viral vector analytical paradigms, platforms and …€¢ infectivity = report results (iu/ml) •...
TRANSCRIPT

Eric Pastor
Principal Scientist, Biopharmaceutics Development, Sanofi US
CASSS CMC Strategy Forum 2017
17 JUL 2017
Viral Vector Analytical Paradigms, Platforms and
Proposed Specifications

Agenda
1. How do we shape and manage a phase appropriate Gene Therapy
viral vector analytical paradigm to support Drug Substance/Drug
Product release and characterization?
• How do we select what tests are on release?
• What attributes do we escalate from characterization to release?
• How do evolve our paradigms throughout drug development?
• Are there platform gaps and how do we manage this?
2. How do we select phase appropriate specifications?
• How many lots do we need?
• What are the appropriate units and ranges for key quality attributes?
Not intending to promote what we do as “right” or “wrong” or
to champion any particular platform.
Objective is to share some strategies and start discussions….

Gene Therapy VV Analytical Paradigm
● What defines a viral vector analytical paradigm?● Critical Quality Attributes
• Most safety, efficacy and immunogenicity scores may be carried over from one AAV product to another if the manufacturing process is platformed
• Host-cell DNA
• PK/PD scores are likely to be product-specific based on change in therapeutic gene
• CQA’s may be introduced/removed based on changes to the purification process
● Regulatory Guidance*• Expectations are continuously increasing and evolving as more companies reach
later stage clinical trials
• Specifications are becoming more stringent, new attributes are emerging
• Specific gene therapy guidance documents are branching off from general biologics (i.e. Potency Assay recommendations)
● Overall goal is to release and characterize a safe and high quality product
Guidance for FDA Reviewers and Sponsors: Content and Review of Chemistry, Manufacturing, and Controls (CMC Information
for Human Gene Therapy Investigational New Drug Applications (INDs), USDHHS/FDA/CBER, 4/2008
Note for Guidance on the Quality, Non-Clinical and Clinical Aspects of Gene Transfer Medicinal Products,
CHMP/GTWP/671639/2008

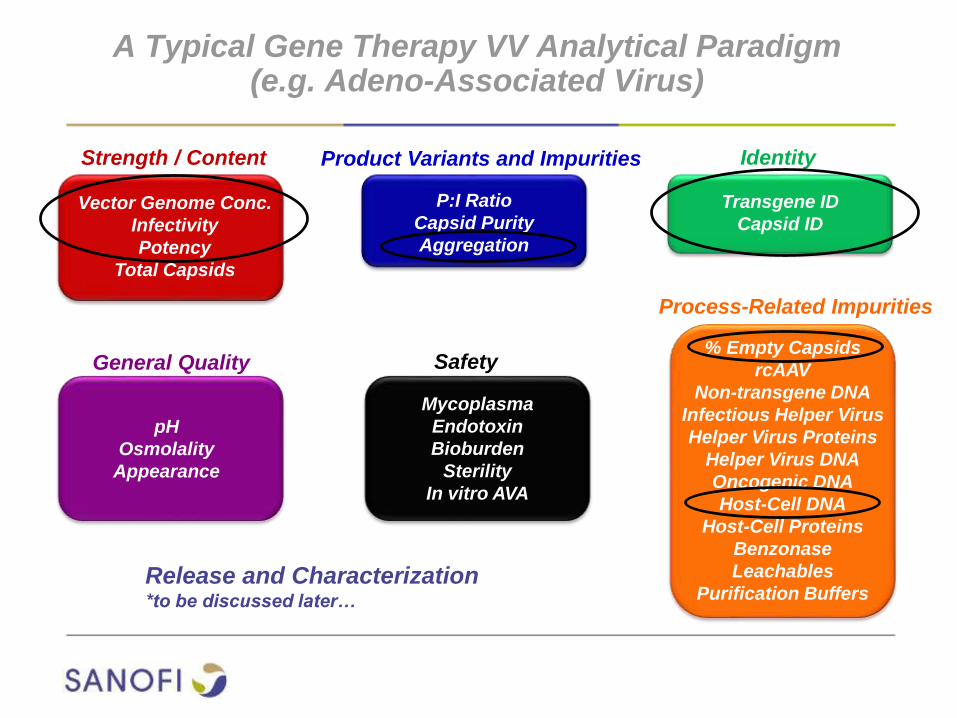
Example Gene Therapy VV Analytical Paradigm(e.g. Adeno-Associated Virus)
Vector Genome Conc.
Infectivity
Potency
Total Capsids
P:I Ratio
Capsid Purity
Aggregation
% Empty Capsids
rcAAV
Non-transgene DNA
Infectious Helper Virus
Helper Virus Proteins
Helper Virus DNA
Oncogenic DNA
Host-Cell DNA
Host-Cell Proteins
Benzonase
Leachables
Purification Buffers
Transgene ID
Capsid ID
Mycoplasma
Endotoxin
Bioburden
Sterility
In vitro AVA
pH
Osmolality
Appearance
Strength / Content Product Variants and Impurities Identity
General Quality Safety
Process-Related Impurities
Release and Characterization*to be discussed later…
A Typical Gene Therapy VV Analytical Paradigm(e.g. Adeno-Associated Virus)
Vector Genome Conc.
Infectivity
Potency
Total Capsids
P:I Ratio
Capsid Purity
Aggregation
% Empty Capsids
rcAAV
Non-transgene DNA
Infectious Helper Virus
Helper Virus Proteins
Helper Virus DNA
Oncogenic DNA
Host-Cell DNA
Host-Cell Proteins
Benzonase
Leachables
Purification Buffers
Transgene ID
Capsid ID
Mycoplasma
Endotoxin
Bioburden
Sterility
In vitro AVA
pH
Osmolality
Appearance
Strength / Content Product Variants and Impurities Identity
General Quality Safety
Process-Related Impurities
Release and Characterization*to be discussed later…

Evolving Assays and ParadigmsEarly Phase to Late Phase
Current Release Methods* Current Characterization Methods
Vector Genome Concentration
Infectivity
Relative Potency
Capsid Purity
Transgene Identity
% Empty Capsids
rcAAV
Infectious Helper Virus
Host-Cell DNA
Host-Cell Protein
Aggregation
Capsid Identity
Non-transgene DNA
Helper Virus Proteins
Helper Virus DNA
Oncogenic DNA
Residual Benzonase
Residual Column Leachables
Residual Purification Buffers
NGS Sequencing
VP Modifications
Infection/Transduction Kinetics
HCP Profiles
*minus safety and compendials
Characterization methods focus on investigative attributes and/or CQA’s where
there are plans to put process controls around. May also include method
platforms that need additional development or are not yet “QC-friendly”
Release methods focus on high CQA’s and required
tests using validate-able method platforms

Current Release Methods* Current Characterization Methods
Vector Genome Concentration
Infectivity
Relative Potency
Capsid Purity
Transgene Identity
% Empty Capsids
rcAAV
Infectious Helper Virus
Host-Cell DNA
Host-Cell Protein
Aggregation
Capsid Identity
Non-transgene DNA
Helper Virus Proteins
Helper Virus DNA
Oncogenic DNA
Residual Benzonase
Residual Column Leachables
Residual Purification Buffers
NGS Sequencing
VP Modifications
Infection/Transduction Kinetics
HCP Profiles
*minus safety and compendials
Evolving Assays and ParadigmsEarly Phase to Late Phase

Current Release Methods* Current Characterization Methods
Vector Genome Concentration
Infectivity
Relative Potency
Capsid Purity
Transgene Identity
% Empty Capsids
rcAAV
Infectious Helper Virus
Host-Cell DNA
Host-Cell Protein
Aggregation
Capsid Identity
Non-transgene DNA
Helper Virus Proteins
Helper Virus DNA
Oncogenic DNA
Residual Benzonase
Residual Column Leachables
Residual Purification Buffers
NGS Sequencing
VP Modifications
Infection/Transduction Kinetics
HCP Profiles
*minus safety and compendials
Evolving Assays and ParadigmsEarly Phase to Late Phase

Current Release Methods* Current Characterization Methods
Vector Genome Concentration
Infectivity
Relative Potency
Capsid Purity
Transgene Identity
% Empty Capsids
rcAAV
Infectious Helper Virus
Host-Cell DNA
Host-Cell Protein
Aggregation
Capsid Identity
Non-transgene DNA
Helper Virus Proteins
Helper Virus DNA
Oncogenic DNA
Residual Benzonase
Residual Column Leachables
Residual Purification Buffers
NGS Sequencing
VP Modifications
Infection/Transduction Kinetics
HCP Profiles
*minus safety and compendials
Evolving Assays and ParadigmsEarly Phase to Late Phase

Strength
Vector Genome Concentration
(Dose-Defining Assay)
Prelim. Spec. = Dose ± 0.5 log
*qPCR on release, ddPCR on
charact. (not yet ready, need
suitable cross-over data and
system confidence, validation)
Infectivity (TCID50)
Plate A 1 2 3 4 5 6 7 8 9 10 11 Ctls.
Plate A A + + + + + + + +
B + + + + + + + +
C + + + + + + + +
D + + + + + + + + +
E + + + + + + + +
F + + + + + +
G + + + + + + +
H + + + + + +
Prelim. Spec.
• Infectivity = Report Results (IU/mL)
• Particle:Infectivity ratio = < X vg/IU*
*P:I will vary depending on serotype.
Arbitrary number, dependent on tropism
to assay cell line. Use for lot-to-lot
consistency and stability
Relative Potency
Prelim. Spec. = 50 – 200 % of Ref.
Measures both gene
expression and biological
activity of expressed gene (e.g.
enzymatic activity)
Reference Standard is from
Engineering Run, prior to GMP
CTM – full characterization of
RS including NGS!
Data courtesy of JD

Strength
Vector Genome Concentration
(Dose-Defining Assay)
Prelim. Spec. = Dose ± 0.5 log
*qPCR on release, ddPCR on
charact. (not yet ready, need
suitable cross-over data and
system confidence, validation)
Infectivity (TCID50)
Plate A 1 2 3 4 5 6 7 8 9 10 11 Ctls.
Plate A A + + + + + + + +
B + + + + + + + +
C + + + + + + + +
D + + + + + + + + +
E + + + + + + + +
F + + + + + +
G + + + + + + +
H + + + + + +
Prelim. Spec.
• Infectivity = Report Results (IU/mL)
• Particle:Infectivity ratio = < X vg/IU*
*P:I will vary depending on serotype.
Arbitrary number, dependent on tropism
to assay cell line. Use for lot-to-lot
consistency and stability
Relative Potency
Prelim. Spec. = 50 – 200 % of Ref.
Measures both gene
expression and biological
activity of expressed gene (e.g.
enzymatic activity)
Reference Standard is from
Engineering Run, prior to GMP
CTM – full characterization of
RS including NGS!
Data courtesy of JD

Strength
Vector Genome Concentration
(Dose-Defining Assay)
Prelim. Spec. = Dose ± 0.5 log
*qPCR on release, ddPCR on
charact. (not yet ready, need
suitable cross-over data and
system confidence, validation)
Infectivity (TCID50)
Plate A 1 2 3 4 5 6 7 8 9 10 11 Ctls.
Plate A A + + + + + + + +
B + + + + + + + +
C + + + + + + + +
D + + + + + + + + +
E + + + + + + + +
F + + + + + +
G + + + + + + +
H + + + + + +
Prelim. Spec.
• Infectivity = Report Results (IU/mL)
• Particle:Infectivity ratio = < X vg/IU*
*P:I will vary depending on serotype.
Arbitrary number, dependent on tropism
to assay cell line. Use for lot-to-lot
consistency and stability
Relative Potency
Prelim. Spec. = 50 – 200 % of Ref.
Measures both gene
expression and biological
activity of expressed gene (e.g.
enzymatic activity)
Reference Standard is from
Engineering Run, prior to GMP
CTM – full characterization of
RS including NGS!
Data courtesy of JD

Identity
Transgene
Prelim. Spec. = Conforms to Ref.
Capsid
Prelim. Spec. = AAVx Confirmed
Do we need both assays on Release?
What if one of the assay platforms does not lend itself well to a QC environment?
Viral vectors have both a nucleic acid and viral shell component• A multi-product facility could have the same capsid serotype, different transgene
AND/OR vice versa
Data courtesy of JD, LL and XJ

Product Variants and ImpuritiesAggregation / Particle Distribution
What is an ideal platform?• Quantifiable
• Monomeric AAV plus other (dimer,
trimer, higher order?)
• Validate-able (ICH Q2(R1))
• QC-friendly, low IVR
What other platforms can be used for
improved detection/quantitation?• Nanosight
• SEC-HPLC
• AUC
How do we report and what are the units for
specifications?• > X % monomer?
• < X % other?
• X – X nm?
• Comparable to Ref?
• No sig. HMWS?
Viral vectors (and their potential aggregates) exist in a nanoparticle to sub-visible particle range
(10 nm – 1 µm)• Faces similar challenges of traditional biologics
Data courtesy of BD

Process-Related Impurities
Is there a regulatory concern with hcDNA levels in rAAV products?
1 cellular genome = 15 pg DNA
~75 pg genomic DNA per 1 copy 18S
1 AAV genome = 4800 bp DNA
~0.0026 fg viral DNA per 1 copy 18S
Fold-difference = ~28,000
Genomic DNA STDs rAAV Test Article (DS)
If all we detect is encapsidated 18S (+/- DNase), can we convert genomic hcDNA to viral DNA?
5.6e5 pg per mL / 0.07519 pg genomic DNA per 18S copy = 7447799 copy per mL
7447799 copy per mL x 0.00000263 pg viral DNA per 18S copy = 19.6 pg/mL
560 ng/dose
(1 mL)
0.02 ng/dose
(1 mL)
WHO Guidance
= < 10 ng/dose

Process-Related Impurities
Empty Capsids / Full Capsids
Multiple platforms available to monitor % full and % empty particles (and % fragmented
genomes?)• Optical Density – Small range, need extinction coefficients, assumes all “full” particles are full length
• Capsid ELISA / qPCR - Combined variability of 2 platforms, “full” particles are those that you detect with
your primers/probes
• AUC – Difficult to validate, but discriminates between all species
• TEM - Difficult to quantify, but discriminates between empty and “other”
What will be the preferred platform?• Any of the ones listed above?
• Another platform? – e.g. HPLC? Cryo TEM?
What will be specifications in early development?• > 50 % full OR < 50 % empty
• Do we need to do in vivo experiments prior to IND-enabling studies?
• % full = 0%, 25%, 50%, 75%, 100%
• Use GLP Tox output as a benchmark? – or can we leverage other products?
• If GLP tox vector is 70% full, is the Phase I release spec = ≥ 70% full (< 30% empty?)
• But what about assay and process variability?
Data courtesy of BB

Questions and Discussion Points
1. What do phase-appropriate analytical paradigms look like for viral vector
gene therapy drug substance/drug product release and characterization?
2. How do we set phase appropriate specifications?

Thank you for your time!

Acknowledgments
Sanofi Biopharmaceutics Development
• Bioanalytics - Analytical Development
• Bioanalytics - Characterization
• Gene Therapy Skill Center
CASSS Organizers