Preferible por vía laparoscópica. Opcional en
casos de contraindicación, la embolización o irradiación
esplénica. Respuestas globales en el esplenectomizado en
torno al 80%
Rituximab
Uso no aprobado en PTI, si bien es un tratamiento con frecuencia utilizado en
enfermedades AI. Tasas de respuestas mantenidas a largo
plazo en torno al 30%
Agentes trombopoyéticos
Aprobados en pacientes refractarios al tratamiento
CTC, en recaída tras esplenectomía o en la que la misma esté contraindicada.
Escasos efectos secundarios, el tratamiento continuado
consigue respuestas mantenidas en torno al 80%
Romiplostim sc Eltrombopag oral

• Se le considera el tratamiento de 2ª línea con mayor tasa de RCduraderas.
• Para algunos pacientes puede considerarse una intervención de altoriesgo, o simplemente que se nieguen a la misma.
• Por tanto, la esplenectomía está indicada como tratamiento de 2ªlínea en pacientes con PTI persistente o crónica grave, en los queno esté contraindicada la cirugía. No se recomienda antes de los 6meses del diagnóstico.– Respuesta aproximada en el 80% de los pacientes, mantienen una
cifra normal de plaquetas a los 5 años un 60% de los mismos. Lamayoría de las recaídas se producen en los dos siguientes años tras lacirugía.
• En aquellos pacientes en los que se contraindica la esplenectomía,la irradiación o embolización puede constituir una alternativa avalorar.– La embolización tiene una tasa de respuesta del 50% con un 30% de
recaídas en los 3 primeros años.

INMUNOGLOBULINA HUMANA NORMAL (IgIV)Las inmunoglobulinas humanas para administración intravascular (IgIV)contienen inmunoglobulina G (IgG) con una distribución de subclasesprácticamente proporcional a la del plasma humano natural.
� Indicaciones:� Tratamiento de reposición en adultos, niños y adolescentes (2-18 años) en:
� Síndromes de inmunodeficiencia primaria con producción de anticuerposdeteriorada
� Hipogammaglobulinemia e infecciones bacterianas recurrentes enpacientes con leucemia
� linfocítica crónica en los que los antibióticos profilácticos no han dadoresultado
� Hipogammaglobulinemia e infecciones bacterianas recurrentes enpacientes con mieloma múltiple en fase estacionaria y que norespondieron a la inmunización neumocócica.
� Hipogammaglobulinemia en pacientes después de un trasplantealogénico de células madre.
� SIDA congénito con infecciones bacterianas recurrentes

� Inmunomodulación en adultos, niños y adolescentes (2-18 años) en:� Trombocitopenia inmune primaria, en pacientes con riesgo elevado de
sufrir hemorragia o en pacientes antes de someterse a cirugía paracorregir el recuento de plaquetas
� Síndrome de Guillain Barré� Enfermedad de Kawasaki
� Se debe administrar como perfusión intravenosa a una velocidad inicial de 0,01a 0,02 ml/kg/min durante 30 min. Si se tolera bien se puede aumentargradualmente hasta un máximo de 0,12 ml/kg/min
� En general se utilizan en combinación con los glucocorticoides en primera líneaterapéutica en pacientes con hemorragia activa grave (OMS >2) en los que serequiere una respuesta rápida.
� Reacciones Adversas:Tienden a estar relacionadas con la dosis y la velocidad de perfusión. Engeneral, se han descrito varios tipos de reacciones alérgicas y dehipersensibilidad como dolores de cabeza, escalofríos, dolor lumbar, dolor enel pecho, fiebre, reacciones cutáneas, vómitos, artralgia, presión arterial baja ynáuseas. Con menor frecuencia (< 0,01%) se han observado trastornosvasculares como trombosis o fallo circulatorio periférico, además de trastornoscardiacos como infarto de miocardio, entre otros

AGONISTAS DEL RECEPTOR DE LA TROMBOPOYETINA
1. ROMIPLOSTIM
� Mecanismo de AcciónRomiplostim es una proteína de fusión que señala y activa las rutas detranscripción intracelular a través del receptor de la trombopoyetina (TPO)(también denominado cMpl) para aumentar la producción de plaquetas. Lamolécula del cuerpo peptídico está formada por un dominio Fc de lainmunoglobulina humana IgG1, con cada subunidad de cadena simple unidamediante enlace covalente en el extremo C a una cadena peptídica quecontiene dos dominios de unión del receptor de la TPO. No existe homologíasecuencial de aminoácidos entre romiplostim y la TPO endógena.
� Indicaciones Terapéuticas� Para pacientes adultos esplenectomizados con Púrpura Trombocitopénica
Inmune (Idiopática) (PTI) crónica que son refractarios a otros tratamientos(por ejemplo, corticosteroides, inmunoglobulinas).
� Como segunda línea de tratamiento en pacientes adultos noesplenectomizados en los que la cirugía esté contraindicada

� Reacciones AdversasLos acontecimientos adversos más frecuentes descritos en los pacientestratados con romiplostim son leves, predominando ligeras cefaleas. En algunospacientes se ha observado un aumento de la reticulina en la médula ósea, quees reversible tras la retirada del fármaco. Otros efectos adversos reportadosfueron diarrea, mareos, dorsalgias y artromialgias, además de fenómenoshemorrágicos probablemente asociados con la enfermedad
� La tasa de respuesta plaquetaria global de romiplostim es del 88% en pacientesno esplenectomizados y del 77% en pacientes esplenectomizados, según sederiva de los resultados de los ensayos fase III aleatorizados. El más recienteestudio aleatorizado publicado ha mostrado que la utilización de romiplostimdurante 52 semanas, en comparación con el tratamiento estándar, se asocia auna menor incidencia de requerimiento de esplenectomía, menos episodioshemorrágicos, menos fallos al tratamiento y mejoría en la calidad de vida.
� El tratamiento a largo plazo, según la experiencia clínica con seguimiento dehasta 5 años, es posible, efectivo y seguro.
� Los efectos adversos asociados al fármaco son, en general, leves, manejables yno aumentan en gravedad ni en frecuencia con la administración continuada delfármaco.
� La cefalea es el efecto adverso más frecuente

� Se administra por vía subcutánea una vez por semana, iniciando eltratamiento con una dosis de 1 μg/kg e incrementándola en 1 μg/kg/semanasi el recuento de plaquetas es <50 x 109/L, sin exceder la dosis máxima de 10μg/kg.
� La máxima respuesta se alcanza a las dos semanas de la primera dosis. Si lacifra de plaquetas en dos semanas consecutivas es >150 x 109/L, debebajarse la dosis en 1 μg/kg. Si la cifra de plaquetas es >250 x 109/L, debesuspenderse el tratamiento temporalmente, para volver a iniciarlo con unadosis reducida en 1μg/kg cuando la cifra de plaquetas sea <150 x 109/L.

2. ELTROMBOPAG
� Mecanismo de AcciónEltrombopag es una fenilhidrazona. Actúa como agonista del receptor detrombopoyetina (RTPO) que interacciona con el dominio transmembrana deéste, iniciando un proceso en cascada similar, pero no idéntico, al generadopor la trombopoyetina endógena, induciendo así la proliferación ydiferenciación de los megacariocitos y dando lugar al consiguiente incrementoen la producción de plaquetas.
� Indicaciones Terapéuticas� en pacientes adultos esplenectomizados con púrpura trombocitopénica
inmune (idiopática) (PTI) crónica que son refractarios a otros tratamientos(por ejemplo, corticosteroides, inmunoglobulinas).
� como segunda línea de tratamiento en pacientes adultos noesplenectomizados en los que la cirugía está contraindicada.

� La eficacia y la seguridad clínicas de eltrombopag han sido adecuadamentecontrastadas para las indicaciones terapéuticas autorizadas, fundamentalmentemediante dos ensayos clínicos de fase 3, multicéntricos, aleatorizados,doblemente ciegos y controlados con placebo. Los resultados del primero de losestudios (TRA100773B; Bussel, 2009) mostraron unos porcentajes de pacientesrespondedores del 59% (eltrombopag, E) vs. 16% (placebo, P). Asimismo, de lospacientes tratados con la dosis inicial de 50 mg de eltrombopag respondieron un44,6% (un 24,3% alcanzaron recuentos de = 200.000 plaquetas/mm3); de los queno respondieron y recibieron dosis de 75 mg/día a partir de la cuarta semana, un29,4% respondió a este incremento de dosis. Los resultados del otro estudio(TRA102537, RAISE; Cheng, 2010) mostraron unos porcentajes de pacientesrespondedores del 49% (eltrombopag, E) vs. 10% (placebo, P) a las cuatrosemanas y del 52% (E) vs. 17% (P) a los seis meses.
� Reacciones AdversasCefalea moderada, alteraciones de la función hepática, aumento de lareticulina en MO. En hepatopatías crónicas, aumento de fenómenostrombóticos

� Se administra por vía oral a la dosis inicial de 50 mg al día, salvo en pacientes deorigen asiático o con insuficiencia hepática grave, en los que se comenzará con 25mg al día.
� Se podrá subir la dosis hasta un máximo de 75 mg al día con el fin de alcanzar unrecuento plaquetario >50 x 109/L. El tratamiento de mantenimiento se realizará deacuerdo a los recuentos plaquetarios; la dosis de eltrombopag oscilará entre 25 y75 mg/día.
� La respuesta máxima se alcanza a las dos semanas de iniciado el tratamiento. Si sesuspende éste, los recuentos de plaquetas vuelven a los valores basales dentro delas 2 semanas siguientes a la suspensión del fármaco.
� La administración conjunta de eltrombopag con antiácidos y productos lácteospuede disminuir su absorción.
� La inhibición por eltrombopag de los transportadores de membrana OATP1B1 yBCRP podría aumentar la exposición de los fármacos que utilizan estostransportadores, como las estatinas.

En aproximadamente el 20% de los pacientes con PTI no se logran respuestassatisfactorias tras tratamientos de primera y segunda línea, incluyendoglucocorticoides, esplenectomía y agentes trombopoyéticos.
Rituximab
� Es un anticuerpo monoclonal quimérico dirigido contra el receptor CD20presente en los linfocitos B. Durante la última década se ha utilizado enpacientes con PTI refractaria.
� Generalmente se administra en infusión intravenosa a la dosis de 375mg/m2/semana, durante 4 semanas, pero también se han propuestodosis más reducidas (100 mg/m2). No obstante, no existen estudiosprospectivos y aleatorizados.
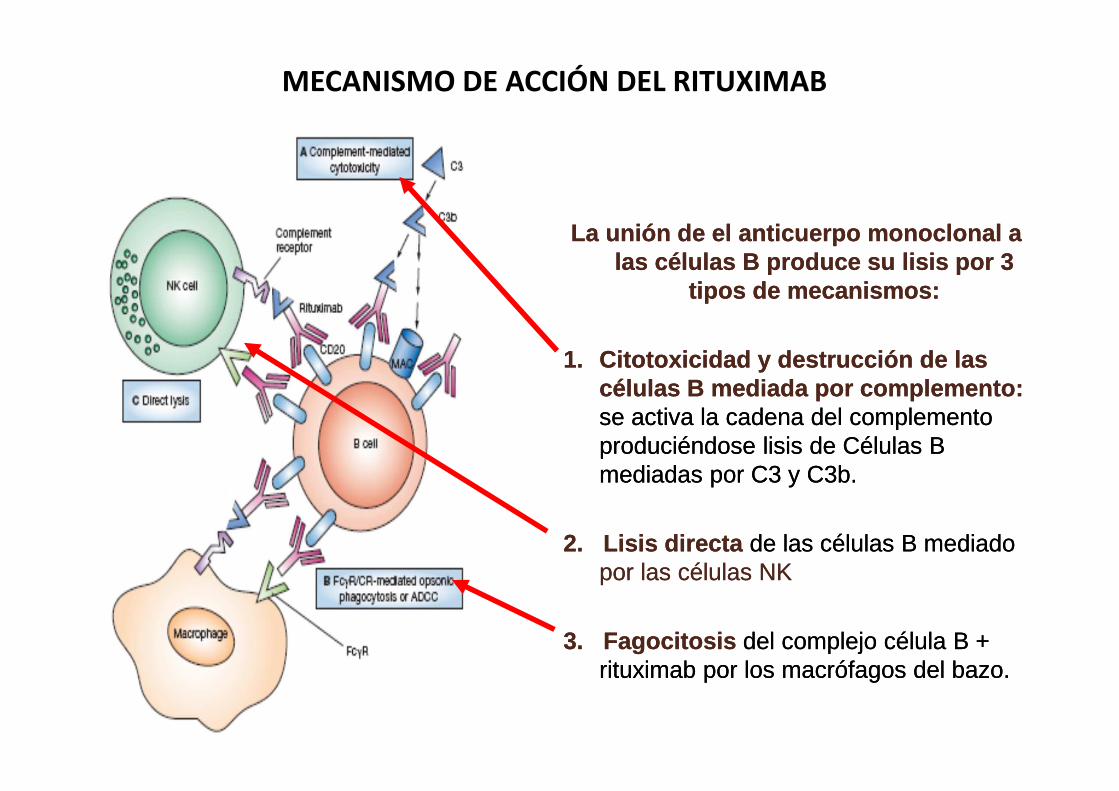
La unión de el anticuerpo monoclonal a las células B produce su lisis por 3
tipos de mecanismos:
1. Citotoxicidad y destrucción de las células B mediada por complemento:se activa la cadena del complemento produciéndose lisis de Células B mediadas por C3 y C3b.
2. Lisis directa de las células B mediado por las células NK
3. Fagocitosis del complejo célula B + rituximab por los macrófagos del bazo.
La unión de el anticuerpo monoclonal a las células B produce su lisis por 3
tipos de mecanismos:
1. Citotoxicidad y destrucción de las células B mediada por complemento:se activa la cadena del complemento produciéndose lisis de Células B mediadas por C3 y C3b.
2. Lisis directa de las células B mediado por las células NK
3. Fagocitosis del complejo célula B + rituximab por los macrófagos del bazo.
MECANISMO DE ACCIÓN DEL RITUXIMAB

� En el único estudio multicéntrico en fase II diponible, se observó que, al año derecibir el tratamiento, el 40% de los pacientes tenían un recuento plaquetario ≥ 50x 10/9L y el 33% mantenían cifras elevadas de plaquetas a los dos años. Se hanpublicado los resultados de un estudio aleatorizado en el que se usó lacombinación de rituximab y dexametasona en pacientes con diagnóstico nuevo dePTI. Estos resultados sugieren que el tratamiento combinado con dexametasona yrituximab puede ser una opción eficaz antes de indicar la esplenectomía.
� Reacciones Adversas:� Son destacables los relacionados con la primera infusión (náuseas,
escalofríos, rinitis, urticaria con sensación de sofoco, hipertensión, erupción,fiebre, prurito, irritación de garganta e hipotensión), generalmente leves ytransitorios, más infrecuentes en las siguientes dosis y que se reducen ylimitan con la premedicación.
� Se ha descrito la reactivación de la infección en los portadores asintomáticosdel virus de la hepatitis B, por lo que es un aspecto que hay que tener encuenta antes de decidir el inicio del tratamiento con rituximab.
� Otra rara pero importante complicación es la reactivación del poliomavirusJC, que causa leucoencefalopatía multifocal progresiva.

Opciones basadas en quimioterapia
Quimioterapia con agentes únicos
� Los más usados han sido los alcaloides de la vinca (p. ej., vincristina, 1mg/m2/semana durante 4 semanas, o vinblastina, 0,1 mg/kg/semana durante 6semanas), la ciclofosfamida (1-3 mg/kg/día) y la azatioprina (150-300 mg/día).
� No se dispone de estudios controlados que avalen y comparen su eficacia. Contodo, cabe decir que, en general, la eficacia es muy limitada y, sobre todo,transitoria. Alguno de estos fármacos puede dar lugar a efectos secundariosimportantes.
Quimioterapia en combinación
� Hay algún estudio en el que se ha ensayado la combinación de agentes conacción inmunosupresora y citotóxica. Un ejemplo es la asociación deciclofosfamida (100-200 mg/día IV), días 1-5 o 1-7 y prednisona (0,5-1mg/kg/día) días 1-7, combinada con vincristina (1-2 mg IV) día 1, y uno de lossiguientes: azatioprina (100 mg/día VO) días 1-5 o 1-7 o etopósido (50 mg/díaVO) días 1-7.
� La respuesta global en 31 pacientes fue del 68% y la respuesta completa del42%. La tolerancia fue buena (6). Otra posible combinación es la de rituximab,ciclofosfamida y dexametasona.

Agentes inmunosupresores
Ciclosporina A y micofenolato
� La ciclosporina A (5 mg/kg/día durante 6 días y 2,5-3 mg/kg/día, con controlesde concentraciones plasmáticas) y el micofenolato (1.000 mg/12 h, durante 3-4 semanas) se han empleado ocasionalmente con una respuesta muy variable,que suele desaparecer tras su suspensión.
� Sus efectos secundarios y la escasa experiencia limitan su uso a casos con faltade respuesta a otras opciones.
Otros fármacos
� El danazol es un derivado semisintético de la progesterona con efecto anabolizante androgénico atenuado.
� Se ha administrado a dosis entre 400 y 800 mg/día durante un mínimo de 8 semanas, aunque se ha sugerido administrarlo entre 3 y 6 meses antes de asegurar su falta de respuesta.
� No está exento de efectos secundarios y los resultados del tratamiento son controvertidos.
� Con la dapsona (75-100 mg) se han conseguido respuestas hasta en el 50% de los pacientes, aunque sus efectos secundarios, sobre todo cutáneos, obligan con frecuencia a suspender su administración. Puede provocar metahemoglobinemia, así como anemia hemolítica en los pacientes con déficit de glucosa 6-fosfato deshidrogenasa.

CONTROVERSIA CLÍNICA
Las opciones terapéuticas para pacientesrefractarios a esplenectomía y a agentestrombopoyéticos son múltiples, pero la tasa derespuesta y su duración son muy variables.Además, no hay estudios controlados queevaluen y comparen su eficacia.

Tratamiento Reacciones Adversas Opciones de Manejo
Corticosteroides Aumento de peso (puede ser muy rápido y llevar arasgos de Cushing)
• Evitar sal para disminuir la retención de líquidos• Evitar alimentos de mucha grasa o azúcar• Comer carbohidratos complejos
Pérdida de músculo • Aumentar el ejercicio
Gastrointestinales • Administrar después del desayuno• Se pueden requerir inhibidores de la bomba de protones
Edema • Usar diuréticos• Evitar exceso de sodio dietético• Utilizar medias de compresión
Cansancio • Ser cuidadoso con las actividades• Tiempo de administración: o muy temprano en la mañana o muytarde en lanoche• Asegurarse que el paciente duerme lo suficiente
Hiperglucemia/diabetes • Modificar dieta• Evitar carbohidratos y azucares• Monitorización regular de niveles de glucosa en sangre• Se pueden requerir hipoglucemiantes orales o insulinasubcutánea
Erupción tipo acné • Usar jabones no irritantes• Se pueden requerir antibióticos tópicos u orales
Inmunosupresión • Monitorización de infecciones• Educar a los pacientes a informar de signos y síntomas
Otras • Explicarle a los pacientes la posibilidad de cambios de humor• Controlar la presion arterial• Monitorización de signos de osteoporosis

Tratamiento Reacciones Adversas Opciones de Manejo
IgIV Reacciones a las infusiones • Velocidad de infusión lenta (sobre todo las dos primeras veces)• Advertir a los pacientes de posibles reacciones alérgicas• Hidratación adecuada antes de iniciar la perfusión de IgIV,monitorizar la diuresis, monitorizar los niveles de creatinina séricay evitar el uso concomitante de diuréticos del asa
Inmunosupresores Inmunosupresión • Monitorización de infecciones• Educar a los paciente a informar de signos y síntomas
Problemas con el funcionamientodel hígado (azatioprina)
• Monitorización de las enzimas del hígado (semanal durante lasprimeras 8 semanas, luego mensual)
Supresión de la médula ósea(azatioprina)
• Monitorización de los niveles de la tiopurina metiltransferasa
Esplenectomía Infecciones • Se pueden requerir antibióticos• Vacunas antes de la esplenectomía y desde entonces cada 5años (contra neumococo y haemophilus)• Si se viaja al extranjero asegurar que las inmunizaciones estánactualizadas
Agonistas de losreceptores de latrombopoyetina
Dolores de cabeza • Usar paracetamol en lugar de aspirina o agentes anti-inflamatorios, ya que éstos pueden interferir con elfuncionamiento de las plaquetas• Vigilar la función hepática
Anticuerpo monoclonalanti-CD20
Problemas inmunoalérgicosRiesgo infeccioso por depleciónprolongada de linfocitos BPosible leucoencefalopatíamultifocal progresiva
• Infusión lenta con premedicación 30 minutos antes de suadministración consistente en paracetamol 1g IV ydexclorfeniramina 5mg IV

Evitar tratamientos innecesarios, potencialmente tóxicos, en pacientesasintomáticos o con descensos moderados de las plaquetas y conseguir unaadecuada calidad de vida con la mínima toxicidad asociada a la terapia.
Contraindicado: el uso de acido acetilsalicilico y derivados. Se debe tenerprecaución y administar, sólo en caso necesario, fármacos que puedan alterar laagregación plaquetaria (antihistamínicos, antiinflamatorios no esteroideos).
Evitar inyectables intramusculares y punciones vasculares en vasos de difícilcompresión.
Deportes: se indicara restricción de la actividad deportiva en función de la clínica y riesgo traumático.

• No existen recomendaciones establecidas y por tanto el seguimiento esvariable. Estas recomendaciones están basadas en la experiencia clínica. Elseguimiento debe ser individual para cada paciente.
• Paciente con recuento de plaquetas estable que no requiere tratamiento:– Hemograma cada 3-6 meses.– Educación del paciente en cuanto a signos de hemorragia, previsión de cirugía
o procedimientos invasivos.– Gestación y seguimiento de sospecha de otras enfermedades autoinmunes.
• Pacientes que requieren tratamiento activo:– Hemogramas a demanda, con valoración máxima 24h desde la extracción.– En pacientes tratados con CTC prolongadamente:
• Control de glucemias, TA, prevención de osteoporosis e infecciones.
– En pacientes tratados con Rituximab:• Función hepática (reactivación VHB), clínica neurológica (leucoencefalopatía multifocal)
– En pacientes tratados con agentes trombopoyéticos:• Hemograma semanal hasta alcanzar dosis de mantenimiento, función hepática, signos de
trombosis…
– En pacientes esplenectomizados:• Prevención y tratamiento temprano de las infecciones. Revacunar cuando se indique.

• Douglas B. Cines, James B. Bussel. How I treat idiopathic thrombocytopenic purpura (ITP).Blood 2005; 106 (7): 2244-2251.
• Waleed Ghanima, Bertrand Godeau, Douglas B. Cines, James B. Bussel. How I treat immunethrombocytopenia: the choice between splenectomy or a medical therapy as a second-linetreatment. Blood 2012; 120 (5): 960-969.
• Miguel A. Sanz, Enric Carreras. Manual práctico de hematología clínica 2012. 4ª ed. Molins deRei: Ediciones Escofet Zamora SL; 2012.
• Miguel A. Sanz, Vicente Vicente. Directrices de diagnóstico, tratamiento y seguimiento de laPTI: Documento de consenso de la SEHH. Madrid: Prodrug Multimedia SL; 2011.
• Ficha Técnica de Flebogamma. http://www.ema.europa.eu/docs/es_ES/document_library/EPAR__Product_Information/human/000781/WC500023473.pdf
• Ficha Técnica de Romiplostim
http://www.ema.europa.eu/docs/es_ES/document_library/EPAR__Product_Information/human/000942/WC500039537.pdf
• Kuter DJ, Rummel M, Boccia R, Macik BG, Pabinger I, Selleslag D, Rodeghiero F, Chong BH,Wang X, Berger DP. Romiplostim or standard of care in patients with immunethrombocytopenia. N Engl J Med 2010; 363: 1889-98.

• Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immunethrombocytopenic purpura: a doubleblind randomised controlled trial. Lancet 2008;371:395-403
• Kuter DJ, Phil D, Rummel M, Boccia R, Macik BG, Pabinger I, et al. Romiplostim or standard of carein patients with immune thrombocytopenia. N Engl J Med 2010;363:1889-1899.
• Ficha Técnica de Eltrombopag
http://www.ema.europa.eu/docs/es_ES/document_library/EPAR__Product_Information/human/001110/WC500089964.pdf
• Bussel JB, Provan D, Shamsi T, Cheng G, Psaila B, Kovaleva L, Salama A, Jenkins JM, RoychowdhuryD, Mayer B, Stone N, Arning M. Effect of eltrombopag on platelet counts and bleeding duringtreatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet. 2009; 373(9664): 641-8
• Cheng G, Saleh MN, Marcher C, Vasey S, Mayer B, Aivado M, Arning M, Stone NL, Bussel JB.Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month,randomised, phase 3 study. Lancet 2011; 377:393-402
• Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab foradults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007; 146: 25-33.
• Godeau B, Porcher R, Fain O, et al. Rituximab efficacy and safety in adult splenectomy candidateswith chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2study. Blood 2008; 112: 999-1004.
• Zaja F, Baccarani M, Mazza P, et al. Dexamethasone plus rituximab yields higher sustainedresponse rates than dexamethasone monotherapy in adults with primary immunethrombocytopenia. Blood 2010; 115: 2755-62.
• Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenancetherapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007; 110:3526-31