aggressive periodontitis ul
TRANSCRIPT
Aggressive Periodontitis (etiology and pathogenesis)
Seminar :
Leena Parmar

Index Introduction
Definition
Classification
Etiology & pathogenesis:-
a) microbiologic factor
b) immunologic factor
c) genetic factor
d) environmental factor

Introduction :-
Aggressive periodontitis may be universally
distinguished from chronic periodontitis by the
age of onset, the rapid rate of disease
progression, the nature and composition of the
associated subgingival microflora, alterations in
the host's immune response, and a familial
aggregation of diseased individuals.

Definition
In1971, Baer' defined it as "a disease of the
periodontium occurring in an otherwise healthy
adolescent which is characterized by a rapid
loss of alveolar bone about more than one
tooth of the permanent dentition. The amount of
destruction manifested is not commensurate
with the amount of local irritants."

Classification and clinical syndromes:-
In the absence of an etiologic classification,
aggressive forms of periodontal disease have been
defined based on the following primary features
(Lang et al. 1999):
• Non-contributory medical history
• Rapid attachment loss and bone destruction
• Familial aggregation of cases.

Secondary features that are considered to be generally
but not universally present are:
•Amounts of microbial deposits inconsistent with the
severity of periodontal tissue destruction.
•Elevated proportions of Actinobacillus
actinomycetemcomitans
(Aggregatibacter actinomycetemcomitans) and, in some
Far East populations, Porphyromonas gingivalis

• Phagocyte abnormalities.
• Hyper-responsive macrophage phenotype,
including elevated production of prostaglandin
E2 (PGE2) and interleukin-1β (IL-1β) in
response to bacterial endotoxins
• Progression of attachment loss and bone loss
may be self-arresting.

The international classification workshop identified clinical
and laboratory features deemed specific enough to allow
subclassification of AgP into localized and generalized
forms (Lang et al. 1999; Tonetti & Mombelli 1999).
The following features were identified:
• Localized aggressive periodontitis (LAP)
• Generalized aggressive periodontitis (GAP)

• Localized aggressive periodontitis (LAP):
Circumpubertal onset.
Localized first molar/incisor presentation with
interproximal attachment loss on at least two permanent
teeth, one of which is a first molar, and involving no more
than two teeth other than first molars and incisors
Robust serum antibody response to infecting agents

• Generalized aggressive periodontitis (GAP):
Usually affecting persons under 30 years of age, but
patients may be older.
Generalized interproximal attachment loss affecting at
least three permanent teeth other than first molars and
incisors.
Pronounced episodic nature of the destruction of
attachment and alveolar bone.
Poor serum antibody response to infecting agents.

Etiology and Pathogenesis :-
Microbiologic Factors Immunologic Factors Genetic Factors Environmental Factors

A) Microbiologic Factors :-
Early studies attempting the identification
of the involved bacteria using culture
techniques were performed by Newman et
al. and by Slots (Newman et al. 1976;
Slots 1976; Newman & Socransky 1977).

Dominant microorganisms in LAP included
Actinobacillus actinomycetemcomitans (A.a., now
termed Aggregatibacter actinomycetemcomitans),
Capnocytophaga sp., Eikenella corrodens,
saccharolytic Bacteroides-like organisms now
classified as Prevotella sp., and motile anaerobic rods
today labeled Campylobacter rectus.
Gram-positive isolates were mostly streptococci,
actinomycetes, and peptostreptococci.

A.a., Capnocytophaga sp., and Prevotella sp. were also
shown to be the most prominent members of the subgingival
microbiota of periodontitis lesions in the primary dentition.
One of these organisms, A. actinomycetemcomitans, a
short, facultatively anaerobic, non-motile, Gram negative
rod, received particular attention and was increasingly
viewed as a key microorganism in LAP.

This view was principally based on four lines of evidence (Socransky &
Haffajee 1992):
1. Association studies, linking the organism to the disease: A.a. was
isolated in periodontal lesions from more than 90% of LAP patients
and was much less frequent in periodontally healthy individuals
(Ashley et al. 1988; Van der Velden et al. 1989; Albandar et al. 1990;
Gunsolley et al. 1990; Slots et al. 1990; Asikainen et al. 1991; Aass et
al. 1992; Ebersole et al. 1994; Listgarten et al. 1995). In some studies
it was possible to demonstrate elevated levels of A.a. in sites showing
evidence of recent or ongoing periodontal tissue destruction (Haffajee
et al. 1984; Mandell 1984; Mandell et al. 1987).

2. Demonstration of virulence factors: A.a. was shown to
produce several potentially pathogenic substances,
including a leukotoxin, was capable of translocating
across epithelial membranes, and could induce disease
in experimental animals and non-oral sites ( Zambon et
al. 1988; Slots & Schonfeld 1991).

3. Findings of immune responses towards this bacterium:
Investigators repeatedly reported significantly elevated
levels of serum antibodies to A.a. in LAP patients
(Listgarten et al. 1981; Tsai et al. 1981; Altman et al. 1982;
Ebersole et al. 1982, 1983; Genco et al. 1985; Vincent et
al. 1985; Mandell et al. 1987; Sandholm et al. 1987). Such
patients were furthermore shown to produce antibodies
locally against this organism at diseased sites (Schonfeld
& Kagan 1982; Ebersole et al. 1985b; Tew et al. 1985).

4. Clinical studies showing a correlation between
treatment outcomes and levels of A.a. after
therapy: unsuccessful treatment outcomes were
linked to a failure in reducing the subgingival load
of A.a. (Slots & Rosling 1983; Haffajee et al.
1984; Christersson et al. 1985; Kornman &
Robertson 1985; Mandell et al. 1986, 1987;
Preus 1988).

Recently six serotypes ( a,b,c,d,e and f ) of A.a have
been described based on the composition of structurally
and antigenically distinct O-polysaccharides of their
lipopolysaccharides. In addition, a novel serotype g has
recently been proposed.

(Zambon et al. 1983, 1996).
In the United States, A serotype-dependent pattern of
association with LAP was found .
Serotype b strains were more often isolated from
patients with localized juvenile periodontitis.
(Asikainen et al. 1991, 1995)
A higher frequency of serotype b strains was also
reported from Finnish subjects with periodontitis

Leukotoxin production
A major virulence factor of A.
actinomycetemcomitans and all strains.
Which makes the bacterium capable of evading the
host response by killing leukocytes.
A highly leukotoxic clonal type of A.
actinomycetemcomitans serotype b was first isolated
, in the early 1980s, from an 8 year old male child
with localized aggressive periodontitis.
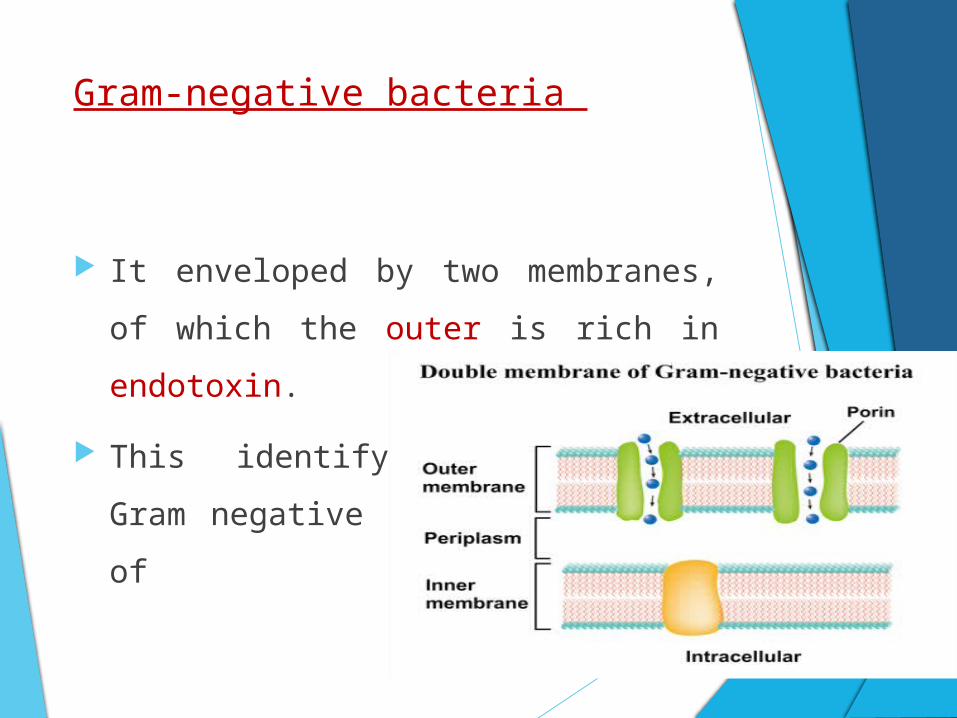
Gram-negative bacteria
It enveloped by two membranes, of which
the outer is rich in endotoxin.
This identifying feature of Gram negative
bacteria consists of
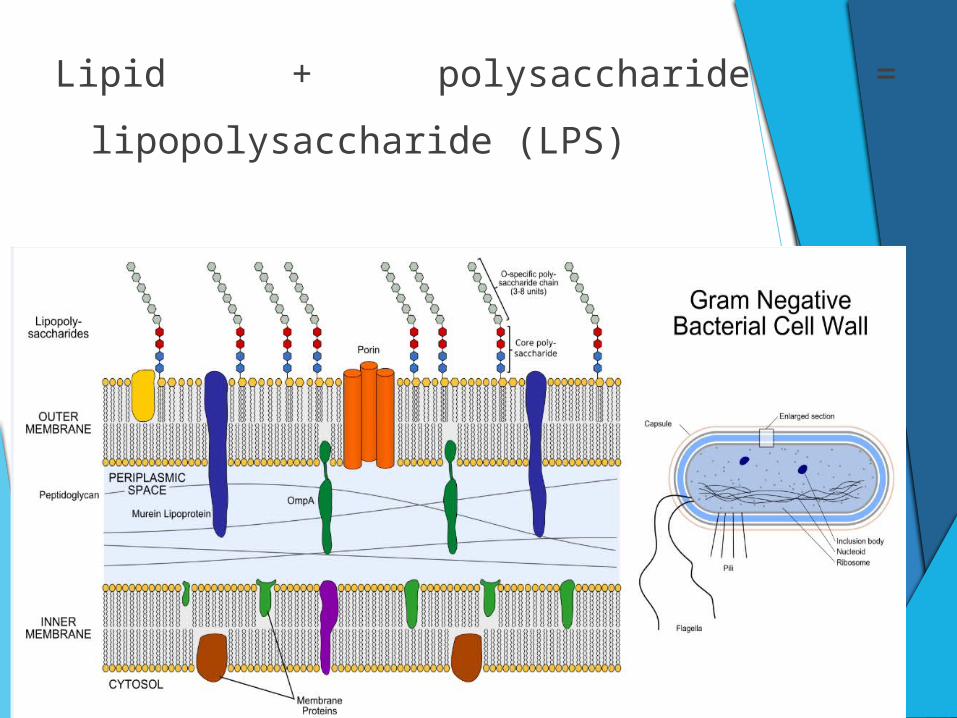
Lipid + polysaccharide = lipopolysaccharide (LPS)
LPS is set free when bacterial cells die or
multiply.
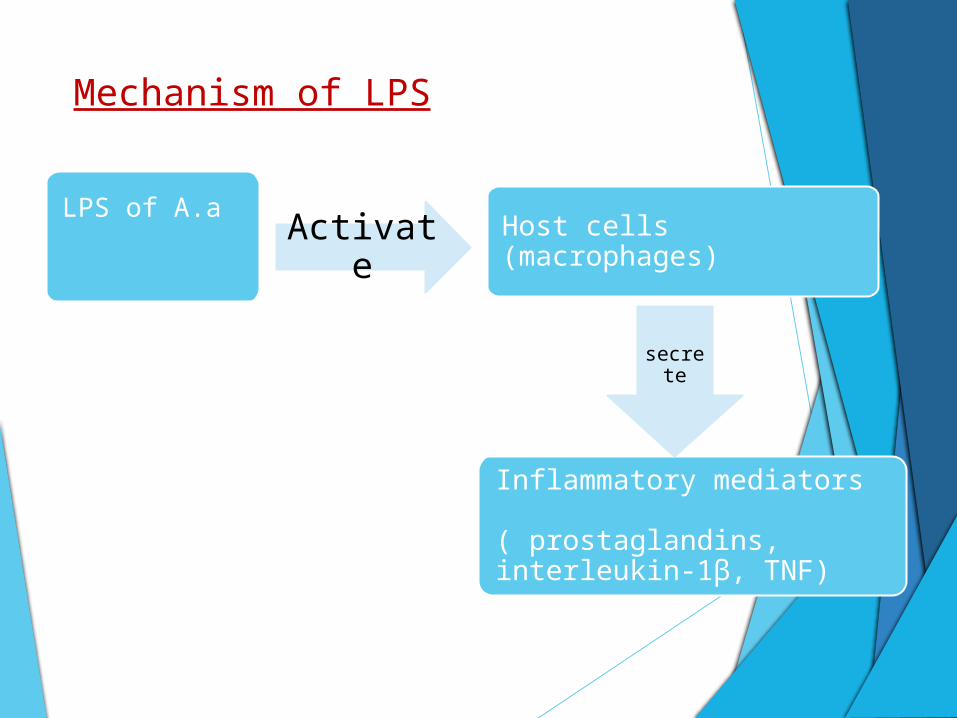
Mechanism of LPS
LPS of A.a
Host cells (macrophages)
Inflammatory mediators ( prostaglandins, interleukin-1β, TNF)
Activate
secrete
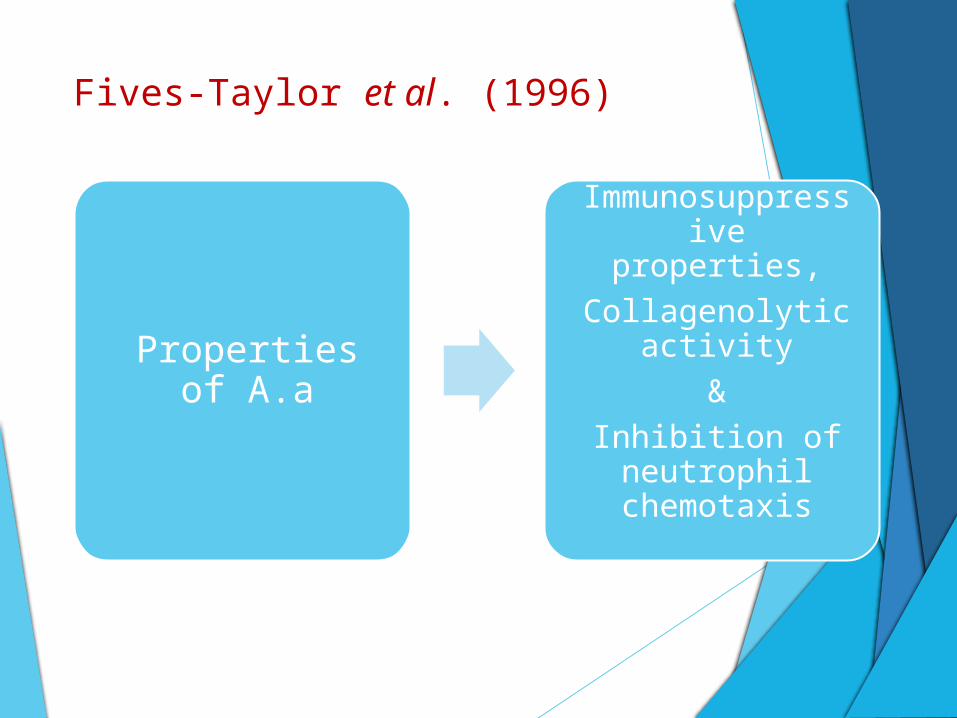
Fives-Taylor et al. (1996)
Properties of A.a
Immunosuppressive properties,
Collagenolytic activity
&
Inhibition of neutrophil
chemotaxis
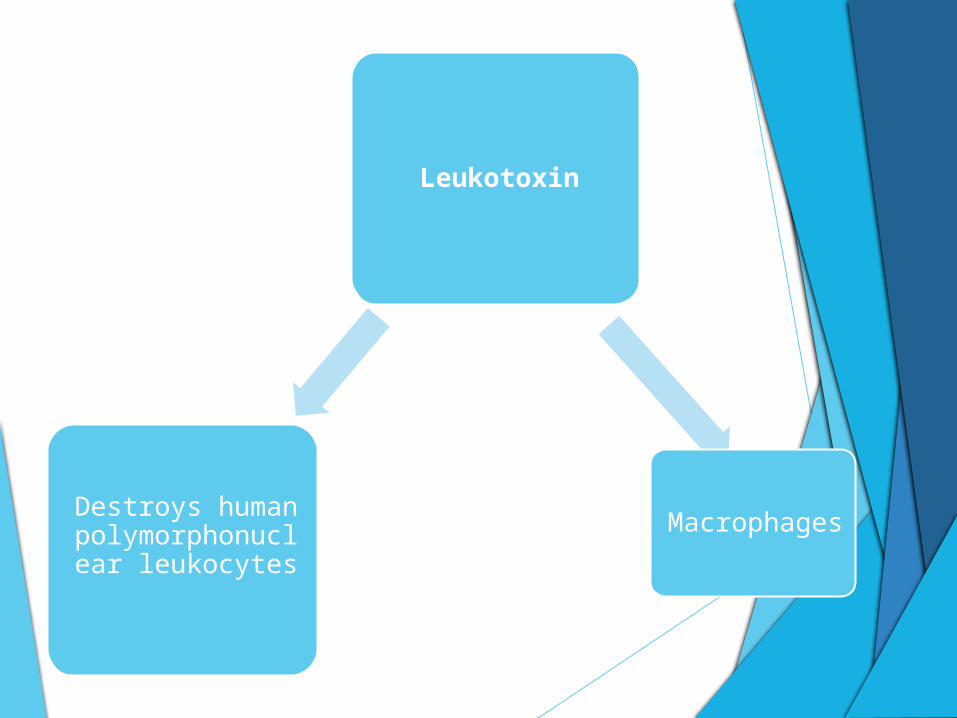
Leukotoxin
Destroys human polymorphonuclear
leukocytes
Macrophages
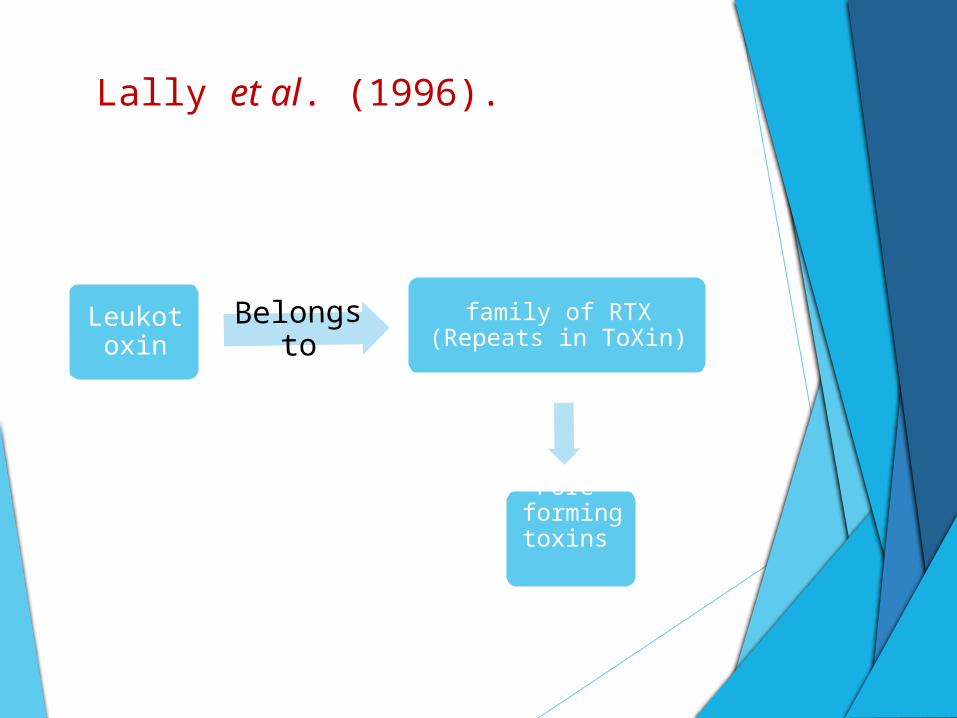
Lally et al. (1996).
Leukotoxin
Belongs to
family of RTX (Repeats in ToXin)
Pore-forming
toxins

Haraszthy et al. (2000); Tan et al. (2001); Cortelli et al.(2005)
JP2 clone Commonly found Aggressive periodontitis patients
(North and West African)

Among potential virulence factors, cytolethal distending toxin,
which seems to be characteristic for A.a, but not for other
periodontal organisms, has gained considerable research
interest.
Outer membrane vesicles of A.a
Cytolethal distending toxin
& Bacterium
Other virulence agents
Host tissue
Transfer of

Determinants of virulence and pathogenic potential of A. actinomycetemcomitans
• Significance
Factor
• Destroys human polymorphonuclear leukocytes and macrophages
Leukotoxin
• Activates host cells to secrete infammatory mediators (prostaglandins, interleukin-1β, tumor necrosis factor-α)
Endotoxin

• May inhibit growth of beneficial species
Bacteriocin
• May inhibit IgG and IgM production
Immunosuppressive
Factors
• Cause degradation of collagen
Collagenases

• May inhibit neutrophil chemotaxis
Chemotactic inhibition
factors

Bacterial damage to the periodontium :-
Disease-associated bacteria are thought to cause destruction of the marginal periodontium via two related mechanisms:
(Tonetti 1993).
Disease-associated bacteria
(Direct) Action of the microorganisms or their products on the host tissues
(Indirect)Eliciting tissue-damaginginflammatory responses
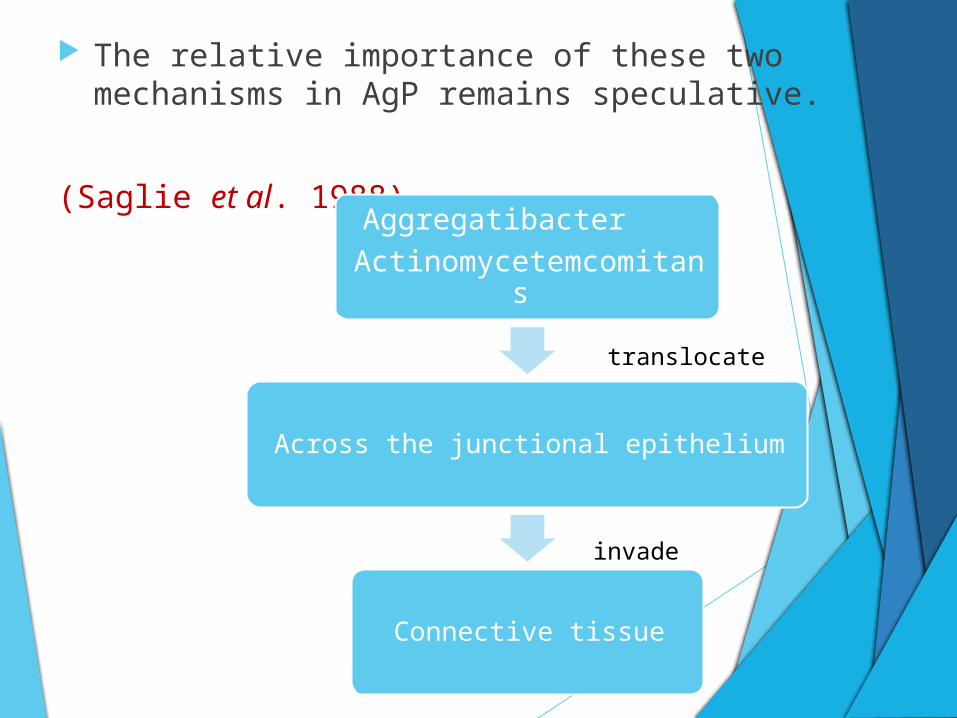
The relative importance of these two mechanisms in AgP remains speculative.
(Saglie et al. 1988)Aggregatibacter
Actinomycetemcomitans
Across the junctional epithelium
Connective tissue
translocate
invade
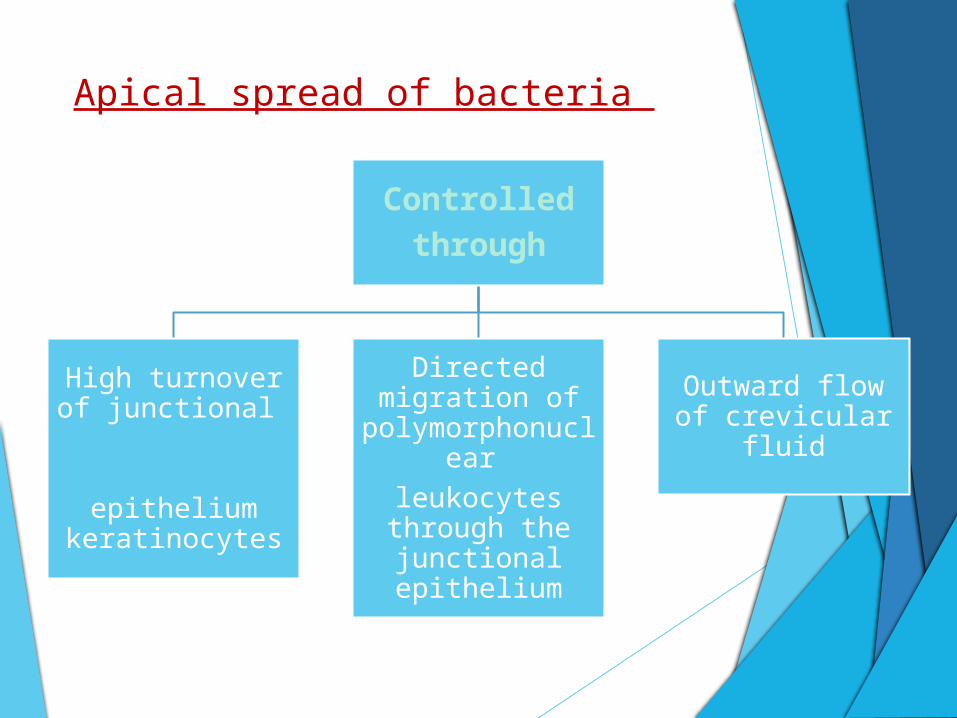
Apical spread of bacteria
Controlled
through
High turnover of junctional
epithelium
keratinocytes
Directed migration of
polymorphonuclear
leukocytes through the junctional epithelium
Outward flow of crevicular fluid
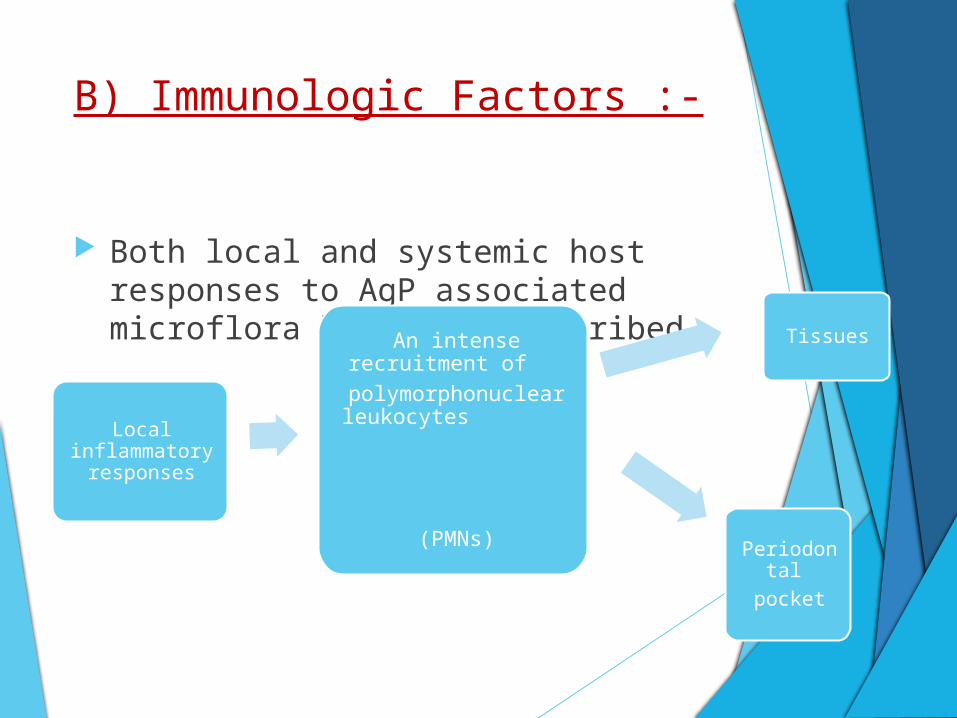
B) Immunologic Factors :-
Both local and systemic host responses to AgP associated microflora have been described.
Local inflammatory
responses
An intense recruitment of
polymorphonuclear leukocytes
(PMNs)
Tissues
Periodontal

Presence of PMNs underlines the importance of these
cells in the local defense against bacterial aggression
and their potential role in host-mediated tissue
destruction.
B cells and antibody-producing plasma cells also
represent a significant component (Liljenberg & Lindhe
1980).
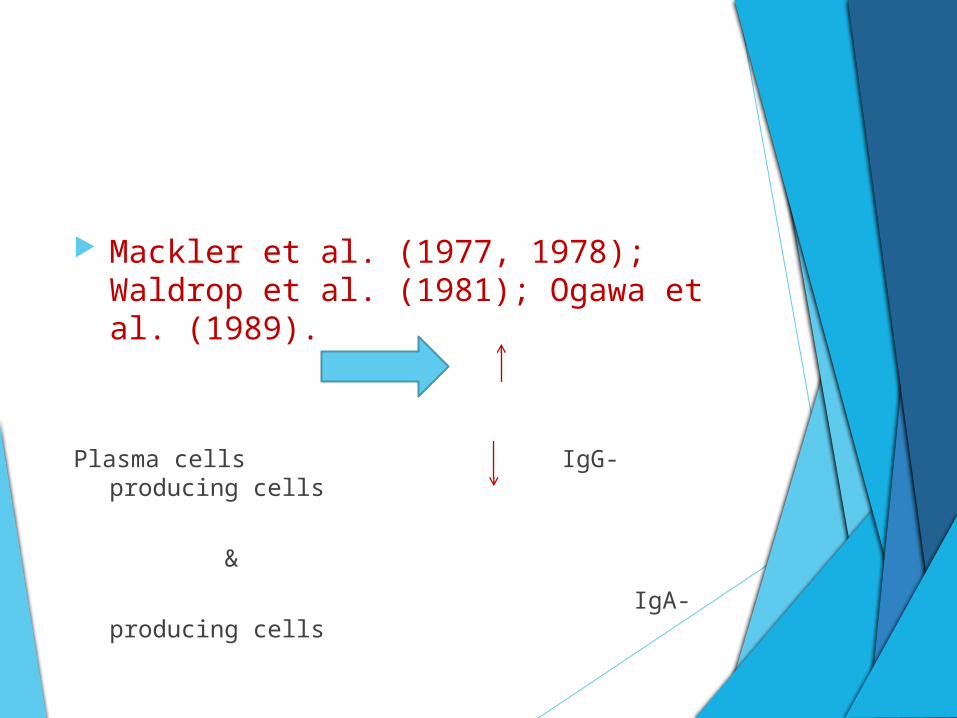
Mackler et al. (1977, 1978); Waldrop et al. (1981); Ogawa et al. (1989).
Plasma cells IgG-producing cells
&
IgA-producing cells

Taubman et al. (1988, 1991).
Important component of the local inflammatory
infiltrate are T cells.
Subset analysis of local T cells has indicated a
depressed T-helper to T suppressor ratio as
compared to both healthy gingival and peripheral
blood.
These findings have been interpreted to suggest
the possibility of altered local immune regulation
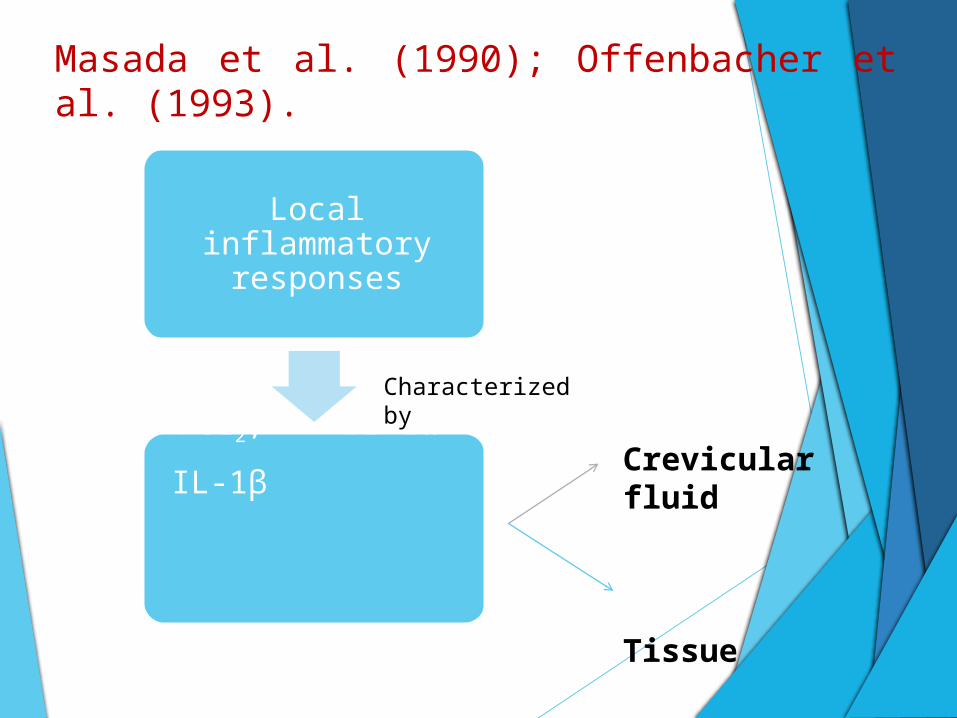
Masada et al. (1990); Offenbacher et al. (1993).
Local inflammatory responses
PGE2, IL-1α
IL-1β
Crevicular fluid
Tissue
Characterized by

Schenkein & Genco (1977); Patters et al. (1989)
Specific antibodies against AgP-associated microorganisms
Crevicular fluid from AgP lesions.
Detected in
Steubing et al. (1982); Hall et al. (1990, 1991, 1994)
Substantial amount of antibodies
against A.a. and P. gingivalis
serum of AgP patients
Detected in

Genco et al. (1980, 1986); Van Dyke et al. (1982, 1986, 1988).
PMNs of some LAP and GAP
Decreased migration
Decreased antibacterial
functions
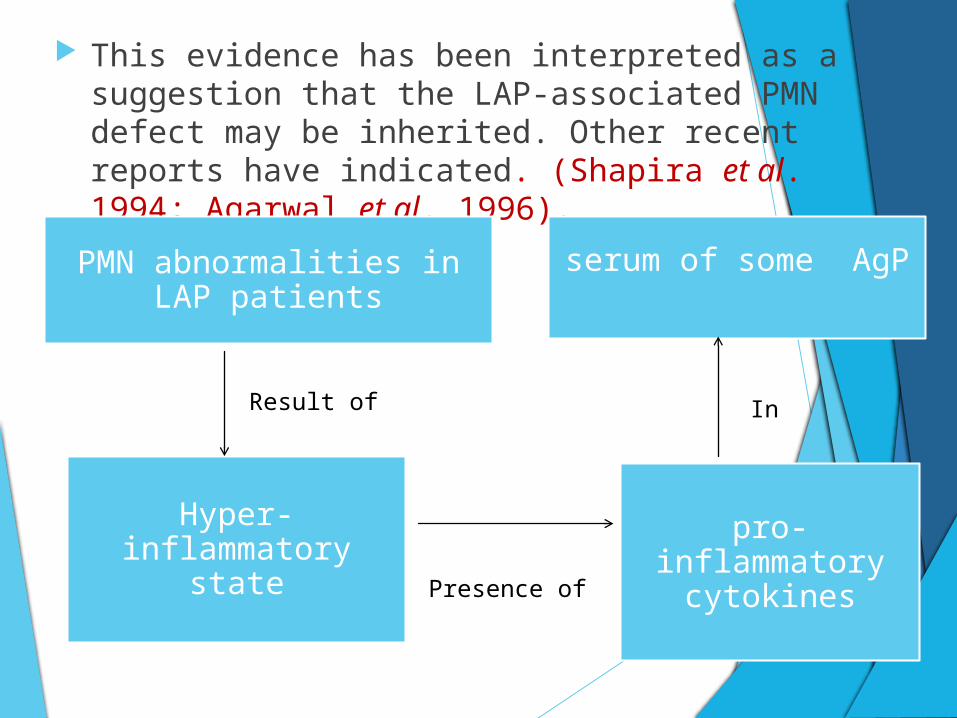
This evidence has been interpreted as a suggestion that the LAP-associated PMN defect may be inherited. Other recent reports have indicated. (Shapira et al. 1994; Agarwal et al. 1996).
PMN abnormalities in LAP patients
serum of some AgP
Hyper-inflammatory state
pro-inflammatory cytokines
Result of
Presence of
In

C) Genetic Factors:-
Periodontitis is a multifactorial disease for which
several risk and susceptibility factors are proposed.
The striking familial aggregation of trait in
Aggressive Periodontitis is consistent with a
significant genetic etiology.
A gene of major effect in Aggressive Periodontitis
appears to be etiologically complex and
heterogenous.

In 1986, Boughman et al reported that a
major gene located on chromosome 4q
was responsible for autosomal dominant
transmission of Localised Aggressive
Periodontitis in an extended family that
also exhibited Dentinogenesis Imperfecta.

It is now established that genetic factors regulate the innate immune system and that certain genetic polymorphism may render the immune system defective.
Genetic factors may play a more significant role in the pathogenesis of AgP.

Formyl Peptide receptors on the cell surface of leukocytes are involved in mediating immune cell responses to infection.
The bacteria derived N-formly-methionyl peptides have high affinity to the N-formly-methionyl peptide cell receptor and after binding to neutrophil receptor the neutrophils get activated.
Thus triggering them to migrate to the site of infection.

Some reports suggest that the abnormal neutrophil chemotactic response to N-formly-methionyl peptides is limited to some cases of AgP.
Early studies suggested that neutrophils from the serum of patients with AgP show impaired chemotaxis to these antigens.
An in-vitro experiment showed that phosphoionositide dependent kinase-1 regulates neutrophil chemotaxis.
This suggests that the expression & activation levels of phosphoionositide dependent kinase-1 which are significantly reduced in AgP may explain the impaired neutrophil chemotaxis in such patients.

Albandar et al reported that the serum levels of IgA reactive to periodontal pathogens were significantly higher in AgP.
Furthermore neutrophils from AgP patients show increased levels of expression of the FcαRI receptor.
Cross-linking of IgA with the Fcα receptor on phagocytes triggers the following host cellular responses such as phagocytosis, antibody-dependent cell mediated cytotoxicity and release of inflammatory mediators.
Hence it concluded that individuals with increased expression levels of FcαRI receptor on phagocytes and elevated levels of IgA reactive to periodontal pathogen, may be at higher risk for AgP.

Papillon-Lefevre syndrome there is a loss of function
mutation affecting the cathepsin C gene on chromosome
11q14.2 and this influences a key enzyme essential in
activation of certain immune cells and in regulation of
epitheial cells.
In Chediak-Higashi syndrome, mutations have been
identified in Lysosomal trafficking regulator ( CHS1/
LYST) gene on chromosome 1q42.3.

D) Environmental Factors:-
In a large study, cigarette smoking was shown to be a risk factor for patients with generalized forms of AgP (Schenkein et al. 1995).
Smokers with GAP had more affected teeth and greater mean levels of attachment loss than patients with GAP who did not smoke.
IgG2 serum levels as well as antibody levels against A.a. are significantly depressed in subjects with GAP who smoke.

References:
Clinical periodontology and implant dentistry -5th edition, Volume-1, Jan Lindhe.
Clinical periodontology -10thedition,Carranza,Neuman,Takei,Klokkevold.
Periodontology 2000,vol-65, 2014.
Assessment of peripheral neutrophil functions in patients with localized aggressive periodontitis in the Indian population. Rahul S. Bhansali, R. K. Yeltiwar, K. G. Bhat, Journal of Indian Society of Periodontology - Vol 17, Issue 6, Nov-Dec 2013.