10. elektroanalytische methoden - moodle.unifr.ch 2014... · das potential für die reduktion von...
TRANSCRIPT

Analytik 10.1
10. Elektroanalytische Methoden 10.1. Elektrogravimetrie Das Metallion wird vollständig elektrolytisch an einer Kathode mit bekannter Masse abgeschieden. Nach der Elektrolyse wird die Kathode gewogen und die Masse des abgeschiedenen Metalls bestimmt.
V: Potentiometer
A: Ampèremeter
Fig. 10.1. Elektrolysezelle zur Elektrogravimetrie. Beispiel: Bestimmung von Cu2+ in einer schwefelsauren CuSO4 Lösung. Die Spannung der Stromquelle wird so lange erhöht, bis ein Strom fliesst, das heisst eine Redox Reaktion abläuft. Die möglichen Reduktionen an der Kathode sind: a) Reduktion von Cu2+
E0 = 0.337 VCu2+ + 2 e- Cu b) Reduktion von H3O+
c)

Analytik 10.2
E0 = 0.00 V2 H3O+ + 2 e- H2 Die möglichen Oxidationen an der Anode sind: a) Oxidation von H2O
E0 = -1.229 V6 H2O O2 + 4 H3O+ + 4 e-
b) Oxidation von SO42-
E0 = -2.01 V2 SO42- S2O8
2- + 2 e-
Die Reaktionen, welche thermodynamisch am günstigsten sind, laufen in der Zelle ab, wenn keine Ueberspannungen auftreten, d.h. an der Kathode wird Cu2+ reduziert und Cu abgeschieden, an der Anode wird H2O oxidiert und es entsteht O2(g). Das Potential, welches an die Zelle angelegt werden muss, ist im allgemeinen grösser als das thermodynamische Potential, welches aus den Halbzellenpotentialen und den Konzentrationen der elektroaktiven Teilchen berechnet wird. Speziell Reaktionen, bei denen mehrere Elektronen übertragen werden und bei denen Gase gebildet werden, sind nicht reversibel und zeigen eine Ueberspannung, so z.B die Oxidation von H2O zu H2 und die Reduktion von H2O zu O2. Die Ueberspannung hängt mit der Aktivierungsenergie der spontanen Reduktion von O2 zu H2O ab, wie das folgende Schema zeigt. Die Ueberspannung wird durch die Beschaffenheit der Elektrodenoberfläche und der Ladungsdichte beeinflusst.

Analytik 10.3 Fig. 10.2 Reaktionsprofil für die Erzeugung von O2 resp. Cl2 an der Anode. Als Folge der Überspannung entsteht an der Anode bei der Elektrolyse einer wässrigen Chlorid-Lösung Cl2 und nicht O2, wie auf Grund der Standard-Reduktions-Potentiale (Thermodynamik) erwartet wird. Man kann daher sagen, dass die Elektrodenreaktion nicht thermodynamisch, sondern kinetisch kontrolliert ist. Zusätzlich besitzt die Zelle einen ohmschen Widerstand, welcher überwunden werden muss, damit der Strom fliesst. Der Widerstand der Zelle ist besonders gross, wenn keine wässrigen Lösungen verwendet werden, resp. wenn die Elektrolytkonzentration sehr klein ist. Das Abscheiden von Metall an der Kathode aus wässriger Lösung wird durch die Reduktion von H3O+ konkurrenziert. Es können daher nur Metalle abgeschieden werden, deren Standard-Reduktionspotential grösser ist als das Potential für die Wasserstoffbildung. Die Reduktion von H3O+ zeigt eine Ueberspannung an den meisten Elektroden. Diese ist stark von der Beschaffenheit der Elektrodenoberfläche abhängig. An einer blanken Pt-Elektrode ist die Ueberspannung ca. 0.1 V. Wird die Pt-Oberfläche elektolytisch mit Cu überzogen, ist die Ueberspannung grösser, 0.7 V. Unter diesen Bedingungen gelingt es daher auch unedle Metalle wie z.B. Zn abzuscheiden. Das Potential für die Reduktion von H2O ist vom pH abhängig. In basischer Lösung wird es kleiner (siehe 8.4). Daher können nicht edle Metalle aus basischen Lösungen abgeschieden werden, wenn kein Hydroxid ausfällt. Ein Beispiel dafür ist das Abscheiden von Zn aus einer stark basischen Lösung. In dieser Lösung liegt Zn2+ als Zn(OH)4
2- vor und fällt daher nicht als Hydroxid aus. Die Konzentration des Metalles, welches abgeschieden wird, nimmt während der Elektrolyse ab. Da das Potential für die Abscheidung proportional zum Logarithmus der Konzentration ist, fällt auch dieses während der Elektrolyse. Wird es kleiner als das Potential der Konkurrenzreaktion, z.B. der Reduktion von H2O, kann kein Metall mehr abgeschieden werden und die Elektrolyse muss abgebrochen werden. Als Kathode verwendet man ein Platinnetz. Das Metall sollte auf dem Netz

Analytik 10.4 möglichst gleichmässig und mechanisch stabil abgeschieden werden. Dies wird am besten erreicht, wenn das elektrische Feld an der Kathode möglichst homogen ist. Daher wird die Kathode zylinderförmig um die Anode angeordnet. Die Feldlinien sind dann radial angordnet und die Distanz zwischen Anode und Kathode ist für die ganze Fläche der Kathode ungefähr gleich gross. 10.2. Coulometrie bei kontrolliertem Potential
Fig. 10.3 Elektrolysezelle für die Coulometrie bei konstantem Potential. Bei der Coulometrie bei konstantem Potential wird die Elektrolyse in einer elektrochemischen Zelle bei konstantem Potential durchgeführt. Das Potential wird so gewählt, dass an der Arbeitselektrode nur die gewünschte Analysereaktion abläuft. Da bei dieser Methode kein Produkt abgeschieden wird, kann die Arbeitselektrode Anode oder Kathode sein. Die Hilfselektrode ist entsprechen Kathode resp. Anode. Als Arbeitselektrode

Analytik 10.5 dient je nach Anwendung ein Platinnetz oder Quecksilber, auf dem Boden der Probezelle. Die Halbzelle mit der Hilfselektrode ist durch eine Fritte von der Probe getrennt, damit die Produkte, welche an der Hilfselektrode gebildet werden, nicht mit den Produkten an der Arbeitselektrode in Kontakt kommen, und die spontane Rückreaktion abläuft. Das Potential zwischen der Probe und der Arbeitselektrode muss mit einer Referenzelektrode gemessen werden, da es nicht möglich ist das Potential zuverlässig zu messen, wenn in einem Stromkreis ein Strom fliesst. Der Widerstand des Potentiometers und der Referenzelektrode muss daher grösser sein, als der Widerstand an der Arbeitselektrode. Die äussere Spannung wird von einem Potentiostaten nach der Spannung geregelt, welche zwischen Arbeitselektrode und Probe gemessen wird. Die Probe wird bei einem vorgegebenen Potential elektrolysiert, bis kein Strom mehr fliesst. Der Stom wird mit einem Coulometer aufintegriert. Die Menge des elektrolysierten Analyten wird dann mit Hilfe der Faradayschen Beziehung berechnet.
Analyt elektrolysiert mol = geflossener Strom Coulomb96 487 Coulomb
mol e- • n mol e-mol Analyt
10.1 n: Anzahl Elektronen die pro Mol Analyt übertragen werden. Der Strom kann elektronisch oder chemisch integriert werden. Zur chemischen Integration wird an Stelle des Coulometers die folgende elektrochemische Zelle in den Stromkreis eingebaut:
Ag(s) {AgBr}s, KBr(0.03 M), K2SO4(0.2 M) Pt
Anodenreaktion:!!!Ag + Br - AgBr + e-
Kathodenreaktion: 2 H 2O + 2 e- H2 + OH- 2
An der Anode dieser Zelle wird Ag zu AgBr oxidiert. An der Kathode Wasser zu H2(g) und OH- reduziert. Nach der Elektrolyse wird die Lösung in der Coulometerzelle mit einer starken Säure zurücktitriert. Für jedes

Analytik 10.6
Elektron, welches durch das Coulometer geflossen ist, wurde ein OH- gebildet. Aus dem Verbrauch an Säure kann der geflossenen Strom berechnet werden und die Menge des elektrolysierten Analyten bestimmt werden.
Analyt elektrolysiert mol = VSäure ml • cSäure mol l -1 • 10-3 l
mln mol e-
mol Analyt 10.2 10.3. Coulometrie bei kontrolliertem Strom
Fig. 10.4. Elektrolysezelle für Coulometrie bei konstantem Strom. Biamperometrische Endpunktsindikation. Die Coulometrie bei kontrolliertem Strom nennt man auch Coulometrische Titration, da dabei die Lösung mit e- aus einer Stromquelle titriert wird. In vielen Fällen wird der Analyt nicht direkt mit e- titriert, sondern das Teilchen, welches mit dem Analyten die Analysereaktion eingeht, wird an der Arbeitselektrode erzeugt und reagiert dann sofort mit dem Analyten.

Analytik 10.7 Ein Beispiel ist die coulometrische Titration von Cr2O7
2- mit Fe2+. Die Reaktion bei dieser Analyse ist:
Das Fe2+, welches mit dem Cr2O72- reagiert, wird durch elektochemische
Reduktion von Fe3+ an der Arbeitselektrode, in diesem Fall die Kathode, erzeugt und reagiert dann sofort mit Cr2O7
2-. Wird die Analyse in einer salzsauren Lösung ausgeführt entsteht an der Hilfselektrode (Anode) Cl2(g). Die Hilfselektrode wird durch eine Fritte von der Arbeitselektrode getrennt, damit die Produkte, welche gebildet werden, nicht mit der Probe reagieren können. Das Hilfsredoxpaar Fe3+/Fe2+ hat bei dieser Analyse zwei Aufgaben. Erstens ist es Katalysator für die Reduktion von Cr2O7
2-. Die direkte Reduktion an einer Pt-Elektrode ist nicht möglich. Zweitens kontrolliert es das Potential nach dem Aequivalenzpunkt. Da die Stromquelle einen konstanten Strom liefert, muss nur die Zeit bis zum Endpunkt der Titration gemessen werden. Aus dem Produkt Strom • Zeit kann die Ladung berechnet werden, welche durch die Zelle geflossen ist. Diese ist durch die Beziehung von Faraday mit der Menge Analyten in der Probe verknüpft.
Analyt elektrolysiert mol = Stomstärke Ampere = Coulomb
sec • Zeit sec
96 487 Coulombmol e- • n mol e-
mol Analyt 10.3 Am Ende der Reaktion ändert das Potential der Probe sprunghaft. Im diskutierten Beispiel, wird vor dem Endpunkt das Potential durch das Redoxpaar Cr2O7
2-/Cr3+ kontrolliert (E0 = 1.33 V), nach dem Endpunkt durch das Paar FeCl4-/Fe2+ (E0 = 0.70 V). Der Endpunkt der Titration wird entweder durch einen Farbumschlag der Probelösung (orange Farbe von Cr2O7
2- verschwindet) angezeigt oder kann mit einem Redoxindikator sichtbar gemacht werden. Oft verwendet man jedoch eine biampero-
Cr2O72- + 6 Fe2+ + 14 H+ 2 Cr3+ + 6 Fe3+

Analytik 10.8 metrische Endpunktsindikation, da diese direkt zu einem elektrischen Signal führt, welches elektronisch verarbeitet werden kann. Biamperometrische Endpunktsindikation: Zwei Indikatorelektroden werden in die Lösung getaucht. Zwischen diesen wird eine kleine Spannung angelegt, ca. 50 - 100 mV und so ein Stomkreis aufgebaut. Enthält die Lösung ein Redoxpaar, welches an den Indikatorelektroden reversibel oxidiert und reduziert werden kann, fliesst im Indikatorstromkreis ein Strom. An der einen Indikatorelektrode wird das Redoxpaar oxidiert, an der andern reduziert. Enthält die Probe nur die oxidierte Form oder die reduzierte Form, kann kein Strom fliessen. Die Stromstärke wird durch die kleinere der beiden Konzentrationen (oxidierte Form, reduzierte Form) bestimmt. Bei der diskutierten Titration von Cr2O7
2- in Anwesenheit von Fe3+ ist nur das Redoxpaar Fe3+/Fe2+ an einer Pt-Elektrode reversibel. Da die Probe vor dem Aequivalenzpunkt praktisch nur Fe3+ enthält, alles Fe2+, welches an der Arbeitselektrode gebildet wird reagiert sofort mit Cr2O7
2-, fliesst kein Strom in Indikatorstromkreis. Nach den Aequivalenzpunkt fliesst ein Strom, da die Lösung nun Fe2+ und Fe3+ enthält. Dabei wird an der einen Indikatorelektrode Fe3+ zu Fe2+ reduziert, an der andern Fe2+ zu Fe3+ oxidiert. Diese Methode den Endpunkt zu bestimmen, kann auch eingesetzt werden, wenn beide Redoxpaare an den Indikatorelektroden reversible Redoxreaktionen eingehen. Unter diesen Bedingungen fällt der Strom vor dem Aequvalenzpunkt, ist am Aequivalenzpunkt minimal und steigt dann wieder an.
__________ Beispiel: Coulometrische Bestimmung von Cl- (Cotlove Chloridimeter) Analysereaktion:
Ag+ + Cl- {AgCl}s

Analytik 10.9 Reaktion an der Arbeitselektrode (Ag, Anode): {Ag}s Ag+ + e-
Biamperometrische Endpunktsindikation:
AgAg+Ag
Sobald die Lösung Ag+ enthält beginnt der Strom im Indikator- stromkreis zu fliessen.
__________ Wasser kann nach Karl-Fischer auch coulometrisch titriert werden. Die Analysereaktion ist: 2 H2O + SO2 + I2 H2SO4 + 2 HI Reaktion an der Arbeitselektrode: 2 I- I2 + 2 e-
An der Arbeitselektrode wird I- elektrolytisch zu I2 oxidiert, um nachher mit SO2 in der Lösung zu reagieren. Der Endpukt kann biamperometrisch angezeigt werden. Im Indikatorstromkreis fliesst ein Strom, sobald die Probe I2 und I- enthält. Coulometrische Titrationsmethoden mit einer biamperometrischen Endpunkts-Indikation sind für Routineanalysen geeignet, da sie leicht automatisiert werden können. Es werden keine Masslösungen gebraucht. Ströme können gut stabilisiert werden und Zeitintervalle können sehr genau gemessen werden. In der folgenden Tabelle sind weitere coulometrische Titrationen zusammengestellt. In allen diesen Analysen wird der Titrand elektrolytisch an der Arbeitselektrode erzeugt.

Analytik 10.10
Tab. 10.1. Einige Beispiele für coulometrische Titrationen. 10.4. Voltammetrie Voltammetrie resp. Polarographie erlauben sowohl eine qualitative, wie auch quantitative Analyse von redoxaktiven Teilchen, z.B. Metallionen. Es handelt sich dabei um sehr empfindliche quantitative Methoden. Nachweisgrenzen von 10-8 - 10-10 mol l-1 sind möglich. Zusätzlich geben diese Methoden einen Einblick in die Mechanismen von Redoxreaktionen in Lösung. Dazu wird die Strom-Spannungskurve der Probelösung in einer elektrochemischen Zelle gemessen.

Analytik 10.11
10.5. Elektrolysezelle für dieVoltammetrie. In einem Voltammogramm, resp. Polarogramm wird der Strom, welcher im Stromkreis Arbeitselektrode-Hilfselektrode fliesst, als Funktion der Spannung zwischen Arbeitselektrode und Referenzelektrode gemessen und aufgetragen. Das Potential wächst im allg. proportional mit der Zeit. Die Potentiale werden gegen eine gesättigte Kalomelelektrode ESCE gemessen. Sie sind durch die folgende Beziehung mit den Potentialen gegen die Wasserstoffelektrode ENHE verknüpft:
Evs SCE = Evs NHE - 0.242 V
Das Potential zwischen Hilfselektrode und Lösung ist nicht kontrolliert.
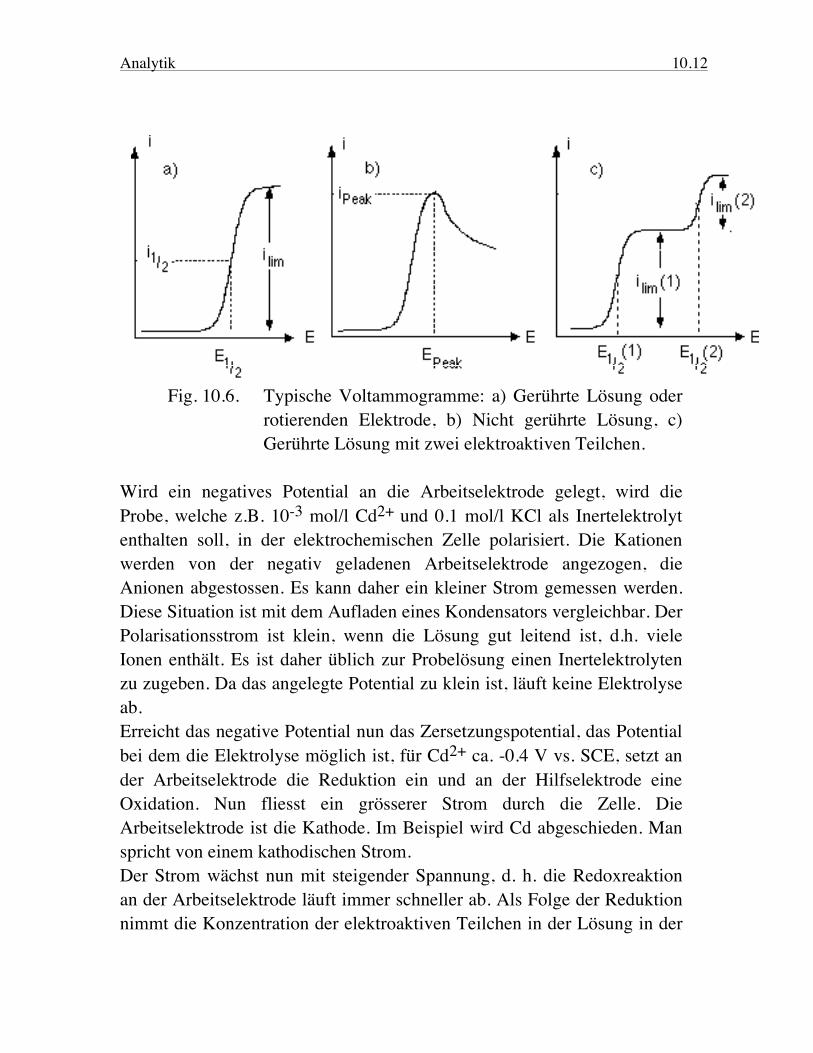
Analytik 10.12
Fig. 10.6. Typische Voltammogramme: a) Gerührte Lösung oder rotierenden Elektrode, b) Nicht gerührte Lösung, c) Gerührte Lösung mit zwei elektroaktiven Teilchen. Wird ein negatives Potential an die Arbeitselektrode gelegt, wird die Probe, welche z.B. 10-3 mol/l Cd2+ und 0.1 mol/l KCl als Inertelektrolyt enthalten soll, in der elektrochemischen Zelle polarisiert. Die Kationen werden von der negativ geladenen Arbeitselektrode angezogen, die Anionen abgestossen. Es kann daher ein kleiner Strom gemessen werden. Diese Situation ist mit dem Aufladen eines Kondensators vergleichbar. Der Polarisationsstrom ist klein, wenn die Lösung gut leitend ist, d.h. viele Ionen enthält. Es ist daher üblich zur Probelösung einen Inertelektrolyten zu zugeben. Da das angelegte Potential zu klein ist, läuft keine Elektrolyse ab. Erreicht das negative Potential nun das Zersetzungspotential, das Potential bei dem die Elektrolyse möglich ist, für Cd2+ ca. -0.4 V vs. SCE, setzt an der Arbeitselektrode die Reduktion ein und an der Hilfselektrode eine Oxidation. Nun fliesst ein grösserer Strom durch die Zelle. Die Arbeitselektrode ist die Kathode. Im Beispiel wird Cd abgeschieden. Man spricht von einem kathodischen Strom. Der Strom wächst nun mit steigender Spannung, d. h. die Redoxreaktion an der Arbeitselektrode läuft immer schneller ab. Als Folge der Reduktion nimmt die Konzentration der elektroaktiven Teilchen in der Lösung in der

Analytik 10.13 Nähe der Arbeitselektrode, im Beispiel Cd2+, ab. Damit weiteres Cd2+ reduziert werden kann, muss dieses zur Arbeitselektrode diffundieren. Da die negative Ladung der Elektrode durch die Kationen des Inertelektrolyten weitgehend abgeschirmt wird, handelt es sich im wesentlichen um thermische Diffusion. Wird die Diffusion langsamer als die Reduktion an der Elektrode, fällt die lokale Konzentration des elektroaktiven Teilchens an der Elektrodenoberfläche ab. Der Strom wird daher kleiner. Er wird von der Geschwindigkeit, mit der Cd2+ an die Elektrode diffundiert, kontrolliert. Man spricht von einem diffusions-kontrollierten Strom. Dieses Abfallen kann daher unterdrückt werden, wenn die Lösung kräftig gerührt wird oder dafür gesorgt wird, dass immer neue Probelösung über die Elektrode fliesst. Der Strom ist bei grosser Spannung durch die Konzentration des Analyten und dessen Transport durch Diffusion oder Konvektion an die Elektrode limitiert (Rotating-Disk-Elektrode). Die Stärke des maximalen Stromes iPeak in einer ungerührten Lösung, resp. des Diffusionstromes ilim in gerührter Lösung ist proportional zur Konzentration des elektroaktiven Teilchens in der Lösung (quantitative Analyse). Das Potential bei halbem Diffusionsstrom, das Halbwellenpotential, E1/2 resp. beim maximalen Strom in einer nicht gerührten Lösung EPeak, ist charakteristisch für das elektroaktive Teilchen (qualitative Analyse). Das Potential kann nun weiter negativ werden ohne dass der Strom wesentlich wächst, bis eine zweite Reduktion einsetzt, resp. das Lösungsmittel oder der Inertelektrolyt an der Arbeitselektrode reagiert. Man beobachtet dann ein weiteres Ansteigen des Stromes. Entsprechend kann auch ein positives Potential angelegt werden. Die Zelle wird dann polarisiert, bis an der Arbeitselektrode eine Oxidation ablaufen kann. Dann beginnt ein Strom zu fliessen. Man spricht in diesem Falle von einem anodischen Strom. Das Voltammogramm wird üblicherweise so dargestellt, dass der kathodische Strom nach oben und das kathodische Potential (negativ) nach rechts zunimmt. Das Halbwellenpotential ist charakteristisch für das elektroaktive Teilchen

Analytik 10.14
+
_
Fig. 10.7. Konvention für die Darstellung eines Voltammo- gramms, resp. Polarogramms. in der Probelösung (siehe Form der voltammetrischen Welle). Es wird durch die Komplexierung der Metallionen in der Lösung beeinflusst. Durch die Komplexbildung wird das solvatisierte Ion stabilisiert. Es ist daher schwieriger dieses zu reduzieren. Das Halbwellenpotential verschiebt sich gegen negativere Werte. Die Verschiebung hängt mit der Grösse der Komplexbildungskonstante zusammen. Für nicht reversible Redoxreaktionen ist das Halbwellenpotential negativer als für reversible Reaktionen. Es muss zusätzlich die Überspannung überwunden werden (vergl. 10.1). Die Ueberspannung ist sehr stark von der Beschaffenheit der Elektrodenoberfäche abhängig.
Fig. 10.8. Potentialfenster für Voltammetrie, resp. Polarographie

Analytik 10.15 in wässriger Lösung. Das Potentialfenster, in dem eine Probe mittels der Voltammetrie unter-sucht werden kann, wird durch die Zersetzungpotentiale für die Reduktion und die Oxidation des Lösungsmittels, resp. des Inertelektrolyten gegeben. Für wässrige Lösungen ist die obere, anodische Grenze des Potential-fensters durch die Oxidation von H2O zu O2 gegeben. Diese läuft an einer Pt-Elektrode bei Potentialen > 1.0 V ab. Die untere, kathodische Grenze wird durch die Reduktion von H2O zu H2 gegeben. Dieses ist an einer Pt-Elktrode < - 0.1 V. Beide Zersetzungsspannungen sind abhängig vom pH. Daher verschiebt sich das Potentialfenster mit steigendem pH gegen negativere Potentiale. Ist die Zersetzungsspannung für das Anion des Inertelektrolyten negativer als diejenige von H2O zu O2, resp. für das Kation positiver als diejenige für H2O zu H2 wird das Fenster durch diese Spannungen bestimmt und ist daher kleiner. An Stelle von Pt-Elektroden verwendet man auch Kohle (Graphit, Glassy Carbon) Elektroden. Die Ueberspannung für die Reduktion von H2O zu H2 ist an dieser Elektrode wesentlich grösser. Die Zesetzung von Wasser beginnt erst bei Potentialen > -1.0 V. Das Potentialfenster an einer Kohle-elektrode ist daher in Richtung kathodischer Potentiale grösser. An einer Hg-Elektrode ist die Ueberspannung für die H2 Entwicklung noch grösser. Die Zersetzungsspannung von H2O liegt bei ca -2.0 V (Polarographie).
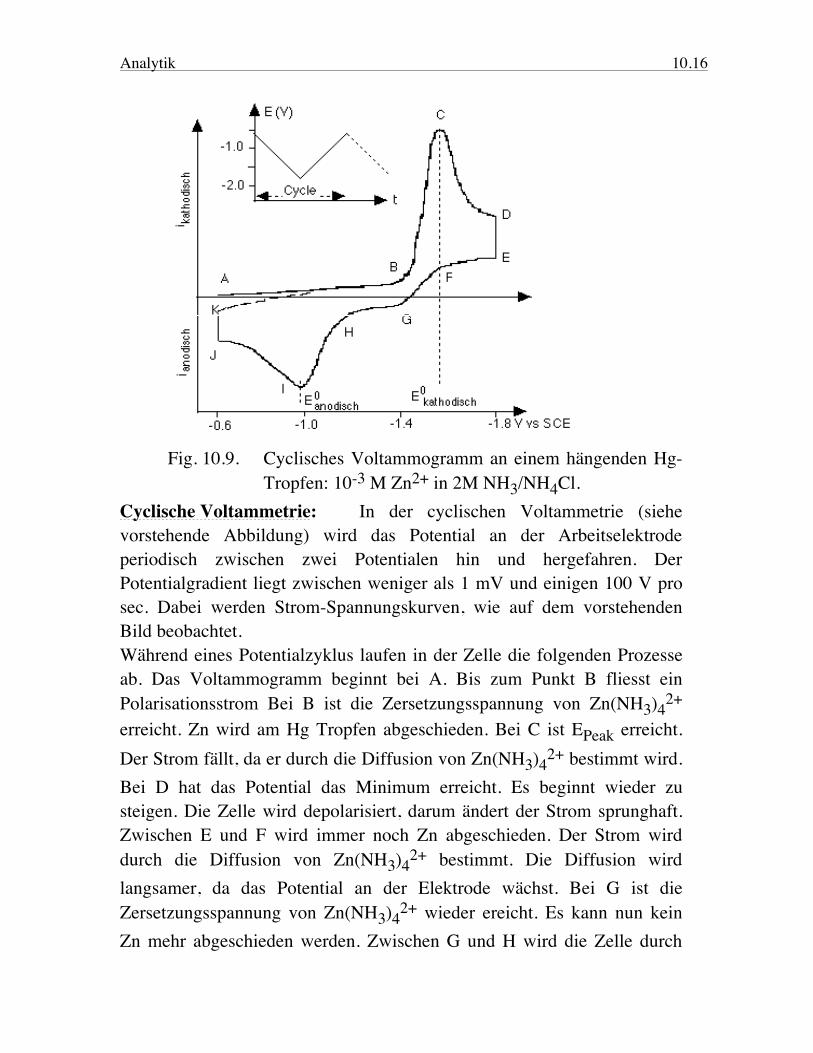
Analytik 10.16
Fig. 10.9. Cyclisches Voltammogramm an einem hängenden Hg- Tropfen: 10-3 M Zn2+ in 2M NH3/NH4Cl. Cyclische Voltammetrie: In der cyclischen Voltammetrie (siehe vorstehende Abbildung) wird das Potential an der Arbeitselektrode periodisch zwischen zwei Potentialen hin und hergefahren. Der Potentialgradient liegt zwischen weniger als 1 mV und einigen 100 V pro sec. Dabei werden Strom-Spannungskurven, wie auf dem vorstehenden Bild beobachtet. Während eines Potentialzyklus laufen in der Zelle die folgenden Prozesse ab. Das Voltammogramm beginnt bei A. Bis zum Punkt B fliesst ein Polarisationsstrom Bei B ist die Zersetzungsspannung von Zn(NH3)4
2+ erreicht. Zn wird am Hg Tropfen abgeschieden. Bei C ist EPeak erreicht. Der Strom fällt, da er durch die Diffusion von Zn(NH3)4
2+ bestimmt wird. Bei D hat das Potential das Minimum erreicht. Es beginnt wieder zu steigen. Die Zelle wird depolarisiert, darum ändert der Strom sprunghaft. Zwischen E und F wird immer noch Zn abgeschieden. Der Strom wird durch die Diffusion von Zn(NH3)4
2+ bestimmt. Die Diffusion wird langsamer, da das Potential an der Elektrode wächst. Bei G ist die Zersetzungsspannung von Zn(NH3)4
2+ wieder ereicht. Es kann nun kein Zn mehr abgeschieden werden. Zwischen G und H wird die Zelle durch

Analytik 10.17 den anodischen Strom polarisiert. Bei H ist die kathodische Zersetzungs-spannung erreicht. Zn aus dem Amalgam, welches bei der Reduktion gebildet wurde, wird oxidiert und geht in Lösung. Bei I ist das EPeak kathodisch erreicht. Die Geschwindigkeit der Elektrodenreaktion wird nun durch die Diffusion von Zn im Amalgam bestimmt. Bei J ist das maximale Potential erreicht. Das Potential beginnt wieder zu fallen. Die Zelle wird depolarisiert, daher ändert der Strom sprunghaft. Der Strom ist immer noch mehr anodisch als im ersten Cyclus, da das Potential noch ausreicht um Zn zu oxidieren. Nachdem das kathodische Zersetzungspotential unterschritten wird, sollten die Kurven des ersten und zweiten Cyclus zusammenfallen. Ist das nicht der Fall, zeigt dies, dass sich während des Cyclus die Oberfläche der Elektrode verändert hat. Es wurde z.B. ein Film abgeschieden, der sich nicht mehr auflösen lässt. Das cyclische Polarogramm ist in diesem Fall nicht symmetrisch, da die Form der Welle durch die Diffusionsgeschwindigkeit der oxidierten Form, Zn(NH3)4
2+ in der Probe, und der reduzierten Form, Zn in Hg, bestimmt wird. Diese Geschwindigkeiten sind sicher verschieden. Laufen bei der Oxidation und Reduktion dieselbe Reaktion in verschiedener Richtung ab, und handelt es sich um reversible Elektronenübertragungsreaktionen, ist die Differenz zwischen E0 kathodisch und E0 anodisch 0.059/n V. Weicht diese Differenz von diesem Wert ab, wird dies als Zeichen für eine nicht reversible Redoxreaktion angesehen. Unterschiede zwischen der kathodischen und der anodischen Welle geben auch direkte Informationen über die Stabilität der oxidierten, resp. reduzierten Form. Laufen Zersetzungsreaktionen ab, bevor das Teilchen wieder reduziert, resp. oxidiert wird, ist die Redoxreaktion nicht reversibel. Wird die Geschwindigkeit des Potentialsweeps variiert, kann Einblick in die Kinetik der Zersetzungreaktionen erhalten werden. 10.5. Polarographie Polarographie ist Voltammmetrie mit einer tropfenden Hg-Elektrode als Arbeitselektrode. Die Methode wurde 1921 durch den tschechischen Elektrochemiker J. Heyrovsky entdeckt und dann weiter entwickelt. Er

Analytik 10.18 erhielt für seine Arbeiten 1952 den Nobelpreis. Eine tropfende Hg-Elektrode ist eine Glaskapillare mit einem Durchmesser von 0.03 - 0.05 mm durch die Hg aus einem Reservoir im Gravitationsfeld ausfliesst. Die Vorteile einer tropfenden Hg Elektrode sind: a) Eine hohe Ueberspannung für die Elektrolyse von Wasser. An einer Hg-Elektrode können sogar auch Alkalimetalle, wie K und Na abgeschieden werden. b) Die Elektrodenoberfläche wird immer wieder erneuert. Sie ist daher gut reproduzierbar. Schwerlösliche Niederschläge und Metall-überzüge, welche die Eigenschaften einer Arbeitselektrode stark be-einflussen können, stören die Messung nicht. Ein Nachteil ist, dass der Strom an einer tropfenden Hg-Elektrode stark schwankt. Der Strom ist abhängig von der Fläche der Elektrode und steigt daher an, während der Hg Tropfen wächst und fällt zusammen, wenn der Tropfen abfällt. Die anodische Grenze des Potentialfensters liegt bei ca. 0.1 V vs. SCE. Sie wird durch die Oxidation von Hg zu Hg2
2+ gegeben. O2 wird an der Hg-Elektrode bei einem Potential von ca -0.1 V zu H2O reduziert. Es ist daher nötig vor der Analyse O2 mit N2 oder Argon aus der Lösung auszutreiben, um die O2-Welle zu eliminieren. Die polarographische Welle an einem Hg-Tropfen zeigt oft ein Strommaximum. Der Grund für diesen Effekt ist nicht klar. Er scheint mit der Benetzung der Elektrodenoberfläche zusammenzuhängen, da er unterdrückt wird, wenn der Probe wenig einer oberfächenaktiven Substanz (Triton X-100) zugegeben wird.
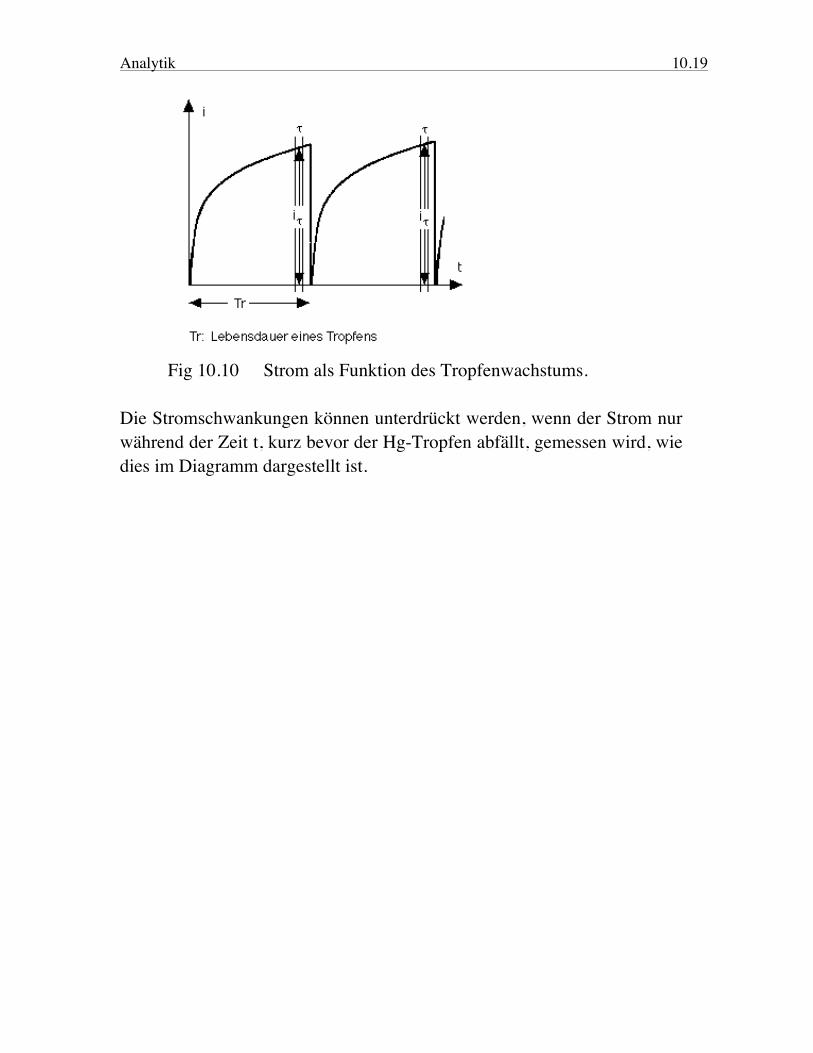
Analytik 10.19
Fig 10.10 Strom als Funktion des Tropfenwachstums. Die Stromschwankungen können unterdrückt werden, wenn der Strom nur während der Zeit t, kurz bevor der Hg-Tropfen abfällt, gemessen wird, wie dies im Diagramm dargestellt ist.
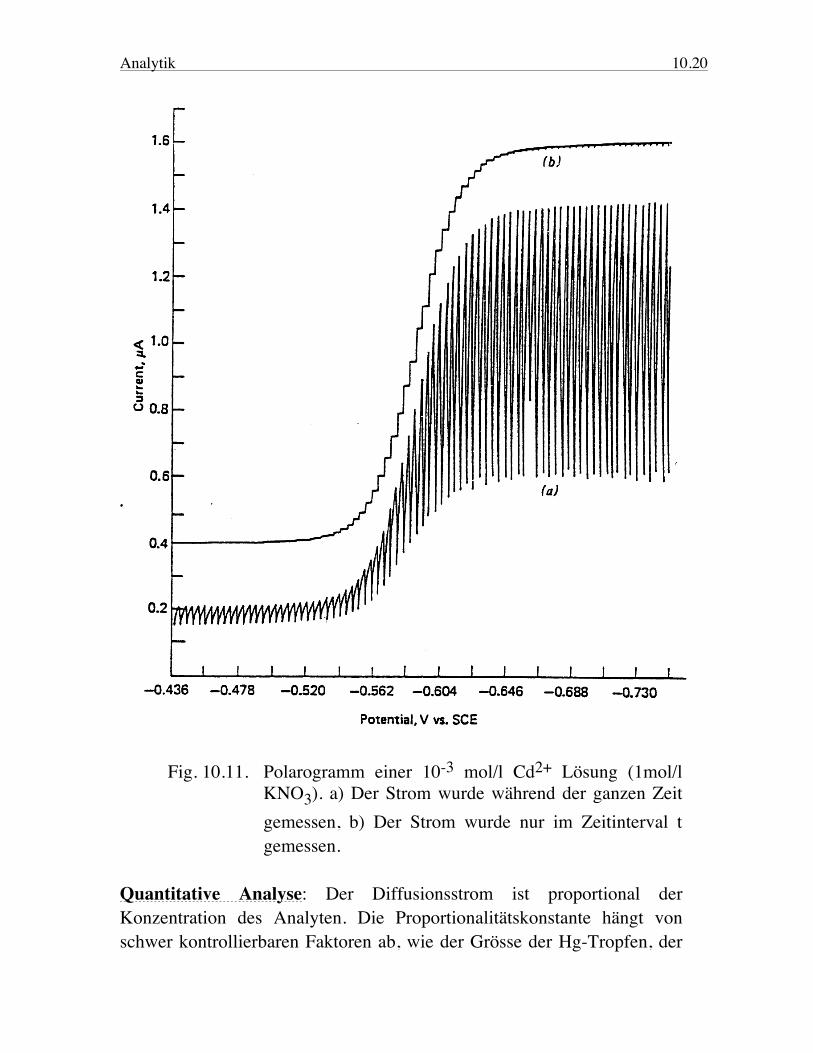
Analytik 10.20
Fig. 10.11. Polarogramm einer 10-3 mol/l Cd2+ Lösung (1mol/l KNO3). a) Der Strom wurde während der ganzen Zeit gemessen, b) Der Strom wurde nur im Zeitinterval t gemessen. Quantitative Analyse: Der Diffusionsstrom ist proportional der Konzentration des Analyten. Die Proportionalitätskonstante hängt von schwer kontrollierbaren Faktoren ab, wie der Grösse der Hg-Tropfen, der

Analytik 10.21 Diffusionsgeschwindigkeit der elektroaktiven Teilchen und der Viskosität der Probelösung. Ein quantitative Analyse wird daher im allg. als eine relative Messung ausgeführt. Gleichzeitig mit der Analyse wird eine Eichlösung gemessen.
ilim(Probe)ilim(ref)
= c(Probe)c(ref)
Fig. 10.12. Schematische Darstellung der polarographischen Konzentrationsbestimmung. Um den Einfluss der Matrix zu eliminieren wird für die Eichung oft die Aufstockmethode verwendet. Die Nachweisgrenze der Methode ist durch den Polarisationsstrom gegeben. Dieser kann klein gehalten werden, wenn die Probe eine hohe elektrische Leitfähigkeit zeigt, d.h. viele Ionen enthält. Der Probe wird daher wenn möglich ein Inertelektrolyt zugegeben. Qualitative Analyse: E1/2 ist charakteristisch für den Analyten in einer bestimmten Matrix. In der folgenden Tabelle sind die Halbwellenpotentiale von einigen Kationen in verschiedenen Inertelektrolyten zusammengestellt. Tab. 10.2. Halbwellenpotentiale für einige Metallionen.

Analytik 10.22
Um die Empfindlichkeit der polarographischen Analysemethoden zu verbessern, wurden einige Varianten entwickelt. Differential Puls Polarographie:
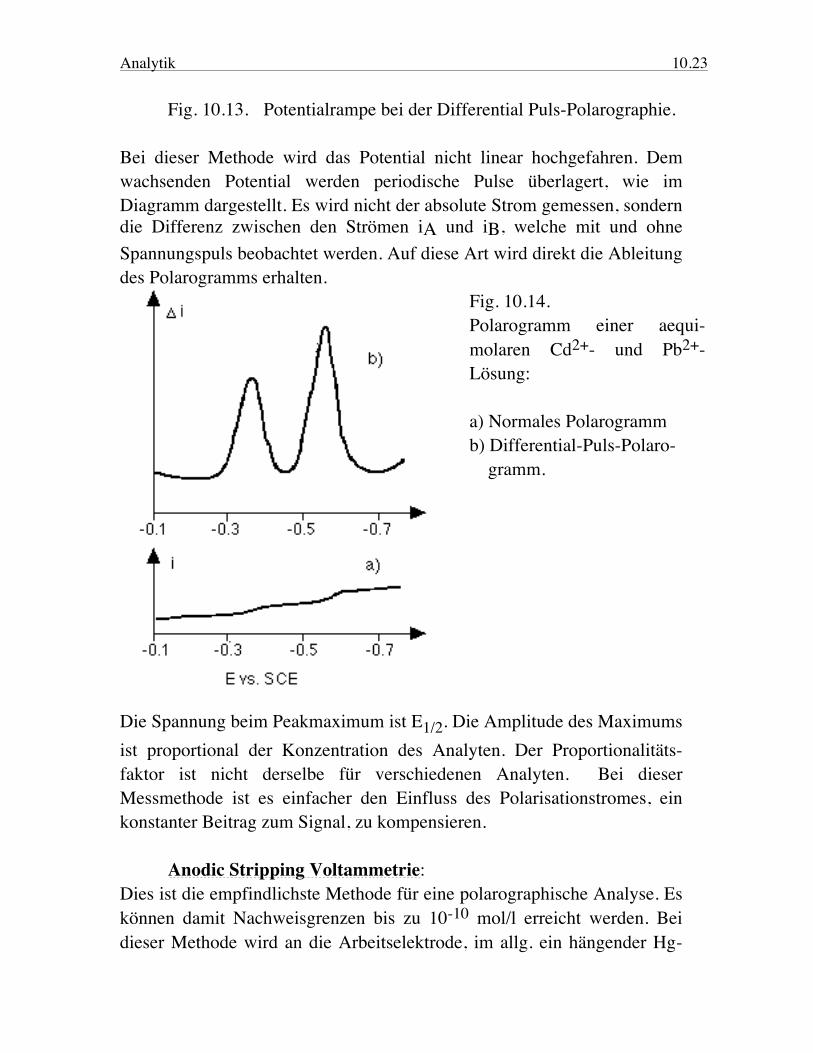
Analytik 10.23 Fig. 10.13. Potentialrampe bei der Differential Puls-Polarographie. Bei dieser Methode wird das Potential nicht linear hochgefahren. Dem wachsenden Potential werden periodische Pulse überlagert, wie im Diagramm dargestellt. Es wird nicht der absolute Strom gemessen, sondern die Differenz zwischen den Strömen iA und iB, welche mit und ohne Spannungspuls beobachtet werden. Auf diese Art wird direkt die Ableitung des Polarogramms erhalten.
Fig. 10.14. Polarogramm einer aequi-molaren Cd2+- und Pb2+- Lösung: a) Normales Polarogramm b) Differential-Puls-Polaro- gramm.
Die Spannung beim Peakmaximum ist E1/2. Die Amplitude des Maximums ist proportional der Konzentration des Analyten. Der Proportionalitäts-faktor ist nicht derselbe für verschiedenen Analyten. Bei dieser Messmethode ist es einfacher den Einfluss des Polarisationstromes, ein konstanter Beitrag zum Signal, zu kompensieren. Anodic Stripping Voltammetrie: Dies ist die empfindlichste Methode für eine polarographische Analyse. Es können damit Nachweisgrenzen bis zu 10-10 mol/l erreicht werden. Bei dieser Methode wird an die Arbeitselektrode, im allg. ein hängender Hg-

Analytik 10.24 Tropfen, über längere Zeit (bis zu einigen Stunden) eine Spannung angelegt, die so gross ist, dass ein kathodischer Strom fliesst und der Analyt abgeschieden wird. Dieser Prozess ist sehr langsam, wenn die Konzentration des Analyten in der Probe sehr klein ist. Der Strom, welcher fliesst, ist daher sehr klein und kann nicht gemessen werden. Nach einer vorgegebenen Zeitspanne wird das Potential an der Arbeitselektrode erhöht, so dass ein anodischer Strom fliesst, und der Analyt, welcher an der Arbeitselektrode akkumuliert wurde, in kurzer Zeit oxidert wird. Der Strom, welcher vorher während langer Zeit (Stunden) geflossen ist, fliesst in kurzer Zeit (sec) zurück. Es ist daher ein Strompeak zu beobachten, dessen Integral proportional zum abgeschiedenen Analyten, resp. proportional zur Konzentration des Analyten in der Probe ist. Polarographische und Voltammetrische Analysenmethoden sind nicht nur auf Metallionen in wässrigen Lösungen beschränkt. Viele organische Verbindungen können an einer Elektrode oxidiert, resp. reduziert werden. Da die reduzierte und die oxidierte Form löslich sind, werden sie im allg. nicht an der Elektrode abgeschieden. Die einzige Voraussetzung ist, dass die Probelösung eine minimale elektrische Leitfähigkeit besitzt. Als Lösungmittel können alle organischen Lösungsmittel verwendet werden, in denen Salze löslich sind und mindest teilweise in Ionen dissozieren. Beispiele sind Dimethylformamid, Dimethylsulfoxid, Alkohole, und Ketone, ev. gemischt mit Wasser, Amine, Nitrile etc. Als Inertelektrolyten dienen im allg. Tertiäralkylammoniumsalze. Matrixeffekte sind in organischen Lösungen viel grösser. Die Zusammensetzung der Probe, pH, Ionenstärke etc. muss daher kontrolliert werden. 10.6. Die Form der voltammetrischen, polarographischen Welle Aus diesen qualitativen Ueberlegungen kann die Form der polarographischen Welle leicht abgeleitet werden. Als Beispiel soll die Reduktion von Cd2+ an einer Hg-Elektrode dienen. Das abgeschiedenen Cd bildet mit der Elektrode ein Amalgam. Das Potential an der Arbeitselektrode ist durch die Nernstsche Gleichung gegeben.

Analytik 10.25
E = E0Cd2+/Cd - R • T
n • F ln Cd(Hg)
Cd2+(s) = E0
Cd2+/Cd - 0.0592
log Cd(Hg)Cd2+(s)
10.4 [Cd2+
(s)]: lokale Cd2+ Konzentration an der Elektrodenoberfläche [Cd(Hg)]: Cd-Konzentration im Amalgam Der Strom wird durch die Diffusion von Cd2+ an die Elektrode bestimmt. Nach dem Fickschen Gesetz ist die Diffusionsgeschwindigkeit proportional zur Konzentrationsdifferenz zwischen der Lösung und der Oberfläche.
i = k • D • Cd2+ - Cd2+(s)
10.5 D: Diffusionskoeffizient des elektroaktiven Teilchens Die Proportionalitätskonstante k wird durch die Geometrie der Elektrode bestimmt. Sie ist unabhängig von der Art der Elektrodenreaktion. Der maximale Strom der fliessen kann, wird erreicht, wenn die lokale Konzentration von Cd2+ an der Elektrodenoberfläche verschwindet, d.h. jedes Cd2+, welches an die Oberfläche diffundiert, wird sofort reduziert. Der diffusionslimitierte Strom ist daher:
ilim = k • D • Cd2+
10.6 Differenz zwischen dem Strom und dem diffusionslimitierten Strom:
Cd2+(s) = ilim - ik • D
10.7 Die Konzentration von Cd im Amalgam ist proportional dem Strom. Cd diffundiert von der Oberfläche in die Hg Elektrode, wo die Konzentration von Cd praktisch null ist.
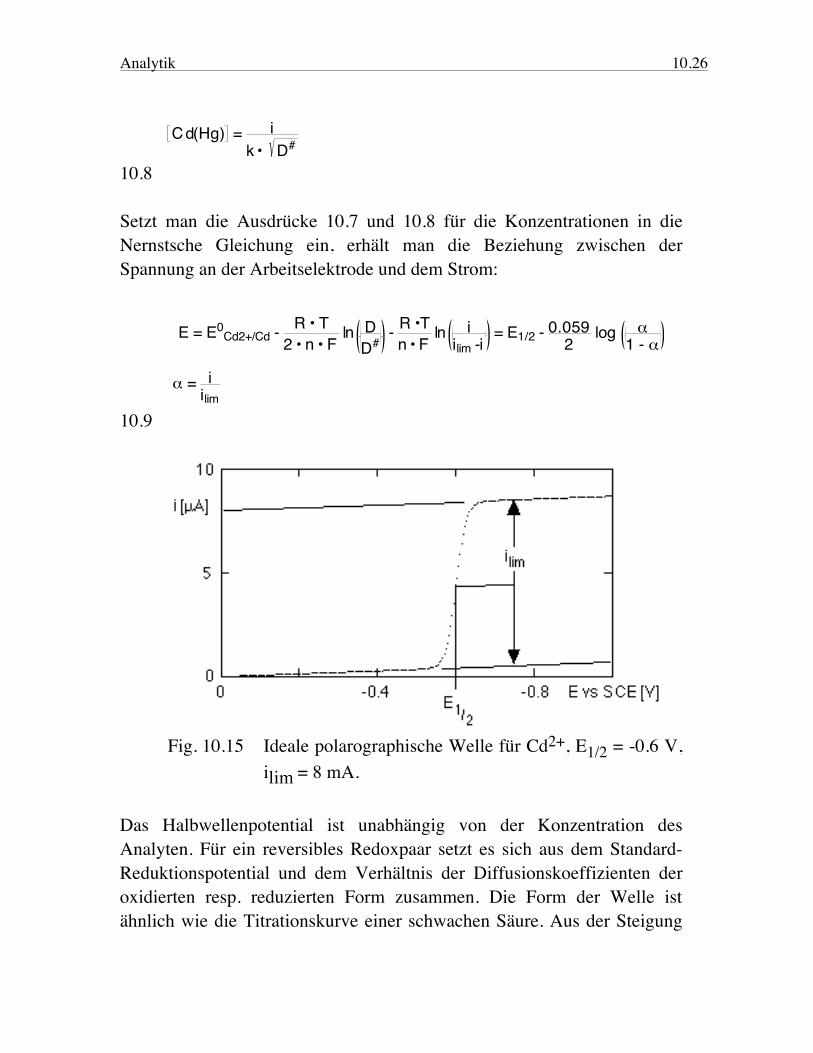
Analytik 10.26
Cd(Hg) = i
k • D# 10.8 Setzt man die Ausdrücke 10.7 und 10.8 für die Konzentrationen in die Nernstsche Gleichung ein, erhält man die Beziehung zwischen der Spannung an der Arbeitselektrode und dem Strom:
E = E0Cd2+/Cd - R • T
2 • n • F ln D
D# - R •T
n • F ln i
ilim -i = E1/2 - 0.059
2 log α
1 - α
α = iilim
10.9
Fig. 10.15 Ideale polarographische Welle für Cd2+, E1/2 = -0.6 V, ilim = 8 mA. Das Halbwellenpotential ist unabhängig von der Konzentration des Analyten. Für ein reversibles Redoxpaar setzt es sich aus dem Standard-Reduktionspotential und dem Verhältnis der Diffusionskoeffizienten der oxidierten resp. reduzierten Form zusammen. Die Form der Welle ist ähnlich wie die Titrationskurve einer schwachen Säure. Aus der Steigung
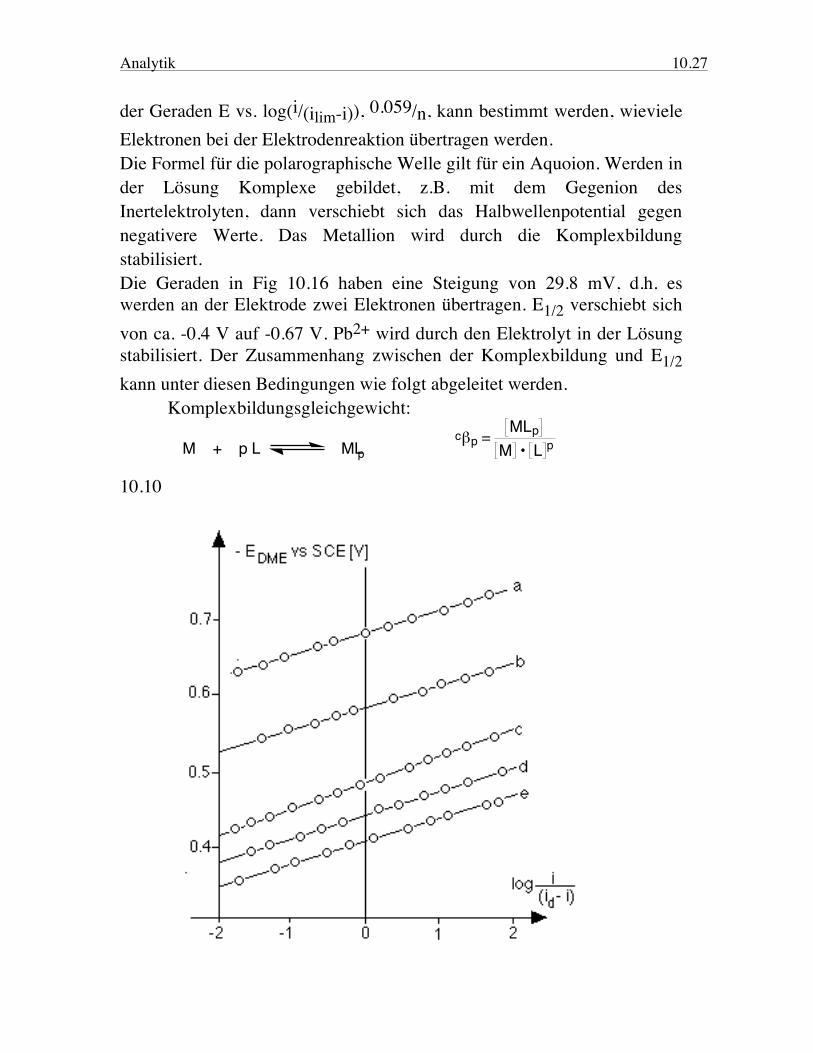
Analytik 10.27
der Geraden E vs. log(i/(ilim-i)), 0.059/n, kann bestimmt werden, wieviele Elektronen bei der Elektrodenreaktion übertragen werden. Die Formel für die polarographische Welle gilt für ein Aquoion. Werden in der Lösung Komplexe gebildet, z.B. mit dem Gegenion des Inertelektrolyten, dann verschiebt sich das Halbwellenpotential gegen negativere Werte. Das Metallion wird durch die Komplexbildung stabilisiert. Die Geraden in Fig 10.16 haben eine Steigung von 29.8 mV, d.h. es werden an der Elektrode zwei Elektronen übertragen. E1/2 verschiebt sich von ca. -0.4 V auf -0.67 V. Pb2+ wird durch den Elektrolyt in der Lösung stabilisiert. Der Zusammenhang zwischen der Komplexbildung und E1/2 kann unter diesen Bedingungen wie folgt abgeleitet werden. Komplexbildungsgleichgewicht:
M + p L MLp
cβp = MLp
M • L p
10.10

Analytik 10.28 Fig. 10.16. EDME vs log [i . ( id - i )] für eine Pb2+ Lösung. a) in 0.1 mol/l NaOH, b) in 1 mol/l K2C2O4, c) in 0.5 mol/l Natrium-Tartrat, d) in 1 mol/l KCl, e) in 1 mol/l KNO3. In einer Lösung, welche einen grossen Überschuss von Ligand enthält, [L]tot >> [M]tot, liegt der grösste Teil des Metallions komplexiert vor, d.h. [MLp] ≈ [M]tot. Der Strom wird daher durch die Diffusion von MLp an die Elektrode bestimmt. Man kann analoge Gleichungen wie 10.6 und 10.7 finden. Der diffusionslimitierte Strom ilim ist auch unter diesen Bedingungen proportional zur Metallkonzentration [M]tot. Durch Substitution von MLp mit Hilfe der Komplexbildung 10.10 erhält man die Beziehung zwischen der Metallkonzentration an der Elektrode und dem Strom i.
ilim - i = k • DKomp • MLp(s) = k • DKomp •cβp • L p • M(s)
10.11
ilim = k • DKomp • MLp ≈ k • DKomp • M tot
10.12 Setzt man diese Ausdrücke in die Nernstsche Gleichung ein, findet man den Zusammenhang zwischen der Komplexbildungskonstanten cβp, der Ligandkonzentration und dem Halbwellenpotential.
E = EM+/M0 - 0.059
2 • n log DKomp
D# - 0.059
n log cβp - 0.059 • pn log L - 0.059
2 log i
ilim - i
E1/2 = E0M+/M - 0.059
2 • n log DKomp
D# - 0.059
n log cβp - p • 0.059n log L
10.13 Unter diesen Bedingungen ist das Halbwellenpotential abhängig vom Logarithmus der Ligandkonzentration und der Komplexbildungs-konstanten. Durch die Variation der Ligandkonzentration kann man die
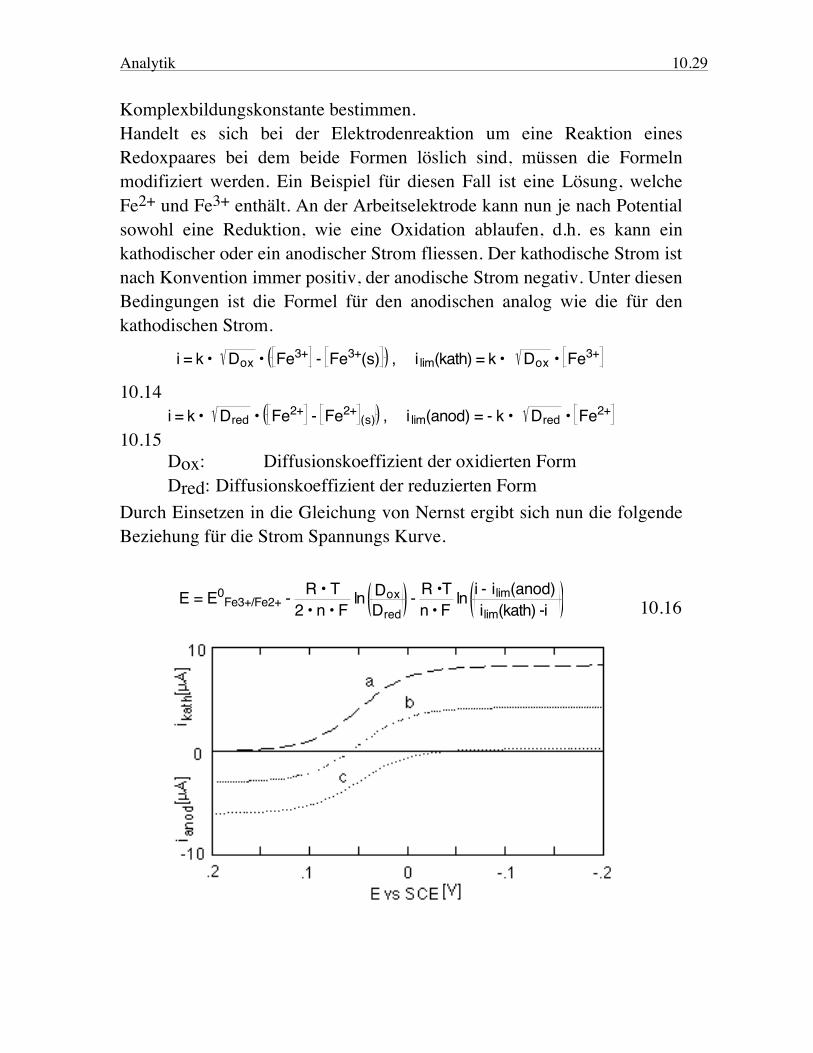
Analytik 10.29 Komplexbildungskonstante bestimmen. Handelt es sich bei der Elektrodenreaktion um eine Reaktion eines Redoxpaares bei dem beide Formen löslich sind, müssen die Formeln modifiziert werden. Ein Beispiel für diesen Fall ist eine Lösung, welche Fe2+ und Fe3+ enthält. An der Arbeitselektrode kann nun je nach Potential sowohl eine Reduktion, wie eine Oxidation ablaufen, d.h. es kann ein kathodischer oder ein anodischer Strom fliessen. Der kathodische Strom ist nach Konvention immer positiv, der anodische Strom negativ. Unter diesen Bedingungen ist die Formel für den anodischen analog wie die für den kathodischen Strom.
i = k • Dox • Fe3+ - Fe3+(s) , i lim(kath) = k • Dox • Fe3+
10.14 i = k • Dred • Fe2+ - Fe2+
(s) , i lim(anod) = - k • Dred • Fe2+ 10.15 Dox: Diffusionskoeffizient der oxidierten Form Dred: Diffusionskoeffizient der reduzierten Form Durch Einsetzen in die Gleichung von Nernst ergibt sich nun die folgende Beziehung für die Strom Spannungs Kurve.
E = E0Fe3+/Fe2+ - R • T
2 • n • F ln Dox
Dred - R •T
n • F ln i - ilim(anod)
ilim(kath) -i 10.16

Analytik 10.30 Fig. 10.17. Idealisiertes Polarogramm einer Lösung, die Fe3+ und Fe2+ enthält. E1/2(Fe3+/Fe2+) = 0.5 V, ilim(Fe3+) = 8 mA, ilim(Fe2+) = 6 mA, a) 1.4 mM Fe3+, b) 0.7 mM Fe2+, 0.7 mM Fe3+, c) 1.4 mM Fe2+. 10.7. Amperometrie In der Amperometrie wird der diffusionslimitierte Strom in einer Zelle bei einem bestimmten Potential gemessen, um so die Konzentration von redoxaktiven Teilchen zu verfolgen. Auf diese Art kann z.B. der Ablauf einer Titration verfolgt werden. Als Beispiel soll die Fällungstitration von Pb2+ mit CrO4
2- diskutiert werden. Die Analysereaktion ist:
Pb2+ + CrO42- {PbCrO4}s
Ksp(PbCrO4) = 1.8 •10-14
Die folgende Figur zeigt die Polarogramme, welche bei den folgenden f Werten (f = [CrO42-]/[Pb]) erhalten werden. Am Anfang der Titration enthält die Lösung nur Pb2+ (E1/2 = -0.5 V vs. SCE). Das Voltamogramm zeigt die charakteristische Welle. Im Laufe der Titation nimmt die Konzentration von Pb2+ ab. Es fällt als PbCrO4 aus. Entsprechend wird die Welle bei -0.5 V kleiner. Am Aequivalenzpunkt verschwindet sie ganz. Nach dem Aequivalenzpunkt steigt die Konzentration von CrO4
2- in der Lösung an. Das Polarogramm zeigt nun die Welle charakteristisch für CrO4
2- bei ca 0.0V vs. SCE. Der Strom il nimmt linear mit dem zugegebenen CrO4
2- zu. Um die Titration auszuwerten, wird der
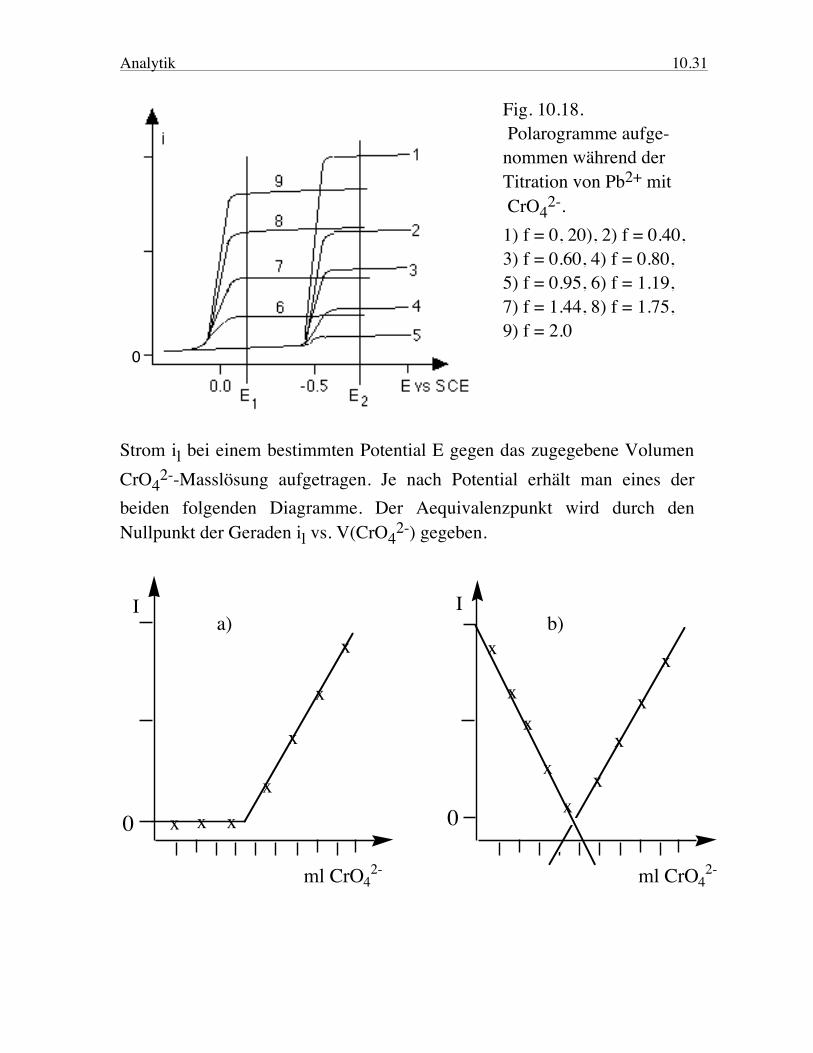
Analytik 10.31
0
Fig. 10.18. Polarogramme aufge- nommen während der Titration von Pb2+ mit CrO4
2-. 1) f = 0, 20), 2) f = 0.40, 3) f = 0.60, 4) f = 0.80, 5) f = 0.95, 6) f = 1.19, 7) f = 1.44, 8) f = 1.75, 9) f = 2.0
Strom il bei einem bestimmten Potential E gegen das zugegebene Volumen CrO4
2--Masslösung aufgetragen. Je nach Potential erhält man eines der beiden folgenden Diagramme. Der Aequivalenzpunkt wird durch den Nullpunkt der Geraden il vs. V(CrO4
2-) gegeben.
x
x
x
x
x x x0
I Ia) b)
x
xx
x
x
x
x
x
x
ml CrO42- ml CrO4
2-
0

Analytik 10.32 Fig. 10.19. Strom, welcher während der Titration von Pb2+ mit CrO4
2- gemessen wird. a) bei -0.2 V vs. SCE, b) bei -0.7 V vs. SCE. Es können auch Messonden (Sensoren) konstruiert werden, welche es erlauben die Konzentration von Teilchen in einer Lösung amperometrisch zu messen. Ein Beispiel ist der Sauerstoffsensor.
10.20. Amperometrischer O2 Sensor. In der Spitze des Sensors ist eine elektrochemische Zelle eingebaut. Diese ist durch eine Teflonmembran, welche durchlässig ist für O2, von der Probe getrennt. Enthält die Probe O2, diffundiert dieser durch die Membran in die Messzelle. An der Arbeitselektrode, in diesem Fall Kathode, wird ein Potential angelegt, so dass der Sauerstoff reduziert werden kann. Der Sauerstoff, welcher in der Messzelle verbraucht wird, muss durch Diffusion ersetzt werden. Das System ist in einem dynamischen Gleichgewicht. Ist die Diffusion durch die Membran langsam im Vergleich zur Elektronen-übertragung an der Arbeitselektrode, ist der Strom proportional zur Diffusionsgeschwindigkeit, resp. proportional zur Menge O2 in der Probe. Die Proportionalitätskonstante wird durch die Geschwindigkeit der Diffusion durch die Membran an die Arbeitselektrode bestimmt. Solche voltammetrischen Messzellen können gut miniaturisiert werden, so dass der

Analytik 10.33 ganze Sensor sehr klein wird. Dieses Messprinzip ist für viele elektroaktiven Substanzen anwendbar. Eine Modifikation davon sind Enzymelektroden. Dabei enthält die Membran ein Enzym. Dieses wandelt das Substrat in ein elektroaktives Teilchen um, welches dann in die Messzelle diffundiert und an der Arbeitselektrode reagiert. Die Menge des elektroaktiven Analyten ist proportional zur Konzentration des Substrates in der Lösung, solange die Membran einen Ueberschuss an Enzym enthält. Ein Beispiel für eine solche Elektrode ist die Glucoseelektrode. Dabei wird die folgende enzymatische Reaktion ausgenützt. Das elektroaktive Teilchen ist H2O2. Glucose + O2 + H2O Glucose Oxidase Gluconsäure + H2O2 10.8. Konduktometrie In der Konduktometrie wird die Leitfähigkeit der Lösung in Abhängigkeit ihrer Zusammensetzung gemessen. Die Ionenleitfähigkeit einer Elektrolyt-lösung ist auf die Beweglichkeit der Ionen zurückzuführen. Die Leit-fähigkeit einer Lösung kann nur mit Wechselstrom gemessen werden, da nur dann in der Zelle keine Redoxreaktion abläuft. Bei einer kondukto-metrischen Messung verwendet man ein Wechselfeld mit einer Frequenz > 1 kHz. Wird die Leitfähigkeit mit Wechselstrom mit einer Frequenz von 1 MHz und mehr bestimmt, spricht man von Oszillometrie. In diesem Falle können auch Informationen über die Beweglichkeit der Ladungsträger in der Lösung erhalten werden. Die Leitfähigkeit einer Lösung setzt sich aus den Beiträgen aller geladenen Teilchen in der Lösung zusammen. Diese hangen von der Konzentration der geladenen Teilchen und deren Beweglichkeit ab. Letztere wird durch die Grösse, die Ladung und die Solvatation der Ionen bestimmt. Im weiteren wird die Beweglichkeit von der Viskosität des Lösungsmittels beeinflusst. Die spezifische Leitfähigkeit einer Lösung wird durch die folgende Gleichung gegeben:
κ = e0 • N+ • z+ • u+ + N- • z- • u-
V = F • ceq
1000 • u+ + u-
10.17 κ: spezifische Leitfähigkeit [(Ω cm)-1 = S(Siemens) cm-1]

Analytik 10.34 N+,N-: Zahl der Kationen resp. Anionen in Lösung n+, n-: Mole Kationen resp. Anionen in Lösung F = NL • e0: Faraday Konstante z+, z-: Ladung der Ionen n+ • z+ = neq: Mole bewegter positiver Einheitsladungen n- • z- = neq: Mole bewegter negativer Einheitsladungen neq • V-1 = ceq Konzentration der bewegten Ladung [mol/l = 10-3.mol.cm-1] u+, u-: Beweglichkeit der Ionen (v • E-1 [cm2 s-1 V-1] Die spezifische Leitfähigkeit ist proportional der Zahl der Ionen in der Lösung, wenn sich die Ionen nicht gegenseitig in ihrer Beweglichkeit beeinflussen. Dies ist nur in idealen, unendlich verdünnten Lösungen der Fall. Daher ist die spezifische Leitfähigkeit einer Elektrolytlösung von der Konzentration abhängig. Bewegen sich die Ladungen schnell im Vergleich zur Aenderung des elektrischen Feldes, wird die Leitfähigkeit nicht durch die Diffusionsgeschwindigkeit der Teilchen bestimmt. Unter diesen Bedingungen gehorcht die Leitfähigkeit dem Ohmschen Gesetz. Der Leitwert der Lösung wird durch folgenden Ausdruck gegeben:
L Siemens = κ • A cm2
l cm = κ • Konst
10.18 A: Fläche der Elektroden l: Abstand der Elektroden Konst: Zellkonstante Die spezifische Leitfähigkeit ist proportional zur Zahl der Ladungen und eignet sich nicht, um die Leitfähigkeit einer unendlich verdünnten Lösung zu charakterisieren. Man führt daher die Aequivalenzleitfähigkeit Λeq, resp die molare Leitfähigkeit Λmol eines Elektrolyten ein. Es handelt sich dabei um die Leitfähigkeit von einem Mol Einheitsladungen, resp. um die Leitfähigkeit von einem Mol eines Salzes.

Analytik 10.35
Λeq = κceq • 1000
Λmol = κceq
• 1000 • z
10.19 Die Aequivalenzleitfähigkeit, resp. die molare Leitfähigkeit ist nicht null für eine unendlich verdünnte, ideale Lösung. Der Grenzwert der Aequivalenzleitfähigkeit für eine unendlich verdünnte Lösung nennt man die Grenzleitfähigkeit. Unter diesen Bedingungen beeinflussen sich die Ionen gegenseitig nicht mehr. Die Grenzleifähigkeit ist daher eine charakteristische Grösse für ein Salz in einem bestimmten Lösungsmittel, unabhängig von der Konzentration. Sie setzt sich aus der Grenzleitfähigkeit der Anionen und der Kationen zusammen.
lim c→0 Λeq = Λ0 = λ+
0 + λ-0
10.20 Der Teil des Stromes, welcher durch die Kationen, resp. die Anionen transportiert wird, nennt man die Ueberführungszahl, n+, resp. n-. Da es möglich ist, diese experimentell zu bestimmen, kann man die Grenz-leifähigkeit eines einzelnen Ions angeben.
n+ = λ+
Λ n+0 = λ
+0
Λ0 n- = λ
-
Λ n-0 = λ-0
Λ0
10.21 Die Grenzleitfähigkeit ist eine charakteristische Grösse des Ions in einem Lösungsmittel und kann tabelliert werden. Mit Hilfe dieser Werte kann die Grenzleitfähigkeit eines Elektrolyten berechnet werden. In der folgenden Tabelle sind die Grenzleitfähigkeiten einige Ionen zusammengestellt. Tab 10.3: Grenzleitfähigkeiten einiger Ionen. Kationen λ+
o[S cm2 mol-
1] Anionen λ-
o[S cm2 mol-1]
H3O+ NH4+ K+
349.8 73.7 73.6
OH- SO42- Br-
197.0 80.8 78.4

Analytik 10.36
Ba2+ Ag+ Ca2+ Mg2+ Na+ Li+
63.2 62.2 59.8 53.1 50.1 38.6
I- Cl- NO3- ClO4- F- CH3COO-
76.5 76.4 71.5 68.0 55.4 40.8
Die Grenzleitfähigkeit für H3O+ und OH- sind grösser als die anderer Ionen. Die Leitfähigkeit für diese Ionen ist nicht nur auf die Diffusion der Ionen zurück zu führen, sondern wird durch das Uebertragen von Protonen von einem Wassermolekül zum anderen ermöglicht. Leitfähigkeit von starken Elektrolyten (vollständig dissoziert):
Λ ≈ Λ0 - A • I =Λ0 - A • 1
2 c+ • z+2 + c- • z-2
10.22 Die Leitfähigkeit eines starken Elektrolyten ist abhängig von der Konzentration des Elektrolyten, resp. der Ionenstärke der Lösung. Der Grund für diesen Effekt ist das elektrostatische Potential zwischen den Ionen. Diese sind nicht homogen in der Lösung verteilt. In der Umgebung der Anionen sind Kationen angereichert und umgekehrt. Die Abweichen von der Idealität kann durch die Theorie von Debye, Hückel und Onsager behandelt werden. Die Abweichung von der idealen Lösung für verdünnte Lösungen (< 0.1 mol l-1) ist proportional zur Wurzel der Ionenstärke I. Leitfähigkeit eines schwachen Elektrolyten (Ostwaldsches Verdünnungsgesetz): Die Äquivalentleitfähigkeit eines schwachen Elektrolyten hängt von dessen Konzentration und dem Dissoziationsgrad ab.
Λ = κ
α • ctot • 1000
α = A+
ctot = B-
ctot 10.23 Die spezifische Leitfähigkeit ist auch für schwache Elektrolyten

Analytik 10.37 proportional zur Konzentration, da diese in unendlich verdünnter Lösung vollständig dissozieren.
lim c→0 α = 1, κ = ctot • Λ • 10-3 = α • ctot • Λ0 • 10-3
10.24 Die Aequivalentleitfähigkeit für einen schwachen Elektrolyten ist proportional zum Dissoziationsgrad α. Der Dissoziationsgrad ist abhängig von der Totalkonzentration ctot und der Dissoziationskonstante Kd.
Kd = A+ • B-
A B = α
2 • ctot1- α α = - Kd + Kd
2 + 4 • Kd • ctot2 • ctot
10.25 Für einen sehr schwachen Elektrolyten α << 1 vereinfacht sich der Ausdruck 10.25:
α ≈ Kd
ctot Λ ≈ Λ0 • Kd
ctot 10.26 Die Aequivalenzleitfähigkeit ist unter diesen Bedingungen umgekehrt proportional zur Wurzel aus ctot. Konduktometrische Titration: Bei der Konduktometrischen Titration wird die Leitfähigkeit der Lösung als Funktion der zugegebenen Masslösung gemessen. Die Leitfähigkeit setzt sich aus den Beiträgen aller Ionen in der Lösung zusammen: a) Titration einer starken Säure mit einer starken Base, HA mit NaOH:
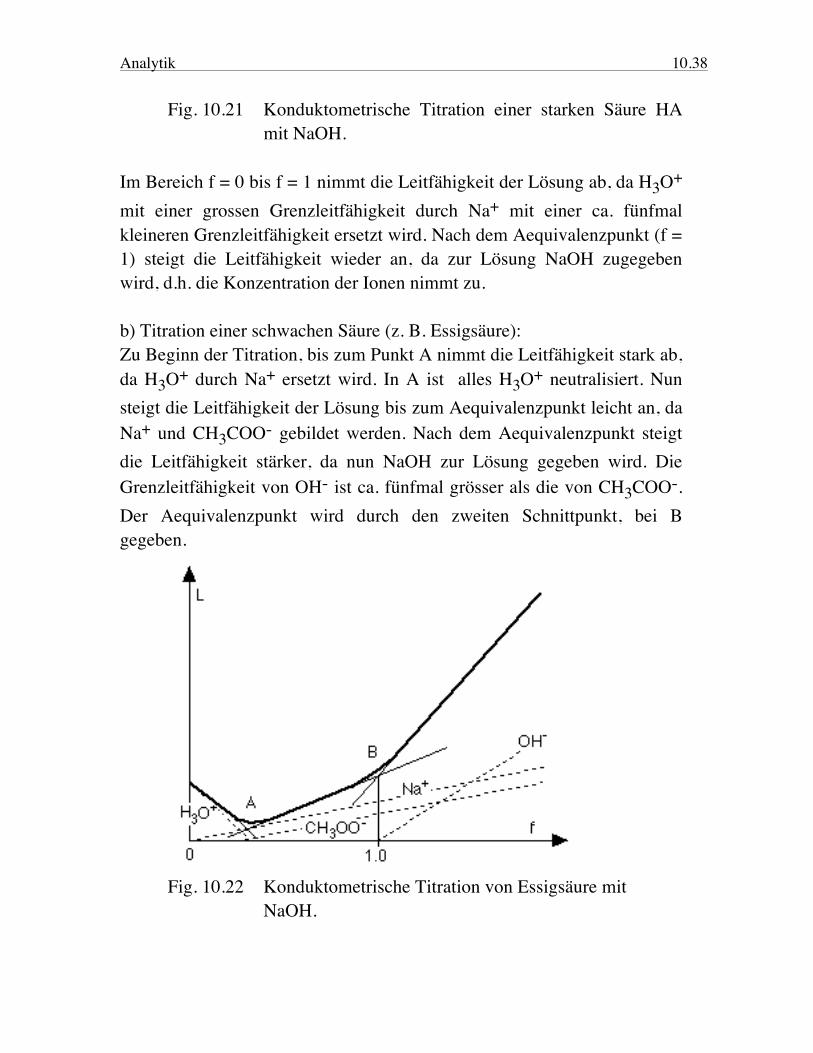
Analytik 10.38 Fig. 10.21 Konduktometrische Titration einer starken Säure HA mit NaOH. Im Bereich f = 0 bis f = 1 nimmt die Leitfähigkeit der Lösung ab, da H3O+ mit einer grossen Grenzleitfähigkeit durch Na+ mit einer ca. fünfmal kleineren Grenzleitfähigkeit ersetzt wird. Nach dem Aequivalenzpunkt (f = 1) steigt die Leitfähigkeit wieder an, da zur Lösung NaOH zugegeben wird, d.h. die Konzentration der Ionen nimmt zu. b) Titration einer schwachen Säure (z. B. Essigsäure): Zu Beginn der Titration, bis zum Punkt A nimmt die Leitfähigkeit stark ab, da H3O+ durch Na+ ersetzt wird. In A ist alles H3O+ neutralisiert. Nun steigt die Leitfähigkeit der Lösung bis zum Aequivalenzpunkt leicht an, da Na+ und CH3COO- gebildet werden. Nach dem Aequivalenzpunkt steigt die Leitfähigkeit stärker, da nun NaOH zur Lösung gegeben wird. Die Grenzleitfähigkeit von OH- ist ca. fünfmal grösser als die von CH3COO-. Der Aequivalenzpunkt wird durch den zweiten Schnittpunkt, bei B gegeben.
Fig. 10.22 Konduktometrische Titration von Essigsäure mit NaOH.
Analytik 10.39
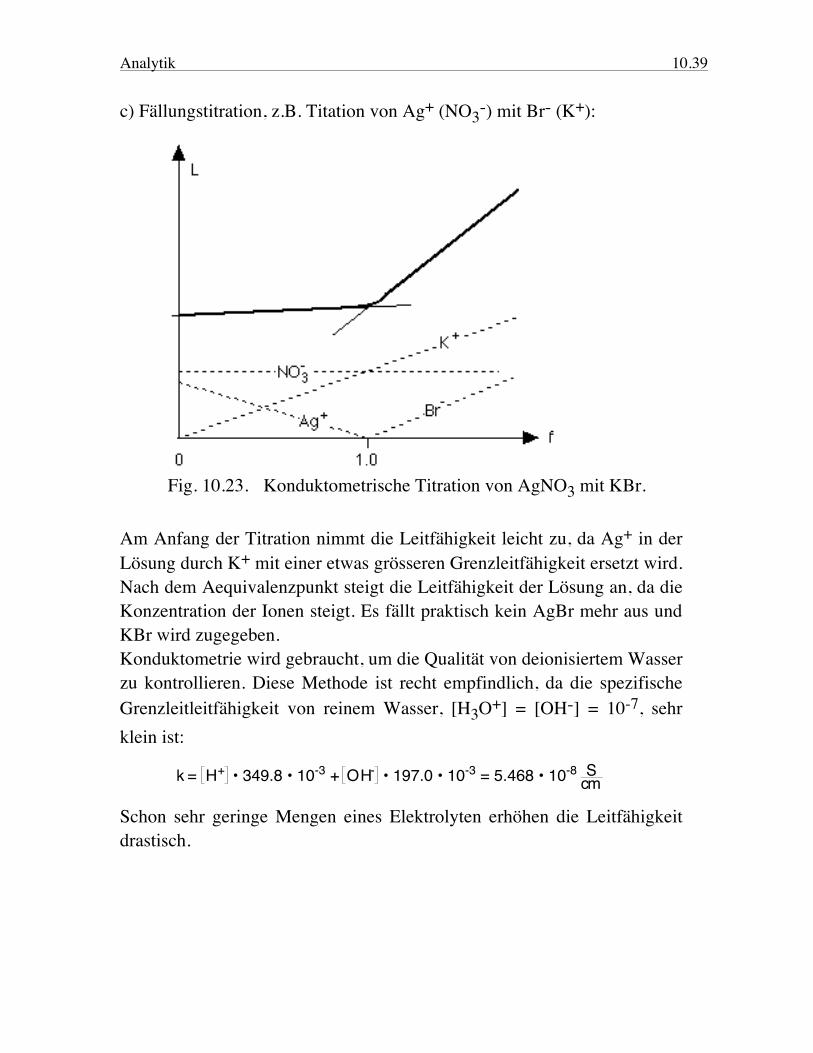
c) Fällungstitration, z.B. Titation von Ag+ (NO3-) mit Br- (K+):
Fig. 10.23. Konduktometrische Titration von AgNO3 mit KBr. Am Anfang der Titration nimmt die Leitfähigkeit leicht zu, da Ag+ in der Lösung durch K+ mit einer etwas grösseren Grenzleitfähigkeit ersetzt wird. Nach dem Aequivalenzpunkt steigt die Leitfähigkeit der Lösung an, da die Konzentration der Ionen steigt. Es fällt praktisch kein AgBr mehr aus und KBr wird zugegeben. Konduktometrie wird gebraucht, um die Qualität von deionisiertem Wasser zu kontrollieren. Diese Methode ist recht empfindlich, da die spezifische Grenzleitleitfähigkeit von reinem Wasser, [H3O+] = [OH-] = 10-7, sehr klein ist:
k = H+ • 349.8 • 10-3 + OH- • 197.0 • 10-3 = 5.468 • 10-8 S
cm Schon sehr geringe Mengen eines Elektrolyten erhöhen die Leitfähigkeit drastisch.