a functional link between store-operated calcium …
TRANSCRIPT

The Pennsylvania State University
The Graduate School
The Huck Institutes of the Life Sciences
A FUNCTIONAL LINK BETWEEN STORE-OPERATED
CALCIUM CHANNELS AND THE ACTIN-BINDING PROTEIN
DREBRIN IN MAST CELLS REVEALED BY 3,5-BIS-
TRIFLUOROMETHYL PYRAZOLE (BTP) COMPOUNDS
A Dissertation in
Immunology and Infectious Diseases
by
Mankit Law
© 2011 Mankit Law
Submitted in Partial Fulfillment
of the Requirments
for the Degree of
Doctor of Philosophy
August 2011

ii
The dissertation of Mankit Law was reviewed and approved* by the following:
Avery August
Professor of Immunology
Dissertation Advisor
Chair of Committee
Pamela A. Hankey
Associate Professor of Immunology
Robert F. Paulson
Associate Professor of Veterinary and Biomedical Sciences
Anthony Schmitt
Assistant Professor of Molecular Immunology and Infectious Diseases
Graham Thomas
Associate Professor of Biology and of Biochemistry and Molecular Biology
Margherita T. Cantorna
Professor of Molecular Immunology
Co-chair of Intercollege Graduate Degree Program in Immunology and Infectious
Diseases
*Signatures are on file in The Graduate School.

iii
ABSTRACT
Calcium ions (Ca2+
) are important secondary messengers in signaling pathways of mast
cell activation. Mast cells are central effector cells of allergic inflammation. Therefore,
impaired calcium signaling, which correlates with decreased activation, in mast cells results in
markedly attenuated allergic responses. Nonetheless, the molecular mechanisms of how calcium
homeostasis is regulated in mast cells remain poorly characterized. In particular, the identity of
the players involved in the gating of store-operated Ca2+
channels has eluded biochemical
definition.
A tool that could potentially elucidate the regulatory mechanisms of store-operated Ca2+
entry (SOCE) is the immunosuppressant compound 3,5-bis-trifluoromethyl pyrazole (BTP).
Recently, our laboratory identified the actin-reorganizing protein drebrin as a target of BTP and
demonstrated a novel role for this protein in the regulation of SOCE in T cells. These findings
implicate the involvement of actin-binding proteins in the regulation of Ca2+
mobilization in
mast cells and downstream allergic responses. Nevertheless, the effects of BTP on mast cells
have yet to be properly characterized, and the role of its target drebrin in the biology of these
immune cells has not been determined.
Here, we demonstrate that treatment with the BTP derivative N-(4-(3,5-
bis(trifluoromethyl)-1H-pyrazol-1-yl)phenyl)-4-methyl-1,2,3-thiadiazole-5-carboxamide (BTP2)
potently inhibits the release of mast cell-specific inflammatory mediators that initiate and
propagate allergic responses. In the context of in vitro and in vivo models, our studies have
determined that BTP2 suppresses the release of preformed mediators, such as histamine, and
cytokines through the attenuation of SOCE. Moreover, our analysis of the structure-activity

iv
relationship of BTP identified the trifluoromethyl group at the carbon 3 (C3) position of the core
BTP pyrazole ring as the chemical moiety principally responsible for its inhibitory activity on
mast cell activation and effector responses.
In corroboration, using a novel knockout murine model, we show that the BTP-targeted
protein drebrin is required for Ca2+
-dependent allergic responses. In vivo, sensitized drebrin-/-
mice release less histamine upon antigenic challenge. Importantly, in drebrin-/-
mice, the
development of mast cells from bone marrow precursors is intact, but the survival of bone
marrow-derived mast cells (BMMCs) is reduced in vitro. Levels of serum immunoglobulin E
(IgE) are normal in these mice, and drebrin-/-
BMMCs express normal levels of surface high-
affinity receptor for IgE (FcεRI). Moreover, drebrin deficiency does not alter phosphorylation
events downstream of the FcεRI. The activation of tyrosine kinases and the mitogen-activated
protein kinases Erk1/2, JNK, and p38 is unchanged. However, drebrin is involved in FcεRI-
induced Ca2+
mobilization. In mast cells from drebrin-/-
mice, FcεRI-induced degranulation, as
well as cytokine secretion, is diminished. Thus, drebrin is selectively required for the mast cell-
mediated allergic response, and it is is a novel player in Ca2+
mobilization and mast cell
activation.
We propose that, upon depletion of intracellular Ca2+
stores, drebrin aggregates into
macromolecular complexes that induce cytoskeletal rearrangement. In conjunction with other
modulators of the cytoskeleton in these complexes, drebrin facilitates the juxtaposition of the
endoplasmic reticulum (ER) and plasma membrane (PM) and the subsequent interaction of
SOCE-associated components and insertion of store-operated Ca2+
channels into lipid raft
domains of the PM. Through this role, drebrin plays an integral part of the calcium signaling
pathway in mast cells.

v
TABLE OF CONTENTS
LIST OF FIGURES……………………………………………………………………..... viii
LIST OF TABLES………………………………………………………………………... xi
ABBREVIATIONS……………………………………………………………………….. xii
ACKNOWLEDGEMENTS……………………………………………………………... xv
CHAPTER 1. Introduction……………………………………………………………… 1
Mast Cell Biology………………………………………………………………….. 3
Morphology and phenotype………………………………………………... 3
Development……………………………………………………………….. 4
Activation…………………………………………………………………... 5
FcεRI pathway……………………………………………………... 7
Mediators and effector function………………………………………….… 10
Preformed mediators………………………………………………. 11
Lipid mediators/Eicosanoids………………………………………. 12
Cytokines and chemokines………………………………………… 12
Role in health and disease…………………………………………………. 13
Ca2+
Signaling……………………………………………………………………… 19
Regulation of Ca2+
homeostasis…………………………………………… 19
Store-operated Ca2+
entry…………………………………………………. 24
Activation of store-operated Ca2+
channels……………………………….. 28
Ca2+
signaling and the actin cytoskeleton…………………………………. 31
Drebrin……………………………………………………………………………... 33
Structure……………………………………………………………………. 33
Function……………………………………………………………………. 35
3,5-bis-trifluoromethyl pyrazole (BTP)……………………………………………. 37
Objective of thesis…………………………………………………………………..40
Hypothesis…………………………………………………………………. 40
CHAPTER 2. Materials and Methods………………………………………………….. 41
Mice………………………………………………………………………………... 42
Cell culture and reagents…………………………………………………… ……... 42
Genotyping of mice………………………………………………………………... 43
Quantitative real-time PCR analysis……………………………………………….. 44
Western blotting……………………………………………………………………. 45
Histological staining……………………………………………………………….. 46
Transmission electron microscopy………………………………………………… 47

vi
Flow cytometry…………………………………………………………………….. 47
Apoptosis assay……………………………………………………………..……… 48
Degranulation assay………………………………………………………………... 48
In vivo histamine release assay…………………………………………………….. 50
Serum IgE ELISA…………………………………………………………….……. 51
Cytokine secretion assay…………………………………………………………… 51
Measurements of intracellular Ca2+
concentration………………………………… 52
Analysis of NFAT localization…………………………………………………….. 54
Statistical analysis………………………………………………………………….. 54
CHAPTER 3. Effect of BTP2 on mast cell biology……………………………………. 55
Rationale…………………………………………………………………………… 56
Effects of BTP2 on Ca2+
mobilization in RBL-2H3 cells and BMMCs…… ……... 57
Effects of BTP2 on FcεRI-mediated signaling in BMMCs………………………... 59
BTP2 inhibits degranulation in RBL-2H3 cells……………………………………. 61
BTP2 inhibits histamine release in mice in response to antigenic challenge……… 64
BTP2 inhibits cytokine secretion from mast cells…………………………………. 66
BTP2 inhibits NFAT nuclear localization in mast cells…………………………… 70
BTP2 inhibits de novo synthesis of cytokines in mast cells……………………….. 70
Discussion………………………………………………………………………….. 73
CHAPTER 4. Structure-Activity Relationship of BTP2……………………………..... 76
Rationale…………………………………………………………………………… 77
The trifluoromethyl group at the C3 position is required for the inhibitory
effect of BTP2 on the degranulation of BMMCs……………………………… 78
Effects of 3T5M-P on Ca2+
mobilization in BMMCs…………………….……....... 79
Discussion………………………………………………………………………….. 83
CHAPTER 5. Role of drebrin in mast cell biology…………………………………….. 85
Rationale…………………………………………………………………………… 86
Generation of Drebrin-/-
mice………………………………………………………. 87
Cellular morphology but not distribution of drebrin-/-
mast cells in skin tissue
is normal………………………………………………………………………...94
Drebrin is not required for development but is necessary for survival of
drebrin-/-
BMMCs in vitro……………………………………………………… 96
Drebrin is required for degranulation of BMMCs…………………………………. 101
Drebrin is required for histamine release in mice in response to antigenic
challenge……………………………………………………………………….. 102
Drebrin is required for cytokine secretion of BMMCs…………………………….. 105
Drebrin-/-
BMMCs exhibit normal levels of FcεRI surface expression……………. 108
Phosphorylation events downstream of FcεRI are normal in drebrin-/-
BMMCs….. 110
Drebrin is required for Ca2+
mobilization in mast cells……………………………. 114

vii
Discussion………………………………………………………………………….. 118
Drebrin regulates the mast cell-mediated allergic response………………. 118
Drebrin is required for FcεRI-mediated Ca2+
mobilization……………….. 122
CHAPTER 6. Conclusion and Future Direction……………………………………...... 124
BIBLIOGRAPHY………………………………………………………………………… 132

viii
LIST OF FIGURES
Figure 1.1. Principal signaling cascade of mast cell activation………………………… 9
Figure 1.2. Early phase of allergic inflammation………………………………………. 15
Figure 1.3. Late phase of allergic inflammation……………………………………….. 16
Figure 1.4. Activation of store-operated Ca2+
entry……………………………………. 21
Figure 1.5. Principal pathways of Ca2+
movement…………………………………….. 22
Figure 1.6. Domain structures of STIM1 and Orai1…………………………………… 25
Figure 1.7. Models of CRAC channel activation………………………………………. 30
Figure 1.8. Domain structure of drebrin………………………………………………... 34
Figure 1.9. Chemical structure of BTP2………………………………………………...39
Figure 3.1. BTP2 blocks intracellular Ca2+
mobilization in RBL-2H3
cells and BMMCs……………………………………………………… 58
Figure 3.2. BTP2 does not affect tyrosine kinase nor MAP kinase activation
following FcεRI triggering…………………………………………….. 60
Figure 3.3. BTP2 inhibits mast cell degranulation in vitro…………………………….. 62
Figure 3.4. BTP2 inhibits mast cell degranulation and release of
β-hexosaminidase in vitro……………………………………………… 63
Figure 3.5. BTP2 inhibits FcεRI-mediated histamine release in vivo………………...... 65
Figure 3.6. BTP2 inhibits cytokine secretion of PMA/ionomycin-activated
BMMCs……………………………………………………………....... 67
Figure 3.7. BTP2 inhibits cytokine secretion of IgE/anti-IgE-activated mast cells……. 68
Figure 3.8. BTP2 inhibits cytokine secretion of IgE/antigen-activated mast cells.......... 69
Figure 3.9. BTP2 inhibits stimulus-induced NFAT nuclear translocation……………... 71
Figure 3.10. BTP2 does not affect preformed cytokine mRNA expression……………...72

ix
Figure 4.1. Chemical structures of BTP analogs……………………………………….. 80
Figure 4.2. The 3-trifluoromethyl group is critical for the inhibitory activity
of BTP2 on BMMC degranulation…………………………………….. 81
Figure 4.3. 3T5M-P blocks intracellular Ca2+
mobilization in BMMCs……………….. 82
Figure 5.1. Strategy for simultaneous inactivation and rapid identification of
the disrupted Dbn1 gene by gene-trapping in mouse ES cells………..... 88
Figure 5.2. Mapping of genomic insertion site of the gene-trap vector in intron
8 of the Dbn1 gene……………………………………………………... 91
Figure 5.3. Verification of genetic disruption of the Dbn1 gene by gene-trap
insertion with RT-PCR………………………………………………… 92
Figure 5.4. Verification of ablation of protein expression of the disrupted Dbn1
gene by western blot…………………………………………………… 93
Figure 5.5. Mast cells retain normal cellular morphology but not distribution in
the skin of drebrin-/-
mice………………………………………………. 95
Figure 5.6. Drebrin-/-
BMMCs and basophils show normal development in
vitro…………………………………………………………………….. 98
Figure 5.7. Drebrin-/-
BMMCs show less viability in vitro….......................................... 100
Figure 5.8. Drebrin-/-
mice exhibit impairment in mast cell degranulation in vitro……. 103
Figure 5.9. Drebrin-/-
mice exhibit impairment in FcεRI-mediated histamine
release in vivo…………………………………………………………... 104
Figure 5.10. Drebrin-/-
BMMCs exhibit impairment in PMA/ionomycin-induced
cytokine secretion…………………………………………………….... 106
Figure 5.11. Drebrin-/-
BMMCs exhibit impairment in FcεRI-mediated cytokine
secretion………………………………………………………………... 107
Figure 5.12. Cell surface expression of FcεRI on drebrin-/-
BMMCs is normal………… 109
Figure 5.13. Tyrosine kinase activation following FcεRI triggering is not affected
in drebrin-/-
BMMCs………………………………………………….... 111

x
Figure 5.14. MAP kinase activation following FcεRI triggering is not affected in
drebrin-/-
BMMCs…………………………………………………….... 113
Figure 5.15. Ca2+
mobilization is impaired in drebrin-/-
BMMCs……………………….. 116
Figure 5.16. Activation of PLCγ1 is not affected in drebrin-/-
BMMCs………………… 117
Figure 5.17. BTP2 inhibits ionomycin-induced degranulation of Drebrin-/-
BMMCs…... 121
Figure 6.1. Model for the involvement of drebrin in store-operated Ca2+
entry……….. 130

xi
LIST OF TABLES
Table 1.1. Mast cell mediators………………………………………………………… 10

xii
ABBREVIATIONS
2-APB- Aminoethoxydiphenyl borate
3T-P- 3-trifluoromethyl pyrazole
3T5M-P- 3-trifluoromethyl-5-methyl pyrazole
3M5M-P- 3,5-bis-methyl pyrazole
5T-P- 5-trifluoromethyl pyrazole
5T3M-P- 5-trifluoromethyl-3-methyl pyrazole
ADF-H- Actin-depolymerizing factor-homology
AP-1- Activator Protein-1
ATP- Adenosine triphosphate
BMMC- Bone marrow-derived mast cell
BTP- 3,5-bis-trifluoromethyl pyrazole
BTP2- N-(4-(3,5-bis(trifluoromethyl)-1H-pyrazol-1-yl)phenyl)-4-methyl-1,2,3-thiadiazole-5-
carboxamide
C3- Carbon at 3’ position
C3a- Complement component 3a
Ca2+
- Calcium ion
CaV channel- Voltage-gated Ca2+
channel
CCR- C-C chemokine receptor
CHO cells- Chinese hamster ovary cells
CXCR- C-X-C chemokine receptor
COX- Cyclooxygenase
CRAC channel- Ca2+
release-activated Ca2+
channel
CsA- Cyclosporin A
CTMC- Connective tissue mast cell
CysLT- Cysteinyl leukotrienes
DAG- Diacylglycerol
Dbn1- Mus musculus drebrin 1
DMSO- Dimethyl sulfoxide
DNP-HSA- Dinitrophenyl-human serum albumin
Drebrin-/-
- Drebrin knockout
Drebrin A- Drebrin adult form
Drebrin E- Drebrin embryonal form
EAE- Experimental autoimmune encephalitis
EB1- Microtubule end-binding protein 1
ER- Endoplasmic reticulum
Erk1/2- Extracellular-regulated kinase 1/2
ES cells- Embryonic stem cells
F-actin- Filamentous actin
FcεRI- High-affinity receptor for IgE
G-actin- Globular actin
GADS- Grb2-related adaptor protein
GAPDH- Glyceraldehyde 3-phosphate dehydrogenase

xiii
GDP- Guanosine diphosphate
GEF- guanine nucleotide-exchange factor
GM-CSF- Granulocyte-macrophage colony stimulating factor
Grb2- Growth factor receptor-bound protein 2
GTP- Guanosine triphosphate
HIP-55- HPK1-interacting Protein of 55 kDa
HPK1- Hematopoietic progenitor kinase 1
HRP- Horseradish peroxidase
IC50- Half maximal inhibitory concentration
ICRAC- Ca2+
release-activated Ca2+
current
IgE- Immunoglobulin E
IL- Interleukin
IL-3R- Interleukin-3 receptor
IFN-γ- Interferon-γ
i.p.- Intraperitoneally
IP3- Inositol-1,4,5-triphosphate
IP3R- IP3 receptor
ITAM- Immunoreceptor tyrosine-based activation motif
i.v.- Intravenously
JNK- JUN amino-terminal kinase
LAT- Linker for Activated T cells
LT- Leukotriene
LTR- Long terminal repeat
mAbp1- Mammalian actin-binding protein 1
MAPK- Mitogen-activated protein kinase
MCT- Tryptase+ mast cell
MCTC- Tryptase+ Chymase
+ mast cell
MFI- Mean fluorescence intensity
MMC- Mucosal mast cell
NCX- Na2+
-Ca2+
exchanger
NF-κB- Nuclear factor-κB
NFAT- Nuclear factor of activated T cells
NFATc1- Nuclear factor of activated T cells, cytoplasmic 1
OST- Omnibank sequence tag
OVA- Ovalbumin
PAF- Platelet-activating factor
PAMP- Pathogen-associated molecular pattern
pFv- Protein Fv
PG- Prostaglandin
PIP2- Phosphoinositol-4,5-bisphosphate
PKC- Protein kinase C
PLA2- Phospholipase A2
PLCγ1- Phospholipase γ1
PM- Plasma membrane
PMA- Phorbol myristic acid

xiv
PMCA pump- Plasma membrane Ca2+
-ATPase pump
PSD-95- Post-synaptic density-95
Pyr- Pyrazole
RBL-2H3 cells- Rat basophil leukemia-2H3 cells
RF- Relative fluorescence
RyR- Ryanodine receptor channel
SCF- Stem cell factor
SCID- Severe combined immunodeficiency
SERCA pump- Sarcoplasmic/endoplasmic reticulum Ca2+
-ATPase pump
SH2- Src-homology 2
SHC- SH2 domain-containing transforming protein C
siRNA- Small interfering RNA
SLP-76- SH2-domain-containing leukocyte protein of 76 kDa
SNARE- Soluble N-ethylmaleimide-sensitive factor attachment proteins receptor
SOCE- Store-operated Ca2+
entry
SOS- Son of Sevenless homologue
STIM1- Stromal interactional molecule 1
Syk- Spleen tyrosine kinase
TCR- T cell receptor
TGF-β- Transforming growth factor-β
Th1/2- T helper 1/2
TLR- Toll-like receptor
TNF- Tumor necrosis factor
TRP- Transient receptor potential
TRPC- Transient receptor potential cation
TRPM4- TRP channel, melastatin subfamily 4
WASP- Wiskott-Aldrich syndrome protein
WAVE-2- WASP family member 2
WT- Wild-type

xv
ACKNOWLEDGEMENTS
First of all, I wish to extend my appreciation to my advisor Avery August for his support
and guidance throughout my years of graduate education. He has provided me with an
outstanding work environment to develop my potential as a scientist. He has been an invaluable
source of knowledge and expertise. Most importantly, he has been a living example of education
through his commitment to mentorship of the next generation of scientists and his solid work
ethic. I will always value him as a role model and friend.
I would also like to thank Andrew Henderson, the Immunology and Infectious Diseases
program chair Margherita Cantorna, and my committee members Robert Paulson, Pamela
Hankey, Anthony Schmitt, and Graham Thomas for their intellectual input into the development
of my thesis project and words of counsel.
I thank Blake Peterson and Laurie Mottram for their collaborative efforts in providing me
with the library of BTP analogs that are utilized in this study. I also thank Gang Ning for his
technical help with histology and electron microscopy. A special thanks is offered to Elaine
Kunze, Susan Magargee, Nicole Bem, Walter Iddings, and Rodman Getchell for their technical
support and assistance during the transition to Cornell.
I would like to acknowledge my colleagues at Penn State and Cornell for providing a
collegial and stimulating workplace. In particular, I would like to thank past and present
members of the August laboratory for their scientific discussion and friendship.
Lastly, I would like to thank my family for their constant encouragement. I thank my
wife Julia for her understanding, patience, and smiles. And, to my parents, I dedicate this thesis
for instilling a love of learning in their children and giving us the courage to pursue our dreams.

1
CHAPTER 1
Introduction

2
In 1878, Paul Ehrlich, a Nobel laureate whose studies pioneered the modern-day medical
sciences of hematology and immunology, publicized his descriptions of the tissue mast cell (1).
For the majority of the time thereafter, mast cells have been exclusively associated with the
mediation of pathological secondary responses to allergens in sensitized hosts. In addition to this
classical role, recent evidence implicates their participation in the regulation of host responses to
pathogens, autoimmune diseases, fibrosis, and wound healing (2, 3). These roles for mast cells
in allergy, infection, autoimmunity, and homeostasis implicate additional pharmaceutical targets
for the prevention of the development of allergic disease, as well as allergic exacerbations of
established disease.
Mast cells utilize diverse signaling mechanisms to transmit information between different
compartments of the cell. These mechanisms integrate the action of a myriad of proteins that
perform enzymatic and structural functions in the signal transduction process. Associated
signaling cascades culminate in the production of cellular changes, which range from
cytoskeletal reorganization to transcriptional activation. Frequently, one messenger is involved
in the activation of a variety of functional outcomes. A rise in intracellular Ca2+
levels triggers
the activation of a disparate array of mast cell responses. Nonetheless, though Ca2+
signaling is
an ancient and conserved mechanism in multicellular organisms, the processes that regulate Ca2+
remain poorly understood.
In this thesis, I present work which incorporates a unique group of immunosuppressant
compounds in the identification of drebrin as an integral component of the cellular processes that
regulate Ca2+
mobilization in mast cells. As a result, I provide insight for the development and
design of new research tools that could facilitate the elucidation of regulatory mechanisms of

3
Ca2+
signaling, and I provide support for the advancement of potential therapeutic strategies for
mast-cell mediated disease.
Mast cell biology
Mast cell: Morphology and phenotype
Based upon his observations from the histological application of basic aniline dyes to
human tissues, Ehrlich first described mast cells as aniline-positive cells with large granules in
connective tissues. With the belief that the granules were involved in the nourishment of
surrounding tissue, he named them “Mastzellen”, wherein the German word “mast” denotes a
“fattening” or “suckling” function. He ascribed mast cells to the nutritional requirements of
tissues in states of chronic inflammation and tumors. In addition, he recognized that mast cells
were located in association with blood vessels in connective tissues but were not part of the
perivascular system. As a whole, his conclusions were partially, but not totally, correct (1).
The current thinking of the scientific community recognizes mast cells as tissue-based
inflammatory cells. Generally, they are localized in association with blood vessels and at
epithelial surfaces. With the capacity to be up to 20 µm in diameter, mast cells are characterized
as ovoid or irregularly elongated cells with an ovoid nucleus. They are rich in cytoplasmic
granules, which can be identified with metachromatic staining due to ample sulfated
proteoglycan content in the granules. With electron microscopy, granules are identified based
upon their crystalline content (2).
Depending upon the protease content of their granules, human mast cells can be classified
into the following 2 major subtypes: 1) tryptase-positive (MCT) or 2) tryptase- and mast cell-

4
specific chymase-positive (MCTC). Each subtype predominates in a distinct set of locations.
MCT are located within the mucosa of the respiratory and gastrointestinal tracts. MCTC are
localized within connective tissues. MCTC are the outstanding mast cell subtype in the dermis,
submucosa of the gastrointestinal tract, heart, conjunctivae, and perivascular tissues (2). In
rodents, 2 similar subtypes of mast cells exist. Mucosal mast cells (MMC) are located in
mucosal tissue; serosal mast cells (CTMC) reside in connective tissue (2, 4).
Canonically, mast cells are identified by surface expression of the receptors c-kit/CD117
and FcεRI. Based upon their stage of differentiation, location, and activation, they can also
express other receptors on their cell surface. For example, in the resting state, mast cells express
the activating IgG receptor FcγRIIa/CD32a. The β2-adrenergic receptor, the adenosine receptor
A2B, and the prostaglandin (PG) E2 receptor EP2 compromise a group of inhibitory G protein-
coupled receptors that can be expressed on mast cells. Mast cells can express the following
cytokine and chemokine receptors: interleukin (IL)-3 receptor (IL-3R), IL-4R, IL-5R, IL-9R,
IL10-R, granulocyte-macrophage colony stimulating factor receptor (GM-CSFR), interferon-γ
receptor (IFN-γR), C-C chemokine receptor 3 (CCR3), CCR5, C-X-C chemokine receptor 2
(CXCR2), and CXCR4. In addition, amongst others, mast cells can express complement
receptors, nerve growth factor receptors, and Toll-like receptors (TLRs) (2, 4).
Mast cell: Development
Mast cells originate from pluripotent hematopoietic stem cells in the bone marrow.
CD34+ mast cell precursors circulate in the blood until they migrate into tissues where they
mature into long-living effector cells. According to common paradigm, maturation of precursors
in the tissues is dependent upon binding of their cell surface-bound c-kit receptors to stem cell

5
factor (SCF). Interaction with fibroblasts, stromal cells, and endothelial cells, which express
SCF on their surface, drives mast cell maturation. Thus, SCF and c-kit signaling are considered
to be central for both human and murine mast cell development (2). Importantly, murine mast
cell hyperplasia requires IL-3 (5). In addition to SCF and IL-3, the cytokines IL-4, IL-9, IL-10,
and IL-13 are regarded as mast cell growth factors. In the presence of SCF or IL-3, they act
synergistically to drive mast cell proliferation and differentiation. These cytokines alone,
however, cannot support the differentiation or survival of mast cells (5).
Hu et al. have determined that, as triggers or regulators, inflammatory mediators and
cytokines function as crucial determinants of mast cell development. As a result, they advance
the idea that mast cell development cannot be defined only in terms of mast cell growth factors
(5). Consistent with this, recent evidence indicates that IL-3 stimulation of bone marrow cells
induces the production of tumor necrosis factor (TNF), an important mast cell survival factor
both in vitro and in vivo (6). Moreover, cytokines, such as IL-4, IL-5, and IFN-γ, influence mast
cell phenotype and behavior. IL-4 upregulates the expression of FcεRI. In the presence of SCF,
IL-5 enhances proliferation, whereas exposure to IFN-γ correlates with a decrease in mast cell
number. Therefore, differential expression of homing receptors, tissue-specific expression of
SCF, and the cytokine milieu together likely define the heterogeneity of differentiation and
distribution of mast cells in specific tissues (2).
Mast cell: Activation
The best characterized pathway of mast cell activation is that ensuing IgE-mediated
crosslinking of the membrane-bound FcεRI. Crosslinking can be mediated by polyvalent antigen
specifically recognized by membrane-bound IgE molecules. Alternatively, unspecific

6
crosslinking can be mediated through interaction with superantigens (4). For example, the
endogenous superallergen protein Fv (pFv), which is a human sialoprotein that is found in
normal liver and largely released in the intestinal tract in patients with viral hepatitis, induces
histamine from human lung mast cells (7). Interestingly, in the presence of increased free IgE
levels or IL-4, the surface expression of FcεRI on mast cells is upregulated, thereby enhancing
the activation of these cells (2). Independent of crosslinking of FcεRIs, mast cell activation
induced by IgE binding alone is a matter of debate (4, 8). Nonetheless, Kalesnikoff et al. have
demonstrated that engagement of a single FcεRI with monomeric IgE stimulates signaling
pathways that induce cytokine production and regulate survival (9). Along with the FcεRI, mast
cells express activating immunoglobulin G (IgG) receptors. In mice, Fcγ receptor-mediated
activation is driven by engagement with primarily IgG1 antibodies. On the other hand, studies
have shown that the IgG receptor FcγRIIB negatively regulates IgE-mediated mast cell
activation. In support of this, FcγRIIB-deficient mice are characterized by increased
anaphylactic reactions and higher susceptibility to allergic rhinitis (4).
In addition to immunoglobulins, mast cells can be activated by exogenous and
endogenous stimuli, such as pFv (7). Mast cells are activated by neurotrophin through the high-
affinity nerve growth factor receptor TRKA and by complement component 3a (C3a) and C5a
through C3a receptor (C3aR) and C5aR. They express TLR-1, -2, -3, -4, -6, -7, and -9 and
consequently are activated by corresponding ligands (2, 4). Based upon the ligand, associated
intensity of signal, and the cytokine milieu, the profile, as well as amount, of mediators released
by mast cells can change drastically. This is exemplified by increased mediator release
associated with the presence of increasing amounts of SCF (2).

7
FcεRI pathway
FcεRI-mediated signaling is integral to the activation of mast cells and downstream
effector functions. The canonical signal transduction pathway originates with the binding of
multivalent antigen to IgE-occupied FcεRIs. Upon engagement of the FcεRI α chain (FcεRIα)
subunit with antigen, Lyn kinase is recruited into closer proximity of the FcεRI. As a result,
phosphorylation of the tyrosine residues of the immunoreceptor tyrosine-based activation motifs
(ITAMs) in the FcεRI β- and γ-chains occurs. These ITAMs are responsible for tethering of
Spleen tyrosine kinase (Syk) to the FcεRI. The Src-homology 2 (SH2) domains of Syk are
involved in this interaction with ITAMs. Subsequently, in addition to phosphorylation by Lyn,
trans- and auto-phosphorylation of the catalytic domain of Syk enhances the catalytic activity of
the kinase. Thereafter, Lyn and Syk phosphorylate the transmembrane adaptor molecule Linker
for Activated T cells (LAT), allowing for the association of a protein complex that includes the
cytosolic adaptor molecules SH2-domain-containing Leukocyte Protein of 76 kDa (SLP-76),
SH2 domain-containing transforming protein C (SHC), Growth factor receptor-bound protein 2
(Grb2), and Grb2-related Adaptor protein (GADS), the guanine nucleotide-exchange factors
(GEFs) Vav and Son of Sevenless homologue (SOS), and the signaling enzyme phospholipase
Cγ1 (PLCγ1). Hydrolysis of phosphoinositol-4,5-bisphosphate (PIP2) by PLCγ1 generates the
secondary messengers inositol-1,4,5-triphosphate (IP3) and diacylglycerol (DAG) that induce the
mobilization of Ca2+
and the activation of Protein Kinase C (PKC), respectively. Together, these
events stimulate degranulation. Concurrently, the activation of the mitogen-activated protein
kinase (MAPK) pathway is stimulated through the GEF-mediated exchange of guanosine
diphosphate (GDP) for guanosine triphosphate (GTP) with the small GTPase Ras. Activated Ras
can then transmit signals to Raf and other downstream elements of the MAPK pathway.

8
Activation of the mitogen-activated protein kinases (MAPKs) extracellular-regulated kinase 1
(Erk1) and Erk2, JUN amino-terminal kinase (JNK), and p38 induce the activation of
phospholipase A2 (PLA2) and transcription factors that are linked to eicosanoid generation and
the production of cytokines, respectively. In addition, increases in intracellular Ca2+
levels can
activate transcription factors, thereby leading to increased cytokine secretion (Fig. 1.1) (10-13).

9
Principal Signaling Cascade of Mast Cell Activation
Figure 1.1. Binding of multivalent antigen to IgE-occupied FcεRIs leads to the activation of a
myriad of proteins. The phosphorylation of LAT allows for the formation of a multi-protein
complex, encompassing cytosolic adaptors, guanine nucleotide exchange factors, and the
signaling enzyme PLCγ1. The hydrolysis of PIP2 by activated PLCγ1 generates secondary
messengers that induce Ca2+
mobilization and the activation of PKC. Thereafter, the effector
response of degranulation follows. In parallel, the GEF-mediated exchange of GDP for GTP
with Ras promotes the activation of Raf and downstream MAPK elements, which are involved in
eicosanoid generation and cytokine production. Reprinted by permission from Macmillan
Publishers Ltd: Nat. Rev. Immunol., Vol. 6, Issue 3, pp. 218-30, copyright 2006.

10
Mast cell: Mediators and effector function
Depending upon the type and strength of stimulation, mast cells release different patterns
of inflammatory mediators. Some of these mediators are stored in cytoplasmic granules. Others
are produced de novo following activation. Overall, mast cell mediators can be categorized into
the following classes: 1) preformed substances, 2) newly synthesized lipid mediators, and 3)
cytokines and chemokines (Table 1.1). Importantly, some cytokines cannot be exclusively
categorized, for they are stored in granules as preformed molecules. For example, TNF-α occurs
both preformed and in a newly synthesized form (2, 4).
Category Specific Molecules
Preformed mediators Histamine
Neutral proteases (tryptase, chymase,
cathepsin G, carboxypeptidase A)
Proteoglycans (heparin and chondroitin
sulfates)
Cytokines (IL-4, TNF-α)
Lipid mediators/Eicosanoids Leukotrienes (LTA4, LTB4, LTC4,
LTD4, and LTE4)
Prostaglandins (PGD2, 9α,11β-PGF2)
Platelet-activating factor
Cytokines/Chemokines Cytokines (IL-3, IL-4, IL-5, IL-6, IL-8,
IL-10, IL-13, GM-CSF, TNF-α)
Chemokines (IL-8/CXCL8, CCL2,
CCL3, CCL5)
Table 1.1. Mast cell mediators. The 3 main categories of mediators (left) are divided into
subclasses of mediators (right). Representative examples of each subclass are presented in
parentheses.

11
Preformed mediators
Proteoglycans, neutral proteases, and amines are stored as preformed mediators in
cytoplasmic granules. Proteoglycans include heparin and chondroitin sulfates. In great quantity
in granules, proteoglycans complex with histamine, proteases, and other granule contents due to
their negative charge. The physiological role of heparin is not well understood. In medicine, it
is generally utilized as an anti-coagulant; however, because anti-coagulation of blood is
principally mediated by endothelial cell-derived heparan sulfate proteoglycans, the exact role of
heparin stored within the granules of mast cells remains poorly defined (14). Recently, a role for
heparin in immune defense against bacterial pathogens and other foreign particles has been
proposed (15). Upon release into the extracellular environment, proteoglycans dissociate from
histamine in the cytoplasmic granules, exchanging sodium ions in its place. Histamine
modulates smooth muscle contraction and mucus secretion. It also has effects on endothelial
cells and nerve endings (2). Depending upon the pattern of histamine receptor expression in T
cells, histamine can stimulate the production of T helper 1 (Th1) cytokines, such as IFN-γ, or
Th2-specific cytokines, such as IL-4 and IL-13. Thus, through its influence on cytokine
production, histamine can have pro-inflammatory, as well as anti-inflammatory, effects.
Interestingly, recent evidence has suggested that mast cell-derived histamine mediates its pro-
inflammatory effects via suppression of CD4+ CD25
+ regulatory T cells (4). Neutral proteases,
which compose the majority of preformed content in granules, include tryptase, chymase,
cathepsin G, and carboxypeptidase. Unlike MCTC, MCT contain granules that are composed of
tryptase alone. In vivo, the function of tryptase has yet to be discovered. Reports have indicated
that, in vitro, it plays a role in the digestion of fibrinogen, fibronectin, prourokinase, pro-matrix
metalloproteinase 3, protease-activated receptor 2, and complement component C3.

12
Additionally, it has the capacity to activate fibroblasts, promote the accumulation of
inflammatory cells, and potentiate histamine-induced airway bronchoconstriction (2).
Lipid mediators/Eicosanoids
Rapidly synthesized proceeding mast cell activation, eicosanoid mediators are generated
from endogenous membrane stores of arachidonic acid, which is released by PLA2. PLA2 is
activated downstream of the MAPK family of serine/threonine kinases (10). Arachidonic acid
can be converted into prostaglandin 2 (PGD2) through the action of cyclooxygenase (COX) and
PGD endoperoxide synthase-1 and -2. PGD2 functions as a bronchoconstrictor and
chemoattractant for eosinophils and basophils. Its metabolite 9α,11β-PGF2 plays a role in the
constriction of coronary arteries. Alternatively, arachidonic acid can be converted into
leukotriene A4 (LTA4) via the 5-lipoxygenase pathway in partnership with 5-lipoxygenase
activating protein. LTA4 can be further metabolized to LTB4 or conjugated with glutathione to
form LTC4. LTC4 is the parent compound of the cysteinyl leukotrienes (CysLTs). The CysLT
subfamily includes LTD4 and LTE4. LTB4 regulates the chemotaxis of neutrophils and effector
T cells. CysLT1 and CysLT2 act as potent bronchoconstrictors. They also increase vascular
permeability, trigger mucus production, and attract eosinophils. All of these effects are mediated
through the binding of LTs to G protein-coupled receptors (2). Collectively, prostaglandins and
LTs are central for the regulation and attraction of immunocompetent cells (Figs. 1.2 and 1.3).
Cytokines and chemokines
As a key effector cell of inflammation, mast cells secrete a spectrum of cytokines and
chemokines. One of the major cytokines stored and secreted by mast cells is TNF-α. Besides its

13
anti-tumor activity, TNF-α enhances bronchial responsiveness and leads to the upregulation of
adhesion molecules on endothelial and epithelial cells (2). In murine models, TNF-α has also
been implicated in the upregulation of mucus gene expression (4). Amongst a multitude of other
cytokines, mast cells secrete IL-3, IL-5, and GM-CSF that contribute to eosinophil development
and survival. They also secrete IL-6, IL-10, and IL-13. CCL3 and CXCL8 are some of the
chemokines produced by mast cells (Figs. 1.2 and 1.3) (2).
Mast cell: Role in health and disease
Mast cells are integral members of the immune system. Mast cells are central effector
cells in allergic inflammation. Central to the pathogenesis of allergic diseases, including
anaphylaxis, allergic rhinitis, and allergic asthma, is mast cell activation through the FcεRI.
Aggregation of FcεRIs by polyvalent antigen recognized by bound IgE activates mast cells,
subsequently initiating an immediate hypersensitivity reaction, as well as a late-phase reaction.
Occurring within minutes, the immediate reaction is determined by the degranulation of
preformed mediators, including histamine, serine proteases (tryptase and chymase),
carboxypeptidase A, and proteoglycans, and the release of rapidly synthesized lipid mediators.
In the extracellular environment, these mediators contribute to a variety of allergic symptoms.
According to the site of the reaction, these symptoms broadly range from erythema and edema to
bronchospasm and mucus production in the lower airways (2, 4, 10, 16, 17). Clinical hallmarks
encompass vasodilation, markedly increased vascular permeability, contraction of bronchial
smooth muscle, and increased mucus secretion. Vasodilation results from the action of
mediators on local nerves, producing erythema of the skin or conjunctiva. Tissue swelling and
tear production in the eyes are consequences of increased vascular permeability. The contraction

14
of bronchial smooth muscle can cause the obstruction of airflow and wheezing. Airflow
obstruction in the lower respiratory tract can be further worsened by increased mucus secretion.
Mediators can also stimulate nociceptors of sensory nerves of the nose, skin, and airways. As a
result, sneezing, itching, and coughing occur respectively (Fig. 1.2) (18). Late-phase reactions
are mediated by cytokines and chemokines and occur 6 to 24 hours after the immediate reaction.
During this step, mast cells also release growth factors. TNF-α, CCL2, CXCL8, and other
chemokines are involved in the recruitment of other immune cells. TNF-α and IL-5 activate
innate immune cells, and TNF-α, IL-10, and transforming growth factor-β (TGF-β) affect many
aspects of the biology of dendritic cells, T cells, and B cells. Nonetheless, other mast cell-
derived products can have anti-inflammatory or immunosuppressive effects. These include IL-
10 and TGF-β. Mast cell-derived products can also alter structural cells, including vascular
endothelial cells, fibroblasts, smooth muscle cells, and nerve cells (18). Characterized by edema
and leukocytic influx, late-phase reactions contribute to persistent asthma (Fig. 1.3) (2).

15
Early Phase of Allergic Inflammation
Figure 1.2. In this model of the early phase of allergen-induced airway inflammation, FcεRI
aggregation mediated by antibody-antigen complexes activates mast cells, thereby inducing the
immediate release of granule-associated preformed mediators and lipid-derived mediators and
the de novo synthesis of cytokines, chemokines, and growth factors. The rapid secretion of
preformed mediators and eicosanoids promotes bronchoconstriction, vasodilation, increased
vascular permeability, and increased mucus production. Mast cells also contribute to the
transition to the late phase reaction (Fig. 1.3) via recruitment of inflammatory leukocytes.
Reprinted by permission from Macmillan Publishers Ltd: Nature, Vol. 454, Issue 7203, pp. 445-
54, copyright 2008.

16
Late Phase of Allergic Inflammation
Figure 1.3. In this model of the late phase of allergen-induced airway inflammation, activated
mast cells mediate many of the features of early phase reactions (Fig. 1.2). Occurring hours after
allergen exposure, this step of allergic reactions is regulated by the influx of immune cells from
the circulation and the secretion of inflammatory mediators by tissue-resident cells. In
particular, mast cell-derived products upregulate adhesion molecules on vascular endothelial
cells and secrete chemotactic mediators and chemokines. In conjunction with cytokines that
regulate survival, these promote further influx of inflammatory leukocytes into the site of allergic
inflammation. Reprinted by permission from Macmillan Publishers Ltd: Nature, Vol. 454, Issue
7203, pp. 445-54, copyright 2008.

17
In addition to this classical role as an effector cell during the allergic response, mast cells
are also implicated in host responses to pathogens (2). Localized preferentially at sites bordering
bodily surfaces, mast cells are ideally positioned to be the first responder cells during attack by
incoming pathogens. As cells of immune surveillance, mast cells recognize microbial pathogens
through a variety of mechanisms. First, mast cells can directly interact with pathogens or their
components through their cell surface receptors that recognize pathogen-associated molecular
patterns (PAMPs). Such receptors include TLRs. Second, complement receptors or
immunoglobulin receptors on mast cells can bind opsonized bacteria or related products. Lastly,
mast cells can detect endogenous peptides derived from host cells that have been infected or
injured. A prime illustration of this is the induction of histamine release from human lung mast
cells by the endogenous superallergen pFv, which is predominantly released in the intestinal tract
in patients diagnosed with viral hepatitis (7). Upon binding to these elements, mast cell
activation occurs. Consequently, cytokines and chemokines are released, thereby facilitating the
recruitment of immune cells to sites of infection and the elimination of invading pathogens. In
support of this, investigations have shown that mast cell-deficient mice have diminished immune
responses in lung infection models that involve Klebsiella pneumoniae and Francisella
tularensis. Mast cells also eliminate pathogens directly. Opsonized bacteria can be endocytosed
and subsequently killed by mast cells. Through the release of antimicrobial peptides, such as
LL37, mast cells can eliminate invading pathogens (19, 20). In addition, mast cells exhibit
extracellular killing activity in the form of extracellular structures that trap bacteria. These
structures consist of DNA, histones, tryptase, and LL37 (21, 22). In summary, through the
recruitment and activation of immunocompetent cells and direct killing of pathogens, mast cells
play critical roles in the innate immune response against infection. In the context of adaptive

18
immunity, mast cells initiate and coordinate this arm of the immune response through the
recruitment and maturation of dendritic cells and the secretion of T cell polarizing cytokines.
The mast cell-derived product TNF-α upregulates dendritic cell expression of CCR7. This
chemokine receptor is central to the homing of dendritic cells to lymph nodes, where its ligands
CCL19 and CCL21 are produced. TNF-α also drives the maturation of dendritic cells.
Similarly, upon activation, mast cells drive the differentiation of Th2 cells through the secretion
of polarizing cytokines, such as IL-4, IL-10, and IL-13. Another potential role for mast cells in
the adaptive immune response involves their ability to interact with T cells as antigen-presenting
cells. However, this possibility is clouded with controversy (4, 19).
Lastly, recent studies have associated mast cells with autoimmune diseases, fibrosis, and
wound healing. In particular, research on experimental allergic encephalomyelitis (EAE), an
animal model of multiple sclerosis, has incited interest in the role of mast cells in the initiation or
propagation of autoimmune disease. Past observations have provided suggestive evidence for a
role for mast cells in autoimmune diseases of the central nervous system. These include data
providing an association between mast cell numbers and distribution with the development of
multiple sclerosis or EAE. However, the association remained indirect until recent studies
performed by Brown and colleagues. This group demonstrated that W/WV mice, which lack
mast cells, develop EAE less severely and later. Moreover, the reconstitution of W/WV mice
with immature mast cells rescued EAE susceptibility. Thus, this line of evidence highly suggests
an essential role for mast cells in the pathogenesis of autoimmune diseases. Because of their
ability to shape the cytokine milieu, mast cells have also been implicated in arthritis and bullous
pemphigoid. As in the case of fibrosis and wound healing, the autoimmune skin disease of

19
bullous pemphigoid requires neutrophil recruitment, which can be regulated by mast cell-derived
cytokines (23, 24).
Ca2+
Signaling
Regulation of Ca2+
homeostasis
A myriad of hormones, neurotransmitters, paracrine signals, and other stimuli impinge on
the surface membrane of cells. A broad array of intracellular secondary messengers is generated
to orchestrate cellular responses induced thereafter. Of these signaling molecules, Ca2+
is a
universal messenger that regulates a variety of physiological responses (25, 26). In immune
cells, calcium signals are involved in the control of cell activation, differentiation, proliferation,
transcriptional programs, and effector functions (27). They can also activate pathways that
culminate in cell death (26). Therefore, the use of Ca2+
for intracellular signaling requires tight
local and global control of cytosolic Ca2+
concentration. Cells have resultantly evolved intricate
cellular mechanisms for maintaining the balance of net intracellular Ca2+
levels (25).
To elucidate these complex systems that maintain a balance of Ca2+
uptake, intracellular
storage, and efflux, proteins involved in Ca2+
transport have been identified (26). In immune
cells, following the engagement of immunoreceptors with antigen or antigen-antibody
complexes, the enzyme PLC is activated and drives the rapid production of IP3. Binding of IP3
to the IP3 receptor (IP3R) localized in the ER stimulates the emptying of ER Ca2+
stores into the
cytosol. This event is correlated to a very small, transient cytosolic Ca2+
rise. SOCE from the
extracellular space occurs following this influx of Ca2+
from intracellular stores. SOCE through
Ca2+
release-activated Ca2+
(CRAC) channels is an important mechanism to sustain an increased

20
intracellular Ca2+
concentration (Fig. 1.4). Importantly, increases in cytosolic Ca2+
are critical
for the activation of the transcription factor Nuclear Factor of Activated T cells (NFAT) and the
altered expression of cytokines, chemokines, and many other NFAT-targeted genes, all of which
are important for the development of a productive immune response (28, 29). In the case of mast
cells, the aggregation of FcεRIs by IgE and antigen initiates the signaling pathway leading to the
activation of PLC. The resultant FcεRI-triggered biphasic increase in cytosolic Ca2+
is an
essential step during mast cell activation and in the generation of productive mast cell responses,
particularly degranulation. The close correlation between Ca2+
mobilization and gene expression
of cytokines and chemokines underscores the importance of cytosolic Ca2+
increases for mast
cell function. Moreover, Ca2+
mobilization is central for driving the degranulation of preformed
mediator-containing vesicles and de novo synthesis of lipid mediators (30-32).
In addition to the aforementioned mechanism of immunoreceptor-induced Ca2+
mobilization, several pathways for Ca2+
movement between the cytoplasm and ER and between
the cytoplasm and extracellular space exist to regulate intracellular Ca2+
concentration. Like
IP3R channels, ryanodine receptor channels (RyR) release Ca2+
from the ER. On the other hand,
sarcoplasmic/endoplasmic reticulum Ca2+
-ATPase (SERCA) pumps take up Ca2+
from the
cytoplasmic space. Ca2+
is also removed from the cytoplasm by plasma membrane Ca2+
-ATPase
(PMCA) pumps and, in select cells, via a Na2+
-Ca2+
exchanger (NCX). Dependent upon cell-
type, Ca2+
can enter the cytoplasm from the extracellular fluid through a wide spectrum of Ca2+
channels. For example, voltage-gated Ca2+
(CaV) channels execute a disparate array of functions
in electrically excitable cells; however, their role in electrically non-excitable cells, such as
lymphocytes and mast cells, remains controversial (Fig. 1.5) (25).

21
Activation of Store-operated Ca2+
Entry
Figure 1.4. Activated cell surface receptors stimulate the activity of PLC, thereby promoting the
hydrolysis of PIP2 and the concomitant generation of IP3. Thereafter, a decrease in ER Ca2+
levels results from the depletion of IP3-sensitive ER Ca2+
stores. This fall in Ca2+
concentration
is sensed by STIM1, which transmits an activation signal to Orai1, the pore-forming subunit of
the CRAC channel. CRAC channels are highly selective for Ca2+
and mediate the influx of Ca2+
from the extracellular space. Reprinted by permission from Macmillan Publishers Ltd: Nat. Rev.
Drug Discov., Vol. 9, Issue 5, pp. 399-410, copyright 2010.

22
Principal Pathways of Ca
2+ Movement
Figure 1.5. Ca2+
release from the ER is released through RyR or physiologically stimulated
IP3R channels. Ca2+
uptake is mediated via SERCA pumps. From the extracellular fluid, Ca2+
flows into the cytoplasmic space through store-operated Ca2+
channels and a variety of other
Ca2+
channels. The removal of Ca2+
occurs through PMCA pumps and, in select cell types, via
NCX. Overall, the generation of IP3 by PLC leads to depletion of ER Ca2+
reserves and the
subsequent activation of store-operated Ca2+
channels. Importantly, the representative Ca2+
transporters participate in modulating the balance between stimulation and inactivation of store-
operated Ca2+
channel activity by shaping the intracellular Ca2+
gradient. Reprinted from Trends
Biochem. Sci., Vol. 32, P.G. Hogan and A. Rao, Dissecting ICRAC, a store-operated calcium
current, pp. 235-45, copyright 2007, with permission from Elsevier.

23
Importantly, the rate at which Ca2+
enters into the cell through open CRAC channels is
determined by the Ca2+
concentration gradient and membrane potential. Various channels can,
therefore, modulate calcium signals through their capacity to change the membrane potential.
Although they lack the ability to directly conduct Ca2+
, K+ channels and the non-selective cation
channel transient receptor potential (TRP) channel, melastatin subfamily 4 (TRPM4) serve such
a purpose. TRPM4 is activated by Ca2+
(33). Mitochondria can also modulate CRAC channel
activity through sequestration of Ca2+
and concomitant buffering of intracellular Ca2+
concentration. In support of this, studies have shown that uptake and release of Ca2+
by
mitochondria are necessary for store depletion-induced sustained increases in intracellular Ca2+
concentration in human T cells. The mitochondrion rapidly withdraws Ca2+
from the cytosol
because of the negative potential across its inner membrane. In particular, recent investigation
proposes that mitochondria facilitate CRAC channel opening by promoting increasing levels of
store depletion and, thereafter, by sustaining the opening of these channels through the
prevention of Ca2+
-dependent inactivation. Potential mechanisms implicate a direct role for
mitochondria in decreasing free Ca2+
in the proximity of the inactivation site or an indirect role
through the local production of adenosine triphosphate (ATP), a Ca2+
buffer (25, 30, 33).

24
Store-operated Ca2+
entry
One of the principal mechanisms of Ca2+
influx into cells of the peripheral immune
system is the cellular process, known as SOCE. In 1986, James Putney proposed that, in non-
excitable cells, the depletion of ER Ca2+
stores stimulates sustained Ca2+
influx across the PM
independently of receptor engagement, production of secondary messengers, or the transient
peak in intracellular Ca2+
concentration induced by the release of Ca2+
from intracellular stores.
Since then, biophysical experiments have characterized the unique electrophysiological profile of
CRAC channels and confirmed their expression in lymphocytes and mast cells. However, for a
long time, the molecular identity of the players which mediate the Ca2+
release-activated Ca2+
current (ICRAC) eluded biochemical definition (33).
In recent years, the identification of Stromal Interaction Molecule 1 (STIM1) and the
Orai1 protein as integral parts of the ER-to-PM signaling system, necessary for SOCE, has
accelerated research in the field of calcium signaling (25). Anchored in the ER, STIM1 is a
single-spanning membrane protein with a Ca2+
-binding EF-hand motif. Investigations indicate
that STIM1 functions as the sensor of ER luminal Ca2+
levels and that its reorganization in the
ER allows it to transduce information directly to the PM. In specific, it migrates within the ER
membrane to sites closely apposed to the PM and reorganizes into punctae that interact with Ca2+
influx channels and activate them. At the PM, its interactions with the CRAC channel open the
gates for SOCE. The Orai1 protein is described as a tetra-spanning PM protein that functions as
the pore-forming subunit of the highly selective CRAC channel in the PM (Fig. 1.6) (25, 26).

25
Domain Structures of STIM1 and Orai1
Figure 1.6. Anchored in the ER, STIM1 contains a single-spanning transmembrane domain.
The luminal portion of STIM1 is characterized by a Ca2+
-binding EF hand sequence, a vestigial
EF hand motif, and a sterile α motif (SAM) domain that is central to STIM1 oligomerization.
The cytoplasmic portion of STIM1 contains a number of predicted functional domains, including
two coiled-coil domains, an ezrin-radixin-moesin (ERM) domain, and serine or proline-rich and
lysine-rich segments. In addition, it contains the CRAC activation domain (CAD) that is
essential for the gating of Orai1. In the case of Orai1, it is a PM-embedded protein with four
transmembrane segments (TM1-TM4). Represented by a purple dot is the point mutation
(R91W) responsible for Severe Combined Immunodeficiency of CRAC channel-deficient
patients. The red dot represents glutamate 106, which has been implicated in ion permeation.
The yellow dots represent aspartate residues 112 and 114 and glutamate residue 190 that are
crucial determinants of ion selectivity. Reprinted by permission from Macmillan Publishers Ltd:
Nat. Rev. Drug Discov., Vol. 9, Issue 5, pp. 399-410, copyright 2010.

26
Despite experimental evidence showing that STIM1 and Orai1 are necessary and
sufficient for SOCE, many questions linger about the details of the coupling mechanism between
these proteins (34). A structural analysis, conducted by Varnai and colleagues, of STIM1-Orai1
interactions has implicated the presence of additional molecular components within the STIM1-
Orai1 complex (35).
To further complicate the matter, recent findings have supported the idea that TRP cation
(TRPC) channels function as store-operated channels. TRPC channels are non-selective, Ca2+
-
permeable cation channels which are activated through stimulation of G protein-coupled
receptors, as well as tyrosine-phosphorylated receptors. Reports have indicated that silencing of
TRPC1 and TRPC3 by antisense RNA correlates with diminished Ca2+
influx under stimulation
conditions of receptor triggering or passive store depletion. Consistent with this, siRNA-
mediated knockdown of these channels in combination or individually markedly inhibits SOCE.
Of interest, Orai1 forms a complex with STIM1 and TRPC1. STIM1 itself binds to TRPC
channels TRPC1, TRPC2, TRPC4, and TRPC5 but not TRPC3, TRPC6, and TRPC7, and it is
involved in the gating of TRPC1. In line with this, knockdown of STIM1 by siRNA inhibits the
activity of TRPC1. Parallel investigation with other TRPC channels demonstrates their
regulation by STIM1 (36-38). Collectively, these data suggest that all TRPC channels, with the
exception of TRPC7, function as store-operated channels (36). Nonetheless, controversy
remains, considering that the properties of native TRPC channels are different from those of the
channels mediating the ICRAC (36-38).
Therefore, the identification of other molecules, which regulate the operation of CRAC
channels, will allow us to better understand how the interactions between STIM1, Orai1, and

27
TRPC channels occur and where they take place within cells. Most importantly, this will allow
us to have an impact on the diseases that associate with malfunctioning states of SOCE.
The absence of Ca2+
influx through CRAC channels can severely compromise immune
cell activation, proliferation, and effector functions. This is underscored by the existence of one
form of severe-combined immunodeficiency (SCID) syndrome, whose pathological roots trace to
defective CRAC channel function. In T lymphocytes from the patients who are affected by this
form of SCID, a missense mutation and an Arginine-to-Tryptophan amino acid (a.a.) substitution
at a.a. position 91 in the first transmembrane domain of the Orai1 protein result in the ablation of
all CRAC channel activity (28, 29). Along with the devastating consequences of CRAC channel
impairment in some patients with severe-combined immunodeficiencies, the significance of
SOCE in the pathogenesis of immune-related disease is underscored by its role in
hypersensitivity disorders of the immune system, particularly with a focus on mast cell activation
and the generation of allergic reactions. In mast cells, FcεRI stimulation induces the liberation of
intracellular Ca2+
stores and the phenomenon called “SOCE”. The consequential rise in
cytoplasmic Ca2+
is central for driving the release of a battery of paracrine signals, chemokines,
and cytokines, which help to sculpt subsequent allergic inflammation. Murine models which
lack SOCE signaling components exhibit defective mast cell function and allergic responses.
For example, mice lacking STIM1 or Orai1 are characterized by severely impaired histamine
release and leukotriene production, reduced TNF-α secretion, and an inability to mount a
subcutaneous anaphylactic response (39, 40).

28
Activation of store-operated Ca2+
channels
As discussed earlier, despite the intensive efforts of early investigations, the identity of
the molecular players that participate in the mechanism of CRAC channel activation have
remained shrouded in mystery until recent years. The identification of STIM1 and Orai1 as
major components of the SOCE pathway has significantly advanced the field of Ca2+
signaling
research (28, 29). It has provided the molecular “bridges” for the search for accessory proteins
that are involved in the molecular communication between STIM1 and Orai1. Moreover, it is
driving researchers to reevaluate some of the models, which were proposed in the period
predating the discovery of STIM1 and Orai1, of CRAC channel activation (41, 42).
To explain the mechanism of communication between intracellular Ca2+
stores and the
PM, the following three models have been proposed: 1) the diffusible messenger hypothesis, 2)
the vesicular fusion hypothesis, and 3) the secretion-like conformational coupling hypothesis
(Fig. 1.7) (41, 42). The diffusible messenger hypothesis proposes the existence of a small
molecular messenger that transmits an activation signal, which links intracellular stores to CRAC
channels in the PM (43-45). Predicted to be stored within the ER, this messenger is released into
the cytosol upon store depletion (41, 43, 44). On the other hand, the vesicular fusion hypothesis
proposes that this activation signal may be synthesized de novo in the cytosol upon store
depletion. This model suggests that, at resting stages, CRAC channels are absent in the PM but,
upon depletion of Ca2+
stores, are integrated into the PM via exocytosis (46, 47). Past evidence
shows that inhibitors of exocytosis block the ICRAC. Nonetheless, recent studies report that Orai1
is constitutively present in the PM and, thus, indicate that trafficking of the CRAC channel to the
PM after the emptying of stores is not essential for the activation of SOCE. Whether the
trafficking of STIM1 or the ER to the PM necessitates the molecular machinery of exocytosis

29
has not been determined though. Strikingly, genetic analyses indicate that the molecular
candidates, which are involved in the inhibition of Ca2+
influx but not ICRAC, are classically
associated with vesicular transport. These potential regulators include soluble N-
ethylmaleimide-sensitive factor attachment proteins receptor (SNARE) proteins, which have
established roles in vesicle transport, membrane docking, and fusion (41, 45-47). Like the
vesicular fusion hypothesis, the secretion-like conformational coupling model implicates a
potential role for SNARE proteins in the activation of CRAC channels. This model proposes
that emptying of intracellular Ca2+
stores triggers a migratory process of the peripheral ER to the
PM (48-52). In early versions of this model, the optimum juxtaposition between the membranes
of these two compartments results in a coupling reaction between IP3Rs in the ER and CRAC
channels in the PM. Recently, some scientists have proposed that STIM1 mediates this coupling
reaction (45). Some have also suggested a role for the peripheral actin cytoskeleton, as well as
SNARE proteins, in the regulation of this coupling reaction (42). Importantly, studies show that
stabilization of the cytoskeleton inhibits the coupling reaction between IP3Rs to Ca2+
channels
but that disruption of the cytoskeleton assists the binding of these molecular components.
Experimental evidence also shows that actin-stabilizing agents interfere with the activation of
SOCE (41). Overall, researchers have demonstrated that the cortical actin network modulates
SOCE in numerous cell types. Consequently, it will be important to test whether the
cytoskeleton prevents the coupling reaction between intracellular Ca2+
stores and the PM or the
fusion of vesicles, harboring CRAC channels, with the PM. In addition, it will be important to
determine if actin-reorganizing proteins and SNARE proteins direct the precise interaction
between the membranes of transported organelles or vesicles and the PM. Defining such roles is
central to the clarification of many aspects of the models presented above (42).

30
Models of CRAC Channel Activation
Figure 1.7. (A) The conformational coupling hypothesis proposes that, upon ER Ca2+
store
depletion, physical interactions occur between the ER and PM, inducing the activation of CRAC
channels. Importantly, researchers have suggested that reorganization of the actin cytoskeleton
could regulate when, where, and how these interactions take place. (B) The vesicular fusion
model postulates that whole CRAC channels or components of them may be sequestered in
cytoplasmic vesicles that are trafficked to the PM either for integration into the PM or for
transient interactions that activate Ca2+
influx upon intracellular Ca2+
store depletion. (C) The
diffusible messenger model hypothesizes that a secondary messenger, which is released from the
ER upon Ca2+
reservoir depletion, diffuses to the PM, where it is involved in the gating of CRAC
channels. [With kind permission from Springer Science+Business Media: <Pflugers Arch., On
the activation mechanism of store-operated calcium channels, Vol. 453, 2006, pp. 303-11, A.B.
Parekh, Fig. 2>.].

31
Ca2+
Signaling and the Actin Cytoskeleton
In the cell, actin exists in two forms: 1) globular (G-actin) and 2) filamentous (F-actin).
In an ATP-dependent manner, monomeric G-actin is polymerized to form microfilaments of F-
actin. Central to the regulation of the actin cytoskeleton is Ca2+
signaling. In the absence of
activated PLC, which can be considered a starter molecule of the Ca2+
mobilization process, PIP2
suppresses the activity of the actin-severing proteins gelsolin and profilin (53-55). In a
potentially related mechanism, the binding of Ca2+
to gelsolin activates this protein’s actin-
severing activity (55). The function of actin-stabilizing proteins, including α-actinin, is
suppressed in the context of higher intracellular Ca2+
levels (56, 57). Also, select interactions
that stable actin filaments are differentially regulated at varying concentrations of intracellular
Ca2+
. For example, low Ca2+
concentrations promote the interaction between caldesmon and
tropomyosin, whereas high concentrations impede the interaction (57). In summary, these data
suggest that the reorganization of the actin cytoskeleton is highly sensitive to localized changes
in intracellular Ca2+
concentration. A prime example of this concept in action is chemotaxis
during which the leading edge of cells is governed by microdomains of high Ca2+
concentration,
which catalyzes the breakdown of actin by actin-severing proteins, and the trailing end of cells
consists of less Ca2+
-concentrated sites, which permit the maintenance of adhesive structures and
overall cell shape (58).
Actin cytoskeletal reorganization has been implicated in models of CRAC channel
regulation. Nonetheless, little is known about actin-modulatory proteins that are involved in this
process or how actin regulates CRAC channel function and, thereby, downstream immune cell
responses. Classic experiments that have analyzed the effect of cytoskeleton disruption on
SOCE have provided conflicting evidence. Though the majority of reports have indicated that

32
actin depolymerization agents do not affect SOCE, studies with cytochalasins have shown that
this group of actin depolymerizing agents attenuates SOCE (59). Equally intriguing, mast cells
of mice deficient in Wiskott-Aldrich syndrome protein (WASP), a key regulatory protein of F-
actin assembly, are characterized by diminished Ca2+
mobilization, degranulation, and cytokine
secretion (60). These results strongly suggest that unidentified actin-binding members of a store-
operated calcium influx complex are waiting to be discovered.

33
Drebrin
Structure of Drebrin
Characterized by unique protein domains and an ability to induce dramatic cytoskeletal
rearrangements, drebrin can be considered one of the molecular candidates, which potentially
plays a role in the communication between STIM1 and Orai1. Most of our knowledge about
drebrin has been gathered from neuroscience studies. Also termed developmentally regulated
brain protein, drebrin is fittingly defined by pronounced expression in neurons. Nonetheless, it is
expressed in numerous non-neuronal cell types. Drebrin is a member of the actin-
depolymerizing factor-homology (ADF-H)/cofilin family of actin-binding proteins. In
mammals, drebrin is found in the following two splice-variant isoforms: 1) an adult form
(Drebrin A) and 2) an embryonal form (Drebrin E). Drebrin A mRNA differs from drebrin E
mRNA only by the presence of an internal 138-nucleotide sequence insert, which is absent from
drebrin E mRNA. As a result, drebrin E is detected at an approximate molecular weight of 115
kilodaltons (kDa), whereas drebrin A is traced at approximately 125 kDa. Drebrin is
distinguished by the following 4 major protein domains/motifs: 1) an amino-terminal ADF-H
domain, which allows it to interact with actin, 2) a proline-rich region, 3) a Homer-binding
domain, and 4) a carboxyl-terminal putative SH2-binding domain. Importantly, a central 85-a.a.
sequence, spanning a.a. residues 233-317, has been attributed with being necessary and sufficient
for drebrin to bind and remodel F-actin (Fig. 1.8) (61). On a related note, drebrin shares
sequence homology with mammalian actin-binding protein 1, which is marked by a Src-
homology 3 domain (62).

34
Domain Structure of Drebrin
Figure 1.8. Drebrin A and E are distinguished by the presence or absence of an internal insert,
respectively. A member of the ADF-H/cofilin family of actin-binding proteins, the protein
drebrin is characterized by an ADF-H domain that mediates binding to F-actin. The actin-
binding domain is a central 85 a.a. sequence that is both sufficient and required for the binding
and remodeling activity of drebrin. Drebrin also contains a proline-rich (P-rich) region, which
may potentially bind the SH3 domains of interacting proteins, and a homer-binding motif.

35
Function of Drebrin
In terms of function, biochemical studies have demonstrated that drebrin attaches to the
side of F-actin. Drebrin blocks the interaction between actin and myosin. In addition, it
competes with tropomyosin, fascin, and α-actinin, decreasing their actin-binding activity.
Fascinatingly, unlike F-actin that is bound to tropomyosin, F-actin which is bound to drebrin is
susceptible to being taken apart by the actin-severing protein gelsolin. This correlates with
observations of drebrin-transfected fibroblasts. The morphology of these cells transforms
dramatically, likely involving an outstanding change in the arrangement of F-actin. In these
cells, F-actin forms thick curving bundles, and exogenous drebrin directly interacts with F-actin.
This evidence supports a role for drebrin in actin dynamics (63). Other studies have established
a role for drebrin in the shaping of neuronal dendrites and in the recruitment of F-actin and post-
synaptic density-95 (PSD-95) to dendritic filopodia (64-71). Dependent upon Ras activation,
drebrin-induced spine destabilization is involved in the maintenance of stability and plasticity of
dendritic spines (72). Decreased drebrin expression correlates with the morphological changes
of spines in neurodegenerative diseases, such as Alzheimer’s disease and Down’s syndrome (73).
Interestingly, drebrin A-specific knockout mice exhibit impairment in context-dependent
freezing after fear conditioning, thereby implicating an important role for drebrin A in learning
behavior and generation of memory (74). Additionally, interactions between drebrin and the
microtubule plus-tip protein EB3 are required for neuritogenesis (75). In this light, the loss of
drebrin expression or function may play a role in the pathogenesis of other neurological
disorders.
Recent evidence demonstrates that drebrin stabilizes Connexin-43-containing gap
junctions at the PM (61, 76, 77). This implicates a potential role for drebrin in the establishment

36
of PM domains. In support of this, through knockdown experiments in T cells, recent
investigation has shown that drebrin participates in the polymerization of actin at the
immunological synapse and recruitment of CXCR4. Resultantly, in the absence of drebrin, IL-2
production is decreased. These data provide strong evidence for a functional role for drebrin
during the generation of an immune response. Additional studies with T cells have highlighted a
role for drebrin in Ca2+
signaling and, therefore, immune cell activation. In past studies, we
identified drebrin as a molecular target of 3,5-bis-trifluoromethyl pyrazole (BTP), a known
inhibitor of SOCE. We showed that BTP inhibits the ability of drebrin-overexpressing Chinese
hamster ovary (CHO) cells to develop long filopodia-like membrane extensions, indicating that
BTP inhibits the ability of drebrin to induce plasticity in the actin cytoskeleton. Moreover, we
demonstrated that loss of drebrin protein expression prevents SOCE in T cells at levels similar to
treatment with BTP. In line with this, small interfering RNA (siRNA)-mediated knockdown of
drebrin expression in T cells correlates with decreased NFAT activation. Our identification of
drebrin as a mediator of SOCE has provided insight into the interaction between actin
rearrangement and stimulus-induced Ca2+
mobilization (78).

37
3,5-bis-trifluoromethyl pyrazole (BTP)
Recently, a class of immunosuppressant compounds, that were termed BTPs, was
discovered. Predominantly, the pharmacological profiling of BTPs has been performed with
focus on the BTP derivative BTP2 (Fig. 1.9). Initial studies showed that BTP2 inhibits T cell
activation. Mechanistic exploration revealed that BTP2 blocks the activation of NFAT though it
does not affect other transcription factors, such as Nuclear Factor-κB (NF-κB) or Activator
Protein-1 (AP-1). Interestingly, unlike other NFAT inhibitors, such as FK506 and cyclosporine
A (CsA), BTP2 does not inhibit NFAT activation through direct ablation of calcineurin
phosphatase activity. Instead, BTP2 modulates CRAC channel activity (79, 80). The functional
outcome of this effect is inhibition of the production of Th1 and Th2 cytokines, particularly IL-2,
IL-5, and IFN-γ (79, 80). Through impeding SOCE, BTP2 also attenuated superoxide anion
production in human neutrophils (81). To further understand the mechanism of action of BTP2,
Takezawa et al. have demonstrated that BTP2 decreases Ca2+
influx by depolarizing
lymphocytes with the enhancement of TRPM4 activity (82). In contrast, He et al. have shown
that BTP2 blocks TRPC3 activity in medium devoid of monovalent cations. They have
concluded that BTP2 does not inhibit divalent cation entry through depolarization mediated by
activated monovalent cation entry channels. Moreover, their recordings of single TRPC3
channels suggested that BTP2 diminishes the probability of the channel to open rather than its
pore properties (83). In addition to these findings, our past studies identified the actin-binding
protein drebrin as a target of BTP. We demonstrated that BTP inhibits actin reorganization
mediated by the actin-binding protein and that loss of drebrin protein expression prevents SOCE,
similar to BTP treatment, in T cells (78).

38
Because of the unique ability of BTP2 to modulate SOCE, researchers have tested it as a
potential therapeutic for allergic disease. Scientists at Astellas Pharma have demonstrated that
BTP2 treatment inhibits the production of Th2 cytokines and leukotrienes, both of which are
important in the induction of allergic responses. They have also shown that BTP2 inhibits
eosinophil infiltration into airways in vivo. Observed clinical manifestations of these effects are
decreased bronchoconstriction and airway hyperresponsiveness in different in vivo models (84,
85).

39
Chemical Structure of BTP2
Figure 1.9. The boxed structural components are shared by all members of the BTP family of
chemical compounds. They include the core BTP ring. The derivatives of BTP, however, differ
in the excluded ring structure. In the case of BTP2, it is a thiadiazole ring.

40
Objective of Thesis:
To carve a niche into the area of allergy-related research, the focus of this study is to
understand the effects of BTP2 and the absence of its target drebrin on the biology of mast cells,
the central effector cells of allergic inflammation. This information will provide insight into the
role of drebrin in biological processes that regulate mast cell activation and, thus, facilitate the
design and development of novel pharmaceuticals for mast cell-mediated disease.
Hypothesis: BTP2 inhibits Ca2+
mobilization and mast cell activation through its effect on
drebrin.

41
CHAPTER 2
Materials and Methods

42
Mice
For BTP-related experiments, C57BL/6 mice were used. In the case of Drebrin knockout
(Drebrin-/-
) mouse-related experiments, 129S6/SvEvTac mice were obtained from Taconic USA
and utilized as wild-type (WT) control mice. Drebrin-/-
mice were generated from the 129/SvEv
gene trap embryonic stem (ES) cell clone OST 7352 for Mus musculus drebrin 1 (Dbn1),
provided by the Texas A&M Institute for Genomic Medicine (TIGM, College Station, TX).
With standard methods, the drebrin-/-
mouse line was generated by microinjection of Omnibank
ES cell clones into host blastocysts. Resultant chimeras were bred to C57BL/6 mice. Mice
heterozygous for the gene-trap mutation (genotype drebrin+/-
) were subsequently intercrossed to
produce drebrin-/-
mice. All experiments were carried out in accordance with the regulations of
the Institutional Animal Care and Use Committee (IACUC) at The Pennsylvania State University
and Cornell University.
Cell Culture and Reagents
Rat basophilic leukemia (RBL)-2H3 cells (American Type Culture Collection, Manassas,
VA, USA) were cultured at 37°C in Dulbecco’s Modified Eagle Medium (DMEM)
supplemented with 15% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin, 100
μg/mL streptomycin, and 100 µM non-essential amino acid solution. Mouse BMMCs were
grown from femoral marrow cells of mice as previously described with a few changes (86). In
brief, bone marrow cells were obtained from 6–10-week-old mice and cultured at 37
oC in
DMEM supplemented with 10% heat-inactivated FBS, 100 U/mL penicillin, 100 μg/mL
streptomycin, 100 µM non-essential amino acids, 1 mM sodium pyruvate, 1 mM glutamine, 50
μM 2-β-mercaptoethanol (2-ME), IL-3 (10 ng/mL), and SCF (50 ng/mL) (Peprotech, Rocky Hill,

43
NJ). In the case of drebrin-/-
mouse studies, DMEM was supplemented with recombinant IL-3
(10 ng/mL, Cell Sciences, Canton, MA) alone. Cells were passaged every 2 d by replating the
cells in fresh medium. BMMCs were used for experiments after 4-8 weeks of culture (>95%
mast cells) and were routinely >95% positive for cell surface expression of FcεRI and c-kit as
determined via flow cytometry. To examine the population growth of BMMC cultures, 106
BMMCs were harvested and cultured for 4 weeks (>95% mast cells) at a concentration of
approximately 106 cells/mL as detailed above. Cells were counted weekly with a
hemacytometer. Weekly, the percentage of c-kit+ FcεRI
+ cells was determined as described
above. Then, the total number of mast cells for individual cultures was determined with the
following formula: (total number of cells in culture at timepoint, determined by counting with a
hemacytometer) (% of c-kit+ FcεRI
+ cells of culture at timepoint, determined by flow cytometry).
BTP2 (YM-58483; N-(4-(3,5-bis(Trifluoromethyl)-1H-pyrazol-1-yl)phenyl)-4-methyl-1,2,3-
thiadiazole-5-carboxamide) and other derivatives were synthesized as previously described (78,
87) or were purchased from Calbiochem. Both 2-Aminoethoxydiphenyl borate (2-APB) and
≥99.9% anhydrous dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. For all
BTP2-based experiments, DMSO was utilized for vehicle control purposes at a maximum
concentration of 1 µL/mL (~14 µM or 1:1000 dilution).
Genotyping of Mice
Oligonucleotide primers (long terminal repeat (LTR) reverse, 5’–
ATAAACCCTCTTGCAGTTGCATC-3’; A, 5’–CTTCATCTTTGTCAGTCACCAGC-3’; B,
5’–CCAGTTCTAGGAGAGCCACTATC-3’) (all from Integrated DNA Technologies, USA)
were used in a multiplex reaction to amplify corresponding Dbn1 alleles on mouse chromosome

44
13. Approximately 100 ng of purified mouse tail or ear genomic DNA was used as a template
for polymerase chain reaction (PCR) in a 50 µL reaction volume. Cycling conditions were 94°C
for 60 sec, 60.2°C for 60 sec, and 72°C for 60 sec (38 cycles). Amplified products were
separated on 2% agarose gels and visualized with a Kodak Gel Logic 2200 Imaging System for
genotyping documentation.
Quantitative Real-Time PCR Analysis
For BTP-related studies, BMMCs (5 x 106 cells) were treated with 1 μM BTP2 or vehicle
and cultured for 2 days. Total RNA was prepared from respective BMMC populations, using a
RNeasy Mini Kit (Qiagen Sciences, MD). Complementary DNA (cDNA) was generated using
Ready-To-Go You-Prime First-Strand Beads (GE Healthcare, Buckinghamshire, UK) per
manufacturer’s protocol. Quantitative PCR was performed with the ABI 7300 Real-Time PCR
System using Taqman gene expression assay probes for IL-4, IL-6, TNF-α, and Dbn1 Exons 8-
10 (probe sequence, 5’– AGAAGTCGGAGTCAGAGGTGGAGG -3’; forward primer, 5’–
GGAGGTTAAAGGAGCAGTCTATC -3’; reverse primer, 5’– CGTGGGTTATCAGGCCG -
3’) with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a housekeeping gene (Applied
Biosystems, Branchburg, NJ). Data was analyzed using the Comparative ΔΔCT (threshold cycle)
method. Expression of IL-4, IL-6, TNF-α, and Dbn1 Exons 8-10 was normalized based on the
levels of mRNA for GAPDH and set relative to a calibrator sample. Fittingly, the normalized
relative gene expression levels of samples were then compared with the expression level of WT
populations or vehicle-treated populations, set as 1.

45
Western Blotting
Neocortical homogenates of 6-10-week-old mice were prepared with a plastic
homogenizer and cell strainer. Then, whole homogenate lysates were prepared with a x10
weight-to-volume ratio of lithium dodecyl sulfate (LDS) sample buffer, consisting of 2x NuPage
LDS Sample Buffer (Invitrogen, USA), 2 protease inhibitor tablets/10 mL total buffer (Roche,
USA), 1 mL mammalian cell culture protease inhibitor/10 mL total buffer, 50 µg 3,4-
dichloroisocoumarin/mL total buffer, 1 mM benzamidine, 1 mM sodium orthovanadate, 5.4 mM
sodium pyrophosphate, 50 mM sodium fluoride, 10% β-mercaptoethanol, and 10 mM
dithiothreitol. With the exception of NuPage LDS Sample Buffer and protease inhibitor tablets,
all sample buffer reagents were purchased from Sigma-Aldrich. Aliquots of whole homogenate
lysates were loaded to approximately 100 µg wet weight of cortical tissue per lane with
polyacrylamide gels and were subsequently subjected to Sodium Dodecyl Sulfate
Polyacrylamide Gel Electrophoresis (SDS-PAGE) and western blotting, as previously described
(86). Blots were probed with anti-pan-drebrin monoclonal antibodies (1:5000 dilution; clone
M2F6; Abcam, USA), followed by horseradish peroxidase (HRP)-linked goat anti-mouse Ig.
Thereafter, blots were stripped and reprobed with anti-β-actin control antibodies. Blots were
developed in chemiluminescent reagents and exposed to x-ray film.
In the case of western blotting with BMMC samples, cells were sensitized with 1 μg
mouse IgE anti-dinitrophenyl (DNP)/mL overnight, washed with phosphate-buffered saline
(PBS) solution once, and finally challenged with 100 ng DNP-human serum albumin (HSA)/mL
for indicated times (0-60 min). After challenge, the cells were washed with cold PBS and, then,
lysed in Radio Immuno Precipitation Assay (RIPA) lysis buffer supplemented with phosphatase
inhibitors on ice. To quantitate the protein concentration of whole cell lysates, the bicinchoninic

46
acid (BCA) protein assay (Pierce Biotechnology, Rockford, IL) was performed. Thereafter,
polyacrylamide gels were equally loaded with 20-25 µg protein per lane (depending upon the
experiment), and SDS-PAGE and western blotting were performed. Blots were probed with the
following antibodies: anti-phospho-Lyn (Tyr507
), anti-phospho-Erk1/2 (Thr202
, Tyr204
), anti-
phospho-SAPK/JNK (Thr183
, Tyr185
), anti-phospho-p38 (Thr180
, Tyr182
), and anti-phospho-
PLCγ1 (Tyr783
) (manufacturer’s recommended dilutions; all antibodies from Cell Signaling
Technology, Beverly, MA), followed by HRP-linked goat anti-mouse or goat anti-rabbit Ig
(Jackson Immunoresearch, USA). HRP-linked anti-phospho-tyrosine was also used. Blots were
stripped and reprobed with anti-β-actin or anti-α-tubulin control antibodies. Blots were
developed in chemiluminescent reagents and exposed to x-ray film. Densitometric analysis of
the western blots was performed with ImageJ software, and the data was standardized to the level
of β-actin or α-tubulin. Data for each lane is reported in the following manner: (fold change of
intensity of probed protein for WT sample at 0 min timepoint)/(fold change of intensity of
probed β-actin or α-tubulin for WT sample at 0 min timepoint).
Histological Staining
Skin tissue of approximately 5-8 mm in diameter was removed from the upper left
quandrant of the back of mice and immediately immersed in fixative solution for 2 h at room
temperature with minimal agitation. The fixative solution was composed of 2%
paraformaldehyde, 2.5% glutaraldehyde, 3 mM calcium chloride in 0.1 M sodium cacodylate
buffer (pH 7.4) (Sigma-Aldrich and Electron Microscopy Sciences, USA). After fixation, tissue
was processed into 1 x 2 mm blocks with the use of razor blades. Blocks were selected based
upon visualization of hardened tissue that was not mechanically damaged by separation between

47
the dermis and epidermis layers of the tissue. Subsequently, tissue blocks were transferred into
fresh fixative solution prior to fixation and embedding in epoxy resin. 0.5 µm thick tissue blocks
were sectioned and placed onto glass slides. Three slides were collected from each block with an
interval of 100 µm of tissue, separating each slide. The sections were stained with 0.1%
toluidine blue in borate buffer for 30 sec at 60°C, immersed in a 70% ethanol/2% acetic acid
solution for 5 sec, and rinsed with double distilled water (ddH2O). The sections were, then,
examined with an Olympus B51 microscope under a 100x magnification oil immersion objective
lens. Mast cells were identified based upon the intensity of metachromatic staining and counted
for quantification of the number of cells per standardized area. Representative images were also
recorded.
Transmission Electron Microscopy
Skin samples were prepared as detailed for histological staining with a few exceptions.
In brief, thin sections of approximately 80 nm in thickness were prepared with a Leica UC-6
ultramicrotome and collected onto 200-mesh hexagonal copper grids. Thereafter, the grids were
stained with 2% aqueous uranyl acetate and lead citrate. The grids were, then, examined with a
JEM-1200 transmission electron microscope at 80 kV. Representative images were documented.
Flow Cytometry
Approximately 5 x 105-1 x 10
6 cells per sample were stained for all assays. In brief,
BMMCs were harvested, washed with 2% FBS in PBS twice, and treated with Mouse BD Fc
Block for 15 min on ice (BD Pharmingen, USA). Then, cells were stained appropriately with
fluorescein isothiocyanate (FITC)-conjugated anti-mouse CD117/c-kit (BD Pharmingen, USA),

48
phycoerythrin (PE)-conjugated anti-mouse FcεRI alpha (FcεRIα), and/or allophycocyanin
(APC)-conjugated anti-IL-3R alpha chain (IL-3Rα) antibodies (eBioscience, USA) for 30 min on
ice. Cells were subsequently washed with 2% FBS in PBS twice, resuspended in PBS, and
monitored for surface expression of fluorescent markers with a FC500 Benchtop Cytometer or
BD LSR II. In the case of observation of IL-3Rα, cells were washed and incubated in the
absence of IL-3 overnight to remove exogenous IL-3 and prevent receptor recycling due to the
presence of stimulatory IL-3. Then, the cells were stained with anti-IL-3Rα antibodies.
Recorded measurements were analyzed with BD FACSDiva and FlowJo software.
Apoptosis Assay
To evaluate the viability of BMMCs by flow cytometry, the LIVE/DEAD Fixable Dead
Cell Red Stain Kit was used according to manufacturer’s instructions (Invitrogen, USA). The
cells were monitored with a BD LSR II, and recorded measurements were analyzed with BD
FACSDiva and FlowJo software.
Degranulation Assay
RBL-2H3 cells were seeded at 2 x 105 per well (in 1 mL) of a 12-well cell culture plate
and cultured overnight. In some cases, the cells were treated with BTP2, 2-APB (at 50 μM),
BTP2 derivatives (at the indicated concentrations or 1 μM), or vehicle for 30 min at 37°C and
subsequently challenged with 500 nM ionomycin for 30 min at 37°C. Alternatively, the cells
were sensitized with 1 μg mouse IgE anti-ovalbumin (OVA)/mL for 3 h at 37°C. The cells were
washed with PBS, treated with BTP2, BTP2 derivatives, or vehicle for 30 min at 37°C, and then
treated with 10 μg rat anti-mouse IgE in a final volume of 1 mL medium, supplemented with

49
BTP2, BTP2 derivatives, or vehicle for 30 min at 37°C. Mast cell degranulation was
quantitatively measured using a flow cytometric annexin-V binding assay (88). After stimulus,
RBL-2H3 cells were washed with pre-chilled PBS twice and subsequently stained with annexin-
V-PE at a dilution of 1:20 in binding buffer (commercially available as a part of the annexin-V-
PE kit; BD Pharmingen) for 15 min at room temperature. The cells were analyzed using a FC500
Benchtop Cytometer. Degranulation was calculated as a percentage of the mean fluorescence
intensity (MFI) of PE fluorescence of live RBL-2H3 cells, challenged in the presence of negative
control (vehicle) medium, after subtracting background release (% degranulation).
Degranulation was also determined by measuring levels of secreted β-hexosaminidase.
In the case of BTP-related experiments, RBL-2H3 cells or BMMCs were treated as described
above, except that after activation, β-hexosaminidase activity in the supernatants was determined
spectrophotometrically. In brief, 50 μL of supernatant samples and 80 μL of p-nitrophenyl-N-
acetyl-β-D-glucosamide (pNAG) solubilized in 0.05 M citrate buffer (pH 4.5) were added to the
wells of a flat-bottom 96-well plate. Color was allowed to develop for 30 min at 37°C. The
enzymatic reaction was terminated with the addition of 200 μL 0.05 M sodium carbonate buffer
(pH 10.0). Absorbances at 415 nm were measured in a microplate reader. Cells were lysed with
1% Triton-X, and the extracts were analyzed for endogenous β-hexosaminidase activity (total -
test). The β-hexosaminidase activity in unstimulated cells (spontaneous) was subtracted from the
enzyme activity of the stimulated cells (test). The percentage of β-hexosaminidase released into
each supernatant (% of total) was calculated using the following formula: β-hexosaminidase
secretion (% of total) = (test - spontaneous)/(total – spontaneous) x 100. Degranulation was
calculated as a percentage of the percentage released into each supernatant of total β-
hexosaminidase (% of total) for BMMCs, challenged in the presence of negative control

50
(vehicle) medium, after subtracting background release (% degranulation). For Drebrin-/-
mouse
experimentation, BMMCs were harvested, washed with Tyrode’s buffer, and sensitized with
anti-DNP IgE (2 µg/mL) in DMEM in the absence of IL-3 at 37°C overnight (4 x 106 cells/mL).
The cells were subsequently washed with Tyrode’s buffer to remove unbound IgE and
resuspended in Tyrode’s buffer at a concentration of 106 cells/60 µL. 10 µL of the cell
suspension was transferred to a 96-well round bottom plate, and cells were challenged with 10
µL of 2x stimulus (for final concentration of 0-1000 ng DNP-HSA/mL or 500 nM ionomycin)
for 1 h at 37°C. After activation, the β-hexosaminidase activity in the supernatants was
determined spectrophotometrically. In brief, 10 μL of supernatant samples and 50 μL of pNAG
solubilized in 0.05 M citrate buffer (pH 4.5) were added to the wells of a flat-bottom 96-well
plate. Color was allowed to develop for 60 min at 37°C. The enzymatic reaction was terminated
with the addition of 150 μL 0.2 M glycine (pH 10.7). Absorbances at 405 nm were measured in
a microplate reader. Cells were lysed with 0.5% Triton-X, and the extracts were analyzed for
endogenous β-hexosaminidase activity (total – test – blank). The β-hexosaminidase activity of
the blank substrate solution (blank) was subtracted from the enzyme activity of the unstimulated
or stimulated cells (test). The percentage of β-hexosaminidase released into each supernatant (%
of total) was calculated using the following formula: β-hexosaminidase secretion (% of total) =
(test - blank)/(total – blank) x 100.
In Vivo Histamine Release Assay
Analysis of in vivo histamine release was determined as previously described (86). In
short, mice (n=3-5 per group, 8 weeks old) were sensitized with 1 μg of mouse IgE anti-DNP via
intraperitoneal (i.p.) injection overnight. 12 h post-sensitization, mice were challenged with 50

51
μg DNP-HSA (solubilized in 100 µL PBS), which was delivered via intravenous (i.v.) injection.
For BTP-related experiments, mice were administered 10 mg BTP2/kg (~50 μL in PBS per
mouse) or vehicle (DMSO, 50 μL in PBS) as appropriate via i.p. injection. Then, 1 h post
treatment, mice were challenged with DNP-HSA as aforementioned. Control mice were either
not manipulated or were sensitized with mouse IgE anti-DNP and subsequently challenged with
PBS alone without exposure to DNP-HSA. In BTP-related experiments, Control mice were
sensitized with mouse IgE anti-DNP, treated with DMSO or BTP2, and were challenged with
PBS alone (i.e. no antigen). 3 min post-challenge, blood was collected by cardiac puncture and
stored on ice until centrifugation was performed for the preparation of serum samples.
Histamine was determined using a Beckman Coulter EIA Histamine Assay. All experiments
were carried out in accordance with the regulations of IACUC at The Pennsylvania State
University and Cornell University.
Serum IgE ELISA
For preparation of serum samples, blood was collected from mice via cardiac puncture
and transferred into a serum separator tube. Thereafter, tubes were centrifuged, and the serum
layer was removed. The IgE concentration of serum samples was assayed with the Mouse IgE
enzyme linked-immunosorbent assay (ELISA) MAX Deluxe Set according to manufacturer’s
instructions (BioLegend, USA).
Cytokine Secretion Assay
Analysis of cytokine secretion by BMMCs was performed as previously described (86).
In the case of BTP-related experiments, cells were incubated in the presence of 1 μM BTP2 or

52
vehicle for 30 min on ice in Tyrode’s buffer prior to stimulation. In brief, cells were washed and
sensitized with or without mouse IgE anti-DNP (1ug/mL) in the absence of IL-3 or SCF
overnight to remove exogenous IL-3. Subsequently, the cells were washed and resuspended in
Tyrode’s buffer. At this point, optionally, cells were incubated in the presence of 1 μM BTP2 or
vehicle for 30 min on ice in Tyrode’s buffer. Cells were then stimulated in medium starved of
IL-3 and SCF in V-bottom 96-well culture plates (2 x 105 cells/150 μL). Cells were stimulated
with 50 nM phorbol myristic acid (PMA) and 500 nM ionomycin. Alternatively, cells were
stimulated with 30 μg rat anti-mouse IgE/mL or 100 ng DNP-HSA/mL. In BTP2-related
experiments, for these steps, stimulation was performed in the presence or absence of 1 μM
BTP2. Cytokines in cell culture supernatants obtained 8 h and 24 h after stimulation were
measured using a Milliplex MAP immunoassay according to the suppliers’ instructions. Within
the assay’s sensitivities, mouse IL-2, IL-3, IL-4, IL-6, IL-13, TNF-α, and GM-CSF were
detected. IL-17 and IFN-γ were not detected. Cytokine concentrations are reported in pg/mL
units.
Measurements of Intracellular Ca2+
Concentration
Intracellular Ca2+
concentration in RBL-2H3 cells was measured using the Ca2+
-reactive
fluorescent probe Fura-2 acetoxymethylester (Fura-2AM) as previously described (86). First,
cells were pretreated with 1 μM BTP2 or vehicle for 1 h at 37⁰C and, thereafter, washed with
Ringer’s Solution (155 mM NaCl, 4.5 mM KCl, 2 mM MgCl2, 10 mM dextrose, 5 mM HEPES,
pH 7.4), supplemented with 1 mM CaCl2. Cells were loaded with 1 μM Fura-2AM at a
concentration of 107 cells/mL in Ca
2+-supplemented Ringer’s Solution for 1 h in the dark. Cells
were then washed, resuspended in Ca2+
-supplemented Ringer’s Solution, and the intracellular

53
Ca2+
concentration of 5 x 105 cells was monitored using a Photon Technology International
Quantamaster Spectrofluorometer. Fluorescence of Fura-2AM was monitored at room
temperature. Intracellular Ca2+
concentration was expressed as the ratio (Relative Fluorescence,
RF) of Fura-2AM fluorescence at 510 nM caused by the two excitation wavelengths (340
nm/380 nm).
Intracellular Ca2+
concentration in BMMCs was measured using the Ca2+
indicator Fluo-4
(Invitrogen, USA). BMMCs were sensitized with mouse IgE anti-DNP at a concentration of 1
μg IgE/2 x 106 cells/mL in medium starved of IL-3 and SCF overnight, washed, and resuspended
in factor-starved media. At this time, optionally, the cells were treated with BTP2 (1 μM),
3T5M-P, or vehicle for 1 hr at 37°C. Afterwards, the cells were loaded with Fluo-4 according to
manufacturer’s instructions. Subsequently, the cell suspension was supplemented with 2 mM
CaCl2. The intracellular Ca2+
concentration of 1.25 x 106 cells was monitored using a Coulter
XL-MCL flow cytometer. Relative fluorescence (RF) of Fluo-4 was measured at 494 nm when
excited by 488 nm of light at room temperature. Intracellular Ca2+
concentration was expressed
as the RF of Fluo-4. Mean calcium post-stimulation was calculated as the mean of intracellular
Ca2+
concentration ratiometric Fura-2AM or Fluo-4 RF measurements post-stimulation after
subtracting the mean of baseline measurements pre-treatment. Peak calcium post-stimulation
was calculated as the maximum intracellular Ca2+
concentration ratiometric Fura-2AM or Fluo-4
RF measurement post-stimulation after subtracting the mean of respective pre-treatment baseline
measurements. The slope of calcium decay was calculated as the slope of the linear regression
line fitted for Fluo-4 RF measurements post-stimulation.

54
Analysis of NFAT Localization
RBL-2H3 cells were cultured in glass bottom 6-well plates (2 x 105 cells/2 mL)
overnight. For DNP-HSA stimulation, cells were sensitized with 1 μg mouse IgE anti-DNP/mL
overnight, washed, and resuspended in factor-starved media. Then, the cells were treated with 1
μM BTP2 or vehicle for 30 min at 37°C, stimulated with 50 nM PMA and 500 nM ionomycin or
100 ng/mL DNP-HSA for 45 min at 37°C, and washed with PBS once. The cells were
immediately fixed and permeabilized in a 50:50 methanol:acetone mixture for, at least, 15 min at
-20°C. The cells were subsequently washed with PBS three times, blocked with normal rat serum
for 2 h at room temperature, and stained with Alexa Fluor 488-conjugated mouse IgG anti-
Nuclear Factor of Activated T cells, cytoplasmic 1 (NFATc1) (Santa Cruz Biotechnology, Inc.)
overnight at 4°C. Then, the cells were stained with TO-PRO-3 for identification of localization
of the nucleus. After staining, cells were observed using an Olympus Fluoview 1000 confocal
microscope for fluorescence at 519 nm with an excitation wavelength of 488 nm. At a 100x
magnification, images were captured at 0.2 μm slices as Z-plane stacks. Data was analyzed with
Autodeblur and Autovisualize X licensed software for deconvolution and 3-D image processing.
Statistical Analysis
Data represent the mean ± standard error mean (SEM) of, at least, three independent
experiments. The concentration causing 50% inhibition (IC50) was calculated using non-linear
regression analysis. The statistical significance was analyzed by Student’s t-test (unpaired t-test,
two-tailed) or by two-way ANOVA. Values of p<0.05 were considered significant. All data
analyses were performed using BD FACSDiva, FlowJo, or GraphPad Prism 5.

55
CHAPTER 3
Effect of BTP2 on Mast Cell Biology

56
Rationale
Much of our initial understanding of the electrophysiological properties of CRAC
channels came from a substantial body of work on T cells and mast cells. The precise molecular
mechanism of CRAC channel gating, however, is still largely unknown. Specific inhibitors of
CRAC channels could facilitate the molecular identification of the elusive key regulatory players
and would be excellent tools to study CRAC channel function. Unfortunately, all CRAC channel
blockers described thus far, including the most potent ones SK&F 96365, econazole, and 2-
aminoethyldiphenyl borate, have IC50 values in the micromolar range and are non-specific.
Djuric and colleagues described pyrazole derivatives that interfere specifically with the
expression of Ca2+
-dependent cytokine production following T cell receptor (TCR) stimulation,
but they could not detect inhibition of TCR-dependent Ca2+
signals in T cells (87). On the
contrary, Ishikawa et al. and we demonstrated that one of the pyrazole derivatives BTP2, also
known as YM-58483, is a potent inhibitor of store-operated influx in Jurkat T cells (78, 79).
Furthermore, we characterized an interaction between BTP2 and the actin-regulating protein
drebrin and identified a novel role for this protein as a regulator of calcium responses (78). We,
however, have not discriminated whether the mechanisms contributing to Ca2+
signals in mast
cells are affected by BTP2.
In this study, the pharmacological profile of BTP2 was investigated in the RBL-2H3 and
BMMC in vitro mast cell models and in vivo in the C57BL/6 murine system to evaluate the
therapeutic potential of this compound for mast cell-mediated diseases (6).

57
Effects of BTP2 on Ca2+
mobilization in RBL-2H3 cells and BMMCs
Increases in intracellular Ca2+
concentration, triggered through the FcεRI by IgE and
allergen, are necessary for the functional responses of mast cells. To analyze the possibility that
BTP2 has the potential to inhibit Ca2+
mobilization in mast cells, Ca2+
signals in the presence or
absence of BTP2 were compared. We first used RBL-2H3 cells which have been used
extensively as a mast cell model to study FcεRI signaling. These cells were pre-treated with
BTP2 (1 μM) or vehicle (DMSO) for 30 minutes and, then, analyzed for calcium responses, as
described in the Materials and Methods, following stimulation. These initial studies with RBL-
2H3 cells involved stimulation with the established Ca2+
ionophore ionomycin, which can
activate mast cell degranulation, as well as cytokine secretion, downstream of the FcεRI in the
presence of the phorbol ester PMA (89-91). We found that BTP2, at a 1 μM concentration,
significantly reduced intracellular Ca2+
following stimulation by ionomycin (Fig. 3.1A, left and
center panel). Moreover, analysis of peak Ca2+
responses showed a significant difference in this
parameter (Fig. 3.1A, right panel). Because ionomycin is not a physiological stimulus, we next
tested whether Ca2+
responses following more physiological stimuli via the FcεRI were also
inhibited by BTP2. In these experiments, we used anti-IgE. We also used antigen-specific IgE
anti-DNP in conjunction with DNP-HSA for the activation of primary sensitized BMMCs. These
BMMCs were pre-treated with BTP2 (1 μM) or vehicle for 30 minutes and then stimulated with
mouse anti-DNP IgE and rat anti-mouse IgE or the antigen DNP-HSA for the indicated time
period in the presence of CaCl2. Data summarized in Fig. 3.1B and 3.1C reveal that BTP2
inhibited the Ca2+
response in these BMMCs following FcεRI aggregation, with the Ca2+
peak
being significantly inhibited regardless of the stimulation method (Fig. 3.1, A-C, right panels).

58
Figure 3.1. BTP2 blocks intracellular Ca2+
mobilization in RBL-2H3 cells and BMMCs.
(A) RBL-2H3 cells were pre-treated with BTP2 (1 μM) or vehicle (DMSO) for 30 min, then
analyzed for calcium responses (as described in the Materials and Methods) following
stimulation with 500 nM ionomycin in the presence of CaCl2 for the indicated time period.
Intracellular Ca2+
concentration was expressed as ratiometric Fura-2AM, and representative data
of 3-5 independent experiments is shown (left panel). Quantification of mean calcium increase
(middle panel); peak calcium increase (right panel), with data expressed as the mean ± SEM of
3-5 independent experiments. *p<0.05 vs. Vehicle. (B) BMMCs were pre-treated with BTP2 (1
μM) or vehicle for 30 min, then stimulated with mouse anti-DNP IgE and rat anti-mouse IgE (10
μg/mL), and analyzed as in (A), except that intracellular Ca2+
concentration was expressed as
Fluo-4 relative fluorescence with representative data of 3-5 independent experiments shown (left
panel). Quantification of mean calcium increase (middle panel); peak calcium increase (right
panel) as in (A). Data are expressed as the mean ± SEM of 3-5 independent experiments.
*p<0.05 vs. Vehicle. (C) BMMCs were pre-treated with BTP2 (1 μM) or vehicle for 30 min,
then stimulated with 100 ng DNP-HSA and analyzed as in (A), except that intracellular Ca2+
concentration was expressed as Fluo-4 relative fluorescence with representative data of 3-5
independent experiments shown (left panel). Quantification of mean calcium increase (middle
panel); peak calcium increase (right panel) as in (A). Data are expressed as the mean ± SEM of
3-5 independent experiments. *p<0.05 vs. Vehicle.

59
Effects of BTP2 on FcεRI-mediated signaling in BMMCs
By contrast, BTP2 did not affect FcεRI-triggered tyrosine phosphorylation of cellular
proteins in BMMCs (Fig. 3.2A). Activation of Erk1/2, JNK, and p38 MAPK pathways were also
not affected by BTP2 treatment (Fig. 3.2, B-D, respectively). Finally, expression of c-Fos,
which is dependent upon the activation of Erk, was not altered by BTP2 treatment (Fig. 2E).
Thus, BTP2 does not grossly affect phosphorylation events immediately downstream of the
FcεRI.

60
Figure 3.2. BTP2 does not affect tyrosine kinase nor MAP kinase activation following
FcεRI triggering. BMMCs were treated with either BTP2 (1 μM) or vehicle (DMSO) for 30
min and then stimulated with mouse IgE anti-DNP and DNP-HSA for the indicated time periods,
lysed, and analyzed by western blot for (A) total phosphotyrosine; (B) phospho-ERK; (C)
phospho-JNK; (D) phospho-p38; or (E) c-Fos expression. Blots were stripped and reprobed with
control antibodies for β-actin. Numbers indicate fold increase corrected for expression levels.

61
BTP2 inhibits degranulation in RBL-2H3 cells
Mast cell degranulation is a highly regulated Ca2+
-dependent process (92). As BTP2
inhibits Ca2+
mobilization in RBL-2H3 cells and BMMCs (Fig. 3.1, A-C), we next determined if
mast cell degranulation could be inhibited by BTP2. For these experiments, we used an annexin-
V binding assay, as described in the Materials and Methods section. RBL-2H3 cells were pre-
treated with BTP2 or vehicle, at a 1 μM concentration, and then stimulated with ionomycin for
30 minutes, followed by analysis of degranulation. We found that BTP2 significantly reduced
ionomycin-induced degranulation (Fig. 3.3A). This inhibition occurred in a dose-dependent
fashion, with an IC50 value of 23 nM (Fig. 3.3B). Comparative analysis of population-based
measurements of the secretion of the granule-derived enzyme β-hexosaminidase between BTP-
treated cells and those that were treated with vehicle alone revealed inhibition of ionomycin-
induced degranulation upon pre-treatment with BTP2, thereby validating the results generated
through the annexin-V binding assay (Fig. 3.4). Furthermore, BTP2 could also significantly
reduce degranulation induced by FcεRI crosslinking with anti-IgE antibodies. In these
experiments, RBL-2H3 cells were sensitized with mouse IgE anti-OVA, pretreated with vehicle
or BTP2 (1 μM) for 30 minutes, and finally stimulated with rat anti-mouse IgE for 30 minutes.
Our results indicate that BTP2 significantly reduced degranulation induced by FcεRI as well
(Fig. 3.3C). BTP2 is therefore a potent inhibitor of mast cell degranulation induced through the
FcεRI.

62
Figure 3.3. BTP2 inhibits mast cell degranulation in vitro. (A) RBL-2H3 cells were pre-
treated with BTP2 or vehicle (DMSO) at 1 μM and then stimulated with ionomycin (500 nM) for
30 min. (B) RBL-2H3 cells were pre-treated with vehicle (DMSO) or BTP2 at the indicated
concentrations for 30 min then stimulated as in (A). (C) RBL-2H3 cells were pre-treated with
BTP2 (1 μM) or vehicle for 30 min, then stimulated with mouse IgE anti-OVA and rat anti-
mouse IgE for 30 min. All cells were analyzed for degranulation via an annexin-V binding
assay. Data are expressed as the mean ± SEM of 3 independent experiments. *p<0.05 vs.
Vehicle.
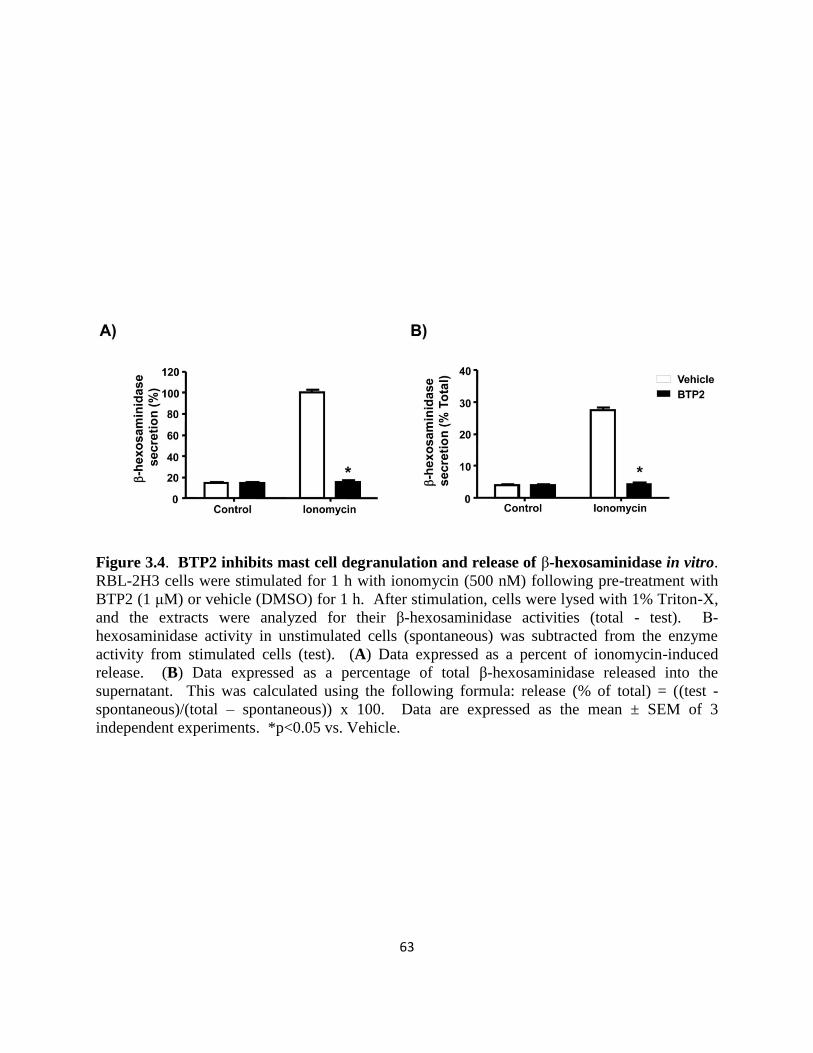
63
Figure 3.4. BTP2 inhibits mast cell degranulation and release of β-hexosaminidase in vitro.
RBL-2H3 cells were stimulated for 1 h with ionomycin (500 nM) following pre-treatment with
BTP2 (1 μM) or vehicle (DMSO) for 1 h. After stimulation, cells were lysed with 1% Triton-X,
and the extracts were analyzed for their β-hexosaminidase activities (total - test). Β-
hexosaminidase activity in unstimulated cells (spontaneous) was subtracted from the enzyme
activity from stimulated cells (test). (A) Data expressed as a percent of ionomycin-induced
release. (B) Data expressed as a percentage of total β-hexosaminidase released into the
supernatant. This was calculated using the following formula: release (% of total) = ((test -
spontaneous)/(total – spontaneous)) x 100. Data are expressed as the mean ± SEM of 3
independent experiments. *p<0.05 vs. Vehicle.

64
BTP2 inhibits histamine release in mice in response to antigenic challenge
To further characterize the effects of BTP2 on the mast cell-dependent physiological
response of degranulation, we analyzed the effect of BTP2 on histamine release in vivo. Mice
were sensitized with mouse IgE anti-DNP (1 μg) 12 hours prior to being pre-treated with 10 mg
BTP2/kg or vehicle that was delivered intraperitoneally. These mice were then challenged with
DNP-HSA an hour later to stimulate mast cell degranulation. 3 minutes post-challenge with
DNP-HSA, mice were sacrificed, and serum was collected and assayed for histamine. Control
mice were exposed to anti-DNP IgE, but they were left unexposed to antigen to ensure that the
measured responses were antigen-specific. The results show that BTP2 significantly reduces
histamine release upon antigenic stimulation in vivo (Fig. 3.5). In summary, this data provides
strong support for the idea that BTP2 is a potent inhibitor of mast cell degranulation in vivo.

65
Figure 3.5. BTP2 inhibits FcεRI-mediated histamine release in vivo. C57BL/6 mice were
sensitized with mouse IgE anti-DNP overnight, then treated with BTP2 (BTP2, 10 mg/kg) or
vehicle (DMSO) for 1 h. Mice, treated with BTP2 or vehicle, were then challenged with DNP-
HSA or PBS (as a control) for 3 min. Serum histamine concentration levels were then
determined and data are expressed as the mean ± SEM, n=8 except for control mice treated with
BTP and no antigen, n=3. *p<0.05 vs. Vehicle.

66
BTP2 inhibits cytokine secretion from mast cells
BTP2 was originally reported to inhibit IL-2 production in activated Jurkat T cells and
human CD4+ T cells, as well as IL-4 and IL-5 production in antigen-stimulated murine Th2 clone
D10.G4.1 cells (79, 80, 85, 87, 93). BTP2 also inhibits NFAT activation in Jurkat T cells and
primary human T cells (79, 80, 87, 93). Because secretion of these cytokines is regulated
differently in mast cells in certain cases (94-97), we tested whether the cytokine secretion of
BMMCs is affected by BTP2. BMMCs were stimulated with PMA and ionomycin in the
presence or absence of BTP2, and supernatants were analyzed for IL-2, IL-3, IL-4, IL-13, TNF-
α, and GM-CSF. As illustrated in Fig. 3.6, at both 8 and 24 hours after stimulation, BTP2
inhibited the secretion of IL-2, IL-3, IL-4, and TNF-α from BMMCs. While little IL-13 was
detected at the 8 hour timepoint, BTP2 inhibited IL-13 secretion by BMMCs at the 24 hour
timepoint (Fig. 3.6). BTP2 also significantly inhibited ionomycin-induced secretion of GM-CSF
at the 24 hour timepoint (Fig. 3.6). We also analyzed cytokine secretion in sensitized BMMCs
stimulated with anti-IgE. We found that there was little production of IL-2 and IL-13 up to 24
hours post-stimulation; however, BTP2 pre-treatment inhibited the secretion of IL-3, IL-4, IL-6,
TNF-α, and GM-CSF (Fig. 3.7). Similar results were obtained for IL-4, IL-6 and TNF-α when
sensitized BMMCs were stimulated with DNP-HSA (Fig. 3.8).

67
Figure 3.6. BTP2 inhibits cytokine secretion of PMA/ionomycin-activated BMMCs.
BMMCs were pre-treated with BTP2 (1 μM) or vehicle (DMSO) for 30 min and, then,
stimulated with PMA and ionomycin. 8 and 24 h after stimulation, the concentration of the
indicated cytokines was determined. Data are expressed as the mean ± SEM of technical non-
independent replicates, representative of 3 independent experiments. **,p = 0.001 – 0.01;
*p<0.05 vs. Vehicle.

68
Figure 3.7. BTP2 inhibits cytokine secretion of IgE/anti-IgE-activated mast cells. BMMCs
were sensitized with mouse anti-DNP IgE, pretreated with BTP2 (1 μM) or vehicle (DMSO) for
30 min, and then stimulated rat anti-mouse IgE. 8 and 24 h after stimulation, the concentration
of the indicated cytokines was determined. Data are expressed as the mean ± SEM of technical
non-independent replicates, representative of 3 independent experiments. *p<0.05 vs. Vehicle.
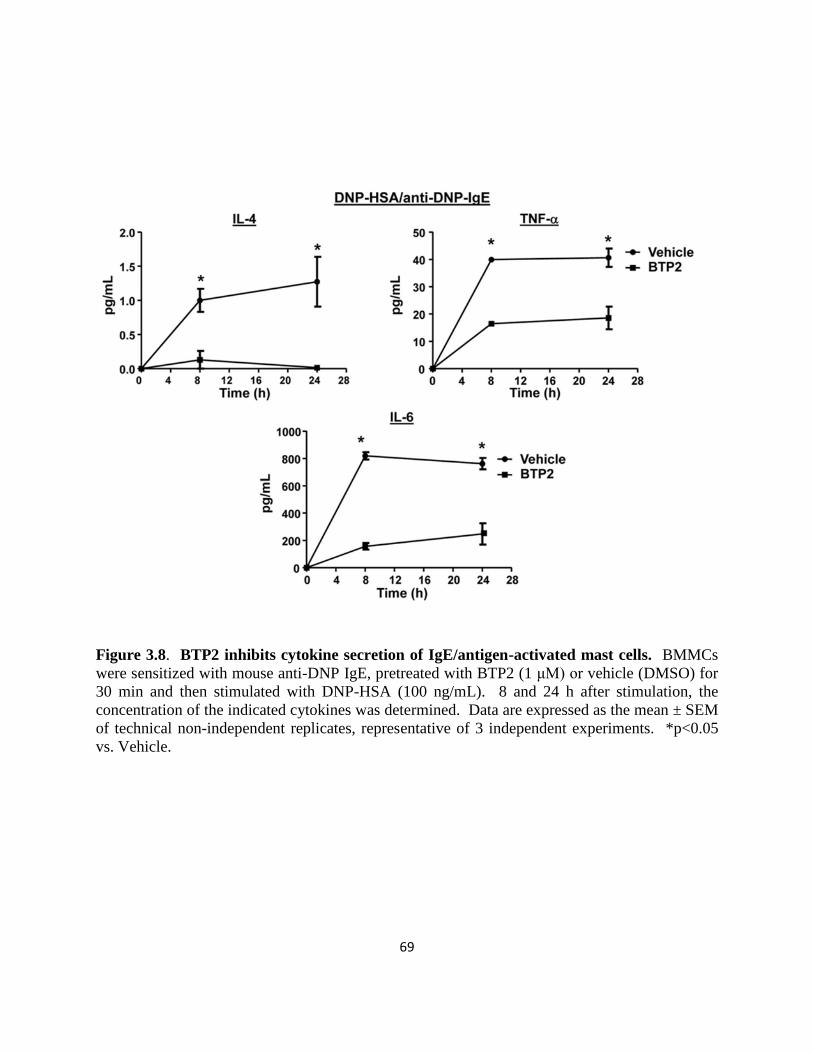
69
Figure 3.8. BTP2 inhibits cytokine secretion of IgE/antigen-activated mast cells. BMMCs
were sensitized with mouse anti-DNP IgE, pretreated with BTP2 (1 μM) or vehicle (DMSO) for
30 min and then stimulated with DNP-HSA (100 ng/mL). 8 and 24 h after stimulation, the
concentration of the indicated cytokines was determined. Data are expressed as the mean ± SEM
of technical non-independent replicates, representative of 3 independent experiments. *p<0.05
vs. Vehicle.

70
BTP2 inhibits NFAT nuclear localization in mast cells
Because the cytokines that were inhibited by BTP2 are regulated, in part, by NFAT (98,
99) and NFAT activation and nuclear translocation is dependent upon increases in cytosolic
Ca2+
, we examined the effect of BTP2 on NFAT localization in stimulated RBL-2H3 cells. Cells
were stimulated with ionomycin, and NFAT localization was examined. Alternatively, sensitized
RBL-2H3 cells were stimulated with DNP-HSA. These experiments show that BTP2 inhibited
ionomycin-induced, as well as antigen-IgE/FcεRI-induced, NFAT nuclear translocation (Fig.
3.9, A-B).
BTP2 inhibits de novo synthesis of cytokines in mast cells
We noted that BTP2 treatment completely suppressed the production of IL-2 and IL-3 but
partially inhibited the secretion of IL-4, IL-6, and TNF-α (Fig. 3.6-8). In mast cells, the
production of IL-2 is controlled almost entirely at the transcriptional level (100). On the
contrary, TNF-α, IL-4, and IL-13 occur both preformed and as newly synthesized molecules (2,
101). To determine whether the mechanism of inhibition responsible for our observation
involved a block in the transcription of preformed cytokines or de novo transcription, we
examined the effect of BTP2 on mRNA transcript levels for IL-4, IL-6, and TNF-α under steady-
state conditions in BMMCs. As illustrated in Fig. 3.10, BTP2 treatment did not significantly
inhibit the levels of preformed mRNA transcripts for these cytokines suggesting that BTP2
inhibits cytokine production primarily via its effects upon de novo transcription that is regulated
by NFAT or other Ca2+
-sensitive transcription factors.

71
Figure 3.9. BTP2 inhibits stimulus-induced NFAT nuclear translocation. (A) RBL-2H3
cells were pre-treated with BTP2 (1 μM) or vehicle (DMSO) for 30 min prior to stimulation for
45 min with ionomycin or (B) mouse anti-DNP IgE and DNP-HSA (100 ng/mL). Cells were
fixed and permeabilized, and NFATc1 localization was determined by confocal microscopy.
The nucleus was identified using TO-PRO-3 staining (red), and nuclear localization of NFAT
was identified with Alexa 488-conjugated anti-NFATc1 staining (green). Colocalization is
evident by yellow nuclear staining (red plus green). White bars indicate 40 μm. Representative
data of 3-5 independent experiments is shown.
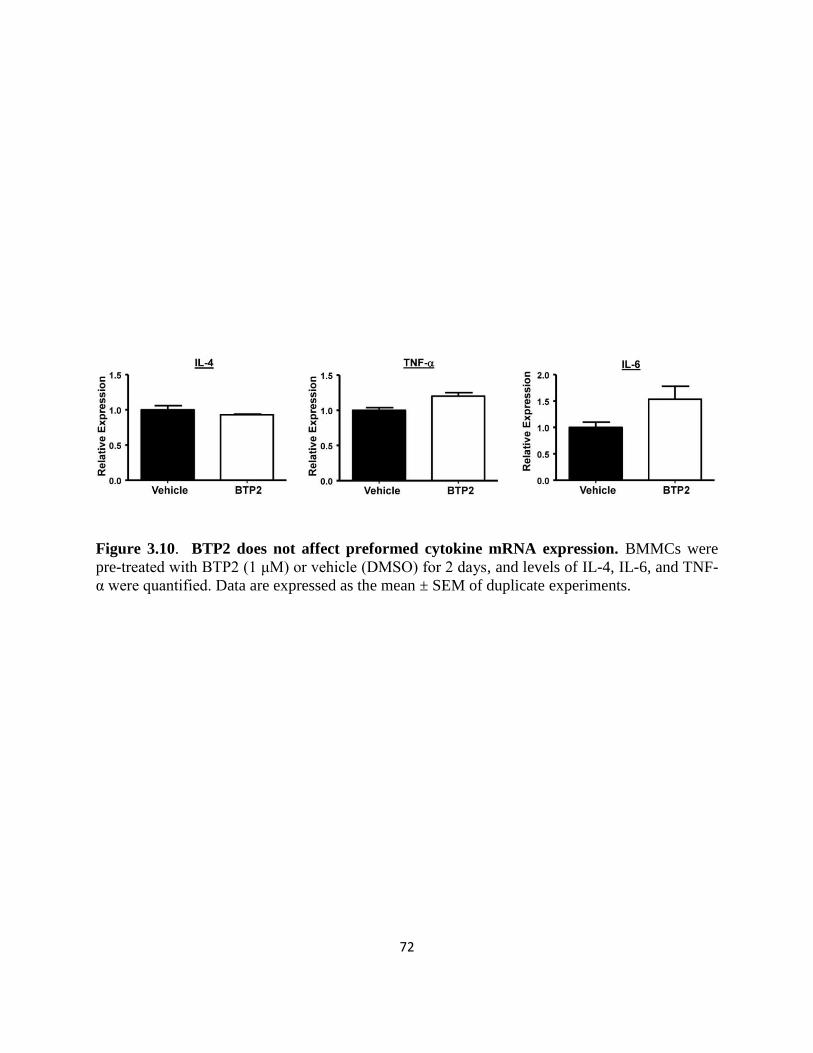
72
Figure 3.10. BTP2 does not affect preformed cytokine mRNA expression. BMMCs were
pre-treated with BTP2 (1 μM) or vehicle (DMSO) for 2 days, and levels of IL-4, IL-6, and TNF-
α were quantified. Data are expressed as the mean ± SEM of duplicate experiments.

73
Discussion
BTP2 has been characterized as a selective blocker of store-operated Ca2+
entry. We
investigated the effect of BTP2 on mast cell function using the RBL-2H3 cell line and BMMCs
in vitro, as well as in in vivo murine models, to evaluate its therapeutic potential in mast cell-
mediated diseases. We show that BTP2 inhibits activation-induced increases in intracellular
Ca2+
responses. In addition, we show that BTP2 inhibits IgE and antigen-induced degranulation
in vitro and histamine release in vivo, as well as activation-induced cytokine secretion in vitro.
Sustained intracellular Ca2+
concentration increases are integral for allergen-induced mast
cell activation. The activation of mast cells leads to the release of performed mediators that are
stored in cytoplasmic granules. These mediators include histamine, serine proteases (tryptase
and chymase), carboxypeptidase A, and proteoglycans (2). The effects of BTP2 on mast cell
degranulation and cytokine secretion in vitro, as well as histamine release in vivo, suggest that
this compound could modulate the effect of histamine on smooth muscle contraction, endothelial
cell function, nerve endings, and mucous secretion. This agrees with other studies suggesting
that BTP2 may be efficacious in models of respiratory anaphylaxis in guinea pigs. In these
models, BTP2 was able to inhibit increased bronchoconstriction and airway hyperresponsiveness
(2, 84). Sustained elevations in intracellular Ca2+
concentration are also essential for the
production and secretion of cytokines by mast cells, which can modulate subsequent immune
responses (102). The ability of BTP2 to inhibit cytokine secretion in BMMCs is likely due to
reduced NFAT nuclear translocation since this transcription factor is essential for the expression
of many cytokine genes in mast cells (96, 103). Interestingly, BTP2 treatment completely
suppressed ionomycin-induced secretion of IL-2 and IL-3 but partially inhibited secretion of IL-4
and TNF-α. In mast cells, TNF-α and IL-4 can be expressed from both preformed mRNA, as

74
well as newly transcribed mRNA (2, 101). Our studies demonstrate that BTP2 treatment did not
affect the levels of preformed IL-4 and TNF-α mRNA. In addition, BTP2 also did not affect
preformed levels of IL-6 mRNA. This suggests that translation of preformed mRNA for TNF-α
and IL-4 in BMMCs is likely not affected by increases in intracellular Ca2+
concentration. On
the other hand, de novo transcription that is regulated by NFAT or other Ca2+
-sensitive
transcription factors is likely affected by BTP2. We found that, though BTP2 could significantly
inhibit FcεRI-mediated GM-CSF secretion, it was not as effective in inhibiting the secretion of
this cytokine when the cells were stimulated with PMA and ionomycin. This suggests that this
cytokine may be regulated differently and that its expression may not have as stringent a
requirement for increases in intracellular Ca2+
concentration. Indeed, analysis of the regulation
of GM-CSF in transgenic mice has revealed multiple cis-acting elements that regulate this
cytokine in T cells and mast cells (94). This analysis reveals that, in addition to NFAT, other
transcription factors, such as AP-1, Sp-1, GATA1, and GATA2 can be differentially used in
different cell types to regulate GM-CSF expression (94). Thus, while both FcεRI- and PMA and
ionomycin-mediated activation of mast cells result in GM-CSF secretion, they may execute this
by activating slightly different combinations of transcription factors, with FcεRI activation being
more Ca2+
-dependent and thus more potently inhibited by BTP2. Similar to BTP2-treated
BMMCs, BMMCs from mice lacking either Orai1 or STIM1 produce weak Ca2+
signals in
response to agonist (27, 39, 40). These BMMCs are characterized by severely impaired
histamine release, decreased leukotriene production, reduced TNF-α secretion, and a
dysfunctional subcutaneous anaphylactic response. Therefore, Ca2+
entry through CRAC
channels is essential for mast cell function (39, 40, 102). Here, our profiling of the effects of
BTP2 on mast cell activation and downstream responses suggests that BTP2 acts in a similar

75
pathway. Our recent characterization of an interaction between BTP2 and the actin-regulating
protein drebrin and the resultant discovery of a novel role for drebrin as a mediator of store-
operated Ca2+
entry further implies such a mechanism of action (78). Further analysis of the role
of drebrin in calcium signaling and mast cell function will reveal insights into the mechanism
behind this process.

76
CHAPTER 4
Structure-Activity Relationship Analysis of BTP2

77
Rationale
Specific inhibitors of the Ca2+
signaling pathway are potential therapeutics for various
immune and allergic diseases. As experimental tools, they could also facilitate molecular
identification of mechanisms of Ca2+
mobilization, particularly those governing CRAC channel
gating. Unfortunately, blockers, such as SK&F 96365, econazole, and 2-APB, have IC50 values
in the micromolar range and are non-specific (80, 104-111).
A number of groups have defined pyrazole derivatives exemplified by BTP2, that
specifically block TCR-induced Ca2+
entry and Ca2+
-dependent cytokine production (78-80, 87,
93). Considering that mast cell activation and degranulation are critically dependent on increases
in intracellular Ca2+
, compounds that inhibit this process may be useful as potential therapeutics
for allergies and asthma. Pyrazole compounds, including BTP2, represent potential leads for
treating these diseases; however, limited work has been performed on the effects of BTP2 on
mast cells (89-91). In addition, little is known in regards to the effects of modifications of the
core pyrazole ring to which the activity of this compound is attributed. The core pyrazole ring is
defined by the attachment of two trifluoromethyl groups and is shared by all members of the
BTP class of compounds. Here, we provide the first structure-activity relationship analysis of
the core pyrazole ring of BTP to define the active portion of the BTP2 parent compound.

78
The trifluoromethyl group at the C3 position is required for the inhibitory effect of BTP2
on the degranulation of BMMCs
To determine which moiety within the BTP2 molecule is important for its activity, we
performed limited structure-activity relationship studies of the BTP2 parent compound. We
synthesized a series of BTP2 derivatives that have retained the core structure shared by all
members of the BTP class of compounds but have been altered by replacing the trifluoromethyl
groups of the BTP ring with less bulky methyl groups (5T3M-P: 5-trifluoromethyl-3-methyl
pyrazole or 3T5M-P: 3-trifluoromethyl-5-methyl pyrazole), deleting a single trifluoromethyl at
either the C5 or C3 position (3T-P or 5T-P), deleting the trifluoromethyl groups entirely
(Pyrazole, Pyr), or replacing both trifluoromethyl groups with methyl groups (3M5M-P) (Fig.
4.1). In addition, the remaining ring of BTP2, which is unique and not shared by all members of
the BTP class of compounds, was replaced with the ring structure characteristic of BTP1 for
these BTP2 derivatives (Fig. 4.1) (112). Accordingly, BTP1 was also tested.
The BTP2 parent compound and its derivatives (1 μM) were tested for their ability to
inhibit degranulation in BMMCs upon ionomycin stimulation. BTP1 and BTP2 were
indistinguishable in their ability to inhibit degranulation (Fig. 4.2A). Pyr did not suppress
degranulation. On the contrary, 5T3M-P and 3T5M-P significantly reduced degranulation in
comparison to vehicle-treated cells (Fig. 4.2A). However, 5T3M-P only inhibited degranulation
by approximately 25%, whereas the level of inhibition linked to 3T5M-P was close in potency to
the level of the parent BTP2 compound (Fig. 4.2A). Our data also showed that the derivative
with a single trifluoromethyl group at the C3 position (3T-P) maintained the capacity to inhibit
degranulation. Nonetheless, the derivative with only a trifluoromethyl group at the C5 (5T-P)
position did not (Fig. 4.2A). Finally, the replacement of both trifluoromethyl groups with methyl

79
groups (in 3M5M-P) produced effects similar to those rendered by derivatives with complete
lack of trifluoromethyl groups (Pyr) (Fig. 4.2A). Control experiments verified that the known
Ca2+
influx inhibitor 2-APB was also able to inhibit this process. In summary, 2-APB confirmed
that BTP1, BTP2, and derivatives that retain the trifluoromethyl group at the C3 position inhibit
degranulation and validated the importance of elevations in intracellular Ca2+
concentration in
the generation of mast cell effector responses (Fig. 4.2A). Furthermore, consistent with this data,
3T5M-P inhibited ionomycin-induced degranulation with an IC50 of 25 nM, which is comparable
to that of the parent BTP2 compound (Fig. 4.2B)
Effects of 3T5M-P on Ca2+
mobilization in BMMCs
In line with the previous findings, 3T5M-P significantly inhibited IgE-DNP-HSA/FcεRI-
induced increases in intracellular Ca2+
concentration in BMMCs (Fig. 4.3, A-B). Overall, these
results suggest that the trifluoromethyl group at the C5 position of the pyrazole compound is
nonessential for the inhibitory activity of BTP2 on the Ca2+
-dependent process of degranulation.

80
Figure 4.1. Chemical structures of BTP analogs. BTP1; BTP2; Pyrazole: Pyr; 3-
trifluoromethyl-5-methyl-pyrazole: 3T5M-P; 3-trifluoromethyl-pyrazole: 3T-P; 5-
trifluoromethyl-3-methyl-pyrazole: 5T3M-P; 5-trifluoromethyl-pyrazole: 5T-P; 3-methyl-5-
methyl-pyrazole: 3M5M-P.

81
Figure 4.2. The 3-trifluoromethyl group is critical for the inhibitory activity of BTP2 on
BMMC degranulation. (A) BMMCs were pre-treated with vehicle, 2-APB (at 50 μM), or BTP
analogs (at 1 μM) and then stimulated with ionomycin. Degranulation was determined (as
indicated for β-hexosaminidase secretion in Materials and Methods). (B) RBL-2H3 cells were
pre-treated with 3T5M-P at the indicated concentrations. Cells were then stimulated with
ionomycin, and degranulation was measured (as indicated for the annexin-V binding assay in
Materials and Methods). Data are expressed as the mean ± SEM of 3 independent experiments.
*p< 0.05 vs. vehicle.

82
Figure 4.3. 3T5M-P blocks intracellular Ca2+
mobilization in BMMCs. (A) BMMCs were
pre-treated with 3T5M-P (1 μM) or vehicle for 30 min, then stimulated with mouse IgE anti-
DNP and DNP-HSA (100 ng/mL). Calcium responses were determined as in Fig. 3.1.
Representative data of 3-6 experiments is shown. (B) Mean calcium increase post-stimulation.
Data are expressed as the mean ± SEM of 3 independent experiments. **,p = 0.001 – 0.01 vs.
Vehicle.

83
Discussion
Our exploration of the structure-activity relationship between BTP2 and mast cell
degranulation has shown that the 3-trifluoromethyl group on the pyrazole ring is critical for the
inhibitory activity of the BTP family of immunosuppressants. Our study indicates that the
trifluoromethyl groups at positions C3 and C5 of the pyrazole compound play critical roles in the
mechanism of action of the BTP2 parent compound, with the trifluoromethyl group at C3 being
more critical for inhibitory activity of BTP2. While the 3T5M-P and 3T-P derivatives inhibited
activation-induced degranulation of BMMCs, the 5T3M-P, 5T-P, Pyr, and 3M5M-P derivatives
were less potent. Additionally, 3T5M-P inhibited FcεRI-triggered Ca2+
mobilization in BMMCs.
These observed activities are similar to those reported by Kiyonaka et al. for pyrazole
compounds that target the TRPC3 channel. This group recently showed that bulky functional
groups at the 3,5-positions of the pyrazole may be important for the inhibition of Ca2+
mobilization via the TRPC3 channel (112). In this work, Kiyonaka et al. characterized the
activity of a BTP derivative that substitutes the 3,5-bis-trifluoromethyl pyrazole group with an
ethyl-3-trifluoromethyl-pyrazole-4-carboxylate group in the BTP2 parent compound (4-(2,3,3-
trichloroacrylamide)phenyl)-5-(trifluoromethyl)-1H-pyrazole-4-carboxylate). This compound
selectively inhibited the TRPC3 channel and was a more potent inhibitor of NFAT in cardiac
myocytes than BTP2. This work also showed that the 3,5-bis-trifluoromethyl pyrazole group or
a trichloroacrylic amide group is critical for the selectivity for TRPC5 or TRPC3, respectively
(112). This suggests that the pyrazole group provides a molecular scaffold with which to
develop inhibitors of calcium signaling (112). These analyses suggest that the attachment of
bulky groups on the pyrazole ring of such molecules, particularly in the C3 position is important
for their ability to inhibit Ca2+
mobilization. All taken together, these studies indicate that BTP2

84
and its relatives provide a solid molecular framework for a new generation of small molecule
inhibitors for the treatment of bronchial asthma, allergies, and other mast-cell mediated diseases.

85
CHAPTER 5
Role of Drebrin in Mast Cell Biology

86
Rationale
Actin cytoskeletal reorganization has been implicated in models of CRAC channel
regulation. Nonetheless, little is known about actin-modulatory proteins that are involved in this
process or how actin regulates CRAC channel function and, thereby, downstream allergic
responses.
Pharmacological tools that could help to elucidate the role of actin-binding proteins in
store-operated channel regulation and allergic reactions are represented by a group of
immunosuppressant compounds that are derived from BTP. Recent evidence suggests that the
BTP derivative BTP2 may be beneficial in the treatment of allergic disorders, particularly
bronchial asthma. Amongst other effects, BTP2 inhibits antigen-induced histamine release from
and leukotriene production in IgE-primed RBL-2H3 cells, a model in vitro cell line for mast cells
(84). In agreement with other published work, we have demonstrated that BTP potently inhibits
immune cell activation via modulation of store-operated channel activity (78). Furthermore, our
past studies identified the actin-binding protein drebrin as a target of BTP. We demonstrated that
BTP inhibits actin reorganization mediated by the actin-binding protein and that loss of drebrin
protein expression prevents SOCE, similar to BTP treatment, in T cells. Our identification of
drebrin as a mediator of SOCE has provided insight into the interaction between actin
rearrangement and stimulus-induced Ca2+
mobilization (78).
All of these results implicate a regulatory role for drebrin in mast cell-mediated disease.
Here, utilizing a novel knockout mouse, we demonstrate that drebrin is required for in vivo
histamine release. Also, we show that drebrin regulates FcεRI-induced degranulation and
cytokine secretion in a Ca2+
-dependent manner in BMMCs. Collectively, these observations

87
provide the first genetic evidence that the actin-binding protein drebrin is required for Ca2+
mobilization in mast cells and has a role in allergic reactions.
Generation of Drebrin-/-
mice
To further understand the function of drebrin in immune cells, we generated drebrin-/-
mice from ES cell clones that we obtained from the TIGM. The TIGM has used high-throughput
gene-trapping with retroviral vectors in mouse ES cells to generate OmniBank, a library of
mutated ES cell clones. In our study, the ES cells were mutated by trapping the Dbn1 gene,
encoding Mus musculus drebrin 1. The gene-trapping cassette consists of a splice acceptor
sequence followed by the promoterless selectable marker β-geo, a functional fusion between the
β-galactosidase and neomycin resistance genes, with a polyadenylation signal. Insertion of the
retroviral vector into an expressed Dbn1 gene results in the splicing of upstream endogenous
exons into the cassette to generate a fusion transcript. Also encoded in the gene-trapping vector
is the mouse phosphoglycerate kinase gene promoter, a promoter active in ES cells, followed by
an exon, encoding puromycin resistance. The exon of this selectable marker in the 3’ trapping
component is upstream of a splice donor signal. Splicing from this signal to the exons
downstream of the insertion site produces a fusion transcript that was used by TIGM to generate
an Omnibank sequence tag (OST) of the trapped Dbn1 gene by Rapid Amplification of cDNA
ends (RACE), as previously described (113, 114). The TIGM has demonstrated that the OST
sequence is a reliable indicator of the genomic location of gene-trap inserts (114). Importantly,
the puromycin resistance exon contains termination codons which prevent the translation of
downstream fusion transcripts (Fig. 5.1).

88
Figure 5.1. Strategy for simultaneous inactivation and rapid identification of the disrupted
Dbn1 gene by gene-trapping in mouse ES cells. SA, splice acceptor sequence; β-Geo, fusion
of β-galactosidase and neomycin resistance genes; pA, polyadenylation signal; Pgk,
phosphoglycerate kinase gene promoter; Puro, puromycin resistance gene; SD, splice donor
sequence.

89
The procedures for infection of mouse ES cells with the described Omnibank gene-
trapping construct VICTR vector 21, selection and growth of ES cell clones, and determination
of the mutation sequence for the ES cell clone were performed by the TIGM, as reported in detail
by Zambrowicz et al. (114). The TIGM selected ES cell clones for microinjection based upon
the confirmation of intronic insertion of the gene-trapping construct. This was determined by
cloning the genomic insertion site with inverse PCR, as previously described (114). The ES cell
clone represented by OST 7352 was selected for the generation of drebrin-/-
mice. Inverse
genomic PCR of DNA isolated from OST 7352 cells confirmed that the retroviral gene-trap
vector had inserted in intron 8 of the mouse Dbn1 gene on chromosome 13 (114). To
specifically amplify the WT allele, PCR-based genotyping employed a primer that flanks the
genomic insertion site of the gene-trap vector (primer B). To amplify the mutated allele,
genotyping employed a LTR reverse primer, complementary to OmniBank vectors (Fig. 5.2).
Mice heterozygous for the mutation were generated by using standard methods of host embryo
microinjection, chimera production, and germ-line transmission (114). Heterozygous
intercrosses yielded homozygous animals, demonstrating that drebrin is not required for
embryonic development.
To demonstrate that trapping the Dbn1 gene led to a decrease in Dbn1 mRNA expression,
Dbn1 mRNA was measured by performing quantitative real-time RT-PCR with TaqMan probes
specific for the region spanning between exons 8-10 of the Dbn1 gene, where genomic insertion
of the gene-trapping cassette occurred. BMMCs of drebrin-/-
mice exhibited approximately a 4-
fold decrease in Dbn1 mRNA levels (Fig. 5.3). Moreover, our western blot analyses
demonstrated the absence of drebrin protein and correlated with the observed mRNA levels in
drebrin-/-
mice. In drebrin-/-
mice, drebrin protein expression was not detected in whole cell

90
lysates of the brain, where drebrin protein is known to be highly expressed. The truncated form
of drebrin protein that could be generated by the insertion-mediated fusion was also not detected
in the brain of drebrin-/-
mice with antibodies (M2F6 clone) (Fig. 5.4). Importantly, the epitope
recognized by the M2F6 clone has yet to be mapped. Collectively, these data suggest that Dbn1
mRNA derived from drebrin-/-
mice is unstable, resulting in a lack of drebrin protein expression
in these mice.

91
Figure 5.2. Mapping of genomic insertion site of the gene-trap vector in intron 8 of the
Dbn1 gene. Genotyping PCR primers A and B flank the genomic insertion site and amplify a
product for the WT allele. The LTR reverse primer was utilized in conjunction with primer B to
specifically amplify the mutated allele. Rev, reverse.

92
Figure 5.3. Verification of genetic disruption of the Dbn1 gene by gene-trap insertion with
RT-PCR. RT-PCR utilized primers that are complementary to exons flanking the insertion site
in the Dbn1 gene in mouse chromosome 13. Levels of drebrin mRNA from BMMCs were
quantified with murine GAPDH as a housekeeping gene. Data was analyzed using the
Comparative ΔΔCT method. Expression of drebrin was normalized based on the levels of
mRNA for GAPDH and set relative to a calibrator sample. The expression level of WT
populations is set as 1. Data are expressed as the mean ± SEM of 3 independent experiments.
*p<0.05 vs. WT.

93
Figure 5.4. Verification of ablation of protein expression of the disrupted Dbn1 gene by
western blot. Whole cell lysates for neocortical homogenates were analyzed by western blot for
pan-drebrin expression. Blots were stripped and reprobed with control antibodies for β-actin.

94
Cellular morphology but not distribution of drebrin-/-
mast cells in skin tissue is normal
Mast cells are characterized as tissue-based inflammatory cells of hematopoietic origin.
They are located primarily in association with blood vessels and at epithelial surfaces (2). To
characterize the mast cells of mice homozygous for the Dbn1 gene-trap mutation, phenotypic
analysis of skin tissue-resident mast cells was performed with toluidine blue staining and with
transmission electron microscopy. Similar to WT mice, the mast cells of drebrin-/-
mice were
identified by their metachromatic granule content, which was visualized as clusters of dark blue
circular elements with toluidine blue staining. The size and shape of drebrin-/-
mast cells
appeared normal. WT and drebrin-/-
mast cells retained similar granule density and shape and
shared similar patterns of tissue distribution (Fig. 5.5A). Quantitation of the density of mast cells
per square mm indicated a significant decrease in drebrin-/-
mice (Fig. 5.5B). Observation with
electron microscopy further corroborated our findings with histological staining (Fig. 5.5C).
Overall, these observations suggest that there is no difference between the cellular morphology
of WT and drebrin-/-
mast cells but that there is a difference in the distribution of skin mast cells
between these two groups in vivo.

95
Figure 5.5. Mast cells retain normal cellular morphology but not distribution in the skin of
drebrin-/-
mice. (A) Toluidine blue-stained skin sections from WT and drebrin-/-
mice (6-8
weeks old). Black arrows identify mast cells. (B) Quantification of the number of skin-resident
mast cells per standardized area. Data are expressed as the mean ± SEM of n = 3 per group, each
representative of the average of individual counts of 3 separate tissue block sections from (A) per
mouse. *p<0.05 vs. WT. (C) Transmission electron micrographs of skin sections from WT and
drebrin-/-
mice (6-8 weeks old). For (A and C), representative images is shown; n = 3 per group.

96
Drebrin is not required for development but is necessary for survival of drebrin-/-
BMMCs
in vitro
Mast cells arise from CD34+ pluripotent progenitor cells in the bone marrow. From the
bone marrow, mast cell precursors migrate into blood circulation and then home to tissues, where
they mature (2). Though both IL-3 and SCF represent critical mast cell growth factors, there are
published findings suggesting that IL-3 alone elicits mast cell growth from unfractionated mouse
bone marrow cells in vitro. This implicates the existence of bone marrow cell subpopulations
that are capable of mast cell growth in response to IL-3 alone (115-117). To determine if
absence of drebrin expression altered the development of mast cells from bone marrow
precursors, the differentiation of drebrin-/-
BMMC cultures in the presence of IL-3 alone was
monitored over a time course of 6 weeks. The percentage of cultured cells that expressed the
cell-surface receptors c-kit and FcεRI, which are canonical markers of fully differentiated mast
cells, was monitored on a weekly basis via flow cytometry. For WT mice, differentiation of
bone marrow precursors into mature mast cells occurred between 2 to 4 weeks and reached a
plateau of >95% c-kit+ FcεRI
+ cells between 5 to 6 weeks in IL-3 supplemented cultures. No
significant difference in the rate of differentiation of drebrin-/-
BMMCs was observed (Fig.
5.6A).
Like mast cells, basophils develop from CD34+ progenitors. After they differentiate and
mature in the bone marrow, they migrate to the periphery where they circulate in the blood.
Basophils are characterized by a c-kit- FcεRI
+ cell surface receptor expression profile. IL-3 is
the dominant cytokine driving basophil differentiation and is sufficient to differentiate stem cells
into basophils (2). Considering that we supplemented our BMMC cultures with IL-3 alone, the
development of basophils in our cultures was evaluated. Throughout 6 weeks of culture,

97
basophils comprised approximately less than 5 percent of our cell cultures. Interestingly, a slight
decrease in the percentage of c-kit- FcεRI
+ cells was observed for drebrin
-/- BMMC cultures
during 2 to 6 weeks of development; however, these differences were not statistically significant
(Fig. 5.6B). Together, these data suggest that the maturation and differentiation of drebrin-/-
BMMCs and basophils occurs normally in the presence of IL-3 in vitro.

98
Figure 5.6. Drebrin-/-
BMMCs and basophils show normal development in vitro. (A) Rate
of differentiation of c-kit+ FcεRI
+ cells in BMMC cultures. Data are expressed as the mean ±
SEM of replicates, representative for BMMC cultures of 8 individual mice per group. (B)
Drebrin-/-
bone marrow-derived basophil development. Rate of differentiation of c-kit- FcεRI
+
cells in BMMC cultures. Data are expressed as the mean ± SEM of replicates, representative for
BMMC cultures of 8 individual mice per group.

99
Next, the population growth of our fully differentiated drebrin-/-
BMMC cultures was
monitored to evaluate the contribution of drebrin to this process in vitro. In short, after 4 weeks
of growth in the presence of IL-3, cultures that consisted of >95% c-kit+ FcεRI
+ BMMCs were
seeded at 106 cells, cultured, and monitored over the course of another 4 weeks, as described in
the Materials and Methods section. A decrease in the total number of c-kit+ FcεRI
+ BMMCs was
observed for drebrin-/-
cultures as early as 1 week after the initiation of the culture. After 4
weeks, an approximate 3-fold difference in the number of BMMCs between WT and drebrin-/-
cultures was observed. This suggests that drebrin-/-
BMMC cultures grow at a slower rate in
vitro (Fig. 5.7A). Because the growth of our cultures was mediated by IL-3, a potential
explanation for the observed differences is that drebrin-/-
BMMCs express lower levels of IL-3
receptor. Flow cytometry showed that IL-3Rα was expressed at similar levels on the surface of
WT and drebrin-/-
BMMCs (Fig. 5.7B). Alternatively, decreased viability of drebrin-/-
BMMCs
could account for the slower rate of growth of respective cultures. The viability of drebrin-/-
BMMCs was evaluated by flow cytometry with LIVE/DEAD dye staining, which can permeate
the compromised membranes of necrotic cells and react with free amines in the interior, as well
as on the cell surface. Unlike WT BMMCs that were relatively dimly stained due to the presence
of only cell surface amines, drebrin-/-
BMMCs were intensely stained. Gating of non-viable cells
demonstrated that approximately 60 percent of drebrin-/-
BMMCs were non-viable in comparison
to approximately 2 percent of WT BMMCs (Fig. 5.7C). Therefore, the decreased viability of
drebrin-/-
cultures suggests that drebrin is required for the survival and growth of BMMCs in
vitro.

100
Figure 5.7. Drebrin-/-
BMMCs show less viability in vitro. (A) Rate of growth of c-kit+
FcεRI+ population in BMMC cultures with >95% c-kit
+ FcεRI
+ cells. (B) BMMCs were
analyzed for cell surface expression of IL-3Rα via flow cytometry. (C) BMMCs were analyzed
for LIVE/DEAD Fixable Dead Cell Red Stain via flow cytometry. The percentage of non-viable
cells was calculated based upon the non-viable cell gate, set for the LIVE/DEAD Fixable Dead
Cell Red Stain histogram. For (A-C), data are expressed as the mean ± SEM of 3 independent
experiments. *p<0.05 vs. WT.

101
Drebrin is required for degranulation of BMMCs
Mast cells respond to signals of innate and adaptive immunity with immediate and
delayed release of inflammatory mediators. The critical mediators of the immediate response are
comprised of preformed mediators that are stored in cytoplasmic granules. They include
histamine, serine proteases (tryptase and chymase), carboxypeptidase A, and proteoglycans.
Upon activation of mast cells, granules fuse with the plasma membrane, and the contents are
released into the extracellular environment within minutes to drive immune responses (2). To
assess if the absence of drebrin affected the mast cell effector response of degranulation, a β-
hexosaminidase secretion assay was first performed to measure the magnitude of degranulation
for drebrin-/-
BMMCs. β-hexosaminidase is a lysosomal enzyme which is secreted
proportionally to the extent of degranulation (118, 119). Initial studies involved stimulation with
the Ca2+
ionophore ionomycin, which can elevate intracellular Ca2+
concentration and, thus,
activate mast cell degranulation downstream of the FcεRI (89-91). In comparison to WT
BMMCs, drebrin-/-
BMMCs exhibited reduced levels of degranulation upon stimulation with
ionomycin. The decrease equalled to an approximate reduction of 20 percent of WT levels (Fig.
5.8A). Because ionomycin is not a physiological stimulus, the degranulation responses of
drebrin-/-
BMMCs following more physiologically relevant stimuli via the FcεRI were also
measured. Drebrin-/-
BMMCs showed significantly reduced levels of degranulation induced by
FcεRI crosslinking with IgE and graded doses of the antigen DNP-HSA. Degranulation
responses steadily increased with increasing amounts of DNP-HSA presumably as the saturation
of FcεRIs with crosslinking antibody-antigen complexes was reached. The optimal dose was
100 ng. Similar to stimulation with ionomycin, challenge with 10 to 1000 ng DNP-HSA
demonstrated diminished degranulation for drebrin-/-
BMMCs to approximately 80 percent of

102
WT levels (Fig. 5.8B). These results show that lack of drebrin expression significantly reduces
degranulation mediated by FcεRI crosslinking with antigen. Drebrin is, therefore, an important
regulator of mast cell degranulation induced through the FcεRI.
Drebrin is required for histamine release in mice in response to antigenic challenge
To further characterize the effects of knockout of drebrin expression on the mast cell-
dependent physiological response of degranulation, the release of histamine in vivo was analyzed
in drebrin-/-
mice. As described in the Materials and Methods section, control mice were either
left untreated or sensitized with IgE but left unexposed to antigen to ensure that the measured
responses were antigen-specific. As expected, histamine release was undetectable under basal
conditions or with sensitization alone in WT and drebrin-/-
mice. Upon antigenic challenge with
DNP-HSA, evoked histamine release was detected in both WT and drebrin-/-
mice. Drebrin-/-
mice exhibited significantly reduced levels of in vivo histamine release (Fig. 5.9A). Varying
levels of serum IgE in drebrin-/-
mice could be responsible for the observed difference. However,
ELISAs showed that levels of serum IgE in drebrin-/-
mice were similar to WT levels (Fig. 5.9B).
In summary, our data provides strong support for the idea that drebrin plays an important role in
potentiating mast cell degranulation in vitro and in vivo independent of serum IgE levels.

103
Figure 5.8. Drebrin-/-
mice exhibit impairment in mast cell degranulation in vitro. (A-B) β-
hexosaminidase secretion of BMMCs. β-hexosaminidase secretion in supernatants was
measured for BMMCs either (A) stimulated with ionomycin (500 nM) for 1h or (B) sensitized
with mouse IgE anti-DNP (2 µg/mL) overnight and subsequently challenged with DNP-HSA
(for final concentration of 0-1000 ng/mL) for 1 h. The β-hexosaminidase activities of the blank
substrate solutions (blank) were subtracted from the enzyme activities of the unstimulated or
stimulated cells (test). After stimulation, cells were lysed with 1% Triton-X, and the extracts
were analyzed for their β-hexosaminidase activities (total – test – blank). The percentage of β-
hexosaminidase released into each supernatant was calculated using the following formula: β-
hexosaminidase secretion (% of total) = (test – blank)/(total – blank) x 100. Data are expressed
as the mean ± SEM of 3 independent experiments. For (A-B), *p<0.05 vs. WT.

104
Figure 5.9. Drebrin-/-
mice exhibit impairment in FcεRI-mediated histamine release in vivo.
(A) In vivo histamine release of WT and drebrin-/-
mice (8 weeks old). Experimental mice were
sensitized with mouse IgE anti-DNP overnight (1 µg) then challenged with DNP-HSA (50 μg)
for 3 min. Control mice were either left untreated (Basal) or sensitized and injected with PBS
alone to avoid exposure to DNP-HSA and ensure antigen dependence for experimental mice.
Thereafter, serum histamine concentration levels were determined. Data are expressed as the
mean ± SEM of n = 3-5 per group. (B) Serum IgE levels of WT and drebrin-/-
mice (8 weeks
old). Data are expressed as the mean ± SEM of n = 3 per group. For (A-B), *p<0.05 vs. WT.

105
Drebrin is required for cytokine secretion of BMMCs
Unlike granule-associated mediators that are immediately released following mast cell
activation, cytokines drive the delayed responses of hypersensitivity immune reactions (2).
Importantly, the release of both groups of inflammatory mediators is regulated by overlapping
intracellular signaling pathways (10). In consideration of this and our observation that the
absence of drebrin diminished mast cell degranulation, the release of cytokines from drebrin-/-
BMMCs was evaluated. Previous investigation has demonstrated that stimulation with
ionomycin can activate mast cell cytokine secretion downstream of the FcεRI in the presence of
the phorbol ester PMA (89-91). At both 8 and 24 hours after stimulation with PMA and
ionomycin, drebrin-/-
BMMCs secreted less IL-2, TNF-α, and GM-CSF. Diminished levels of
IL-3, IL-4, and IL-13 secreted by drebrin-/-
BMMCs at 8 and 24 hour time points were also
observed; however, these differences were not statistically significant with the exception of that
observed for IL-13 at 24 hours post-stimulation (Fig. 5.10). Under conditions of physiological
stimulation with crosslinking anti-IgE antibodies, FcεRI triggering resulted in decreased IL-2
and GM-CSF secretion by drebrin-/-
BMMCs. The difference observed for IL-2 at 24 hours post-
stimulation was not statistically significant. In addition, though TNF-α was not detected for WT
nor drebrin-/-
BMMCs at 8 hours after challenge with anti-IgE antibodies, a significant reduction
in TNF-α secretion at the 24 hour time point was monitored for drebrin-/-
BMMCs (Fig. 5.11).
No to little secretion of IL-3, IL-4, and IL-13 was observed for both WT and drebrin-/-
BMMCs
with FcεRI crosslinking. Thus, mast cell cytokine secretion that is initiated through the FcεRI
necessitates the presence of drebrin.

106
Figure 5.10. Drebrin-/-
BMMCs exhibit impairment in PMA/ionomycin-induced cytokine
secretion. BMMCs were stimulated with PMA (50 nM) and ionomycin (500 nM). 8 and 24 hr
after stimulation, the concentration of the indicated cytokines was determined through
measurement via Milliplex MAP immunoassay. Data are expressed as the mean ± SEM of
technical non-independent replicates, representative of 3 independent experiments. *p<0.05 vs.
WT.

107
Figure 5.11. Drebrin-/-
BMMCs exhibit impairment in FcεRI-mediated cytokine secretion.
BMMCs were sensitized with mouse IgE anti-DNP and then stimulated with rat anti-mouse IgE
(30 µg/mL). 8 and 24 hr after stimulation, the concentration of the indicated cytokines was
determined through measurement via Milliplex MAP immunoassay. Data are expressed as the
mean ± SEM of technical non-independent replicates, representative of 3 independent
experiments. *p<0.05 vs. WT.

108
Drebrin-/-
BMMCs exhibit normal levels of FcεRI surface expression
To elucidate potential mechanisms responsible for the differences observed for drebrin-/-
mast cell effector responses, biochemical analysis of cellular signaling pathways leading to mast
cell activation was performed. As shown in Fig. 5.9B, serum titers of IgE were comparable
between WT and drebrin-/-
mice, thereby eliminating the possibility that varying levels of IgE
were altering the ability of exogenous antigen-specific IgE to activate drebrin-/-
mast cells in
vitro and in vivo. On the other hand, an alternative explanation for our observed phenotypical
differences is that drebrin-/-
BMMCs could express lower levels of FcεRI on their surface and
resultantly modulate activation signal strength. As determined by flow cytometry, surface
expression of the FcεRIα chain, which is responsible for binding IgE, was slightly decreased for
drebrin-/-
BMMCs; however, this difference was not statistically significant (Fig. 5.12). Drebrin
is, ergo, not required to maintain normal levels of FcεRI surface expression.

109
Figure 5.12. Cell surface expression of FcεRI on drebrin-/-
BMMCs is normal. (A) BMMCs
were analyzed for FcεRI via flow cytometry. Data are expressed as the mean ± SEM of triplicate
experiments. *p<0.05 vs. WT.

110
Phosphorylation events downstream of FcεRI are normal in drebrin-/-
BMMCs
Phosphorylation events triggered by FcεRI engagement with antibody-antigen complexes
relay signals required for the activation of degranulation and cytokine secretion. In brief, upon
crosslinking of FcεRIs, Lyn kinase is brought into closer proximity of the FcεRI where it can
phosphorylate the tyrosine residues of ITAMs in the FcεRI β- and γ-chains. These ITAMs
provide docking sites for the tethering of Lyn and Syk and subsequent activation of Syk. Syk,
then, phosphorylates the transmembrane adaptor molecule LAT, coordinating the association of
a multimolecular complex that consists of cytosolic adaptor molecules, GEFs, and signaling
enzymes (Fig. 1.1). Integral to the assembly of this macromolecular complex is tyrosine
phosphorylation (10). To evaluate tyrosine kinase activity in drebrin-/-
BMMCs, global patterns
of tyrosine phosphorylation for WT and drebrin-/-
BMMCs that were challenged with antigen
were monitored over a time course of 1 hour post-stimulation. For both WT and drebrin-/-
BMMCs, overall tyrosine phosphorylation peaked at 2 minutes post-challenge and declined
thereafter. Densitometric analysis showed no major differences between WT and drebrin-/-
BMMCs for this parameter (Fig. 5.13A). Moreover, the phosphorylation of Lyn in drebrin-/-
BMMCs was further assessed because this kinase has been shown to be a negative regulator of
degranulation for BMMCs derived from mice of the 129/Sv genetic background (120-126). No
substantial difference in Lyn phosphorylation, which reached its highest levels at 2 minutes post-
challenge and gradually decreased over the remaining time course, was observed in drebrin-/-
BMMCs. As expected, densitometric analysis of the intensity of corresponding bands revealed
no differences (Fig. 5.13B).

111
A)
B)
Figure 5.13. Tyrosine kinase activation following FcεRI triggering is not affected in
drebrin-/-
BMMCs. BMMCs were stimulated with mouse IgE anti-DNP and DNP-HSA for the
indicated time periods, lysed, and analyzed by western blot for (A) total phosphotyrosine; or (B)
phospho-Lyn. Blots were stripped and reprobed with control antibodies for either β-actin or α-
tubulin. Densitometric values indicate fold increase that has been corrected to account for
expression levels.

112
Occurring in parallel to the canonical mast cell activation pathway described above, the
MAPK pathway can be activated through GEF-mediated exchange of GDP for GTP with the
small GTPase Ras. Activated Ras can subsequently activate the serine/threonine kinase family
of MAPK proteins by transmitting signals to Raf and other downstream elements, leading to
eicosanoid generation and the production of cytokines (10). To address whether the absence of
drebrin affects MAPK activation, phosphorylation of Erk1/2, JNK, and p38 was monitored.
Peak phosphorylation for Erk1/2 and p38 was detected at 2 minutes post-stimulation (Fig. 5.14A
and 14C, respectively). In the case of JNK, phosphorylation reached its highest level at the 10
minute time point post-challenge (Fig. 5.14B). Phosphorylation of the MAPKs declined after
time points of peak phosphorylation as determined by densitometric analysis. No major
differences were observed between WT and drebrin-/-
BMMCs (Fig. 5.14, A-C). Collectively,
these results suggest that drebrin does not regulate mast cell effector responses via modulation of
tyrosine and serine/threonine kinase activity downstream of FcεRI triggering.

113
A)
B)
C)
Figure 5.14. MAP kinase activation following FcεRI triggering is not affected in drebrin-/-
BMMCs. BMMCs were stimulated with mouse IgE anti-DNP and DNP-HSA for the indicated
time periods, lysed, and analyzed by western blot for (A) phospho-ERK1/2; (B) phospho-JNK;
or (C) phospho-p38 expression. Blots were stripped and reprobed with control antibodies for
either β-actin. Densitometric values indicate fold increase that has been corrected to account for
expression levels.

114
Drebrin is required for Ca2+
mobilization in mast cells
As detailed previously, increases in intracellular Ca2+
concentration that are triggered
through the FcεRI by IgE-antigen complexes are necessary for the functional responses of mast
cells. To evaluate the potential that drebrin may have a role in the mobilization of Ca2+
in mast
cells, Ca2+
signals generated in WT and drebrin-/-
BMMCs upon activation of FcεRI-mediated
signaling were compared. Utilizing the calcium-sensitive dye Fluo-4, changes in intracellular
Ca2+
concentration were monitored. Upon physiological stimulation of sensitized BMMCs with
the antigen DNP-HSA, drebrin-/-
BMMCs exhibited impaired Ca2+
mobilization (Fig. 5.15A).
Importantly, diminished Ca2+
flux was more pronounced in the latter phase of Ca2+
mobilization
than in the initial response. The initial phase of Ca2+
mobilization corresponds to the depletion
of intracellular stores. This step is primarily driven by the generation of IP3 by activated PLCγ
and the subsequent binding of IP3 to IP3Rs on the surface of the ER. To investigate whether this
step was affected in the absence of drebrin, the phosphorylation of PLCγ1 in drebrin-/-
BMMCs
was monitored over a time course of 1 hour after stimulation with DNP-HSA. Based upon the
profile of phosphorylation for PLCγ1 in WT and drebrin-/-
BMMCs, activation of the signaling
enzyme peaked at 2 minutes post-antigenic challenge and remained in the active state until
approximately the 10 minute time point. In comparison to WT BMMCs, no major difference in
the level of phosphorylation of PLCγ1 for drebrin-/-
BMMCs was shown according to
densitometric analysis (Fig. 5.16). This data, therefore, implicates a role for drebrin in the latter
phase of Ca2+
mobilization. The latter phase of Ca2+
mobilization is due almost entirely to the
uptake of extracellular Ca2+
through CRAC channels. To quantitate the differences observed in
the latter phase, we determined mean intracellular Ca2+
concentration levels post-stimulation, as
well as the slope of calcium decay. Both parameters showed a significant difference in the

115
magnitude of Ca2+
mobilization in drebrin-/-
BMMCs (Fig. 5.15, B-C). In summary, these results
provide the first genetic evidence for a novel role for drebrin in the regulation of SOCE and
downstream Ca2+
-dependent mast cell effector responses.
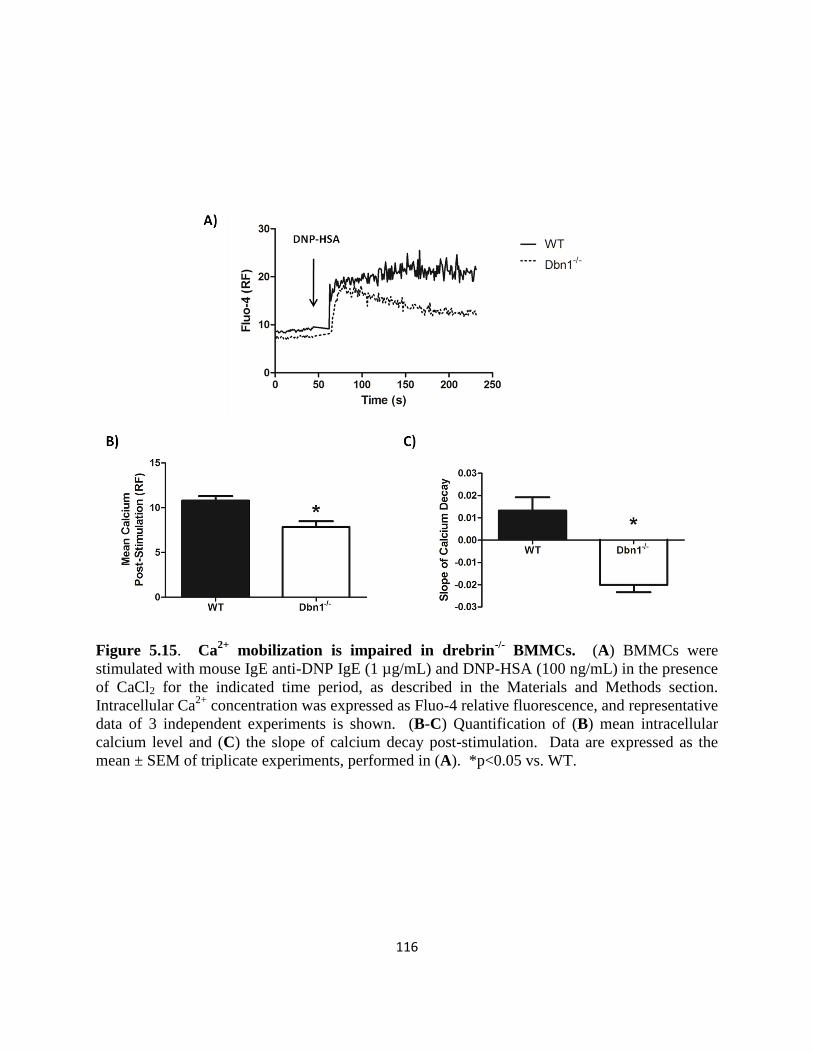
116
Figure 5.15. Ca2+
mobilization is impaired in drebrin-/-
BMMCs. (A) BMMCs were
stimulated with mouse IgE anti-DNP IgE (1 µg/mL) and DNP-HSA (100 ng/mL) in the presence
of CaCl2 for the indicated time period, as described in the Materials and Methods section.
Intracellular Ca2+
concentration was expressed as Fluo-4 relative fluorescence, and representative
data of 3 independent experiments is shown. (B-C) Quantification of (B) mean intracellular
calcium level and (C) the slope of calcium decay post-stimulation. Data are expressed as the
mean ± SEM of triplicate experiments, performed in (A). *p<0.05 vs. WT.

117
Figure 5.16. Activation of PLCγ1 is not affected in drebrin-/-
BMMCs. BMMCs were
stimulated with mouse IgE anti-DNP and DNP-HSA for the indicated time periods, lysed, and
analyzed by western blot for phospho-PLCγ1. Blots were stripped and reprobed with control
antibodies for β-actin. Densitometric values indicate fold increase that has been corrected to
account for expression levels.

118
Discussion
Drebrin regulates the mast cell-mediated allergic response.
We demonstrate that an actin-binding protein drebrin plays a crucial role in mast cell
activation and mast cell-mediated allergic responses. Acknowledged as the central effector cell
of allergic inflammation, the mast cell is integral to the pathogenesis of IgE-associated disorders,
such as anaphylaxis and allergic asthma (2, 127). In support of this, characterization of
genetically engineered mast cell-deficient KitW
/KitW-V
or KitW-sh
/KitW-sh
mice has demonstrated
the requirement for mast cells in the IgE-mediated immediate-type allergic response. This
immediate response is prompted by degranulation of preformed mediators and synthesis of
arachidonic acid metabolites (prostaglandins, leukotrienes) and platelet-activating factor (PAF)
(128-130). The contribution of mast cells in the immediate allergic reaction has, therefore, been
well-documented. Beyond this conventional role, ample investigations have shown that mast
cells participate in delayed-type allergic responses and chronic inflammatory diseases, including
arthritis, EAE, colitis, and Chediak-Higashi syndrome (131-134). A myriad of mast cell-derived
cytokines and chemokines presumably play an important role in delayed-type allergic reactions
and chronic inflammatory diseases. Promoting this idea, experiments have shown that EAE can
be rescued in mast cell-defective KitW
/KitW-V
mice, which do not develop EAE, by the transfer
of WT BMMCs into these mice; however, transfer of IL-4-deficient BMMCs does not rescue the
defect (135). This evidence indicates that mast cell-derived cytokines are crucial determinants of
inflammatory disease.
Here, we demonstrated that drebrin-/-
mast cells had an impaired degranulation process,
as well as dysfunctional cytokine secretion. Under different activation stimuli, drebrin-/-

119
BMMCs exhibited diminished levels of degranulation. Secretion of IL-2, IL-3, IL-4, IL-13,
TNF-α, and GM-CSF by drebrin-/-
BMMCs was reduced in response to stimulation with PMA
and ionomycin. A similar trend was monitored upon FcεRI triggering with crosslinking antibody
complexes; however, no to little IL-3, IL-4, and IL-13 was detected in this scenario. In mast
cells, the production of IL-2 is controlled almost entirely at the transcriptional level (100). On
the contrary, TNF-α, IL-4, and IL-13 occur both preformed and as newly synthesized molecules
(2, 101). To decipher the molecular mechanism responsible for reduced cytokine secretion by
drebrin-/-
BMMCs, future research efforts should be invested in determining whether preformed
levels of TNF-α, IL-4, and IL-13 are reduced in drebrin-/-
BMMCs or whether de novo
transcription is modulated differently in drebrin-/-
BMMCs. This will elucidate whether the
diminished secretion of select cytokines by drebrin-/-
BMMCs is attributed to dysfunctional
transcription and ensuing translation of preformed mRNA in these cells. Based upon our
observations for IL-2 secretion by drebrin-/-
BMMCs, it is reasonable to predict that, at least,
drebrin is important for de novo transcription of cytokines whose production is stimulated by
FcεRI triggering and subsequent mast cell activation. In addition, drebrin-/-
mice were
characterized by defective histamine release upon antigenic challenge. Importantly, histamine is
derived from mast cells and basophils. Like mast cells, basophils express FcεRI on their cell
surface. Ensuing the aggregation of IgE-bound FcεRIs by multivalent antigen, basophils are
activated. Granule exocytosis and release of mediators follows (2). Because of parallel
mechanisms of activation for mast cells and basophils, basophils from drebrin-/-
mice may
potentially have diminished degranulation. In combination with decreased mast cell
degranulation, this may have exaggerated the reduction in histamine release responses observed
for drebrin-/-
mice in our study. Further investigation will need to be performed to explore this

120
possibility. It will also be important to evaluate whether the reduction in histamine release is
associated with lower numbers of drebrin-/-
skin mast cells in vivo, as shown by histology.
Nonetheless, our histological staining and TEM analysis implicates that the granule content of
drebrin-/-
mast cells is not different than that of WT mast cells. These results suggest that
histamine is stored in the cytoplasmic granules of drebrin-/-
mast cells at normal levels.
However, as determined by our in vivo assay, drebrin-/-
mast cells release histamine in lower
quantities. Our in vitro characterization of degranulation for drebrin-/-
BMMCs provides further
evidence for the concept that mast cell degranulation is compromised in the absence of drebrin.
In summary, our results implicate that the drebrin-/-
mouse will be a valuable tool for analyzing
the pathological status of a variety of immune-related diseases (134).
Yet, the incomplete impairment of effector responses of drebrin-/-
BMMCs suggests the
possibility that other molecular players may have a compensatory role for drebrin and vice-versa.
Importantly, our recent studies have indicated that BTP has off-target effects. When drebrin-/-
BMMCs were treated with the BTP derivative BTP2, already diminished levels of ionomycin-
induced degranulation were further decreased (Fig. 5.17). It is, therefore, likely that other
molecules play a part in calcium responses and are also targeted by BTP. Further exploration
should determine the existence of proteins with redundant immunomodulatory roles.

121
Figure 5.17. BTP2 inhibits ionomycin-induced degranulation of Drebrin-/-
BMMCs. β-
hexosaminidase secretion in supernatants was measured for Drebrin-/-
BMMCs that were pre-
treated with BTP2 (1 µM) or vehicle (DMSO) and then either left unstimulated or stimulated
with ionomycin (500 nM) for 1h. The β-hexosaminidase activities of the blank substrate
solutions (blank) were subtracted from the enzyme activities of the unstimulated or stimulated
cells (test). After stimulation, cells were lysed with 1% Triton-X, and the extracts were analyzed
for their β-hexosaminidase activities (total – test – blank). The percentage of β-hexosaminidase
released into each supernatant was calculated using the following formula: β-hexosaminidase
secretion (% of total) = (test – blank)/(total – blank) x 100. Data are expressed as the mean ±
SEM of 6 independent experiments.

122
Drebrin is required for FcεRI-mediated Ca2+
mobilization.
Our results revealed that drebrin is important for FcεRI-mediated degranulation and
cytokine secretion. As drebrin-/-
mice exhibited diminished levels of histamine release upon
antigenic challenge, we first examined whether drebrin-/-
mice were defective in IgE production.
Serum IgE titers were normal, indicating that IgE production was unaffected by drebrin
deficiency in these mice. We also observed normal surface expression of FcεRI on drebrin-/-
BMMCs. This observation is consistent with our results which demonstrate normal FcεRI-
induced activation of tyrosine kinases and the serine/threonine kinase family of MAPKs.
Collectively, these results led to the conclusion that phosphorylation events downstream of the
FcεRI occur normally in drebrin-/-
mast cells. Our results, however, indicate that drebrin is
involved in the regulation of Ca2+
mobilization.
FcεRI-mediated Ca2+
mobilization was impaired in drebrin-/-
BMMCs. Our previous
work has demonstrated that loss of drebrin protein expression via drebrin-specific siRNA
treatment blunts the latter phase of stimulus-induced Ca2+
mobilization, which corresponds to
extracellular Ca2+
influx, in Jurkat T cells (78). Likewise, in drebrin-/-
BMMCs, the reduction in
Ca2+
flux was more pronounced in the latter phase of Ca2+
mobilization than in the initial
response to physiological stimulus. The diminished Ca2+
response was quantitated to be
significant based upon measurements of population-based mean Ca2+
values over a time course
proceeding stimulation. The slope of calcium decay further corroborated our assessment of
impaired Ca2+
mobilization in drebrin-/-
BMMCs. We, therefore, conclude that drebrin is
selectively required for SOCE in mast cells. In line with this, western blot analysis showed that
the phosphorylation of PLCγ1 in drebrin-/-
BMMCs was unaffected. Because activation of
PLCγ1 is essential for the generation of IP3 and successive liberation of intracellular Ca2+
stores,

123
this result indicates that intracellular Ca2+
store depletion is unaffected in drebrin-/-
BMMCs.
Collectively, our analysis suggests that SOCE and downstream activation of NFAT and other
Ca2+
-dependent transcription factors are impaired in the absence of drebrin.
FcεRI-mediated Ca2+
mobilization is indispensible for degranulation (92). Because the
expression of cytokines depends upon the activation of transcription factors, such as NFAT,
which are Ca2+
-dependent, it also plays a key role in cytokine secretion. Unlike FcεRI-induced
degranulation which occurs within 5 to 30 minutes of antigen binding, the generation of
cytokines and chemokines resulting from increased gene expression takes place within the
delayed-phase of mast cell activation. This occurs 2 to 6 hours post-stimulation (129). Thus,
while both degranulation and cytokine secretion processes necessitate an increase in intracellular
Ca2+
concentration, their requirements for calcium signals may slightly differ because of their
temporal dynamics. The immediate response of degranulation may not have as stringent a
requirement for sustained elevations in intracellular Ca2+
concentration in comparison to the
delayed responses linked to cytokine production. This may resolve the discrepancies in the
degree of impairment of effector responses of drebrin-/-
BMMCs, which exhibit partial decreases
in degranulation and more marked reductions in cytokine secretion. This would fit the
hypothesis that the role of drebrin in Ca2+
mobilization is limited to SOCE.

124
CHAPTER 6
Conclusion and Future Direction

125
These studies provide strong evidence for the capacity of BTP2 and its relatives to act as
potent inhibitors of FcεRI-triggered Ca2+
mobilization. Through this mechanism of action, BTP2
can modulate the potentiation of mast cell activation and downstream effector responses. It,
therefore, holds promise as a potential therapeutic for allergies and other mast-cell mediated
diseases. Interestingly, knockout of expression of the BTP-targeted protein drebrin in our novel
genetically engineered murine model leads to unique mast cell-specific phenotypes that mimic
treatment with BTP2. Consequently, we conclude that our chemico-genetic studies identify a
functional link between the actin-binding protein drebrin and store-operated Ca2+
channels in
mast cells. We hypothesize that BTP interferes with this key interaction, which plays an integral
role in regulatory mechanisms of store-operated Ca2+
entry. Nonetheless, we understand that
careful review of our investigation can introduce caveats to our story.
As previously discussed, the incomplete impairment of effector responses of drebrin-/-
BMMCs suggests the possibility that other molecular players may have a compensatory role for
drebrin and vice-versa. The actin-binding adaptor protein Hematopoietic Progenitor Kinase 1
(HPK1)-interacting Protein of 55 kDa (HIP-55; also called drebrin-like protein, mammalian
Actin-binding protein 1 (mAbp1), and SH3P7) is a good candidate (136). HIP-55 shares
sequence homology with drebrin. In particular, like drebrin, it contains an actin-binding domain
that allows it to control the dendritic spine morphology of neurons (137). BTP interacts with this
domain in drebrin to inhibit its functional role in actin dynamics and, presumably, in Ca2+
mobilization. Importantly, our recent studies have indicated that BTP has off-target effects. As
demonstrated earlier, when drebrin-/-
BMMCs were treated with the BTP derivative BTP2,
already diminished levels of ionomycin-induced degranulation were further decreased (Fig.
5.17). It is, ergo, plausible that HIP-55 plays a part in calcium responses and is also targeted by

126
BTP. Interestingly, analogous to the phenotypes of drebrin-/-
BMMCs, T cells from HIP-55
knockout mice develop normally but display defective proliferation and decreased cytokine
production (138). To a degree, these immunomodulatory roles for HIP-55 have been attributed
to its involvement in TCR expression (138, 139). There has been confusion, however, regarding
its requirement in this process. Experiments have shown that overexpression of HIP-55 down-
regulates TCR expression (139). Nevertheless, a slight increase in TCR internalization in T cells
derived from HIP-55 knockout mice has also been reported (138). In terms of signaling, HIP-55
deficiency in T cells is linked to defective LAT and PLCγ1 phosphorylation, as well as decreased
HPK1 and JNK activation (136, 138). Unfortunately, the role of HIP-55 in SOCE has not been
evaluated. Further exploration with HIP-55 knockout mice and the generation of HIP-55/drebrin
double-knockout mice should determine if the proteins have redundant immunomodulatory roles.
Together, our previous and present findings confirm a novel role for the actin-binding
protein drebrin in the regulation of SOCE in T cells and mast cells (78). Using actin- and
microtubule-depolymerizing agents, others have firmly established a connection between
cytoskeletal rearrangement and the regulation of extracellular Ca2+
influx (50, 140, 141).
Nonetheless, the molecular details of how actin-regulating proteins, such as drebrin, participate
in the regulation of SOCE remain poorly defined. The formation of ER-PM junctions would
necessitate the coordinated effort of a myriad of proteins, ranging from cytoskeletal elements to
anchoring and targeting proteins, in order to adequately dock all of the elements involved in
SOCE. The identification of STIM1 as a microtubule-tracking molecule further advocates this
hypothesis. Recent evidence indicates that dissociation of STIM1 from the microtubule end-
binding protein 1 (EB1) is a prerequisite for the initiation of STIM1 puncta formation and the
activation of SOCE (59, 142). Nonetheless, the purpose of the association between STIM1 and

127
EB1 is not well understood. The identification of binding partners of EB1 and related protein
family members could provide leads to understanding the role of the STIM1-EB1-microtubule
complex in SOCE (59). Of interest, drebrin interacts with a related protein family member EB3.
The direct interaction between drebrin and EB3 is required for neuritogenesis (75). More
investigation is necessary to evaluate the relevance of this interaction in Ca2+
influx and the
assembly of SOCE-associated complexes and to identify possible yet undiscovered ones. On a
related note, in T cells, the actin-modulating protein WASP family member 2 (WAVE-2)
regulates store-operated channel activity. This promotes the idea that modulating the actin
cytoskeleton modulates store-operated channels and related Ca2+
influx (143). Similar to our
work with drebrin, reduction in the expression of WAVE-2 results in a selective block in Ca2+
mobilization at a point distal to PLCγ1 activation and associated IP3-mediated intracellular store
release (143). Therefore, our data, which implicates the involvement of drebrin in store-operated
channel regulation, should provide novel insight into the relationship between the actin
cytoskeleton and the activation of store-operated channels.
Drebrin may regulate extracellular Ca2+
influx by facilitating actin rearrangement that
accompanies the activation of store-operated channels following the depletion of intracellular
Ca2+
reserves. This may be mediated by formation of a macromolecular complex at the IP3R
where drebrin could interact with Homer adaptor proteins, which have been shown to bind both
IP3R and drebrin, as well as the TRPC family of ion-channels (144, 145). Drebrin has recently
been shown to exist in a complex and co-immunoprecipitate with TRPC5 and TRPC6 (146).
Both TRPC5 and TRPC6 are Ca2+
-permeable non-selective cation channels (147, 148).
Interestingly, it has been reported that BTP2 facilitates the activity of TRPM4. As a result, Ca2+
influx and cytokine production are inhibited in BTP2-treated lymphocytes (82). In BMMCs

128
from TRPM4-/-
mice, antigen stimulation leads to more Ca2+
entry as a result of a lack of control
on the driving force for Ca2+
influx. Consequently, the degranulation of TRPM4-/-
BMMCs is
augmented. They release more histamine, leukotrienes, and TNF (149). However, BMMCs
from TRPM4-/-
mice show defects in the highly Ca2+
-dependent process of migration. Shimizu
et al. conclude that TRPM4 controls this process by setting the threshold for Ca2+
-dependent
actin cytoskeleton rearrangements upon FcεRI aggregation (150). It will be intriguing to check
whether TRPM4 activity is affected in drebrin-/-
BMMCs. By recruiting drebrin to such
complexes, the associated actin structure could be induced to rearrange, thereby regulating store-
operated channel function.
We propose a role for drebrin in a revised version of the conformational coupling model.
In our model, when intracellular Ca2+
stores are full, drebrin participates in a macromolecular
complex at the IP3R where it associates with Homer adaptor proteins. Drebrin is tethered to the
IP3R via interaction with Homer. In this state, drebrin is sequestered within the cytoplasmic
space. Simultaneously, STIM1 is associated with EB1 and travels through the ER constantly
(Fig. 6.1). Following store depletion, a conformational change in STIM1 induces its dissociation
from EB1 and the initiation of puncta formation. EB1 and related family members, then, bind to
drebrin. EB proteins direct ER microtubule protrusions toward cortical sites, where drebrin
reorganizes cortical actin promoting the juxtaposition of the ER with the PM. The
rearrangement of actin allows for STIM1 to interact with Orai1 and consequently stimulates Ca2+
influx. Coincidently, in conjunction with STIM1, drebrin drives the fusion of tubular cargos,
containing the TRPC family of ion channels, into lipid raft domains at the PM, where they insert
and activate Ca2+
influx. The IP3R-associated complex participates in the controlled insertion of
TRPC channels into lipid rafts. Drebrin specifically promotes the formation of new cytoskeletal

129
structures that act as physical forces involved in the relocation of TRPC channels into the lipid
raft domains of the PM (Fig. 6.1). In support of this, pharmacological studies have shown that
nocodazole, a microtubule-depolymerizing agent, prevents the triggering of SOCE (151).
Moreover, recent evidence has shown that STIM1 co-immunoprecipitates with Orai1 and TRPC1
(59). In activated mast cells, STIM1 forms puncta associated with microtubules in protrusions
(152). Recent studies also demonstrate that STIM1 governs the insertion of TRPC1 into lipid
raft domains at the PM (153). BTP may inhibit the function of drebrin in this process by either
blocking the interaction between drebrin and this complex or blocking the ability of drebrin to
induce cytoskeletal changes. In summary, this model proposes a critical role for drebrin in
protein anchoring and transport during the assembly of a store-operated Ca2+
influx-associated
complex at the ER-PM junction.

130
Figure 6.1. Model for the involvement of drebrin in store-operated Ca2+
entry. When
intracellular Ca2+
stores are filled, drebrin is tethered to the IP3R via interaction with Homer.
Upon store depletion, EB1 proteins and related family members interact with drebrin and direct
it to the PM, where it reorganizes cortical actin to facilitate the juxtaposition of the ER with the
PM. This promotes the interaction between STIM1 and Orai1 and the insertion of TRPC
channels into the lipid raft domains of the PM, thereby activating Ca2+
influx.

131
Further research dissecting macromolecular complexes in which drebrin participates will
lead to a better understanding of the mechanisms regulating store-operated channel function in
mast cells. Most importantly, it will illuminate the “big picture” of the sophisticated and highly
regulated Ca2+
-dependent mechanisms governing mast cell activation and allergic responses.
In consideration of the role of drebrin in the highly conserved mechanism of SOCE
activation, characterization of the development, activation, and function of other immune cell
populations in the drebrin-/-
mouse will provide general insights into signal transduction, immune
cell function, and mechanisms that contribute to inflammatory disorders and autoimmunity. For
the long term, it will provide a stronger basis for the targeting of drebrin in the design of
pharmaceuticals for immune-related disease.

132
BIBLIOGRAPHY
1. Beaven, M. A. 2009. Our perception of the mast cell from Paul Ehrlich to now. Eur. J.
Immunol. 39: 11-25.
2. Stone, K. D., C. Prussin, and D. D. Metcalfe. 2010. IgE, mast cells, basophils, and eosinophils.
J. Allergy Clin. Immunol. 125: S73-80.
3. Weller, C. L., S. J. Collington, T. Williams, and J. R. Lamb. 2011. Mast cells in health and
disease. Clin. Sci. (Lond) 120: 473-484.
4. Reuter, S., M. Stassen, and C. Taube. 2010. Mast cells in allergic asthma and beyond. Yonsei
Med. J. 51: 797-807.
5. Hu, Z. Q., W. H. Zhao, and T. Shimamura. 2007. Regulation of mast cell development by
inflammatory factors. Curr. Med. Chem. 14: 3044-3050.
6. Wright, H. V., D. Bailey, M. Kashyap, C. L. Kepley, M. S. Drutskaya, S. A. Nedospasov, and
J. J. Ryan. 2006. IL-3-mediated TNF production is necessary for mast cell development. J.
Immunol. 176: 2114-2121.
7. Patella, V., A. Giuliano, J. P. Bouvet, and G. Marone. 1998. Endogenous superallergen protein
Fv induces IL-4 secretion from human Fc epsilon RI+ cells through interaction with the VH3
region of IgE. J. Immunol. 161: 5647-5655.
8. Pandey, V., S. Mihara, A. Fensome-Green, S. Bolsover, and S. Cockcroft. 2004. Monomeric
IgE stimulates NFAT translocation into the nucleus, a rise in cytosol Ca2+, degranulation, and
membrane ruffling in the cultured rat basophilic leukemia-2H3 mast cell line. J. Immunol. 172:
4048-4058.
9. Kalesnikoff, J., M. Huber, V. Lam, J. E. Damen, J. Zhang, R. P. Siraganian, and G. Krystal.
2001. Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine
production and cell survival. Immunity 14: 801-811.
10. Gilfillan, A. M. and C. Tkaczyk. 2006. Integrated signalling pathways for mast-cell
activation. Nat. Rev. Immunol. 6: 218-230.
11. Gilfillan, A. M. and J. Rivera. 2009. The tyrosine kinase network regulating mast cell
activation. Immunol. Rev. 228: 149-169.
12. Metcalfe, D. D., R. D. Peavy, and A. M. Gilfillan. 2009. Mechanisms of mast cell signaling
in anaphylaxis. J. Allergy Clin. Immunol. 124: 639-46; quiz 647-8.

133
13. Wilson, B. S., J. R. Pfeiffer, and J. M. Oliver. 2002. FcepsilonRI signaling observed from the
inside of the mast cell membrane. Mol. Immunol. 38: 1259-1268.
14. Marcum, J. A., J. B. McKenney, S. J. Galli, R. W. Jackman, and R. D. Rosenberg. 1986.
Anticoagulantly active heparin-like molecules from mast cell-deficient mice. Am. J. Physiol.
250: H879-88.
15. Nader, H. B., S. F. Chavante, E. A. dos-Santos, T. W. Oliveira, J. F. de-Paiva, S. M.
Jeronimo, G. F. Medeiros, L. R. de-Abreu, E. L. Leite, J. F. de-Sousa-Filho, R. A. Castro, L.
Toma, I. L. Tersariol, M. A. Porcionatto, and C. P. Dietrich. 1999. Heparan sulfates and
heparins: similar compounds performing the same functions in vertebrates and invertebrates?
Braz. J. Med. Biol. Res. 32: 529-538.
16. Minai-Fleminger, Y. and F. Levi-Schaffer. 2009. Mast cells and eosinophils: the two key
effector cells in allergic inflammation. Inflamm. Res. 58: 631-638.
17. Hamid, Q. and M. Tulic. 2009. Immunobiology of asthma. Annu. Rev. Physiol. 71: 489-507.
18. Galli, S. J., M. Tsai, and A. M. Piliponsky. 2008. The development of allergic inflammation.
Nature 454: 445-454.
19. Hofmann, A. M. and S. N. Abraham. 2009. New roles for mast cells in modulating allergic
reactions and immunity against pathogens. Curr. Opin. Immunol. 21: 679-686.
20. Kumar, V. and A. Sharma. 2010. Mast cells: emerging sentinel innate immune cells with
diverse role in immunity. Mol. Immunol. 48: 14-25.
21. von Kockritz-Blickwede, M. and V. Nizet. 2009. Innate immunity turned inside-out:
antimicrobial defense by phagocyte extracellular traps. J. Mol. Med. 87: 775-783.
22. von Kockritz-Blickwede, M., O. Goldmann, P. Thulin, K. Heinemann, A. Norrby-Teglund,
M. Rohde, and E. Medina. 2008. Phagocytosis-independent antimicrobial activity of mast cells
by means of extracellular trap formation. Blood 111: 3070-3080.
23. Benoist, C. and D. Mathis. 2002. Mast cells in autoimmune disease. Nature 420: 875-878.
24. Christy, A. L. and M. A. Brown. 2007. The multitasking mast cell: positive and negative
roles in the progression of autoimmunity. J. Immunol. 179: 2673-2679.
25. Hogan, P. G. and A. Rao. 2007. Dissecting ICRAC, a store-operated calcium current. Trends
in Biochemical Sciences 32: 235-45.
26. Parekh, A. B. 2010. Store-operated CRAC channels: function in health and disease. Nat. Rev.
Drug Discov. 9: 399-410.

134
27. Baba, Y. and T. Kurosaki. 2009. Physiological function and molecular basis of STIM1-
mediated calcium entry in immune cells. Immunol. Rev. 231: 174-188.
28. Feske, S. 2007. Calcium signalling in lymphocyte activation and disease. Nature Reviews 7:
690-702.
29. Gwack, Y., S. Feske, S. Srikanth, P. G. Hogan, and A. Rao. 2007. Signalling to transcription:
store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium 42: 145-56.
30. Di Capite, J. and A. B. Parekh. 2009. CRAC channels and Ca2+ signaling in mast cells.
Immunol. Rev. 231: 45-58.
31. Scharenberg, A. M. and J. P. Kinet. 1998. PtdIns-3,4,5-P3: a regulatory nexus between
tyrosine kinases and sustained calcium signals. Cell 94: 5-8.
32. Turner, H. and J. P. Kinet. 1999. Signalling through the high-affinity IgE receptor Fc
epsilonRI. Nature 402: B24-30.
33. Hogan, P. G., R. S. Lewis, and A. Rao. 2010. Molecular basis of calcium signaling in
lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 28: 491-533.
34. Hewavitharana, T., X. Deng, J. Soboloff, and D. L. Gill. 2007. Role of STIM and Orai
proteins in the store-operated calcium signaling pathway. Cell Calcium 42: 173-82.
35. Varnai, P., B. Toth, D. J. Toth, L. Hunyady, and T. Balla. 2007. Visualization and
manipulation of plasma membrane-endoplasmic reticulum contact sites indicates the presence of
additional molecular components within the STIM1-Orai1 Complex. J. Biol. Chem. 282: 29678-
29690.
36. Yuan, J. P., W. Zeng, G. N. Huang, P. F. Worley, and S. Muallem. 2007. STIM1
heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat.
Cell Biol. 9: 636-645.
37. Worley, P. F., W. Zeng, G. N. Huang, J. P. Yuan, J. Y. Kim, M. G. Lee, and S. Muallem.
2007. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 42: 205-211.
38. Huang, G. N., W. Zeng, J. Y. Kim, J. P. Yuan, L. Han, S. Muallem, and P. F. Worley. 2006.
STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 8:
1003-1010.
39. Baba, Y., K. Nishida, Y. Fujii, T. Hirano, M. Hikida, and T. Kurosaki. 2008. Essential
function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat.
Immunol. 9: 81-88.

135
40. Vig, M., W. I. DeHaven, G. S. Bird, J. M. Billingsley, H. Wang, P. E. Rao, A. B. Hutchings,
M. H. Jouvin, J. W. Putney, and J. P. Kinet. 2008. Defective mast cell effector functions in mice
lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels.
Nature Immunology 9: 89-96.
41. Parekh, A. B. 2006. On the activation mechanism of store-operated calcium channels.
Pflugers Arch 453: 303-11.
42. Rosado, J. A., P. C. Redondo, S. O. Sage, J. A. Pariente, and G. M. Salido. 2005. Store-
operated Ca2+ entry: vesicle fusion or reversible trafficking and de novo conformational
coupling? Journal of Cellular Physiology 205: 262-9.
43. Parekh, A. B. and R. Penner. 1997. Store depletion and calcium influx. Physiological
Reviews 77: 901-30.
44. Randriamampita, C. and R. Y. Tsien. 1993. Emptying of intracellular Ca2+ stores releases a
novel small messenger that stimulates Ca2+ influx. Nature 364: 809-14.
45. Vig, M. and J. P. Kinet. 2007. The long and arduous road to CRAC. Cell Calcium 42: 157-
62.
46. Yao, Y., A. V. Ferrer-Montiel, M. Montal, and R. Y. Tsien. 1999. Activation of store-
operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger.
Cell 98: 475-85.
47. Fasolato, C., M. Hoth, and R. Penner. 1993. A GTP-dependent step in the activation
mechanism of capacitative calcium influx. The Journal of Biological Chemistry 268: 20737-40.
48. Berridge, M. J. 1995. Capacitative calcium entry. The Biochemical Journal 312 ( Pt 1): 1-11.
49. Irvine, R. F. 1990. 'Quantal' Ca2+ release and the control of Ca2+ entry by inositol
phosphates--a possible mechanism. FEBS Letters 263: 5-9.
50. Patterson, R. L., D. B. van Rossum, and D. L. Gill. 1999. Store-operated Ca2+ entry:
evidence for a secretion-like coupling model. Cell 98: 487-499.
51. Rosado, J. A., S. Jenner, and S. O. Sage. 2000. A role for the actin cytoskeleton in the
initiation and maintenance of store-mediated calcium entry in human platelets. Evidence for
conformational coupling. The Journal of Biological Chemistry 275: 7527-33.
52. Rosado, J. A. and S. O. Sage. 2000. Coupling between inositol 1,4,5-trisphosphate receptors
and human transient receptor potential channel 1 when intracellular Ca2+ stores are depleted.
The Biochemical Journal 350 Pt 3: 631-5.

136
53. Janmey, P. A., K. Iida, H. L. Yin, and T. P. Stossel. 1987. Polyphosphoinositide micelles and
polyphosphoinositide-containing vesicles dissociate endogenous gelsolin-actin complexes and
promote actin assembly from the fast-growing end of actin filaments blocked by gelsolin. J. Biol.
Chem. 262: 12228-12236.
54. Janmey, P. A. and T. P. Stossel. 1987. Modulation of gelsolin function by
phosphatidylinositol 4,5-bisphosphate. Nature 325: 362-364.
55. Lassing, I. and U. Lindberg. 1988. Specificity of the interaction between phosphatidylinositol
4,5-bisphosphate and the profilin:actin complex. J. Cell. Biochem. 37: 255-267.
56. Rosenberg, S., A. Stracher, and K. Burridge. 1981. Isolation and characterization of a
calcium-sensitive alpha-actinin-like protein from human platelet cytoskeletons. J. Biol. Chem.
256: 12986-12991.
57. Burridge, K. and J. R. Feramisco. 1981. Non-muscle alpha actinins are calcium-sensitive
actin-binding proteins. Nature 294: 565-567.
58. Gilbert, S. H., K. Perry, and F. S. Fay. 1994. Mediation of chemoattractant-induced changes
in [Ca2+]i and cell shape, polarity, and locomotion by InsP3, DAG, and protein kinase C in newt
eosinophils. J. Cell Biol. 127: 489-503.
59. Vaca, L. 2010. SOCIC: the store-operated calcium influx complex. Cell Calcium 47: 199-
209.
60. Pivniouk, V. I., S. B. Snapper, A. Kettner, H. Alenius, D. Laouini, H. Falet, J. Hartwig, F. W.
Alt, and R. S. Geha. 2003. Impaired signaling via the high-affinity IgE receptor in Wiskott-
Aldrich syndrome protein-deficient mast cells. Int. Immunol. 15: 1431-1440.
61. Majoul, I., T. Shirao, Y. Sekino, and R. Duden. 2007. Many faces of drebrin: from building
dendritic spines and stabilizing gap junctions to shaping neurite-like cell processes.
Histochemistry and Cell Biology 127: 355-61.
62. Xu, W. and M. Stamnes. 2006. The actin-depolymerizing factor homology and
charged/helical domains of drebrin and mAbp1 direct membrane binding and localization via
distinct interactions with actin. J. Biol. Chem. 281: 11826-11833.
63. Hayashi, K., R. Ishikawa, R. Kawai-Hirai, T. Takagi, A. Taketomi, and T. Shirao. 1999.
Domain analysis of the actin-binding and actin-remodeling activities of drebrin. Experimental
Cell Research 253: 673-80.
64. Hayashi, K. and T. Shirao. 1999. Change in the shape of dendritic spines caused by
overexpression of drebrin in cultured cortical neurons. J Neurosci 19: 3918-25.

137
65. Ishikawa, R., K. Hayashi, T. Shirao, Y. Xue, T. Takagi, Y. Sasaki, and K. Kohama. 1994.
Drebrin, a development-associated brain protein from rat embryo, causes the dissociation of
tropomyosin from actin filaments. The Journal of Biological Chemistry 269: 29928-33.
66. Sasaki, Y., K. Hayashi, T. Shirao, R. Ishikawa, and K. Kohama. 1996. Inhibition by drebrin
of the actin-bundling activity of brain fascin, a protein localized in filopodia of growth cones.
Journal of Neurochemistry 66: 980-8.
67. Shirao, T., K. Hayashi, R. Ishikawa, K. Isa, H. Asada, K. Ikeda, and K. Uyemura. 1994.
Formation of thick, curving bundles of actin by drebrin A expressed in fibroblasts. Experimental
Cell Research 215: 145-53.
68. Takahashi, H., Y. Sekino, S. Tanaka, T. Mizui, S. Kishi, and T. Shirao. 2003. Drebrin-
dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic
density-95 and dendritic spine morphogenesis. J Neurosci 23: 6586-95.
69. Toda, M., T. Shirao, and K. Uyemura. 1999. Suppression of an actin-binding protein,
drebrin, by antisense transfection attenuates neurite outgrowth in neuroblastoma B104 cells.
Brain Research 114: 193-200.
70. Sekino, Y., N. Kojima, and T. Shirao. 2007. Role of actin cytoskeleton in dendritic spine
morphogenesis. Neurochemistry International 51: 92-104.
71. Mizui, T., H. Takahashi, Y. Sekino, and T. Shirao. 2005. Overexpression of drebrin A in
immature neurons induces the accumulation of F-actin and PSD-95 into dendritic filopodia, and
the formation of large abnormal protrusions. Molecular and Cellular Neurosciences 30: 630-8.
72. Biou, V., H. Brinkhaus, R. C. Malenka, and A. Matus. 2008. Interactions between drebrin
and Ras regulate dendritic spine plasticity. Eur. J. Neurosci. 27: 2847-2859.
73. Kojima, N. and T. Shirao. 2007. Synaptic dysfunction and disruption of postsynaptic drebrin-
actin complex: a study of neurological disorders accompanied by cognitive deficits.
Neuroscience Research 58: 1-5.
74. Kojima, N., K. Hanamura, H. Yamazaki, T. Ikeda, S. Itohara, and T. Shirao. 2010. Genetic
disruption of the alternative splicing of drebrin gene impairs context-dependent fear learning in
adulthood. Neuroscience 165: 138-150.
75. Geraldo, S., U. K. Khanzada, M. Parsons, J. K. Chilton, and P. R. Gordon-Weeks. 2008.
Targeting of the F-actin-binding protein drebrin by the microtubule plus-tip protein EB3 is
required for neuritogenesis. Nat. Cell Biol. 10: 1181-1189.
76. Butkevich, E., S. Hulsmann, D. Wenzel, T. Shirao, R. Duden, and I. Majoul. 2004. Drebrin is
a novel connexin-43 binding partner that links gap junctions to the submembrane cytoskeleton.
Curr Biol 14: 650-8.

138
77. Giepmans, B. N. 2006. Role of connexin43-interacting proteins at gap junctions. Advances in
Cardiology 42: 41-56.
78. Mercer, J. C., Q. Qi, L. F. Mottram, M. Law, D. Bruce, A. Iyer, J. L. Morales, H. Yamazaki,
T. Shirao, B. R. Peterson, and A. August. 2010. Chemico-genetic identification of drebrin as a
regulator of calcium responses. Int. J. Biochem. Cell Biol. 42: 337-345.
79. Ishikawa, J., K. Ohga, T. Yoshino, R. Takezawa, A. Ichikawa, H. Kubota, and T. Yamada.
2003. A pyrazole derivative, YM-58483, potently inhibits store-operated sustained Ca2+ influx
and IL-2 production in T lymphocytes. J. Immunol. 170: 4441-4449.
80. Zitt, C., B. Strauss, E. C. Schwarz, N. Spaeth, G. Rast, A. Hatzelmann, and M. Hoth. 2004.
Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the
pyrazole derivative BTP2. The Journal of Biological Chemistry 279: 12427-37.
81. Steinckwich, N., J. P. Frippiat, M. J. Stasia, M. Erard, R. Boxio, C. Tankosic, I. Doignon,
and O. Nusse. 2007. Potent inhibition of store-operated Ca2+ influx and superoxide production
in HL60 cells and polymorphonuclear neutrophils by the pyrazole derivative BTP2. Journal of
Leukocyte Biology 81: 1054-64.
82. Takezawa, R., H. Cheng, A. Beck, J. Ishikawa, P. Launay, H. Kubota, J. P. Kinet, A. Fleig,
T. Yamada, and R. Penner. 2006. A pyrazole derivative potently inhibits lymphocyte Ca2+
influx and cytokine production by facilitating transient receptor potential melastatin 4 channel
activity. Mol. Pharmacol. 69: 1413-1420.
83. He, L. P., T. Hewavitharana, J. Soboloff, M. A. Spassova, and D. L. Gill. 2005. A functional
link between store-operated and TRPC channels revealed by the 3,5-bis(trifluoromethyl)pyrazole
derivative, BTP2. The Journal of Biological Chemistry 280: 10997-1006.
84. Ohga, K., R. Takezawa, T. Yoshino, T. Yamada, Y. Shimizu, and J. Ishikawa. 2008. The
suppressive effects of YM-58483/BTP-2, a store-operated Ca2+ entry blocker, on inflammatory
mediator release in vitro and airway responses in vivo. Pulm. Pharmacol. Ther. 21: 360-369.
85. Yoshino, T., J. Ishikawa, K. Ohga, T. Morokata, R. Takezawa, H. Morio, Y. Okada, K.
Honda, and T. Yamada. 2007. YM-58483, a selective CRAC channel inhibitor, prevents antigen-
induced airway eosinophilia and late phase asthmatic responses via Th2 cytokine inhibition in
animal models. European Journal of Pharmacology 560: 225-33.
86. Iyer, A. S. and A. August. 2008. The Tec family kinase, IL-2-inducible T cell kinase,
differentially controls mast cell responses. J. Immunol. 180: 7869-7877.
87. Djuric, S. W., N. Y. BaMaung, A. Basha, H. Liu, J. R. Luly, D. J. Madar, R. J. Sciotti, N. P.
Tu, F. L. Wagenaar, P. E. Wiedeman, X. Zhou, S. Ballaron, J. Bauch, Y. W. Chen, X. G. Chiou,
T. Fey, D. Gauvin, E. Gubbins, G. C. Hsieh, K. C. Marsh, K. W. Mollison, M. Pong, T. K.
Shaughnessy, M. P. Sheets, M. Smith, J. M. Trevillyan, U. Warrior, C. D. Wegner, and G. W.

139
Carter. 2000. 3,5-Bis(trifluoromethyl)pyrazoles: a novel class of NFAT transcription factor
regulator. J. Med. Chem. 43: 2975-2981.
88. Demo, S. D., E. Masuda, A. B. Rossi, B. T. Throndset, A. L. Gerard, E. H. Chan, R. J.
Armstrong, B. P. Fox, J. B. Lorens, D. G. Payan, R. H. Scheller, and J. M. Fisher. 1999.
Quantitative measurement of mast cell degranulation using a novel flow cytometric annexin-V
binding assay. Cytometry 36: 340-348.
89. Gertler, R. and I. Pecht. 1988. Ionic signalling in mast cells; antigen and ionophore induced
changes in cytosolic pH. Mol. Immunol. 25: 1087-1092.
90. Gomperts, B. D., S. Cockcroft, J. P. Bennett, and C. M. Fewtrell. 1980. Early events in the
activation of Ca2+ dependent secretion: studies with rat peritoneal mast cells. J. Physiol. (Paris)
76: 383-393.
91. Plaut, M., J. H. Pierce, C. J. Watson, J. Hanley-Hyde, R. P. Nordan, and W. E. Paul. 1989.
Mast cell lines produce lymphokines in response to cross-linkage of Fc epsilon RI or to calcium
ionophores. Nature 339: 64-67.
92. Melicoff, E., L. Sansores-Garcia, A. Gomez, D. C. Moreira, P. Datta, P. Thakur, Y. Petrova,
T. Siddiqi, J. N. Murthy, B. F. Dickey, R. Heidelberger, and R. Adachi. 2009. Synaptotagmin-2
controls regulated exocytosis but not other secretory responses of mast cells. J. Biol. Chem. 284:
19445-19451.
93. Trevillyan, J. M., X. G. Chiou, Y. W. Chen, S. J. Ballaron, M. P. Sheets, M. L. Smith, P. E.
Wiedeman, U. Warrior, J. Wilkins, E. J. Gubbins, G. D. Gagne, J. Fagerland, G. W. Carter, J. R.
Luly, K. W. Mollison, and S. W. Djuric. 2001. Potent inhibition of NFAT activation and T cell
cytokine production by novel low molecular weight pyrazole compounds. The Journal of
Biological Chemistry 276: 48118-26.
94. Bert, A. G., B. V. Johnson, E. W. Baxter, and P. N. Cockerill. 2007. A modular enhancer is
differentially regulated by GATA and NFAT elements that direct different tissue-specific
patterns of nucleosome positioning and inducible chromatin remodeling. Mol. Cell. Biol. 27:
2870-2885.
95. Monticelli, S., D. U. Lee, J. Nardone, D. L. Bolton, and A. Rao. 2005. Chromatin-based
regulation of cytokine transcription in Th2 cells and mast cells. Int. Immunol. 17: 1513-1524.
96. Monticelli, S., D. C. Solymar, and A. Rao. 2004. Role of NFAT proteins in IL13 gene
transcription in mast cells. J. Biol. Chem. 279: 36210-36218.
97. Solymar, D. C., S. Agarwal, C. H. Bassing, F. W. Alt, and A. Rao. 2002. A 3' enhancer in the
IL-4 gene regulates cytokine production by Th2 cells and mast cells. Immunity 17: 41-50.

140
98. De Boer, M. L., V. A. Mordvinov, M. A. Thomas, and C. J. Sanderson. 1999. Role of nuclear
factor of activated T cells (NFAT) in the expression of interleukin-5 and other cytokines
involved in the regulation of hemopoetic cells. Int. J. Biochem. Cell Biol. 31: 1221-1236.
99. Savignac, M., B. Mellstrom, and J. R. Naranjo. 2007. Calcium-dependent transcription of
cytokine genes in T lymphocytes. Pflugers Arch. 454: 523-533.
100. Jain, J., C. Loh, and A. Rao. 1995. Transcriptional regulation of the IL-2 gene. Curr. Opin.
Immunol. 7: 333-342.
101. Gessner, A., K. Mohrs, and M. Mohrs. 2005. Mast cells, basophils, and eosinophils acquire
constitutive IL-4 and IL-13 transcripts during lineage differentiation that are sufficient for rapid
cytokine production. J. Immunol. 174: 1063-1072.
102. Di Capite, J. and A. B. Parekh. 2009. CRAC channels and Ca2+ signaling in mast cells.
Immunol. Rev. 231: 45-58.
103. Klein, M., S. Klein-Hessling, A. Palmetshofer, E. Serfling, C. Tertilt, T. Bopp, V. Heib, M.
Becker, C. Taube, H. Schild, E. Schmitt, and M. Stassen. 2006. Specific and redundant roles for
NFAT transcription factors in the expression of mast cell-derived cytokines. J. Immunol. 177:
6667-6674.
104. Braun, F. J., O. Aziz, and J. W. Putney Jr. 2003. 2-Aminoethoxydiphenyl Borane Activates
a Novel Calcium-Permeable Cation Channel. Mol. Pharmacol. 63: 1304-1311.
105. Chung, S. C., T. V. McDonald, and P. Gardner. 1994. Inhibition by SK&F 96365 of Ca2+
current, IL-2 production and activation in T lymphocytes. Br. J. Pharmacol. 113: 861-868.
106. Franzius, D., M. Hoth, and R. Penner. 1994. Non-specific effects of calcium entry
antagonists in mast cells. Pflugers Arch. 428: 433-438.
107. Ma, H. T., K. Venkatachalam, J. B. Parys, and D. L. Gill. 2002. Modification of store-
operated channel coupling and inositol trisphosphate receptor function by 2-
aminoethoxydiphenyl borate in DT40 lymphocytes. J. Biol. Chem. 277: 6915-6922.
108. Peppiatt, C. M., T. J. Collins, L. Mackenzie, S. J. Conway, A. B. Holmes, M. D. Bootman,
M. J. Berridge, J. T. Seo, and H. L. Roderick. 2003. 2-Aminoethoxydiphenyl borate (2-APB)
antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has
a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell
Calcium 34: 97-108.
109. Prakriya, M. and R. S. Lewis. 2001. Potentiation and inhibition of Ca(2+) release-activated
Ca(2+) channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP(3)
receptors. J. Physiol. 536: 3-19.

141
110. Schindl, R., H. Kahr, I. Graz, K. Groschner, and C. Romanin. 2002. Store depletion-
activated CaT1 currents in rat basophilic leukemia mast cells are inhibited by 2-
aminoethoxydiphenyl borate. Evidence for a regulatory component that controls activation of
both CaT1 and CRAC (Ca(2+) release-activated Ca(2+) channel) channels. J. Biol. Chem. 277:
26950-26958.
111. Wu, J., N. Kamimura, T. Takeo, S. Suga, M. Wakui, T. Maruyama, and K. Mikoshiba.
2000. 2-Aminoethoxydiphenyl borate modulates kinetics of intracellular Ca(2+) signals mediated
by inositol 1,4,5-trisphosphate-sensitive Ca(2+) stores in single pancreatic acinar cells of mouse.
Mol. Pharmacol. 58: 1368-1374.
112. Kiyonaka, S., K. Kato, M. Nishida, K. Mio, T. Numaga, Y. Sawaguchi, T. Yoshida, M.
Wakamori, E. Mori, T. Numata, M. Ishii, H. Takemoto, A. Ojida, K. Watanabe, A. Uemura, H.
Kurose, T. Morii, T. Kobayashi, Y. Sato, C. Sato, I. Hamachi, and Y. Mori. 2009. Selective and
direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound.
Proc. Natl. Acad. Sci. U. S. A. 106: 5400-5405.
113. Stanford, W. L., J. B. Cohn, and S. P. Cordes. 2001. Gene-trap mutagenesis: past, present
and beyond. Nat. Rev. Genet. 2: 756-768.
114. Zambrowicz, B. P., A. Abuin, R. Ramirez-Solis, L. J. Richter, J. Piggott, H. BeltrandelRio,
E. C. Buxton, J. Edwards, R. A. Finch, C. J. Friddle, A. Gupta, G. Hansen, Y. Hu, W. Huang, C.
Jaing, B. W. Key Jr, P. Kipp, B. Kohlhauff, Z. Q. Ma, D. Markesich, R. Payne, D. G. Potter, N.
Qian, J. Shaw, J. Schrick, Z. Z. Shi, M. J. Sparks, I. Van Sligtenhorst, P. Vogel, W. Walke, N.
Xu, Q. Zhu, C. Person, and A. T. Sands. 2003. Wnk1 kinase deficiency lowers blood pressure in
mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc. Natl.
Acad. Sci. U. S. A. 100: 14109-14114.
115. Gurish, M. F., N. Ghildyal, H. P. McNeil, K. F. Austen, S. Gillis, and R. L. Stevens. 1992.
Differential expression of secretory granule proteases in mouse mast cells exposed to interleukin
3 and c-kit ligand. J. Exp. Med. 175: 1003-1012.
116. Ihle, J. N., J. Keller, S. Oroszlan, L. E. Henderson, T. D. Copeland, F. Fitch, M. B.
Prystowsky, E. Goldwasser, J. W. Schrader, E. Palaszynski, M. Dy, and B. Lebel. 1983. Biologic
properties of homogeneous interleukin 3. I. Demonstration of WEHI-3 growth factor activity,
mast cell growth factor activity, p cell-stimulating factor activity, colony-stimulating factor
activity, and histamine-producing cell-stimulating factor activity. J. Immunol. 131: 282-287.
117. Yuan, Q., M. F. Gurish, D. S. Friend, K. F. Austen, and J. A. Boyce. 1998. Generation of a
novel stem cell factor-dependent mast cell progenitor. J. Immunol. 161: 5143-5146.
118. Gondokaryono, S. P., H. Ushio, F. Niyonsaba, M. Hara, H. Takenaka, S. T. Jayawardana, S.
Ikeda, K. Okumura, and H. Ogawa. 2007. The extra domain A of fibronectin stimulates murine
mast cells via toll-like receptor 4. J. Leukoc. Biol. 82: 657-665.

142
119. Naal, R. M., J. Tabb, D. Holowka, and B. Baird. 2004. In situ measurement of
degranulation as a biosensor based on RBL-2H3 mast cells. Biosens. Bioelectron. 20: 791-796.
120. Yamashita, Y., N. Charles, Y. Furumoto, S. Odom, T. Yamashita, A. M. Gilfillan, S.
Constant, M. A. Bower, J. J. Ryan, and J. Rivera. 2007. Cutting edge: genetic variation
influences Fc epsilonRI-induced mast cell activation and allergic responses. J. Immunol. 179:
740-743.
121. Odom, S., G. Gomez, M. Kovarova, Y. Furumoto, J. J. Ryan, H. V. Wright, C. Gonzalez-
Espinosa, M. L. Hibbs, K. W. Harder, and J. Rivera. 2004. Negative regulation of
immunoglobulin E-dependent allergic responses by Lyn kinase. J. Exp. Med. 199: 1491-1502.
122. Nishizumi, H. and T. Yamamoto. 1997. Impaired tyrosine phosphorylation and Ca2+
mobilization, but not degranulation, in lyn-deficient bone marrow-derived mast cells. J.
Immunol. 158: 2350-2355.
123. Kawakami, Y., J. Kitaura, A. B. Satterthwaite, R. M. Kato, K. Asai, S. E. Hartman, M.
Maeda-Yamamoto, C. A. Lowell, D. J. Rawlings, O. N. Witte, and T. Kawakami. 2000.
Redundant and opposing functions of two tyrosine kinases, Btk and Lyn, in mast cell activation.
J. Immunol. 165: 1210-1219.
124. Parravicini, V., M. Gadina, M. Kovarova, S. Odom, C. Gonzalez-Espinosa, Y. Furumoto, S.
Saitoh, L. E. Samelson, J. J. O'Shea, and J. Rivera. 2002. Fyn kinase initiates complementary
signals required for IgE-dependent mast cell degranulation. Nat. Immunol. 3: 741-748.
125. Hernandez-Hansen, V., G. A. Mackay, C. A. Lowell, B. S. Wilson, and J. M. Oliver. 2004.
The Src kinase Lyn is a negative regulator of mast cell proliferation. J. Leukoc. Biol. 75: 143-
151.
126. Hernandez-Hansen, V., A. J. Smith, Z. Surviladze, A. Chigaev, T. Mazel, J. Kalesnikoff, C.
A. Lowell, G. Krystal, L. A. Sklar, B. S. Wilson, and J. M. Oliver. 2004. Dysregulated
FcepsilonRI signaling and altered Fyn and SHIP activities in Lyn-deficient mast cells. J.
Immunol. 173: 100-112.
127. Galli, S. J., M. Tsai, and A. M. Piliponsky. 2008. The development of allergic
inflammation. Nature 454: 445-454.
128. Miyajima, I., D. Dombrowicz, T. R. Martin, J. V. Ravetch, J. P. Kinet, and S. J. Galli. 1997.
Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and Fc gammaRIII.
Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with
active or IgE- or IgG1-dependent passive anaphylaxis. J. Clin. Invest. 99: 901-914.
129. Metcalfe, D. D., R. D. Peavy, and A. M. Gilfillan. 2009. Mechanisms of mast cell signaling
in anaphylaxis. J. Allergy Clin. Immunol. 124: 639-46; quiz 647-8.

143
130. Wershil, B. K., Y. A. Mekori, T. Murakami, and S. J. Galli. 1987. 125I-fibrin deposition in
IgE-dependent immediate hypersensitivity reactions in mouse skin. Demonstration of the role of
mast cells using genetically mast cell-deficient mice locally reconstituted with cultured mast
cells. J. Immunol. 139: 2605-2614.
131. Galli, S. J., A. M. Dvorak, and I. Hammel. 1993. Mast cell abnormalities in the Chediak-
Higashi syndrome. Int. Arch. Allergy Immunol. 100: 89-92.
132. Lee, D. M., D. S. Friend, M. F. Gurish, C. Benoist, D. Mathis, and M. B. Brenner. 2002.
Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 297: 1689-
1692.
133. Araki, Y., A. Andoh, Y. Fujiyama, and T. Bamba. 2000. Development of dextran sulphate
sodium-induced experimental colitis is suppressed in genetically mast cell-deficient Ws/Ws rats.
Clin. Exp. Immunol. 119: 264-269.
134. Nishida, K., A. Hasegawa, S. Nakae, K. Oboki, H. Saito, S. Yamasaki, and T. Hirano. 2009.
Zinc transporter Znt5/Slc30a5 is required for the mast cell-mediated delayed-type allergic
reaction but not the immediate-type reaction. J. Exp. Med. 206: 1351-1364.
135. Gregory, G. D., S. S. Raju, S. Winandy, and M. A. Brown. 2006. Mast cell IL-4 expression
is regulated by Ikaros and influences encephalitogenic Th1 responses in EAE. J. Clin. Invest.
116: 1327-1336.
136. Han, J., R. Kori, J. W. Shui, Y. R. Chen, Z. Yao, and T. H. Tan. 2003. The SH3 domain-
containing adaptor HIP-55 mediates c-Jun N-terminal kinase activation in T cell receptor
signaling. J. Biol. Chem. 278: 52195-52202.
137. Haeckel, A., R. Ahuja, E. D. Gundelfinger, B. Qualmann, and M. M. Kessels. 2008. The
actin-binding protein Abp1 controls dendritic spine morphology and is important for spine head
and synapse formation. J. Neurosci. 28: 10031-10044.
138. Han, J., J. W. Shui, X. Zhang, B. Zheng, S. Han, and T. H. Tan. 2005. HIP-55 is important
for T-cell proliferation, cytokine production, and immune responses. Mol. Cell. Biol. 25: 6869-
6878.
139. Le Bras, S., I. Foucault, A. Foussat, C. Brignone, O. Acuto, and M. Deckert. 2004.
Recruitment of the actin-binding protein HIP-55 to the immunological synapse regulates T cell
receptor signaling and endocytosis. J. Biol. Chem. 279: 15550-15560.
140. Hao, S. and A. August. 2005. Actin depolymerization transduces the strength of B-cell
receptor stimulation. Mol. Biol. Cell 16: 2275-2284.

144
141. Smyth, J. T., W. I. DeHaven, G. S. Bird, and J. W. Putney Jr. 2007. Role of the microtubule
cytoskeleton in the function of the store-operated Ca2+ channel activator STIM1. J. Cell. Sci.
120: 3762-3771.
142. Sampieri, A., A. Zepeda, A. Asanov, and L. Vaca. 2009. Visualizing the store-operated
channel complex assembly in real time: identification of SERCA2 as a new member. Cell
Calcium 45: 439-446.
143. Nolz, J. C., T. S. Gomez, P. Zhu, S. Li, R. B. Medeiros, Y. Shimizu, J. K. Burkhardt, B. D.
Freedman, and D. D. Billadeau. 2006. The WAVE2 complex regulates actin cytoskeletal
reorganization and CRAC-mediated calcium entry during T cell activation. Curr. Biol. 16: 24-34.
144. Shiraishi, Y., A. Mizutani, H. Bito, K. Fujisawa, S. Narumiya, K. Mikoshiba, and T.
Furuichi. 1999. Cupidin, an isoform of Homer/Vesl, interacts with the actin cytoskeleton and
activated rho family small GTPases and is expressed in developing mouse cerebellar granule
cells. J. Neurosci. 19: 8389-8400.
145. Yuan, J. P., K. Kiselyov, D. M. Shin, J. Chen, N. Shcheynikov, S. H. Kang, M. H. Dehoff,
M. K. Schwarz, P. H. Seeburg, S. Muallem, and P. F. Worley. 2003. Homer binds TRPC family
channels and is required for gating of TRPC1 by IP3 receptors. Cell 114: 777-789.
146. Goel, M., W. Sinkins, A. Keightley, M. Kinter, and W. P. Schilling. 2005. Proteomic
analysis of TRPC5- and TRPC6-binding partners reveals interaction with the plasmalemmal
Na(+)/K(+)-ATPase. Pflugers Arch. 451: 87-98.
147. Blair, N. T., J. S. Kaczmarek, and D. E. Clapham. 2009. Intracellular calcium strongly
potentiates agonist-activated TRPC5 channels. J. Gen. Physiol. 133: 525-546.
148. Dietrich, A. and T. Gudermann. 2007. Trpc6. Handb. Exp. Pharmacol. (179): 125-141.
149. Vennekens, R., J. Olausson, M. Meissner, W. Bloch, I. Mathar, S. E. Philipp, F. Schmitz, P.
Weissgerber, B. Nilius, V. Flockerzi, and M. Freichel. 2007. Increased IgE-dependent mast cell
activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation
channel TRPM4. Nat. Immunol. 8: 312-320.
150. Shimizu, T., G. Owsianik, M. Freichel, V. Flockerzi, B. Nilius, and R. Vennekens. 2009.
TRPM4 regulates migration of mast cells in mice. Cell Calcium 45: 226-232.
151. Thompson, M. A., C. M. Pabelick, and Y. S. Prakash. 2009. Role of STIM1 in regulation of
store-operated Ca2+ influx in pheochromocytoma cells. Cell. Mol. Neurobiol. 29: 193-202.
152. Hajkova, Z., V. Bugajev, E. Draberova, S. Vinopal, L. Draberova, J. Janacek, P. Draber,
and P. Draber. 2011. STIM1-directed reorganization of microtubules in activated mast cells. J.
Immunol. 186: 913-923.

145
153. Alicia, S., Z. Angelica, S. Carlos, S. Alfonso, and L. Vaca. 2008. STIM1 converts TRPC1
from a receptor-operated to a store-operated channel: moving TRPC1 in and out of lipid rafts.
Cell Calcium 44: 479-491.

VITA
Name Mankit Law
Education
2005-2011 Doctor of Philosophy in Immunology and Infectious Diseases
The Pennsylvania State University
University Park, PA.
1999-2004 Bachelor of Science in Biotechnology
The Pennsylvania State University
University Park, PA.
Research Experience
2005-2011 The Huck Institutes of the Life Sciences
The Pennsylvania State University
Advisor: Avery August
Dissertation Title: A Functional Link Between Store-Operated
Calcium Channels and the Actin-Binding Protein Drebrin in Mast
Cells Revealed by 3,5-Bis-Trifluoromethyl Pyrazole (BTP)
Compounds
Teaching Experience
Fall 2006 Teaching Assistant- Principles of Immunology (MICRB 410)
Publications 1. M. Law and A. August. “p130Cas, a multi-function scaffold for diverse signals.”
Adaptor Proteins and Cancer, 2008. Ed. Maria-Magdalena Georgescu.
2. J.C. Mercer*, Q. Qi*, L.F. Mottram*, M. Law, D. Bruce, A. Iyer, J.L. Morales, H.
Yamazaki, T. Shirao, B.R. Peterson, and A. August. “Chemico-genetic identification of
drebrin as a regulator of calcium responses.” (2010) Int. J. Biochem. & Cell Biol. 42:
337-345. (*equal contributors)
3. M. Law, J.L. Morales, L.F. Mottram, A. Iyer, B.R. Peterson, and A. August. “Structural
requirements for the inhibition of calcium mobilization and mast cell activation by the
pyrazole derivative BTP2.” (2011) Int. J. Biochem. & Cell Biol. 43: 1228-1239.