an intron transcriptional enhancer element regulates il-4 gene
TRANSCRIPT

of April 5, 2018.This information is current as
Mast CellsRegulates IL-4 Gene Locus Accessibility in An Intron Transcriptional Enhancer Element
Hock and Melissa A. BrownJohn A. Hural, Millie Kwan, Greg Henkel, M. Benjamin
http://www.jimmunol.org/content/165/6/3239doi: 10.4049/jimmunol.165.6.3239
2000; 165:3239-3249; ;J Immunol
Referenceshttp://www.jimmunol.org/content/165/6/3239.full#ref-list-1
, 29 of which you can access for free at: cites 80 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2000 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

An Intron Transcriptional Enhancer Element Regulates IL-4Gene Locus Accessibility in Mast Cells1
John A. Hural,2* Millie Kwan,* Greg Henkel, † M. Benjamin Hock,‡ and Melissa A. Brown3*‡§
The cell type-specific expression of a gene is dependent on developmentally regulated modifications in chromatin structure thatallow accessibility of basal and inducible transcription factors. In this study, we demonstrate that acis-acting element in the secondintron of the murine IL-4 gene has a dual function in regulating transcription in mast cells as well as chromatin accessibility ofthe IL-4 gene locus through its influence on the methylation state of the gene. Previous studies have shown that mast cell-restrictedtranscription factors GATA-1/2 and PU.1 associate with the intron element and regulate its activity. In this study, we use DNaseI footprinting and mutational analyses to identify two additional sites that contribute to the element’s ability to enhance tran-scription. One of these sites associates preferentially with STAT5a and STAT5b. We also demonstrate that deletion of the elementor mutation of the GATA binding site in the context of a stably integrated IL-4 genomic construct prevents maintenance of ademethylated locus in IL-4-producing mast cells. These data indicate that, analogous to Ig and TCR intron regulatory elements,the intron enhancer has an essential role in maintaining developmentally regulated demethylation at the IL-4 gene locus. Inaddition, they indicate that members of the GATA family of transcription factors likely play an important role in theseprocesses. The Journal of Immunology,2000, 165: 3239–3249.
I nterleukin-4 is a pleiotropic cytokine that plays a central rolein immunoregulation (for review, see Refs. 1 and 2). It is bestknown for its ability to induce selective Ig isotype switching
in B lymphocytes and to promote the development of Ag-specificTh2 cells from naive CD41 precursors. In mice, strain-specificdifferences in the levels of IL-4 produced in infections such asLeishmania majorandSchistosoma mansonicontribute to the vari-able Th2 response that influences disease course (3–8). However,IL-4 also has profound effects on the activation and/or growth ofmany other target cells, including endothelial cells, neutrophils,mast cells, eosinophils, and CD81 T cells. The dysregulation ofIL-4 production has been linked to allergic disease and autoim-munity in both human and murine models, attesting to its impor-tance (9, 10). While most studies have focused on IL-4 productionby select subsets of activated T lymphocytes, including Th2,gd1,and NK1.11 cells (2, 11, 12), mast cells also express IL-4 and arelikely to provide a significant source of this cytokine in vivo (13,14). In addition to de novo inducible synthesis, mast cells containstores of preformed IL-4 that are released immediately upon acti-vation (15). Mast cells are most prevalent at sites of Ag entry, suchas the skin, respiratory tract, and gastrointestinal tract (16, 17),where mast cell-derived IL-4 can initiate inflammatory reactions ofthe innate immune response. Activated mast cells can also migrate
to local lymph nodes (18). This observation supports the idea thatmast cells can impact adaptive immune responses by localizing tosites of initial T cell activation and, through elaboration of cyto-kines such as IL-4, directly influence Th development.
IL-4 production by naive T cells is dependent on the delivery ofat least two signals: cross-linkage of the TCR and costimulationthrough CD28 and/or CD40 ligand (19, 20). In contrast, mast cellsare activated through Fc receptor interactions with IgE or IgG (21,22), as well as via direct interactions with bacterial and parasiteproducts, complement components, and neuropeptides such asSubstance P (16, 23–27). We previously hypothesized that the dif-ferences in the ligand-receptor interactions leading to IL-4 produc-tion in these cell types would be reflected in distinct nuclear sig-naling events. There is now substantial evidence to support thishypothesis. For example, GATA-3 and Maf, transcription factorsthat are essential for maximal IL-4 gene expression by Th2 cells(28, 29), are not expressed in mast cells (30, 31). Intact STAT6signaling pathways are also required for Th2 differentiation (32–34), yet mast cells exhibit STAT6-independent IL-4 production:bone marrow-derived mast cell precursors from STAT62/2 micecan differentiate into mature cells that express IL-4 levels compa-rable to those of wild-type cells (35). Finally, although IL-4 genetranscription is NF-AT-dependent in both mast cells and T cells,there is evidence that each cell type uses distinct NF-AT isoforms(36–39).
A transcriptional regulatory element located in the second intronof the IL-4 gene may also contribute to cell type-specific expres-sion patterns (40, 41). Several features of this element suggest thatit is mast cell specific. Its location was defined by a DNase I hy-persensitive site in chromatin examined from a variety of trans-formed and nontransformed mast cell lines but absent in EL-4 Tcells. It exhibits mast cell-specific activity in enhancer-reporterassays, and sequences that contribute to its activity are the bindingtargets of factors selectively expressed in mast cells and not Tcells. These factors include PU.1, an ets family member that isessential for mast cell development, and GATA-1 and -2. How-ever, simultaneous mutation of both GATA and PU.1 binding sites
*Graduate Program in Immunology and Molecular Pathogenesis, Emory University,Atlanta, GA 30322;†Aurora Biosciences Corporation, San Diego, CA 92121;‡Grad-uate Program in Genetics and Molecular Biology, and§Department of Pathology,Emory University, Atlanta, GA 30322
Received for publication March 21, 2000. Accepted for publication July 5, 2000.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by National Institutes of Health Grants AI 34040 andCA47992 and a scholarship from the Leukemia Society of America (to M.A.B.).2 Current address: Infectious Disease Research Institute, Corixa Corporation, 1124Columbia Street, Suite 200, Seattle, WA 98104.3 Address correspondence and reprint requests to Dr. Melissa A. Brown, Emory Uni-versity, Department of Pathology, 1639 Pierce Drive, Atlanta, GA 30322. E-mailaddress: [email protected]
Copyright © 2000 by The American Association of Immunologists 0022-1767/00/$02.00
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

reduces enhancer activity by;50%, suggesting other regions alsocontribute to the enhancer activity.
In this study, we have further examined the molecular require-ments for full enhancer activity of the intron regulatory element inmurine mast cells. We demonstrate that a consensus STAT site thatpreferentially associates with STAT5 contributes to its enhanceractivity in mast cells. We also provide evidence that the intronelement plays a role in acquiring and/or maintaining the IL-4 genelocus in a demethylated state in IL-4-producing cells. An intactGATA binding site is essential for this function. Thus, analogousto the enhancer sequences identified in the intervening sequencesof Ig and TCR genes, this element has a dual function: it acts as atranscriptional enhancer in transient transfection assays and alsoplays a role in conferring developmentally regulated demethyl-ation to the IL-4 gene locus.
Materials and MethodsCell lines
CFTL-15 is a nontransformed murine mast cell line derived from fetal liverand is IL-3 dependent (42). These cells express low levels of IL-4 mRNAand protein constitutively and up-regulate this expression initiated in re-sponse to activation by cross-linking of the high affinity Fce receptor orcalcium mobilization by treatment with ionomycin (14). CFTL-15 cellswere cultured in complete RPMI 1640 containing 10% FCS in the presenceof 25% WEHI-3B cell supernatants as a source of IL-3. Bone marrow-derived mast cells were derived from bone marrow taken from the femursof 6- to 8-wk-old C57BL/6 mice and cultured under the same conditions asCFTL-15 mast cells except that recombinant murine stem cell factor (12.5ng/ml; R&D Systems, Minneapolis, MN) was added during the first 2 wkof culture. In some experiments, ABFTL-3, a growth factor-independentAbelson murine leukemia virus-transformed murine mast cell line that ex-presses IL-4 constitutively, was used (13). M12 B cells have been de-scribed previously (43).
DNase I footprinting
In vitro DNA footprinting was conducted as described (44). Briefly, 10mgof crude mast cell nuclear extracts were incubated with 0.5 ng (10,000–15,000 cpm) of a32P-labeled DNA sequence corresponding to base pairs353–554 of the 683-bpBglII fragment in the second intron of the IL-4gene. Five nanograms of DNase I (Life Technologies) was added to thereaction, and the digestion was stopped after 60 min. A control reactionwas conducted containing no nuclear extract. Following precipitation, thedigested DNA fragments were resolved on a 6% denaturing polyacryl-amide gel, and results were assessed by autoradiography. The final reactionconditions described above were determined by titrating concentrations ofDNase I, protein, and competitor DNA, as well as varying digestion times.The sequence used in these experiments (bp 353–554) was amplified byPCR and subcloned into pGEM-3 (Promega, Madison, WI). The probe wasprepared by linearizing pGEM-3 at the 39or 59 end of the probe, dephos-phorylating that end, and then cutting at the other end to release the frag-ment. The dephosphorylated end was labeled with [g-32P]ATP using T4kinase. Sequence homology searches were performed using the Transfacdatabase (Ref. 45; http://transfac.gbf.de/TRANSFAC/).
Reporter gene constructs
The murine IL-4 intron enhancer-reporter plasmid used in these studieswas constructed by deleting aBglII/PstI fragment corresponding to basepairs 1–254 of the originally defined 683-bp enhancer inp-chloramphen-icol acetyltransferase (CAT)4 promoter (Promega) (41). This subsequencecorresponds to base pairs 255–683.
Site-directed mutagenesis
Mutations were introduced into the IL-4 intron/pCAT promoter reporterconstruct by oligonucleotide-directed mutagenesis using the either theQuick Change system (Stratagene, La Jolla, CA) or the Sculptor M13 kit(Amersham, Arlington Heights, IL; indicated by an asterisk). All mutationswere verified by sequencing. The mutant forms of the oligonucleotides areas follows (altered nucleotides are underlined): m426, 415–446, 59-GGGAGGGGACTCGATCGACAGGCTGATAGTGC*; m475, 491–454,
59-GCTATTGATACACCTGCAGCAAGTCATGTGTTTGTCA; m482,498–469, 59-GCACAAAGCTACCTGCAGAGCATAGCCAAG; m491,478–511, 59-GCTGTATCAATAGCGATCGACATTTCAGTTCCTG*; m504,520–490, 59-CCATGAAAACAGGCCTGCAGATGCACAAAGC;m514 (STAT), 495–433, 59-GTGCATTTCAGTTCCTGTTGGCATGGAAACACACCACTG*; m523, 508–546, 59-CCTGTTTTCATGGAACGATCGCACTGAGAATGAAAGGCC*; m531, 519–548, 59-GGAAACACACCATGGCCAATGAAAGGCCCC; m538, 528–543, 59-CCACTGAGAAGAGTCGGCCCCAAAG; m545, 533–563, 59-GAGAATGAAAGGGTCGACCGTCTTGACTTAC; m553, 543–568, 59-GGCCCCAAAGCCCGAGCTTACCAGTG.Derivation of the reporter constructs containingthe GATA and PU.1 mutations have been previously described (41).
Transient transfections and CAT reporter gene assays
A total of 5 3 106 CFTL-15 mast cells were transfected by electroporation(Bio-Rad Gene Pulser at 425V and 400mF; Richmond, CA) using 25mgof reporter plasmid. After 24 h, cells were divided into two aliquots, andone aliquot was stimulated with 1mg/ml ionomycin (Sigma). The cellswere harvested 18 h poststimulation. Equal amounts of cell extracts wereassayed for CAT activity using the scintillation diffusion assay as previ-ously described (40). In some experiments, CAT data were normalized tothe relative expression of a cotransfected pSV-b-galactosidase expressionvector (Promega) to correct for variations in transfection efficiency.
EMSA
Nuclear extracts of CFTL-15 mast cells were prepared using the methoddescribed by Dignam et al. or Fiering et al. (46, 47) with high concentra-tions of protease inhibitors (Boehringer Mannheim, Indianapolis, IN). Pro-tein concentrations of the nuclear extracts were determined with the Bio-Rad protein assay kit. EMSAs were performed as previously described (38,48). Briefly, 5mg of crude nuclear extract was incubated with 0.1 ng (FP2,see below and Fig. 2A) or 1 ng (intron enhancer STAT (iSTAT), see belowand Fig. 2A)32P-labeled oligonucleotide for 1 h. Binding reactions with theFP2 probe were performed in a buffer containing 10 mM Tris (pH 7.5), 45mM KCl, 1 mM EDTA, 0.1% Triton X-100, 12.5% glycerol, 0.5 mM DTT,and 20mg/ml poly(dI-dC). Reactions with the iSTAT probe were con-ducted in 20 mM HEPES (pH 7.9), 40 mM KCl, 1 mM MgCl2, 0.1 mMEDTA, 4% Ficoll, 0.5 mM DTT, 1.2 mg/ml BSA, and 100mg/ml poly(dI-dC). The protein/DNA complexes were separated on a 5% polyacryl-amide gel in a buffer consisting of 25 mM Tris, 190 mM glycine, and 1 mMEDTA (for FP2 EMSAs) or 10 mM Tris, 10 mM boric acid, and 2 mMEDTA (for STAT EMSAs). For all competition experiments, a 100-foldmolar excess of unlabeled competitor oligonucleotide was used.
Oligonucleotide probe and competitor sequences
The following oligonucleotide probe and competitor sequences were used:intron enhancer STAT site probe (iSTAT), 59-TGTTTTCATGGAAACACA; FP2 region probe (FP2), 59-CATGACTTGGCTATGCTGTATCAATAGCTTTGTG; mutated intron enhancer STAT site (altered nucle-otides are underlined) (miSTAT), 59-TGTGGGCATGGAAACACA;b-casein IFN-g activation site element (GAS), 59-AGATTTCTAGGAATTCAAATC; IL-4 promoter STAT site –154 to –132 of the murine IL-4 59region (48) (IL-4 STAT), 59-TGATTTCACAGGAAAATT; consensus Pit1, 59-TGTCTTCCTGAATATGAATAAGAAATA; consensus Oct 1 (Oct),59-TGTCGAATGCAAATCACTAGAA; consensus AP-1, 59-CGCTTGATGAGTCAGCCGGAA; FP2 m475 (m475), 59-CATGACTTGCTGCAGGTGTATCAATAGCTTTGTG; FP2 m482 (m482), 59-CATGACTTGGCTATGCTCTGCAGGTAGCTTTGTG.
Western blot analyses
Western blot analyses were performed essentially as described (44). Pro-teins were separated on 10% SDS-PAGE gels and electro-blotted to nitro-cellulose membrane (Schleicher and Schuell, Keene, NH). The membraneswere blocked with 5% nonfat dry milk in 10 mM Tris (pH 8.0), 150 mMNaCl, and 0.5% Tween 20. Anti-Pit 1 and anti-NF-kB Abs (rabbit poly-clonal Ig) (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-STAT1,STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6 mouse mAbs(Zymed, San Francisco, CA) were used as the primary Ab at 1:1000 di-lution. HRP-conjugated goat anti-rabbit secondary Ab (Amersham) wasused at a 1:5000 dilution. The blots were visualized using the RenaissanceWestern blot chemiluminescence reagent (NEN, Boston, MA).
Oligonucleotide affinity precipitation
Large-scale (200–500 fold) EMSA binding reactions were conducted usingthe indicated 59biotinylated probe. The resulting protein/DNA complexeswere precipitated with streptavidin-agarose (Sigma, St. Louis, MO) and
4 Abbreviations used in this paper: CAT, chloramphenicol acetyltransferase; iSTAT,intron enhancer STAT; GAS, IFN-g activation site; GFP, green fluorescent protein.
3240 DUAL FUNCTIONS FOR AN IL-4 GENE INTRON REGULATORY ELEMENT
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

washed extensively with binding buffer. The specifically bound proteinswere eluted with SDS-PAGE loading dye and separated on a 10% SDS-PAGE gel. The presence of specific proteins was visualized by Westernblot analysis as described above.
Stable cell transfections
Two versions of an IL-4 genomic construct, with (gIL-4) and without(gDIL-4) the intron enhancer sequences, were stably transfected intoCFTL-15 mast cells and M12 B cells together with pEGFP-N1, a greenfluorescent protein (GFP)/neomycin-resistance gene expression vector(Clontech, Palo Alto, CA) at a 10:1 ratio using a Bio-Rad Gene Pulser at425 V and 400mF. The wild-type construct contains the complete IL-4genomic sequence (gIL-4) from 797 bp 59of the transcriptional start site to;2000 bp 39of the fourth exon. It was made by ligating theHindIIIfragment of the IL-4 genomic clone, pMIL-5 (49), which contains exons I,II, and III, with a HindIII fragment from pMIL-1 (49) into pUC18. Theenhancerless version of this construct was assembled from PCR-generatedfragments that lack the 683-bpBglII intron enhancer element. Mutations inthe GATA, ets, FP2 (m482), and STAT sites were made in the context ofpMIL-5 (48), a construct containing exons I, II, and III using the QuikChange mutagenesis system (Stratagene). After transfection, the cells werecultured in medium containing 400–600mg/ml G418 for at least 4 wk.Cells were cloned by limiting dilution. Integration of the transfected plas-mids was monitored by flow cytometry and by PCR using genomic DNAas a template and vector- and IL-4 exon-specific primers.
Determination of the methylation status of the IL-4 gene locus
Genomic DNA was isolated from stably transfected and untransfected cellswith DNAzol (Life Technologies, Rockville, MD) using the manufactur-er’s suggested protocol. DNA was digested withHindIII followed by di-gestion with eitherMspI (methylation insensitive) orHpaII (methylationsensitive) (Life Technologies). The digested DNA was separated on a 1%agarose-TBE gel and transferred to nitrocellulose membrane by the South-ern blot protocol as described (44). The blot was probed with a32P-radio-labeledStuI/EcoRI fragment from the second intron of IL-4 (see Fig. 5A).
ResultsIn vitro footprinting reveals three regions of protein bindingwithin the IL-4 intron enhancer
Previous analyses of the sequences that comprise the IL-4 geneintron enhancer demonstrated that at least two elements, corre-sponding to consensus PU.1 (ets) and GATA binding sites, con-tribute to the enhancer’s activity (41). Because the simultaneousmutation of both these sites does not completely eliminate en-hancer activity, it is likely that additional regions are involved. Asa first step toward defining such potential sites, in vitro footprintingexperiments were performed using mast cell nuclear extracts and aDNA probe comprising a 428-bp intron sequence. This probe (basepairs 255–683 of the 683-bpBglII fragment, see Fig. 1A) corre-sponds to a sequence that confers full enhancer activity in reporterconstruct assays (41). These assays revealed protein binding tothree distinct regions, termed FP1, FP2, and FP3 (Fig. 1,B–D).FP1 encompasses a previously defined SP-1 site and the GATAbinding site, while FP3 is adjacent to the PU.1 site (Fig. 2A).However, the sequences within FP2 and FP3 have not been pre-viously analyzed for regulatory activity.
Additional sequences contribute to IL-4 enhancer activity
To examine the ability of sequences within FP1, FP2, and FP3 toconfer enhancer activity, mutations were introduced into se-quences that span the three footprinted regions of the intron se-quence within the context of a CAT reporter construct (Fig. 2A).Transient transfection assays were performed in CFTL-15 mastcells to assess the consequence of these mutations on enhanceractivity. As shown in Fig. 2B, the mutations designated m482,
FIGURE 1. In vitro DNase I footprinting revealsthree regions of protein binding within the intronenhancer.A, Schematic of the IL-4 gene indicatingthe 683-bpBglII (B) fragment in the second intronthat contains the regulatory element. HS denotes theposition of the mast cell-specific DNase I hypersen-sitive site that first indicated the presence of a reg-ulatory element within the second intron of the IL-4gene (40). Numbering of the nucleotides within thisfragment is based on designating the first base of theBglII site as 1.B–D, In vitro DNase I footprinting ofthe IL-4 gene intron enhancer region. Mast cell nu-clear extracts were incubated with PCR-generated32P-end-labeled DNA fragments corresponding tobase pairs 353–554 of theBglII enhancer fragment.The protein-DNA complexes were treated withDNase I, and fragments were resolved on a 6% de-naturing polyacrylamide gel.B andC represent twodifferent experiments with the probe labeled at the 59end and the gel run for different lengths of time.D shows an experiment with the probe labeled fromthe 39end, and arrows designate hypersensitive re-gions. The three regions of protein binding (foot-prints) are labeled FP1, FP2, and FP3. Ø indicateslanes in which no protein was added. A-T lanes rep-resent dideoxy sequencing reactions of the intronfragment that are included as a sequence reference.
3241The Journal of Immunology
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

within FP2, and m514, within FP3, had the most profound effecton enhancer activity. Each mutation alone caused an 80% reduc-tion in CAT activity relative to the wild-type levels. A transcrip-tion factor binding site homology search using the Transfac data-base (45) revealed that the m482 mutation alters a site resemblinga consensus Pit 1 binding site. The mutation m514 disrupts a con-sensus STAT binding site (50, 51).
The site defined by m482 within FP2 forms specific DNA-proteincomplexes in mast cells
Specific DNA-protein interactions at regions defined by footprintanalysis were first assessed by EMSA using an oligonucleotideprobe corresponding to sequences in the FP2 region. A single com-plex forms using both unstimulated (data not shown) and stimu-lated CFTL15 nuclear extracts (Fig. 3A). Competition experimentswith mutant forms of unlabeled FP2 demonstrate that the se-
quences defined by m482, but not m475, contribute to mast cellprotein binding.
A protein antigenically related to Pit 1 is expressed in mastcells
Based on computer analyses (45), m482 disrupts a site resemblinga Pit 1 binding site. Pit 1 was originally described as a pituitary-specific protein belonging to the POU domain family of transcrip-tion factors (52). Of potential interest is the finding that Pit 1 spe-cifically interacts with another IL-4 intron binding protein,GATA-2, to regulate development of distinct pituitary cell types(53). Its expression in mast cells has not been previously evalu-ated. Western blot analysis of CFTL-15 and bone marrow-derivedmast cell nuclear extracts was performed using commerciallyavailable anti-Pit 1 Abs. The results indicate that a protein of 38
FIGURE 2. Mutations in the Pit 1-like and STAT binding sites within the intron element substantially reduce enhancer activity.A, Mutations introducedinto the footprinted regions of the IL-4 gene intron enhancer within the context of a CAT IL-4 gene intron enhancer-reporter construct (base pairs 255–683).The numbering of mutations corresponds to the first disrupted base pair for each mutation in the 683-bpBglII fragment within the second intron. Mutationsare indicated by gray boxes, consensus transcription factor binding sites are designated with light brackets, and footprinted regions (FP1–3) are indicatedwith heavy brackets. The sequences corresponding to the probe used for STAT binding analyses are also shown (iSTAT).B, CFTL-15 mast cells weretransfected with equimolar amounts of the indicated IL-4 intron enhancer constructs. After an overnight incubation, cultures were stimulated with iono-mycin, harvested 48 h posttransfection, and assayed for CAT activity as previously described (40). The data are compiled from at least four independenttransfection experiments in which the wild-type control data was considered 100% activity. The activity of other plasmids is expressed relative to thewild-type activity. Error bars represent SD. In some experiments, the IL-4 reporter constructs were cotransfected with ab-galactosidase expression construct(85) allowing normalization of the CAT activity to the transfection efficiency of each electroporation as previously described (40).
3242 DUAL FUNCTIONS FOR AN IL-4 GENE INTRON REGULATORY ELEMENT
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

kDa, the reported molecular mass of Pit 1, is expressed constitu-tively in both mast cell lines but not in M12 B cells (Fig. 3B) or Tcells (data not shown).
Pit 1 does not associate with the intron enhancer element
The apparent constitutive and selective expression of this tran-scription factor in mast cells, together with protein binding to thePit 1-like sequence in the FP2 region, suggests that Pit 1 maycontribute to the activity of this element. To examine the possibleinvolvement of Pit 1 in the FP2 complex, several experiments wereperformed. A Pit 1 consensus site oligonucleotide was used as aprobe in EMSA experiments. Proteins in mast cell nuclear extractsform specific complexes with oligonucleotides containing the Pit 1binding site (Fig. 3C). Of note, these complexes migrate with adistinct mobility compared with the FP2 complexes. Unlabeledoligonucleotides corresponding to the Pit 1 sequence or to an Oct1 binding sequence compete for protein binding in competitionexperiments. However, protein binding to this sequence is notcompeted by a 100-fold molar excess of FP2 oligonucleotides.Likewise, competitor Pit 1 and Oct 1 consensus oligonucleotidesdo not disrupt FP2 DNA-protein complexes (data not shown). Nei-ther the Pit 1 nor the FP2 protein-DNA complex were supershiftedby the commercially available Pit 1 antisera (data not shown).
Oligonucleotide affinity precipitation assays were also per-formed. Large-scale binding reactions were performed using mastcell nuclear extracts and a biotinylated FP2 probe. DNA-proteincomplexes were precipitated with streptavidin-agarose beads. Af-ter extensive washing, specifically bound proteins were eluted andsubjected to Western blot analysis using Pit 1 antisera. As shownin Fig. 3D, there is no evidence that anti-Pit 1-reactive proteins arespecifically eluted from the FP2 probe. These data indicate thatdespite the strong constitutive expression of this protein in mastcells, Pit 1 does not associate with this IL-4 gene regulatory site.
We speculate that a related, “Pit 1-like” factor, perhaps anothermember of the POU domain family, binds to this site in vivo.
STAT proteins bind the intron STAT element in mast cells
The functional region defined by m514 flanks the previously de-fined PU.1 binding site and directly disrupts a STAT consensussite (TTTCATGGAA) that could potentially bind either STAT6(TTN6AA) or other STAT family members (TTN5AA) (50, 51).We first asked if this site could function as a bona fide STAT6binding site. Extracts from IL-4-stimulated B cells were used inEMSA binding reactions with probes derived from the FP3 region,designated iSTAT (Fig. 2A). Two complexes are observed, both ofwhich are competed by “self” (lane 2) but not by the unrelatedAP-1 oligonucleotides (lane 4). A conventional STAT6 oligonu-cleotide corresponding to sequences present in the murine IL-4promoter competes for proteins binding to the slower mobilitycomplex (lane 3). STAT6-specific antisera can also supershift thiscomplex (lane 5). These data indicate that STAT6 as well as otherfactors can bind to the iSTAT binding site. Similar experimentswere conducted with extracts from unstimulated and IL-4- or iono-mycin-stimulated mast cells. As shown in Fig. 4B, IL-4 induces theassociation of a single complex (lane 2). This complex contains amast cell-specific isoform of STAT6 that lacks the C-terminalamino acids and fails to react with antisera that can supershiftSTAT6 in B and T cells (48). EMSA experiments with ionomycin-stimulated mast cell extracts reveal that several activation-depen-dent DNA-protein complexes form with the iSTAT probe (lane 3).Notably, the number and mobility of complexes formed with iono-mycin-activated mast cell extracts are distinct when comparedwith the IL-4-induced complex.
The specificity of the ionomycin-inducible protein-DNA inter-actions is demonstrated in cold competition experiments (Fig. 4C).Competition with a 100-fold molar excess of unlabeled iSTAT
FIGURE 3. Sequences defined by the m482 muta-tion are critical for protein binding in the FP2 region.A, EMSAs were performed with nuclear extracts iso-lated from ionomycin-stimulated CFTL-15 mast cellsand the FP2 DNA probe. Where indicated, a 100-foldmolar excess of unlabeled wild-type (self), mutated(m482 and m475), or unrelated (UR) competitor (anAP-1 consensus site) oligonucleotide was added to thestandard binding reaction mixture.B, Pit-1 expressionin mast cells. One hundred micrograms of crude nu-clear extract from unstimulated M12 B cells, CFTL-15, or bone marrow-derived mast cells (BMMC) wasanalyzed by Western blot techniques usinga-Pit-1 an-tisera.C, The binding of mast cell nuclear proteins toa consensus Pit 1 binding site was assayed usingEMSA analysis. Where indicated, a 100-fold molarexcess of unlabeled wild-type (self), mutated (m482and m475), Oct binding site, or unrelated (UR) com-petitor (an AP-1 consensus site) oligonucleotide wasadded to the standard binding reaction mixture.D, Analysis of Pit 1 binding to FP2 sequences usingDNA oligonucleotide affinity precipitation. Biotinyl-ated oligonucleotides corresponding to the FP2 regionof protein binding were incubated with crudeCFTL-15 mast cell extract under the same bindingconditions used for the EMSA experiments. After pre-cipitation with streptavidin-agarose beads, the boundproteins were analyzed by Western blot with anti-Pit-1 antisera. Crude extracts and unbound fractionswere also analyzed.
3243The Journal of Immunology
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
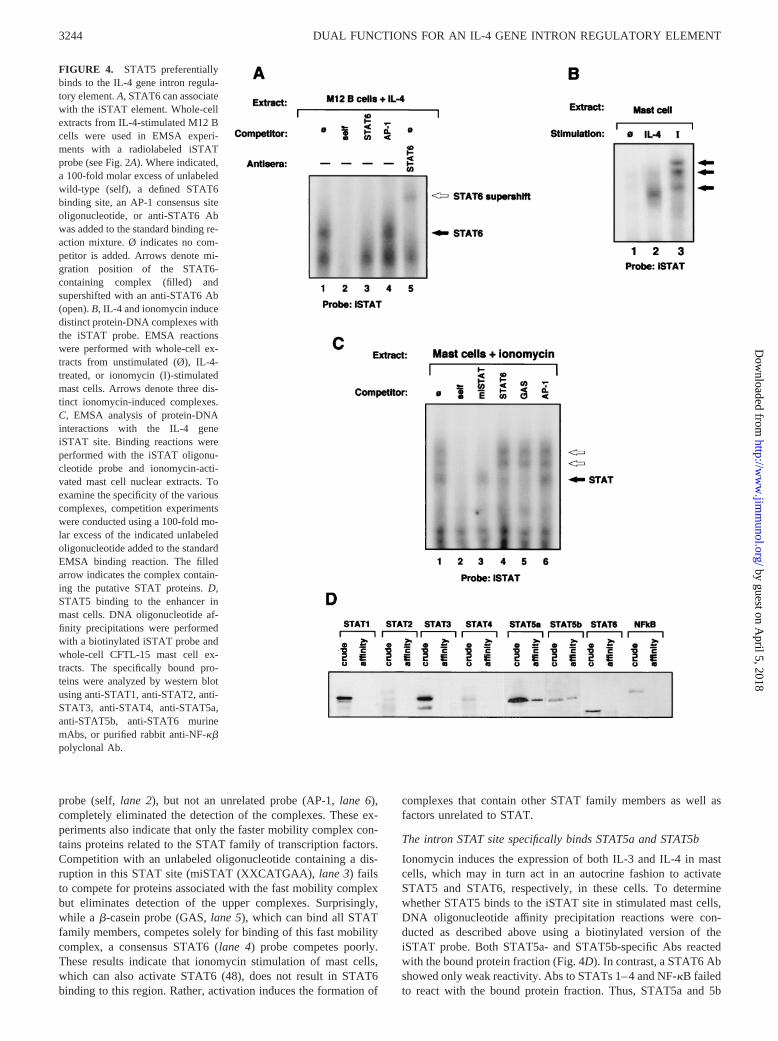
probe (self,lane 2), but not an unrelated probe (AP-1,lane 6),completely eliminated the detection of the complexes. These ex-periments also indicate that only the faster mobility complex con-tains proteins related to the STAT family of transcription factors.Competition with an unlabeled oligonucleotide containing a dis-ruption in this STAT site (miSTAT (XXCATGAA),lane 3) failsto compete for proteins associated with the fast mobility complexbut eliminates detection of the upper complexes. Surprisingly,while a b-casein probe (GAS,lane 5), which can bind all STATfamily members, competes solely for binding of this fast mobilitycomplex, a consensus STAT6 (lane 4) probe competes poorly.These results indicate that ionomycin stimulation of mast cells,which can also activate STAT6 (48), does not result in STAT6binding to this region. Rather, activation induces the formation of
complexes that contain other STAT family members as well asfactors unrelated to STAT.
The intron STAT site specifically binds STAT5a and STAT5b
Ionomycin induces the expression of both IL-3 and IL-4 in mastcells, which may in turn act in an autocrine fashion to activateSTAT5 and STAT6, respectively, in these cells. To determinewhether STAT5 binds to the iSTAT site in stimulated mast cells,DNA oligonucleotide affinity precipitation reactions were con-ducted as described above using a biotinylated version of theiSTAT probe. Both STAT5a- and STAT5b-specific Abs reactedwith the bound protein fraction (Fig. 4D). In contrast, a STAT6 Abshowed only weak reactivity. Abs to STATs 1–4 and NF-kB failedto react with the bound protein fraction. Thus, STAT5a and 5b
FIGURE 4. STAT5 preferentiallybinds to the IL-4 gene intron regula-tory element.A, STAT6 can associatewith the iSTAT element. Whole-cellextracts from IL-4-stimulated M12 Bcells were used in EMSA experi-ments with a radiolabeled iSTATprobe (see Fig. 2A). Where indicated,a 100-fold molar excess of unlabeledwild-type (self), a defined STAT6binding site, an AP-1 consensus siteoligonucleotide, or anti-STAT6 Abwas added to the standard binding re-action mixture. Ø indicates no com-petitor is added. Arrows denote mi-gration position of the STAT6-containing complex (filled) andsupershifted with an anti-STAT6 Ab(open).B, IL-4 and ionomycin inducedistinct protein-DNA complexes withthe iSTAT probe. EMSA reactionswere performed with whole-cell ex-tracts from unstimulated (Ø), IL-4-treated, or ionomycin (I)-stimulatedmast cells. Arrows denote three dis-tinct ionomycin-induced complexes.C, EMSA analysis of protein-DNAinteractions with the IL-4 geneiSTAT site. Binding reactions wereperformed with the iSTAT oligonu-cleotide probe and ionomycin-acti-vated mast cell nuclear extracts. Toexamine the specificity of the variouscomplexes, competition experimentswere conducted using a 100-fold mo-lar excess of the indicated unlabeledoligonucleotide added to the standardEMSA binding reaction. The filledarrow indicates the complex contain-ing the putative STAT proteins.D,STAT5 binding to the enhancer inmast cells. DNA oligonucleotide af-finity precipitations were performedwith a biotinylated iSTAT probe andwhole-cell CFTL-15 mast cell ex-tracts. The specifically bound pro-teins were analyzed by western blotusing anti-STAT1, anti-STAT2, anti-STAT3, anti-STAT4, anti-STAT5a,anti-STAT5b, anti-STAT6 murinemAbs, or purified rabbit anti-NF-kbpolyclonal Ab.
3244 DUAL FUNCTIONS FOR AN IL-4 GENE INTRON REGULATORY ELEMENT
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

constitute the major components of the faster migrating induciblemast cell iSTAT complex.
The intron regulatory element influences the methylation state ofthe IL-4 gene in murine mast cells
Several cell type-restricted regulatory elements have been identi-fied in the intervening sequences of both the Ig and TCR genes (forreview see Refs. 54 and 55). Originally defined as conventionalenhancer sequences in reporter assays, they appear to have an ad-ditional function in vivo. They have a major influence on chroma-tin accessibility of these loci. For example, deletion of them or kIg enhancer element in the context of a stably transfected recom-bination substrate results in a loss of the ability to undergo recom-bination and methylation of the introduced sequences in appropri-ate cell types, indicative of an inaccessible chromatin configuration(56, 57).
Based on this paradigm, we asked whether the defined mast cellintron enhancer could also influence chromatin accessibility of theIL-4 gene. It is well established that methylated cytosines withinchromosomal DNA are associated with gene inactivation, whereasdemethylated regions correlate with transcriptionally active or in-
ducible genes (58, 59). However, until recently, the relationship ofDNA methylation state to the conventional markers of chromatinaccessibility such as DNase I hypersensitivity and local histoneacetylation has been less clear. The recent discovery of MeCp2, aprotein that binds methylated CpG dinucleotides and recruitsdeacetylase activity to specific chromosomal regions, provides adirect link between methylation of DNA and histone deacetylationand, therefore, chromatin accessibility (60, 61). Thus, we askedwhether the methylation state of integrated IL-4 gene contructs isinfluenced by the intron regulatory element. Two versions of anIL-4 genomic construct, with (gIL-4) and without (gDIL-4) theintron enhancer sequences (Fig. 5A), were stably transfected intoCFTL-15 mast cells and M12 B cells, a non-IL-4-producing cellline, along with a GFP/neomycin-resistance gene expression vec-tor. After at least 4 wk of selection in G418, genomic DNA wasisolated from each transfectant culture.
We first verified that the appropriate constructs were integratedin each cell population using PCR. Primers that hybridize topUC18 vector sequences 59of the IL-4 gene cloning site as well asto sequences just 39of the intron element were used to amplify thetransfected IL-4 gene sequences (Fig. 5A). As shown in Fig. 5B,
FIGURE 5. The intron enhancer region is required for the demethylation of the IL-4 locus in mast cells.A, Schematic of IL-4 genomic constructs usedin transfection experiments.HindIII sites are indicated by H. The location ofHpaII/MspI sites within the second intron are designated byu. Light bracketsindicate theStuI/EcoRI fragment used as a probe in Southern blot analysis. The approximate position of PCR primers used to verify the integration of thetransfected constructs is also shown by arrows.B, PCR using vector- and gene-specific primers (seeA) and genomic DNA isolated from M12 B cells andCFTL-15 mast cells that were transfected with the indicated plasmids. gIL-4 designates DNA isolated from cells transfected with the wild-type IL-4genomic construct; gDIL-4 designates DNA isolated from cells transfected with the enhancerless IL-4 genomic construct. Ø designates control cellstransfected with the GFP/neomycin vector alone.C, Analysis of the methylation status at the IL-4 gene locus in transfectants carrying stably integratedcopies of IL-4 genomic constructs with and without the intron enhancer. Genomic DNAs from M12 B cells and CFTL-15 mast cells were subjected toSouthern blot analysis. DNA was digested with eitherHindIII alone (2) (lanes 1,4, and7) or with HindIII followed by eitherMspI (M) (lanes 2,5, and8) or HpaII (H) (lanes 3,6, and9). Blots were probed with a32P-labeledStuI-EcoRI fragment from the second intron of IL-4 (seeA above). Ø designatescells that received the GFP/neomycin vector alone; gIL-4 designates DNA isolated from cells transfected with the wild-type IL-4 genomic construct; gDIL-4designates DNA isolated from cells transfected with the enhancerless IL-4 genomic construct. Arrow indicates position ofHpaII-insensitiveHindIIIfragment.
3245The Journal of Immunology
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

DNA from cells transfected with either the wild-type IL-4 con-struct or the deleted IL-4 construct but not those transfected withthe GFP/neomycin vector alone served as successful templates andyielded a PCR product of the expected size. These data confirmthat the wild-type and enhancerless IL-4 genomic constructs arestably integrated into both M12 B cells and CFTL-15 mast cells.
To assess the chromatin state, the methylation status of thetransfected sequences was then examined by Southern blot anal-ysis using a pair of enzymes whose recognition site is CCGG:HpaII, which is sensitive to methylated CpGs, andMspI, whichcuts irrespective of the methylation state. Our hypothesis predictsthat both the transfected wild-type and enhancerless genes will bemethylated in non-IL-4-producing cells, but only the deleted con-struct will be methylated in IL-4-producing mast cells. As ex-pected, both versions of the transfected IL-4 gene constructs aremethylated in M12 cells (Fig. 5C, lanes 6and 9, left), as is theendogenous gene, reflecting the inactivity of the IL-4 gene in Bcells. The wild-type construct is demethylated in bulk cultures oftransfected CFTL-15 mast cells (Fig. 5C,lane 6, right). In contrast,the integrated, enhancerless construct is methylated in IL-4-pro-ducing cells (as indicated by its resistance toHpaII digestion, Fig.5C, lane 9, right), despite the demethylated state of the endoge-nous IL-4 locus. We speculate that the relatively light intensity ofthe HpaII-resistant band reflects the fact that fewer cells in thebulk-transfected culture contain the integrated transgene, whereasall contain the endogenous IL-4 gene. Using a probe correspondingto 2800 to160 of the IL-4 gene with the same DNA samples, weconfirmed that demethylation of the two upstreamHpaII sitespresent in the transfected construct (at;749 and between intron Iand II, see Fig. 5A) is likewise dependent on the presence of theintron regulatory element (data not shown).
Individual clones were also isolated from mast cell transfectantsusing limiting dilution cloning techniques. The analyses of twoclones of each type of transfectant are presented in Fig. 6,A andB,and are representative of our analysis of at least four clones of eachtype. As shown in Fig. 6A, PCR analyses confirmed the appropri-ate IL-4 gene constructs are integrated into these clonal popula-tions. In addition, the methylation status of the transfected con-structs was identical with that observed in the bulk CFTL-15 mastcell cultures (Fig. 6B). The integrated enhancerless construct isresistant toHpaII digestion (lanes 15and18) in gDIL-4-transfec-tant clones, indicating a failure to become demethylated. It is no-table that the intensity of theHpaII-resistant band is much strongerthan that observed in experiments with the bulk-transfected cells,which likely reflects the higher proportion of cells containing theintegrated transgene in these cloned lines. Taken together, theseresults strongly support the hypothesis that the intron sequenceshave an important role in regulating cell type-specific accessibilityof the IL-4 gene in murine mast cells by influencing the methyl-ation status of the locus.
Mutations in the intron GATA binding sites interfere with theability to acquire and/or maintain a demethylated locus
We next asked whether the functionally important sites defined inenhancer assays could contribute to the ability of the intron ele-ment to influence IL-4 gene methylation status. Southern blot anal-yses were performed as described above using DNA isolated fromcells stably transfected with IL-4 genomic constructs (see Fig. 7A)containing mutations in the GATA, ets, and STAT sites as well asthe site defined by m482. PCRs with vector- and gene-specificprimers were conducted to confirm integration of the constructs intransfected cells (Fig. 7B). As shown in Fig. 7C, DNA from themGATA transfectants was unable to achieve a completely demeth-ylated state as shown by its partial resistance toHpaII (lane 9).
The same results were obtained in several independent experi-ments even under conditions in which the DNA samples weresubjected to multiple treatments of excess enzyme to assure thatcomplete digestion occurred. The STAT and ets site mutations, butnot m482, also reproducibly resulted in decreased demethylation atsome sites within the intron region of the transgene; however, theeffect was less marked than that observed in the mutant GATAtransfectants. Thus, association of GATA, and perhaps to a lesserextent STAT and PU.1, with an element in the second intron of themouse IL-4 gene influences its methylation state.
DiscussionWhile most previous studies have focused on thecis- andtrans-acting factors that initiate IL-4 gene transcription in activated cells,there is clearly an additional level of regulation. Studies in T cells(62, 63) as well as those in mast cells by our laboratory (S. K. Leeand M. A. Brown, manuscript in preparation) have established arole for developmentally regulated chromatin accessibility in IL-4gene expression. DNase I hypersensitivity and demethylationwithin the IL-4 gene locus are observed at an early stage in thecommitment to a Th2 or mast cell lineage. Importantly, these hall-marks of “open” chromatin are evident in both cell types, inde-pendent of active IL-4 transcription. These observations demon-strate that the acquisition of an IL-4-producing phenotype occursin two steps: 1) developmental signals confer locus opening; and2) activation signals in differentiated cells promote transcriptionalactivation of the IL-4 gene.
FIGURE 6. Clonal populations of mast cell transfectants exhibit thesame dependence on the intron regulatory element for demethylation.A, PCR using genomic DNA from cloned CFTL-15 mast cell transfectants.Primers that hybridize with the pUC18 vector in region 39of the intronenhancer sequence are shown in Fig. 5A. Ø designates control cells trans-fected with the GFP/neomycin vector alone. gIL-4 designates DNA iso-lated from cells transfected with the wild type IL-4 genomic construct;gDIL-4 designates DNA isolated from cells transfected with the enhanc-erless IL-4 genomic construct.B, Analysis of the methylation status of theIL-4 gene locus in two different cloned CFTL-15 mast cell transfectantscarrying stably integrated copies of IL-4 genomic constructs with and with-out the intron enhancer. Genomic DNA from cloned cells was subjected toSouthern blot analysis as described above (Fig. 5C). M denotes samplesdigested withHindIII plus MspI, H denotes samples digested withHindIIIplus HpaII, and – denotes samples digested withHindIII alone. Arrowindicates position ofHpaII-insensitiveHindIII fragment.
3246 DUAL FUNCTIONS FOR AN IL-4 GENE INTRON REGULATORY ELEMENT
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

In this study, we provide evidence that the regulatory elementdefined within the second intron of the murine IL-4 gene plays arole in both these processes in mast cells. First, it functions as aposition- and orientation-independent transcriptional enhancer inconventional reporter gene assays and can act with both the nativeIL-4 gene promoter as well as heterologous promoters (40). En-hancer activity is dependent on sequences forming binding sitesfor GATA-1 and GATA-2, PU.1, STAT5, and a Pit 1-related fac-tor. Second, this element appears to play a role in mediating ormaintaining locus opening. Both the endogenous IL-4 gene and astably transfected wild-type IL-4 chromosomal gene construct areappropriately demethylated in IL-4-producing mast cells, indica-tive of an open locus. However, in IL-4-producing mast cells, themethylated state of a transfected construct lacking the intron reg-ulatory element strongly suggests that these sequences are neces-
sary to achieve and/or maintain an accessible chromatin configu-ration. The dependence on cell type-specific factors for this effectis demonstrated by the inability of either construct to exhibit ademethylated state in B cells.
This proposed “dual-function” element located within an IL-4gene intron has many parallels to elements that influence devel-opmentally regulated TCR and Ig gene expression in T and Blymphocytes (54, 55). Several studies of the TCRa, b, and dgenes, as well as Ig heavy and light chain genes, demonstrate thatsequences defined as enhancers in transient transfection assaysalso influence processes such as VDJ and VJ recombination aswell as somatic mutation of the Ig genes. It is likely that theseelements act to influence chromatin accessibility that allows therecombination machinery access to the region. In support of thisidea, in vivo and in vitro studies of thek- andm-chain enhancersconfirm their role in demethylation of the Ig locus. For example, atransfectedm-chain gene containing the intronm heavy chain en-hancer is hypomethylated in pre-B cells, correlating with expres-sion of the heavy chain gene. Deletion of them enhancer leads totranscriptional inactivation and de novo methylation (64). Similartypes of analyses have shown that thek light chain enhancer alsohas a role in inducing demethylation of the locus in a lineage- anddevelopmental stage-specific manner (65, 66). Genetic deletion ofthe TCRa andb enhancers also negatively affects recombination(67–69). The apparent commonality of mechanisms that operateon such diverse gene systems such as the Ig, TCR, and IL-4 genesto regulate accessibility suggests that similar regulatory events oc-cur at other gene loci. Indeed, these data may necessitate the de-velopment of a new paradigm for the in vivo function of manyother gene regulatory elements, first defined as transcriptional en-hancers, located in introns or elsewhere.
Our analysis of specific sites and proteins that associate with theintron element and mediate its activity also supports the idea thatthis element has dual function. Although little is known about themechanisms through which enhancers act to influence locus open-ing, it is striking that many of the factors that associate with thefunctional elements of the IL-4 enhancer have been implicated ininfluencing chromatin accessibility at loci encoding other celltype-specific genes. These include GATA-1, PU.1, STAT5, andPOU domain family factors. For example, it has been shown thatGATA-1, expressed in myeloid lineage cells, is able to bind toGATA binding sites in nucleosomal DNA and cause extensive andcooperative breakage of the DNA/histone contacts (70). Thus, thisfactor likely plays an important role in initiating locus-openingevents. PU.1, a factor whose expression is limited to B cells, mac-rophages, neutrophils, and mast cells (41, 71–73), can bind to plas-mids containing them enhancer (71, 74). Binding by PU.1 does notalter the nucleosomal array that assembles around the transfectedplasmid, but does increase the restriction enzyme accessibilitywithin the enhancer (74). In addition, ectopically expressed PU.1cooperates with other ets family members to induce Im transcrip-tion, a marker of locus opening that precedes VDJ recombination,and to increase chromatin sensitivity to restriction enzyme diges-tion in stably transfected NIH 3T3 and pro-T cells. Finally, it hasbeen established that STAT family members can interact with tran-scriptional coactivators such as CBP/p300 and PCAF, proteins thatexpress intrinsic histone acetyltransferase activity (75–79). Localhistone acetylation is also associated with gene transcriptional ac-tivity and is another marker of accessible chromatin (80, 81). It hasbeen proposed that through their interaction with these coactiva-tors, transcription factors direct this histone acetyltransferase ac-tivity to specific gene regulatory regions. STAT5, in particular,activated through IL-7 signaling, can interact with p300/CBPthrough an adapter protein termed Nmi (77) and has been shown to
FIGURE 7. Mutations within intron transcription factor binding sitesalso disrupt the ability of the IL-4 gene locus to maintain a demethylatedstate.A, Schematic of mutant IL-4 genomic constructs used in transfectionexperiments.HindIII sites are indicated by H. The location ofHpaII/MspIsites within the second intron are designated I as is theStuI/EcoRI fragmentused as a probe in Southern blot analysis. The approximate position of PCRprimers used to detect the integration of the transfected constructs is alsoshown by arrows.B, PCR using vector- and gene-specific primers (seeA)and genomic DNA isolated from CFTL-15 mast cells that were transfectedwith the IL-4 genomic constructs containing the indicated mutations withinthe intron element. WT designates DNA isolated from cells transfectedwith the wild-type IL-4 genomic construct.C, Analysis of the methylationstatus at the IL-4 gene locus in transfectants carrying stably integratedcopies of IL-4 genomic constructs with mutations in intron transcriptionfactor binding sites. DNAs from transfected CFTL-15 mast cells were sub-jected to Southern blot analysis. DNA was digested with eitherHindIIIalone (2) (lanes 1,4, and7) or with HindIII followed by eitherMspI (M)(lanes 2,5, and8) or HpaII (H) (lanes 3,6, and9). Blots were probed witha 32P-labeledStuI-EcoRI fragment from the second intron of IL-4 (seeAabove). WT designates DNA from cells that were transfected with thewild-type IL-4 gene construct.
3247The Journal of Immunology
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

regulate the accessibility of the TCRg locus (82, 83). The estab-lished roles for these IL-4 intron binding factors in chromatinopening is consistent with the idea that they play similar roles inmodifying IL-4 gene chromatin in IL-4-producing cells.
It is still unclear whether the intron element mediates mast cell-specific effects on IL-4 gene expression. GATA-1/2, Pit 1, andPU.1 are not expressed in T cells, and we have been unable todemonstrate conventional enhancer activity in Th2-derived celllines (J. Hural and M. A. Brown, manuscript in preparation), sup-porting the idea that this element acts as a mast cell-specific en-hancer. However, this intron region is also demethylated duringthe course of T cell differentiation (63), and Agarwal and Raorecently demonstrated that the appearance of intron DNase I hy-persensitive sites are associated with the development of an IL-4-producing phenotype in Th2 cells (62). Of note, these sites appearto be in close proximity to, but spatially distinct from, those iden-tified in mast cells. These findings raise the possibility that despiteits lack of enhancer activity, the second intron is essential for reg-ulating IL-4 transcription in both cell lineages. In T cells, it mayonly be involved in processes that mediate chromatin remodeling.Just as there are distinct cell type-specific factors that regulatetranscriptional activation of the IL-4 gene, different developmentalpathways leading to mast cell vs T cell development may neces-sitate the use of distinct subsets of factors and sites for locus open-ing. Related, but distinct factors that impact Th2 development,such as GATA-3 and STAT6 (28, 34), may act on this element inT cells and serve such a function. This possibility is currentlybeing examined.
Many questions remain regarding the relative in vivo contribu-tion of this enhancer element and its associatedtrans-acting factorsin directing transcriptional enhancement vs induction and/or main-tenance of locus accessibility. Because these elements are gener-ally defined in transient transfection assays where the native chro-mosomal context of a sequence is not preserved, we cannotexclude the possibility that it does not act in direct transcriptionalenhancement in vivo. The observed enhancer activity in such as-says may merely be a marker of its influence on transcription butis unrelated to its physiologic role. However, if the intron doesmediate both functions in mast cells, determining whether thesame set of transcription factors mediates both effects will be ofgreat interest. The range of influence that the intron regulatoryelement exerts on the accessibility of the entire IL-4 gene locus isalso undefined. It will be important to determine whether the intronelement exerts only a local influence or also regulates the acces-sibility of distal sequences, such as those comprising the IL-13/IL-4 intergenic region. This region is postulated to play a role incoordinately regulating the expression of cytokine genes that arelinked on chromosome 11 in mice and chromosome 5 in humans(84). Future studies using transgenic mice as well as those withtargeted deletions in genes encoding the implicated factors willaddress these issues.
AcknowledgmentsWe thank Drs. Jerry Boss, Melanie Sherman, and Virginia Secor, as wellas Susan Lee, for critical reading of the manuscript and helpful discussions.We are also grateful to Doris Powell who contributed to the generation ofthe IL-4 genomic constructs and analysis of the transfectants, TammyNachman who provided important assistance with cell culture and cloning,and Janice Moser who assisted with flow cytometric analysis oftransfectants.
References1. Brown, M. A., and J. Hural. 1997. Functions of IL-4 and control of its expression.
Crit. Rev. Immunol. 17:1.
2. Paul, W. E. 1991. Interleukin 4: a prototypic immunoregulatory cytokine.Blood77:1859.
3. Sadick, M. D., F. P. Heinzel, B. J. Holaday, R. T. Pu, R. S. Dawkins, andR. M. Locksley. 1990. Cure of murine leishmaniasis with anti-interleukin-4monoclonal antibody: evidence for a T cell-dependent, interferong-independentmechanism.J. Exp. Med. 171:115.
4. Locksley, R. M., F. P. Heinzel, M. D. Sadick, B. J. Holaday, and K. D. Gardner,Jr. 1987. Murine cutaneous leishmaniasis: susceptibility correlates with differen-tial expansion of helper T cell subsets.Ann. Inst. Pasteur Immunol. 138:744.
5. Launois, P., K. G. Swihart, G. Milon, and J. A. Louis. 1997. Early production ofIL-4 in susceptible mice infected withLeishmania majorrapidly induces IL-12unresponsiveness.J. Immunol. 158:3317.
6. Chatelain, R., K. Varkila, and R. L. Coffman. 1992. IL-4 induces a Th2 responsein Leishmania major-infected mice.J. Immunol. 148:1182.
7. Pearce, E., and E. Sabin. 1995. Early IL-4 production by non-CD41 cells at thesite of antigen deposition predicts the development of a T helper 2 cell responseto Schistosoma mansonieggs.J. Immunol. 155:4844.
8. Scott, P., E. Pearce, W. Cheer, R. I. Coffman, and A. Sher. 1989. Role of cyto-kines and CD41 T cell subsets in the regulation of parasite immune disease.Immunol. Rev. 112:161.
9. Ricci, M. 1994. IL-4: a key cytokine in atopy.Clin. Exp. Allergy. 24:801.10. Schattner, A. 1994. Lymphokines in autoimmunity—a critical review.Clin. Im-
munol. Immunopath. 70:177.11. Paul, W. E., and R. A. Seder. 1994. Lymphocyte responses and cytokines.Cell
76:241.12. Ferrick, D. A., M. D. Schrenzel, T. Mulvania, B. Hsieh, W. G. Ferlin, and
H. Lepper. 1995. Differential production of interferon-g and interleukin-4 in re-sponse to Th1- and Th2-stimulating pathogens bygd T cells in vivo. Nature373:255.
13. Brown, M. A., J. H. Pierce, C. J. Watson, J. Falco, J. N. Ihle, and W. E. Paul.1987. B cell stimulatory factor-1/interleukin-4 mRNA is expressed by normal andtransformed mast cell lines.Cell 50:809.
14. Plaut, M., J. H. Pierce, C. J. Watson, J. Hanley-Hyde, R. P. Nordon, andW. E. Paul. 1989. Mast cell lines produce lymphokines in response to cross-linkage of FceRI or to calcium ionophores.Nature 339:64.
15. Bradding, P., I. H. Feather, P. H. Howarth, R. Mueller, J. A. Roberts, K. Britten,J. P. A. Bews, T. C. Hunt, Y. Okayama, C. H. Heusser, et al. 1992. Interleukin-4is localized to and released by human mast cells.J. Exp. Med. 176:1381.
16. Abraham, S. N., and R. Malavia. 1997. Mast cells in infection and immunity.Infect. Immun. 65:3501.
17. Galli, S., and B. Wershil. 1996. The two faces of the mast cell.Nature 381:21.18. Wang, H.-W., N. N. Tedia, A. R. Lloyd, D. Wakefield, and H. P. McNeil. 1998.
Mast cell activation and migration to lymph nodes during induction of an immuneresponse in mice.J. Clin. Invest. 102:1617.
19. Mueller, D. L., M. K. Jenkins, and R. H. Schwartz. 1989. Clonal expansionversus functional clonal inactivation: a costimulatory signalling pathway deter-mines the outcome of T cell antigen receptor occupancy.Annu. Rev. Immunol.7:445.
20. Bluestone, J. 1995. New perspectives of CD28–B7 mediated T cell costimula-tion. Immunity 2:555.
21. Ravetch, J. V., and J. P. Kinet. 1991. Fc receptors.Annu. Rev. Immunol. 9:457.22. Beaven, M. A., and H. Metzger. 1993. Signal transduction by Fc receptors: the
FceRI case.Immunol. Today 14:222.23. Malavija, R., T. Ikeda, E. Ross, and S. Abraham. 1996. Mast cell modulation of
neutrophil influx and bacterial clearance at sites of infection through TNF-a.Nature 381:77.
24. Bidri, M., I. Vouldoukis, M. D. Mossalayi, P. Debre, J.-J. Guillosson, D. Mazier,and M. Arock. 1997. Evidence for direct interaction between mast cells andLeishmaniaparasites.Parasite Immunol. 19:475.
25. Bischoff, S. C., T. Brunner, A. L. de Weck, and C. A. Dahinden. 1990. Interleukin5 modifies histamine release and leukotriene generation by human mast cells inresponse to diverse antagonists.J. Exp. Med. 172:1577.
26. Bischoff, S. C., and C. A. Dahinden. 1992. Effect of nerve growth factor on therelease of inflammatory mediators by mature human mast cells.Blood 79:2662.
27. Ansel, J., J. R. Brown, D. G. Payan, and M. A. Brown. 1993. Substance Pselectively activates TNF-a gene expression in murine mast cells.J. Immunol.150:4478.
28. Zheng, W., and R. A. Flavell. 1997. The transcription factor GATA-3 is neces-sary and sufficient for Th2 cytokine gene expression in CD4 T cells.Cell 89:587.
29. Ho, I.-C., M. Hodge, J. W. Rooney, and L. H. Glimcher. 1996. The proto-onco-gene c-mafis responsible for tissue-specific expression of interleukin-4.Cell85:973.
30. Zon, L. I., M. F. Gurish, R. L. Stevens, C. Mather, D. S. Reynolds, K. F. Austen,and S. H. Orkin. 1991. GATA-binding transcription factors in mast cells regulatethe promoter of the mast cell carboxypeptidase A gene.J. Biol. Chem. 266:22948.
31. Sherman, M. A., T. Y. Nachman, and M. A. Brown. 1999. Cutting edge: IL-4production by mast cells does not requirec-maf. J. Immunol. 163:1733.
32. Shimoda, K., J. Deursen, M. Sangster, S. Sarawar, R. Carson, R. Tripp, C. Chu,F. Quelle, T. Nosaka, D. Vignale, et al. 1996. Lack of IL-4-induced Th2 responseand IgE class switching in mice with disrupted Stat6 gene.Nature 380:630.
33. Takeda, K., T. Tanaka, W. Shi, M. Matsumoto, M. Minami, S. Kashiwamura,K. Nakanishi, N. Yoshida, T. Kishimoto, and S. Akira. 1996. Essential role ofStat6 in IL-4 signalling.Nature 380:627.
34. Kaplan, M., U. Schindler, S. Smiley, and M. Grusby. 1996. Stat6 is required formediating responses to IL-4 and for the development of Th2 cells.Immunity4:313.
3248 DUAL FUNCTIONS FOR AN IL-4 GENE INTRON REGULATORY ELEMENT
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

35. Sherman, M. A., V. H. Secor, S. K. Lee, R. D. Lopez, and M. A. Brown. 1999.STAT6-independent production of IL-4 by mast cells.Eur. J. Immunol. 29:1235.
36. Rooney, J. W., M. R. Hodge, P. G. McCaffrey, A. Rao, and L. H. Glimcher. 1994.A common factor regulates both Th1 and Th2-specific cytokine gene expression.EMBO J. 13:625.
37. Rooney, J. W., T. Hoey, and L. H. Glimcher. 1995. Coordinate and cooperativeroles for NF-AT and AP-1 in the regulation of the murine IL-4 gene.Immunity2:473.
38. Tara, D., D. L. Weiss, and M. A. Brown. 1995. Characterization of the consti-tutive and inducible components of a T cell activation responsive element.J. Im-munol. 154:4592.
39. Weiss, D., J. Hural, D. Tara, L. Timmerman, G. Henkel, and M. Brown. 1995.Nuclear factor of activated T cells is associated with a mast cell interleukin-4transcription complex.Mol. Cell. Biol. 16:228.
40. Henkel, G., D. L. Weiss, R. McCoy, T. Deloughery, D. Tara, and M. A. Brown.1992. A DNase I hypersensitive site defines a mast cell enhancer.J. Immunol.149:323.
41. Henkel, G., and M. A. Brown. 1994. PU.1 and GATA: components of a mastcell-specific interleukin 4 intronic enhancer.Proc. Natl. Acad. Sci. USA 91:7737.
42. Pierce, J. H., P. O. DiFiore, S. A. Aaronson, M. Potter, J. Pumphry, A. Scott, andJ. Ihle. 1985. Neoplastic transformation of mast cells by Abelson MuLV: abro-gation of IL-3 dependence by a nonautocrine mechanism.Cell 41:685.
43. Rothman, P., S. C. Li, B. Gorham, L. Glimcher, F. Alt, and M. Boothby. 1991.Identification of a conserved lipopolysaccharide-plus-interleukin-4-responsive el-ement located at the promoter of germ linee transcripts.Mol. Cell. Biol. 11:5551.
44. Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman,J. A. Smith, and K. Struhl. 1987.Current Protocols in Molecular Biology, Vol.1. John Wiley and Sons, New York.
45. Wingender, E., X. Chen, R. Hehl, H. Karas, I. Leibich, V. Matys, T. Meinhardt,M. Pruss, I. Reuter, and F. Schacherer. 2000. TRANSFAC: an integrated systemfor gene expression regulation.Nucleic Acids Res. 28:316.
46. Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcriptioninitiation by RNA polymerase II in a soluble extract from isolated mammaliannuclei.Nucleic Acids Res. 11:1475.
47. Fiering, J., J. P. Northrop, G. P. Nolan, P. S. Mattila, G. R. Crabtree, andL. A. Herzenberg. 1990. Single cell assay of a transcription factor reveals athreshold in transcription activated by signals emanating from the T-cell antigenreceptor.Genes Dev. 4:1823.
48. Sherman, M. A., V. Secor, and M. A. Brown. 1999. IL-4 preferentially activatesa novel STAT6 isoform in mast cells.J. Immunol. 162:2703.
49. Otsuka, T., D. Villaret, T. Yokata, Y. Takebe, F. Lee, N. Arai, and K. Arai. 1987.Structural analysis of the mouse chromosomal gene encoding interleukin 4 whichexpress B cell, T cell and mast cell stimulating activities.Nucleic Acids Res.15:333.
50. Schindler, U., P. Wu, M. Rothe, M. Brasseur, and S. L. McKnight. 1995. Com-ponents of a Stat recognition code: evidence for two layers of molecular selec-tivity. Immunity 2:659.
51. Seidel, H. M., L. H. Milocco, P. Lamb, J. E. Darnell, S. R. B., and J. Rosen. 1995.Spacing of palindromic half sites as a determinant of selective STAT (signaltransducers and activators of transcription) DNA binding and transcriptional ac-tivity. Proc. Natl. Acad. Sci. USA 92:3041.
52. Ryan, A. K., and M. G. Rosenfeld. 1997. POU domain family: flexibility, part-nerships and developmental codes.Genes Dev. 11:1207.
53. Dasen, J. S., S. M. O’Connell, S. E. Flynn, M. Treier, A. S. Gleiberman,D. P. Szeto, F. Hooshmand, A. A. K., and M. G. Rosenfeld. 1999. Reciprocalinteraction of Pit 1 and GATA 2 mediate signaling gradient-induced determina-tion of pituitary cell types.Cell 97:587.
54. Lewis, S. M. 1994. The mechanism of V(D)J joining: lessons from molecular,immunological, and comparative analyses.Adv. Immunol. 56:27.
55. Clevers, H., and P. Ferrier. 1998. Transcriptional control during T-cell develop-ment.Curr. Opin. Immunol. 10:166.
56. Serwe, M., and F. Sablitzky. 1993. V(D)J recombination in B cells is impairedbut not blocked by targeted deletion of the immunoglobulin heavy chain intronenhancer.EMBO J. 12:2321.
57. Demengeot, J., E. M. Oltz, and F. W. Alt. 1995. Promotion of V(D)J recombi-national accessibility by the intronic Ek element: role of thekB motif. Int. Im-munol. 7:1995.
58. Kass, S. U., D. Pruss, and A. P. Wolffe. 1997. How does DNA methylationrepress transcription?Trends Genet. 13:444.
59. Mostoslavsky, R., and Y. Bergman. 1997. DNA methylation: regulation of geneexpression and role in the immune system.Biochim. Biophys. Acta 1333:F29.
60. Nan, X., H. Ng, C. A. Johnson, C. D. Laherty, B. M. Turner, R. N. Eiseman, andA. Bird. 1998. Transcriptional repression by the methy-CpG-binding proteinMeCps involves a histone deacetylase complex.Nature 393.
61. Jones, P. L., G. J. Veenstra, P. A. Wade, D. Verrmaak, S. U. Kass,N. Landsberger, J. Strouboulis, and A. P. Wolffe. 1998. Methylated DNA andMeCP2 recruit histone deacteylase to repress transcription.Nat. Genet. 19:187.
62. Agarwal, S., and A. Rao. 1998. Modulation of chromatin structure regulatescytokine gene expression during T cell differentiation.Immunity 9:765.
63. Bird, J. J., D. R. Brown, A. C. Mullen, N. H. Moskowitz, M. A. Mahowald,J. R. Sider, T. F. Gajewski, C.-R. Wang, and S. L. Reiner. 1998. Helper T celldifferentiation is controlled by the cell cycle.Immunity:229.
64. Grosschedl, R., and M. Marx. 1988. State of an immunoglobulinm gene requirescontinuous enhancer function.Cell 55:645.
65. Kirillov, A., B. Kistler, R. Mostoslavsky, H. Cedar, T. Wirth, and Y. Bergman.1996. A role of nuclear NF-kB in B-cell-specific demethylation of theIgk locus.Nat. Genet. 13:435.
66. Lichtenstein, M., G. Keini, H. Cedar, and Y. Bergman. 1994. B cell-specificdemethylation: a novel role for the intronick enhancer.Cell 76:913.
67. Sleckman, B. P., C. G. Bardon, R. Ferrini, L. Davidson, and F. W. Alt. 1997.Function of the TCRa enhancer ina/b andg/d T cells. Immunity 4:505.
68. Bories, J. C., J. Demengeot, L. Davidson, and F. Alt. 1996. Gene-targeted dele-tion and replacement mutations of the T-cell receptorb-chain enhancer: the roleof enhancer elements in controlling V(D)J recombination accessibility.Proc.Natl. Acad. Sci. USA 93:7871.
69. Bouvier, G., F. Watrin, M. Naspetti, C. Verthuy, P. Naquet, and P. Ferrier. 1996.Deletion of the mouse T-cell receptorb enhancer blocksab T cell development.Proc. Natl. Acad. Sci. USA 93:7877.
70. Boyes, J., J. Omichinski, D. Clark, M. Pikaart, and G. Felsenfeld. 1998. Pertur-bation of nucleosome strucutre by the erythroid transcription factor GATA-1.J. Mol. Biol. 279:529.
71. Nelsen, B., G. Tian, B. Erman, J. Gregoire, R. Maki, B. Graves, and R. Sen. 1992.Regulation of lymphoid-specific immunoglobulinm heavy chain gene enhancerby ets-domain proteins.Science 261:82.
72. Hromas, R., A. Orazi, R. S. Neiman, R. Maki, C. Van Beveran, J. Moore, andM. Klemsz. 1993. Hematopoietic lineage- and stage-restricted expression of theets oncogene family member PU.1. Blood 82:2998.
73. Galson, D. L., J. O. Hensold, T. R. Bishop, M. M. Schalling, A. D. D’Andrea,C. Jones, P. E. Auron, and E. D. Housman. 1993. Mouseb-globin DNA-bindingprotein B1 is identical to a proto-oncogene, the trancription factor Spi/Pu.1, andis restricted in expression to hematopoietic cells and the testis.Mol. Cell. Biol.13:2929.
74. Nikolajczyk, B. S., J. A. Sanchez, and R. Sen. 1999. ETS protein-dependentaccessibility changes at the immunoglobulinm heavy chain enhancer.Immunity11:11.
75. Gingras, S., J. Simard, B. Groner, and E. Pfitzner. 1999. p300/CBP is required fortranscriptional induction by interleukin-4 and interacts with Stat6.Nucleic AcidsRes. 27:2722.
76. Pfitzner, E., R. Jahne, M. Wissler, E. Stoecklin, and B. Groner. 1998. p300/CREB-binding protein enhances the prolactin-mediated transcriptional inductionthrough direct interaction with the transactivation domain of Stat5, but does notparticipate in the Stat 5-mediated suppression of the glucocorticoid response.Mol. Endocrinol. 12:1582.
77. Zhu, M., S. John, M. Berg, and W. J. Leonard. 1999. Functional association ofNmi with Stat5 and Stat1 in IL-2- and IFN-g-mediated signaling.Cell 96:121.
78. Paulson, M., S. Pisharody, L. Pan, S. Guadagno, A. L. Mui, and D. E. Levy. 1999.Stat protein transactivation domains recruit p300/CBP through widely divergentsequences.J. Biol. Chem. 274:25343.
79. Horvai, A. E., L. Xu, E. Korzus, G. Brard, D. Kalafus, T. M. Mullen, D. W. Rose,M. G. Rosenfeld, and C. K. Galss. 1997. Nuclear integration of JAK/STAT andRas/AP-1 signaling by CBP and p300.Proc. Natl. Acad. Sci. USA 94:1074.
80. Grunstein, M. 1997. Histone acetylation in chromatin structure and transcription.Nature 389:349.
81. Kuo, M.-H., J. Zhou, P. Jambeck, M. E. A. Churchill, and C. D. Allis. 1998.Histone acetyltransferase activity of yeast Gcn5p is required for the activation oftarget genes in vivo.Genes Dev. 12:627.
82. Ye, S. K., K. Maki, T. Kitamura, S. Sunaga, K. Akashi, J. Domen,I. L. Weissman, T. Honjo, and K. Ikuta. 1999. Induction of germline transcriptionin the TCRg locus by Stat5: implications for accessibility control by the IL-7receptor.Immunity 11:213.
83. Durum, S. K., S. Candeias, H. Nakajima, W. J. Leonard, A. M. Baird, L. J. Berg,and K. Muegge. 1998. Interleukin 7 receptor control of T cell receptorg generearrangement: role of receptor-associated chains and locus accessibility.J. Exp.Med. 188:2223.
84. Loots, G. G., R. M. Locksley, C. M. Blankespoor, Z. E. Wang, W. Miller,E. M. Rubin, and K. A. Frazer. 2000. Identification of a coordinate regulator ofinterleukins 4, 13, and by cross-species sequence comparisons.Science 288:136.
85. Price, J., D. Turner, and C. Cepko. 1987. Lineage analysis in the vertebratenervous system by retrovirus-mediated gene transfer.Proc. Natl. Acad. Sci. USA84:156.
3249The Journal of Immunology
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from