209607orig1s000 - food and drug administration...avid malignant and/or recurrent pheochromocytoma or...
TRANSCRIPT

CENTER FOR DRUG EVALUATION AND RESEARCH
APPLICATION NUMBER:
209607Orig1s000
MULTI-DISCIPLINE REVIEW
Summary Review Office Director Cross Discipline Team Leader Review Clinical Review Non-Clinical Review Statistical Review Clinical Pharmacology Review

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
1Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
NDA/BLA Multi-Disciplinary Review and EvaluationApplication Type NDA
Application Number 209607Priority or Standard Priority
Submit Date 10-31-2017Received Date 10-31-2017
PDUFA Goal Date 7-30-2018Division/Office DOP2/OHOP
Review Completion Date 7-24-2018Established Name 131I Iobenguane
(Proposed) Trade Name AZEDRAPharmacologic Class Radiopharmaceutical
Code nameApplicant Progenics Pharmaceuticals Inc.
Formulation SolutionDosing Regimen Dosimetric dose: 5 – 6 mCi (0.1 mCi/kg for patients ≤ 50 kg)
Therapeutic dose: 500 mCi (8 mCi/kg for patients ≤ 62.5 kg) every 12 weeks for two doses
Applicant Proposed Indication
Treatment of patients age years and older with iobenguane avid malignant and/or recurrent pheochromocytoma or paraganglioma
Recommendation on Regulatory Action
Approval
Recommended Indication Treatment of patients age 12 years and older with iobenguane scan positive, unresectable, locally advanced or metastatic pheochromocytoma or paraganglioma who require systemic anticancer therapy
Reference ID: 4297867
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
2Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Table of Contents
Reviewers of Multi-Disciplinary Review and Evaluation.................................................................9
Additional Reviewers of Application ..............................................................................................9
Glossary ........................................................................................................................................10
1 Executive Summary ...............................................................................................................12
1.1 Product Introduction ..........................................................................................................12
1.2 Conclusions on the Substantial Evidence of Effectiveness .................................................14
1.3 Benefit-Risk.........................................................................................................................16
1.4 Patient Experience Data .....................................................................................................22
2 Therapeutic Context ..................................................................................................................24
2.1 Analysis of Condition ..........................................................................................................24
2.2 Analysis of Current Treatment Options ..............................................................................25
3 Regulatory Background .............................................................................................................27
3.1 U.S. Regulatory Actions and Marketing History..................................................................27
3.2 Summary of Presubmission/Submission Regulatory Activity .............................................27
4 Significant Issues from Other Review Disciplines Pertinent to Clinical Conclusions on Efficacy and Safety .............................................................................................................................31
4.1 Office of Scientific Investigations (OSI)...............................................................................31
4.2 Product Quality...................................................................................................................31
4.3 Clinical Microbiology ..........................................................................................................31
4.4 Devices and Companion Diagnostic Issues .........................................................................31
4.5 Division of Medical Imaging Products.................................................................................32
5 Nonclinical Pharmacology/Toxicology.......................................................................................33
5.1 Executive Summary ............................................................................................................33
5.2 Referenced NDAs, BLAs, DMFs ...........................................................................................35
5.3 Pharmacology .....................................................................................................................35
5.4 ADME/PK ............................................................................................................................44
5.5 Toxicology...........................................................................................................................46
5.5.1 General Toxicology ..................................................................................................46
5.5.2 Genetic Toxicology...................................................................................................48
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
3Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
5.5.3 Carcinogenicity ........................................................................................................49
5.5.4 Reproductive and Developmental Toxicology .........................................................49
5.5.5 Other Toxicology Studies .........................................................................................50
6 Clinical Pharmacology................................................................................................................51
6.1 Executive Summary ............................................................................................................51
6.2 Recommendations..............................................................................................................51
6.3 Postmarketing Requirements and Commitments .............................................................52
6.4 Summary of Clinical Pharmacology Assessment ................................................................52
6.5 Pharmacology and Clinical Pharmacokinetics ....................................................................52
6.6 General Dosing and Therapeutic Individualization.............................................................53
6.6.1 General Dosing ........................................................................................................53
6.6.2 Therapeutic Individualization ..................................................................................53
6.6.3 Outstanding Issues...................................................................................................54
6.6.4 Summary of Labeling Recommendations ...............................................................55
6.7 Comprehensive Clinical Pharmacology Review..............................................................55
6.7.1 General Pharmacology and Pharmacokinetic Characteristics .................................55
6.7.2 Clinical Pharmacology Questions............................................................................57
[Source: NDA 209607/SDN 2 – FDA’s IRT-QTc Review, PP 2] ...........................................63
7 Sources of Clinical Data and Review Strategy............................................................................71
7.1 Table of Clinical Studies ......................................................................................................71
7.2 Review Strategy ..................................................................................................................73
8 Statistical and Clinical and Evaluation .......................................................................................74
8.1 Review of Relevant Individual Trials Used to Support Efficacy...........................................74
8.1.1 MIP-IB12B: A Phase II Study Evaluating the Efficacy and Safety of 131I-MIBG in Patients with Malignant Relapsed/Refractory Pheochromocytoma/Paraganglioma.......74
8.1.2 MIP-IB12B Study Results..........................................................................................81
8.1.3 Study MIP-IB12: A Phase I Study Evaluating the Maximum Tolerated Dose, Dosimetry, Safety and Efficacy of Ultratrace Iobenguane I 131 in Patients with Malignant Pheochromocytoma/ Paraganglioma ...............................................................................97
8.1.4 Assessment of Efficacy Across Trials......................................................................100
8.1.5 Integrated Assessment of Effectiveness ................................................................101
8.2 Review of Safety ..............................................................................................................101
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
4Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
8.2.1 Safety Review Approach ........................................................................................102
8.2.2 Review of the Safety Database ..............................................................................103
8.2.3 Adequacy of Applicant’s Clinical Safety Assessments............................................104
8.2.4 Safety Results.........................................................................................................106
8.2.5 Analysis of Submission-Specific Safety Issues........................................................127
8.2.6 Clinical Outcome Assessment (COA) Analyses Informing Safety/Tolerability .......129
8.2.7 Safety Analyses by Demographic Subgroups.........................................................130
8.2.8 Specific Safety Studies/Clinical Trials.....................................................................131
8.2.9 Additional Safety Explorations...............................................................................131
8.2.10 Safety in the Postmarket Setting .........................................................................133
8.2.11 Integrated Assessment of Safety .........................................................................133
10 SUMMARY AND CONCLUSIONS.............................................................................................135
10.1 Statistical Issues..............................................................................................................135
10.2 Conclusions and Recommendations...............................................................................135
11 Advisory Committee Meeting and Other External Consultations .........................................137
12 Pediatrics ...............................................................................................................................138
13 Labeling Recommendations ..................................................................................................138
13.1 Prescription Drug Labeling..............................................................................................138
14 Risk Evaluation and Mitigation Strategies (REMS).................................................................140
15 Postmarketing Requirements and Commitments .................................................................141
16 Division Director (DHOT) .......................................................................................................142
17 Division Director (OCP) ..........................................................................................................143
18 Division Director (OB) ............................................................................................................144
19 Division Director (Clinical) .....................................................................................................145
21 Appendices ............................................................................................................................150
21.1 References ......................................................................................................................150
21.2 Financial Disclosure ........................................................................................................153
21.3 Nonclinical Pharmacology/Toxicology............................................................................154
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
5Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
21.4 OCP Appendices (Technical documents supporting OCP recommendations)..........154
21.5 Additional Clinical Outcome Assessment Analyses ..................................................154
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
6Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
List of Tables
Table 1: Biodistribution of carrier-added and no-carrier-added 131-I MIBG in tissues from xenograft implanted nude mice as a percentage of the injected dose per gram of tissue ..........36Table 2: Increased Activity in 131I-MIBG Treated Mice Implanted with Pheochromocytoma .....41Table 3: Males-Percent Changes from Control in Cardiac Parameters (MIBG only) ....................43Table 4: IC50 s for inhibitions of MIBG uptake by SK-N-SH cells of selected adrenergic ligands...43Table 5: Summary of Blood PK Parameters for 131I-MIBG in 11 Patients .....................................58Table 6: Summary of Urinary Excretion Data of Radioactivity in 11 Patients...............................59Table 7: Metabolic Profiling in Urine (% Injected Dose) ...............................................................59Table 8: Reduction in Use of Antihypertensive Medication Following 131I-MIBG – All treated 68 evaluable PPGL Patients ...............................................................................................................61Table 9: Reduction in Use of Antihypertensive Medication Following 131I-MIBG by Number of Therapeutic Doses Administered .................................................................................................61Table 10: Duration of clinical Benefit in patients who Achieved the Primary Endpoint...............62Table 11: Incidence of TEAEs occurring in ≥10% of 74 evaluable patients...................................62Table 12: The Point Estimates and the 90% CIs of of ΔQTcF Corresponding to the Largest Upper Bounds for All-Treated 131I-MIBG Group (FDA Analysis for Study MIP-IB12B) .............................63Table 13: Comparison of Mean Radiation Absorbed Dose Estimates for Patients with Normal Renal Function and Patients with Mild-to-Moderate Renal Impairment .....................................64Table 14: Patients with Reduced Therapeutic Dose Based on Critical Organ Exposure Estimates from Study MIP-IB12B (Reviewer’s Table)....................................................................................64Table 15: Projected Maximum Therapeutic Dose in Individual Patients from Study MIP-IB11 (mCi)* ...........................................................................................................................................66Table 16: Radiographic Response by RECIST in Patients Receiving One 131I-MIBG Therapeutic Dose in Studies MIP-IB12 AND MIP-IB12B ...................................................................................66Table 17: Summary of Inhibition of CYP-Enzyme Activities by MIBG in Pooled Human Hepatic Microsomes ..................................................................................................................................67Table 18: Effect of MIBG on CYP Enzyme Activities in Human Hepatocytes ................................68Table 19: Apparent Permeability and Efflux Ratios from Caco-2 Cells .........................................69Table 20: Clinical Studies Included in NDA 209607 ......................................................................71Table 21: Patient Disposition, Study MIP-IB12B...........................................................................82Table 22: Major Protocol Deviations During Study MIP-IB12B.....................................................84Table 23: Demographic Characteristics of all Enrolled Patients, Study MIP-IB12B ......................85Table 24: Baseline Characteristics, Study MIP-IB12B ...................................................................87Table 25: Results of the Primary Endpoint (FAS) ..........................................................................89Table 26: Results of Sensitivity Analyses ......................................................................................90Table 27: Duration of Response for Patients Who Attained at Least 50% Reduction in Antihypertensive Medications......................................................................................................90Table 28: Patients with a Decrease in Systolic Blood Pressure > 20 mm Hg, Study MIP-IB12B ...92Table 29: Changes in Systolic Blood Pressure During Study MIP-IB12B .......................................92Table 30: Best Confirmed Overall Tumor Response and Duration of Response ..........................93Table 31: Best Confirmed Overall Tumor Response and Duration of Benefit ..............................94
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
7Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Table 32: Best Confirmed Overall Tumor Response and Duration of Benefit ..............................95Table 33: Primary Endpoint in Subgroups ....................................................................................96Table 34: Distribution of 131I-MIBG Administrations in the MIP-IB12/MIP-IB12B Safety Population (N=88) ......................................................................................................................103Table 35: Serious Adverse Events, Study MIP-IB12/MIP-IB12B Safety Population, Primary Analysis.......................................................................................................................................110Table 36: Adverse Events of Special Interest, Studies MIP-IB12/MIP-IB12B, Primary Analysis..114Table 37: Cases of Renal Failure in the MIP-IB12/MIP-IB12B Safety Population .......................116Table 38: Treatment Emergent Adverse Events in at Least 5% of Patients, Studies MIP-IB12/MIP-IB12B Safety Population, Primary Analysis ................................................................117Table 39: Laboratory Abnormalities, Studies MIP-IB12B & MIP-IB12 ........................................121Table 40: International Normalized Ratio Abnormalities, Study MIP-IB2B ................................122Table 41: Activated Partial Thromboplastin Time Abnormalities, Study MIP-IB2 ......................122Table 42: The Point Estimates and the 90% CIs of ΔQTcF Corresponding to the Largest Upper Bounds for All-Treated 131I-MIBG Group (FDA Analyses for Study IB12B)..................................126Table 43: Completion Rates by Visit ...........................................................................................155
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
8Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
List of Figures
Figure 1: Selective Uptake of MIBG in Cells Expressing Noradrenaline Transporter....................36Figure 2: Radioactive vs. MIBG Exposure in High vs Low Specificity Activity Radiolabeled MIBG Products........................................................................................................................................37Figure 3: The Effect of Reserpine on the Retention of I125 MIBG by SK-N-SH (top) and PC-12 (bottom) Cells ...............................................................................................................................38Figure 4: Cytotoxic Effects of 100 mCi of I-125 MIBG and I-131 MIBG with Equivalent Specific Activity on SK-N-LO and SK-N-SH Cells .........................................................................................38Figure 5: Calculated Values of Absorbed Radiation Dose per Unit of Injected Radioactivity (Gy/MBq) to Tumor and Normal Organs for Non-Carrier-Added (White Bars) and Exchange Preparation (Black Bars) of 131I MIBG ...........................................................................................39Figure 6: Specific Uptake by SK-N-BE(2c) Neuroblastoma Cells of No Carrier-Added 131I-MIBG (closed circles) and a Commercial Carrier-Added Preparation (open circles) ..............................40Figure 7: Increased Exposure in Pheochromocytoma versus Neuroblastoma Implants ..............40Figure 8: Dose Dependent Increase in 131I-MIBG-Mediated Anti-Tumor Activity in PC-12 Implanted Animals........................................................................................................................41Figure 9: Change In Tumor Volume and Days of Tumor Doubling Time after Administration of 131I Ultratace and 131I Comparison product ..................................................................................42Figure 10: Tumor Doubling Times After 131I-MIBG Administration ..............................................42Figure 11: Distribution of Total Injected Dose and Occurrence of DLTs Across Sequential Dose Cohorts .........................................................................................................................................60Figure 12: Swimmer Plot for the Patients Who had Reduction in Antihypertensive Medications by at Least 50% for at Least 6 Months .........................................................................................91Figure 13: Mean Score of PF by Visit ..........................................................................................156Figure 14: Mean Percentage* Change in PF Score from Baseline over Visit ..............................157
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
9Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Reviewers of Multi-Disciplinary Review and Evaluation
Additional Reviewers of Application
OPQ Sithamalli Chandramouli and Dhanalakshmi KasiMicrobiology Julie NemecekFacilities Rebecca DombrowskiOPDP Carole BroadnaxOSI David MenschikOSE/DEPI Carolyn McCloskeyOSE/DMEPA Janine StewartOSE/DRISK Till OlickalDMIP Stanley Stern
OPQ=Office of Pharmaceutical QualityOPDP=Office of Prescription Drug PromotionOSI=Office of Scientific InvestigationsOSE= Office of Surveillance and EpidemiologyDEPI= Division of EpidemiologyDMEPA=Division of Medication Error Prevention and AnalysisDRISK=Division of Risk ManagementDMIP=Division of Medical Imaging Products
Regulatory Project Manager Sharon SickafuseNonclinical Reviewer Dubravka KufrinNonclinical Team Leader Whitney HelmsOffice of Clinical Pharmacology Reviewer(s) Safaa BurnsOffice of Clinical Pharmacology Team Leader(s) Jeanne Fourie ZirkelbachClinical Reviewer Diana BradfordClinical Team Leader Suzanne DemkoStatistical Reviewer Xiaoping (Janet) JiangStatistical Team Leader Lisa RodriguezCross-Disciplinary Team Leader Suzanne DemkoDivision Director (DHOT) John LeightonDivision Director (OCP) Nam Atiqur RahmanDivision Director (OB) Rajeshwari SridharaAssociate Division Director (OHOP) Steven Lemery
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
10Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Glossary
AC advisory committeeADME absorption, distribution, metabolism, excretion AE adverse eventBLA biologics license applicationBPCA Best Pharmaceuticals for Children ActBRF Benefit Risk FrameworkCBER Center for Biologics Evaluation and ResearchCDER Center for Drug Evaluation and ResearchCDRH Center for Devices and Radiological HealthCDTL Cross-Discipline Team LeaderCFR Code of Federal RegulationsCMC chemistry, manufacturing, and controlsCOSTART Coding Symbols for Thesaurus of Adverse Reaction TermsCRF case report formCRO contract research organizationCRT clinical review templateCSR clinical study reportCSS Controlled Substance StaffDDI Drug-Drug InteractionsDHOT Division of Hematology Oncology ToxicologyDMC data monitoring committeeECG electrocardiogrameCTD electronic common technical documentETASU elements to assure safe useFDA Food and Drug AdministrationFDAAA Food and Drug Administration Amendments Act of 2007FDASIA Food and Drug Administration Safety and Innovation ActGCP good clinical practiceGRMP good review management practiceICH International Conference on HarmonizationIND Investigational New DrugISE integrated summary of effectivenessISS integrated summary of safetyITT intent to treatIV intravenousMedDRA Medical Dictionary for Regulatory ActivitiesMIBG metaiodobenzylguanidine mITT modified intent to treatNCI-CTCAE National Cancer Institute-Common Terminology Criteria for Adverse Event
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
11Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
NDA New Drug ApplicationNME New Molecular EntityOCS Office of Computational ScienceOPQ Office of Pharmaceutical QualityOSE Office of Surveillance and EpidemiologyOSI Office of Scientific InvestigationPBRER Periodic Benefit-Risk Evaluation ReportPD PharmacodynamicsPI prescribing informationPK PharmacokineticsPMC Postmarketing CommitmentPMR Postmarketing RequirementPP per protocolPPGL pheochromocytoma and paragangliomaPPI patient package insertPREA Pediatric Research Equity ActPRO patient reported outcomePSUR Periodic Safety Update reportREMS risk evaluation and mitigation strategySAE serious adverse eventSAP statistical analysis planSGE special government employeeSOC standard of careTEAE treatment emergent adverse event
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
12Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
1 Executive Summary
1.1 Product Introduction
On October 31, 2017, Progenics Pharmaceuticals, Inc. (Progenics) submitted a New Drug Application (NDA) under 505(b)(1) of the Federal Food, Drug, and Cosmetic Act for 131I-iobenguane (metaiodobenzylguanidine, 131I-iobenguane, iobenguane I-131, 131I-MIBG, Azedra), a radiopharmaceutical.Iobenguane is a guanethidine derivative that resembles norepinephrine in structure; it acts as a substrate for the norepinephrine transporter (NT) expressed on neuroendocrine cell surfaces. It can be labeled with radioactive isotopes of iodine for diagnostic or therapeutic applications. Conventional formulations of MIBG are known as the “carrier-added” or “carrier-containing” forms of MIBG, because a significant quantity of non-radioactive (or “cold”) carrier MIBG is present in the iobenguane I-131 dose administered to patients. In therapeutic administrations of carrier-added iobenguane I-131, the molar ratio of nonradioactive to radioactive MIBG molecules approaches . This imbalance of “cold” to radioactive MIBG can lead to pressor effects and reduction in efficacy in patients.
The carrier molecule iobenguane is a biogenic amine that interferes with the reuptake of norepinephrine, resulting in increased noradrenergic activity. When administered at high-mass doses, carrier-added MIBG can cause hypertension and other cardiovascular toxicity. The selective active uptake by the NT expressed on neuroendocrine cell surfaces is a competitive process; therefore, the presence of cold iobenguane molecules in the infusion solution can decrease the uptake in target neuroendocrine tumor cells.
FDA has approved carrier-added iobenguane as a diagnostic agent for use in localization of pheochromocytoma and neuroblastoma, and it has been studied as treatment for patients with advanced pheochromocytoma and paraganglioma (PPGL) and other neuroendocrine malignancies.
Progenics has developed a no-carrier-added version of iobenguane I-131 and the final product formulation . The final formulation is an injection containing 555 MBq/mL (15 mCi/ml) at TOC as a clear solution in a single-dose vial. The intended dosimetric dose is 5 – 6 mCi for patients weighing 50.0 kg or more, and 0.1 mCi/kg for those weighing less than 50.0 kg. The intended therapeutic dose is 500 mCi (or 8 mCi/kg for patients weighing 62.5 kg or less, and an iobenguane chemical mass of ).
The current application proposes an indication for 131I-iobenguane for the treatment of patients with iobenguane-avid metastatic or recurrent pheochromocytoma and paraganglioma (PPGL). The indication to be granted is “…for the treatment of adult and pediatric patients 12 years and older with iobenguane scan positive, unresectable, locally advanced or metastatic pheochromocytoma or paraganglioma who require systemic anticancer therapy.”
Reference ID: 4297867
APPEARS THIS WAY ON ORIGINAL
(b) (4)
(b) (4)
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
13Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Reference ID: 4297867
APPEARS THIS WAY ON ORIGINAL

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
14Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
1.2 Conclusions on the Substantial Evidence of Effectiveness
The recommendations for approval of Azedra are supported primarily by one single-arm, multicenter trial entitled, “A Phase II Study Evaluating the Efficacy and Safety of 131I-MIBG in Patients with Malignant Relapsed/Refractory Pheochromocytoma/Paraganglioma,” Study MIP-IB12B. The trial was conducted in patients greater than 12 years of age who were diagnosed with pheochromocytoma or paraganglioma (PPGL) and who were ineligible for curative therapy. Patients had also progressed on prior therapy or were not candidates for chemotherapy. Other eligibility criteria required tumors to have definitive iobenguane avidity, at least one tumor site identified by computed tomography (CT), magnetic resonance imaging (MRI), or iobenguane I 131 scan, Karnofsky performance status greater than 60, no active central nervous system lesions, and no changes to their antihypertensive regimen in the 30 days prior to the first therapeutic dose.
Iobenguane I 131 was administered in two therapeutic doses at 500 mCi each (8 mCi/kg, for patients weighing 62.5 kg or less) approximately three months apart. The doses were administered after an imaging dose and dosimetry. A total of 74 patients received the dosimetric dose. Following dosimetry, 68 patients received at least one therapeutic dose, and 50 patients received two therapeutic doses, administered at least 90 days apart. Among the 68 patients who received at least one dose, the median age was 55 years (16 to 72 years), 57% were male, 75% were White, 21% were Black, 4% were Asian and the remainder had no race or ethnicity reported. For the primary tumor diagnosis, 78% had pheochromocytoma, 21% had paraganglioma, and 1% had both. Fifty percent (50%) of patients with evaluable imaging studies had lung or liver metastases and 61% had bone metastases at baseline. Eighty-eight percent (88%) underwent prior surgery, 50% received prior external beam radiation.
The primary endpoint of the trial was the proportion of patients with a reduction by at least 50%, or discontinuation, of all antihypertensive medications for at least six months. The secondary objectives of the trial included assessment by RECIST for overall tumor response (ORR) to include complete response (CR) and partial response (PR), ORR including CR, PR, and moderate response (MR)—defined as decrease in the sum of the longest diameters of the target lesions of 15-30%, with no evidence of progressive disease (PD) in non-target lesions—and overall survival (OS) up to 5 years post-treatment. Other secondary endpoints were assessment of bone lesion status on the Soloway Scale, tumor marker response, quality of life (QOL) changes, changes in analgesic and pain medication use, and safety.
A Special Protocol Assessment (SPA) agreement was issued for Study MIP-IB12B on March 6, 2009, with the foregoing endpoints. The primary endpoint, reduction in antihypertensive medication of at least 50% for at least 6 months, is a new endpoint in oncology
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
15Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
that was considered specifically for the treatment of patients with PPGL. The endpoint was chosen as a measure of antitumor activity of 131I-iobenguane because detecting a reduction in hypertension, a key contributor to morbidity associated with PPGL, appears to correlate with decreased tumor activity. It is known that severe hypertension in patients with PPGL is caused by the underlying tumor; however, to provide further evidence that 131I-iobenguane demonstrated anti-tumor activity, and not merely antihypertensive activity, the primary endpoint was supported by an evaluation of ORR by established response criteria, i.e. radiologic response by RECIST.
The primary endpoint for Study MIP-IB12B was achieved by 25% of patients (95% CI 16.2% – 36.5%), and antitumor activity of 131I-Iobenguane was demonstrated with 22.1% of patients having a confirmed, centrally reviewed PR (95% CI: 13.6%, 32.7%).
A durable response in the reduction of hypertension as measured by the primary endpoint plus the confirmed ORR are measures of direct clinical benefit in this population of patients with a serious, life threatening, and rare disease. Based upon the foregoing, as well as the entirety of the reviews performed for this application, I am in full agreement with the review teams that 131I-iobenguane (Azedra) has demonstrated substantial evidence of effectiveness.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
16Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
1.3 Benefit-Risk
Reference ID: 4297867
APPEARS THIS WAY ON ORIGINAL

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
17Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Benefit-Risk Summary and Assessment
The effectiveness of 131I-iobenguane is supported by the results from Study MIP-IB12B, a multicenter, open-label, single arm trial conducted in 68 patients (68 patients received at least one therapeutic dose, and 50 patients received two therapeutic doses) 12 years and older with a diagnosis of unresectable pheochromocytoma or paraganglioma. Patients were at least 12 years of age and were ineligible for curative therapy. Patients had also progressed on prior therapy for PPGL or were not candidates for chemotherapy.
The major efficacy outcome measure was the proportion of patients with a reduction (including discontinuation) of all antihypertensive medication by 50% or greater for at least six months after receiving two therapeutic doses each at 500 mCi (or 8 mCi/kg, for patients weighing 62.5 kg or less) of Ultratrace Iobenguane I 131 administered approximately three months apart. The secondary objectives included the proportion of patients with overall tumor response of CR or PR per RECIST criteria, the proportion of patients with overall tumor response of CR, PR or MR per RECIST, and overall survival up to 5 years post-treatment. The study consisted of a screening and dosimetry phase, a 12-month efficacy phase, and a four-year long-term follow-up phase.
The clinical benefit determination of 131I-MIBG for patients with unresectable, locally advanced or metastatic PPGL who require systemic anticancer therapy is based on the results of Study MIP-IB12B. The primary endpoint, a 50% reduction (including discontinuation) of antihypertensive medications for at least 6 months, was a new endpoint in the PPGL population. Given the morbidity associated with hypertension in this population, and the direct contribution of the tumor to hypertension through secretion of catecholamines, this endpoint was designed as a measure of direct clinical benefit. The primary endpoint was achieved by 25% of patients (95% CI: 16% – 37%). The antitumor activity of 131I-MIBG was confirmed by traditional tumor response criteria, i.e. RECIST v. 1.0 For ORR, 22.1% of the evaluable population (15 patients) demonstrated a confirmed, centrally reviewed response of PR (95% CI: 14%, 33%). There were 53% of patients who responded to therapy who maintained a duration of response at least six months.
There were no major statistical or other issues that alter the overall conclusions from Study MIP-IB12B. It is noted, however, that the limitation of single arm trial design prevents the study from providing adequate interpretation of the results of time to event endpoint such as OS, a secondary endpoint of the trial.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
18Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Combined, durable response in hypertension reduction as measured by the primary endpoint coupled with confirmed objective tumor response are a measure of direct clinical benefit in this population.
Safety was evaluated in 88 patients who received at least one dose of 131I-MIBG in two trials, the trial supporting the effectiveness of the drug, Study MIP-IB12B, and Study MIP-IB12, a multi-site, open-label, dose-escalation study of 131I-MIBG in patients with PPGL. The primary objective of Study MIP-IB12 was to determine the maximum tolerated dose of 131I-MIBG in patients with malignant PPGL. The data included in labeling in the warnings and precautions section of labeling comes from a pooled analysis of both trials.
The primary safety risks of 131I-MIBG are related to the radiation exposure of the product. Myelosuppression and gastrointestinal adverse reactions were among the most common. The risk of myelodysplastic syndrome and leukemia was less common, occurring in 7% of the pooled safety population. Although these events were largely confounded by prior, potentially leukemogenic therapy, this observation remains a significant and serious risk of 131I-MIBG and requires further study. A greater percentage of patients who received two doses of 131I-MIBG experienced cytopenias, sialoadenitis, and renal failure. The incidence of hypothyroidism (4% vs. 2.6%) was similar, irrespective of number of doses received. All patients who developed leukemia or MDS had received two doses of 131I-MIBG on study. The most common severe (Grade 3-4) adverse reactions of treatment with 131I-MIBG were lymphopenia (78%), neutropenia (59%), thrombocytopenia (50%), fatigue (26%), anemia (24%), and nausea (16%). Twelve percent of patients discontinued treatment due to adverse reactions (thrombocytopenia, anemia, lymphopenia, nausea and vomiting, and multiple hematologic adverse reactions).
The serious nature of the aforementioned risks supports the use of 131I-MIBG only in patients with unresectable, locally advanced or metastatic PPGL who require systemic anticancer therapy disease and who have no other known curative options.
Based on the rare incidence of this disease, demonstrated clinical benefit, and lack of approved therapeutic options, the review teams have recommended regular approval for 131I-MIBG and this application. A risk evaluation and mitigation strategy (REMS) has not been recommended given the widespread experience with NA-131I-MIBG, and the need for delivery of the product in centers certified to deliver radioactive therapies. A postmarketing requirement (PMR) will be completed to fully characterize the risk of developing secondary malignancies in patients treated with 131I-MIBG. Risk management will also include product labeling and routine pharmacovigilance to ensure the safe and effective use of 131I-MIBG.
Reference ID: 4297867
NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
19Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Patients with iobenguane scan positive, unresectable, locally advanced or metastatic PPGL who require systemic anticancer therapy represent a population with a serious and life-threatening rare disease for which there is no FDA-approved therapy and no known curative therapy. Although some patients demonstrate responses to off-label use of cytotoxic chemotherapy, complete responses are rare. Off-label use of NA-131I-MIBG is a standard therapy for PPGL that cannot be cured by surgical resection alone. The safety profile of this radiopharmaceutical is consistent with other radiolabeled products and acceptable for a population of patients with PPGL as defined in the product label. The benefit: risk for 131I-iobenguane weighs in favor of benefit for patients who have no other FDA-approved therapies and who need systemic treatment.
Dimension Evidence and Uncertainties Conclusions and Reasons
Analysis of Condition
Pheochromocytomas and paragangliomas (PPGLs) are neuroendocrine tumors typically manifesting with clinical signs and symptoms related to catecholamine excess, such as headache, palpitations, diaphoresis, or life-threatening hypertensive crisis.
Hypertension is the most common sign, observed in more than 95% of functional tumors.
Surgical resection may be curative; however, in patients with unresectable primary tumors, surgical debulking may be indicated to reduce tumor burden and decrease catecholamine levels.
Control of hypertension is critical in malignant PPGL due to life-threatening acute hypertensive emergencies, as well as clinical consequences of long-lasting hypertension, which may result in devastating effects on multiple body systems leading to death if untreated.
A small reduction in blood pressure in hypertensive patients can reduce cardiovascular complications and improve overall survival.
Another major cause of death in patients with PPGLs is metastatic disease. The incidence of malignant pheochromocytoma is unclear. The sponsor
estimated an incidence of 60 to 120 per year in the US. The highest reported incidence of pheochromocytoma in the literature was 0.8 per
Patients with PPGLs have serious and rare diseases with a high degree of morbidity and mortality, especially in the case of metastatic and malignant disease. A durable decrease in the incidence and severity of hypertension, the most common sign of the disease, in conjunction with an observed durable decrease in tumor burden represents a true benefit for these patients who have no FDA-approved treatments.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
20Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Dimension Evidence and Uncertainties Conclusions and Reasons
100,000 person-years; however, most of these patients with pheochromocytoma have non-malignant disease (and would not receive systemic treatment).
Clinical manifestations of malignant PPGL are like those of benign tumors. Malignant PPGLs are generally resistant to chemotherapy; treatment is primarily aimed at palliative control of symptoms.
Survival for patients with metastatic PPGL depends upon the location of metastases. Patients with metastases to the lung and liver have a poor prognosis with survival of less than 2 years, while patients with metastases only to bone may survive more than 20 years after diagnosis. Estimates of the overall 5-year survival rate range from 20 – 60%.
Current Treatment
Options
Surgical removal or cytoreduction is the mainstay of treatment for PPGL. There are no FDA-approved therapies for the treatment of iobenguane
avid metastatic and/or recurrent PPGL. The standard of care for locally advanced unresectable PPGLs includes
symptomatic treatment with alpha blockade with or without alpha-methytyrosine, cytoreductive surgery, radiation therapy, or 131 I-metaiodobenzylguanidine (MIBG) therapy.
For patients with distant metastasis, systemic chemotherapy (e.g. dacarbazine, cyclophosphamide, and vincristine or temozolomide), palliative radiotherapy for bone metastases, or non-AZEDRA 131 I-MIBG (NA- 131I-MIBG) therapy are potential options, and clinical trials may be appropriate.
In the medical literature, various meta-analyses of the effects of combination chemotherapy, and NA-131 I-MIBG reported complete response rates of up to 4 percent and partial response rates of up to 37 percent.
131I-MIBG provides a meaningful clinical benefit for a population of patients with a serious and life-threatening rare tumor for which there are no FDA-approved treatments.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
21Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Dimension Evidence and Uncertainties Conclusions and Reasons
Benefit
Study MIP-IB12B was a multicenter, open-label, single arm study conducted in 68 patients 12 years and older with unresectable pheochromocytoma or paraganglioma that was conducted under SPA and achieved its primary endpoint.
The primary endpoint, a 50% reduction (including discontinuation) of antihypertensive medications for at least 6 months, was a new endpoint for oncology in this population.
Given the morbidity associated with hypertension for patients with PPGL and the direct contribution of the tumor to hypertension through secretion of catecholamines, the primary endpoint was deemed a measure of direct clinical benefit.
The primary endpoint was achieved by 25% of patients (95% CI 16.2% – 36.5%).
The antitumor activity of 131 I-MIBG was confirmed by RECIST with 22.1% of patients demonstrating a confirmed, centrally reviewed response of PR (95% CI: 13.6%, 32.7%).
A secondary OS endpoint result was not interpretable in this single arm trial.
Patients with PPGL have serious and rare diseases with a high degree of morbidity and mortality, especially in the case of metastatic disease. A decrease in the incidence and severity of hypertension represented by a 50% or greater decrease in antihypertensive medications for at least 6 months experienced by 25% of patients, in conjunction with an observed antitumor response of 22%, represents a clinically meaningful benefit for patients with PPGL who require therapy have no FDA-approved treatments.
Risk and Risk Management
The primary safety risks of 131I-MIBG are related to the radiation exposure of the product.
A greater percentage of patients who received two doses of 131I-MIBG experienced cytopenias, sialoadenitis, and renal failure.
There was no significant difference in the development of hypothyroidism.
Myelosuppression and gastrointestinal adverse reactions were the most common.
The risk of myelodysplastic syndrome and leukemia was less common, occurring in 7% of the pooled safety population.
In addition to labeling, a postmarketing requirement under section 505(o) will be included with the approval to follow late toxicities from the drug, specifically the development of myelodysplastic syndrome, acute leukemia, and other secondary malignancies. Refer to section 15 of this review for the specifics of the PMR.
Reference ID: 4297867
NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
22Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Dimension Evidence and Uncertainties Conclusions and Reasons
All patients (100%) who developed leukemia or MDS received two doses of 131I-MIBG.
The serious nature of these risks supports the use of 131I-MIBG only in patients with unresectable, locally advanced or metastatic PPGL who require systemic anticancer therapy disease and who have no other known curative options.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
23Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
1.4 Patient Experience Data
Patient Experience Data Relevant to this Application (check all that apply)x The patient experience data that was submitted as part of the application, include:
EORTC Quality of Life; National Institutes of Health (NIH) Quality of Life and Symptoms Questionnaire for PPGL
Section where discussed, if applicable8.2.6
x Clinical outcome assessment (COA) data, such asUse of tumor pain medication
Section 8.1 Study endpoints
X Patient reported outcome (PRO)EORTC Quality of Life; National Institutes of Health (NIH) Quality of Life and Symptoms Questionnaire for PPGL;
Section 8.2.6, Clinical Outcome Assessment Analyses Informing Safety/Tolerability
□ Observer reported outcome (ObsRO)
□ Clinician reported outcome (ClinRO)
□ Performance outcome (PerfO)
□ Qualitative studies (e.g., individual patient/caregiver interviews, focus group interviews, expert interviews, Delphi Panel, etc.)
□ Patient-focused drug development or other stakeholder meeting summary reports [e.g., Section 2.1 Analysis of Condition]
□ Observational survey studies designed to capture patient experience data
□ Natural history studies
□ Patient preference studies (e.g., submitted studies or scientific publications)
□ Other: (Please specify)
□ Patient experience data that was not submitted in the application, but was considered in this review.
Suzanne G. Demko
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
24Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Cross-Disciplinary Team Leader
Reference ID: 4297867
APPEARS THIS WAY ON ORIGINAL

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
25Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
2 Therapeutic Context
2.1 Analysis of Condition
Pheochromocytomas and paragangliomas (PPGLs) are neuroendocrine tumors with an incidence of two to eight cases per million per year in the United States.(National Cancer Institute, 2018)] Pheochromocytomas arise from chromaffin cells in the adrenal medulla that commonly produce one or more catecholamines: epinephrine, norepinephrine, and dopamine. Paragangliomas are derived from extra-adrenal chromaffin cells and characterized by either parasympathetic-associated chromaffin tissues (most commonly along cranial and vagus nerves) or sympathetic-associated chromaffin tissues (often designated as extra-adrenal pheochromocytomas). Approximately 80-85% of chromaffin cell tumors are pheochromocytomas; 15-20% of chromaffin cell tumors are paragangliomas.
PPGLs are diagnosed most frequently in adults between 40 and 50 years of age and typically manifest with clinical signs and symptoms related to catecholamine excess, such as headache, palpitations, diaphoresis, or life-threatening hypertensive crisis. Hypertension is the most common sign, observed in more than 95% of functional tumors. Surgical resection may be curative. In patients with unresectable primary tumors, surgical debulking may be indicated to reduce tumor burden which may decrease catecholamine levels. Control of hypertension is critical in malignant PPGL due to life-threatening acute hypertensive emergencies, as well as clinical consequences of long-lasting hypertension, which may result in devastating effects on multiple body systems leading to death if untreated. A small reduction in blood pressure in hypertensive patients may reduce cardiovascular complications and improve overall survival. (Ayala-Ramirez, 2012)
In addition to complications of hypertension, another major cause of death in patients with PPGLs is metastatic disease. According to the sponsor, approximately 10-20% of PPGLs are malignant, defined by the World Health Organization classification as the presence of distant metastases, not local invasion. The incidence of pheochromocytoma is unclear, but based on literature reports on the incidence of PPGL, there are approximately, 2 to 8 cases per million person years. A recent report estimated (approximately) 10% of cases to be malignant (National Cancer Institute, 2018; Park, 2011). Clinical manifestations of malignant PPGL are like those of benign tumors; however, metastatic lesions can be life threatening (due to inability to control catecholamine excess or due to metastatic spread). Malignant PPGLs are generally resistant to chemotherapy; treatment is primarily aimed at palliative control of symptoms. Survival for patients with metastatic PPGL depends upon the location of metastases. Patients with metastases to the lung and liver have a poor prognosis with survival of less than 2 years, while patients with metastases only to bone may survive more than 20 years after diagnosis. (Pacak,
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
26Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
2007) Estimates of the overall survival 5-year survival rate range from 20 – 60%. (Pacak, 2007) Thus, while some patients, including some with metastatic disease, have a relatively long life-expectancy, some patients have a poor life expectancy and require systemic therapy.
Iobenguane, or metaiodobenzylguanidine (MIBG), is a norepinephrine analog, and is taken up by norepinephrine transporters; thus, radiolabeled MIBG (123I-MIBG and 131I-MIBG) has been used for diagnostic purposes in tumors arising from the sympathetic nervous system, including pheochromocytoma, paraganglioma and neuroblastoma. Most pheochromocytomas are iobenguane avid, or are detectable via 131I-MIBG or 123I-MIBG scintigraphy; the sensitivity of these methods for detection of pheochromocytoma has been reported as 77 – 90% and 85 – 100%, respectively, though false negatives may occur in patients with tumors which are dedifferentiated or have undergone necrosis. (Pacak, 2007)
2.2 Analysis of Current Treatment Options
Surgical removal or cytoreduction is the mainstay of treatment for PPGL. There are no FDA-approved therapies for the treatment of iobenguane avid metastatic and/or recurrent PPGL in the United States. The standard of care for locally advanced unresectable PPGLs includes symptomatic treatment with alpha blockade with or without alpha-methytyrosine, cytoreductive surgery, radiation therapy, or 131I-metaiodobenzylguanidine (MIBG) therapy. For patients with distant metastasis, systemic chemotherapy (e.g. dacarbazine, cyclophosphamide, and vincristine or temozolomide), palliative radiotherapy for bone metastases, or non-AZEDRA 131I-MIBG (NA- 131I-MIBG) therapy are potential options, and clinical trials may be appropriate.(Lenders, 2005) A 2014 meta-analysis of the effect of cyclophosphamide, vincristine, and dacarbazine in 50 patients with malignant PPGL demonstrated complete responses in 4% of patients (95% CI: 1 – 15%), partial response in 37% of patients (95% CI: 25 – 51%), and stable disease in 14% of patients (95% CI: 7 – 27%). Duration of response was reported in two of the four studies included in this meta-analysis as 20 and 40 months.(Lenders, 2005) A meta-analysis of 243 patients with malignant PPGL treated with NA-131I-MIBG demonstrated that 3% of patients achieved a complete response and 27% of patients demonstrated a partial response.(van Hulsteijn, 2014)
Reviewer note: Non-AZEDRA 131I-MIBG (NA- 131I-MIBG) is not approved for the treatment of PPGL, but is part of the standard of care for PPGL. Patients have generally received 131I-MIBG through multiple expanded access programs. The product evaluated in this review will be referred to by the chemical name, 131I-MIBG, whereas products not manufactured by Progenics will be referred to collectively as NA-131I-MIBG.
NA-131I-MIBG has been studied in patients with neuroblastoma and patients with pheochromocytoma and paraganglioma. Adverse effects associated with NA- 131I-MIBG therapy reported in the literature include myelosuppression, nausea, elevated liver enzymes, renal toxicity, sialadenitis, and hypothyroidism. Veno-occlusive disease has been reported following administration of NA-131I-MIBG in combination with chemotherapy in patients with
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
27Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
neuroblastoma (these patients receive a higher dose). Ten percent of patients with pheochromocytoma may experience a transient exacerbation of symptoms related to catecholamine excess such as headache, palpitations, diaphoresis, and/or hypertension following infusion.(Pacak, 2007; Gonias, 2009) Hypertensive adverse events following administration of NA-131I-MIBG have also been reported in patients with neuroblastoma. (Kosmin, 2012)Infrequent pulmonary complications have occurred following administration of NA-131I-MIBG, including acute respiratory distress syndrome and bronchiolitis obliterans organizing pneumonia.(Gonias, 2009) Solid tumors, myelodysplastic syndrome and leukemia have been reported in patients who have received NA-131I-MIBG. (DuBois, 2013)
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
28Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
3 Regulatory Background
3.1 U.S. Regulatory Actions and Marketing History
131I Iobenguane is not currently marketed in the U.S. 131Iobenguane sulfate was previously approved on March 25, 1994, for the imaging of neuroendocrine tumors. The NDA has since been withdrawn. 131I Iobenguane is not currently approved by any foreign regulatory agency.
3.2 Summary of Presubmission/Submission Regulatory Activity
January 10, 2006: Molecular Insight Pharmaceuticals (MIP) submitted a new IND for 131I Iobenguane. The IND was deemed safe to proceed on February 10, 2006.
January 18, 2006: MIP received orphan drug designation for 131I Iobenguane for the treatment of neuroendocrine tumors, including pheochromocytoma.
March 8, 2006: Fast Track designation was granted for 131I Iobenguane for the treatment of neuroendocrine tumors.
March 6, 2009: A Special Protocol Assessment (SPA) agreement was issued for Study MIP-IB12B, a single-arm, open-label study in 75 patients with relapsed/refractory metastatic pheochromocytoma or paraganglioma. The primary objective of the study as agreed upon in the SPA was to determine the proportion of patients with a 50% reduction (or discontinuation) of all hypertensive medication for six months or two cycles. Secondary objectives included overall tumor response, assessment of bone lesion status on the Soloway Scale, tumor marker response, quality of life (QOL) changes, changes in analgesic and pain medication use and safety.
Reviewer note: The development program as agreed to in the SPA uses an unconventional endpoint to evaluate clinical benefit of 131I-MIBG in a rare population. As noted in section 2.1, the incidence of pheochromocytoma is poorly defined, but it is estimated that 70-290 cases occur per year in the United States. As with other rare diseases, the small population presents numerous challenges in the recruitment for and conduct of clinical trials. PPGL may progress slowly, and overall have a variable natural history, which makes the use of a traditional endpoint such as overall survival difficult and time-consuming. The endpoint in this development program was chosen to evaluate a key cause of morbidity in PPGL and thereby reflect direct clinical benefit.
December 21, 2011: A pre-NDA meeting was held to discuss preclinical, clinical and CMC information for a proposed Subpart H Accelerated Approval NDA. FDA suggested a separate CMC meeting, and stated that the non-clinical program appeared adequately complete to support filing based on the meeting package, but that a final decision would be made following review of the data submitted with the NDA. At that time, the FDA suggested re-opening the trial, which had closed due to financial reasons, to collect data
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
29Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
on a total of 58 patients. The plan to address renal impairment was discussed; FDA stated that MIP’s plan appeared reasonable. A potential future trial to support an application was also discussed.
January 18, 2013: MIP was acquired by Progenics Pharmaceuticals, LP.
October 21, 2013: A Type C CMC Meeting was held. FDA agreed that a radiochemical purity specification of greater than or equal to % appeared reasonable based on the data provided, and that a determination of acceptability would be determined during review of the NDA.
July 26, 2015: FDA granted a Breakthrough Therapy Designation for “Ultratrace Iobenguane 131I” for the treatment of patients with iobenguane-avid metastatic and/or recurrent pheochromocytoma and paraganglioma (PPGL) based on preliminary clinical evidence of efficacy obtained in Study MIP1B12B.
January 22, 2016: A Type B meeting was held to discuss the development program following Breakthrough Therapy designation. The agency agreed that a future NDA would receive rolling review on the basis of the Fast Track and Breakthrough Therapy designations. FDA agreed that the NDA for 131I Iobenguane would support approval of a treatment regimen that included both imaging and dosimetric doses. The agency stated that the endpoint in Study MIP-IB12B constituted a clinical benefit, provided the data are supported by an assessment that the product provides a significant advance over alternative therapy and the natural history of the disease. FDA stated that a determination of whether additional, randomized studies would be required would be made during review of the planned NDA. FDA also noted that analyses of time-to-event endpoints such as overall survival are not interpretable in a single arm study, and thus would not be included in product labeling. Agreement was reached that drug interaction studies in humans were not necessary, and that clinical pharmacology studies in patients with organ impairment were not necessary; however, the agency stated that the need for these studied would be evaluated during review of the data included in the original NDA submission.
April 11, 2016: FDA granted conditional acceptance of the proprietary name AZEDRA following review.
October 6, 2016: During a pre-NDA CMC meeting it was determined that 6-month stability data (long-term, intermediate and accelerated) for the 3 Drug Substance
commercial batches would be submitted in the NDA and that 12 months of stability data (long-term and intermediate) will be submitted during NDA review (November 2017); it was agreed that 24 months of supportive long-term storage data from the clinical Good Manufacturing Practice (GMP) batch would also be submitted in the NDA. FDA and MIP agreed that release data, stability data, executed batch records and information on synthesis equipment would be submitted for the 3 validation batches manufactured at , 3 validation batches manufactured at the
, and three recent clinical batches
Reference ID: 4297867
(b) (4)
(b) (4) (b) (4)
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
30Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
manufactured at . It was determined that in-use stability data, representing simulated conditions at the maximum dilution, would be included in the NDA. It was agreed that data from an extractable/leachable study, conducted at ambient room temperature for 24 hours in the primary container/closure system, using a cold formulation of 131I-MIBG, would be included in the NDA. It was determined that justification and documentation that the container/closure components are resistant to radiolysis would be submitted in the NDA, and that release data, including impurities, for manufactured from the would be included in the NDA.
September 9, 2016: A Written Response Only letter was issued regarding content and format of the Integrated Summary of Safety, datasets, and presentation of efficacy data.
October 26, 2016: FDA agreed with Progenics’s proposal, submitted August 22, 2016, providing justification for not conducting a thorough QT study with Iobenguane I 131.
November 18, 2016: MIP issued a response to FDA’s Written Response Only (WRO) letter of September 9, 2016. MIP proposed to provide Analysis Dataset Model [10] datasets for studies MIP-IB12B and MIP-IB13, but not for MIPIB-11 and MIP-IB12 as these studies were started in 2006 and 2007, respectively, and were previously completed and submitted to the IND in legacy format. FDA recommended that the Sponsor contact [email protected] to request a waiver to the proposed partial non-CDISC submission for studies MIP-IB11 and MIP-IB12 that are unsupported or retired by December 17, 2016. The request for the waiver was submitted on December 7, 2016.
January 17, 2017: A pre-NDA meeting was held. MIP sought feedback regarding the content and format of the intended NDA. FDA stated that it was amenable to receiving sections of the NDA on a rolling basis, and that the need for a REMS was not anticipated, but advised MIP to submit a detailed risk-benefit profile to the NDA. FDA requested that the 120-day safety update be submitted 90 days after NDA submission if possible. Progenics stated that it planned to submit one study report (pivotal study MIP-IB12B) in granular eCTD format and three study reports (MIP-IB11, MIP-IB12, and MIP-IB13) in legacy electronic format. FDA agreed that the plan was acceptable, however noted that the waiver regarding formatting was under review. MIP proposed a revised definition of primary outcome responder as follows (new wording underlined):
FDA stated that the revised definition was not acceptable, as it could result in classification of some patients as responders who would not be appropriate. FDA noted
Reference ID: 4297867
(b) (4)
(b) (4)(b) (4)
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
31Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
that changes in tumor size and catecholamine levels as evidence of an antitumor response to Iobenguane 131I will also be considered during review of the NDA.
FDA stated that Progenics’s plan to provide validation reports for LC/MS/MS bioanalytical methods, but not direct gamma counting using an automated well gamma counter, was acceptable. FDA also agreed that geometric means for the PK parameter data would be provided in Modules 2.7.1 and 2.7.2, but would not be available in the previously submitted MIP-IB11 report.
January 26, 2016 and February 6, 2017: Emails from FDA clarified the expectations for submission of data sets.
February 9, 2017: FDA granted Rolling Review for the NDA.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
32Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
4 Significant Issues from Other Review Disciplines Pertinent to Clinical Conclusions on Efficacy and Safety
4.1 Office of Scientific Investigations (OSI)
DOP2 consulted the Office of Scientific Investigation (OSI) on July 28, 2017, to perform an audit of three clinical trial sites (site #2; site #5; site #11) and the Applicant, Progenics Pharmaceuticals, Inc. The Division, in consultation with OSI, selected clinical sites for inspection using manual assessment of the trends in screening and enrollment characteristics, patterns of protocol violations reported for the sites, patterns of efficacy reporting, and patterns of serious adverse event (SAE) reporting. The three inspected sites were selected largely due to having the highest enrollment, covering 65% of enrolled patients. In general, the inspectional findings supported validity of data as reported by the Applicant under this NDA. FDA form 483, with voluntary action indicated (VAI) was issued to the Applicant for failure to ensure proper monitoring of the study due to a gap of monitoring visits from 7/2013 through 4/2014. A response from the Applicant indicated that this corresponded to the time shortly after Progenics acquired Molecular Insight Pharmaceuticals, the former sponsor, and that there was no gap in safety surveillance. Otherwise, there were no notable inspectional observations. OSI concluded that although regulatory violations related to failure to ensure proper monitoring were noted, the observations are unlikely to significantly impact primary safety and efficacy analyses.
See OSI review for additional details regarding site inspections and findings.
4.2 Product Quality
Please see the FDA CMC review by Dhanalakshmi Kasi for details regarding 131I-MIBG quality.
4.3 Clinical Microbiology
Please see FDA product quality microbiology review by Julie Nemecek for further details.
4.4 Devices and Companion Diagnostic Issues
There is no device or companion diagnostic test for review in support of this NDA.
4.5 Division of Medical Imaging Products
DOP2 consulted the Division of Medical Imaging Products (DMIP) to review the radiation dosimetry, and expected organ and bone marrow exposure from treatment with 131I-MIBG. DMIP concluded that the prescribing information provided by the Applicant insufficiently
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
33Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
described the risks associated with radiation. In particular, DMIP provided feedback regarding the potential risks of secondary malignancies and other acute and chronic radiation toxicities. DMIP suggested a different approach to dose-adjustment than that recommended by the Applicant. Rather than dose-adjustment based on specified absorbed activity thresholds in three specific organs, (the kidney, liver, and lungs), DMIP recommended dose-adjustment based on provider-identified critical organs. The specified absorbed activity thresholds were removed, to allow provider use of the best available clinical practice guidelines, and allow individualization of the prescribed absorbed dose-limits for each patient. DMIP provided a table including absorbed-dose thresholds for radiation toxicity in critical organs, based on the International Commission on Radiological Protection’s Publication 118, for inclusion in the product labeling. Section 13.1 provides specific labeling changes made in collaboration with DMIP and the Applicant.
DMIP suggested inclusion of the following sentence in section 5.1: “Radiation epidemiological studies of survivors of the atomic bombing in Japan and studies of survivors of medical radiation therapy suggest that a 12-year-old child undergoing a full course of AZEDRA radiotherapy is likely to develop a secondary cancer sometime later in life as a result of the radiation exposure. An adult undergoing AZEDRA radiotherapy would face a less-than-50% chance for incidence of secondary cancer from the radiation.” DOP 2 agreed with inclusion of a statement that the risks of radiation associated with the use of 131I-MIBG are greater in pediatrics than in adults, but removed reference to atomic bomb survivors for the sake of brevity, and specific reference to secondary malignancy in this section given the dedication of section 5.3 to this topic. The concern for greater radiation-associated risks in pediatric patients due to greater absorbed radiation doses and potential longer life expectancy was reiterated in section 8.4 of the product labeling.
DMIP provided literature references indicating that, for some patients with tumors under 5.5 cm, the five-year overall survival rate ranged between 94.7 and 100%; the longer life expectancy of these patients would require additional consideration in the benefits and risks of a treatment associated with long-term radiation toxicities including secondary malignancies. Therefore, DMIP agreed with DOP2 that the indication should be restricted to a population with a reduced life expectancy, given the risks of long-term toxicities, which is reflected in the revised product labeling. DMIP discussed the potential risk of cardiovascular and cerebrovascular disease 10 – 15 years after exposure to irradiation with 131I-MIBG. This risk was not specifically addressed in product labeling because it was not observed in the clinical development program. In addition, DOP2 noted that the population of patients with unresectable, locally advanced or metastatic PPGL had a relatively shorter life expectancy, and would not be expected to live 10 -15 years without further therapy. A 5-year survival of 95 – 100% is not expected in the population specified by the indication statement. Refer to the clinical reviewer’s discussion of prognosis and treatment options in Section 2 Therapeutic Context
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
34Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
DMIP also noted that prediction of radiation absorbed dose for two subsequent therapeutic doses based on a single dosimetric assessment had some limitations. DMIP concluded that an assumption of proportionality of radiation absorbed dose between dosimetric dose and therapeutic dose, as well as consistency in absorbed dose between therapeutic doses, was reasonable but not certain based on the data provided. Changes to product labeling were not suggested based on this observation.
5 Nonclinical Pharmacology/Toxicology
5.1 Executive Summary
131I Meta-iodobenzylguanidine (MIBG) is a radioactive therapeutic agent; it is a guanethidine analog, structurally similar to norepinephrine, that shares the same uptake, storage, and release mechanisms as norepinephrine. Due to this structural similarity, MIBG concentrates in highly sympathetically innervated tissues with high levels of norepinephrine transporter (NET), including the adrenal gland and heart. In addition, MIBG can concentrate in neuroendocrine tumors that have high levels of NET, including pheochromocytoma and paraganglioma (PPGL), which can take up MIBG and store it in neurosecretory granules. Iodine 131 is a beta and gamma emitting isotope with a half-life of 8 days and penetration length of 0.6 to 2 mm that induces cellular damage by the production of free radicals.
Pharmacology studies submitted in the form of published articles demonstrated the shared uptake mechanism of MIBG and biogenic amines through NET. Studies showed differences in MIBG uptake and storage in tumors of different origins. Uptake of radiolabeled MIBG was saturable in both PC-12 rat pheochromocytoma and SK-N-SH neuroblastoma cells, but in the pheochromocytoma cells MIBG was stored in vesicular granules, leading to longer exposures. Due to their origin, neuroblastoma cells do not have vesicular granules. In SK-N-SH cells, but not in some other neuroblastoma cell lines, MIBG was able to concentrate in the extra-vesicular space resulting in limited long term retention. Differences in uptake and retention between NET positive tumors of different origins may help account for differences in clinical activity to 131I MIBG therapy.
The Applicant presented data showing that 131I MIBG administration resulted in anti-tumor activity in mice implanted with PC-12 pheochromocytoma cells and in some sensitive neuroblastoma cell lines. Generally, radiolabeled MIBG preparations with high specific activity led to better anti-tumor activity. High specific activity MIBG preparations also resulted in modest increases in radioactivity exposure in organs with cells that have NET expression, but lower levels of cold MIBG exposure in these organs. Lower exposure to MIBG itself may lead to fewer MIBG-related toxicities, particularly pharmacologically-mediated increases in blood pressure.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
35Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Consistent with its mechanism of action, clear increases in arterial pressure with reflex bradycardia occurred in a cardiovascular study in dogs at unlabeled MIBG doses ≥ of 0.3 mg/kg; this dose is approximately 60 times higher than the amount of MIBG given at the radiotherapeutic dose of Azedra. MIBG exhibited low potential for hERG-mediated QTc prolongation with an IC50 of 93.6 M, which is about 200-fold higher than the maximum theoretical blood concentration of MIBG at the therapeutic dose of Azedra in humans.Consistent with this finding, there were no QT interval effects at MIBG doses of up to 3 mg/kg in dogs or at a cold-labeled MIBG (127I MIBG) dose of 1.085 mg/kg.
Pharmacokinetic and distribution studies assessed the difference in pharmacokinetics and tissue distribution of Ultratrace Iobenguane 131I and CIS-US Iobenguane 131I. The Ultratrace product employs a solid state labeling technique resulting in less than 0.0028 mg carrier Iobenguane /mCi while the CIS technique uses a carrier added labeling approach resulting in ~ 0.3 mg carrier Iobenguane /mCi with lower specific activity. Regardless of labelling method, the kidney was the predominant organ for MIBG clearance, and 131I-MIBG cleared with elimination half-lives of approximately 16 hours in rat, and 32 hours in dogs. In rats administered with equivalent doses of radioactivity, the tissue distribution and exposure of Ultratrace 131I-MIBG and commercially available carrier-added 131I-MIBG were highly similar. At the single IV dose of 40 mCi/kg of Ultratrace 131I-MIBG in rats, the highest levels of accumulated radioactivity occurred in the lungs, heart and adrenals (≥223 nCi/g) followed by the kidneys, salivary glands, thyroid ( ≥ 100 nCi/g), liver, small intestine, adipose tissue and bone (51- 80 nCi/g). Based on the literature, bone marrow (34 nCi/g in this assessment) is the dose limiting organ for 131I-MIBG.
In a 28-day repeat IV dose toxicity study in dogs Ultratrace 127I-MIBG (cold product) was tolerated at doses up to 1.085 mg/kg (21700 g/m2), the highest dose tested. Findings in this study were limited to injection site reactions. There were no major target organs of MIBG toxicity. Toxicities caused by radiation are well described and are expected following administration of Azedra in patients with malignant recurrent pheochromocytoma and paraganglioma (PPGL).
The Applicant did not conduct carcinogenicity studies and these studies are not required to support the marketing application for a drug intended to treat advanced cancer. The cold 127I-MIBG was not mutagenic in the Ames bacterial mutagenicity assay and was negative for clastogenic activity in vitro in mouse lymphoma cells and in an in vivo mouse bone marrow micronucleus assay. As 131I-MIBG is a radioactive product, it is both genotoxic and potentially carcinogenic. Due to its genotoxicity, and consistent with the ICH S9 Guidance, no reproductive toxicology studies were conducted or required to support the marketing of 131I-MIBG. The risk of radiopharmaceuticals to a developing fetus is well-established in the scientific literature.
The nonclinical team recommends a warning for the potential for embryo-fetal toxicity and the inclusion of advice for females and males of reproductive potential to use contraception during
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
36Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
and for 7 and 4 months, respectively, following the last dose of Azedra. In addition, because this is an 131I-labeled product, women are advised not to breastfeed during and for 80 days following the last dose of Azedra. There are no outstanding issues from a pharmacology/toxicology perspective that would prevent approval of the Azedra for treatment of patients years and older with Iobenguane avid malignant, recurrent, or unresectable pheochromocytoma and paraganglioma therefore the pharmacology/toxicology team recommends approval.
5.2 Referenced NDAs, BLAs, DMFs
None.
5.3 Pharmacology
Primary pharmacologyA. In Vitro Studies
The Applicant demonstrated the affinity and selective uptake of MIBG into cells that express noradrenaline transporter (NET) using a published study by Boyd et al 2004. Investigators compared the effects of incubation of a radiation-resistant human glioma cell line (UVW), either stably expressing NET or untransformed, with 7 kBq of 131I-MIBG (specific activity 45-65 MBq/mg) in the presence or absence of 1.5mM desmethylimipramine (DMI), a potent NET inhibitor.
In vitro, cells expressing NET had ~27-fold higher uptake of radioactive MIBG compared to uptake in cells without NET expression. Similar differences in uptake occurred between NET positive cells in the absence or presence of DMI (Figure 1). Additionally, in an in vivo experiment, nude mice were implanted with each of the two cell types and later treated with no-carrier added 131I-MIBG or a carrier-added form of the drug to compare the biodistribution in both xenografted tumors and mouse tissues. The carrier added vs. non-carrier added represent two manufacturing process resulting in different specific activities for the radiolabeled drug. NET positive tumors had significantly higher concentrations of either formulation of the drug than NET negative tumors. Overall distribution was similar in all tissues for each of the drug formulations with slightly increased levels of radioactivity in high NET expressing tissues for the no carrier added product. Tissues with the highest levels of radioactivity included the adrenal gland, heart, liver, lung, and kidney (Table 1).
Reference ID: 4297867
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
37Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Figure 1: Selective Uptake of MIBG in Cells Expressing Noradrenaline Transporter
(Excerpted from Boyd et al. 2004)
Table 1: Biodistribution of carrier-added and no-carrier-added 131-I MIBG in tissues from xenograft implanted nude mice as a percentage of the injected dose per gram of tissue
(Excerpted from Boyd et al. 2004)
The Applicant Barrett et al. 2010, describes a
solid labeling approach to make a non-carrier-added radiolabeled MIBG. Solid labeling with 123I or 131I resulted in higher specific activity of radioactive iodine labeled MIBG with less unlabeled MIBG compared to other methods. Investigators then assessed the impact of cold carrier MIBG versus other radiolabeling methods on tissue distribution of 123/131I MIBG in the conscious rat. The carrier free Ultratrace MIBG radiolabeled with either 123I or 131I exhibited similar radioactive tissue distribution as the carrier added radiolabeled MIBG following administration of the drug on a μCi/kg basis, but the higher specific activity of the Ultratrace product resulted in much lower MIBG exposures compared to the carrier added product (Figure 2). In the heart, with high expression of NET, the higher specific activity of the ultratrace preparation resulted in some
Reference ID: 4297867
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
38Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
increases in radioactivity exposure compared to preparations with higher amounts of unlabeled MIBG.
The investigators also showed a dose-dependent increase in arterial pressure concomitant with a reflex bradycardia in conscious instrumented dogs treated with unlabeled MIBG. Mean arterial pressure increases of up to 45% occurred in dogs at the IV administered unlabeled MIBG dose of 3000 g/kg, accompanied by heart rate decreases of up to 43%, whereas arterial pressure increases at the 30 μg/kg dose were only 13%. Based on these findings, radiolabeled MIBG products with higher specific activity and, therefore, lower doses of unlabeled MIBG may lead to relative decreases in the cardiovascular side effects of hypertension and bradycardia associated with MIBG exposure compared to low specific activity MIBG products.
Figure 2: Radioactive vs. MIBG Exposure in High vs Low Specificity Activity Radiolabeled MIBG Products
Radioactive Exposure MIBG Exposure
(Adapted from Barrett et al., 2011)
To explore the uptake and retention of MIBG in neural crest tumor cell lines including both pheochromocytoma and neuroblastoma cells, the Applicant referenced Smets et al., (Cancer Research 1989, 49:2941-2944). The authors measured uptake of extracellular MIBG in cell cultures of SK-N-SH or several other human neuroblastoma cells incubated for 2 hours with 125I MIBG (10-8 M) in the presence or absence of imipramine, a NET blocker, or in the presence or absence of excess unlabeled MIBG or NE. In the SK-N-SH cells, but not other neuroblastoma cell lines, there was evidence of active uptake of MIBG (decreased cell associated radioactivity in the presence of imipramine). Passive diffusion occurred in all cell lines tested.
In chromaffin derived cells like the PC-12 pheochromocytoma cell line, MIBG is stored in secretory granules. By blocking intracellular transport into storage granules with reserpine, the authors showed decreases in MIBG retention of up to 50% in the PC-12 cells, but not in SK-N-SH cells (Figure 3). This difference in retention between different types of neural crest derived tumors along with differences in uptake in different neuroblastoma cell lines may explain differences in sensitivities of these NET-positive types of tumors to treatment with 131I-MIBG.
Reference ID: 4297867
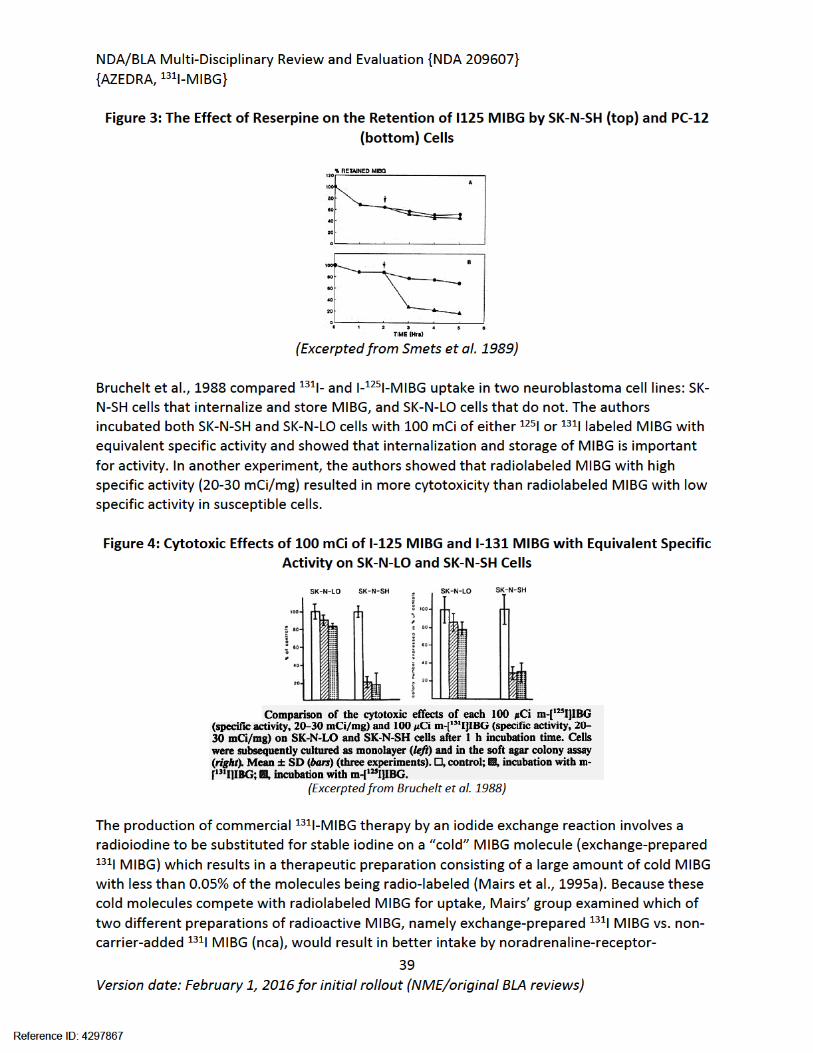

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
40Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
expressing neuroblastoma cells when compared to normal tissues that accumulate MIBG by passive diffusion in tumor-bearing mice.
Figure 5: Calculated Values of Absorbed Radiation Dose per Unit of Injected Radioactivity (Gy/MBq) to Tumor and Normal Organs for Non-Carrier-Added (White Bars) and Exchange
Preparation (Black Bars) of 131I MIBG
T=tumor; M=muscle; Li=liver; Sp=spleen; SK=skin; Lu=lung; H=heart; K=kidney; Thy=thyroid; B=blood; A=adrenal
(Excerpted from Mairs et al. 1995a)
Absorption was calculated by estimating the areas under the time-activity curves, corrected for radionuclide decay, and multiplying by the equilibrium dose constant for 131I. In practice, injected radiolabeled MIBG activity is usually limited by bone marrow toxicity. Based on the above results, if the absorbed dose to the bone marrow is directly proportional to blood dose, the non-carrier-added preparation would deliver a slightly reduced bone marrow dose of radioactivity, suggesting that the higher specific activities of non-carrier-added preparations could result in better therapy. In addition, these results suggest that a higher tumor-radiation absorption dose is possible, but with increases of radioactivity to the heart and adrenals as well.
Radioactive uptake by SK-N-BE (2c) active transport competent neuroblastoma cells was higher at higher doses for the exchange-labeled non-carrier added 131I-MIBG when compared to a commercial carrier-added preparation (Figure 6). Carrier-added 131I-MIBG accumulation plateaued at 100 kBq, while nca 131I-MIBG continued to accumulate even at levels at which there was no further accumulation with the carrier added product. Mairs and collaborators (Mairs et al., 1995b) also showed that A2780 control cells which only take up MIBG by passive activity incorporated equivalent amounts of non-carrier added (nca) and carrier added (ca) 131I-MIBG (data not shown).
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
41Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Figure 6: Specific Uptake by SK-N-BE(2c) Neuroblastoma Cells of No Carrier-Added 131I-MIBG (closed circles) and a Commercial Carrier-Added Preparation (open circles)
(Excerpted from Mairs et al., 1995b)
B. In Vivo StudiesStudies by Rutgers et al. (2000a and 2000b) demonstrated the activity of 131I-MIBG in the treatment of mice implanted with PC-12 rat pheochromocytoma cells or SK-N-SH human neuroblastoma cells. While both cell types express NET and actively take up MIBG, the storage mechanisms for MIBG differ between the cell types. Implanted PC12 tumors had more extensive accumulation and higher MIBG exposure than the SK-N-SH tumors following treatment of mice with 131I-MIBG (Figure 7), resulting in a relative increase in tumor growth delay in the pheochromocytoma line compared to the neuroblastoma line Figure 8). 131I-MIBG also showed some anti-tumor activity in a model of metastatic pheochromocytoma in these experiments.
Figure 7: Increased Exposure in Pheochromocytoma versus Neuroblastoma Implants
(Excerpted from Rutgers et al. 2000a)
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
42Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Figure 8: Dose Dependent Increase in 131I-MIBG-Mediated Anti-Tumor Activity in PC-12 Implanted Animals
(Excerpted from Rutgers et al. 2000a)
Table 2: Increased Activity in 131I-MIBG Treated Mice Implanted with Pheochromocytoma
(Excerpted from Rutgers et al. 2000a)
An additional study by Rutgers (Rutgers et al. 2000b) suggested that there was no clear advantage in administering 131I-MIBG as a divided fraction rather than a single dose.
Study: RPT-2200-09-004 Comparison of anti-tumor effects of Ultratrace Iobenguane I-131 and Iobenguane I 131 from Amersham Bioscience in the human neuroblastoma SK-N-BE\(2c\) mouse xenograft model
CD/1 nu/nu athymic mice were subcutaneously implanted with SK-N-BE(2c) human neuroblastoma cells (2x106 cells) and assigned to treatment groups (10 mice/group) of saline, Ultatrace 131I-MIBG, or a lower specific activity 131I-labeled product (Amersham) at doses of 81, 270 and 810 μCi. Treatment began after tumors reached an average size of 390 mm3.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
43Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
In the saline control group tumor size increased 4-fold, with a tumor doubling time of 7 days. Treatment with either 131I-MIBG product resulted in delays in tumor growth at dose levels ≥ 270 μCi. At 270 and 810 μCi the Amersham Bioscience 131I-MIBG delayed the tumor growth doubling rate to 9 and 18 days, respectively. Treatment with Ultratrace 131I-MIBG at 81, 270 and 810 μCi delayed tumor doubling times to 8, 13, and 31 days, respectively.
Figure 9: Change In Tumor Volume and Days of Tumor Doubling Time after Administration of 131I Ultratace and 131I Comparison product
(Applicant Figure reproduced from Study RPT-2200-09-004)
Figure 10: Tumor Doubling Times After 131I-MIBG Administration
(Applicant Figure reproduced from Study RPT-2200-09-004)
Safety PharmacologyStudy 071206NJQ Effects of 2503 (127I-iobenguane drug product) on cloned hERG potassium channels expressed in human embryonic kidney cells
In GLP-compliant Study # 071206NJQ, HEK293 cells stably expressing the human hERG potassium channel were incubated with MIBG at 10, 30, and 95 mM, 0.1% DMSO (negative control), or terfenadine at 60 nM (positive control) followed by measurement of potassium current using the patch-clamp technique. Negative and positive controls resulted in expected outcomes. MIBG inhibited the hERG potassium current with an IC50 value of 93.6 M suggesting low risk of MIBG-mediated QT prolongation by this mechanism in humans.
Reference ID: 4297867
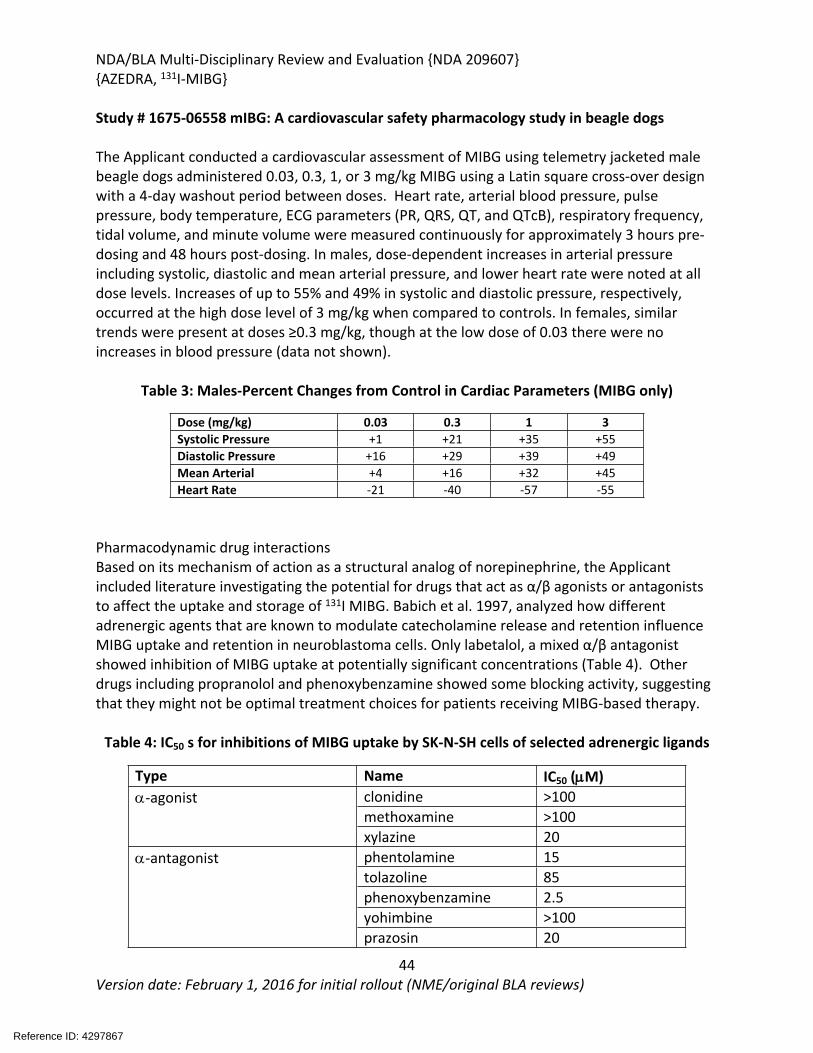
NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
44Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Study # 1675-06558 mIBG: A cardiovascular safety pharmacology study in beagle dogs The Applicant conducted a cardiovascular assessment of MIBG using telemetry jacketed male beagle dogs administered 0.03, 0.3, 1, or 3 mg/kg MIBG using a Latin square cross-over design with a 4-day washout period between doses. Heart rate, arterial blood pressure, pulse pressure, body temperature, ECG parameters (PR, QRS, QT, and QTcB), respiratory frequency, tidal volume, and minute volume were measured continuously for approximately 3 hours pre-dosing and 48 hours post-dosing. In males, dose-dependent increases in arterial pressure including systolic, diastolic and mean arterial pressure, and lower heart rate were noted at all dose levels. Increases of up to 55% and 49% in systolic and diastolic pressure, respectively, occurred at the high dose level of 3 mg/kg when compared to controls. In females, similar trends were present at doses ≥0.3 mg/kg, though at the low dose of 0.03 there were no increases in blood pressure (data not shown).
Table 3: Males-Percent Changes from Control in Cardiac Parameters (MIBG only)
Dose (mg/kg) 0.03 0.3 1 3Systolic Pressure +1 +21 +35 +55Diastolic Pressure +16 +29 +39 +49Mean Arterial +4 +16 +32 +45Heart Rate -21 -40 -57 -55
Pharmacodynamic drug interactionsBased on its mechanism of action as a structural analog of norepinephrine, the Applicant included literature investigating the potential for drugs that act as α/β agonists or antagonists to affect the uptake and storage of 131I MIBG. Babich et al. 1997, analyzed how different adrenergic agents that are known to modulate catecholamine release and retention influence MIBG uptake and retention in neuroblastoma cells. Only labetalol, a mixed α/β antagonist showed inhibition of MIBG uptake at potentially significant concentrations (Table 4). Other drugs including propranolol and phenoxybenzamine showed some blocking activity, suggesting that they might not be optimal treatment choices for patients receiving MIBG-based therapy.
Table 4: IC50 s for inhibitions of MIBG uptake by SK-N-SH cells of selected adrenergic ligands
Type Name IC50 (M)clonidine >100methoxamine >100
-agonist
xylazine 20phentolamine 15tolazoline 85phenoxybenzamine 2.5yohimbine >100
-antagonist
prazosin 20
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
45Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Type Name IC50 (M)propranolol >10-antagonistatenolol >100isoprenaline >100-agonistsalbutamol >100
Mixed / antagonist labetalol 0.65Neuronal blocking agent guanethidine 4
(Adapted from Babich et al. 1997)
Solanki et al. 1992 published a pharmacological guide to medicines which interfere with biodistribution of radiolabeled meta-iodobenzylguanidine (MIBG). Studies, including this study, have been previously reviewed under NDAs for other MIBG products. Recommendations have been reviewed by the clinical pharmacology review team.
5.4 ADME/PK
Study Major FindingsPK, Distribution
MIP-120-05-001 Pharmacokinetics and tissue distribution of Ultratrace Iobenguane I 131 and CIS-US Iobenguane I 131 in the conscious rat
No major differences in the mean blood clearance values between two different manufacturing procedures of Iobenguane,
Heart and kidney uptake was similar for both products
Blood Heart Marrow Kidney LiverCmax (nCi/g)Ultratrace 21 304 18 144 46CIS-US 23 342 21 108 50AUC0-inf
(hr*nCi/mL)Ultratrace 286 2794 202 758 368CIS-US 323 2460 332 702 428Clearance (L/hr/kg)Ultratrace 0.10 0.01 0.20 0.05 0.1CIS-US 0.10 0.02 0.10 0.06 0.09Vss (L/kg)Ultratrace 3.1CIS-US 2.4MRT (hr)Ultratrace 22.2 12.1 12.3 17.9 21.9CIS-US 19.3 11.7 9.8 14.4 22.2
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
46Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
MIP-120-05-003 The impact of 46.5 and 93 mg/mCi carrier Iobenguane pharmacokinetics and tissue distribution of Ultratrace Iobenguane I 131 in the conscious rat
Ultratrace (0.0028 mg/mCi) exposure to heart and kidney was higher when given alone than when carrier Iobenguane was added to Ultratrace 131I-MIBG at 46.5 and 93 mg/mCi, consistent with the hypothesis that unlabeled (carrier) Iobenguane competed for target binding.
Blood Heart Marrow Kidney LiverCmax (nCi/g)Ultratrace 21 304 18 144 46Ultratrace + 46.5 21 183 40 67 87Ultratrace + 93 13 141 25 44 69AUC0-inf
(hr*nCi/mL)Ultratrace 286 2794 202 758 368Ultratrace + 46.5 286 1131 396 496 452Ultratrace + 93 227 860 322 496 354MRT (hr)Ultratrace 22.2 12.1 12.3 17.9 21.9Ultratrace + 46.5 11.1 12 6.8 11.3 11.4Ultratrace + 93 14 12.2 10.9 9.9 11.3
RPT-2200-09-002 A1 Pharmacokinetics and tissue distribution of Ultratrace 131 I Iobenguane and Draximage 131I-iobenguane in the conscious rat
At the same radioactive dose of 40 mCi/kg of Ultratrace 131I Iobenguane and Draximage 131I Iobenguane, the tissue distribution and exposure of the two drugs were highly comparable.
Utratrace 131I MIBG at the dose of 10 mCi reached Cmax at 14.8 hr. Mean volume of distribution ranged from 533 to 609 mL, and the product cleared from the blood in ~24 hr. Uptake of 131I MIBG was highest in the lungs and heart and adrenals (395, 346 and 223 nCi/g). Kidneys, salivary glands and thyroid had concentrations of ≥ 100 nCi/g. Liver, small intestine, adipose tissue, and bone concentrations ranged from 51 to 80 nCi/g. Based on the literature, bone marrow is dose limiting organ for 131I MIBG; the concentration in rat bone marrow was 34 nCi/g in this assessment.
8204237 PK, distribution, metabolism, and excretion of I-131 MIBG following IV administration to rats
After single IV administration of 6.89 mCi/kg (0.819 mg/kg) 131I MIBG to rats, animals were observed for mortality, clinical signs, general health (appearance), PK assessment, distribution and metabolism. No specific toxicity was noted in rats throughout the study. The maximum concentration of radioactivity in blood occurred 5 minutes’ post-dose, with plasma concentration reaching comparable levels to the blood concentrations by 24 hours after dosing.
Elimination half-lives were ~18 and ~16 hours for males and females in plasma, respectively, and 16.5 and 15 hours in blood. The main route of elimination was via urine, with a mean of ~50% being excreted over the
Reference ID: 4297867
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
47Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
first 24 hr postdose, regardless of sex. Elimination via feces accounted for ~12% over the first 168 hours’ post dose regardless of sex.
8204231 PK, distribution, metabolism, and excretion of I-131 MIBG following IV administration to dogs
A single IV dose of 0.058 mg/kg was administered to dogs to assess PK, metabolism and excretion, while animals were observed daily for clinical signs, mortality and activity. All dogs appeared healthy with no abnormal clinical signs during the length of this study. Maximum concentration of radioactivity in blood occurred ~1 hour post-dose; blood concentration was continuously higher than that in plasma indicating retention of MIBG in RBC. The main route of radioactivity elimination was in urine: 80.0 ± 2.2% (male) and 86.9 ± 1.3% (female) over the first 168 h postdose. Excretion of radioactivity via feces accounted for <2% of the administered dose. The total recovery of radioactivity was 91% in male and 92% in female dogs.
MIP-IB11, 8204231, and 8204237 Metabolic Pathways of MIBG in Rats, Dogs, and Humans (summary schema)
No unique metabolites were observed in humans.
(Applicant Figure Excerpted from Pharmacokinetic Summary)
5.5 Toxicology
5.5.1 General Toxicology
Study title/ number: A 4-week Intravenous Bolus Injection Toxicity and Toxicokinetic Study with I-127 Iobenguane in Beagle Dogs with a 4-week Recovery Period / Study Number: 7736-128.
Key Study Findings
Reference ID: 4297867
(b) (4)
(b) (4)
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
48Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
IV administration of I-127 MIBG at all dose levels resulted in transient, irregular respiration and increased heart rate during dosing.
Conducting laboratory and location
GLP compliance: Yes
MethodsDose and frequency of dosing: Saline, vehicle, 0.22, 0.66, and 1.085 mg/kg/day,
once daily bolus injection for 29 days with 28 days recovery period
Route of administration: IVFormulation/Vehicle: 0.9% Sodium ChlorideSpecies/Strain: Beagle dogNumber/Sex/Group: Main:7/sex/group; Recovery: 3/sex/groupAge: 7-8 monthsSatellite groups/ unique design: NoDeviation from study protocol affecting interpretation of results:
No
Observations and Results: changes from control Parameters Major findingsMortality NoneECG Minimally lowered heart rate of males at 1.085 mg/kg (↓~30% compared to
saline or vehicle males) Organ weights Liver and
gallbladderVehicle control and HD (1.085 mg/kg)
↓mean and absolute weight when compared to LD (0.22 mg/kg) and MD (0.66 mg/kg)
Absolute brain weight
↑ significantly in LD females
Main salivary gland-to-body weight
↑ in all MIBG-administered females compared to controls
Histopathology All findings related to IV infusion, regardless to test-article IV or its concentrationIntravenous infusion site findings:
chronic/chronic active inflammation, fibrosis, edema, and hemorrhage of the perivascular/subcutaneous area, endothelial/medial proliferation and adventitial inflammatory reaction as
a vascular responseDermal/subdermal findings:
Reference ID: 4297867
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
49Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
acanthosis, epidermal surface necrotic cell debris, chronic inflammation and folliculitis in all study animals
All findings persisted in recovery with a trend towards recovery Toxicokinetic Exposure to 127I-Iobenguane increased with the increase in dose level from 0.22
to 1.085 mg/kg/day. The increases in Cmax and AUC 0-24 were dose proportional. Possible accumulation occurred in dosed animals.
General toxicology; additional studies
5.5.2 Genetic Toxicology
In Vitro Reverse Mutation Assay in Bacterial Cells [10]Study title/ number: Bacterial Reverse Mutation Assay with a Confirmation Assay /
Study No. 7736-130.
Key Study Findings: 127I-iobenguane at concentrations up to 5000 g/plate did not show genotoxic activity in Salmonella typhimurium strains (TA1535, TA1537, TA98, TA100) and Escherichia coli strain WP2uvrA in the presence and absence of S9 mix. Standard positive controls confirm the sensitivity and validity of the assay.
GLP compliance: YesTest system: salmonella strains TA1535, TA1537, TA98, TA100 and E. coli Wp2uvrA; up to 5000 ug/plate; +/- S9Study is valid: Yes
In Vitro Chromosomal Aberration Assay in Cultured Human Peripheral Blood LymphocytesStudy title/ number: Chromosomal Aberration in Cultured Human PeripheralBlood Lymphocytes / Study No. 7736-132
Reference ID: 4297867
(b) (4)
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
50Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Key Study Findings: 127I-iobenguane at concentrations of up to 900 g/mL did not show any evidence of
genotoxic activity in the in vitro mammalian chromosome aberration test in the absence or presence of S9 mix.
The positive controls caused substantial increases in the proportion of aberrantmetaphases in each phase of the study, confirming the sensitivity and validity of the testsystem.
GLP compliance: YesTest system: human peripheral blood lymphocytes; up to 900 g/mL; +/-S9 Study is valid: Yes
In Vivo-Mouse Micronucleus AssayStudy title/ number: In Vivo Mouse Bone Marrow Micronucleus Assay / Study No. 7736-131.Key Study Findings:
Oral administration of 127I-iobenguane at ≤ 9 mg/kg (2.25, 4.5, and 9 mg/kg did not show evidence of dose-dependent chromosomal damage (no micro nucleated polychromatic erythrocytes (PCE), no decreases in the PCE: NCE (normochromatic erythrocytes) ratios)
GLP compliance: YesTest system: mouse, bone marrow micronuclei; one IV doses of 2.25, 4.5, and 9 mg/kgStudy is valid: Yes
Other Genetic Toxicity Studies: None
5.5.3 Carcinogenicity
None submitted or required for the proposed indication.
5.5.4 Reproductive and Developmental Toxicology
None submitted or required as 131I MIBG is a radioactive product with an understood risk to development.
5.5.5 Other Toxicology Studies
Study 7736-129- 12-day intravenous injection T-cell dependent antibody response (TDAR) assay using sheep red blood cells with 127-Iobenguane in ratsCrl:CD (SD) rats were assigned to six groups with 10 animals/sex in control groups (saline-Group 1, vehicle-Group 2, 25 mg/kg cyclophosphamide-Group 6), and 12 animals/sex in 127I
Reference ID: 4297867
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
51Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Iobenguane treatment groups at dose levels of 0.65, 1.9, or 4.5 mg/kg/day (Group 3, 4, and 5). 127I MIBG was administered IV at a dose volume of 0.5 mL/kg followed by an approximate 0.5 mL saline flush via IV injection. All animals were immunized with ~2x108 sheep red blood cells (SRBC)/animal by IV injection at a dose volume of 0.5 mL/animal four days prior to respective scheduled sacrifice. Assessment of TDAR response and toxicity was based on antibody-forming cells (AFCs) in response to SRBC in the agarose plaque forming assay.
Negative and positive controls resulted in expected findings, with splenocytes from cyclophosphamide-treated animals showing a clear decrease in AFCs. There were deaths on Day 1 of the dosing phase in two females at the 127I-MIBG dose of 4.5 mg/kg/day 4 hours after toxicokinetic blood collection; these animals were replaced. One female at the 127I-MIBG dose level of 0.65 mg/kg/day died after toxicokinetic blood collection on Day 7 of the dosing phase, with no gross anatomy findings or clinical signs noted. All three deaths were attributed to blood collection methods. There were no macroscopic findings or organ weight changes in test-article administered animals and no test-article related effects on immune function as determined by the TDAR assay at following administration of 127I IMBG at doses of up to 4.5 mg/kg/day for 12 consecutive days.
Study 8225771- 12-day intravenous injection T cell dependent antibody response (TDAR) assay using sheep red blood cells with 127-Iobenguane in rats Study 8225771 recapitulated the study-design of Study 7736-129, but resulted in no TK-animal deaths. There were no test-article related defects in immune function as determined by the TDAR assay at doses up to 4.5 mg/kg/day of 127I IMBG.
X X
Primary Reviewer Team Leader
6 Clinical Pharmacology
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
52Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
6.1 Executive Summary
The Applicant is seeking the approval of AZEDRA (131I-iobenguane, 131I-MIBG) based on evidence from the multicenter, single-arm, activity-estimating Study MIP-IB12B which demonstrated a clinically significant proportion of patients (what proportion?) who experienced a 50% or greater reduction of all antihypertensive medication for at least six months. The proposed therapeutic dose of 131I-MIBG in the current submission is 500 mCi (or 8 mCi/kg for patients weighing <62.5kg), given approximately 90 days apart. Prior to the first therapeutic dose, a dosimetric dose (5 to 6 mCi or 0.1 mCi/kg for patients weighing < 50 kg) of 131I-MIBG is to be administered and the results from this dosimetry step are to be used to evaluate tumor avidity and to allow for individual dose adjustment, if needed, to protect from radiation overexposure to critical organs.
This clinical pharmacology review evaluated the acceptability of the proposed dosage regimen, as well as the need for dose adjustments for drug-drug interactions and organ impairment.
6.2 Recommendations
The proposed dosing regimen for 131I-iobenguane treatment is adequately justified. It is based results from Study MIP-IB12B, showing a clinically significant proportion of patients experienced a 50% or greater reduction of all antihypertensive medication for at least six months for effectiveness and an acceptable safety profile. The dosing regimen is further supported by results from the dose finding Study MIP-IB12. From a Clinical Pharmacology standpoint, the NDA is approvable provided the Applicant and the FDA reach an agreement regarding the labeling language.
Review Issues Recommendations and CommentsSupportive evidence of effectiveness
A phase 2 single-arm study provided the primary evidence of efficacy in a total 68 evaluable patients with PPGL. Dosimetry was used to decide the therapeutic doses in patients weighing > 62.5 kg and those weighing > 62.5 kg as per the Medical Internal Radiation Dose (MIRD) schema utilizing the Adult Male or Female anthropomorphic phantom of the OLINDA/EXM software.
General dosing instructions
The dosimetric dose (185 to 222 MBq for patients weighing > 50 kg and 3.7 MBq/kg for patients weighing less than or equal to 50 kg) is to be administered as an intravenous injection in approximately 6 mL over 60 seconds. Following 131I-MIBG dosimetric injection, three whole body imaging scans are to be performed within one hour of injection (Day 0; Scan 1), on Day 1-2 post-injection (Scan 2) and on Day 2-5 post-injection (Scan 3) to assess biodistribution over time. The radiation dose to normal organs and tissues per mCi of administered dose is to be calculated using data extracted from these three scans in accordance with the Medical Internal Radiation Dose (MIRD) schema utilizing the Adult Male or Female anthropomorphic phantom of the OLINDA/EXM® software. The
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
53Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
therapeutic dose is to be adjusted proportionally if the radiation dose estimates calculated from dosimetry data for a specific patient exceeds the maximum allowable radiation dose per Emami limits for kidney, liver and lung (less than or equal to 23 Gy; less than or equal to 30 Gy; and less than or equal to 17.5 Gy, respectively).
Dosing in patient subgroups (intrinsic and extrinsic factors)
No dosing adjustment is needed in patients with mild and moderate renal impairment or with concomitant medications as 131I-MIBG dose is to be administered to each individual patient based on dosimetric calculations. Patients with severe renal impairment have not been studied. Formal renal impairment or DDI studies have not been conducted as agreed upon with the Agency during the Type B meeting dated 1/22/2016.
Labeling The review team provided extensive modifications to all sections of the proposed labeling as the content and format provided by the Applicant were inconsistent with current labeling practices. Significant additions to the label by the FDA included radiation risk assessment and QT prolongation information.
Bridge between the to-be marketed and clinical trial formulations
The to-be-marketed formulation was administered in Study MIP-IB12B that demonstrated effectiveness and safety of 1131I-iobenguane for the proposed indication.
6.3 Postmarketing Requirements and Commitments
No PMRs were requested by OCP.
6.4 Summary of Clinical Pharmacology Assessment
The Clinical Pharmacology Section of the NDA includes single-dose pharmacokinetic (PK) studies and biodistribution studies of 131I-iobenguane based on dosimetry scans from multiple images of the whole body, kidney, heart, liver and bone marrow, QT/QTc prolongation study, in vitro protein binding and in vitro CYP450 and transporters inhiation/induction studies.
6.5 Pharmacology and Clinical Pharmacokinetics
The PK parameters were generated by noncompartmental analyses with intensive PK sampling in Study MIP-IB11 and measured by total radioactivity in blood assuming this represents the parent compound only in 11 patients. The mean blood exposure (AUC0-inf) of 131I-iobenguane at a dosimetric dose of single 5 mCi (185 MBq) is 1.08 µCi.h/mL [coefficient of variation [10] 33 %] with the mean maximum blood concentration (Cmax) of 0.064 µCi/mL (CV 36%), which generally occurred at the end of the infusion.
Distribution: Mean±SD (%CV) steady-state volume of distribution (Vss) is 2.9±0.59 L/kg (20%). 131I-MIBG is 62% bound to human plasma proteins.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
54Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Metabolism: 131I-MIBG undergoes minimal metabolism. The injected radioactive dose is primary eliminated by the kidneys via glomerular filtration.
Elimination: Following a single IV dosimetric dose of 5 mCi (185 MBq) in 11 patients (N=7 with carcinoid tumors and N=4 with PPGL), the radioactivity declined in a biphasic fashion with a mean±SD (%CV) distribution half-life (t1/2,α) of 0.37 ± 0.22 hour (60%) and terminal half-life (t1/2,β) of 35.3 ± 14.0 (40%). The mean±SD (%CV) total CL and elimination t1/2 of 131I-MIBG are 62.5±24.1 mL/h/kg (38%) and 40.8±10.4 hours (25%) respectively. Approximately 80% of injected dose recovered in the urine over 120 hours after injection. The unchanged I-131 MIBG is the predominate radioactivity in urine (25%) over the first 6 hours after dosing. Minor metabolites detected in few patients included free iodide (1%), metaiodohippuric acid (MIHA) (0.5%) and metaiodobenzylbisguanidine (MIBBG) (0.7%).
6.6 General Dosing and Therapeutic Individualization
6.6.1 General Dosing
The proposed dosing regimen for 131I-MIBG is:
One dosimetric dose administered via intravenous (IV) bolus injection: 5 to 6 mCi (185 to 222 MBq) for patients weighing > 50 kg0.1 mCi/kg (3.7 MBq/kg) for patients weighing ≤ 50 kg
Two therapeutic Doses administered IV over 30 minutes and separated by 3 months apart:500 mCi (18.5 GBq) for patients weighing > 62.5 kg8 mCi/kg (296 MBq/kg) for patients weighing ≤ 62.5 kg
The proposed dosing regimen is supported by the clinically significant proportion of patients who experienced a 50% or greater reduction of all antihypertensive medication for at least six months and an acceptable safety profile observed in the phase 2, single-arm Study MIP-IB12B. The Applicant submitted PK data from 11 patients in Study MIP-IB11 who were administered a single 5.0-mCi (185 MBq) dosimetric dose of 131I-MIBG, and therefore, no exposure-response analysis for either efficacy or safety were performed. The MTD determined in the dose escalation Study MIP-IB12 in 21 patients support the proposed dosing regimen.
6.6.2 Therapeutic Individualization
Specific Populations
No formal studies were conducted for 131I-MIBG in patients with renal impairment. Dose adjustment for patients with mild to moderate renal impairment is adequately addressed used the proposed dosing regimens with the built-in dosimetric steps. In Study MIP-IB12B, dosimetry data were analyzed to estimate the radioactivity exposure in patients with normal and mild-to-moderate renal impairment. Patients were stratified into two groups by their creatinine clearance (CrCl) as calculated using Cockcroft-Gault equation (patients with normal renal function with CrCl > 90 mL/min, n=31 and patients with mild-to-moderate renal
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
55Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
impairment with CrCL=30-90 mL/min, n=43). The results demonstrated that only 8 out of 43 patients (18%) with mild-to-moderate renal impairment required reduction to their therapeutic dosage. The rest of the patients with mild-to-moderate renal impairment (n = 35) received administered the full therapeutic dosage. Thus, the dosimetry step allows for an individualized dose adjustment and serves as a safety check point for patients with mild-to- moderate renal impairment.
Patients with severe renal impairment (CrCl < 30 mL/min) were not enrolled in any of the clinical trials conducted by the Applicant. Based on the Information Request dated 1/31/2018, the Applicant performed a post-hoc analysis to simulate and predict the exposure of 131I-MIBG in patients with severe renal impairment using dosimetry data from patients in Study MIP-IB11. The dosimetry results from study MIP-1B11 showed that 27% (3 of 11) of patients were indicated to receive a therapeutic dose (mCi) reduction. Under the assumption of zero renal clearance, the simulated absorbed radioactive doses to the kidneys, liver, and lungs increased by 42%, 41%, and 53%, respectively, and thus, 100% (11 of 11) of patients would be predicted to have a therapeutic dose reduction. In addition, the efficacy data from Studies MIP-IB12 and MIP-IB12B were used to determine the best overall confirmed response of progressive disease (PD), stable disease (SD), partial response (PR), or complete response (CR). In Study MIP-IB12, patients received a single 131I-MIBG therapeutic dose ranging from 325 mCi to 696 mCi and the lowest administered therapeutic doses associated with radiologic responses of PR and SD were 571 mCi and 325mCi, respectively. In Study MIP-IB12B, 10 patients, who received one 131I-MIBG therapeutic dose that ranged from 380 mCi to 539 mCi, achieved SD responses.
DDI Potential
No formal in vivo DDI studies were conducted for 131I-MIBG. Dose adjustment for concomitant medications is adequately addressed used the proposed dosing regimens with the built-in dosimetric steps. The potential for a DDI between 131I-MIBG and other concomitant mediations is not expected as 131I-MIBG undergoes minimal metabolism and eliminated primary by the kidneys. In addition, the results from in vitro studies conducted by the Applicant indicate that 131I-MIBG does not inhibit or induce any major CYP isozymes at clinically relevant concentrations, and it is not a substrate or an inhibitor of P-glycoprotein (P-gp). Thus, the potential for PK-based interactions is unlikely in vivo. Per agreement with the Agency at the Type B meeting on 1/22/2016, clinical DDI studies with 131I-MIBG are not necessary. However, a potential for pharmacodynamic DDIs may exist between 131I-MIBG (a substrate of norepinephrine transporter [NET]) and drugs that inhibit NET (e.g., amitryptiline, imipramine, antidepressants), drugs that deplete norepinephrine stores (e.g., antihypertensives) and drugs that inhibit reuptake at NET sites (e.g., sympathomimetric amines and cocaine). This risk is adequately addressed in the Applicant’s proposed labeling under the Drug Interactions section.
6.6.3 Outstanding Issues
There are no outstanding issues identified from a Clinical Pharmacology perspective.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
56Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
6.6.4 Summary of Labeling Recommendations
Specific Populations: 131I-MIBG is cleared by glomerular filtration. The radiation dose to patients with severe renal impairment may be increased due to the delayed elimination of the drug. Individualized therapeutic dose adjustment based on radiation exposure estimates from the dosimetry step is recommended for patients with mild to moderate renal impairment (creatinine clearance 30-89 mL/min). In Study IB12B, of the 43 patients with mild-to-moderate renal impairment (CLcr 30-90 mL/min using the Cockcroft Gault calculation), 8 (18.6%) required therapeutic dose reductions based on radiation dose estimates to critical organs exceeding Emami limits (absorbed dose exceeding 23 Gy). The pharmacokinetics of iobenguane I-131 has not been studied in patients with severe renal impairment (creatinine clearance less than 30 mL/min), end stage renal disease, or hepatic impairment.
DDIs: Labetalol, a combined alpha and beta blocker, has been reported to reduce iobenguane uptake and deplete intracellular storage in patients. Labetalol is to be discontinued for at least 5 biological half-lives prior to and following 131I-MIBG administration and other appropriate alpha- or beta-blockers are to be used to control hypertension.
Severe treatment-related hematological adverse reactions including thrombocytopenia may be exacerbated in patients receiving medications that interfere with platelet function or coagulation.QT/QTc: Intense monitoring and assessments of patient ECGs were performed throughout the 131I-MIBG clinical development program, and no clinically meaningful changes in QT prolongation or arrhythmias was observed.
6.7 Comprehensive Clinical Pharmacology Review
6.7.1 General Pharmacology and Pharmacokinetic Characteristics
PHYSICOCHEMICAL PROPERTIESChemical structure and molecular formula & weight
131I- iobenguane (131I-MIBG)
Aqueous solubility Insoluble in water and all organic solventsPHARMACOLOGYMechanism of action PPGL tumors exhibit high levels of expression of norepinephrine
reuptake transporter (NET) on their cell surface. 131I-MIBG is known to be a substrate for this transporter. It is actively transported by NET and accumulates within PPGL cells. The ionizing radiation resulting from the radioactive decay of I-131 causes cell death and tumor necrosis.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
57Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Active moiety 131I-MIBG, no major metabolite(s)
QT/QTc prolongation There were no clinically meaningful changes in QTcB interval prolongation in Study MIP-IB12B. Most patients had QTcB interval values within the baseline values (≤ 480 msec) after Therapeutic Dose 1 and after Therapeutic Dose 2 within the first 24 hours after each dose. The mean (90% CI) change in QTcF is 3.7 (-1.6,9.0) ms after the dosimetric dose (N=38), 0.9 (-4.6, 6.3) ms after Therapeutic Dose 1 (N=51) and 8.3 (2.1, 14.5) ms after Therapeutic Dose 2 (N=41) within the first 24 hours after dosing. For more details, see Table 8 below.
GENERAL INFORMATIONBioanalytical assay In Study MIP-IB11, validated radiometric assay methods were used
to measure the radioactivity concentrations in whole blood, plasma and urine samples (See Appendix 19.4).
Patient PK vs. healthy subject PK
Not intended to be administered to healthy subjects
Steady-state exposure at the proposed dosing regimen
No Sparse PK samples collected in the pivotal phase 2 Study MIP-IB12B; PK parameters were determined only after single dose administration in Study MIP-IB11.
Minimal effective dose or exposure
Based on the PK simulations from Study MIP-IB11, the minimum dose required for efficacy is 325 to 380 mCi (submission dated 2/16/2018, SDN 12).
Maximum tolerated dose or exposure
Based on the dose-limiting toxicity data from the dose-ranging Study MIP-IB12, the MTD assessed in in 21 patients with PPGL is 8 mCi/kg (588 mCi) (maximum dose tested = 9 mCi/kg (680 mCi).
Dose proportionality Not determined. Only one single 5 mCi (185 MBq) dosimetric dose was administered in Study MIP-IB11.
Accumulation No accumulation is expected as 131I-MIBG is to be administered as two therapeutic doses 90 days apart (t1/2=40 hours).
Variability In Study MIP-IB11, the inter-patient variability (CV%) is 35% and 30% for Cmax and AUC0-inf, respectively, after single 5 mCi (185 MBq) dosimetric dose of 131I-MIBG.
ABSORPTIONTMAX Mean TMAX after IV infusion of the therapeutic dose is 30 minutes. The
dosimetric dose is to be given by IV bolus.DISTRIBUTIONVolume of distribution
Mean±SD (%CV) volume of distribution at steady-state (Vss) is 2.9±0.59 L/kg (20%) following a single 5 mCi (185 MBq) dosimetric dose of 131I-MIBG in 11 patients. 131I-MIBG is 62% bound to human plasma proteins. Protein binding was concentration-independent (0.05 to 5 μM).
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
58Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Substrate of CYP/ transporter systems
In vitro studies conducted by the Applicant indicate that: MIBG reversibly inhibited CYP2D6 activity with an estimated IC50
value of 3.57 μM which is 8.5 times greater than the theoretical maximum instantaneous blood level for a therapeutic dose of 131I-MIBG (0.42 μM).
MIBG is not an inducer of any CYP enzymes. MIBG is neither a substrate nor an inhibitor of P-gp.
ELIMINATIONTerminal elimination half-life and clearance
Following a single 5 mCi (185 MBq) dosimetic dose of 131I-MIBG in 11 patients, mean±SD (%CV) systemic clearance (CL) and elimination half-life (t1/2) of 131I-MIBG are 62.5±24.1 mL/h/kg (38%) and 40.8±10.4 hours (25%), respectively (Study MIP-IB11).
Metabolism 131I-MIBG undergoes minimal metabolism. The injected radioactive dose is primary eliminated by the kidneys via glomerular filtration.
Excretion Approximately 80% of injected dose recovered in the urine over 120 hours after injection (mean±SD [%CV]=79.9±9.6 [12%], n=11). The radioactivity recovered in urine over 6 hours after injection consisted predominately of unchanged 131I-MIBG (25.9 ± 8.0%, n=11). Other components were detected in urine at low levels in some patients including free 131I (1.2±0.4%, n=6), metaiodohippuric acid (MIHA), a reported metabolite of 131I-MIBG (0.5%, n=1) and
an impurity present in the dose formulation) ( n=1).
Drug interaction (DDI) liability
The potential for PK interaction in vivo is unlikely as 131I-MIBG undergoes minimal metabolism and is not a substrate, inhibitor or an inducer of any CYP enzyme at clinically relevant concentrations of 131I-MIBG. A potential for PD interaction may exist with concomitant use of drugs that inhibit the NET uptake and this will be addressed in the product’s labeling.
6.7.2 Clinical Pharmacology Questions
Does the clinical pharmacology program provide supportive evidence of effectiveness?
Evidence of effectiveness in this NDA submission was obtained from the pivotal phase 2, single-arm Study MIP-IB12B in 68 patients with PPGL as demonstrated in the sections below of this review. The PK of 131I-MIBG was determined in 11 patients (7 with carcinoid tumors and 4 with PPGL). Each patient received a single intravenous (IV) bolus dose of 131I-MIBG at a target level of 5.0 mCi (1.85 GBq) supplemented with the mass of 0.185 mg cold MIBG equivalent to the maximum projected therapeutic dose. The radioactivity in blood, plasma and urine samples collected up to 120 hours after dosing. Fecal samples were not collected in this study as only negligible amounts of injected dose (<1% of) have been excreted in the feces following IV dosing of 131I-MIBG (Mangner et al., Metabolism of Iodine-131 Metaiodobenzylguanidine in
Reference ID: 4297867
(b) (4)
(b) (4)



NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
61Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
0.1 mCi/kg (3.7 MBq/kg) for patients weighing ≤ 50 kg
Therapeutic Doses: (To be administered IV over 30 minutes)
500 mCi (18.5 GBq) for patients weighing > 62.5 kg 8 mCi/kg (296 MBq/kg) for patients weighing ≤ 62.5 kg
Administration of therapeutic doses approximately 90 days apart is recommended. Prior to the first therapeutic dose of 131I-MIBG, a dosimetry dose is administered for treatment planning. Results from this step are used to confirm tumor avidity and to allow for individual therapeutic dose adjustment, if necessary, to protect from radiation overexposure to critical organs.
The rationale for the therapeutic dose of 500 mCi (18.5 GBq) was based on the results from the phase 1 dose-escalation Study MIP-IB12 in 21 patients with PPGL. Initially, all patients received a single 5 mgCi dosimetric dose of 131I-MIBG supplemented with 0.185 mg of cold MIBG to confirm uptake of radioactivity by at least one known tumor on the two or 3 whole body scans performed 2 to 7 days after dosing. When the uptake of radioactivity by at least one tumor was confirmed, three patients then received a starting therapeutic IV dose of 6 mCi/kg. This starting dose was then escalated in cohorts of 3-6 patients to 7 mCi/kg, 8 mCi/kg and 9 mCi/kg until the MTD was established. The maximum injected dose was not to exceed that of a 75- kg patient (i.e., 450, 525, 600, or 675 mCi for the 6, 7, 8 and 9 mCi/kg dose groups, respectively).
The distribution of total administered dose (mCi/GBq) and occurrence of DLTs across all cohorts is summarized in Applicant’s Figure 11 below. The DLTs observed in this study included thrombocytopenia, febrile neutropenia, decreased platelet count and low absolute neutrophil count in 4 patients (actual administered radioactivity of 524 mCi (7 mCi/kg), 588 mCi (8 mCi/kg), 662 mCi (9 mCi/kg), and 680 mCi (9 mCi/kg) in each patient). Based on the occurrence of two DLTs (two events of low absolute neutrophil count [ANC]) at the 9 mCi/kg dose, the MTD was determined to be 8 mCi/kg (588 mCi).
Figure 11: Distribution of Total Injected Dose and Occurrence of DLTs Across Sequential Dose Cohorts
[Source: NDA 209607/SDN 2, Clinical Pharmacology Summary, PP 16]
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
62Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Based on the results from this study, a total administered dose of 500 mCi (18.5 GBq) or 8 mCi/kg (0.296 GBq/kg) if body weight was less than 62.5 kg was chosen as the therapeutic dose in the pivotal phase 2 Study MIP-IB12B.
Exposure-Response (E-R) Relationships for Efficacy, Safety and QTc
No sparse PK samples were collected during Study MIP-IB12B and thus, exposure-response relationships for efficacy and safety could not be explored.
Efficacy Analysis
The evidence of efficacy was demonstrated in a phase 2, single-arm study (MIP-IB12B) in patients with iobenguane-avid malignant unresectable PPGL. The primary endpoint was met, and the efficacy results are described in Section 7 in details of this multidisciplinary review. The efficacy results are summarized in the Applicant’s Table 8 and Table 9 below in all treated patients.
Table 8: Reduction in Use of Antihypertensive Medication Following 131I-MIBG – All treated 68 evaluable PPGL Patients
[Source: NDA 209607/SDN 2, final study report for Study MIP-IB12B, PP 110]
Table 9: Reduction in Use of Antihypertensive Medication Following 131I-MIBG by Number of Therapeutic Doses Administered
[Source: NDA 209607/SDN 2, final study report for Study MIP-IB12B, PP 111]
As seen in the tables above, the primary endpoint was met in 17 patients out of 68 evaluable patients (25% of treated patients), sixteen (32%) out of 50 patients who received two therapeutic doses and one (5.6%) of 18 patients who received only one therapeutic dose
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
63Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
showed a clinical benefit response Table 10). The reduction of antihypertensive medication was sustained with a median duration of 13.3 months (range 8 to 50 months, Table 10).
Table 10: Duration of clinical Benefit in patients who Achieved the Primary Endpoint
[Source: NDA 209607/SDN 2, final study report for Study MIP-IB12B, PP 112]
Safety Analysis
The Applicant’s table (Table 11) below summarizes the most common treatment-emergent adverse events (TEAEs) which occurred in ≥10% of patients in Study MIP-IB12B. The most commonly reported TEAEs included nausea (71.6%), thrombocytopenia (66%), anemia (58%), leukopenia (55%), fatigue (55%), neutropenia (53%), vomiting (48%), dry mouth (38%), dizziness (38%) and headache (28%).
Table 11: Incidence of TEAEs occurring in ≥10% of 74 evaluable patients
[Source: NDA 209607/SDN 2, final study report for Study MIP-IB12B, PP 140]
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
64Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
QT/QTc Prolongation
As per the FDA’s IRT-QT review, the results from Study IB12B indicate no large mean increases in the ΔQTc interval (i.e., 20 ms) with the proposed therapeutic dosing regimen of 131I-MIBG, based on central tendency analysis (largest 90% CI upper bound was 14.5 ms at 1 h post-dose after administration of second therapeutic dose). None of the patients had QTcF >500 ms and only one patients had ΔQTcF >60 ms. There was no placebo or positive control in the study. In this open-label, multicenter, single-arm, Phase 2 study, 74 patients received dosimetric doses (3-6 mCi by IV injection) and 68 patients received at least one therapeutic dose of AZE131I-MIBG DRA (500 mCi for patients > 62.5 kg and 8 mCi/kg for patients ≤ 62.5 kg; 2 therapeutic doses 3 months apart). Overall summary of findings is presented in Table 12 below.
Table 12: The Point Estimates and the 90% CIs of ΔQTcF Corresponding to the Largest Upper Bounds for All-Treated 131I-MIBG Group (FDA Analysis for Study MIP-IB12B)
[Source: NDA 209607/SDN 2 – FDA’s IRT-QTc Review, PP 2]
Is an alternative dosing regimen or management strategy required for subpopulations based on intrinsic patient factors?
Dose adjustment for patients with mild to moderate renal impairment is adequately addressed used the proposed dosing regimen with the built-in dosimetric step.
The current proposed dosing regimen allows for individualized dose adjustment for patients with renal impairment based on the dosimetric step. Therefore, no formal PK studies were needed to assess the influence of organ impairment on 131I-MIBG exposure, as agreed upon with the Agency during the Type B meeting dated 1/22/2016.
In Study MIP-IB12B, the dosimetry data were analyzed to estimate the radioactivity exposure in patients with renal impairment stratified into two groups by their creatinine clearance (CrCl) calculated using Cockcroft-Gault equation (patients with normal renal function with CrCl > 90 mL/min, n=31 and patients with mild-to-moderate renal impairment with CrCL=30-90 mL/min, n=43). The results showed that a total of 12 patients required a dose reduction based on radiation dose estimates of the critical organs that exceeded the theoretical limits published by Emami (See Applicant’s Table 13 and Reviewer’s Table 14). Only 8 out of 43 patients (18%) with mild-to-moderate renal impairment required a reduction of the therapeutic dosage. The rest of the patients (n=35) could be administered the full therapeutic dosage based on the estimated radioactivity exposure. Thus, the dosimetry step allows for an individualized dose adjustment in
Reference ID: 4297867


NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
66Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Patients with severe renal impairment (CrCl < 30 mL/min) have not been enrolled in any of the clinical trials conducted by the Applicant. Information Request was sent on 1/31/2018 requesting the Applicant to perform some analyses to predict the exposure of 131I-MIBG in patients with severe renal impairment, to determine the minimum dose required for efficacy based on experience in PPGL and to assess whether the predicted dose in patients with severe renal impairment would lead to exposures that fall below this threshold. A post-hoc analysis was performed to simulate severe renal impairment using dosimetry data from patients in Study MIP-IB11 in 11 patients. Simulations were performed to generate absorbed radiation dose estimates and maximum absorbed radiation dose (Gy) to the critical organs (kidneys, liver, and lungs) per Emami limit in the worst-case scenario of extreme renal impairment, i.e. no renal clearance. The dosimetry results showed that 27% (3 of 11) of patients were indicated to receive a therapeutic dose (mCi) reduction. Under the assumption of zero renal clearance, the simulated absorbed radioactive doses to the kidneys, liver, and lungs increased by 42%, 41%, and 53%, respectively, and thus, 100% (11 of 11) of patients would be predicted to have therapeutic dose reduction. In cases of severe renal impairment, the absorbed radioactive doses to the critical organs will increase and therapeutic dose reductions will be required. Based on simulation results of no renal function using the dosimetry dataset from Study MIP-IB11, the projected maximum therapeutic dose ranged from 493 mCi to 993 mCi (Applicant’s Table 15). In addition, the efficacy data from Studies MIP-IB12 and MIP-IB12B were used to determine the best overall confirmed response of progressive disease (PD), stable disease (SD), partial response (PR), or complete response (CR). In Study MIP-IB12, patients received a single 131I-MIBG therapeutic dose ranging from 325 mCi to 696 mCi (Applicant’s Table 16), and the lowest administered therapeutic doses associated with radiologic responses of PR and SD were 571 mCi and 325 mCi, respectively. In Study MIP-IB12B, 10 patients, who received one 131I-MIBG therapeutic dose that ranged from 380 mCi to 539 mCi, achieved SD responses (Applicant’s Table 16). All responders with confirmed radiographic responses of PR received two therapeutic doses of 131I-MIBG at levels ranging from 891 mCi to 1010 mCi.
Reference ID: 4297867



NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
69Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
MIBG is not an inducer of any CYP enzyme at clinically relevant concentrations:
The induction potential of MIBG on the activities of CYP 1A, 2B6, 2C9, 2C19 and 3A was evaluated in human hepatocytes at concentrations of 0.5, 1.5, or 5 μM. Exposure of the MIBG, at any in vitro concentration tested, did not elicit substantial increases in CYP1A, CYP2B6, CYP2C9, CYP2C19, or CYP3A enzyme activities. In addition, the increases in these enzyme activities were <40% compared to the induction by probe inducers. Thus, in vivo drug-drug interactions resulting from the induction of CYP 1A, 2B6, 2C9, 2C19, or 3A enzyme activities are unlikely (See Applicant’s Table 18 below).
Table 18: Effect of MIBG on CYP Enzyme Activities in Human Hepatocytes
MIBG is neither a substrate nor an inhibitor of P-glycoprotein (P-gp):
The potential interactions between MIBG with P-gp was evaluated in Caco-2 cell model at concentrations of 1, 3, and 10 μM. Inhibition of P-gp mediated efflux of vinblastine was evaluated at MIBG concentrations of 0.1, 1, 3, 10, and 30 μM. MIBG was found to have low mean apparent permeability in Caco-2 cell monolayers that was independent of the direction of transport evaluated (≤2.89 x 10-6 cm/s). In addition, the apparent permeability of vinblastine in the presence of various MIBG concentrations ranged from 14.2 to 23.1 indicating lack of inhibition of P-gp by MIBG. Thus, MIBG was determined to be a low permeability compound that was neither a P-gp substrate nor a P-gp inhibitor (see Applicant’s Table 19 below).
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
70Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Table 19: Apparent Permeability and Efflux Ratios from Caco-2 Cells
No formal clinical DDI studies were conducted for 131I-MIBG as agreed during the Type B meeting dated 1/22/2016. Drugs that are combined alpha and beta adrenergic receptors blockers (labetalol) or drugs that deplete norepinephrine (reserpine) may interfere with the efficacy of MIBG. In an article by Jacobson and Travin, it was reported that labetalol has consistently been shown to significantly interfere with MIBG uptake due to its combined significant beta- and alpha-receptor antagonistic activity (Arnold F. Jacobson, AF and Travin, MI: Impact of medications on MIBG uptake, with specific attention to the heart: Comprehensive review of the literature. J Nucl Cardiol 2015; 22:980 – 993). Evidence also indicates that reserpine interferes with MIBG uptake due to NE depletion through irreversible inactivation of vesicular monoamine transporter, thereby preventing norepinephrine storage in vesicles and increasing the rate of enzymatic degradation and diffusion from nerve terminals. As per the authors, there is significant in vitro and in vivo evidence for inhibition of MIBG uptake for only labetalol and reserpine. Based on these data, it is recommended that labetalol and reserpine should be discontinued prior to and following 131I-MIBG administration. The results of the present review are summarized in Authors’ table below. Accordingly, the Applicant revised the package insert originally submitted on 1/12/2018 to include this information and resubmitted the package insert on 2/9/2018.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
71Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Question on clinically relevant specifications
None
X X
Primary Reviewer Team Leader
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
72Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
7 Sources of Clinical Data and Review Strategy
7.1 Table of Clinical Studies
Table 20: Clinical Studies Included in NDA 209607
Trial ID
Trial Design Regimen/ schedule/ route
Study Endpoints Treatment Duration/ Follow Up
No. of patients enrolled
Study Population No. of Centers
and Countries
Controlled Studies to Support Efficacy and SafetyMIP-IB12B
Single arm, open label
Primary study supporting efficacy
Dosimetric dose: 5.0 mCi 131I-MIBG with 185 mcg cold MIBG
Therapeutic dose: ~500 mCi 131I-MIBG IV approximately 90 days apart
Primary: decrease in antihypertensive medications by 50% for 6 months
Two therapeutic doses ~90 days apart/ 5 years (ongoing)
N=68 received at least one therapeutic dose
Patients with malignant and/or recurrent PPGL
10 US sites
MIP-IB 12
Single arm, open label, dose-escalation
Dosimetric dose: 5.0 mCi 131I-MIBG with 185 mcg cold MIBGTherapeutic dose: planned as 6, 7, 8 or 9 mCi/kg 131I-MIBG IV, 7-28 days following dosimetric dose
Some patients not dosed
Safety, maximum tolerated dose, dosimetry, efficacy, and quality of life
Single therapeutic dose / 1 year following completion of 12-month efficacy phase
N=21 received therapeutic dose
Patients with PPGL
3 US sites
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
73Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
as planned, so grouped for analysis as ≤500 mCi or >500 mCi 131I-MIBG
Studies to Support SafetyMIP-IB11
Single arm, open label
5.0 mCi 131I-MIBG with 185 mcg cold MIBG
Safety, dosimetry and PK of single imaging dose of AZEDRA
Single imaging dose / 2 weeks
N=11 Patients with PPGL and metastatic carcinoid tumor
1 US site
MIP-IB13
Single arm, open label
Study conducted in conjunction with NANT* consortium
Dosimetric dose: 0.1 mCi/kg 131I-MIBG (maximum 5.0 mCi) with 185 mcg cold MIBGTherapeutic dose: 11.2, 15.5, 18.2 mCi/kg IV, 7-28 days following dosimetric dose
Safety, maximum tolerated dose, dosimetry, efficacy, and quality of life
Single therapeutic dose/ 60 days
N=15 Patient with relapsed/refractory high risk neuroblastoma
5 US sites enrolled patients (9 sites initiated study)
* NANT: New Approaches to Neuroblastoma Therapy
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
74Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
7.2 Review Strategy
The FDA statistical and clinical BLA review team consisted of one primary statistical reviewer of efficacy and one primary clinical reviewer of safety and efficacy. The NDA submission contained data from four studies, listed in Table 20, and was primarily supported by one single-arm multicenter trial entitled “A Phase II Study Evaluating the Efficacy and Safety of 131I-MIBG in Patients with Malignant Relapsed/Refractory Pheochromocytoma/Paraganglioma,” Study MIP-IB12B. The clinical and statistical review of efficacy focused on Study MIP-IB12B.
The statistical and clinical review of safety and effectiveness included the following:• Review of the current literature on PPGL natural history and treatment and the
Applicant’s orientation materials• Review of Study MIP-IB12B, including the clinical study report (CSR), protocol, protocol
amendments, SAP and SAP amendments• Review of Study MIP-IB12, including the CSR, protocol, and protocol amendments• Review and assessment of Applicant analyses of 131I-MIBG safety and efficacy in the
clinical study reports and the ISS• Review of datasets submitted as SAS transport files• Review of patient narratives of SAEs, deaths, and adverse events of special interest
(AESI)• Review of minutes of key meetings conducted during the 131I-MIBG development
program for PPGL• Review and assessment of the Module 2 summaries including the Summary of Clinical
Efficacy and the Summary of Clinical Safety • Review of the 90-day safety update and proposed labeling modifications• Review of consultation reports from the Interdisciplinary Review Team for QT Studies
(QT IRT), Office of Scientific Investigation (OSI), Office of Prescription Drug Promotion (OPDP) and Patient Labeling Team (PLT)
• Review of Applicant responses to multiple FDA requests for additional analyses and clarifications throughout the review
• Formulation of the benefit-risk analysis and recommendations• Review, evaluation, and revision of proposed labeling
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
75Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
8 Statistical and Clinical and Evaluation
8.1 Review of Relevant Individual Trials Used to Support Efficacy
8.1.1 MIP-IB12B: A Phase II Study Evaluating the Efficacy and Safety of 131I-MIBG in Patients with Malignant Relapsed/Refractory Pheochromocytoma/Paraganglioma
Trial Design
Study MIP-IB12B is the primary study used to demonstrate efficacy. MIP-IB12B is a multicenter, open-label, single arm study conducted in patients 12 years and older with a diagnosis of unresectable pheochromocytoma or paraganglioma. The primary objective was to determine the proportion of patients with a reduction (including discontinuation) of all antihypertensive medication by at least 50% for at least six months, from two therapeutic doses each at 500 mCi (or 8 mCi/kg, for patients weighing 62.5 kg or less) of Ultratrace iobenguane I 131 administered approximately three months apart. The secondary objectives included 1) assessing the proportion of patients with overall tumor response of CR or PR per RECIST criteria, 2) assessing the proportion of patients with overall tumor response of CR, PR or MR (moderate response, i.e., decrease in the sum of the longest diameters of the target lesions of 15-30%, with no evidence of PD in non-target lesions) per RECIST criteria, and 3) assessing overall survival, up to 5 years post-treatment.
The study consists of a screening and dosimetry phase, a 12-month efficacy phase, and a four-year long-term follow-up phase. Approximately 75 patients were expected to enroll to ensure that 58 patients were evaluable for safety and efficacy. Eligible patients receive two doses of 131I-MIBG (500 mCi, or 8mCi/kg for patients <62.5 kg, per dose), three months apart, administered after an imaging dose and dosimetry.
Patients enrolled on this study were required to be ineligible for curative surgery for PPGL, and had either failed a prior therapy for PPGL, or were not candidates for chemotherapy. Patients were required to be on a stable antihypertensive medication regimen for tumor-related hypertension for at least 30 days prior to the first therapeutic dose, and had at least one tumor site by computed tomography (CT), magnetic resonance imaging (MRI), or MIBG scan. Patients’ tumors were required to have definitive MIBG avidity and an expected survival of at least 6 months, as predicted by the physician. Exclusion criteria included previous systemic radiotherapy resulting in marrow toxicity within three months of study entry, receipt of prior whole-body irradiation, or external beam radiotherapy to >25% of bone marrow. In addition, patients receiving a medication which inhibited tumor uptake of iobenguane I 131 were excluded.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
76Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Potassium iodide was administered from 24 hours prior to 131I-MIBG and continued for 10 days. A dosimetric dose of 5 to 6 mCi (or 0.1 mCi/kg for patients weighing <50 kg) was administered, followed by three Iobenguane I131 scans of the entire body to confirm uptake of the agent, assess biodistribution, and determine dosimetry. The dosimetry scans were reviewed to ensure that the planned therapeutic doses would not deliver greater than 23 Gray absorbed dose to the kidney, 17.5 Gy to the lungs, or 30 Gy to the liver. Following therapeutic dose administration, patients had a complete body scan within 7 days to assess biodistribution.
Hematologic recovery was required prior to administration of the second dose. The second dose could be delayed from 12 to up to 24 weeks after the first therapeutic dose. In the event of severe hematologic toxicity (febrile neutropenia, Common Terminology Criteria for Adverse Events [CTCAE] Grade 3 thrombocytopenia with active bleeding or CTCAE Grade 4 hematologic toxicity of lasting more than 1 week), the protocol required a dose reduction to 425 mCi or 7mCi/kg for patients ≤62.5 kg.
Patients were followed twice weekly during the first 6 weeks after each dose, and once a week for remaining weeks through week 24; they were then followed monthly (months 7 – 12) and every 6 months during long term follow-up. Tumor response was evaluated with CT or MRI scans obtained every 3 months for 1 year. Overall tumor response at 3, 6, 9 and 12 monthsper RECIST criteria was assessed centrally by independent, blinded readers. Tumor markers (serum chromogranin A, plasma-free metanephrines, plasma catecholamines, 24-hour urinary vanillylmandelic acid and catecholamines, and urinary metanephrines) were measured every 2 weeks for 24 weeks, and every month during months 7 to 12.
Safety was evaluated by analysis of treatment-emergent adverse events (TEAEs), serious adverse events (SAEs), adverse events of special interest (AESIs), baseline and pre- versus post-administration electrocardiograms (ECGs), physical examinations, vital signs, laboratory assessments, and human radiation absorbed dose estimates to target lesions and normal organs. Following the 12-month assessment, patients entered a 4-year long term follow-up (LTFU) phase to assess OS, tumor markers, and use of antihypertensive medication and concomitant medications, until the patient experienced disease progression, started another anticancer therapy, or died.
Study Endpoints
As agreed upon in the SPA, the primary endpoint for this study is the proportion of patients who received at least one therapeutic dose with a reduction (including discontinuation) of all antihypertensive medications by at least 50% for at least six months, beginning during the 12-month efficacy phase. Secondary endpoints are as follows:
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
77Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Secondary endpoints (efficacy): Proportion of patients with overall tumor response of complete response (CR) or partial
response (PR) per Response Evaluation Criteria in Solid Tumors (RECIST) v 1.0
Proportion of patients with overall tumor response of CR, PR or moderate response (MR) per modified RECIST
Bone lesion response per the Soloway Scale
Tumor marker response in 24-hour urine and other serum/plasma tumor markers associated with PPGL
Patient Quality of Life (QoL) per the recommended guidelines from the European Organization for Research and Treatment of Cancer (EORTC) QLQ-C30 manual
Symptoms as evaluated through the National Institutes of Health (NIH) Quality of Life and Symptoms Questionnaire for pheochromocytoma and paraganglioma
Change in use of analgesics and pain medications
Karnofsky Performance Status post-treatment
Overall survival, defined as the time from the date of first therapeutic dose to the date of death from any cause (censored at the last date the patient was known to be alive when confirmation was absent or unknown)
Secondary endpoints (safety and tolerability): Incidences of treatment-emergent adverse events (TEAEs), serious adverse events
(SAEs), adverse events of special interest (AESIs), deaths, and discontinuations due to AEs, status of hypertension, changes in blood pressure (BP), and changes in the use of analgesics and pain medications
Clinical laboratory measurements (including hematology, serum chemistry, and urinalysis), physical examinations, vital signs, ECG measurements, and concomitant medications
Human radiation absorbed dose estimates to target lesions and normal organs were also assessed in this study
Statistical Analysis Plan
There was no formal hypothesis testing planned in Study MIP-IB12B; therefore, no statistical inference is drawn based on data from the study. Per the statistical analysis plan
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
78Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
(SAP), the duration of benefit (duration with a reduction, including discontinuation, of all antihypertensive medications by at least 50% for at least six months) in the definition of the primary endpoint means the maximum of all durations of consecutive months of reduction should be at least six months. The 50% reduction in the definition of the primary endpoint is determined separately for each baseline medication, based on the total daily dose of the antihypertensive medication(s) on the day of the first therapeutic dose. To meet the primary endpoint, a patient was required to meet the following criteria:
Receive an Imaging Dose
Receive at least one Therapeutic Dose
Have a reduction of each pre-therapeutic dosing (baseline) antihypertensive medication ≥50% for a minimum of six consecutive months beginning during the 12-Month Efficacy Phase of the study, during which no new, long-term antihypertensive medication is introduced and maintained for longer than 14 days.
The duration of the primary endpoint response began on the date during the 12-MonthEfficacy Phase when all baseline antihypertensive medications reduced by at least 50% and any new medications are discontinued (the date post- therapeutic dosing when the reduction of the last antihypertensive medication reduced by at least 50% or the new medication is discontinued). The end date for which the duration of the ≥50% reduction of all antihypertensive medications was determined as the earlier of:
a) the date that the patients no longer meets the criteria of having a ≥50% reduction of allbaseline antihypertensive medications (i.e., the starting date of a new long-term continuing antihypertensive medication or a dose change in an existing regimen of antihypertensive medication that exceeds the ≥50% reduction of the baseline dose that is greater than 14 days in duration and continues without a reduction to the ≥50% level)
OR
b) the date of the patient’s last applicable recorded study-related disposition date. If the patient had discontinued the study prior to the end of the 12-Month Efficacy Phase due to any of the following reasons:
• Withdrew consent
• Was lost to follow-up
• Death
• Withdrew due to progressive disease
• Received other anticancer therapy
• Experienced an adverse event resulting in study discontinuation
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
79Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
The full analysis set (FAS) was defined as patients who received at least one therapeutic dose of 131I-MIBG. The Per Protocol Set (PPS) is a subset of the Full Analysis Set, excluding patients who received only one therapeutic dose, who did not attend the 3 Month and 6 Month Efficacy Visits, or with major protocol violations. The Dosimetry Set and Safety Set include all patients who received the dosimetric dose of 131I-MIBG. Per the SAP, the primary analysis of the primary endpoint was to calculate a point estimate with a two-sided 95% confidence interval (CI) using the Agresti-Coull method. Assuming the proportion of patients experiencing a reduction (including discontinuation) of all antihypertensive medications by at least 50% for at least six months or two cycles was 0.25, a sample size of 58 would provide 90% power to exclude 10% at a one-sided significance level of 0.025. The trial was considered a success if the lower bound of the CI from the FAS analysis exceeded 0.10.
Tumor responses were required to be confirmed by repeat assessment at least 4 weeks after first meeting response criteria. Overall tumor response at 3, 6, 9 and 12 months per RECIST criteria were assessed centrally by independent, blinded readers. A frequency distribution table would be provided showing, by visit, target, non-target and overall response.
Adverse events were coded according to System Organ Class (SOC) and Preferred Term by the Medical Dictionary for Regulatory Activities (MedDRA) Version 19.1, and were graded according to the CTCAE v. 3.0. Treatment-emergent adverse events were defined as AEs with onset date on or after the first administration of any amount of study drug to the end of 15 weeks after the administration of the last therapeutic dose of 131I-MIBG. Incidences of TEAEs were summarized by maximum relationship to study drug and by maximum severity. Incidences of SAEs, hematologic AEs, and AESIs were also presented. In addition, all deaths and TEAEs resulting in study drug discontinuation were presented. Shift tables were prepared for laboratory parameters that had CTCAE v. 3 criteria. Incidences of potentially clinically significant abnormalities in ECG parameters were also provided.
Quality of life (QoL) was evaluated by the EORTC QLQ-C30 v.3. The data were summarized as follows: Global Health Status/Quality of Life, Functional Scales, and Symptom scales. The results of QoL and changes from baseline were summarized by visit and domain across the Full Analysis Set. A symptom response instrument was used to evaluate symptoms specific to NIH Quality of Life and Symptoms Questionnaire for Pheochromocytoma and Paraganglioma as a secondary endpoint. Changes in symptoms over time were evaluated by patients and across the FAS in aggregate. Changes in total doses of single medications to control tumor pain, and changes in pain medications, were evaluated by patient and across the FAS. The Kaplan-Meier method was used to analyze overall survival (OS).
Statistical Reviewer’s Comments: OS was one of the secondary endpoints in Study MIP-IB12B. Because the analysis of a time
to event endpoint is not interpretable in a single arm study, the OS result will not be evaluated in this review.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
80Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Besides evaluating the overall tumor response, duration of response should be evaluated as a measure of the antitumor activity. However, no plan for evaluation of duration of tumor response was provided in the protocol or the SAP.
QoL data were analyzed by the clinical and statistical reviewers; however, these analyses were considered exploratory in the context of a single-arm trial.
Missing, Unused, and Spurious DataThe SAP stated that no imputation of values for missing data would be performed in any primary analysis. Patients who received the dosimetric dose or a therapeutic dose but had no follow-up for safety were not included when computing percentages of patients experiencing adverse events or laboratory toxicities. Data that are potentially spurious or erroneous were examined using standard data management operating procedures, prior to database lock and statistical analysis.
Incomplete medication start and end dates were handled as follows: a missing start month wasimputed as June, and a missing end month was imputed as December. Missing start and end dates were imputed as the last day of the month. If only the year was provided, the start date was imputed as June 30, and the end date was imputed as December 31. If the assignment of amissing start or end date conflicted with the available reported data, the assigned dates weremodified. For antihypertensive medications, if the start year could not be determined, the medication was considered a baseline medication unless the end year was present and prior to the first therapeutic dose of 131I-MIBG.
Protocol Amendments
The protocol was issued on January 14, 2009 and was amended three times (Amendment 1: March 31, 2009; Amendment 2: January 19, 2010; Amendment 3: March 21, 2014). The first enrolled patient was consented (June 4, 2009) under Amendment 1, March 31, 2009. There were no changes to the inclusion/exclusion criteria between Amendment 2 and Amendment 3. Notable changes to the protocol made by Amendment 2 that affected data collection included the following:
Changes were made to indicate that if a bone scan performed at screening/baseline indicated if metastatic disease is observed, additional bone scans would be performed at Months 3, 6, 9, and 12.
Amendment 2 specified that AEs (including post radiation toxicity reported as AESIs) would be collected specifically after the Safety Phone Call at 16 weeks after the last therapeutic dose of 131I-MIBG.
Revisions to select eligibility criteria:
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
81Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
o The inclusion criteria were revised to indicate that in addition to having failed a prior therapy for PPGL, potential study patients could not be candidates for chemotherapy or other curative therapies.
o The exclusion criteria were revised to indicate that potential study patients could not have an active malignancy (other than PPGL) requiring additional treatment during the active phase or follow-up period of the 131I-MIBG trial, in addition to not having received any previous systemic radiotherapy resulting in marrow toxicity within 3 months of study entry, or chemotherapy within 30 days of study entry.
o The inclusion criteria were revised to allow enrollment of patients who were unable to use toilet facilities without assistance, required assistance with feeding or had mobility issues.
Changes were made to specify that imaging performed after each therapeutic dose was performed within 7 days, rather than at 5 days, post-infusion only.
The amendment clarified that CT or MRI scans could be taken and that for CT or MRI scans of the chest, abdomen and pelvis, IV contrast did not have to be used if medical condition or allergy prevented its use.
For the imaging parameters, it was specified that anatomical volumes could be measured for organs and tissues to further evaluate absorbed dose.
The required interval between each of the 3 image acquisitions made after the dosimetric dose was revised from at least 1-day to at least 18 hours.
Testing for tumor markers was specified to occur after the first therapeutic dose, specifically starting at Week 2, and that monthly testing occurred during Months 7 to 12 rather than Months 6 to 12.
The efficacy portion of the study was defined as the time of signed informed consent through 12 months.
The amendment specified that patients who discontinued during long term follow-up would only be followed for AESIs, and that the follow-up could occur via telephone for patients who were followed only for AESIs.
The amendment specified that the dosimetric dose would be administered within 30 days prior to the first therapeutic dose, rather than 7 days.
The protocol specified that if screening/pre-screening procedures were completed as standard of care within 48 hours prior to obtaining informed consent, these procedures would not be repeated as screening assessments specifically for this study.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
82Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
The amendment specified that all antihypertensive medication ongoing/ending within 30 days prior to informed consent were recorded in the eCRF.
The amendment added and clarified the timing of blood draws and urine collections performed for laboratory analyses.
The amendment specified that the physical exam and Karnofsky Performance Status scale performed 6 weeks after each therapeutic dose could occur within ± 10 days of the Week 6 window to accommodate potential blood count nadir or travel restrictions.
The Quality of Life and Symptoms Questionnaires were revised.
The list of AESI was revised.
8.1.2 MIP-IB12B Study Results
Compliance with Good Clinical Practices
The Applicant stated the following in the Study Report for Study MIP-IB12B (Module 5.3.5.2): “The Investigators agreed to conduct the study according to the principles of the ICH E6 Guideline for Good Clinical Practice (GCP) and the ethical principles that have their origins in the Declaration of Helsinki. The Investigators conducted all aspects of this study in accordance with all national, state, and local laws or regulations. To ensure compliance, the Investigators agreed, by written consent to this protocol, to fully cooperate with compliance checks by allowing access to all study documentation by authorized individuals.”
DOP2 consulted the Office of Scientific Investigation (OSI) on July 28, 2017, to perform an audit of select clinical sites. Sites were selected primarily based on the number of patients enrolled, as the majority of patients for study MIP-IB12B were enrolled at 3 sites. These three domestic sites were chosen for inspection. In general, the inspectional findings supported validity of data as reported by the Applicant under this NDA. FDA form 483, with voluntary action indicated (VAI) was issued to the Applicant for failure to ensure proper monitoring of the study due to a gap of monitoring visits from 7/2013 through 4/2014. OSI concluded that although regulatory violations related to failure to ensure proper monitoring were noted, the observations are unlikely to significantly impact primary safety and efficacy analyses.
The facility which conducted central imaging review was recently inspected by OSI, therefore re-inspection of the site was deemed unnecessary.
Financial Disclosure
In accordance with 21 CFR 54, the Applicant submitted a financial disclosure certification document in module 1.3.4. The document includes a table listing all investigators who participated in the four covered studies supporting NDA 209607. Only sites, and corresponding
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
83Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
investigators, which accrued patients on studies are included. Box 3 is checked on Form 3454, which states that the Applicant has not entered into any financial arrangements with the listed clinical investigators whereby the value of compensation to the investigator could be affected by the outcome of the study as defined in 21 CFR 54.2(a). The text under box 3, Form 3454, further states that the investigators were required to disclose whether they had a proprietary interest in the product or a significant equity in the Applicant as defined in 21 CFR 54.2(b), and that no clinical investigator disclosed such interests, and that no listed investigator was the recipient of significant payments of other sorts as defined in CFR 54.2(f).
Patient Disposition
Eighty-one patients were enrolled in study MIP-IB12B; two patients were enrolled a second time, after failing screening with initial enrollment. A total of 74 patients received a dosimetric dose, 18 received one therapeutic dose, and 50 received 2 dosimetric doses. Forty-five patients received both therapeutic doses, attended the Month 3 and Month 6 efficacy visits, and had no major protocol deviations, comprising the “Per protocol set” (PPS). Sixty-eight patients received at least one therapeutic dose and comprise the full analysis set (FAS) for the study. By March 10, 2017, the last patient reached their 12 Month Efficacy Visit and the date of data cutoff for the primary analysis, 18 patients remained in long-term follow-up.
At the time of data cut-off for the 90-day safety update, 11 patients were enrolled on the MIP-IB12B expanded access program (EAP) which had similar inclusion and exclusion criteria to the study MIP-IB12B, and delivered the same treatment.
Table 21: Patient Disposition, Study MIP-IB12B
Disposition 131I-MIBG n (%)Number screened 81a
Reasons for exclusion during screening: Not meeting eligibility criteria 7Number receiving dosimetric dose 74
Number receiving at least one therapeutic doseb 68Number receiving two therapeutic doses 50Discontinuation during 12-month efficacy phase 23Reasons for discontinuation during efficacy phase Progressive disease Adverse eventc
Deathd
Lost to follow-up Alternative anti-cancer therapy
68324
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
84Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Disposition 131I-MIBG n (%)Patients in long-term follow-up (LTFU) phase Completed LTFU phase Currently in LTFU phase Discontinued LTFU phase
51829
Reasons for discontinuation during LTFU phase Deathe
Adverse eventsf
Progressive disease Alternative anti-cancer therapy Patient withdrew consent
62
1083
Source: adsl.xpt aTwo patients were screened twice: patient (who failed screening after receiving a dosimetric dose) and patient (who failed screening prior to receiving dosimetric dose). Both patients later enrolled and successfully received at least one therapeutic dose. bFull Analysis Set (FAS) cAdverse events causing discontinuation prior to second therapeutic dose: thrombocytopenia (2), anemia (2), decreased white blood cells (2), nausea and vomiting (1), multiple hematologic adverse events (thrombocytopenia, leukopenia, neutropenia, hemolytic anemia) (1).dCauses of death during efficacy phase: sepsis (1), metabolic acidosis (1), bowel perforation (1).e Causes of death during LTFU: progressive disease (4), acute myeloid leukemia (1), “hematologic decompensation” (1).fAdverse events: myelodysplastic syndrome (1), metastatic colon cancer (1).
An IR was sent to clarify the reason for study discontinuation for patients who withdrew consent; reasons for discontinuation included desire not to travel to the clinical site, withdrawal to seek additional treatment, and an unspecified reason.
Protocol Violations/Deviations
Major protocol deviations were pre-specified and included those pertaining to study eligibility criteria, those that harmed or posed a significant risk of harm to the patient, compromised the scientific integrity of the data, demonstrated a willful or knowing breach of human subject protection, regulations, policies, or procedures on part of the investigator, those involving a serious or continuing noncompliance with federal, state, local or institutional human patient protection regulations, policies, or procedures. Protocol deviations in general were categorized as one of the following: patient eligibility criteria, AE reporting, required protocol procedure not completed or incorrectly performed, variation in use of investigational product from protocol requirements, use of non-approved concomitant medication, out-of-window trial visit or procedure, and other. The Applicant notes a total of 21 major protocol deviations occurred related to 19 patients.
Reference ID: 4297867
(b) (6)
(b) (6)


NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
86Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
PATIENT ID* PROTOCOL DEVIATION
Received second therapeutic dose outside of allowed window (33 weeks instead of 24 weeks).
Deviations in Adverse Event reporting
SAE not reported within 24 hours.
SAEs (acute renal failure, sepsis, and disseminated intravascular coagulation) not reported within 24 hours.SAE (thrombocytopenia) not reported within 24 hours, SAE (leukopenia) not reported within 24 hours.
SAE (acute myeloid leukemia) not reported within 24 hours.
SAE (febrile neutropenia) not reported within 24 hours.
SAE not reported within 24 hours.
Source: DV.xpt, Study MIP-IB12B; *Patients counted once within each category if they had more than one deviation.
Reviewer comment: These protocol deviations were clearly described in the dataset and do not substantially impact the integrity of the study or the reliability of the study results for conducting the safety and efficacy reviews.
Table of Demographic Characteristics
A total of 68 patients received at least one therapeutic dose of 131I-MIBG on Study MIP-IB12B. Of these 68 patients, 39 (57%) were male and 29 (43%) were female. Fifty-one patients (75%) were White, 14 (20.6%) were African American, and 3 (4.4%) were Asian. Eleven (16.2%) were 65 years or older, and no patients were 75 years or older.
Table 23: Demographic Characteristics of all Enrolled Patients, Study MIP-IB12B
Demographic Characteristics131I -MIBG (N=68)Mean (SD) or n (%)
Age 50.9 (13.9)
Age Group
<17 years 1 (1.5%)
≥ 17 - < 65 years 56 (82.4%)
> 65 - < 75 years 11 (16.2%)
Reference ID: 4297867
(b) (6)
(b) (6)


NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
88Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Other Baseline Characteristics (e.g., disease characteristics, important concomitant drugs)
All patients had a diagnosis of malignant or relapsed/refractory pheochromocytoma or paraganglioma, and were ineligible for curative surgery. Most patients had one or more prior cancer therapies: 52.9% had prior radiation, 51.9% had prior drug or biologic therapy, and 88.2% had prior surgery. Approximately one third (30.9%) of patients received prior NA- 131I-MIBG therapy. Two patients (2.9%) had no prior therapy, 23.5% of patients (n=16) had surgery as their only prior therapy, and 4.4% of patients (n=3) had received chemotherapy or NA- 131I-MIBG therapy as their only prior therapy. Half of the patients (50%) had lung or liver metastases, and 60% had bone metastases.
Thirteen patients had a prior history of cancer. Two patients had a history of multiple cancers (basal cell carcinoma, papillary thyroid carcinoma; atypical carcinoid tumor, endometrial cancer). There were two cases each of basal cell carcinoma and neuroendocrine tumors, and one case each of the following: B-cell lymphoma, breast cancer, carcinoid tumor, colon cancer, endometrial cancer, ovarian cancer, prostate cancer, skin cancer, testicular cancer, and papillary thyroid carcinoma.
Table 24: Baseline Characteristics, Study MIP-IB12B
Characteristic131I -MIBG (N=68)
n (%)
Presence of visceral metastatic disease 47 (53)
Primary diagnosis
Pheochromocytoma 53 (77.9%)
Paraganglioma 14 (20.6%)
Both pheochromocytoma and paraganglioma 1 (1.5%)
Prior NA-131I-MIBG therapy
Yes 21 (30.9%)
No 47 (69.1%)
Types of Prior Anticancer Therapy
Radiation 36 (52.9%)
Prior drug or biologic treatment for cancer 35 (51.5%)
Surgery 60 (88.2%)
Location of Metastases*
Reference ID: 4297867


NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
90Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Antihypertensive medications administered on the day following 131I-MIBG administration are of special interest given the potential for hypertensive crisis with 131I-MIBG administration. Five patients had a change in antihypertensive medication within 24 – 48 hours of the first 131I-MIBG therapeutic dose administration. All but one had an increase in the dose of an existing antihypertensive medication; one received a single intravenous dose of enalapril. The administration of increased doses of antihypertensive medications may indicate higher blood pressures, or health care provider anticipation of higher blood pressures, immediately following 131I-MIBG administration. Section 8.2.4 contains a complete analysis of vital signs in patients receiving 131I-MIBG.
Efficacy Results – Primary Endpoint
Table 25 summarizes the applicant’s and the reviewer’s analyses of the primary endpoint. Among the 68 patients who received at least one therapeutic dose of 131I-MIBG, 17 (25%) had a reduction in the total dose of antihypertensive medications by at least 50% for at least 6 consecutive months.
Table 25: Results of the Primary Endpoint (FAS)
FAS population (n=68)
Proportion of patients who had reduction (including discontinuation) of all antihypertensive medications by at
least 50% maintained for at least 6 months 25.00%95% CI (Agresti-Coull) 16.2%, 36.5%
Source: Adapted from the CSR Table 17.
Statistical Reviewer’s Comments:The study met the its primary objective because the lower bound of the 95% confidence interval (CI) of the primary endpoint was greater than 10%. The exact 95% CI: (16.0%, 35.9%) calculated by the statistical reviewer is consistent with the applicant’s 95% CI obtained using the Agresti-Coull method specified in the SAP.
The applicant conducted two sensitivity analyses to evaluate the robustness of the primary analysis result. In their sensitivity analysis 1, patients were considered non-responders if the 6 months of antihypertensive medication reduction was completed outside of the 12-month Efficacy Phase. While in sensitivity analysis 2, patients who began a new non-transient antihypertensive medication (i.e., used for more than 14 days) following therapeutic dosing were considered non-responders. Table 26 summarizes the two sensitivity analyses.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
91Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Table 26: Results of Sensitivity Analyses
Sensitivity Analysis 1
(n=68)Sensitivity Analysis 2
(n=68)Proportion of patients who had reduction (including discontinuation) of all antihypertensive medications by at least 50% maintained for at least 6 months95% CI (Agresti-Coull)
20.6%
(12.6%, 31.8%)
23.5%
(14.9%, 35.0%)Source: Adapted from CSR Tables 14.2.1.6 and 14.2.1.7.
Statistical Reviewer’s Comments:The results of the two sensitivity analyses are consistent with the result of the primary analysis.
Among the 68 patients who received at least one therapeutic dose of 131I-MIBG, there are 33 patients who attained at least 50% reduction in all antihypertensive medications. Table 27
summarizes the statistical reviewer’s analysis of duration of response for those patients who attained at least 50% reduction in all antihypertensive medications.
Table 27: Duration of Response for Patients Who Attained at Least 50% Reduction in Antihypertensive Medications
>=6 months < 6 months Total#Patients attained at least 50% reduction in all antihypertensive medications 17 16 33
Median duration in months (min, max) 13.3 (8.0, 60.2) 1.79 (0.1, 5.0) 8.0 (0.1, 60.2)
Figure 12 displays a swimmer plot for the 17 patients who had reduction (including discontinuation) of all antihypertensive medications by at least 50% maintained for at least 6 consecutive months.
Reference ID: 4297867


NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
93Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Table 28: Patients with a Decrease in Systolic Blood Pressure > 20 mm Hg, Study MIP-IB12B
Time Point N* Patients with Decrease in Systolic Blood Pressure M> 20 mm Hg (%)
12 weeks after Therapeutic Dose 1
56 14 (25%)
12 weeks after Therapeutic Dose 2
35 7 (20%)
Source: Table 49, CSR MIP-IB12B*Number of patients with a blood pressure measurement at the specified time point.
Table 29: Changes in Systolic Blood Pressure During Study MIP-IB12B
Time Point N Median Systolic Blood Pressure, mm Hg (Range)
Change from Baseline. mmHg (Range)
Baseline 73 132 (95 – 211.0) -
12 weeks after Treatment Dose 1
56 135.25 (91 – 177) +3 (-37, 61)
12 weeks after Treatment Dose 2
35 125 (78 – 157) -4 (-61, 38)
Source: Table 14.5.1.3, Clinical Study Report, Study MIP-IB12B
Data Quality and Integrity
A study monitor maintained contact with the investigator and conducted initiation and closeout visits. The Applicant states that all data required by the protocol were reported accurately on the CRF and had to be consistent with other documents. Source documents were available for inspection. Data were captured using an electronic data capture (EDC); electronic case report forms (eCRFs) were designed for the capture of all relevant clinical data for the study. Quality control checks of documentation and data recording were carried out on behalf of the Applicant in accordance with standard operating procedures. Quality was assured through regular monitoring of the documentation, study center, facilities, and quality control (QC) audit of the database. To ensure collection of accurate and complete data, all eCRF data were the subject of 100% source data verification.
The quality of the data was sufficient to allow the statistical reviewer to reproduce the results of the primary analysis and other major submitted efficacy analyses. However, the result and data of duration of tumor response for patients whose best confirmed tumor response were CR
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
94Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
or PR were not included in the original NDA submission. This data was requested by FDA to understand consistency of the primary endpoint with traditional endpoints, e.g., overall tumor response and duration of response.
Efficacy Results – Secondary and other relevant endpoints
Best confirmed overall tumor response was a secondary endpoint in Study MIP-IB12B. A confirmed response was defined as a response of CR, or PR that was confirmed by repeat assessment performed no less than 4 weeks after the criteria for response were first met. By the time of data cutoff for the analysis, there were 15 patients who had a PR. Among 15 responders, 13 had ongoing responses with durations ranging from 2.3+ to 9.4+ months. Table 30 summarizes the applicant’s and reviewer’s analyses of best confirmed overall tumor response and duration of response. The reviewer’s result was based on the data of duration of response for patients whose best confirmed tumor response were CR or PR, which were requested by FDA.
Table 30: Best Confirmed Overall Tumor Response and Duration of Response
Reviewer’s Result
(n=68)Applicant Result
(n=64)
Best confirmed response
Responders, n 15 15 CR 0 0 PR 15 15
Response rate (95%CI*) 22.1% (13.6%, 32.7%) 23.4% (14.5%, 34.6%)
Duration of response (DOR) n=15
Median in Months (95%CI) 8.7 (6.4, NE*) 6.5
Responders with DOR >=6 months**, n 8 (53.3%) 9 (60.0%)Source: The applicant’s result was adapted from the table in mib-ib12b-tumrespduration (DUR).pdf which was submitted in response to FDA’s request.* NE=Not estimable due to few events. **Month=28 days in the applicant’s result and Month=30.4375 days in the reviewer’s results.
Statistical Reviewer’s Comments: In Table 30, the applicant’s result was based on 64 patients whose last available best
response was assessed within the 12-month efficacy phase. The reviewer’s result was based on 68 patients in the FAS. Patients in the FAS and whose last available best response assessed was outside of the 12-month Efficacy Phase were considered as non-responders in the reviewer’s result.
In Table 30, the reviewer’s result shows that 8 out of 15 responders had at least 6 months
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
95Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
duration of response including 6 responders with DOR longer than 6 months.
Results and data related to duration of tumor response were not provided in the original NDA submission, and no plan for evaluation of duration of tumor response was provided in the protocol or the SAP. The applicant submitted the results of duration of response for patients whose best confirmed tumor response was CR or PR per FDA request. As shown in Table 24, there are discrepancies between the statistical reviewer’s DOR results and the applicant’s results. The discrepancies are due to the following reasons:
1) definition of month o 30.4375 days per month (regular conversion) used in the reviewer’s analysis o 28 days per month (defined in the protocol) used in the applicant’s analysis
2) analysis method o The reviewer used the Kaplan-Meier method and censored the DOR for the
responders who maintained their response by the date of the data cut-off o The applicant used the regular summary method without censoring the responders
who maintained their response as the date of the data cut-off. The applicant’s method is not appropriate for summarizing the duration of response data where some responders still maintained their response by the time of the analysis.
3) denominator for response rateo The reviewer used the 68 patients in the FAS. For primary and key secondary results,
all patients in FAS should be included.o The applicant used only the 64 patients assessed for response within the 12-month
efficacy phase.
To evaluate if the benefit of patients who met the primary endpoint can translate to the benefit of the tumor response, the statistical reviewer conducted two analyses to explore the relationship between patients who met the primary endpoint and the responders per RECIST. Table 31 and Table 32 summarize these analyses. As shown in Table 31, 7 out of 17 patients who met the primary endpoint had best confirmed response of PR.
Table 31: Best Confirmed Overall Tumor Response and Duration of Benefit
Best Confirmed Overall Tumor Response per RECIST
Patients met primary endpoint PD PR SD UE TotalNo 3 8 34 2 47Yes 0 7 10 0 17Total 3 15 44 2 64
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
96Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
On the other hand, 7 out of 15 responders who had a response by RECIST met the primary endpoint, as shown in Table 32.
Table 32: Best Confirmed Overall Tumor Response and Duration of Benefit
Best Confirmed Overall Tumor Response*
Duration of Benefit** <6 months
Duration of Benefit** >=6 Months (met primary endpoint) Total
CR 0 0 0
PR 8 7 15*Patient’s last available best response was assessed within the 12-month Efficacy Phase **Duration of benefit: duration of attaining at least 50% reduction in all antihypertensive medication beginning the 12-Month Efficacy Phase (months).
Dose/Dose Response
Eighteen patients on study MIP-IB12B received only one therapeutic dose of 131I-MIBG, and 50 patients received two therapeutic doses. A greater percentage (32%) of patients who received two doses demonstrated a response for the primary endpoint, compared to patients who received only one dose (5.6%). This analysis is limited by the small number of patients involved in each subgroup. In addition, additional confounding factors may be present; patients who had more aggressive disease may have progressed quickly and been unable to receive a second dose, for example. Limited conclusions can be drawn from the analysis of response by number of doses received.
Durability of Response
The duration of response in reduction of antihypertensive medications is summarized in Table 27: Duration of Response for Patients Who Attained at Least 50% Reduction in Antihypertensive Medications. Among primary endpoint responders (n=17), the median duration of reduction of antihypertensive medications by at least 50% was 13.3 months (range 8.0 – 60.2 months). An additional 16 patients achieved an equivalent reduction in antihypertensive medications for a shorter period (median 1.79 months, range 0.1 – 5.0 months). The duration of response, as determined by the primary endpoint, was sufficient to be considered of meaningful clinical benefit. The median duration of tumor response as measured by RECIST 1.0 was 8.7 months, with 40% of responders maintaining a response for at least 6 months (see Table 24). The duration of tumor response as measured by RECIST was also considered clinically meaningful.
Persistence of Effect
As the treatment consisted of a defined number of doses, a discussion of persistence of effect is duplicative of the discussion of durability of response. See above.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
97Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Efficacy Results – Secondary or exploratory COA (PRO) endpointsThe statistical reviewer conducted primary endpoint analyses in the subgroups defined by age (greater than 65, less than or equal to 65 years), gender (male, female), race (White vs. non-White) location of metastases and number of therapeutic doses. Because Study IB12B was conducted in US, subgroup analysis based on region is not applicable. Table 33 summarizes the reviewer’s analyses of subgroups.
Table 33: Primary Endpoint in Subgroups
SubgroupResponders*/Total Patients, n (%)
Age <65 14/57 (24.6)>=65 3/11 (27.3)
GenderFemale 10/29 (34.5)Male 7/39 (18.0)
RaceWhite 17/51 (33.3)Non-White 0/17 (0)
Location of metastasesLung/liver metastases 8/32(25.0)Bone-only metastases 6/19 (31.6)
Number of therapeutic doses1 1/18 (5.6)2 16/50 (32.0)Prior 131I-MIBG therapy Yes 4/21 (19.0)No 13/47 (27.6)
*Patients who had reduction (including discontinuation) of all antihypertensive medications by at least 50% maintained for at least 6 months.
Statistical Reviewer’s Comments:The subgroup analyses results are considered exploratory, results should not be over interpreted given the small samples sizes in some of the subgroups, and no conclusions may be drawn. The statistical reviewer’s analyses of PRO data are provided in the appendix.Clinical Reviewer’s Comments: The clinical reviewer agrees that the subgroup analyses are considered exploratory, and that caution should be exercised in interpretation. Further analyses of the clinical benefit of 131I-MIBG
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
98Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
in patients by race revealed that non-white patients (both African American and Asian patients) did experience clinical benefit, including tumor response as measured by RECIST 1.1. White patients demonstrated a response rate by RECIST of 23.5% (n=51), while response rates in African Americans and Asians were 14.3% (n=14) and 33.3% (n=3), respectively. Non-white patients also demonstrated decreases in blood pressure or in the use of antihypertensive medications, but did not meet the specific parameters defined by the primary endpoint. For example, One Asian patient had a temporary reduction in blood pressure medication use. Five African American patients had either decreases in blood pressure or decreases in blood pressure medications which did not qualify for the primary endpoint.
Given the extremely small number of patients in some subgroups and the exploratory nature of these analyses, the response rates estimated for the primary endpoint alone are not expected to be predictive of clinical benefit by race.
Additional Analyses Conducted on the Individual Trial
Not applicable.
Integrated Review of Effectiveness
See discussion in 8.1.5 Integrated Assessment of Effectiveness
8.1.3 Study MIP-IB12: A Phase I Study Evaluating the Maximum Tolerated Dose, Dosimetry, Safety and Efficacy of Ultratrace Iobenguane I 131 in Patients with Malignant Pheochromocytoma/ Paraganglioma
Trial Design and ObjectiveStudy MIP-IB12 was a multi-site, open-label, dose-escalation study of 131I-MIBG in patients with PPGL. The primary objective was to determine the maximum tolerated dose of 131I-MIBG in patients with malignant PPGL. The study used a traditional 3+3 dose escalation design.
Patients were 18 years or older with metastatic or recurrent, iobenguane-avid PPGL. The study administered a dosimetric dose of 5 mCi 131I-MIBG supplemented with 185 mcg of cold MIBG, followed by a single therapeutic dose of 6, 7, 8 or 9 mCi/kg 131I-MIBG within 4 weeks of the dosimetric dose. The maximum dose administered was not to exceed that of a 75 kg patient. Patients received saturated solution of potassium iodide (SSKI) to block thyroid uptake from 1 day pre-dose to 3 days post-dose. Tumor response was assessed using CT or MRI scans every 3 months for 1 year. Objective tumor response was evaluated by review by a blinded, independent review committee according to the RECIST criteria.
Safety was assessed by evaluation of treatment-emergent adverse events, electrocardiograms, physical examinations, vital signs, and laboratory assessments. Following the 12-month visit,
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
99Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
patients entering a 1-year long-term follow-up phase to assess disease progression, overall survival, serious adverse events and late radiation toxicity at 18 and 24 months post-treatment. Tumor response data was collected per institutional standard during the long-term follow-up phase.
Study EndpointsThe primary endpoint was the maximum tolerated dose (MTD) of Ultratrace iobenguane I 131 in patients with malignant PPGL. Objective tumor response per Response Evaluation Criteria in Solid Tumors (RECIST) was evaluated as a secondary efficacy endpoint. The best overall response endpoint was defined as the best overall response recorded from the start of treatment until completion of the 12-month efficacy visit, disease progression or recurrence.
Statistical Analysis Plan
Standard descriptive statistics were presented. At 3, 6, 9 and 12 months post therapy, the proportion of patients who had overall tumor response CR or PR with a 95% confidence interval for the proportion was provided. All AEs observed were summarized by type, severity (by National Cancer Institute [NCI] Common Terminology Criteria for Adverse Events [CTCAE]), attribution, duration, and outcome.
Protocol AmendmentsThere were 4 amendments to the original study protocol dated December 13, 2006. Important changes are summarized by amendment as follows:
Amendment 1 The definition of DLT was updated; DLTs were reviewed and adjudicated by a Dose
Escalation Safety Cohort Committee. An exclusion criterion also was added to exclude patients with evidence of altered
biodistribution of Ultratrace iobenguane I 131 or evidence of renal obstruction. As well, the imaging assessment section was amended to state that patient-specific kidney measurements would be used for dosimetry estimates.
The drug administration section was updated to recommend the use of a drug infusion system developed by the sponsor for the therapeutic dose. The infusion system was developed to help reduce study drug administration variability between study centers.
The option of performing LTFU visits via telephone was removed. Onlyoverall survival data could potentially be gathered via telephone.
Amendment 2 No critical changes; procedures in the protocol were clarified.
Amendment 3 The study was amended to remove the Phase 2 portion of the study, and end the
study upon determination of the MTD. Study MIP-IB12B was established as a separate study in lieu of continued enrollment of the Phase 2 portion of MIP-IB12.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
100Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
The protocol was clarified to indicate that patients would be monitors for AESIs, including post radiation toxicity, during LTFU annually.
Amendment 4 The LTFU portion of the study was shortened from 5 years to 2 years following
therapeutic dose administration (1 year following the efficacy phase).
Study Results
Compliance with Good Clinical PracticeThe Applicant stated the following in the CSR for Study MIP-IB12 (Module 5.3.5.2): “All considerations regarding the protection of human patients were carried out in accordance with the Declaration of Helsinki. The investigator agreed to conduct the study according to the principles of the ICH E6 Guideline for Good Clinical Practice (GCP) and the ethical principles that have their origins in the World Medical Association Declaration of Helsinki. The investigator(s) conducted all aspects of this study in accordance with all national, state, and local laws or regulations. To ensure compliance, the investigator(s) agreed, by written consent to this protocol, to fully cooperate with compliance checks by allowing access to all study documentation by authorized individuals.”
Financial DisclosureIn accordance with 21 CFR 54, the Applicant submitted a financial disclosure certification document in module 1.3.4. The document includes a table listing all investigators who participated in the four studies supporting NDA 209607. Only sites, and corresponding investigators, which accrued patients on studies are included. Box 3 is checked on Form 3454, which states that the Applicant has not entered into any financial arrangements with the listed clinical investigators whereby the value of compensation to the investigator could be affected by the outcome of the study as defined in 21 CFR 54.2(a). The text under box 3, Form 3454, further states that the investigators were required to disclose whether they had a proprietary interest in the product or a significant equity in the Applicant as defined in 21 CFR 54.2(b), and that no clinical investigator disclosed such interests, and that no listed investigator was the recipient of significant payments of other sorts as defined in CFR 54.2(f).
Patient Disposition Twenty-four patients were enrolled, and twenty-one patients were treated (ITT population). Fourteen of the treated patients (67%) completed the 12-month efficacy phase. Reasons for discontinuation during the efficacy phase include receipt of an alternative anticancer therapy (n=4), death (n=3), and other (n=1). Ten patients completed the Month 24 assessment, and six patients were followed until 3 years post-treatment.
Protocol DeviationsA complete listing of protocol deviations for the study was provided. The majority of protocol deviations were related to missed protocol assessments or assessments which occurred outside of the specified window. Five patients did not receive the specified schedule of SSKI drops; this
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
101Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
was considered a minor deviation. Organ dosimetry did not occur for one patient, because the patient did not have an imaging standard present during the whole-body imaging sessions. The remainder of the protocol deviations were reviewed, and do not appear to substantially impact the integrity of the study or the reliability of the study results for conducting the safety and efficacy reviews.
Demographic CharacteristicsThe median age of patients at enrollment was 49 (range 30 – 72). Thirteen (61.9%) were male and 8 (38.1%) were female. Two (9.5%) were Asian, 3 (14.3%) were African American, and 16 (76.2%) were white. Ten (47.6%) had a primary diagnosis of pheochromocytoma and eleven (52.4%) had a diagnosis of paraganglioma.
Efficacy ResultsAs noted above, the primary endpoint of this study was determination of the MTD. Assessment of efficacy through determination of overall response by RECIST v 1.0 was a secondary endpoint. Four of 21 patients (19%, 95% CI 0.00, 0.38) in the ITT population achieved a response of PR; there were no CRs.
8.1.4 Assessment of Efficacy Across Trials
The efficacy of 131I-MIBG is supported primarily by Study MIP-IB12B, with supporting evidence derived from the dose-escalation study MIP-IB12.
Primary Endpoints
The primary endpoint of Study MIP-IB12B, reduction of antihypertensive medications by 50% for a duration of 6 months, is a new endpoint considered specifically for the treatment of patients with PPGL. The endpoint was chosen, and agreed upon by FDA, as a measure of antitumor activity of 131I-MIBG by means of detecting a reduction in hypertension, which is a key contributor to morbidity associated with PPGL. Severe hypertension in patients with PPGL is caused by the underlying tumor; however, to provide evidence that 131I-MIBG demonstrates anti-tumor activity, and not merely antihypertensive activity, the response in the primary endpoint was supported by an established response criteria, radiologic response by RECIST v1.0.
As discussed in section 8.1.2 MIP-IB12B Study Results, 25% of patients in study MIP-IB12B achieved a 50% reduction in antihypertensive medications for at least 6 months. The duration of response, as determined by the primary endpoint, was sufficient to be considered of meaningful clinical benefit. Among primary endpoint responders (n=17), the median duration of reduction of antihypertensive medications by at least 50% was 13.3 months (range 8.0 – 60.2 months). An additional 16 patients achieved an equivalent reduction in antihypertensive medications for a shorter period (median 1.79 months, range 0.1 – 5.0 months). As a high degree of morbidity in PPGL arises from the complications of hypertension, this represents a significant clinical benefit; however, this does not definitively indicate that this benefit is
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
102Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
achieved through treatment of the tumor, and thus, additional evidence in the form of radiologic response was also considered.
Secondary and Other Endpoints
In Studies MIP-IB12B and MIP-IB12, response as evaluated by RECIST v1.0 was a secondary objective. Among all patients treated with 131I-MIBG on Study MIP-IB12B, 22.1% (95% CI: 13.6 – 32.7%) demonstrated a response by RECIST; these responses were maintained for a median of 8.7 months. In Study MIP-IB12, 4/21 patients (19%, 95% CI: 0 - 38%) demonstrated a partial response. In Study MIP-IB12B, not all primary endpoint responders were also responders by RECIST; 41% of responders by the primary endpoint achieved a response of PR, and the remainder experienced a best response of SD. The clinical meaningfulness of achieving a radiologic response of stable disease in this population is unclear, as PPGL may be slow-growing.
Subpopulations
Exploratory subgroup analyses were performed with regards to response by the primary endpoint in Study MIP-IB12B. The subpopulation analyses are limited by the small size of the study, and by the post-hoc nature of these analyses. A significantly greater percent of responders to the primary endpoint were among those patients who received two doses of 131I-MIBG; 5% (1/18) of patients who received one dose of 131I-MIBG demonstrated a response, compared to 32% of patients (16/50) who received two doses. This finding could indicate that the higher cumulative dose is associated with improved response, or merely that patients with more severe disease and correspondingly poor performance status were less likely to recover sufficiently to receive the second dose. Given the small sample size, limited conclusions can be drawn from this finding.
Additional Efficacy Considerations
Overall survival (OS) was evaluated in Study MIP-IB12B as a secondary endpoint. Given the inability to interpret OS results in a single arm trial, an analysis of this measure is not included in this review. Additional secondary efficacy endpoints, such as tumor marker response and patient-reported outcome measures, were also included in the study as secondary endpoints but not analyzed as part of the efficacy evaluation given the unclear reliability in assessing clinical benefit.
8.1.5 Integrated Assessment of Effectiveness
The Applicant has established that 131I-MIBG demonstrates substantial evidence of effectiveness in patients with locally advanced or metastatic, recurrent or unresectable PPGL, through demonstration of a sustained, substantial reduction in antihypertensive medications, as well as a durable radiologic response. 8.2 Review of Safety
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
103Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
8.2.1 Safety Review Approach
The safety of I131-MIBG in PPGL was primarily evaluated in 68 patients in study MIP-IB12B who received at least one therapeutic dose of I131-MIBG. Fifty patients received the planned two therapeutic doses and 18 patients received one therapeutic dose, which consisted of 500 mCi or 8 mg/kg for patients weighing less than 62.5 kilograms. In total, 74 patients received a dosimetric dose. Database lock occurred after 68 patients completed the 12-month efficacy evaluation (12 months from first therapeutic dose). At the time of database lock, 18 patients remained in long term follow-up (LTFU). An additional 11 patients were enrolled on an expanded access study for study MIP-IB12B, which provided treatment identical to the treatment described above.
Twenty-one patients who received one dosimetric and one therapeutic dose of I131-MIBG in study MIP-IB12 were also analyzed. Patients received 6, 7, 8 or 9 mCi/kg I131-MIBG based on assigned dose cohort. Patients were followed during a 12-month efficacy phase and subsequent 1-year LTFU phase. In total, between studies MIP-IB12 and MIP-IB12B, 88 unique patients received at least one therapeutic dose. Of note, one patient (MIP-IB11- / MIP-IB12B- ) received therapeutic doses on both studies MIP-IB12 and MIP-IB12B. One additional patient enrolled on both studies MIP-IB12 and MIP-IB12B (MIP-IB12 /MIP-IB12B ), but did not receive a therapeutic dose on MIP-IB12B, and therefore is not duplicated in the main safety population.
The Applicant provided additional safety information in the 90-day safety update regarding the 18 patients remaining on study MIP-IB12B, and patients enrolled on the MIP-IB12B Expanded Access Program (n=11). This information was not included in calculations of adverse event incidences, but all serious adverse events were reviewed, and several key adverse events included in the 90-day update are discussed in this review.
A pooled population of patients (N= 118) was analyzed by the Applicant as the main safety population, and included patients from study MIP-IB11 and MIP-IB13 in addition to MIP-IB12 and MIP-IB12B. Though included in the analysis provided by the Applicant, studies MIP-IB11 and MIP-IB13 were excluded from the primary pooled safety analysis by this reviewer. Study MIP-IB11 administered only a dosimetric dose of 5mCi 131I-MIBG with 185 mcg cold MIBG, and thus would not be expected to provide meaningful safety information regarding administration of therapeutic doses of 131I-MIBG. Study MIP-IB13 administered 131I-MIBG to patients with refractory neuroblastoma, a substantially different patient population; in addition, patients received significantly higher doses of 131I-MIBG (dosimetric dose of 0.1 mCi/kg 131I-MIBG with 185 mcg cold MIBG, followed by a 11.2, 15.5, or 18.2 mCi/kg therapeutic dose 7-28 days later), and all received autologous stem cell rescue. The follow-up period for this study was 60 days, thus precluding comprehensive collection of expected late radiation toxicities. The substantial differences in dose administered, study population, the use of stem cell rescue, and the short
Reference ID: 4297867
(b) (6) (b) (6)
(b) (6) (b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
104Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
duration of follow-up decrease the ability to extrapolate safety information from this study to the indicated population in this application.
Safety issues identified prior to the review period notably included the development of myelodysplastic syndrome and acute leukemia. An analysis and discussion of these events is included in Section 8.2.5.
8.2.2 Review of the Safety Database
Overall Exposure
131I-MIBG was administered by infusion in a monitored setting. In the safety population (MIP-IB12 and MIP-IB12B), 88 patients received at least one therapeutic dose in addition to the dosimetric dose. Forty-nine patients (55.6%) received two therapeutic doses. One patient enrolled on both Study MIP-IB12 and MIP-IB12B, and received a total of three therapeutic doses. The administered activity in the safety population ranged from 107.4 mCi to 1101.59 mCi, with a median of 897.1 mCi. The administered activity in Study MIP-IB12B ranged from 101.5 mCi to 1095.8 mCi, with a median of 984.8 mCi. On Study MIP-IB12B, one patient received a significantly lower dose than the proposed dose of 500 mCi or 8 mCi/kg for patients <62.5 kg. This patient’s extensive disease prevented an accurate dosimetry calculation and a lower dose of 101.5 mCi (1.8 mCi/kg) was given rather than the body-weight based dose of 8 mCi/kg (442 mCi). This patient withdrew from the study prior to receipt of a second dose. Although 8 other patients received doses <500 mCi/kg, these patients either received doses within 10% of the recommended dose of 500 mCi or were administered an appropriate body weight-based dose. The study required dose reduction of the second dose in patients who experienced severe hematologic toxicity. Seven patients met criteria for dose reductions due to hematologic or renal toxicity, though five patients did not receive the protocol-required dose reductions.
Table 34: Distribution of 131I-MIBG Administrations in the MIP-IB12/MIP-IB12B Safety Population (N=88)
Number of Administrations
(therapeutic)
PatientsN (%)
Mean total dose (mCi)
Minimum total dose
(mCi)
Maximum total dose
(mCi)1 38 (43.2%) 507.5 107.4 700.72 49 (55.6%) 981.6 735.2 1101.63 1 (1.1 %) 1613.4 - -
Source: ADSL.xpt
Relevant characteristics of the safety population:
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
105Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
The safety population from Studies MIP-IB12B and MIP-IB12 consisted of patients with malignant pheochromocytoma or paraganglioma who were ineligible for curative surgery for PPGL, and either failed a prior therapy for PPGL, or were not candidates for chemotherapy. Consistent with the epidemiology and natural history of PPGL, the median age was 54 years. Approximately 15.7% (N=14) of patients were over the age of 65, and none were over the age of 75. The Applicant provided subgroup safety analyses to evaluate the impact of age on adverse event frequency in Study MIP-IB12B and the pooled population. Additional exploratory analyses were conducted based on gender, race, ethnicity, and history of prior treatment with NA 131I-MIBG. Limitations of these subgroup analyses are the small sample sizes and lack of internal control in both studies
Adequacy of the safety database:
Overall, the safety database submitted by the applicant was adequate in the setting of a rare disease. The primary study, MIP-IB12B, included adequate long-term follow-up to assess late radiation-induced toxicities of a radioactive product.
8.2.3 Adequacy of Applicant’s Clinical Safety Assessments
Issues Regarding Data Integrity and Submission Quality
The data submitted was well-organized and the quality was adequate to perform a complete review of the safety of 131I-MIBG. Multiple information requests were sent to the Applicant during the review of safety to confirm data, request additional data, narratives and case report forms, request alternative presentations of per patient safety data or clarify minor discrepancies in the pooled database. The Applicant provided sufficient responses including additional analyses and clarifications as required.
A Data Fitness assessment was conducted in collaboration with the Office of Computational Science (OCS). Issues identified included: missing standard toxicity grade for 187 events in Study MIP-12B; missing end date or start date for some adverse events in all studies; missing records in Inclusion/Exclusion criteria domain for some screen failures in studies 12, 12B, and 13; missing reference range variables for some laboratory records in all studies; and inconsistent value for standard units for some tests in Studies 11 and 13. These issues did not substantially affect the ability of the reviewer to conduct a thorough safety review.
Categorization of Adverse Events
The Applicant coded verbatim AE terms for Study MIP-IB12B and the integrated database using MedDRA version 19.0 for the primary analyses. Study MIP-IB12 was coded using MedDRA version 10.0. Adverse events in the ISS are coded using MedDRA 19.0. Adverse events in the ISS ADAE dataset match 100% to MedDRA 19.0. Treatment-emergent adverse events (TEAEs) were defined as defined as AEs with onset dates on or after the dosimetric dose date to 15 weeks after the administration of the last therapeutic dose of 131I-MIBG. Notably, the definition of
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
106Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
TEAE used in IB12B was different from that used in the ISS, in which TEAEs are defined as AEs with onset dates on or after the date of the (first) dosimetric dose. AEs of special interest (AESIs) were defined as any serious or non-serious event of scientific or medical concern specific to 131I-MIBG reported on or beyond 16 weeks after the last therapeutic dose, and known to be associated with long-term radiation toxicity. CTCAE Version 3.0 was used for toxicity grading.
The OCS review assessed the adequacy of the Applicant’s mapping of AE verbatim terms to MedDRA preferred terms (PTs) for the Study MIP-IB12B primary dataset. Of the 219 distinct reported terms in the dataset, the reviewer used matching of identical verbatim and MedDRA PTs (n=85 terms) as well as manual evaluation of the remaining verbatim terms (n=134 terms). Most nonidentical terms were due to spelling differences (e.g., anemia versus anaemia), abbreviations and full text (WBC vs. white blood cell) and verbatim terms that included descriptors (e.g., slight weakness versus weakness/asthenia). Overall, the MedDRA PTs listed in the dataset adequately represented the verbatim terms from the CRFs.
Routine Clinical Tests
In study MIP-IB12B, baseline evaluations included history and physical examination with vital signs, laboratory evaluations (chemistry, complete blood count, urinalysis, thyroid function testing), renal function testing (glomerular filtration rate or 24-hour creatinine clearance), tumor markers, and imaging. Vital signs were recorded within 4 hours prior and 4 hours following each 131I-MIBG administration. Twelve-lead ECGs were obtained at the time of dosimetric dose (prior to, up to 2 minutes post dose, and at 10, 15, and 20 minutes post dose, then within 15 minutes prior to the second and third I131 Iobenguane imaging scans) and within 7 days after each therapeutic dose, and 24-hour Holter monitor evaluation was performed at the time of therapeutic dose administration. Pregnancy testing was done at baseline and within 48 hours prior to each dosimetric or therapeutic dose. Blood pressure and heart rate assessment, as well as hematology, chemistry, and urinalysis, were performed weekly during weeks 2-24 post therapeutic dose monthly during months 7-12. Blood pressure and heart rate were also collected twice weekly in the first 6 weeks after each therapeutic dose. Thyroid function was repeated at month 12 and renal function testing was repeated at months 6 and 12. Clinical evaluation for dry mouth was performed at 12 months. Long-term follow-up evaluations performed every 6 months included tumor markers and a CT or MRI of the chest, abdomen and pelvis, but were not required to include laboratory monitoring or physical examination. However, adverse events of special interest (AESIs), which includes events identifying potential late radiation toxicity, were collected during LTFU. The monitoring for immediate and long-term adverse effects was adequate.
In study MIP-IB12, similar evaluations were performed, though the study administered only one therapeutic dose, and the long-term follow-up period, as defined in the most recent amendment, was 1 year after the initial 12-month efficacy phase (2 years total). Vital signs were taken 15 minutes prior to and 15 minutes after each infusion, and within 15 minutes prior to the Day 2-4 scan; blood pressure was not measured on a twice weekly or weekly basis as in
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
107Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Study MIP-IB12B. Laboratory analyses were performed on a similar schedule. The monitoring for immediate side effects in this study is adequate; as the long-term follow-up period was revised to 1 year in the amended protocol, information on late radiation effects is necessarily limited in this study.
8.2.4 Safety Results
Deaths
There were 36 deaths among the 88 patients who received a therapeutic dose of 131I-MIBG on Study MIP-IB12B and Study MIP-IB12. Two of these patients had discontinued Study MIP-IB12 to receive other treatment at the time of death. There were 4 deaths in patients who received 131I-MIBG on Study MIP-IB13, none of which were related to treatment, and no deaths on Study MIP-IB11.
Twenty-three of the 36 deaths in the MIP-IB12/MIP-IB12B safety population were attributed to disease progression. Ten deaths were attributed to adverse events not attributed to 131I-MIBG, and included hepatic failure due to metastatic pheochromocytoma, spinal disorder, thalamic infarction, intestinal perforation leading to sepsis, metastatic colon cancer, pulmonary embolism, peripheral arterial ischemia, sepsis, pneumonia, and metabolic acidosis. Most adverse events leading to death were attributed to complications from metastatic disease.
Reviewer note: The safety narratives for all deaths were reviewed and additional information was requested when the assessment of causality was unclear. Except as noted below, this reviewer agrees with the assessment of causality listed by the Applicant.
Patient MIP-IB12 experienced hepatic failure which the Applicant attributed to metastatic pheochromocytoma. Elevated AST and alkaline phosphatase were first documented 1 month after infusion of the patient’s only dose of 131I-MIBG. The patient experienced hypoglycemia and bleeding (note that the patient was on anticoagulation therapy), and reported a 30- to 45-pound weight loss over the past few months. One month after the initial presentation, she was found unresponsive and admitted with hepatic encephalopathy; during admission, liver imaging demonstrated multiple hepatic masses with right portal vein reversal of flow. She was later diagnosed with a urinary tract infection and treated for sepsis and acute organ failure. Despite aggressive treatment, the patient continued to decline, with metabolic acidosis, persistent thrombocytopenia, and coagulopathy. Ventilator support was withdrawn, and the patient died the following day. The Applicant stated that a role of 131I-MIBG could not be completely ruled out, but stated that the patient’s urosepsis and acute organ failure precipitated the death.
Reviewer note: The patient experienced hepatic failure after receipt of a therapeutic dose of 131I-MIBG, though the case is confounded by apparently progressive metastatic liver disease. The event of sepsis and acute organ failure appears to be the ultimate cause of death, though underlying liver failure likely contributed.
Reference ID: 4297867
(b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
108Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
One cause of death (patient MIP-IB12B- ) was listed as “hematologic decompensation” or “blood disorder” which occurred 1.5 years following treatment with one therapeutic dose of 131I-MIBG. The patient received one therapeutic dose of 131I-MIBG on study in and had severe hematologic toxicity precluding administration of a second dose. The Applicant states that the patient demonstrated slow recovery of bone marrow function. She was later treated with NA-131I-MIBG by her physician off-study in . The patient developed severe hematologic toxicity including Grade 4 thrombocytopenia, Grade 3 anemia and Grade 3 leukopenia. The patient died after a complicated 41-day hospital stay; the cause of death was described by a family member as “full blown hematologic decompensation.” The Principal Investigator made several attempts to obtain additional information from the treating physician and hospital regarding this case without response. The event was considered related to the patient’s off-protocol therapy and not related to study therapy.
Reviewer note: An information request was sent to the Applicant to request additional information about this patient, as the case raises suspicion of an additional case of myelodysplastic syndrome. However, records of the patient’s subsequent off-protocol treatment were unable to be obtained. Without additional information, it is most reasonable to attribute the patients’ decompensation and death to the additional off-study therapy with 131I-MIBG.
Two patients died due to therapy-related myelodysplastic syndrome (MDS, n=1) or acute myeloid leukemia (n=1). These cases were attributed to treatment with 131I-MIBG. Additional cases of MDS and leukemia were non-fatal; a complete discussion of the incidence treatment-related MDS/AML is found in section 0. Serious Adverse Events
There were 142 serious adverse events which occurred in 60 patients in the Study MIP-IB12B and MIP-IB12 Safety Population. Forty-seven (47, 33.1%) were assessed by the Applicant as related (including possibly or probably related) to treatment. Two were fatal, related to AML and MDS (see discussion in section 0). The most common SAE was disease progression. The most common related SAEs by system organ class were blood and lymphatic system disorders (anemia, febrile neutropenia, leukopenia, neutropenia, pancytopenia, thrombocytopenia; N=29) followed by neoplasms (ALL, AML, MDS, N=5). Other SAEs assessed by the Applicant as related to the study product included dehydration (N=3), pulmonary embolism (N=2), hypothyroidism (N=1), constipation (N=1), dyskinesia (N=1), dyspnea (N=1), Aspartate aminotransferase increased (N=1) and acute cholangitis (N=1).
This reviewer reviewed all SAE narratives to assess potential relation to investigational product. Attention was paid to reported adverse events associated with NA-131I-MIBG in the literature, including myelosuppression, treatment-related leukemia or MDS, hypothyroidism, pulmonary toxicity, renal toxicity, and liver toxicity.
Narratives of serious adverse events assessed as related to the investigational product
Reference ID: 4297867
(b) (6)
(b) (6)
(b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
109Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
MIP-IB12B- : acute cholangitis, Grade 3A 22-year-old black male with pheochromocytoma and a history of sickle cell anemia experienced Grade 3 acute cholangitis approximately 2 months after receipt of a therapeutic dose of 131I-MIBG; the patient underwent livery biopsy and was diagnosed with acute cholangitis, percholangitis, and cholecystitis.
The patient developed Grade 1 anemia; the Applicant states that the study medication was withdrawn due to this adverse event, and a second therapeutic dose was not administered. The patient subsequently entered the long-term follow-up phase. Reviewer comment: The event of cholangitis is attributed to the study medication; however, the patient’s sickle cell anemia is a predisposing factor for cholecystitis and the development of acute cholangitis, and may be a contributing factor.
MIP-IB12B- : pulmonary embolism, Grade 4A 57-year-old black male with metastatic pheochromocytoma and history of notable for obesity, hypertension, and prior thrombus of the tibial vein, developed a pulmonary embolism approximately 6 weeks after receipt of the first therapeutic dose of 131I-MIBG. The patient received enoxaparin prophylaxis given his history of prior thrombus and thrombocytosis recognized prior to receipt of 131I-MIBG. The investigator assessed the event as possibly related to 131I-MIBG given the temporal relationship.
Reviewer comment: Multiple confounding factors, including prior history of thrombus, obesity, and underlying malignancy, likely contributed to the event.
MIP-IB12B- : pulmonary embolism, Grade 4A 56-year-old white male with pheochromocytoma metastatic to mediastinal and retroperitoneal lymph nodes, also with paraplegia, diabetes, and dyslipidemia developed a right lower lobe pulmonary embolism 3 months after receiving his only therapeutic dose of 131I-MIBG. The event was attributed as related to the study medication.
Reviewer comment: Multiple confounding factors, including the patient’s widely metastatic disease, vascular risk factors, and paraplegia, likely contributed to the event.
MIP-IB12B- : dyskinesia, Grade 3A 59-year-old white male with a right adrenal pheochromocytoma developed involuntary movements of his left arm approximately 16 days after receiving the first therapeutic dose of 131I-MIBG; the event recurred and prompted hospitalization, during which time it was noted that the patients’ blood pressure was fluctuating greatly, with measurements of 99-185 systolic and 69 – 94 diastolic. A workup including MRI, CT, and ECG did not demonstrate a definitive etiology of the neurologic symptoms, and it was hypothesized that the blood pressure fluctuation was responsible for the neurologic symptoms.
Reference ID: 4297867
(b) (6)
(b) (6)
(b) (6)
(b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
110Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Reviewer comment: Catecholamine-related cardiovascular events such as hypertension are well-documented in the literature following administration of NA-131I-MIBG in patients with pheochromocytoma. This reviewer agrees with the assessment of causality as potentially related to the study medication.
MIP-IB12B- : Dyspnea, Grade 4; aspartate aminotransferase increased, Grade 3A 67-year-old white male with pheochromocytoma metastatic to the lung, liver, and bone, and history of diabetes, right nephrectomy, as well as baseline dyspnea developed Grade 4 dyspnea the day following infusion of 131I-MIBG; Grade 3 AST elevation was noted during the hospitalization, and improved during the hospitalization. The patient also developed lower extremity edema and renal injury. The patient’s renal injury was attributed to diuresis in the setting of presumed congestive heart failure and volume overload, and peripheral edema related to lymphatic obstruction due to abdominal tumor burden. Imaging demonstrated widely metastatic progressive disease.
Reviewer comment: Though the events are attributed to 131I-MIBG, the case is highly confounded and the patient’s underlying medical condition could have contributed to the observed serious adverse events.
MIP-IB12B EAP_ : Pneumonitis, Grade 5; pulmonary embolism, Grade 3A 69-year-old male with metastatic pheochromocytoma enrolled in the MIP-IB12B Expanded Access Program (EAP) developed pneumonitis and a pulmonary embolism 9 weeks after receiving a single dose (521.9 mCi) of 131I-MIBG. The patient was treated with steroids as well as antifungal, antiviral, and antibiotic medications, and was placed on a ventilator. A CT scan demonstrated a ground-glass appearance throughout both lungs with focal areas of consolidation deemed most likely of infectious etiology per the narrative provided by the Applicant. The patient did not have evidence of pulmonary metastatic disease. The patient subsequently developed a right pneumothorax and deteriorated. Twelve days after the initial diagnosis of pneumonitis, the patient developed hypotension and asystole during a central line replacement, and was unable to be resuscitated. An autopsy was not performed. The investigator assessed the event of pneumonitis as related to study drug, and the Applicant assessed the event as possibly related. Pulmonary embolism was assessed by the Applicant as possibly related, and pneumothorax was assessed as not related. Reviewer note: This reviewer agrees with the assessment of the event of pneumonitis as possibly related to 131I-MIBG. Serious lung toxicity including acute respiratory distress syndrome and bronchiolitis obliterans organizing pneumonia has been described in the acute and subacute setting in relation to NA-131I-MIBG therapy in the literature.[7] Radiation therapy in general is also known to cause pneumonitis. As noted in regard to previously discussed cases, there is not a recognized mechanism for the development of pulmonary embolism as a direct result of 131I-MIBG therapy, and the patient’s metastatic cancer poses a clear risk factor. This case occurred on the MIP-IB12B EAP; as such, the event is not included in summary tabulations of adverse events which occurred in the MIP-IB12B/MIP-IB12 safety population.
Reference ID: 4297867
(b) (6)
(b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
111Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Serious pulmonary adverse events were identified in a small number of patients in the primary safety population and one patient in the expanded access program (MIP-IB12B EAP_ , pneumonitis). Reported preferred terms included acute respiratory failure, pulmonary embolism, dyspnea, pleural effusion, pneumothorax, pleural effusion, pneumonia, and pulmonary edema. With the exception of the case of pneumonitis already discussed which occurred in the expanded access program, these cases were attributed to disease (lung metastases associated with pneumothorax or malignant pleural effusion) or distinct disease-related processes (worsening of congestive heart failure, acute-on-chronic respiratory failure, infectious process). Given the presence of additional disease processes that may account for the observed pulmonary events, this reviewer agrees with the assessment that these cases of severe pulmonary adverse events are unlikely to be related to the study product.
Two cases of acute renal failure were reported as serious adverse events; both events were Grade 3 and demonstrated recovery. Neither was assessed as related to the study product, and both were associated with confounding factors (hypertensive urgency and diuresis) that render attribution to 131I-MIBG unlikely. Renal failure is a concern based on published literature of NA-131I-MIBG, and an integrated analysis of renal toxicity in the safety population is found in section 8.2.5.
Table 35: Serious Adverse Events, Study MIP-IB12/MIP-IB12B Safety Population, Primary Analysis
Preferred Term All Grades (N=88)
All Grades %
Grade ≥3(N=88)
Grade ≥3 %
Disease progression 16 18.18 16 18.18Thrombocytopenia 13 14.77 13 14.77Anemia 4 4.55 4 4.55Febrile neutropenia 4 4.55 3 3.41Leukopenia 4 4.55 4 4.55Neutropenia 4 4.55 4 4.55Malignant neoplasm progression
4 4.55 4 4.55
Constipation 3 3.41 2 2.27Dehydration 3 3.41 2 2.27Pathological fracture 3 3.41 3 3.41Pheochromocytoma 3 3.41 3 3.41Dyspnea 3 3.41 2 2.27Pulmonary embolism 3 3.41 3 3.41Pancytopenia 2 2.27 2 2.27Abdominal pain 2 2.27 2 2.27Non-cardiac chest pain 2 2.27 1 1.14Pneumonia 2 2.27 2 2.27
Reference ID: 4297867
(b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
112Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Myelodysplastic syndrome 2 2.27 2 2.27Renal failure acute 2 2.27 2 2.27Hypertension 2 2.27 2 2.27Hypertensive crisis 2 2.27 2 2.27Blood disorder 1 1.14 1 1.14Angina pectoris 1 1.14 0 0.00Cardiomyopathy 1 1.14 1 1.14Cardio-respiratory arrest 1 1.14 1 1.14Tachycardia 1 1.14 1 1.14Ventricular tachycardia 1 1.14 0 0.00Adrenal insufficiency 1 1.14 1 1.14Hypothyroidism 1 1.14 0 0.00Diarrhea 1 1.14 1 1.14Gastrointestinal obstruction 1 1.14 1 1.14Ileus 1 1.14 1 1.14Intestinal perforation 1 1.14 1 1.14Chest pain 1 1.14 1 1.14Cholangitis acute 1 1.14 1 1.14Hepatic failure 1 1.14 1 1.14Cellulitis 1 1.14 1 1.14Infection 1 1.14 1 1.14Localized infection 1 1.14 1 1.14Sepsis 1 1.14 1 1.14Staphylococcal infection 1 1.14 0 0.00Staphylococcal sepsis 1 1.14 1 1.14Radius fracture 1 1.14 1 1.14Subdural hematoma 1 1.14 1 1.14Tendon injury 1 1.14 1 1.14Aspartate aminotransferase increased
1 1.14 1 1.14
Blood pressure abnormal 1 1.14 0 0.00Neutrophil count decreased 1 1.14 1 1.14Platelet count decreased 1 1.14 1 1.14White blood cell count decreased
1 1.14 1 1.14
Metabolic acidosis 1 1.14 1 1.14Groin pain 1 1.14 1 1.14Muscular weakness 1 1.14 1 1.14Spinal disorder 1 1.14 1 1.14Spinal instability 1 1.14 1 1.14Acute lymphocytic leukemia 1 1.14 1 1.14Acute myeloid leukemia 1 1.14 1 1.14
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
113Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Bladder cancer 1 1.14 1 1.14Colon cancer metastatic 1 1.14 1 1.14Lung adenocarcinoma 1 1.14 0 0.00Metastases to bone 1 1.14 1 1.14Tumor pain 1 1.14 1 1.14Convulsion 1 1.14 1 1.14Dyskinesia 1 1.14 1 1.14Seizure 1 1.14 1 1.14Spinal cord compression 1 1.14 1 1.14Thalamic infarction 1 1.14 1 1.14Mental status changes 1 1.14 1 1.14Suicide attempt 1 1.14 1 1.14Hydronephrosis 1 1.14 1 1.14Kidney fibrosis 1 1.14 1 1.14Nephrolithiasis 1 1.14 0 0.00Acute respiratory failure 1 1.14 1 1.14Pleural effusion 1 1.14 1 1.14Pneumothorax 1 1.14 0 0.00Pulmonary edema 1 1.14 1 1.14Accelerated hypertension 1 1.14 1 1.14Deep vein thrombosis 1 1.14 0 0.00Hypotension 1 1.14 0 0.00Peripheral ischemia 1 1.14 1 1.14
Source: ADAE.xpt
Dropouts and/or Discontinuations Due to Adverse Effects
As only one therapeutic dose was administered in Study MIP-IB12, a discussion of therapeutic discontinuation is not meaningful, and this discussion is confined to study MIP-IB12B. As discussed in Patient Disposition, Section 0, 18 of 68 patients (26.5%) in this study received only one therapeutic dose. Reasons for discontinuation during the efficacy phase included progressive disease (n=6), adverse events (n=8), death (n=3), lost to follow-up (n=2) and receipt of an alternative anticancer therapy (n=4). Adverse events causing discontinuation prior to the second therapeutic dose were primarily hematologic toxicities, and included thrombocytopenia (n=2), anemia (n=2), decreased white blood cells (n=2), nausea and vomiting (n=1), and multiple hematologic adverse events (thrombocytopenia, leukopenia, neutropenia, hemolytic anemia; n=1). Causes of death which prevented continued trial participation included renal failure secondary to disease/ascites (n=1), metabolic acidosis (n=1), and bowel perforation (n=1). Twenty-nine patients discontinued the study during the long-term follow-up phase; reasons included death (n=2), adverse events (n=6), progressive disease (n=10), receipt of an alternative anticancer therapy (n=8) and patient withdrawal of consent (n=3).
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
114Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Causes of death during long-term follow-up included progressive disease (n=4), acute myeloid leukemia (n=1), and “hematologic decompensation” (n=1). Please see discussion of these cases under Section 8.4.1. Adverse events causing discontinuation during long-term follow-up included myelodysplastic syndrome (n=1) and metastatic colon cancer (n=1).
Significant Adverse Events
Adverse events of special interest (AESIs) were defined as AEs that are related to the acute and chronic effects of radiation toxicity seen at any time post dosimetric dose during the study. The Applicant defined AESIs using an extensive list of adverse events, notably endocrine (hypothyroidism), gastrointestinal (abdominal pain, malabsorption), genitourinary (elevated creatinine, elevated urea, renal failure), hepatic (elevated liver function tests, hepatic insufficiency), malignancies (sarcomas, leukemias, myelodysplastic syndrome), and pulmonary (bronchiolitis, pulmonary fibrosis). Additional preferred terms, such as those representing salivary gland toxicities (dry mouth, salivary gland pain, salivary gland enlargement) and additional renal toxicities (kidney fibrosis, proteinuria, and glomerular filtration rate decreased) were added by this reviewer.
Hypothyroidism occurred in 3 patients (3.4%); all patients were treated with iodine for thyroid protection. One case occurred within the first month of treatment in a patient previously treated with NA-131I-MIBG. The remaining cases occurred 108 and 536 days after administration of the first therapeutic dose of 131I-MIBG.
Myelosuppression was common following administration of 131I-MIBG; blood count nadirs (median) occurred 4 – 8 weeks following administration of therapeutic doses of 131I-MIBG. The median time to platelet count nadir was 4.3 weeks following therapeutic dose 1 and 5 weeks for therapeutic dose 2. Median times to neutrophil count and hemoglobin nadir were 5.4 weeks and 6.7 weeks, respectively following therapeutic dose 1; following therapeutic dose 2, the median times to nadir were 6 weeks and 8 weeks, respectively. The median time to recovery from nadir was 2.1 weeks for platelets and neutrophils following therapeutic dose 1, and approximately 3 weeks following therapeutic dose 2. On Study MIP-IB12B, 24% (n=16) of patients received one or more red blood cell transfusion during the study (1 – 6 transfusions), and 16% (n=11) of patients received platelet transfusions (1 – 5 transfusions per patient). Approximately 9% (n=6) of patients received granulocyte colony stimulating factor (G-CSF) and 3% (n=2) received erythropoietin.
Two new primary solid malignancies have been diagnosed in patients exposed to 131I-MIBG; this excludes a patient who was diagnosed with bladder cancer during screening. One patient, a 56-year-old female, developed adenocarcinoma of the lung 815 days (>2 years) after administration of the first therapeutic dose of 131I-MIBG; prior therapy included only surgical resection, and she received 2 doses of 131I-MIBG on study. The event of metastatic colon cancer, which occurred in a different 54-year-old female, was recorded 551 days (18 months) after administration of the first therapeutic dose; the patient who experienced this event also developed myelodysplastic syndrome (diagnosed within months of the diagnosis of colon
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
115Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
cancer). The patient’s prior therapy included resection, 3 rounds of EBRT as well as doxorubicin. The patient received two therapeutic doses of 131I-MIBG on study.
Secondary malignancies in patients who have received 131I-MIBG are reported in the literature, and largely consist of myelodysplastic syndrome and leukemia (see section 8.2.5). The case of adenocarcinoma of the lung occurred in a patient who did not have prior treatment-related risk factors such as radiation; it is unclear whether additional risk factors such as smoking were present. Non-small cell lung cancer has not been reported in the literature among patients who have received 131I-MIBG according to literature searches performed by the Applicant and by this reviewer, though a theoretical risk remains given the radiation exposure associated with the product. 131I-MIBG could potentially have contributed to the development of the case of colon cancer described above, though, similarly, this second malignancy is not reported in the literature. However, the latter patient’s prior therapy with multiple courses of radiation is the more likely culprit.
Factors limiting the analysis of secondary malignancy with 131I-MIBG include the small population studied and limited follow-up. Study MIP-IB12 did not include long-term follow-up, thus second malignancy data is available primarily for patients treated on study MIP-IB12B (n=68). In addition, discontinuations due to progressive disease or for other reasons limited collection of long-term adverse event data; while a 5-year long-term follow-up was intended, the median duration of follow-up for patients on study MIP-IB12B was 1.6 years. While the lack of long-term follow-up data could artificially lower the estimate of secondary malignancies associated with 131I-MIBG, the heavily pre-treated population could lead to overestimation of the incidence of secondary malignancies due to 131I-MIBG. The incidence of second malignancies in patients receiving 131I-MIBG should therefore be followed prospectively to attain a more accurate understanding of this risk.
Table 36: Adverse Events of Special Interest, Studies MIP-IB12/MIP-IB12B, Primary Analysis
Preferred Term All Grades (N = 88)
All Grades %
Grades ≥3(N=88)
Grades ≥3%
Blood and lymphatic system disordersThrombocytopenia 59 67.0% 31 35.2%Anemia 51 58.0% 15 17.0%Leukopenia 50 56.8% 33 37.5%Neutropenia 48 54.5% 32 36.4%Lymphopenia 11 12.5% 5 5.7%Febrile neutropenia 4 4.5% 3 3.4%Pancytopenia 4 4.5% 4 4.5%Anemia macrocytic 1 1.1% 0 0.0%Blood disorder 1 1.1% 1 1.1%Gastrointestinal disorders
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
116Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Nausea 69 78.4% 14 15.9%Vomiting 49 55.7% 8 9.1%Dry mouth 42 47.7% 2 2.3%Diarrhea 22 25.0% 3 3.4%Salivary gland pain 19 21.6% 0 0.0%Abdominal pain 17 19.3% 5 5.7%Constipation 17 19.3% 6 6.8%Abdominal distension 5 5.7% 0 0.0%Abdominal pain upper 4 4.5% 0 0.0%Retching 4 4.5% 2 2.3%Salivary gland enlargement 3 3.4% 0 0.0%Stomatitis 3 3.4% 0 0.0%General disorders and administration site conditionsFatigue 55 62.5% 22 25.0%Asthenia 18 20.5% 7 8.0%Decreased activity 1 1.1% 1 1.1%InvestigationsWeight decreased 14 15.9% 1 1.1%Endocrine disordersHypothyroidism 3 3.4% 0 0.0%Neoplasms benign, malignant and unspecified (including cysts and polyps)Malignant neoplasm progression
4 4.5% 4 4.5%
Cancer pain 3 3.4% 0 0.0%Pheochromocytoma 3 3.4% 3 3.4%Myelodysplastic syndrome 2 2.3% 2 2.3%Acute lymphocytic leukemia 1 1.1% 1 1.1%Acute myeloid leukemia 1 1.1% 1 1.1%Bladder cancer 1 1.1% 1 1.1%Colon cancer metastatic 1 1.1% 1 1.1%Lung adenocarcinoma 1 1.1% 0 0.0%Metastases to bone 1 1.1% 1 1.1%Tumor pain 1 1.1% 1 1.1%Renal and Urinary Disorders (includes renal/urinary investigations)Proteinuria 12 13.6 0 0.0Blood creatinine increased 8 9.1 0 0.0Renal failure* 6 6.8 2 2.3Blood urea increased 4 4.5 0 0.0Acute kidney injury** 1 1.1 1 1.1Hematuria 4 4.5 0 0.0Blood urine present 2 2.3 0 0.0Kidney fibrosis 1 1.1 1 1.1
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
117Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Nephrolithiasis 1 1.1 0 0.0Urinary bladder hemorrhage 1 1.1 0 0.0Urinary incontinence 1 1.1 0 0.0Glomerular filtration rate decreased
1 1.1 0 0.0
Source: adae.xpt
Renal failure (preferred terms “renal failure” and “renal failure acute”) was observed in 6.8% (n=6) of patients in the MIP-IB12/MIP-IB12B safety population, demonstrated in Table 37: Cases of Renal Failure in the MIP-IB12/MIP-IB12B Safety Population. Of these cases, 2 were assessed as related to the study product (event Grades 1 and 2). Of these cases, one (a Grade 2 event which occurred 22 days after treatment) was not resolved and resulted in a dose reduction. Among the cases assessed as not related to 131I-MIBG, two were Grade 3. Narratives for these events were reviewed and this reviewer agrees with the Applicant’s assessment that these cases were likely not related to 131I-MIBG, as significant confounding factors (hypotension, concomitant medications, progressive disease) existed. Four of the six cases occurred in patients with baseline impairment in GFR (<90 mL/min/1.73 m2).
Among the additional reported renal adverse events, several (glomerular filtration rate decreased, hematuria) have a potential relationship to radiation toxicity. Renal fibrosis is a potential complication of radiation exposure; however, the narrative provided by the Applicant for the event kidney fibrosis suggests that this condition pre-dated administration of 131I-MIBG. This reviewer agrees with that assessment. It is notable that several patients experienced a decrease in GFR that was not reported as an adverse event, thus the incidence of “glomerular filtration rate decreased” indicated in the table above, based on dataset analysis, is incorrect. A discussion of these findings is found later in this review, Section 8.2.4: Laboratory Analyses.
Table 37: Cases of Renal Failure in the MIP-IB12/MIP-IB12B Safety Population
Patient ID Grade Relatedness (Applicant
assessment)
Recovery Notes
3 Not related Yes Occurred 4 days after therapeutic dose. Confounded by hypotension,
significant tumor burden and baseline mild renal impairment.
2 Related No Occurred 22 days after therapeutic dose, subsequent dose reduced.
2 Not related Yes Occurred 4 years after first therapeutic dose, during treatment for acute lymphoblastic leukemia.
Confounded by leukemia and
Reference ID: 4297867
(b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
118Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
subsequent chemotherapy. Agree with Applicant’s assessment.
1 Related Unknown Occurred on day 391 post treatment, unclear etiology and
recovery status unknown. 1 Not related No Occurred on day 195 post
treatment with unclear etiology, not resolved at time of death due
to PD. Patient experienced hydronephrosis due to progressive retroperitoneal lymphadenopathy,
which was recorded as resolved prior to onset of renal
insufficiency.3 Not related Yes Occurred 9 months after first
therapeutic dose,concurrent with hypertensive
urgency (likely etiology)
Treatment Emergent Adverse Events and Adverse Reactions
Treatment emergent adverse events were consistent with the expected adverse events associated with 131I-MIBG based on prior experience with NA-131I-MIBG and experience with other radiotherapeutics. The gastrointestinal adverse events observed could be associated with the product (as suspected in the case of nausea and vomiting) or with the underlying disease (in the cases of constipation and abdominal pain), or both. Myelosuppression was common, with a delayed onset as discussed in detail above. Salivary gland toxicity was also common, with 40.1% of patients experiencing symptoms of sialoadenitis and 47.7% of patients experiencing dry mouth. Myelosuppression and sialoadenitis are recognized adverse events associated with NA 131I-MIBG based on published experience.
Table 38: Treatment Emergent Adverse Events in at Least 5% of Patients, Studies MIP-IB12/MIP-IB12B Safety Population, Primary Analysis
Preferred Term All Grades (N = 88)
All Grades %
Grades ≥3(N=88)
Grades ≥3%
Nausea 69 78.4% 14 15.9%Fatiguea 62 70.5% 23 26.1%Thrombocytopenia 59 67.0% 31 35.2%Anemia 51 58.0% 15 17.0%Leukopenia 50 56.8% 33 37.5%
Reference ID: 4297867
(b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
119Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Vomiting 49 55.7% 8 9.1%Neutropenia 48 54.5% 32 36.4%Dry mouth 42 47.7% 2 2.3%Sialoadenitisb 36 40.1% 1 1.1%Dizziness 30 34.1% 11 12.5%Headache 28 31.8% 5 5.7%Decreased appetite 26 29.5% 4 4.5%Diarrhea 22 25.0% 3 3.4%Abdominal painc 22 25.0% 5 5.7%Hypotension 21 23.9% 4 4.5%Asthenia 18 20.5% 7 8.0%Hypertensiond 18 20.5% 10 11.4%White blood cell count decreased
18 20.5% 11 12.5%
Constipation 17 19.3% 6 6.8%Cough 16 18.2% 0 0.0%Disease progression 16 18.2% 16 18.2%Dyspnea 16 18.2% 6 6.8%Back pain 15 17.0% 2 2.3%Dysgeusia 15 17.0% 1 1.1%Neutrophil count decreased
15 17.0% 11 12.5%
Platelet count decreased
15 17.0% 10 11.4%
Dehydration 14 15.9% 4 4.5%Weight decreased 14 15.9% 1 1.1%International normalized ratio increased
13 14.8% 1 1.1%
Pain in extremity 13 14.8% 0 0.0%Oropharyngeal pain 12 13.6% 0 0.0%Pyrexia 12 13.6% 2 2.3%Aspartate aminotransferase increased
11 12.5% 2 2.3%
Hemoglobin decreased 11 12.5% 3 3.4%Lymphopenia 11 12.5% 5 5.7%Edema peripheral 11 12.5% 1 1.1%Urinary tract infection 10 11.4% 1 1.1%Alopecia 9 10.2% 0 0.0%Blood alkaline phosphatase increased
9 10.2% 1 1.1%
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
120Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Dyspepsia 9 10.2% 0 0.0%Hyperhidrosis 9 10.2% 0 0.0%Injection site pain 9 10.2% 0 0.0%Tachycardia 9 10.2% 3 3.4%Epistaxis 8 9.1% 0 0.0%Insomnia 8 9.1% 0 0.0%Orthostatic hypotension
8 9.1% 0 0.0%
Palpitations 8 9.1% 1 1.1%Proteinuria 8 9.1% 0 0.0%Prothrombin time prolonged
8 9.1% 0 0.0%
Arthralgia 7 8.0% 0 0.0%Blood creatinine increased
7 8.0% 0 0.0%
Chills 7 8.0% 0 0.0%Dry skin 7 8.0% 0 0.0%Hematocrit decreased 7 8.0% 1 1.1%Neck pain 7 8.0% 0 0.0%Red blood cell count decreased
7 8.0% 1 1.1%
Alanine aminotransferase increased
6 6.8% 2 2.3%
Dysphagia 6 6.8% 0 0.0%Infusion site pain 6 6.8% 0 0.0%Lymphocyte count decreased
6 6.8% 1 1.1%
Nasal congestion 6 6.8% 0 0.0%Pain in jaw 6 6.8% 0 0.0%Petechiae 6 6.8% 2 2.3%Renal failuree 6 6.8% 2 2.3%Abdominal distension 5 5.7% 0 0.0%Ageusia 5 5.7% 0 0.0%Candida infection 5 5.7% 1 1.1%Chest pain 5 5.7% 1 1.1%Contusion 5 5.7% 3 3.4%Gastroesophageal reflux disease
5 5.7% 0 0.0%
Muscle spasms 5 5.7% 0 0.0%Nasopharyngitis 5 5.7% 1 1.1%Sinusitis 5 5.7% 0 0.0%
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
121Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Syncope 5 5.7% 3 3.4%Upper respiratory tract infection
5 5.7% 1 1.1%
a Fatigue is a composite term and includes “fatigue” and “asthenia.”b Sialoadenitis is a composite term and includes “sialoadenitis,” “salivary gland pain,” and “salivary gland enlargement.” c Abdominal pain includes the preferred terms “abdominal pain,” abdominal pain upper,” and “abdominal pain lower.” d Hypertension includes “blood pressure increased” and “hypertension.”e Renal failure includes preferred terms “renal failure” and “renal failure acute”.
An analysis of treatment-emergent adverse events by number of therapeutic doses administered was conducted. At the system organ class level, the incidence of most TEAEs was comparable between patients who received one dose compared to those who received two doses. The exception was in the class ‘infections and infestations,’ which occurred in 38.5% of patients who received one dose and 64.0% of patients who received two doses; this difference is likely related to the increased total duration of myelosuppression experienced by patients who received two doses of 131I-MIBG. The most common infections experienced included urinary tract infection, upper respiratory tract infection, candida infection, and sinusitis.
As expected, a greater percentage of patients who received two doses of 131I-MIBG experienced cytopenias (preferred terms: thrombocytopenia, anemia, leukopenia, neutropenia). A greater percentage of patients who received two doses of 131I-MIBG also experienced sialoadenitis (48% vs. 33.3%, preferred terms: sialoadenitis, salivary gland pain, and salivary gland enlargement). Renal failure (preferred terms: renal failure and renal failure acute) was also more common amongst patients who received two doses of 131I-MIBG (10% vs. 2.6%). There was no significant difference in the development of hypothyroidism (4% vs. 2.6%). All patients (100%) who developed leukemia or MDS had received two doses of 131I-MIBG on study. The analysis of adverse events by number of doses of 131I-MIBG administered confirmed an increased incidence of sialoadenitis, renal failure, cytopenias, MDS and leukemias.
Laboratory Findings
On Study MIP-IB12B, laboratory tests to assess hematology parameters (complete blood count), liver function (alkaline phosphatase, ALT, AST, bilirubin), chemistries (sodium, potassium, chloride, BUN, creatinine, glucose), and minerals (phosphorus, calcium) were measured every week from weeks 2-24, and weekly for 12 weeks following the administration of the second dose. On study MIP-IP12B, patients were excluded if they had AST or ALT ≥2.5 times the upper limit of normal (ULN), bilirubin > 1.5 x ULN, platelets ≤80,000/µL, or absolute neutrophil count ≤1,200/µL.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
122Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
The reviewer analyzed the ADLB.xpt data from the baseline and subsequent measurements. Table 39 summarizes the common laboratory abnormalities for patients in Studies MIP-IB12 and MIP-IB12B. Grade 3 or 4 chemistry abnormalities in more than 2% of patients included elevations in serum glucose, alkaline phosphatase (4.5%), alanine aminotransferase (2.3%), aspartate aminotransferase (2.3%), potassium (4.5%), and bilirubin (3.4%). Changes in hematologic parameters were very common in the study. Grade 3-4 anemia was experienced by 23.9% of patients. Grade 3-4 thrombocytopenia was seen in 50% of patients, with Grade 3-4 neutropenia and lymphopenia occurring in 59.1% and 77.3% of patients, respectively. Thirty-three percent of patients demonstrated Grade 4 thrombocytopenia, and Grade 4 neutropenia and anemia were experienced by 16% and 7% of patients, respectively. In Study IB12B following the first therapeutic dose, patients who experienced Grade 4 neutropenia reached neutrophil nadir at a median of 36 days (27 – 55 days) and remained at nadir for a median of 12 days (8 – 22 days) until recovery to less than or equal to Grade 3. Following the second dose, patients who experienced Grade 4 neutropenia reached nadir at a median of 43 days (38 – 47 days) and remained at nadir for a median of 18.5 days (8 – 31 days) until recovery to less than or equal to Grade 3.
Table 39: Laboratory Abnormalities, Studies MIP-IB12B & MIP-IB12
Category Laboratory Parameter All Grades All Grades %
Grade ≥3 Grade ≥3 % (N=88)
Lymphopenia 84 95.5% 68 77.3%Leukopenia 83 94.3% 54 61.4%Anemia 82 93.2% 21 23.9%Thrombocytopenia 80 90.9% 44 50.0%
HEMATOLOGY
Neutropenia 74 84.1% 52 59.1%Hyperglycemia 83 94.3% 16 18.2%Hypertriglyceridemia 58 65.9% 1 1.1%Hypercholesterolemia 53 60.2% 0 0.0%Increased Alkaline Phosphatase
47 53.4% 4 4.5%
Increased Aspartate Aminotransferase
44 50.0% 2 2.3%
Increased Alanine Aminotransferase
38 43.2% 2 2.3%
Increased Creatinine 34 38.6% 0 0.0%Hyperkalemia 16 18.2% 4 4.5%Increased Bilirubin 13 14.8% 3 3.4%Hypercalcemia 7 8.0% 1 1.1%
CHEMISTRY
Hypernatremia 4 4.5% 0 0.0%Source: adlb.xpt, primary analysis
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
123Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Coagulation parameters were not consistently measured across the pooled safety population. The international normalized ratio (INR) was measured only in Study MIP-IB12B (n=68), and activated partial thromboplastin time (aPTT) was measured only in study MIP-IB12 (n=21). Therefore, these tables are presented separately below. Prothrombin time was measured, but is not graded in CTCAE version 3.
Table 40: International Normalized Ratio Abnormalities, Study MIP-IB2B
Laboratory Parameter All Grades All Grades %
Grade 3* Grade 3 % (N=68)
International Normalized Ratio 58 85.3% 12 17.6%*CTCAE version 3 reports only Grades 0 – 3 for INR; there were no Grade 4 events.
Table 41: Activated Partial Thromboplastin Time Abnormalities, Study MIP-IB2
Laboratory Parameter All Grades All Grades %
Grade 3* Grade 3 % (N=21)
Activated Partial Thromboplastin Time
14 66.7% 4 19.0
*CTCAE version 3 reports only Grades 0 – 3 for aPTT; there were no Grade 4 events.
Despite the high incidence of laboratory evidence of coagulopathy, there were no serious bleeding events attributed to treatment with 131I-MIBG in the pooled safety population. One patient developed a subdural hematoma three years after administration of the second therapeutic dose of 131I-MIBG in the setting of thrombocytopenia (platelet count 24 x 103/uL) while receiving warfarin. This event was attributed to disease progression.
Reviewer comment: This reviewer agrees that this event was unlikely to be attributable to 131I-MIBG. The high incidence of low-grade elevation of clotting parameters may be related to nutritional deficiency of vitamin K in the setting of poor oral intake in patients with nausea and vomiting, and the use of anticoagulation as either prophylaxis or treatment of thromboses.
Laboratory Evidence of Renal ImpairmentThirty-eight percent of patients experienced an increase in creatinine, all of which were Grade 1 or 2. Of these 34 events, all but 7 returned to baseline. Decrease in creatinine clearance (CrCl) or glomerular filtration rate (GFR) is not graded in the CTCAE independently of chronic renal failure and thus is not reflected in Error! Reference source not found.. This reviewer performed an analysis of GFR decreases among patients on Study MIP-IB12B, as GFR was not measured on Study MIP-IB12, but was extrapolated from serum creatinine measurements. GFR was measured by 99mTc diethylenetriaminepentaacetic acid (DPTA) analysis or approximated by 24-hour creatinine clearance at 6 months and 12 months on study. This analysis did not include decreases in GFR which remained in the same range of severity, i.e., mild impairment (60 – 89 ml/min/1.73m2), moderate impairment (30 – 59 ml/min/1.73m2), or severe impairment (15 –
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
124Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
29 mL/min/1.73m2), and excluded decreases of < 10% given the uncertain clinical significance of these small changes. Overall, 16 of 68 patients (24%) demonstrated a significant decrease in GFR which resulted in a shift in range of severity at either the 6-month or 12-month assessment; it should be noted that, due to study discontinuation, not all patients had two GFR measurements or estimates available for comparison. Six patients with normal GFR at baseline had a mildly decreased GFR at 12-month follow-up, and two had a moderate decrease. Seven patients with a mildly decreased GFR had a moderate decrease at 12 months, and one patient with a moderately decreased GFR at baseline had a severely decreased GFR at 12 months. No patients required dialysis or demonstrated a GFR of <15 mL/minute. Only three of the 16 patients identified in this analysis had a reported adverse event of renal failure.
All patients who demonstrated a decrease in GFR had at least one additional risk factors for renal impairment. Seven of these 16 patients (44%) had a history of type 1 or type 2 diabetes mellitus, including the patient who progressed to a severely decreased CrCl; all had a history of hypertension. As GFR was only collected at baseline, 6 months, and 12 months on Study MIP-IB12B, in many cases it is impossible to determine whether the decrease in renal function was recovered. Seven of 16 patients demonstrated a decreased GFR at 6 months. Of these, 3 did not have subsequent 12 month measurements. The remaining 4 had subsequent measurements; two demonstrated recovery to baseline range of severity, and two demonstrated improvement but incomplete recovery at 12 months. In most (9/16) cases the decrease in GFR or calculated creatinine clearance was identified at the 12-month measurement, and no follow-up measurement was required per protocol.
A limitation to the analysis of renal failure is the lack of laboratory data beyond 12 months. Neither GFR nor serum creatinine were required to be measured beyond 12 months. Renal failure, renal dysfunction, and elevated creatinine were among AESIs required to be reported during long-term follow-up, but it is possible that renal dysfunction occurring beyond 12 months was unrecognized if no overt clinical signs of renal failure were present and laboratory evaluations were not performed. A universal confounding factor in the assessment of renal dysfunction in this population is the presence of hypertension, often longstanding, which itself may contribute to the observed renal impairment.
Hepatic Toxicity and Hy’s law Two patients experienced laboratory findings consistent with Hy’s law restricting to patients with concurrent elevation of AST or ALT ≥ 3 times the upper limit of normal (ULN) and bilirubin ≥2 x ULN. A causality analysis was conducted for each of these patients; however, due to other factors, neither clearly fit all criteria for Hy’s law. Brief narratives are provided below.
Patient MIP-IB12B- : A 67-year-old white male with pheochromocytoma metastatic to the liver and bone and baseline liver dysfunction (Grade 1 total bilirubin and Grade 1 AST and ALT elevations) experienced Grade 3 AST and total bilirubin elevation two days following the administration of the first therapeutic dose. The patient experienced Grade 4 dyspnea and right upper quadrant pain, in association with liver enlargement and metastatic pulmonary disease,
Reference ID: 4297867
(b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
125Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
beginning on the first day following the therapeutic dose. The patient was hospitalized and treated supportively. The laboratory abnormalities started to normalize within one week and did return to baseline (Grade 1). The patient died of progressive disease prior to receipt of a subsequent dose.
Reviewer comment: This event may represent a case of drug-induced liver injury. The liver injury was reversible, and occurred in the setting of liver metastases.
Patient MIP-IB12- : A 72-year-old black female with pheochromocytoma experienced Grade 5 hepatic failure attributed by the Applicant to metastatic pheochromocytoma 2.5 months after receipt of the only therapeutic dose. Elevated AST (Grade 1) and alkaline phosphatase (Grade 2) were first documented at baseline. Four weeks after infusion of the patient’s only dose of 131I-MIBG, the patient demonstrated elevated prothrombin time (PT) and international normalized ratio (INR), as well as persistently elevated AST, alkaline phosphatase, and ALT (Grade 1). The patient experienced hypoglycemia and bleeding while taking warfarin for a deep venous thrombosis, and reported a 30- to 45-pound weight loss over the past few months. One month after the initial presentation, she was found unresponsive and admitted with hepatic encephalopathy; during the admission, liver imaging demonstrated multiple hepatic masses with right portal vein reversal of flow. Despite aggressive treatment, the patient continued to decline, with metabolic acidosis, persistent thrombocytopenia, and coagulopathy. The peak laboratory abnormalities were: alkaline phosphatase Grade 3, AST Grade 2 and total bilirubin Grade 3; it is notable that these values were last reported approximately 6 days prior to the patients’ death. The patient died 2.5 months after receipt of the therapeutic dose of 131I-MIBG after being diagnosed with sepsis due to a urinary tract infection and acute organ failure.
Reviewer comment: The liver injury in this case is likely multifactorial. The patient appears to have had progressive metastatic disease with liver involvement. Thus, though 131I-MIBG therapy may have contributed to the liver injury given the timing and a potential mechanism, it is unlikely that 131I-MIBG is solely responsible for the liver injury. The patient’s death appears to be due to sepsis and acute organ failure.
Vital Signs
Vital signs were assessed prior to and within 4 hours following each dosimetric and therapeutic 131I-MIBG infusion. Patients enrolled on Study MIP-IB12B had additional vital sign assessments compared to patients enrolled on Study MIP-IB12, at 24 and 48 hours following each dosimetric dose, and 12 weeks after each therapeutic dose.
Fever was not common following 131I-MIBG infusions. One patient had a fever (temperature 38.3oC, increase of 1.3 o C from baseline) after completion of 131I-MIBG infusion.
Heart rate was considered abnormal if it met one of the following criteria: a) <55 beats per minute, b) >110 beats per minute, or c) represented a change of 20 beats per minute from the
Reference ID: 4297867
(b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
126Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
patient’s baseline. Thirteen (13, 14.7%) of 88 patients met one of these criteria at the post-infusion assessment, with 11 of these representing changes in heart rate of 20 beats per minute or more. Only one patient experienced a heart rate of >110 beats per minute.
The period immediately following 131I-MIBG infusion was examined for the occurrence of significant increases in blood pressure. Ten patients (11.4%) of the 88 patients experienced an increase in blood pressure immediately following a therapeutic 131I-MIBG administration, defined as one of the following: 1) a systolic blood pressure of ≥160 mm Hg and an increase of at least 20 mm Hg from pre-treatment baseline or 2) a diastolic blood pressure of ≥100 mm Hg and an increase of at least 10 mm Hg from pre-treatment baseline. No patients experienced systolic blood pressure >200 mm Hg or diastolic blood pressure greater than 110 mm Hg following infusion. As noted previously, five patients had a change in antihypertensive medication within 24 – 48 hours of the first 131I-MIBG therapeutic dose administration. All but one had an increase in the dose of an existing antihypertensive medication; one had a single intravenous dose of enalapril.
Reviewer note: Increases in blood pressure within the first 24 hours following infusion of therapeutic doses of 131I-MIBG were observed in a small percentage of patients. Given the nearly universal presence of hypertension (controlled or uncontrolled) in this population, close monitoring is warranted to avoid sequelae of severe, uncontrolled hypertension. See Section 13 for labeling recommendations.
Electrocardiograms (ECGs)
During Study MIP-IB12B, three 12-lead electrocardiograms (ECGs) were obtained at least 5 minutes apart within 2 hours prior to the dosimetric dose, then at 0-2 minutes, 10 ±2 minutes, 15 ±2 minutes, and 20 ±2 minutes post dosimetric dose. Additional 12-lead ECGs were obtained within 15 minutes prior to the second and third imaging I-131 scans and within 7 days after each therapeutic dose.
There were three arrhythmias reported as serious adverse events: ventricular tachycardia, tachycardia, and cardio-respiratory arrest (n= 1 each). All were assessed as not related to the study product and were reported as resolved; none of these events occurred in proximity to an infusion of 131I-MIBG, with times to onset from the most recent therapeutic infusion of 23 – 118 days. Seven non-serious events of tachycardia were assessed as possibly or probably related, or related to 131I-MIBG; these events occurred between 3 – 105 days after administration of 131I-MIBG and were mostly Grade 1 – 2 (one Grade 3). Given the pathophysiology of the underlying disease (with catecholamine excess), assessment of the etiology of these events is difficult. Events that occurred in proximity to an infusion are more likely to be related; only one event occurred < 1 week after a therapeutic infusion.
QT
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
127Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
In the MIP-IB12/12B safety population, there were no AEs with the preferred term of “ECG QT prolonged” or “Torsades de pointes” reported for the primary safety population. In the exposure-QTc analysis, there was no evident relationship between 131I-MIBG concentrations and ΔQTc. See the Clinical Pharmacology Review and QT-IRT consult for additional information regarding QT abnormalities and an assessment of the exposure-QTc analysis. A summary copied (with minor edits) from the QT-IRT consult, is included in the following paragraph.
Study IB12B supports no large mean increases in the ΔQTc interval (i.e., 20 ms) with the proposed therapeutic dosing regimen of 131I-MIBG, based on central tendency analysis (largest 90% CI upper bound was 14.5 ms at 1 h post-dose after administration of second therapeutic dose). None of the patients had QTcF >500 ms and only one patients had ΔQTcF >60 ms. There was no placebo or positive control in the study. In this open-label, multicenter, single-arm, Phase 2 study, 74 patients received dosimetric doses (3-6 mCi by IV injection) and 68 patients received at least one therapeutic dose of 131I-MIBG (500 mCi for patients > 62.5 kg and 8 mCi/kg for patients ≤ 62.5 kg; 2 therapeutic doses 3 months apart). Overall summary of findings is presented in Table 42.
Table 42: The Point Estimates and the 90% CIs of ΔQTcF Corresponding to the Largest Upper Bounds for All-Treated 131I-MIBG Group (FDA Analyses for Study IB12B)
The dose used in Study IB12B is the proposed therapeutic dose for the oncology indication and is the MTD. The drug elimination is primarily by the renal route, and renal impairment could likely increase the exposures. 131I-MIBG is rapidly cleared via the kidney; by 24 and 120 hours post-administration, ~50% and 80% of the injected dose was present in the urine, respectively, as per the proposed label. Thus, no accumulation is anticipated with the second therapeutic dose, as the therapeutic treatment consists of 2 single doses given at least 3 months apart. Because, the Study IB12B did not assess the PK, a concentration-QTc relationship was not evaluated for the product. The therapeutic dose of 131I-MIBG (500 mCi with μg MIBG) has comparable ( chemical mass dose range of MIBG as the two approved conventional MIBG imaging products, GE AdreView® (Iobenguane I-123 Injection) and Pharmalucence (Iobenguane Sulfate I-131 Injection). The literature data for commercial experience of these products do not suggest a cardiovascular risk.
Reference ID: 4297867
(b) (4)
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
128Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Immunogenicity
Not applicable.
8.2.5 Analysis of Submission-Specific Safety Issues
Leukemia and Myelodysplastic Syndrome
There were 6 reported cases of leukemia or myelodysplastic syndrome (MDS) which occurred following administration of a therapeutic dose of 131I-MIBG in the development program. The cases are presented below.
MIP-IB12B : This is a 56-year-old white female diagnosed with paraganglioma in 1996 who developed myelodysplastic syndrome 9 months after the first dose of Iobenguane I131. Her prior therapy included brachytherapy, 2500cGy external beam radiotherapy (EBRT) to the L1 vertebrae, and 2 prior doses of NA- 131I-MIBG. During study MIP-IB12B, she received 2 doses of Iobenguane I131. Bone marrow biopsy 9 months after the initial dose demonstrated refractory anemia with excess blasts, and cytogenetic analysis showed loss of chromosomes 3 and 5, gain of 22, and dicentric 17:20 translocation resulting in loss of 17p and 20q with a minor clone 13q deletion. She died approximately 6 weeks after the diagnosis of MDS.
MIP-IB12B- : This is a 52-year-old white female diagnosed with pheochromocytoma in February 2007 and metastatic pheochromocytoma in who developed MDS 21 months from the first dose of Iobenguane I131. Her prior therapy included resection, 3 rounds of EBRT ( ) as well as doxorubicin (unspecified dose, ). She received two therapeutic doses of 131I-MIBG in . The patient was diagnosed with metastatic colon cancer in and discontinued from the study. In , the patient was diagnosed with myelodysplastic syndrome. The bone marrow biopsy revealed “early stage MDS” and chromosome analysis was “consistent with myeloid malignancies.” The patient died from progression of metastatic colon cancer in
.
MIP-IB12B- : This is a 24-year-old Asian male diagnosed with pheochromocytoma in . His prior therapy included several cycles of cyclophosphamide, vincristine, and dacarbazine, as well as radiation therapy. He received two therapeutic doses of 131I-MIBG on study in
. He was treated with hydroxyurea for Sweet’s syndrome from . He was diagnosed with myelodysplastic syndrome in , 3 years and 5 months
after receipt of his first therapeutic dose of 131I-MIBG. The MDS was assessed as Grade 4 and related to study medication. The patient later died due to progressive disease.
MIP-IB12B- : This is a 47-year-old white male diagnosed with pheochromocytoma in who was diagnosed with acute lymphoblastic leukemia 4 years and 3 months from first dose of 131I-MIBG. The patient’s prior therapy for pheochromocytoma included right adrenalectomy
and NA- 131I-MIBG , total 556 mCi from 3 infusions). During
Reference ID: 4297867
(b) (6)
(b) (6)
(b) (6)
(b) (6) (b) (6)
(b) (6)
(b) (6)
(b) (6)
(b) (6)
(b) (6)
(b) (6)
(b) (6)
(b) (6)
(b) (6)
(b) (6)
(b) (6) (b) (6)
(b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
129Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
the study, the patient received two doses of 131I-MIBG in . In , the patient was diagnosed with acute lymphocytic leukemia. A bone marrow
biopsy and aspirate revealed remission of ALL as of . The event was assessed as related to study medication.
Reviewer note: While therapy-related AML is a well-established entity, therapy-related ALL is rarely reported. Giri et al report that therapy-related ALL comprises an estimated 1.2 – 2.5% of all cases of ALL. (Giri, 2015) Therefore, while the attribution to the study therapy is far less certain than for the cases of MDS/AML given that the relationship between prior chemo- or radiotherapy and ALL is less well-established, this reviewer agrees that the event of ALL is possibly related to the use of 131I-MIBG.
MIP-IB12B- : This is a 57-year-old Asian male diagnosed with pheochromocytoma in and experienced a recurrence in . The patient was diagnosed with acute myeloid
leukemia 17 months after the first dose of 131I-MIBG. He was treated with NA- 131I-MIBG in (3 doses), and (3 doses); not all doses are quantified, but the total
for 6 of 8 doses was 1209.4 mCi. Prior therapy also included received external beam radiation therapy (EBRT) in (dose not provided), chemotherapy with cisplatin, vincristine and doxorubicin ( ); cisplatin, bleomycin and etoposide ( ), capecitabine and temozolomide ) and sunitinib ( ). The patient received two therapeutic doses of 131I-MIBG ( ). The patient was diagnosed with acute myeloid leukemia in . He refused chemotherapy and chose to enter hospice. The patient died in during the LTFU phase of the study.
MIP-IB12B- : This is a 66-year-old male with pheochromocytoma who developed acute myeloid leukemia 7 years after the first dose of 131I-MIBG on study MIP-IB12B. He was previously treated with 2 courses of NA-131I-MIBG approximately 7.5 years prior to development of AML. The patient received two therapeutic doses of 131I-MIBG on study. Cytogenetic analysis of the myeloblasts was not provided. The event of Grade 4 AML was not resolved at the time of the 90-day safety report provided by the Applicant, and was assessed as related to 131I-MIBG.
Reviewer note: This case was provided as part of the 90-day safety update and is not be reflected in cumulative analyses derived from datasets (i.e., Table 30), but is included in FDA revisions to product labeling and text portions of this review.
Summary: AML, ALL and MDS occurred in 6 patients on study MIP-IB12B. All patients had received prior therapy, with 4 patients receiving prior NA-131I- MIBG, 4 patients receiving prior radiation therapy, and 3 patients receiving prior chemotherapy. All patients had received at least one of these treatment modalities. AML and MDS developed 9 months to 7 years from the first dose of 131I-MIBG, and the case of ALL developed >4 years from the first dose of 131I-MIBG. The incidence of myelodysplastic syndrome and acute myeloid leukemia following administration of NA- 131I-MIBG has been reported in the literature. Given the histories of
Reference ID: 4297867
(b) (6)
(b) (6)
(b) (6)
(b) (6) (b) (6)
(b) (6)
(b) (6) (b) (6)
(b) (6)
(b) (6) (b) (6)
(b) (6) (b) (6)
(b) (6)
(b) (6)
(b) (6)
(b) (6)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
130Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
substantial prior therapy in the patients treated on this study, the contribution of I131-MIBG
alone is impossible to determine, but it is likely to have contributed to the development of MDS and leukemia in these patients.
Renal Failure
Analysis of adverse event narratives for reported cases of renal failure are discussed in “Significant Adverse Events” in Section 8.2.4, and laboratory evidence of renal impairment is discussed under the sub-heading “Laboratory Analysis.” Sixteen patients enrolled on Study MIP-12B demonstrated a significant decrease in creatinine clearance or GFR. Six patients had a reported event of renal failure. Nineteen patients (28%) had either a decrease in GFR, a reported event of renal failure, or both. Of these patients, all had an additional risk factor of hypertension, and 8/19 (42%) had diabetes mellitus. Of the reported adverse events of renal failure or acute kidney injury, most were mild and recovered. Although some cases of decreased GFR in which subsequent measurements were available demonstrated recovery, most cases lacked subsequent measurements and it is unclear whether the decreases in GFR were reversible.
As previously noted, a limitation to the analysis of renal failure is the lack of laboratory data beyond 12 months. Neither GFR nor serum creatinine were required to be measured beyond 12 months. Renal failure, renal dysfunction, and elevated creatinine were among AESIs required to be reported during long-term follow-up, but it is possible that renal dysfunction occurring beyond 12 months was unrecognized if no overt clinical signs of renal failure were present and laboratory evaluations were not performed. A universal confounding factor in the assessment of renal dysfunction in this population is the presence of hypertension, often severe and longstanding, which itself contributes to renal impairment.
A determination of the degree of renal impairment attributable to 131I-MIBG is thus impossible to make. Given these observations and the potential for underlying renal impairment in this population, close monitoring of renal function warrants inclusion in product labeling.
8.2.6 Clinical Outcome Assessment (COA) Analyses Informing Safety/Tolerability
Analysis of specific quality of life evaluations was a secondary objective of Study MIP-IB12B. The European Organization for Research and Treatment of Cancer (EORTC) Quality of Life (QLQ-C30v.3) and National Institutes of Health (NIH) Quality of Life and Symptoms Questionnaire for Pheochromocytoma and Paraganglioma were administered in study MIP-IB12B at baseline and at the following time points: 5 – 7 days after each therapeutic dose; between weeks 1 – 6; at weeks 8, 10, and 12 following each 131I-MIBG dose; and monthly thereafter until 12 months after the first therapeutic dose. Karnofsky performance score (KPS) was recorded by the investigator at baseline, 6 weeks, and 3, 6, 9 and 12 months following the first therapeutic dose.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
131Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
The NIH Quality of Life and Symptoms Questionnaire includes questions eliciting potentially disease-related symptoms (anxiety, sweating, flushing, palpitations, tremors, etc.), as well as patient-reported changes in blood pressure and heart rate. The EORTC QLQ-C30 elicits changes in exercise tolerance or activities of daily living, as well as the patient’s overall quality of life.
Patient-rated overall health and quality-of life was rated from 1 (very poor) to 7 (excellent) on the EORTC QLQ-C30. An average of these two values for each patient was converted to a percentage in the datasets. Over the course of the study, the value of patient-rated quality of life remained relatively stable. At baseline, the median was 58.3% (range 12.5 – 100%); at 12 weeks after therapeutic dose 1, the median was 66.7% (range 16.7 – 100%); at 12 months after therapeutic dose 1, the median was 66.7% (range 16.7 – 100%). It should be noted that the number of patients for whom data was available was not 100%, and decreased from 57 to 48 over the 12-month period described above. The increase of <10% is unlikely to be clinically meaningful, but may represent stability in overall health and quality of life. KPS during the same time period demonstrated no change in median value (median = 90). Limited conclusions can be drawn from the COA data. Additional exploratory analyses were performed by the statistical reviewer in Section 21.5.
Statistical Reviewer’s Comments:The statistical reviewer’s analysis of QLQ-C30 scores, including completion rates, physical functioning score over time plots and mean score change from baseline over time plots are provided in Section 21.5. These analyses were considered exploratory.
8.2.7 Safety Analyses by Demographic Subgroups
Analyses of treatment-emergent adverse events were conducted by demographic subgroup, including age, sex, racial and ethnic subgroups. These analyses are all limited by the size of the safety population and relevant subgroups. An analysis of the safety population by age revealed that patients >65 years of age experienced greater percentages of most adverse events. For example, while 64% of patients of all age groups experienced a treatment-emergent adverse event (TEAE) in the gastrointestinal disorders system organ class (SOC), 100% of patients over 65 years of age experienced such a TEAE. There were no significant trends noted in the analysis of TEAEs by race or by gender. The distribution of TEAEs by ethnicity was analyzed, though the analysis was limited by the presence of only one patient in the Hispanic or Latino subgroup.
Patients who had received 131I-MIBG therapy prior to study entry experienced somewhat higher incidences of cytopenias when compared with patients who had not received prior 131I-MIBG therapy. For example, 72% of patients with prior 131I-MIBG experienced treatment-emergent anemia, compared to 50% of those without prior 131I-MIBG exposure. Among patients who developed leukemia or myelodysplastic syndrome, 60% of patients had had prior 131I-MIBG, compared to 40% who had not.
8.2.8 Specific Safety Studies/Clinical Trials
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
132Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Not applicable.
8.2.9 Additional Safety Explorations
Human Carcinogenicity or Tumor Development
The applicant did not perform carcinogenicity studies in support of this application.
Human Reproduction and Pregnancy
There were no reported exposures during pregnancy. Female patients were required to have a negative pregnancy test prior to administration of 131I-MIBG, and all patients were required to use an acceptable form of birth control during the study and for 6 months following administration of 131I-MIBG.
Pediatrics and Assessment of Effects on Growth
131I-MIBG has not been studied in a dedicated pediatric study in patients with pheochromocytoma. Study MIP-IB12B allowed enrollment of patients aged 12 years and older, and one patient under 18 (age 16 years) was treated on this study. A small dose-finding study of 131I-MIBG was conducted in 15 patients with neuroblastoma. Given the unmet medical need for adolescents with advanced PPGL, and the treatment of one adolescent on Study MIP-IB12B, the indication was extended to include the treatment of adolescent patients aged 12 years and older. Special consideration regarding radiation exposure in pediatric patients, namely greater absorbed radiation dose and a potential longer life expectancy, was included in the product labeling.
131I-MIBG was granted orphan designation for the treatment of neuroendocrine tumors and is therefore exempt from the Pediatric Research Equities Act (PREA) requirements for this indication.
Overdose, Drug Abuse Potential, Withdrawal, and Rebound
No studies have been conducted to evaluate the abuse potential with 131I-MIBG. There isno evidence that suggests a risk for dependence on 131I-MIBG. No cases of withdrawalsymptoms were reported during human clinical trials. This radiotherapeutic requires special licensing for handling, and will be administered in centers with specialized experience in the administration of radioactive products. Therefore, it is unlikely that drug abuse or any attendant medical issues will occur.
No clinical data regarding overdose was provided. Overdose is unlikely with proper administration since measurement of radioactivity is required prior to administration as defined in the product labeling. An overdose of the product would likely result in radiation-related toxicities consistent with those described in the product label, of potentially increased severity or duration dependent upon the dose administered.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
133Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
8.2.10 Safety in the Postmarket Setting
Safety Concerns Identified Through Postmarket Experience Not applicable. 131I-MIBG has not been approved for treatment purposes. 131I-MIBG was previously approved for use as an imaging agent in 1994 with a different formulation and dosage; the product has since been discontinued. Expectations on Safety in the Postmarket Setting Analysis of patients enrolled in studies MIP-IB12B and MIP-IB12 revealed several patients who developed myelodysplastic syndrome or acute leukemias. In addition, a small number of patients developed a new primary malignancy after receiving 131I-MIBG. The majority of cases were highly confounded by prior therapies with known associations with secondary malignancies including leukemia and myelodysplasia. Therefore, the true contribution of 131I-MIBG in the development of these events, and expected incidence in a less-heavily pretreated population, is unknown. A post-marketing study to evaluate the incidence of secondary malignancies will be conducted by the Applicant; refer to Section 15 Postmarketing Requirements and Commitment, for details.
8.2.11 Integrated Assessment of Safety
The evaluation of the safety of 131I-MIBG in patients with PPGL was primarily based on Study MIP-IB12B, an open-label, multicenter, single arm trial in 68 patients with PPGL who were not eligible for curative surgery and had either failed a prior therapy or were not eligible for chemotherapy. A pooled population of patients from Study MIP-IB12B and the dose escalation study in PPGL, Study MIP-IB12, was analyzed as the main safety population. The size of the pooled safety database was considered adequate to characterize the safety profile of 131I-MIBG.
During Study MIP-IB12B, the most significant safety signal was the development of secondary malignancies including acute leukemias and myelodysplastic syndrome. In the pooled safety population, 7% (n=6) of patients developed acute myeloid leukemia, acute lymphoblastic leukemia, or myelodysplastic syndrome. Two patients developed new solid tumors. An association between the development of MDS and acute leukemia and NA-131I-MIBG has been established in the literature. The confounding factor of exposure to prior therapy with known potential to cause secondary malignancy in all cases of acute leukemia or MDS obscures the magnitude of the risk to patients receiving 131I-MIBG, but a significant risk exists. Thus, a careful consideration of the potential benefits of treatment with 131I-MIBG in light of this and other significant risks, is warranted. The risk for development of secondary malignancies will be included in product labeling, and a post-marketing requirement to further assess the risk of secondary malignancy will be performed by the Applicant.
Myelosuppression was also a prominent, and expected, safety signal in the pooled safety population. Most patients experienced a decrease in one or more cell lines, and severe decreases in platelets and neutrophils were common (50% and 59%, respectively). Myelosuppression was delayed, with blood count nadirs occurring approximately 4 – 8 weeks
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
134Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
after administration of a therapeutic dose, with variable recovery times for neutrophils and platelets of 1 – 35 weeks. Ten percent (n=7) of patients on Study MIP-IB12B discontinued the study prior to receipt of the second therapeutic dose due to hematologic adverse events.
Renal failure was also observed in the study. Initial therapeutic doses of 131I-MIBG were adjusted based on dosimetry, which accounts for renal clearance; one patient required a dose reduction based on renal injury following administration of the first therapeutic dose. Adverse events of renal failure (6.8%) did not exceed CTCAE Grade 3 in severity and usually occurred in the presence of significant confounding factors (i.e., sepsis, dehydration, etc.). Sixteen patients (24%) demonstrated significant decreases in renal function as measured by GFR or creatinine clearance during the study; the contribution of 131I-MIBG as compared to hypertension (100% of patients) or diabetes (44% of patients) is unclear. No patients required dialysis.
Finally, one patient in the expanded access program for study MIP-IB12B developed fatal pneumonitis 9 weeks after treatment with a single dose of 131I-MIBG. The patient did not have pulmonary metastases, which would be expected to increase the risk of radiation toxicity to the lungs. Limited information regarding the expected incidence of or risk factors for radiation pneumonitis can be extrapolated from this single case. Severe pulmonary sequelae, such as ARDS and BOOP, have been reported using non-Azedra formulations of 131I-MIBG.
Other known radiation-related toxicities were not prominent signals in the safety database, but warrant consideration in the overall benefit-risk assessment of 131I-MIBG given the mechanism of action. Hypothyroidism occurred in 2% (n=2) of patients; there were no reports of thyroid neoplasia, though this has been reported in the literature. Hypothyroidism is managed with appropriate monitoring and thyroid hormone replacement. Severe liver sequelae, such as veno-occlusive disease, has also been reported with NA-131I-MIBG preparations. Clinical or laboratory events suggestive of liver disease or injury in the 131I-MIBG development program were confounded by metastatic disease.
Overall, the safety review of the 131I-MIBG database did not reveal new safety concerns, but confirmed expected safety signals based on published reports of NA-131I-MIBG, adverse events reported to the IND, and based on the mechanism of action. The safety data from Study MIP-IB12B and MIP-IB12 do not change with favorable benefit-risk assessment for 131I-MIBG for the treatment of patients with iobenguane scan positive, unresectable, locally advanced or metastatic PPGL who require systemic anticancer therapy.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
135Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
10 SUMMARY AND CONCLUSIONS
10.1 Statistical Issues
There is no major statistical issue that impacts the overall conclusions of Study MIP-IB12B. However, the limitation of single arm design prevents the study from providing adequate interpretation of the results of a time to event endpoint such as OS.
10.2 Conclusions and Recommendations
Patients with iobenguane scan positive, unresectable, locally advanced or metastatic PPGL who require systemic anticancer therapy represent a population with a serious and life-threatening rare disease for which there is no FDA-approved therapy and no known curative therapy. Although some patients demonstrate responses to off-label use of cytotoxic chemotherapy, complete responses are rare. Off-label use of NA-131I-MIBG is a standard therapy for PPGL which cannot be cured by surgical resection alone.
The clinical benefit of 131I-MIBG for patients with unresectable, locally advanced or metastatic PPGL who require systemic anticancer therapy is based on the results of Study MIP-IB12B. The primary endpoint, a 50% reduction (including discontinuation) of antihypertensive medications for at least 6 months, was a novel endpoint in the PPGL population. Given the morbidity associated with hypertension in this population, and the direct contribution of the tumor to hypertension through secretion of catecholamines, this endpoint was designed as a measure of direct clinical benefit. The study was conducted under an SPA, and the endpoint and design of the study were agreed to by FDA. The primary endpoint was achieved by 25% of patients (95% CI: 16.2% – 36.5%). The antitumor activity of 131I-MIBG was confirmed by traditional tumor response criteria, RECIST v. 1.0; 22.1% of the evaluable population demonstrated a confirmed, centrally reviewed response of PR (95% CI: 13.6%, 32.7%).
Combined, durable response in hypertension as measured by the primary endpoint and confirmed objective response are measures of direct clinical benefit in this population.
The primary safety risks of 131I-MIBG are related to the radiation exposure of the product. Myelosuppression and gastrointestinal adverse reactions were the most common adverse effects by system organ class. The risk of myelodysplastic syndrome and leukemia was less common, occurring in 7% of the pooled safety population. Although these events were largely confounded by prior, potentially leukemogenic therapy, this remains a significant and serious risk of 131I-MIBG and requires further study. The serious nature of these risks supports the use of 131I-MIBG only in patients with unresectable, locally advanced or metastatic PPGL who require systemic anticancer therapy disease and who have no other known curative options.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
136Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Based on the rarity of this disease, demonstrated clinical benefit, and lack of approved therapeutic options, the reviewers recommend regular approval of the product. The clinical and statistical reviewers do not recommend a REMS be implemented for 131I-MIBG given widespread experience with NA-131I-MIBG and the necessity of delivering this product in centers certified to deliver radioactive therapies. However, a PMR will be completed to fully characterize the risk of developing secondary malignancies in patients treated with 131I-MIBG. Risk management will also include product labeling and routine pharmacovigilance to ensure the safe and effective use of 131I-MIBG.
X X
Primary Statistical Reviewer Statistical Team Leader
X X
Primary Clinical Reviewer Clinical Team Leader
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
137Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
11 Advisory Committee Meeting and Other External Consultations
The Division did not obtain the advice of the Oncologic Drug Advisory Committee (ODAC) for this application, as there were no public health issues raised that would benefit from a public discussion or that required the expert opinions of the Committee. In addition, the safety profile of the drug is deemed acceptable for the indicated population of patients.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
138Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
12 Pediatrics
This application is exempt from the requirements under the Pediatric Research Equity Act. 131I-MIBG (Azedra) received orphan designation for the treatment of neuroendocrine tumors in January 2006.
The safety and maximum tolerated dose of 131I-MIBG, in the setting of planned autologous stem cell rescue, were explored in a multicenter, single arm, open-label dose escalation study in fifteen patients with relapsed or refractory high-risk neuroblastoma. Patients enrolled on this study were 3 – 30 years old, with 10 patients ≤ 10 years old. Patients received doses of approximately 11, 15 and 18 mCi/kg. The study was not designed to assess radiation toxicities occurring beyond 60 days, precluding a complete safety review of this study.
The safety and effectiveness of 131I-MIBG was assessed in patients with unresectable PPGL, who had either failed a prior therapy or were ineligible for curative therapy, ages 12 and older on Study MIP-IB12B. The youngest patient to enroll was 16 years old. This patient was a primary endpoint responder, and did not demonstrate a response by RECIST. This patient experienced no serious adverse events on study. Though a limited adolescent population enrolled on Study MIP-IB12B, the pathophysiology of the disease is not substantially different in adolescents compared to adult patients. Given the substantial unmet medical need in this rare population, the product should be indicated for patients ages 12 and older.
13 Labeling Recommendations
13.1 Prescription Drug Labeling
Labeling negotiations were ongoing at the time of completion of this review. The table below summarizes significant changes to the proposed label made by FDA prior to negotiations. The package insert for Azedra will include the final prescribing information.
Summary of Significant Labeling Changes (High level changes and not direct quotations)Section Proposed Labeling Approved Labeling
1 Indications and Usage Proposed indication: AZEDRA is
indicated for the treatment of patients years older with iobenguane
unresectable pheochromocytoma
FDA revised the indication statement to include patients age 12 and older. FDA also revised the indication statement to include patients with unresectable, locally advanced or metastatic PPGL which requires systemic
Reference ID: 4297867
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
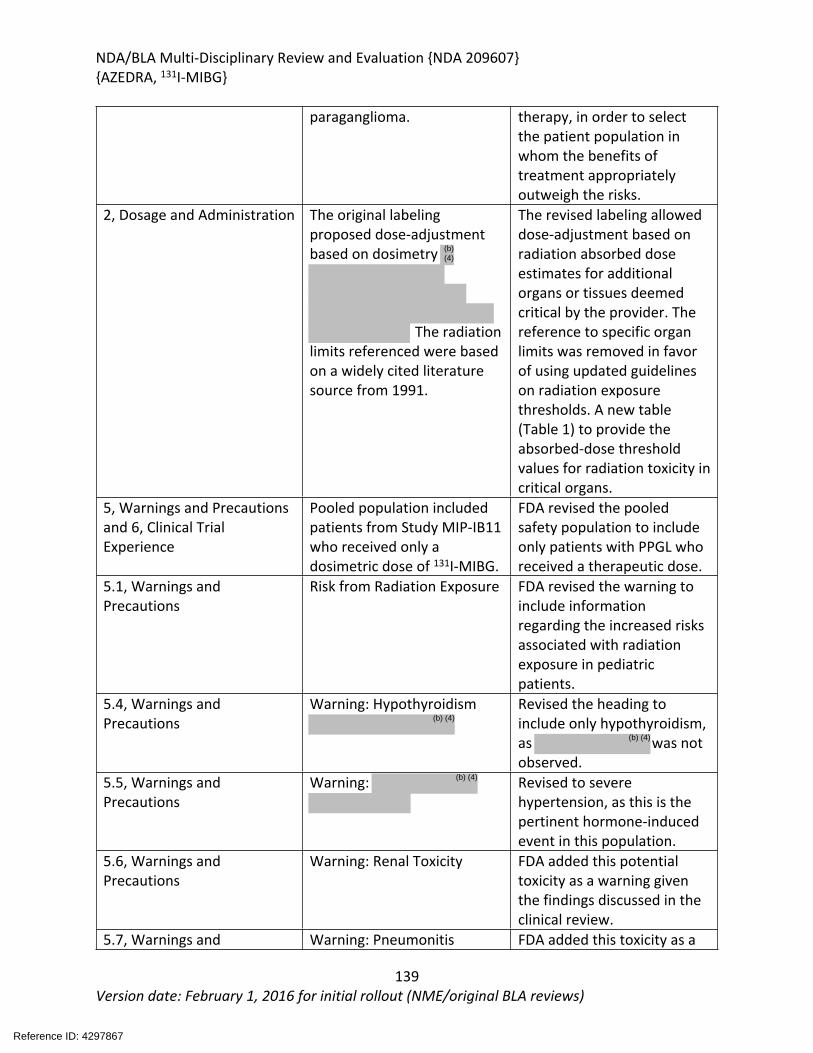
NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
139Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
paraganglioma. therapy, in order to select the patient population in whom the benefits of treatment appropriately outweigh the risks.
2, Dosage and Administration The original labeling proposed dose-adjustment based on dosimetry
The radiation limits referenced were based on a widely cited literature source from 1991.
The revised labeling allowed dose-adjustment based on radiation absorbed dose estimates for additional organs or tissues deemed critical by the provider. The reference to specific organ limits was removed in favor of using updated guidelines on radiation exposure thresholds. A new table (Table 1) to provide the absorbed-dose threshold values for radiation toxicity in critical organs.
5, Warnings and Precautions and 6, Clinical Trial Experience
Pooled population included patients from Study MIP-IB11 who received only a dosimetric dose of 131I-MIBG.
FDA revised the pooled safety population to include only patients with PPGL who received a therapeutic dose.
5.1, Warnings and Precautions
Risk from Radiation Exposure FDA revised the warning to include information regarding the increased risks associated with radiation exposure in pediatric patients.
5.4, Warnings and Precautions
Warning: Hypothyroidism Revised the heading to include only hypothyroidism, as was not observed.
5.5, Warnings and Precautions
Warning: Revised to severe hypertension, as this is the pertinent hormone-induced event in this population.
5.6, Warnings and Precautions
Warning: Renal Toxicity FDA added this potential toxicity as a warning given the findings discussed in the clinical review.
5.7, Warnings and Warning: Pneumonitis FDA added this toxicity as a
Reference ID: 4297867
(b) (4)
(b) (4)
(b) (4)
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
140Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Precautions warning given the occurrence of a fatal case of pneumonitis in the IB12B expanded access program.
8.4, Pediatric Use The label stated that safety and effectiveness of Azedra
FDA revised this section to include patients 12 years and older. Study MIP-IB12B was open to patients ages 12 and older. Exposure, safety and effectiveness are likely to be the same in adolescent patients.
14 Risk Evaluation and Mitigation Strategies (REMS)
No Risk Evaluation and Mitigation Strategy was deemed necessary for 131I-MIBG. 131I-MIBG willbe administered by health care professionals with experience in managing radiolabeledproducts in settings where these products are handled and administered routinely (i.e., licensedcenters). None of the safety signals identified during the review require a Risk Evaluation andMitigation Strategy. The USPI as well as all controlling regulations for the shipping and handlingof radiolabeled products include sufficient information and requirements for safety.
Reference ID: 4297867
(b) (4)

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
141Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
15 Postmarketing Requirements and Commitments
The following post marketing requirements under 505(o) are recommended.
CLINICAL
1. Submit cumulative, integrated safety analyses after 5 and after 10 years of follow-up of patients from an adequate number of clinical trials to identify and characterize the risks of myelodysplastic syndrome, acute leukemia and other secondary malignancies with Azedra; include incidence rates, time to onset, predisposing factors, and outcomes. These safety evaluations should be adequate to inform labeling of patient populations at highest risk and to provide evidence-based dose modifications and monitoring recommendations.
Proposed PMR Milestone Dates: Final Analysis Plan: January 2019Interim Report: April 2024Final Report: April 2029
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
142Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
16 Division Director (DHOT)
X
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
143Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
17 Division Director (OCP)
X
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
144Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
18 Division Director (OB)
X
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
145Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
19 Division Director (Clinical)
I agree with the review teams regarding the approval of this application. Nevertheless, I will summarize my thoughts regarding certain pertinent aspects of the application.
Orphan Indication:Malignant pheochromocytoma (and paraganglioma) is an ultrarare disease that can result in morbidity or mortality either due to the underlying tumor burden or due to excess catecholamine hormone production. The specific incidence of malignant PPGL is hard to quantify. The applicant estimated (approximately) 60 to 120 cases per year in the US. The upper estimate of the reported incidence of pheochromocytoma (0.8 per 100,000 person-years) was described in a paper from the Mayo clinic from 1983 (with data from 1950 to 1979). In the report (Beard et al., Mayo Clin Proc, 1983), five of the eleven cases were initially diagnosed at autopsy. A more modern analysis (data from 2007 to 2015 and published in abstract form) based on a Danish registry provided for an estimate of 4.65 cases per million person years. The report (Ebbehoj et al., Endocrine Abstracts, 2017) stated that the incidence is increasing; however, nearly 60% of cases from 2007 to 2015 were incidentally discovered while the patients were undergoing imaging.
The Danish paper did not describe the percentage of cases of PPGL determined to be malignant. The applicant provided for an estimate of 10 to 20%; however, the literature report provided by the applicant listed this estimate in the background of the report (and I could not determine how these data were obtained). A retrospective record review of 152 patients from Korea found 11% of patients to have metastatic disease with a mean follow-up of 41.5 months.
Clearly, there are limitations to the data providing for estimates of malignant PPGL. If the applicant’s estimate is approximate to the true incidence, it shows the rarity of the disease, and the difficulty in conducting studies in this disease. For example, logistically, to enroll a patient into a trial, a patient would have to live in proximity to a study site (or have the resources to travel multiple times to the site) and would have to meet eligibility criteria for entry into the trial. For the pivotal trial conducted by the applicant, ten centers in the US enrolled patients (generally in major academic centers in large cities). It took from June 2009 to January 2016 to enroll 68 patients who received at least one dose of I-131 MIBG.
Furthermore, due to compounding or expanded access, patients have received I-131 MIBG in the US outside of the Progenics protocol. For example, the 2013 Consensus Guidelines for Neuroendocrine Tumors (Kunz et al., Pancreas, 2013) list I-131 MIBG as an option to consider in patients who need systemic treatment. In Europe, I-131 MIBG is authorized for diagnostic or therapeutic uses, depending on the country (http://www.ema.europa.eu/docs/en GB/document). Ultimately, these factors underscore the
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
146Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
difficulty in conducting investigations in pheochromocytoma, particularly with an I-131 MIBG product.
Unmet NeedAlthough some patients with malignant pheochromocytoma can have long survival, the majority who cannot be successfully treated with locoregional therapies and who require systemic therapy will ultimately die from their disease. In the IB12B trial conducted by Progenics, median survival of all patients who received at least 1 dose was approximately three years (note this was time from treatment, not time from diagnosis). Cytoreductive surgery or local therapies are standard treatment options in patients with malignant PPGL. Most patients in Study IB12B underwent prior adrenalectomy, tumor excision, mass excision, or lymphadenectomy. About half of patients received prior radiation and about half received prior drug or biologic therapy. Clearly, there is a patient population who could potentially benefit from effective systemic therapy. To date, there are limited options for such patients (e.g., either compounded I-131 MIBG or off-label cytotoxic chemotherapy). Some of the patients in the IB12B trial had received prior I-131 MIBG. Importantly, the patient population appropriate for systemic therapy differs from the population described in the DMIP consult (the consult cited a report with data limited to a subset of patients with PPGL who had germline mutations in SDHB [these patients are generally younger] and the report did not describe the extent of metastatic disease or whether patients underwent successful metastatectomy).
Endpoints and Clinical EffectsFDA previously agreed to the primary endpoint in reduction in anti-hypertensive medication use (for at least six months) under a Special Protocol Assessment (in 2009). At the time, this endpoint was considered by FDA to provide a benchmark to assess clinical benefit in the proposed single arm trial while also assessing for an effect on tumor size. I believe as a single endpoint, the reduction in blood pressure medication use (for at least six months) in an unblinded single arm trial could be problematic. First, it represents a surrogate for blood pressure reduction (with elevations in blood pressure one of the causes of morbidity in PPGL); second, the decision to adjust blood pressure medication (increase or decrease) may be influenced by patient- or physician-factors; and third, the perceived clinical benefit of blood pressure reduction may be highly dependent upon the regimen the patient is receiving. To highlight the later example, a patient on a single daily dose of medication who decreases the dose by half would be perceived to have less benefit than a patient on four drugs who can discontinue three or four of them. In Study IB12B, 25% of the patients met the pre-defined endpoint. The point estimate for the response rate was higher (32%) among the 50 patients who could receive two doses. Additional patients also had reductions in blood pressure medications but not for the required six-month duration. As such, the milestone (six month) analysis provided an arbitrary cut-point and probably was less informative than a traditional KM analysis (that shows durability of treatment effect in all responders).
Assessments of the effects of an anti-cancer drug on blood pressure in an oncology trial are difficult based on the methods or frequency of blood pressure monitoring and because blood
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
147Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
pressure can be affected by changes in anti-hypertensive medications (i.e., in addition to I-131 MIBG). Nevertheless, a total of 20-25% of patients had reductions in systolic blood pressure of at least 20 mmHg (described in clinical review above). This is like the proportion of patients who met the primary endpoint and the proportion of patient who had a tumor response per RECIST.
Overall 22% of patients experienced a response per RECIST criteria (per FDA’s analysis). This shows anti-tumor effects of I-131 MIGB; however, I acknowledge that this is a modest effect. A similar modest effect on response rate was observed in a different neuroendocrine tumor population (midgut neuroendocrine tumors) with a different radiotherapeutic (Lutathera) and this translated into a large effect on PFS. Nevertheless, I cannot determine based on the data in this application whether the effects of the two drugs are analogous.
Other clinical effects, indicative of anti-tumor activity (but not necessarily clinical benefit) were also observed during treatment with I-131 MIBG. These included reductions in urine and blood norepinephrine levels. Also, in exploratory PRO analyses, global health status appeared to increase in patients treated with I-131 MIBG; however, these data are difficult to interpret in single arm trials.
Risk-BenefitOverall, I agree with the clinical team regarding the risk-benefit determination. The most important safety risks of I-131 MIBG are related to radiation exposure with myelosuppression and gastrointestinal adverse reactions being the most common toxicities. Myelodysplastic syndrome and leukemia are important risks with 131 I-MIBG. It was difficult to isolate the effect of a single course of I-131 MIBG on MDS/leukemia risk as patients who developed MDS or leukemia had been exposed to other therapies that increase the risk of MDS/leukemia (including prior I-131 MIBG). Nevertheless, I believe that I-131 MIBG will cause MDS and leukemia in some patients. The most important factor regarding the maximization of risk and benefit in PPGL will be patient selection. In labeling, I agree with the recommendation to highlight that the indication for I-131 MIBG will be limited to those patients where systemic therapy is needed. Patients amenable to local therapies or a watch and wait approach would generally be less appropriate for I-131 MIBG given that these patients may have a long-life expectancy where MDS/leukemia (or other delayed radiation effects) may cause significant morbidity or mortality. Importantly, given the complexity of patient management in PPGL, multidisciplinary teams are common which should optimize treatment selection and informed consent for most patients.
Although modest, I-131 MIBG demonstrated anti-tumor effects which may provide benefit for the properly selected patient. Patients with PPGL have other tumor-related symptoms due to catecholamine excess which plausibly might be effected by I-131 MIBG (given reductions on catecholamine levels, blood pressure, and tumor size). This assessment is also made in the specific context of a disease setting where there are no effective systemic treatments and in an ultrarare disease.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
148Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
My approval recommendation of this application is context-specific, considering the rare disease, specific challenges in developing an I-131 MIBG therapy in this disease, the prior SPA agreement, and the lack of available systemic therapies for patients with PPGL (unmet need).
X
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
149Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
This application was reviewed by the Oncology Center of Excellence (OCE) per the OCE Intercenter Agreement. My signature below represents an approval recommendation for the clinical portion of this application under the OCE.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
150Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
21 Appendices
21.1 References
Ayala-Ramirez M, Feng L, Habra M, Rich T, Dickson P, Perrier N, Phan A, Waguespack S, Patel S, Jimenez C, Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas. Cancer, 2012. 118(11): p. 2804-2812.
Babich JW, Graham W, Fischman AJ., Effect of adrenergic receptor ligands on metaiodobenzylguanidine uptake and storage in neuroblastoma cells. Eur J Nucl Med. 1997;24(5): p. 538-543.
Barrett JA, Joyal JL, Hillier SM et al., Comparison of high-specific-activity ultratrace 123/131I MIBG and carrier-added 123/131I-MIBG on efficacy, pharmacokinetics, and tissue distribution. Cancer Biother Radiopharm. 2010;25(3): p. 299-308.
Boyd M, Ross S, Owens J et al., Preclinical evaluation of no-carrier added 131I meta iodobenzyl guanidine for the treatment of tumors transfected with the noradrenaline transporter gene. Lett Drug Design Discov. 2004;1: p. 50-57.
Bruchelt G, Girgert R, Buck J et al., Cytotoxic Effects of m-[131I]-and m- [125I]iodobenzylguanidine on the human neuroblastoma cell lines SK-N-SH and SK-NLO1.Cancer Res. 1988;48: p. 2993-2997.
DuBois SG Matthay KK, 131I-Metaiodobenzylguanidine therapy in children with advanced neuroblastoma. Q J Nucl Med Mol Imaging, 2013. 57(1): p. 53-65.
Giri S, Chi M, Johnson B, McCormick D, Jamy O, Bhatt V, Martin M, Secondary acute lymphoblastic leukemia is an independent predictor of poor prognosis. Leukemia Research, 2015. 39(12): p. 1342-1346.
Gonias S, Goldsby R, Mattha K, Hawkins R, Price D, Huberty J, Lloyd D, Linker C, Sznewajs A, Shiboski P, Fitzgerald P, Phase II study of high-dose [131I]metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J Clin Oncol, 2009. 27(25): p. 4162-8.
Kosmin MA, Bomanji JB, Cork NJ, Shankar A, Gaze MNHypertension complicating 131I-meta-iodobenzylguanidine therapy for neuroblastoma. European Journal of Nuclear Medicine and Molecular Imaging, 2012. 39(4): p. 597-601.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
151Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Lenders JWM Eisenhofer G, Mannelli M, Pacak K, Phaeochromocytoma. The Lancet, 2005. 366(9486): p. 665-675.
Mairs RJ, Russell J, Cunningham S. et al., Enhanced tumour uptake and in vitro radiotoxicity of no-carrier-added [1311] metaiodobenzylguanidine: implications for the targeted radiotherapy of neuroblastoma. Eur J Cancer. 1995a;31A(4): p. 576-581.
Mairs RJ, Cunningham SH, Russell J et al., No-carrier-added Iodine-131-MIBG: evaluation of a therapeutic preparation. J Nucl Med. 1995b;36(6): p. 1088-1095.
National Cancer Institute, Pheochromocytoma and Paraganglioma Treatment-for health professionals (PDQ®). PDQ Cancer Information Summaries 31 January 2018 ; Available from: https://www.cancer.gov/types/pheochromocytoma/hp/pheochromocytoma-treatment-pdq.
Niemeijer ND, Ablas G, van Hulsteijn LT, Dekkers OM, Corssmit EPM., Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis. Clinical Endocrinology, 2014. 81(5): p. 642-651.
Pacak K, Lenders JWM, Eisenhofer G, Pheochromocytoma: Diagnosis, localization, and treatment. 2007, Malden, MA: Blackwell Publishing 184.
Park J, Song C, Park M, Yoo S, Park SJ, Hong S, Hong B, Kim CS, Ahn H, Predictive Characteristics of Malignant Pheochromocytoma. Korean Journal of Urology. 2011; 52(4): 241 – 246.
Rutgers M, Buitenhuis CK, Hoefnagel CA et al., Targeting of meta-iodobenzylguanidine to SK-N-SH human neuroblastoma xenografts: tissue distribution, metabolism and therapeutic efficacy. Int J Cancer. 2000a;87(3): p. 412-422.
Rutgers M, Buitenhuis CK, van der Valk MA et al., [(131)I] and [(125)I] metaiodobenzylguanidine therapy in macroscopic and microscopic tumors: a comparative study in SK-N-SH human neuroblastoma and PC12 rat pheochromocytoma xenografts. Int J Cancer. 2000b;90(6): p. 312-25.
Smets LA, Loesberg C, Janssen M et al., Active uptake and extravesicular storage of miodobenzylguanidine in human neuroblastoma SK-N-SH cells. Cancer Res. 1989;49(11): p. 2941-2944.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
152Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Solanki KK, Bomanji J, Moyes J, Mather SJ, Trainer PJ, Britton KE, A pharmacological guide to medicines which interfere with the biodistribution of radiolabeled metaiodobenzylguanidine (MIBG). Nucl Med Commun. 1992;13: p. 513–521.
van Hulsteijn LT,Niemeijer ND, Dekkers OM, Corssmit EPM, 131I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: systematic review and meta-analysis. Clinical Endocrinology, 2014. 80(4): p. 487-501.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
153Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
21.2 Financial Disclosure
Covered Clinical Study (Name and/or Number): MIP-IB12B
Was a list of clinical investigators provided: Yes No (Request list from Applicant)
Total number of investigators identified: 136
Number of investigators who are Sponsor employees (including both full-time and part-time employees): 0
Number of investigators with disclosable financial interests/arrangements (Form FDA 3455): 0
If there are investigators with disclosable financial interests/arrangements, identify the number of investigators with interests/arrangements in each category (as defined in 21 CFR 54.2(a), (b), (c) and (f)):
Compensation to the investigator for conducting the study where the value could be influenced by the outcome of the study:
Significant payments of other sorts:
Proprietary interest in the product tested held by investigator:
Significant equity interest held by investigator in S
Sponsor of covered study:
Is an attachment provided with details of the disclosable financial interests/arrangements:
Yes No (Request details from Applicant)
Is a description of the steps taken to minimize potential bias provided:
Yes No (Request information from Applicant)
Number of investigators with certification of due diligence (Form FDA 3454, box 3) 0
Is an attachment provided with the reason:
Yes No (Request explanation from Applicant)
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
154Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
21.3 Nonclinical Pharmacology/Toxicology
21.4 OCP Appendices (Technical documents supporting OCP recommendations)
21.5 Additional Clinical Outcome Assessment Analyses
In Study IB12B, EORTC-QLQC30 (QLQ-C30) was used to measure patients’ Quality of Life. There are 30 questions in QLQ-C30. In the NDA submission, the applicant provided descriptive results of scores and change from baseline for 5 functional scales, symptom scale, and Global Health Status/Quality of Life of QLQ-C30 from baseline to the visit of month 12 post therapeutic dose 1. Physical functioning (PF) functional scale is one of 5 functional scales in QLQ-C30 instrument. PF can be defined as a person’s assessment of his/her ability to carry out important and meaningful day-to-day activities (e.g., self-care, domestic) that require physical effort (Painter, P. & Marcus, R.L. (2013)). PF domain in QLQ-C30 can be used to further explore patients’ physical function. In QLQ-C30, the PF score for a patient is derived from at least 3 non-missing responses to the first 5 questions in QLQ-C30 (EORTC QLQ-C30 Scoring Manual, 3th edition, 2001). PF was the primary Quality of Life measure of interest and results are presented in this section.
The following table summarizes the statistical reviewer’s results of completion rates at each visit until the visit of month 12 post therapeutic dose 1. The completion rate for each visit is defined as the number of the patients who completed all 30 questions divided by the patients at the visit. QLQ-C30 completion rate in Study IB12B ranges from 58.1% to 96.1% and completion rate for the PF functional scale ranges from 62.8% to 100%. Both completion rates are greater than 82.4% except the visit of Month 6 Post Therapeutic Dose 1 (Days 169 - 198).
Reference ID: 4297867
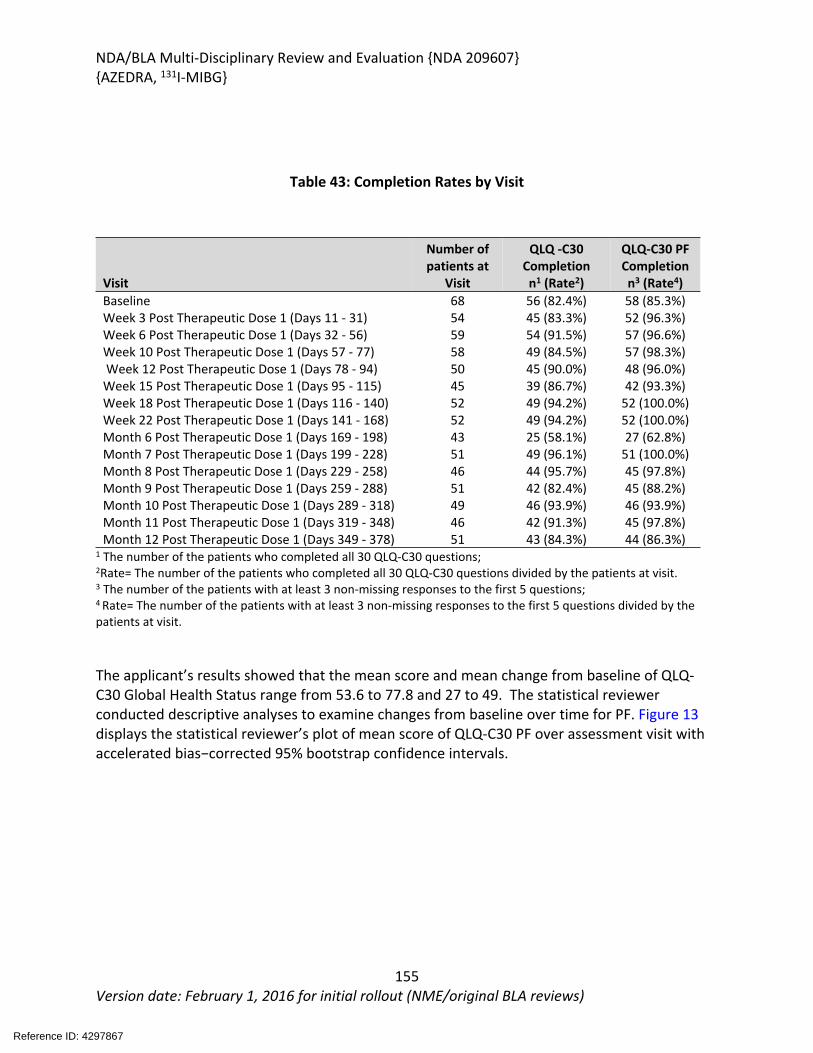
NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
155Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Table 43: Completion Rates by Visit
Visit
Number of patients at
Visit
QLQ -C30 Completionn1 (Rate2)
QLQ-C30 PF Completionn3 (Rate4)
Baseline 68 56 (82.4%) 58 (85.3%)Week 3 Post Therapeutic Dose 1 (Days 11 - 31) 54 45 (83.3%) 52 (96.3%)Week 6 Post Therapeutic Dose 1 (Days 32 - 56) 59 54 (91.5%) 57 (96.6%)Week 10 Post Therapeutic Dose 1 (Days 57 - 77) 58 49 (84.5%) 57 (98.3%) Week 12 Post Therapeutic Dose 1 (Days 78 - 94) 50 45 (90.0%) 48 (96.0%)Week 15 Post Therapeutic Dose 1 (Days 95 - 115) 45 39 (86.7%) 42 (93.3%)Week 18 Post Therapeutic Dose 1 (Days 116 - 140) 52 49 (94.2%) 52 (100.0%)Week 22 Post Therapeutic Dose 1 (Days 141 - 168) 52 49 (94.2%) 52 (100.0%)Month 6 Post Therapeutic Dose 1 (Days 169 - 198) 43 25 (58.1%) 27 (62.8%)Month 7 Post Therapeutic Dose 1 (Days 199 - 228) 51 49 (96.1%) 51 (100.0%)Month 8 Post Therapeutic Dose 1 (Days 229 - 258) 46 44 (95.7%) 45 (97.8%)Month 9 Post Therapeutic Dose 1 (Days 259 - 288) 51 42 (82.4%) 45 (88.2%)Month 10 Post Therapeutic Dose 1 (Days 289 - 318) 49 46 (93.9%) 46 (93.9%)Month 11 Post Therapeutic Dose 1 (Days 319 - 348) 46 42 (91.3%) 45 (97.8%)Month 12 Post Therapeutic Dose 1 (Days 349 - 378) 51 43 (84.3%) 44 (86.3%)
1 The number of the patients who completed all 30 QLQ-C30 questions; 2Rate= The number of the patients who completed all 30 QLQ-C30 questions divided by the patients at visit.3 The number of the patients with at least 3 non-missing responses to the first 5 questions; 4 Rate= The number of the patients with at least 3 non-missing responses to the first 5 questions divided by the patients at visit.
The applicant’s results showed that the mean score and mean change from baseline of QLQ-C30 Global Health Status range from 53.6 to 77.8 and 27 to 49. The statistical reviewer conducted descriptive analyses to examine changes from baseline over time for PF. Figure 13 displays the statistical reviewer’s plot of mean score of QLQ-C30 PF over assessment visit with accelerated bias−corrected 95% bootstrap confidence intervals.
Reference ID: 4297867
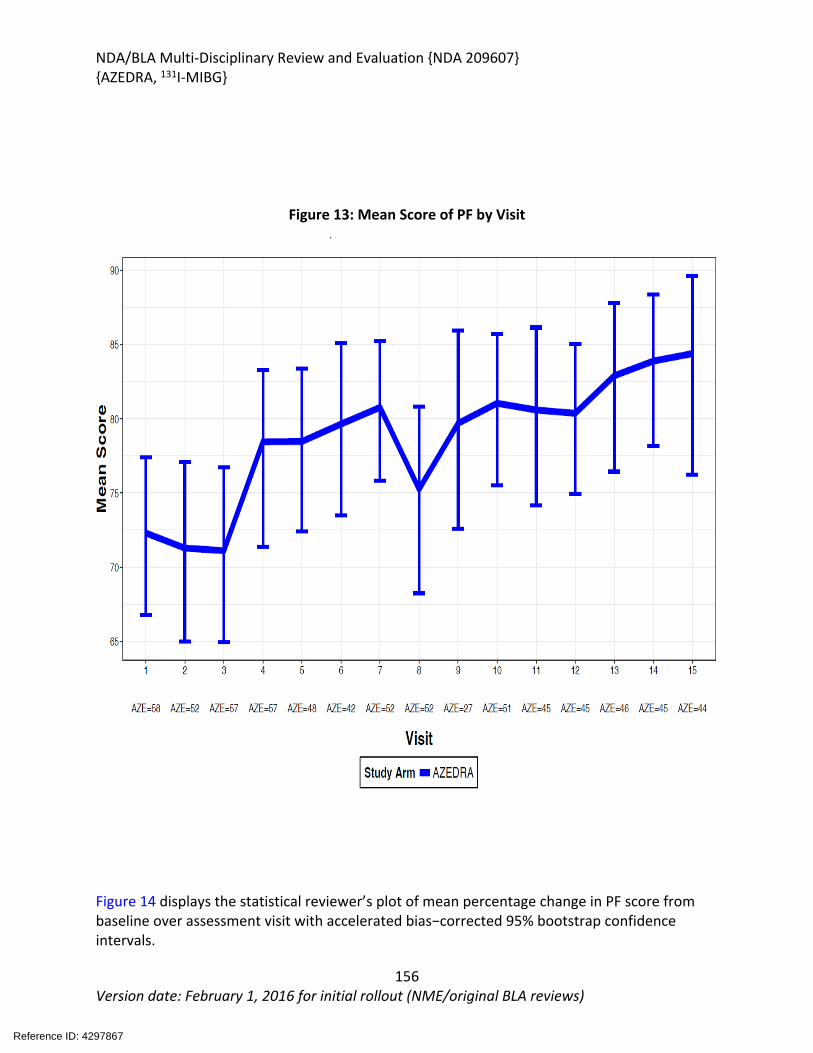
NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
156Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Figure 13: Mean Score of PF by Visit
Figure 14 displays the statistical reviewer’s plot of mean percentage change in PF score from baseline over assessment visit with accelerated bias−corrected 95% bootstrap confidence intervals.
Reference ID: 4297867

NDA/BLA Multi-Disciplinary Review and Evaluation {NDA 209607}{AZEDRA, 131I-MIBG}
157Version date: February 1, 2016 for initial rollout (NME/original BLA reviews)
Figure 14: Mean Percentage* Change in PF Score from Baseline over Visit
*mean percent change in Score from baseline to cycle m = mean ([Cycle m Score] − [Baseline Score])/ [Baseline Score]) *100
Reference ID: 4297867

--------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically. Following this are manifestations of any and allelectronic signatures for this electronic record.--------------------------------------------------------------------------------------------/s/------------------------------------------------------------
SHARON K SICKAFUSE07/26/2018
DENALI D KUFRIN07/27/2018
JOHN K LEIGHTON on behalf of WHITNEY S HELMS07/27/2018
JOHN K LEIGHTON07/27/2018
SAFAA BURNS07/27/2018
HONG ZHAO on behalf of JEANNE FOURIE ZIRKELBACH07/27/2018Sign for Jeanne Fourie
NAM ATIQUR RAHMAN07/27/2018I concur.
XIAOPING JIANG07/27/2018
LISA R RODRIGUEZ07/27/2018
RAJESHWARI SRIDHARA07/27/2018
DIANA L BRADFORD07/27/2018
SUZANNE G DEMKO07/27/2018
Signature Page 1 of 2
Reference ID: 4297867

STEVEN J LEMERY07/30/2018
Signature Page 2 of 2
Reference ID: 4297867