vascular endothelial growth factor in renal cell …144292/...v jacobsen j, grankvist k, rasmuson t,...
TRANSCRIPT

UMEÅ UNIVERSITY MEDICAL DISSERTATIONS
New Series No. 1006 ISSN 0346-6612 ISBN 91-7264-029-4
Departments of Surgical and Perioperative Sciences, Urology and Andrology;
Medical Biosciences, Clinical Chemistry; Radiation Sciences, Oncology, and Medical
Biosciences, Pathology, Umeå University, Umeå, Sweden
Vascular endothelial growth factor
in renal cell carcinoma
Jan Jacobsen
Umeå 2006

Copyright © by Jan Jacobsen New Series No. 1006 ISSN 0346-6612 ISBN 91-7264-029-4-
Printed by Hemströms Offset-Boktryck
Härnösand, Sweden, 2006

3
This thesis is dedicated to Ida and Heini
– Den lige vej er den närmeste – men oftest når man den kun ad lange omveje. Robert Storm Petersen
Danish artist (1882 - 1949)

4
ABSTRACT
VASCULAR ENDOTHELIAL GROWTH FACTOR IN RENAL CELL CARCINOMA
Jan Jacobsen, Departments of Surgical and Perioperative Sciences, Urology and Andrology, Medical Biosciences, Clinical Chemistry; Radiation Sciences, Oncology, and Medical Biosciences, Pathology, Umeå University Medical Dissertations. New Series No. 1006 ISSN 0346-6612 ISBN 91-7264-029-4. Background. Angiogenesis is essential for tumour growth. Vascular endothelial growth factor (VEGF) and its isoforms were investigated in relation to the clinical course in a large number of patients with renal cell carcinoma (RCC). Methods. RCC subtypes and behaviour were established by clinicopathological criteria and surveillance. VEGF expression was analysed in serum by enzyme-linked immuno-sorbent assay (ELISA) and in tumour tissue by reverse transcription polymerase chain reaction (RT-PCR), immunohistochemistry (IHC), and Western blot (WB). Results. Serum VEGF (S-VEGF) was increased in RCC compared to control group. S-VEGF correlated with tumour stage and grade and was associated with survival in men but not in women. S-VEGF correlated with blood platelet counts, which were inversely correlated to increasing age in women, and they were decreased in chronically medicated patients, particularly in men. In contrast to S-VEGF, platelet counts associated with survival only in patients free of medication and chronic diseases. RT-PCR showed a correlation between VEGF121/VEGF165 mRNA and between VEGF165/VEGF-R1 mRNA. There was no association between different VEGF mRNA isoforms and S-VEGF. Conventional renal cell carcinoma (CRCC) had higher VEGF165, VEGF121, and VEGF-R1 mRNA levels compared with papillary renal cell carcinoma (PRCC). IHC VEGF staining was strong in kidney cortex. Kidney tumour showed a considerable variation in cytoplasmatic VEGF expression, which correlated with tumour size. Although, there was no difference in VEGF expression between the RCC types, VEGF expression was associated with survival only in CRCC. WB showed a strong protein expression of both VEGF189 and VEGF165 in kidney cortex. In kidney tumour, expression of VEGF189 varied the most, VEGF165 less so, and VEGF121 was rarely detected. Both CRCC and PRCC expressed low levels of VEGF189 and VEGF165 compared with kidney cortex. Chromophobe renal cell carcinoma (ChRCC) expressed VEGF189 levels comparable to those from kidney cortex, while VEGF165 was lower. In PRCC and ChRCC, VEGF189 levels correlated inversely with advancing tumour stage, and in PRCC, VEGF165 levels correlated inversely with increasing tumour size. VEGF189 was an independent prognostic factor for survival in patients with PRCC. Conclusions. S-VEGF has a stronger association to progression in RCC than platelet count. CRCC showed high levels of VEGF mRNA isoforms and VEGF-R1 mRNA compared to PRCC. VEGF mRNA isoforms expression decreased with advancing stage. IHC demonstrated VEGF expression in cell cytoplasm related to tumour growth, particular in CRCC. Different VEGF isoform patterns were found in different RCC types. Protein VEGF189 expression was associated with tumour stage and was an independent prognostic factor in PRCC. Protein VEGF165 expression was generally low and had no prognostic value. The trend for decreasing levels of VEGF isoforms in advanced tumour stages may indicate that angiogenic activity is an early event in tumour growth induced by VEGF, but that during later tumour progression the role of VEGF is less clear. Key words: VEGF, isoforms, quantitative RT-PCR, immunohistochemistry, tissue microarray, Western blot, stage, survival, renal cell carcinoma.

5
ORIGINAL PAPERS
This thesis is based on the following papers, which are referred to in the text by their roman numerals: I Jacobsen J, Rasmuson T, Grankvist K and Ljungberg B.
Vascular endothelial growth factor as prognostic factor in renal cell carcinoma. Journal of Urology 2000; 163(1): 343-7. Reprinted with permission from American Urological Association.
II Jacobsen J, Grankvist K, Rasmuson T, Ljungberg B. Prognostic importance of serum vascular endothelial growth factor in relation to
platelet and leukocyte counts in human renal cell carcinoma. European Journal of Cancer Prevention 2002; 11(3): 245-52. Reprited with
permission from Lippincott Williams & Wilkins.
III Ljungberg B, Jacobsen J, Häggström-Rudolfssson S, Rasmuson T, Lindh G, Grankvist K. Tumour vascular endothelial growth factor (VEGF) mRNA in relation to serum VEGF protein levels and tumour progression in human renal cell carcinoma. Urological Research 2003; 31(5): 335-40. Reprinted with kind permission of Springer Science and Business Media.
IV Jacobsen J, Grankvist K, Rasmuson T, Bergh A, Landberg G, Ljungberg B. Expression of vascular endothelial growth factor protein in human renal cell carcinoma. British Journal of Urology International 2004; 93(3): 297-302. Reprinted with kind permission of Blackwell Publishing.
V Jacobsen J, Grankvist K, Rasmuson T, Ljungberg B. Different isoform patterns for vascular endothelial growth factor between clear cell and papillary renal cell carcinoma. British Journal of Urology International, accepted for publication 2005. Printed with kind permission of Blackwell Publishing.

6
CONTENTS
ABSTRACT .......................................................................................................................4
ORIGINAL PAPERS ........................................................................................................5
CONTENTS .......................................................................................................................6
ABBREVIATIONS............................................................................................................8
INTRODUCTION ...........................................................................................................10 Background ...................................................................................................... 10 Incidence .......................................................................................................... 10 Environmental risk factors ................................................................................ 10 Inherited genetic vulnerability........................................................................... 11 Clinical and pathological prognostic factors ..................................................... 13 Histopathological grading and classification .................................................... 13 Angiogenesis in cancer .................................................................................... 16 Tumour secreted VEGF-A................................................................................ 17 Synergistic role of bFGF................................................................................... 18 Miscellaneous role of PDGF............................................................................. 18
AIMS OF THE INVESTIGATION ...............................................................................20
MATERIAL AND METHODS ......................................................................................21 Patients............................................................................................................. 21 Tumour staging ................................................................................................ 21 Morphologic classification ................................................................................ 21 Methods............................................................................................................ 22 Blood analysis .................................................................................................. 22 Serum analysis................................................................................................. 23 Tissue collection and preparation .................................................................... 24 Competitive quantitative RT-PCR .................................................................... 24 Immunohistochemestry .................................................................................... 26 Protein extraction ............................................................................................. 28 Western blotting ............................................................................................... 28 Statistical methods ........................................................................................... 29
RESULTS AND COMMENTS.......................................................................................31 Paper I .............................................................................................................. 31 Comments on Paper I ................................................................................... 31 Paper II ............................................................................................................. 31 Comments on Paper II .................................................................................. 32 Paper III ............................................................................................................ 33 Comments on Paper III ................................................................................. 33 Paper IV............................................................................................................ 34 Comments on paper IV ................................................................................. 34

7
Study V............................................................................................................. 35 Comments on paper V .................................................................................. 35
FUTURE DIRECTIONS.................................................................................................37
CONCLUSIONS..............................................................................................................39 Paper I .............................................................................................................. 39 Paper II ............................................................................................................. 39 Paper III ............................................................................................................ 39 Paper IV............................................................................................................ 39 Paper V............................................................................................................. 39
ACKNOWLEDGEMENTS ............................................................................................40
REFERENCES ................................................................................................................42

8
ABBREVIATIONS
AJCC American Joint Committee on Cancer Bcl-2 Gene encodes inhibitors of apoptosis Bcl-2 Protein encoded of Bcl-2 gene bFGF (basic) Fibroblast Growth Factor BHD Birt Hogg Dub’e syndrome cDNA (complementary) Deoxyribonucleic Acid ChRCC Chromophobe Renal Cell Carcinoma CRCC Conventional Renal Cell Carcinoma CT Computerised Tomography Cyclins D and E Promote cyclin-dependent kinases to activate cell cycle DNA Deoxyribonucleic Acid ECM Extra Cellular Matrix EGF Epidermal Growth Factor EGF-R Epidermal Growth Factor Receptor ELISA Enzyme Linked Immuno Sorbent Assay erb-B Gene encodes EGF-R erb-B Protein encoded by erb-B gene ESR Erythrocyte sedimentation rate FGF Fibroblast Growth Factor FH Gene encodes Fumarate Hydratase involved in citric acid cycle FH Protein encoded by FH gene HER-2 Synonymous to erb-B gene encodes EGF-R HER-2 Synonymous to erb-B HIF Hypoxia Inducible Factor HLRCC Hereditary Leiomyoma Renal Cell Carcinoma HPRC Hereditary Papillary Renal Cell Carcinoma IGF Insulin like Growth Factor IHC Immunohistochemistry IS Internal Standard bp Base Pairs kDA (kilo) Dalton met Gene encodes receptor with tyrosine kinase activity met Protein encoded by met gene MMP Matrix Metallo Proteinase MRI Magnetic Resonance Imaging mRNA (messenger) Ribonucleic Acid MVD Micro Vessel Density myc Gene encodes transcription activators and stimulating growth
signals

9
myc Protein encoded by myc gene p27 Gene encodes transcription inhibitors of cell cycle p53 Gene encodes transcription inhibitors of cell cycle and activates
apoptosis p53 Protein encoded by p53 gene PAD Pathological Anatomical Diagnosis PBS Phosphate Buffered Saline PDGF Platelet Derived Growth Factor pRb Gene encodes nuclear transcription factor once activated
stimulates cell cycle PRCC Papillary Renal Cell Carcinoma pVHL (protein) VHL RCC Renal Cell Carcinoma RNA Ribonucleic Acid RT-PCR Reverse Transcription Polymerase Chain Reaction SDS Sodium Dodectyl Sulfate TGF Transforming Growth Factor TMA Tissue Micro Array TNF Tumour Necrosis Factor TNM Tumour Node Metastasis TS Tissue Section or Slide UICC Union Internationale Contre le Cancer VEGF Vascular Endothelial Growth Factor VEGF-R1 VEGF receptor 1 (flt-1) VEGF-R2 VEGF receptor 2 (KDR/flk-1) VHL von Hippel-Lindau VHL Gene encodes VHL promoting transcription of VEGF VHL Protein encoded by VHL gene WB Western Blot

10
INTRODUCTION
Background Renal cell carcinoma (RCC) is a
heterogeneous group of tumours, which demonstrates wide variation in histo-pathological features, as well as clinical diversity, with unpredictable tumour behaviour. The mortality-incidence ratio is higher in RCC than in other urological malignancies [1]. In only 50-60% of RCC tumours which are localized to kidney is surgical treatment curative [2, 3]. New approaches in cancer treatment are needed, and through genetic and molecular biolo-gical research novel molecular therapeutic targets have been identified [4, 5]. Tumours seem to be influenced by serial gene mutations and methylation with phenoltypical expression as dysfunctional cell signalling, cell proliferation, inhi-bition of apoptosis, and promotion of angiogenesis during the growth [6]. Molecular targeting of dysfunction in cell signalling or angiogenesis is a potential novel therapy, and the knowledge is essential in order to design new anti-angiogenic therapies for patients with RCC.
Incidence RCC constitutes 2-3% of adult
malignancies [7]. Almost 900 new cases of RCC are observed annually in Sweden, with 600 deaths each year attributed to RCC. Men are affected more commonly than women, with a ratio of 3:2. RCC is primarily a disease of the elderly, and is usually detected in the sixth or seventh decade of life [1]. Worldwide, there is im-proved and earlier detection of localised tumours, largely due to expanded access to better quality in radiological imaging
techniques such as ultrasonography, com-puterised tomography (CT), and magnetic resonance imaging (MRI) [8-10].
Environmental risk factors
Several epidemiologic studies have demonstrated an association between RCC and tobacco smoking and obesity [11, 12], factors which have been asso-ciated in 40% of RCC cases in our high-risk society [13]. In rat experiments, the constituents from tobacco smoke, which are excreted to the urine are thought to cause RCC by VHL-mutations [14, 15]. Moreover, gene-environment interactions have been recognized among smokers who have slow acetylator genotype for N-acetyl-transferase, where there is an increased risk for development of RCC [16]. A high body mass index or high body weight has been consistently linked to RCC [17, 18], predominantly in women [19]. Obesity itself or factors associated with obesity may have a stressful effect on renal cell metabolism including through insulin resistance, elevated growth factors (EGF), free circulating estrogens, and insulin-like growth factor (IGF-I), all of which may be involved in development of RCC [20, 21]. Hyper-tension seems to be associated with RCC, but difficulties in separate possible side effects from medication prevents classi-fying hypertension as an independent risk factor [22, 23]. Use of diuretics as well as beta-blockers have been implicated as promoting the development of RCC, though it remains unclear whether the medications or hypertension itself is the active factor [22, 23]. The combination of

11
obesity and hypertension [24] might stress the kidney and lead to renal damage with proliferation. Metabolic or functional changes in renal tubules may result in genetic alteration caused by exposure and increasing susceptibility to carcinogens. Other environmental risk factors are less well defined, including occupational exposure to trichloroethelene, nutritional factors, chronic renal failure, urinary tract infection, use of analgesics or reproduce-tive hormones, and alcohol [13, 20, 22, 25-29].
Inherited genetic vulnerability The genetic alterations in RCC may be
a consequence of activation of oncogenes or inactivation of tumour suppressor gene. Since both copies of genes must be dis-torted in order for a tumour to develop, a series of events are necessary [6].
Oncogenes are mutated proto-oncogenes that normally code for proteins involved in control of cell division and differ-rentiation. Intensified signalling in these pathways can lead to uncontrolled cell division and make the cell become cancerous. An activated oncogene that codes for erbB receptors, for example, can cause an over-receptive condition for growth factors as an early stage event in RCC [30]. The myc oncogene facilitates signal transcription in the nucleus and have been found to be elevated as a late stage event in RCC [6]. On the other hand, lack of growth factors lead to apoptosis [31]. The oncogene that code for met tyrosine kinase receptors generate anti-apoptotic signalling by mitogen-activated protein kinase pathways [32-36]. Several oncogenes involved sig-nalling pathways for cell growth control have been reported in RCC [37-40].
Tumour suppressor genes are genes
that code for proteins involved in re-cognition and restitution of the damaged DNA. They also initiate cell apoptosis. Otherwise, continuous cell division would transmit genetic defects, which could be intensified by increasing susceptibility due to carcinogenic exposure. The p53 gene codes for the protein p53, which when inactivated or impaired makes cells less able to restore damaged DNA [41, 42]. Inactivation of the VHL gene induces mRNA transcription of growth factor, which generates anti-apoptotic signalling and promotes angiogenesis [43, 44]
New cytogenetic and molecular gene-tic data have established the association between genetic characterisation and histopathological classification of RCC [45]. Four histopathological types of RCC have been described and linked to genetic diseases such as von Hippel-Lindau (VHL) disease, hereditary papil-lary renal carci-noma (HPRC), Birt-Hogg-Dubé (BHD) syndrome, and hereditary leiomyoma RCC (HLRCC) [46]. Hence, RCC can be divi-ded into predominantly sporadic and non-inherited form or an hereditary form, which accounts only a fraction 2-5% of diagnosed RCC [47-50].
Clear cell or conventional renal cell carcinoma (CRCC) is the most pre-dominate renal tumour and accounts for about 70% of diagnosed RCC tumours [45, 51]. The sporadic type of CRCC is genetically characterised by deletion of chromosome 3p, which is believed to be the early-onset event in renal carcino-genesis [45]. Inactivation of both VHL alleles is mandatory for development of CRCC [50]. Studies have shown that this mutation occurs in about 50% followed soon after by loss of heterozygoty 10-20% and then, as a late event, silencing of the second VHL allele by hypermethylation

12
20% [51-53]. Other genetic alterations, 5q duplication as well as 6q, 8p, 9p, and 14q deletion, have been implicated in carcino-genesis of sporadic CRCC [53-55]. Loss of the tumour suppressor gene (VHL) increases expression of transforming growth factor (TGF-α and β) and vascular endo-thelial growth factor (VEGF). Increased expression of onco-gene myc with ab-normal receptor signalling, despite the lack of growth factors, has also been rela-ted to CRCC [56]. The age-related inci-dence of CRCC increases after 35 years of age and peaks in the sixth decade. Early-onset of CRCC indicates an here-ditary cause [57]. A familiar form of CRCC has been described where there is early-onset with balanced 3p translocation but no traceable VHL changes, which ex-cludes mutations in the VHL gene as the mechanism for cancer development [56, 58-60]. However, a majority of familiar CRCC is associated with von Hippel-Lindau syndrome. Inheritance is autosomal-dominant with high penetrance, with an incidence of 1 in 36,000 live births [61, 62]. The mutation affects the tumour suppressor gene (VHL) located on the short arm of chromosome 3. Up to 45% of these patients develop CRCC, and often the early-onset form [52].
Papillary renal cell carcinoma (PRCC) is the second most frequent type of carcinoma, and accounts for about 10-15% of diagnosed RCC [45, 51]. Papillary tumours are characterised by chromo-somal trisomies, most often 3q, 7, 8, 12, 16,17, and 20, with lost of the Y chromo-some [45, 51, 53, 56, 63]. Hereditary PRCC is a very rare, and is characterized as an autosomal dominate inheritance with high penetrance [64, 65]. The muta-tion affects proto-oncogene (met) loca-ted on chromosome 7q31. About 80% of
families have mutations in this proto-oncogene with late-onset. Histopatho-logically, the familiar and commonly sporadic form of PRCC are both referred to as type I PRCC [53]. The hereditary leiomyomatosis syndrome (HLRCC) was recently characterised as an autosomal dominant tumour syndrome caused by germline mutations in the fumarate hydra-tase (FH) gene on 1q42 [66]. Benign leiomyomas of the skin and uterus are often seen with predisposition to early-onset type 2 PRCC in addition to uterine leiomyosarcoma [67]. In contrast to other PRCCs, the HLRCC tumours are usually solitary and unilateral. The morphological characteristics of type 2 PRCC are inde-pendently associated with poorer survival [68, 69].
Chromophobe renal cell carcinoma (ChRCC) is a third carcinoma originated from tubular epithelium, and accounts for 5% of diagnosed RCC [45, 51]. It has a better prognosis than other variants of RCC [70]. A low chromosome number is characteristics of ChRCC, due to frequent occurrence of loss of chromosomes 1, 2, 6, 10, 13, 17, and 21 [45, 55, 71]. The Birt-Hogg-Dubé (BHD) syndrome shows autosomal dominant inheritance for benign cutaneous tumours with predisposition for ChRCC and oncocytomas [72]. Pulmo-nary cysts are seen causing spontaneous pneumothorax in 25% of patients [73]. The germline mutation gene is located at chromosome 17p11 [74].
Renal cell carcinoma, unclassified, is a diagnostic category used when a RCC does not fit into one of the others cate-gories. This group represent about 4-5% of diagnosed RCC [45, 51]. No consistent pattern of genetic abnormalities has been established [53]. Due to variability in morphological patterns this might also

13
include the collecting duct carcinoma, which accounts for less than 1% of renal neoplasms [45, 51, 53].
Clinical and pathological
prognostic factors Patient-related factors such as gender
and age have no predictive importance [6]. Inherited genetic diseases that pre-dispose for RCC are rare [49]. Biological parameters and serum factors such as ESR, C-reactive protein, haptoglobin, and ferritin have been correlated to survival, but only ESR has been shown to have independent prognostic value [75, 76]. In some studies abnormal hemoglobin va-lues, thrombocytosis, and elevated liver function tests have been correlated to a worse prognosis [77]. Patient performance status together with tumour presentation at the time of diagnosis is more relevant, however, for prediction of prognosis [76, 78].
Tumour related prognostic factors include macroscopic and histologic fea-tures. The use of TNM (tumour-node-metastasis) classification as proposed by Union International Contre Le Cancer (UICC) and American Joint Committee on Cancer (AJCC) [79] is the most important tool to predict clinical be-haviour and outcome for RCC [77]. It takes into account the size of the tumour, growth beyond the renal capsule, venous involvement, adrenal metastasis, lymph node metastases, and distant metastases (Table 1). After partial or radical nephrec-tomy [76, 80-83], the prognosis for survival at 5 years is 90-100% for TNM stage I, 75-95% for TNM stage II, 60-70% for TNM stage III, and less than 10% for distant metastatic RCC [2, 3]. Although the 5-year-survival rate is very favourable for patients within TNM stage
I, a significant number of late recurrences was observed when 10- and 15-years sur-vival rates are taken into account [84]. After such a long follow-up periods, no significant differences in survival has been observed between TNM stages I and II or II and pT3b [81, 84]. The clinical (TNM) and pathological (pTNM) classi-fication was modified in 2002 [85] so as to better stratify patients with favourable prognosis [86] for the clinical setting of nephron-sparing surgery [87]. The T1 category was then subdivided into T1a (less than 4 cm) and T1b (4 to 7 cm) [87, 88]. However, tumour size alone is not a definite predictor of prognosis. Sarco-matoid variants seen in all histological subtypes of RCC are associated with a markedly worse prognosis, even if the primary tumour is small [51, 89]. In ChRCC, the probability of metastatic spread is very low, and in PRCC the lower stages tends to have a better prognosis than CRCC [90].
Histopathological grading and
classification The nuclear grading system in multi-
variate analysis has proved to be the second most important classification (after TNM) for predicting the clinical outcome for RCC. Particular in low stages (T1-T2) the nuclear grading system was found to be an independent prognostic factor for survival [91-95]. Different grading systems have been proposed for RCC [91, 96]. Most evaluations empha-size the nuclear features and not the morphology of cytoplasm or histological formation. The worst area of the tumour, rather than over-all impression, defines the grade. Both approaches are taken into account in the Skinner and Fuhrman systems [91, 96] but the Fuhrman grading

14
system is currently the most used system [91, 97] (Table 2). Both systems suffer from interobserver variability and pro-blems in reproducibility [98-100]. The 5-year survival rates for Fuhrman grades 1–4 are 65–76% in grade 1, 30–72% in grade 2, 20–50% in grade III and 10–35%
in grade IV [3]. New cytogenetic and molecular gene-
tic data have established the histologic classification of RCC [51, 79, 101] which was presented at a consensus meeting in Heidelberg in 1996 [45] and approved by the UICC and AJCC in 1997 [51].
Table 1. TNM classification and stage grouping of RCC [85]
Primary Tumor (T)
TX Primary tumor cannot be assessed. T0 No evidence of primary tumor. T1 Tumor 7 cm or less in greatest dimension, limited to the kidney. T1a Tumor 4 cm or less in greatest dimension, limited to the kidney.
T1b Tumor more than 4 cm but not more than 7 cm in greatest dimension; limited to the kidney.
T2 Tumor more than 7 cm in greatest dimension; limited to the kidney.
T3 Tumor extends into major veins or invades adrenal gland or perinephric tissues, but not beyond Gerota’s fascia.
T3a Tumor directly invades adrenal gland or perirenal and/or renal sinus fat, but not beyond Gerota´s fascia.
T3b Tumor grossly extends into the renal vein or its segmental (muscle-containing) branches, or vena cava below the diaphragm.
T3c Tumor grossly extends into vena cava above diaphragm or invades the wall of the vena cava.
T4 Tumor invades beyond Gerota’s fascia.
Regional Lymph Nodes (N)*
NX Regional lymph nodes cannot be assessed. N0 No regional lymph node metastases. N1 Metastases in a single regional lymph node. N2 Metastasis in more than one regional lymph node.
Distant Metastasis (M)
MX Distant metastasis cannot be assessed. M0 No distant metastasis. M1 Distant metastasis.
Stage grouping
Stage I T1 N0 M0 Stage II T2 N0 M0 Stage III T1, T2 N1 M0 T3 N0, N1 M0 Stage IV T4 N0, N1 M0 Any T N2 M0 Any T Any N M1
* Not affected by laterality. Regional lymph nodes includes renal hilar, paracaval, aortic (para-aortic, peri-aortic).

15
Table 2. Nuclear grading systems
Fuhrman grading system [91]
G1 – Nuclei are small, round and uniform (10 μm), with inconspicuous or absent nucleoli.
G2 – Nuclei are slightly irregular (15 μm), with small nucleoli.
G3 – Nuclei are very irregular (20 μm), with large and prominent nucleoli.
G4 – Nuclei exhibit large and pleomorphic often poly-lobed and bizarre (> 20 μm).
Skinner grading system [96]
G1 – Nuclei are small, indistinguishable from those seen in normal tubular cells
G2 – Nuclei are slightly irregular and frequently pyknotic without abnormal nucleoli
G3 – Nuclei are irregular, enlarged and pleomorphic with prominent nucleoli
G4 – Nuclei are extremely giant and bizarre
Clear cell or conventional renal cell carcinoma (CRCC) that appears in light microscopy with a lucid cytoplasm appears more or less empty after staining with hemotoxylin and eosin (H&E). This is an effect of intense intracytoplasmatic accu-mulation of glycogen and phospholipids due to increased glucose-6-phosphate levels caused by activated glycolysis and reduced gluconeogenesis [102, 103]. The nuclei of well-differentiated tumour cells are condensed, while in less differentiated tumour cells the nuclei demonstrate poly-morphism and prominent nucleoli [104]. Another variant of CRCC is the eosino-philic or granular appearance of the cyto-plasm, due to augmentation of mito-chondria. The architecture is characteris-tically variable within the same tumour, with acinar or tubular growth patterns associated with poor defined stroma de-spite being surrounded by rich branching of delicate vasculature. [51, 89, 105]. There is a positive correlation between the extent of lymphocytic infiltration and in-creasing grade of malignancy [106].
Papillary renal cell carcinoma (PRCC) is characterized by a distinct papillary
growth pattern that can become solid in undifferentiated tumour areas. The papil-lary structure consists of delicate fibro-vascular cores with focal lipid-loaded macrophages which are covered by single layer of neoplastic cells [90, 107]. Type 1 PRCC exhibits a faint basophilic stained cytoplasm with small centrally located nuclei [108]. The cytoplasm is pre-dominantly surrounded by endoplasmic reticulum. Type 2 PRCC represents a more aggressive form [68] with enlarged and prominent nucleoli in addition to a eosinophilic and granular cytoplasm rela-ted to accumulation of mitochondria [109]. This histological sub-typing of PRCC has prognostic value since the morphological characteristics of type-2 is independently associated with poor sur-vival [69, 109].
Chromophobe renal cell carcinoma (ChRCC) is characterised by large poly-gonal cells with transparent cytoplasm and prominent cell membranes [110, 111]. The cytoplasm is filled with glycol-gen deposits and numerous microvesicles. Some tumour cells have an eosinophilic or granular cytoplasm due to an accu-

16
mulation of mitochondria [111]. Nuclear size and shape varies, with lack of cyto-plasm colouring by routine dyes, which have been described as perinuclear ‘halos’. The microvesicles can be stained blue by the Hale colloidal iron technique [89]. The architectural pattern is usually solid [51]. ChRCC has a better prognosis than other variants of RCC [89, 105, 107, 110].
Collecting duct RCC has a tubular struc-ture combined with a microcytic, pseudo-papillary, and solid growth patterns asso-ciated with an indefinite stroma and gra-nulocytic infiltration. It has a clinically aggressive course, often demonstrating metastases at presentation and rapid pro-gression [112, 113]. No specific genetic alterations have been described in the rare collecting duct RCC [113]. Unclassified RCC is a category containing a hetero-geneous group of tumours that cannot be classified as any of the previously mentioned subtypes [51]. Sarcomatoid changes can occur in all subtypes of RCC [51, 89].
The distinct histological subtypes have different biological and clinical behaviour [114-116]. The most common type is CRCC, which has a 55-60% 5-year survival rate [117, 118]. PRCC has a 80-90% 5-year survival rate [90, 107, 109]. ChRCC is less common and therefore survival data are limited, but ChRCC is overall associated with favourable prognosis [70]. Collecting duct RCC show an extremely aggressive behaviour with no reported 5-year survivors [112, 113]. However by using multivariate analysis, the histo-logical subtype was not found to be an independent prognostic factor for survival [119, 120]. Recent studies indicated a clinical utility for classifying RCC in different histological subtypes since they
may respond differently to treatment [121].
Angiogenesis in cancer Angiogenesis is the formation of new
capillaries by outgrowth of endothelial cells from pre-existing blood vessels. Folkman and coworkers demonstrated that tumours could only grow up to 1.75 mm when nutrition was obtained solely by diffusion. Growth beyond this size required a supply by new blood vessels [122]. The onset of angiogenesis depends on a shift in the equilibrium in extra-cellular matrix (ECM) created by numerous of inhibitors and stimulators towards an activation of angiogenesis [123]. Once activated, a sequence of events is required in order for formation of new blood vessels to occur [124]. First, a local de-gradation of basement mem-brane is required to facilitate the migration of endothelial cells, and this is followed by alignment of proliferated endothelial cells which form new ca-pillaries towards an angiogenic stimulus [124]. A growing tumour needs to recruit additional blood supply in order to maintain sufficient oxygen and other nutrient availability for rapidly dividing cells. This onset of angiogenesis may be brought about by a hypoxic or hypo-glycaemic condition, or may be caused by increased angiogenic stimulators due to genetic faults [123, 125]. Several growth factors with angiogenic signalling have been identified [126]. These include basic fibroblast growth factor (bFGF) and platelet-derived growth factors (PDGF), which have signalling activity for several different types of cells, whereas vascular endothelial growth factor (VEGF) affects primarily vascular endothelial cells [127].

17
Tumour secreted VEGF-A
VEGF is a disulfide-bonded dimeric glycoprotein with a molecular mass 34-46 kDa, and it is the most potent of all growth factors that stimulate vascular permeability and endothelial cell proliferation [128-131]. Structurally, VEGF exhibits an sequence amino acid homo-logy to platelet-derived growth factor (PDGF) [132-134]. In cultured vascular smooth muscle cells, three natural occur-ring isoforms of VEGF-A have been iden-tified through analysis of isolated cDNA clones to predict sequences of 189, 165 and 121 amino acids [135]. Applying the reverse transcription polymerase chain reaction (RT-PCR) technique to different carcinoma cell lines has helped to identify additional VEGF isoforms [136]. To date, 7 different mRNA forms (VEGF121, 145, 165,
183, 189, and 206) have been described [136-139]. All VEGF isoforms are generated from the same gene, which is located on the short arm of chromosome 6 [140]. Recently, Bates et al. described a variant of VEGF with 165 amino acid isoforms termed VEGF165b [141]. The VEGF165b demonstrated inhibitory properties on pro-liferation, migration, and vasodilatation in endothelial cells. This indicates that alternative spliced variants of VEGF have different functions or activities. All VEGF isoforms exhibit exons 1-5 and a terminal exon 8 although altered in VEGF165b [141]. Exons 1-5 and terminal exon 8 encodes VEGF121. Inclusion of exon 7 or exons 6 and 7 encodes VEGF165 and VEGF189, respectively [139]. The biolo-gical differences between VEGF isoforms are dependent on incorporated exons. Exon 1 encodes the signal sequence, exon 2 the N terminus, and exon 3 the dimeri-sation domain [142]. Exons 3 and 4 encodes binding domain to VEGF-R1 and VEGF-
R2, respectively [138]. Exons 6 and 7 provide the binding affinity to heparin or heparan sulphate proteoglycans [143]. In addition, exon 7 encodes the binding domain to receptor of Neurophilin-1 [144]. Exon 8 are required for the stimulation of mito-sis [141,145]. Thus, alternative splicing of VEGF mRNA generates forms that differ in solubility, and longer forms are retained by proteoglycans at cell surface or extracellular matrix [143].
Alterations in the extracellular en-vironment can cause affected cells to release VEGF, and extracellular stimuli include, for example, hypoxia [146], and hypoglycaemia [147], the presence of in-flammatory cytokines [148], platelet-derived growth factor (PDGF) [149], basic fibroblast growth factor (bFGF) [150], epidermal growth factor (EGF) [151], and insulin-like growth factor (IGF) [152, 153]. The mechanisms behind these different growth factors leading to VEGF promotion of angiogenesis are unknown. Uncontrolled cell proliferation is a causal event to produce VEGF and allow cell survival. Uncontrolled cell pro-liferation may be driven by an activated oncogene [154] or though series of gene-tic events due to inactivated or impaired tumour suppressor gene [155], by ab-normal cell signalling in response to growth factors (myc) [6, 153], amplifying growth recaptor signalling (erb-B/EGF-R, met) [30, 32-36]., interfering in cell cycle regulation (Cyclins D and E, p27, p53, pRb) [37-40, 156, 157], or preventing apoptosis (p53, Bc12)[158, 159]. In contrast, an inactivated or impaired tumour suppressor gene (VHL) leads tumours to consistent production of VEGF [160]. These tumours have an impaired von Hippel-Lindau protein (pVHL), which cause an accumulation of hypoxia indu-

18
cible factor (HIF-1α), and, as a con-sequence, release of several growth factors such as VEGF, PDGF and TGF-α into the extracellular matrix [150]. Co-expression between growth factors might have an autocrine role as well as a paracrine function [161].
All isoforms of VEGF-A are able to stimulate endothelial cells through two receptor tyrosine kinases VEGF-R1 [162, 163] and VEGF-R2 [164, 165]. Structural dimerisation of VEGF is essential for biological activity, and particularly for VEGF165, where the ligand pairing has a high affinity to VEGF-R2 [142, 144]. Several members of the VEGF family (VEGF-A, -C, -D, and -E) can activate VEGF-R2 and promote endothelial cells to proliferate, migrate, and differentiate [166-169]. On the other hand, VEGF-R1, activated by VEGF-A, -B, and PDGF stimulates endothelial cells to migrate [170-172]. VEGF and PDGF demonstrate an amino acid sequence homology which may in heterodimerisation provide a potential diversity in signal transduction through ligand-mediated paring with receptors VEGF-R1 or PDGF-R [132, 171]. The functional consequences of hetero-dimerisation between the growth factors are not quite understood. Thus, VEGF-R1 regulates structural organisation and sta-bility of new-formed vessels, whereas VEGF-R2 regulates proliferation of endo-thelial cells and appears to have a more prominent role of promoting angio-genesis [144, 169, 170]. The other family members in VEGF (VEGF-C, and -D) are involved in regulation of endothelial cells during formation of new lymphatic vessels [166, 172, 173]. VEGF, PDGF, and bFGF stimulates endothelial cells to release proteases such as matrix metallo-proteinases (MMP), which degrade the
EMC and basement membrane, creating a window through which endothelial cells can migrate [174-176].
Synergistic role of bFGF bFGF (18 kDa) was earlier described
as an important angiogenic factor in-volved in the angiogenic switch during early tumourigenesis [177], though re-cently VEGF has been advocated as responsible [178]. bFGF is produced by a variety of cells including tumour cells, endothelial cells, fibroblasts, and macro-phages [179, 180]. Most bFGF that is secreted from these cells is sequestered by heparan sulphate in the extracellular matrix, with the matrix serving as a reservoir for growth factors [174, 179-181]. bFGF activates additional proteases from endothelial cells to dissolve the basement membrane (ECM) and thus facilitate and induce proliferation and migration of endothelial cells [127, 179, 182]. bFGF is not as potent as VEGF in stimulating endothelial cells [127]. There is evidence that VEGF and bFGF act synergistically to promote angiogenesis [182]. In serum, bFGF has been found at significantly higher concentration levels in patients with RCC than in patients without tumours [183-185]. RCC some-how leads to secretion of bFGF [186], but no differences in expression could be observed between tumour cells and normal renal tissue [187, 188]. bFGF was found localised to ECM, whereas a small number of tumour cells expressed bFGF [189]. The expression of bFGF in general has been associated with poor survival, but was not an independent factor related to survival in RCC [189].
Miscellaneous role of PDGF PDGF (30 kDa) is stored in the plate-

19
let granules, and is released with platelet activation [190]. It can also be produced by a variety of cells, including activated macrophages, endothelial cells, smooth muscle cells as well as tumour cells [191]. PDGF is responsible for migration and proliferation of fibroblasts, smooth muscle
cells, and monocytes [192]. Although PDGF does not appear to be important for early formation of blood vessels, it may play a role at later stages through its ability to recruit pericytes and stimulate development of vascular smooth muscle cells [4, 191].

20
AIMS OF THE INVESTIGATION
I. To analyse the prognostic value of VEGF in preoperatively collected
sera from patients with RCC.
II. To investigate whether intercurrently diseases and medications
affects VEGF in serum as a prognostic factor in patients with RCC.
III. To investigate the prognostic impact of VEGF mRNA isoforms and
VEGF receptor mRNA in RCC
IV. To evaluate the clinical impact of immunohistochemical detected
VEGF expression in RCC.
V. To investigate the prognostic impact of VEGF protein isoforms in
different RCC types.

21
MATERIAL AND METHODS
Patients The study included a total of 265
patients with histopathologically verified RCC after surgery between August 1982 and December 1999. Each patient parti-cipated after informed consent and the ethics committee at the Umeå University approved the studies. The patients were randomly assigned to the individual stu-dies as indicated in figure 1. There were 156 men and 109 women, with a median age of 66.0 years (mean 65.4; range 25 – 87 years). Among the 265 patients, 256 were operated with nephrectomy, seven with partial, and two with combined partial and radical nephrectomy due to bilateral RCC. None of the patients had been treated with radiation, chemotherapy, or immunotherapy before surgery. Most of patients were followed-up according to clinical routine [83] with clinical and radiological examinations at regular inter-vals. Survival was determined from time of nephrectomy to the latest follow-up, where information was obtained from hospital records or from the Cancer Registry of Sweden [1].
At the latest follow-up, 65 patients were alive with a median follow-up time 133 months, (range 26 – 236 months), 138 had died of RCC with median survival time of 17 month (range 1 – 168 months), and 62 had died of unrelated causes with 55 month median survival time (range 1 – 203 months). After radical or partial nephrectomy the survival rate at 5-years was 95% for TNM stage I, 80% for TNM stage II, 45% for TNM stage III, and 5% for TNM stage IV. The distinct histological subtypes showed different biological and clinical behaviour [116]. CRCC had a 35% 15-year survival rate,
PRCC had a 47% 15-year survival rate and the less common ChRCC had an 80% 15-year survival rate. Histological un-classified RCC had an extremely aggres-sive behaviour as no survival could be reported at 10-year follow-up. The 15-year survival rates for Skinner grades 1– 4 was 88% in grade 1, 75% in grade 2, 32% in grade III, and 18% in grade IV.
Tumour staging
Tumour staging was performed according to the TNM stage classification system 1997 (Paper I-III) and 2002 (Paper IV-V) [85]. TNM classification included microscopic evaluation for tumour pre-sence in regional lymph node and peri-nephric tissues as well as tumour invasion through the renal capsule into peri-renal fat or into major renal veins at the renal hilum. There were 78 stage I tumours (29.4%), 37 stage II (14%), 57 stage III (21.5%), and 93 stage IV tumours (35.1%). Among the 57 stage III tumours, 12 invaded peri-nephric tissue or adrenal gland (pT3a, pN0, M0), 23 major veins only (pT3b-c, pN0, M0) and 22 invaded peri-nephric tissues and major veins (pT3a-b, pN0-1, M0). One patient with stage III tumour had metastasis in a single regional lymph node. There was no appa-rent shift in tumour stage distribution during the 20 years of sample collection. Tumour size was measured as the maxi-mal diameter on the surgical specimen or by computerized tomography. The me-dian tumour diameter was 75 mm (mean 80; range 10 – 220 mm).
Morphologic classification Histopathologic nuclear grading was
performed according to Skinner and co-

22
No. patients 233 164 161 96 61Paper IV I II V III
Indi
vidu
alca
ses
No. patients 233 164 161 96 61Paper IV I II V III
Indi
vidu
alca
ses
Figure 1. Depiction of the different studies in which 265 patients were included. The patients were arranged according number of assigned studies and not by time of nephrectomy. workers [96], characterized on the worse histologic features, by pathologists at Umeå University Hospital (Table 3). Eleven (4%) tumours were classified as grade 1, 55 (21%) were grade 2, 132 (50%) were grade 3, and 67 (25%) were grade 4 (Table 4). RCC tumour type was histo-pathologic examined according to the Heidelberg classification system [45]. More than 75% tubulopapillary architec-ture was used for designation of tumour as a papillary RCC [193, 194]. There were 203 (77%) conventional (clear cell), 36 (13%) papillary and 15 (6%) chromo-phobe RCCs while 6 (2%) tumours were not amenable to morphological classi-fication. Five (2%) tumours were missing and not available for second microscopic review according to Heidelberg classi-
fication system by Professor G Kovacs [45].
Methods The methods used in this thesis are
described in material and methods sec-tions of the individual papers. A more general description is presented below.
Blood analysis Preoperative blood samples were
obtained as a routine before 10 a.m. The blood cell composition was analysed in the Clinical Chemistry Laboratory with standard Coulter counter technique (Coulter counters S-Plus STKR, Coulter Elec-tronics, Luton, England, or Sysmex SE-9000, Toa Medical Electronics Co., Kobe, Japan). Patients receiving preoperatively intravenous treatment were excluded.

23
Table 3. Pathologists who participated in the clinical examination of microscopic sections in this thesis.
Nuclear grading according to Skinner [96] Grade 1 Grade 2 Grade 3 Grade 4
R Stenling 9 44 113 58 84.5 % L Boquist 1 3 7 2 4.9 % U Gerdes 3 3 2.3 % O Hassler 2 3 1.9 % J Vasko 2 2 1,5 % S Cajander 1 1 1 1.1 % F Bergman 2 0.8 % B Lundskog 2 0.8 % L Bjersing 1 1 0.8 % B Bozoky 1 1 0.8 % G Hallmans 1 0.4 % A Bergh 1 0.4 %
Table 4. Clinicopatholgoical characteristics of different RCC types involved in this thesis.
Conventional Papillary Chromophope Not classified Not accessible
Skinner Grade 1 5 5 1 Grade 2 35 11 6 1 2 Grade 3 101 18 9 3 1 Grade 4 62 2 1 2
TNM Stage I 57 14 5 1 1 Stage II 23 5 6 1 2 Stage III 47 4 3 2 1 Stage IV 76 13 2 2
Serum analysis
The samples were routinely obtained from peripheral veins in patients. The recommendation was 5 min horizontal bed resting and food restriction before sample taking [195]. Samples were collected in tubes without additive and allowed to coagulate for 30 min and centrifuged (1500 x g) for 15 min at room temperature. Obtained serum samples were stored at -80°C until analysis. Serum
VEGF was analysed by quantitative sand-wich enzyme immunoassay tech-nique (Quantikine, Human VEGF immuno-assay, R & D Systems, Minneapolis, MN). A microplate precoated with mouse mono-clonal antibody against human VEGF, immobilized free VEGF in samples. After a new wash, an enzyme-linked secondary goat polyclonal antibody against VEGF was added to the wells. After additional wash, a substrate solution was added to the wells and colour developed in

24
proportion to the amount of VEGF bound in initial step. Optical density was determined on a microtiter plate reader (Multiskan MCC/340, Lab Systems, Stockholm, Sweden). The results were measured in duplicate, and a deviation more than 10% between measurements led to re-measurement.
Tissue collection and preparation Tumour and kidney cortex tissue
samples were obtained from the surgical specimen as described by Ljungberg et al [196]. Each sample was divided into smaller pieces. One part was snap frozen in liquid nitrogen and stored in –80°C and the other part was formalin fixed and paraffin embedded for immunohisto-chemical staining and morphologic exa-mination.
Competitive quantitative RT-PCR Frozen tumour samples were homo-genised and total RNA was isolated using the TRIzol method (Life Technologies, Stockholm, Sweden). Total RNA concen-trations were measured spectrophoto-metrically at 260 nm (Lambda 2 UV/VIS, Perkin Elmer, Stockholm, Sweden). The mRNA levels of all isoforms of VEGF-A, VEGF-R1 and cyclophilin were quanti-fied by competitive RT-PCR, as described previously [197]. Primer sequences were designed to quantify all isoforms simul-taneously from the human genes of VEGF-A (5´-ATC TTC AAG CCG TCC TGT GTG C-3´ and 5´- TCA CCG CCT CGG CTT GTC ACA T-3´) and flt-1 (5´-AGG AGA GGA CCT GAA ACT GTC TT-3´and 5´-ATT CCT GGC TCT GCA GGC ATA G-3´). Briefly, 50 ng of total RNA was reverse-transcribed together with appropriate amounts of internal RNA-standards (IS), meaning truncated
RNA of VEGF, flt-1, and cyclophilin. Each RNA sample was titrated with three different concentrations of IS for the respective gene (in duplicates). During 30 cycles of PCR (94°C, 30 s; 61°C, 30 s; 72°C, 45 s) templates were competitively amplified with cDNA for corresponding IS. VEGF primers used in the PCR reaction were designed for the simul-taneous amplification of all VEGF iso-forms (Figure 2). The PCR products were analysed by a laser fluorescence system (ABI PRISM 377 DNA sequencer, Perkin Elmer), and processed by the ABI PRISM GenScan software (Perkin Elmer) (Figure 3). Messenger RNA levels were calcu-lated from the linear regression by extra- polation at equivalent templates to IS signals as previously described. The VEGF and flt-1 values were corrected for the corresponding cyclophilin values in each RNA sample and expressed as relative levels (fmol/amol cyclophilin). The measurements were performed twice, and a deviation more than 10% between measurements led to re-measurement.
VEGF-A is a 34-46 kDa peptide for-med by two subunits (monomers) organi-zed in anti parallel (homodimeric) manner by disulfide linked bridges. VEGF exists in different isoforms due to alternative splicing after signal sequence cleavages of transcript encode, mRNA [198]. The human VEGF gene is structured as 8 exons, separated by 7 introns. The basic sequence of mRNA consists of 8 exons and encodes the longest variant of VEGF189 with 189 amino acid. The other isoforms arise from alternative splicing of mRNA regarding sequence of exons 6 and 7. VEGF165 lacks exon 6 and VEGF121 lacks exons 6 and 7. Sense primer 5´ - ATC TTC AAG CCG TCC TGT GTG C - 3´ corresponds with exon 3, and anti-
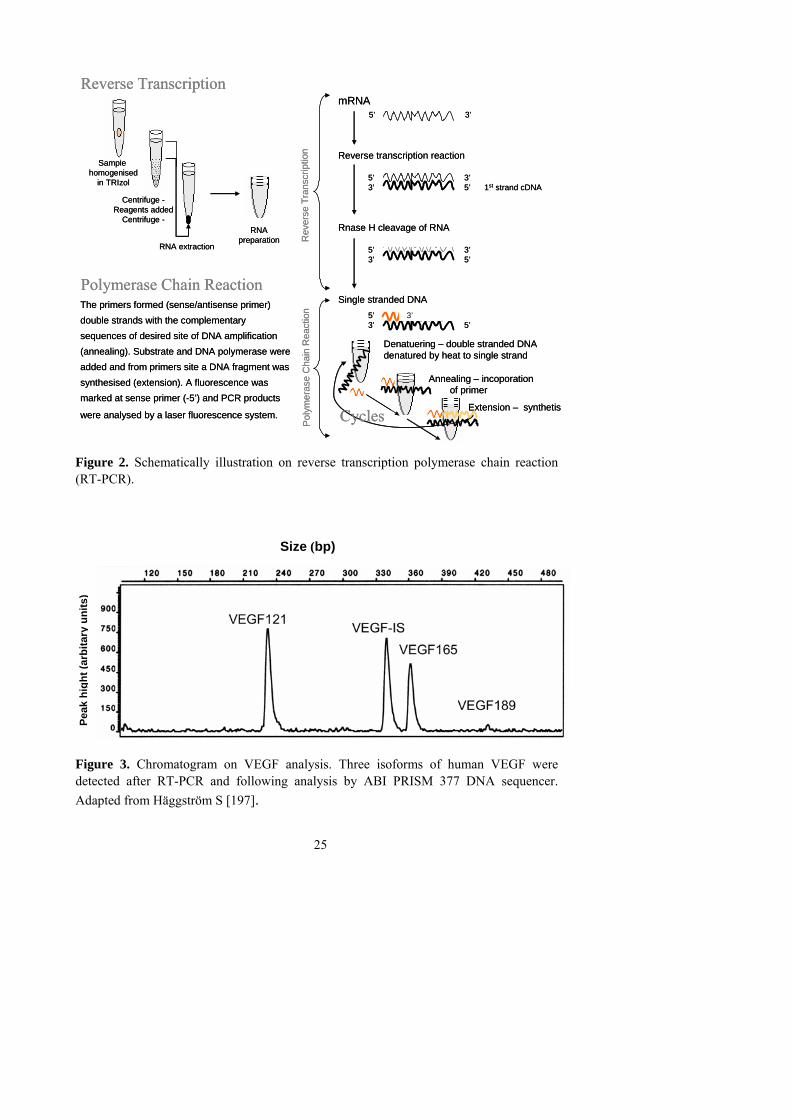
25
Sample homogenised
in TRIzol
RNA extraction
Centrifuge -Reagents added
Centrifuge -RNA
preparation
Poly
mer
ase
Cha
in R
eact
ion
Rev
erse
Tra
nscr
iptio
n The primers formed (sense/antisense primer) a
double strands with the complementary a
sequences of desired site of DNA amplification a
(annealing). Substrate and DNA polymerase werea
added and from primers site a DNA fragment was a
synthesised (extension). A fluorescence was a
marked at sense primer (-5’) and PCR products a
were analysed by a laser fluorescence system.
Polymerase Chain Reaction
Reverse Transcription
Denatuering – double stranded DNA denatured by heat to single strand
Annealing – incoporation of of of primer…
Extension – synthetisCycles
mRNA5’ 3’
Reverse transcription reaction
5’ 3’3’ 5’ 1st strand cDNA
Rnase H cleavage of RNA
5’ 3’3’ 5’
Single stranded DNA5’ 3’3’ 5’
Sample homogenised
in TRIzol
RNA extraction
Centrifuge -Reagents added
Centrifuge -RNA
preparation
Poly
mer
ase
Cha
in R
eact
ion
Rev
erse
Tra
nscr
iptio
n The primers formed (sense/antisense primer) a
double strands with the complementary a
sequences of desired site of DNA amplification a
(annealing). Substrate and DNA polymerase werea
added and from primers site a DNA fragment was a
synthesised (extension). A fluorescence was a
marked at sense primer (-5’) and PCR products a
were analysed by a laser fluorescence system.
Polymerase Chain Reaction
Reverse Transcription
Denatuering – double stranded DNA denatured by heat to single strand
Annealing – incoporation of of of primer…
Extension – synthetisCycles
mRNA5’ 3’
Reverse transcription reaction
5’ 3’3’ 5’ 1st strand cDNA
Rnase H cleavage of RNA
5’ 3’3’ 5’
Single stranded DNA5’ 3’3’ 5’
Figure 2. Schematically illustration on reverse transcription polymerase chain reaction (RT-PCR). Size (bp)
Figure 3. Chromatogram on VEGF analysis. Three isoforms of human VEGF were detected after RT-PCR and following analysis by ABI PRISM 377 DNA sequencer. Adapted from Häggström S [197].
Peak
hig
ht (a
rbita
ry u
nits
)

26
sense primer 5´ - TCA CCG CCT CGG CTT GTC ACA T -3´ with exon 8 of human cDNA.
Immunohistochemestry
Representative paraffin tumour blocks were selected by primary evaluation of haematoxylin/eosin- stained slides before tissue microarray (TMA) preparation. Two tissue cores were taken with a sample needle (0.6 mm in diameter) from each tumour block and placed in a new recipient paraffin-block containing 98 tissue cores with 0,8 mm space between (Beecher Instruments, USA). For micro-scopic evaluation 4-μm thick paraffin sections was slized. The slides were
treated with standard procedures for de-parafinating, rehydrating, microwave hea-ting and immunohistochemical (IHC) staining. Different antibodies were tested and a mouse monoclonal anti-human VEGF antibody (NeoMarkers, Lab Vision Corporation, Fremont, CA; VEGF Ab-3) was selected. The remains of antibody bound to antigen through washing were visualized through a colour reaction with biotinylated multilink secondary antibody (Vectastain Elite ABC kit, Vector Labora-tories, Inc., Burlingame, CA). Most favourable antibody was chosen on three tissue slides selected by presence of kidney tissue, adjacent to tumour tissue, which contained poor, intermediary or strong IHC VEGF expression. (Table 5).
Table 5. An overwiew of primary antibodies that have been used in this thesis.
Applications Antibody Manufacture Reactivity/Epitope
Serum Mouse, monoclonal antibody, anti- VEGF
Quantikine, Human VEGF immunoassay, R & D Systems, Minneapolis, MN
Human VEGF165 and VEGF121 homodimer (VEGF165/hPDGF heterodimer exhibit 20 % cross-reactivity)
Goat polyclonal antibody, anti- VEGF
WB Rabbit, polyclonal antibody, anti-VEGF (A-20)
Santa Cruz, Biotechnology Inc, Santa Cruz, CA
Human (Mouse and Rat) VEGF / N-terminus of VEGF
WB* Rabbit, polyclonal antibody, anti-VEGF (Ab-1)
NeoMarkers, Lab Vision Corporation, Fremont, CA
Human (Mouse, and Rat) VEGF / Not determined
IHC Mouse, monoclonal antibody, anti-VEGF (Ab-3)
NeoMarkers, Lab Vision Corporation, Fremont, CA
Human (Rabbit) VEGF / Not determined
IHC* Rabbit, polyclonal antibody, anti- VEGF (147)
Santa Cruz, Biotechnology Inc, Santa Cruz, CA
Human (Mouse, and Rat) VEGF / Antibody raised against a peptide corresponding to amino acids 1-140 of VEGF
* Not applicable to this study.

27
A B
Figure 4. These images illustrate the evaluation of different profiles using a special grid to calculate percentage of fields with positive IHC staining in cells emphasized by presence of nucleus and the overlaying chosen area. Profiles intersected by inclusions-edges were counted in addition to enclosed ones. A. Cytoplasmic VEGF staining. B. Cell surface VEGF staining. The positive and uniform IHC VEGF expressions in normal kidney tubular cells were used as control for adequate immuno-histochemical application for evaluation positive IHC VEGF expression in tumour
series. An additional control to confirm IHC VEGF expression was performed with the absence of the primary antibody and by a neutralizing primary VEGF antibody with recombine VEGF peptide
A B
Nucleus
Nucleus

28
(Recombinant human VEGF, 293 VE; R&D Systems Inc., Minneapolis, MN) before incubation of the slides. For quantifying IHC VEGF staining, a square-latitude mounted in the eyepiece of a light microscope was used to evaluate the density of VEGF stained tumour cells. Within framework of 121-point eyepiece graticule at 200x magnification, the den-sity was defined as number of manually counted fields with cellular stained VEGF and overlaying chosen area (Figure 4). In TMA the sampling area was demarcated within framework of a 100-square grati-cule at 200x magnification, and area density of VEGF expression was defined as maximal score of two samples from each tumour. For tissue section (TS), the slides were examined at low magnify-cation (100x), and three areas containing the highest VEGF expression were chosen as hot spot areas and then evaluated at 200x magnification. The VEGF expression in the tissue sections was defined as mean score of the volume density from the three hot spot areas. In TS, the staining intensity of IHC VEGF was assessed according to a three-graded scale: weak staining- when visible at 200x magni-fication, moderate intensity- when visible at 100x, and strong staining- when visible at 20x. Staining intensity was evaluated separately and independently three times, and the quantification of volume density was also evaluated three times by one observer without knowledge to preceding results. Any discordance was resolved by re-examined by a pathologist.
Protein extraction Imprints were made from frozen tissue
and stained with Giemsa to verify pre-sence of tumour cells. Frozen tissue samples were homogenized with a micro-
dismembrator for 2 x 10 sec., and after slight thawing, the samples were sus-pended in chilled Tris lysis buffer con-taining protease inhibitors. The homogenates were then centrifuged (13000 x g) for 30 min at 4 Co. Protein in the supernatant was quantified by bicinchoinic acid assay (Pierce, Illinois). A standard curve created on absorbance of the control series was used to set the concentration of protein extractions. These measurements were performed in duplicate, a deviation be-tween measurements of more than 10% led to re-measurement.
Western blotting
A Western blot (WB) procedure was performed on 30 µg protein extracts to which was added an equal volume of electrophoresis sample buffered with reducing agents to accomplish a mono-meric form of VEGF. The samples were electrophoresed on 12% SDS poly-acrylamide gel and then transferred to a nitrocellulose membrane. Staining with Ponceau-red was performed to ensure a total transfer of protein extract to membrane. The membrane was either blocked in 10% horse serum or in 5% milk powder and 2.5% bovine calf serum with PBS, 0.1% Tween-20 prior to incubation with antibodies. The mem-brane was primarily incubated with a polyclonal rabbit antibody to human VEGF, and secondarily incubated with horseradish peroxidase (HRP) labeled anti-rabbit antibody. VEGF was visua-lised by enhanced chemiluminescence (ECL Plus, Amersham Int., England) and recorded on Hyperfilm-ECL (Amersham Int., England) (Figure 5). Protein ex-pression was quantified using Fluor-S Multi Imager scanning and Quantity One software analysis (Bio-Rad Laboratories,

29
Peroxide
2 H2O
O2
+
Luminol
+
Enhancer
LightPrimary
antibody
Secondary
HRP-labelled antibody
VEGF
Oxidized
productH
ybon
dEC
L W
este
rn
nitro
lulo
sem
embr
ane
Det
ectio
nby
Hyp
erfil
m E
CL
Peroxide
2 H2O
O2
+
Luminol
+
Enhancer
LightPrimary
antibody
Secondary
HRP-labelled antibody
VEGF
Oxidized
productH
ybon
dEC
L W
este
rn
nitro
lulo
sem
embr
ane
Det
ectio
nby
Hyp
erfil
m E
CL
Figure 5. The membrane was soaked with ECL Plus detection reagent. Where the HRP-labeled secondary antibody was bound, a peroxidase-catalyzed oxidation of luminol was induced leading to chemiluminescence. This resulting light was detected on hyperfilm ECL Western.
Hercules, California). Relative protein ex-pression was evaluated against a linear standard curve obtained by different dilutions on extracts of a standard tumour used in each gel (Figure 6). Detected protein bands were controlled by not adding primary antibody or by primary antibody was blocked with human VEGF peptide before incubation. At start of the study, different primary VEGF antibodies were evaluated (Table 5). The molecular weights of detected proteins were verified by comparison with pre-stained SDS-PAGE standards (161-0372, Bio-Rad Laboratories, Hercules, California). These measurements were performed in dupli-cate, and a deviation of more than 10% led to re-measurement. Equal loading of protein extracts was confirmed by the actin expression. The band at 28-kDa was defined as VEGF189, the band at 22-23-kDa as VEGF165 and the band at 18-kDa was defined as VEGF121 [21, 22].
Statistical methods The Mann-Whitney U test was used to
identify differences in non-parametic
variables for two independent groups, and the Kruskal-Wallis test was used for comparison of more than two groups. Spearman rank correlation test was used to compare relationship between sets of non-parametric variables that did not demonstrate a linear relation. Chi-Square test was used to evaluate differences in proportions of observations between in-dependent groups. Fisher’s exact test was used when the sample size was too small to use the chi-square test. The median value was chosen as the cut-off value. Survival data was analysed using the Kaplan-Meier method, and comparison of survival times for groups was performed with the log-rank test. The variables were dichotomously tested, and analysed as continuous in a univariate Cox regression analysis. Multivariate analysis was per-formed according to Cox proportional hazard model. Survival time was mea-sured from the date of nephrectomy to date of death, or latest follow-up. In all tests, a two-tailed significance level was set to < 0.05.

30
Figure 6. Using Fluor-S Multi Imager scanning and Quantity One software analysis facilitate the determination of protein density from Hyperfilm ECL. The relative protein concentration was evaluated against a linear standard curve obtained by different dilutions and extracts of tumour loaded in each gel.

31
RESULTS AND COMMENTS
Paper I Serum VEGF concentration (S-VEGF)
was significantly increased in patients with RCC compared to a control group. No alteration in S-VEGF was observed due to storage time of the samples. There were no differences in distribution of tumour stage, grade, and size related to gender or age. Increasing S-VEGF corre-lated with higher tumour stage and grade. Localized tumours (stages I-II) had low S-VEGF compared to tumours with vein invasion or extracapsular dissemination (stage III) as well as tumours with distant metastasis (stage IV). There was no relation between S-VEGF and extent of tumour-thrombus in vena renalis (pT3b) or vena cava (pT3c). A weak correlation was observed between S-VEGF and size of localized tumours (stages I-II). There was no difference in S-VEGF between PRCC and CRCC, although ChRCC results showed lower S-VEGF compared to CRCC but not PRCC. Increasing S-VEGF correlated with shorter survival in male patients, but not in female patients. In multivariate analysis, S-VEGF was not an independent prognostic factor.
Comments on Paper I Considerable efforts have been made
to find biomarkers for progression of disease in order to monitor effect of cancer treatment. Measuring circulating biomarkers in peripheral blood samples has practically appeal since access is easy and results reliable when uniform handling of blood samples is performed [199]. We demonstrated a significant correlation between S-VEGF levels and tumour stage, and that high S-VEGF levels were associated with poor outcome
in patients with RCC. When Paper I was accepted for publication, there was dis-cussion regarding the value of measuring VEGF in sera [199-201]. Peripheral ve-nous blood samples showed higher con-centration of VEGF in serum compared with matched plasma samples [200, 202-207] due to VEGF release related to platelet activation and some contribution from white cells [200, 202-204, 207, 208]. RCCs are thought to affect the bone marrow leading to increased platelet counts [208, 209]. Platelet activation in tumours [210-213] may lead to elabora-tion of several angiogenic growth factors, including VEGF [200]. The theoretical advantage of analysing VEGF in plasma has limitations since circulating free VEGF in vivo is subject to degradation as well as binding to VEGF-binding protein levels, and may not directly reflect tumour secretion of VEGF [201]. S-VEGF levels differ from plasma VEGF also due to the fact that platelets deliver VEGF to tumours [214-216]. It has been suggested that PDGF reflects tumour biological activity [217], and that serum samples are more useful for measurement circulating growth factors [205]. VEGF165 is the predominant isoform in serum [218] and is also the isoform predominantly secreted by a variety of cancer types [219]. In Papers I-II, analysed levels of S-VEGF165 reflect also the biological effects of VEGF121 and VEGF165/PDGF hetero-dimer occurring in serum after platelet agglutination.
Paper II Approximately two-thirds of patients
with RCC were concurrently treated with long-term medication because of chronic

32
diseases. No differences in distribution of tumour stage, grade, or size were ob-served related to gender, age group, or presence of long-term medication. The proportion of patients with long-term medication increased with age for both genders. No difference in S-VEGF was found in relation to gender, age group or presence of long-term me medication. Increasing S-VEGF was correlated with higher tumour stage and grade, as well as higher platelet and leukocyte counts. No difference in blood platelet counts was found between male and female patients. However, platelet counts showed an in-verse correlation to age, particularly in women. In male patients, platelet counts were lower in those on long-term medication compared to those without. Univariate analysis showed that increased S-VEGF and platelet counts correlated with shorter survival especially in male patients. In contrast to S-VEGF, higher blood platelet counts were only associated to shorter survival in patients with RCC with no intercurrent chronic disease or long-term medication. Leukocyte counts were not associated with prognosis. Age, gender, or long-term medication had no association with survival time in patients with RCC. Using Cox proportional hazard step-wise elimination, VEGF levels were the last variable to be excluded, and only stage and grade remained as independent prognostic factors.
Comments on Paper II This paper demonstrated a correlation
between VEGF levels and blood platelet and leukocyte counts, as reported in other malignancies [202]. Platelets have been revealed as the main source of VEGF in serum [200, 207, 210, 211, 220]. However, the lack of relationship between S-VEGF
levels and blood platelet counts in sur-vival curves may be due to peripheral distribution of larger immature platelets with higher quantities of VEGF [207] or caused by other factors as acquired dys-function of platelets commonly associated with medication in elderly people [221]. Moreover, decreased blood platelet counts have been associated with medication and advancing age [222-224]. Epidemiologic studies have shown that established risk factors such as obesity and cigarette smoking can be confounding factors when examining effects of medication [225]. In addition, anti-hypertensive drugs and analgesics have been proposed as in-dependent risk factors for RCC develop-ment [225, 226]. This paper showed that patients with RCC had decreased platelet counts that were associated with inter-current chronic disease, presence of long-term medications, and advancing age. In women, blood platelet counts were not affected by chronic diseases or medica-tions but decreased with advancing age. In addition blood platelet counts had no predictive value for survival for women, regardless of presence or absence of chronic decease or long-term medications. In men with chronic diseases and long-term medications, blood platelet counts were decreased, and no predictive value for survival was found in those subgroups of patients. It is noteworthy that RCC is most common in patients at their sixth to seventh decades of life [1], but this ratio between gender tends to decrease with increasing age [227]. Whether or not other health risk factors may initiate conversion of tumour dormancy to angio-genic phenotype is not clear [228]. S-VEGF levels in peripheral blood samples suggest that S-VEGF has much of its origin from platelets and to some extent

33
from white blood cells. However, the tu-mour angiogenesis process may be dependent on platelets that deliver VEGF to tumours. Still, S-VEGF levels have been reported to be significantly higher in samples from tumour-bearing renal veins compared with samples from peripheral veins [229]. We found that S-VEGF levels were unaffected by intercurrent chronic diseases or medications. In addi-tion, there were no differences in S-VEGF levels in relation to age or gender. In contrast to men, S-VEGF levels had no predictive value for survival in women with RCC, and the reason for this is unclear. Our results indicate that S-VEGF is more useful as a prognostic indicator or biomarker, than platelet or leukocyte counts. Measurement of VEGF in serum may have value for monitoring of therapeutic effect or choice of therapeutic modality [230].
Paper III The RCC samples demonstrated a
wide range of mRNA levels for VEGF121, VEGF165, and Flt-1 (VEGF-R1). Other VEGF isoforms were not be detected. A signi-ficant correlation was observed between mRNA levels for VEGF121 and VEGF165 and between mRNA levels for VEGF165 and VEGF-R1. RCC confined to the kidney (pT1-2 N0 M0) had higher mRNA levels for VEGF121 compared with locally advanced tumours (pT3 N0-1 M0). On the other hand, S-VEGF was lower in loca-lised tumours (pT1-2 N0 M0) compared with locally advanced tumours (pT3 N0-1 M0). There was no association between different isoform VEGF mRNA levels and S-VEGF. CRCC had significantly higher VEGF121 and VEGF-R1 mRNA levels compared with PRCC. In patients with locally advanced tumours (pT3a-c N0-1 M0) increased VEGF121 mRNA
levels correlated with adverse survival. For VEGF165 and VEGF-R1 mRNA levels no predictive information was found.
Comments on Paper III RT-PCR method allows detection and
quantification of small amounts of speci-fic mRNA in RCC samples. Inappropriate preparation of tissue samples can lead to invalid results due to tiny amounts DNA introduced through external contamina-tion. Optimal preservation, storing and handling of samples are important to avoid mRNA degradation. Despite ade-quate execution of the RT-PCR method, the interpretation of analysed mRNA involves a mixture of cells from different origin that includes tumour cells. The surgical procedure, with ligation of the blood supply during nephrectomy and time to excision of tumour samples, inevitable effects VEGF expression. However, previous studies have shown that when handling tissue samples, HIF-1α levels remain constant up to 60 min after clamping of the renal artery [43]. Moreover, no significant up-regulation of mRNA VEGF or changes in protein VEGF content has been demonstrated during 20 minutes of ischemic conditions in kidney tissue [231]. Although the patient sample sizes was small in Paper III we demonstrated that VEGF mRNA and VEGF-R1 mRNA levels were higher in CRCC compared with PRCC, and the appearances of diversity between VEGF mRNA and S-VEGF levels should be further examined. VEGF-R1 mRNA le-vels had no predictive information, which could be explained by endothelial cell activation of VEGF-R1 brought about by several growth factors, including VEGF-A, VEGF-B as well as PDGF released at different stages in RCC.

34
Paper IV Positive immunohistochemical (IHC)
VEGF staining was observed in both RCC and kidney cortex samples. Kidney cortex usually showed a strong IHC VEGF ex-pression in cytoplasm of tubular cells. While samples from RCC showed considerable variation in IHC VEGF expression on cell membranes as well as in cytoplasm of tumour cells. IHC VEGF expression at cell membrane was affected by the storage time of paraffin embedded tumour specimens. Hence, membranous VEGF expression was not further eva-luated. There were no differences in cytoplasm VEGF expressions between gender or different age groups. A signi-ficant correlation was found between VEGF expression in cytoplasm and the size of tumours. In tumours smaller than 7 cm, increasing VEGF expression was observed between stage I (pT1 N0 M0), stage III (pT3a-c N0-1 M0), and stage IV (N2 M1), but no such difference was found in VEGF expression at tumours larger than 7 cm. Tumours invading through the renal capsule (pT3a, N0-1, M0) had higher VEGF expression compared with tumours confined to kidney (pT1-2, N0, M0) or tumours invading veins only (pT3b-c, N0, M0). No difference in VEGF expression was recognized between differrent RCC types. In univariate ana-lysis, increased VEGF expression corre-lated with adverse survival in patients with CRCC. No correlation was found between VEGF expression and survival time in PRCC or in ChRCC. In multi-variate analysis the cytoplasmatic IHC VEGF expression was not an independent prognostic factor.
Comments on paper IV Immunohistochemistry allows identi-
fying also the localization of protein expressed within tissue sections. However, the outcome of immunohistochemical examination is dependent on the quality of the biopsy sample, presence of tissue necrosis, antigen loss due to delay in fixation of samples, type of fixative, dura-tion of fixation, processing of samples, choice of antibody, and sensitivity of detections method for antigen-antibody reaction [232]. Fixation prevents the degradation of tissues, preserves the mor-phology, and stabilizes cells and tissues. On the other hand, fixation causes a cross-linking within protein molecules, which alters the structure of the proteins, possi-bly altering the epitope and masking the antigen against potential antibody bin-ding. Heat-induced antigen retrieval may disrupt these cross-links but recovery of native molecule structure may depend on achieved temperature and length of incubation. Complete recovery of native molecule structure is particularly im-portant for monoclonal antibodies, which recognise a single epitope, whereas poly-clonal antibodies recognise a number of different epitopes. Polyclonal antibodies may express increased sensitivity to target but may also contain increased risk for cross-reactivity. Monoclonal antibodies on the other hand are targeted at a single epitope on the antigen and are therefore highly specific. However, a monoclonal antibody might fail to bind the epitope if the antigen is vulnerable and unstable due to the fixation process. It has been demon-strated that the antigenicity is inversely correlated to duration of exposure to formaldehyde [233]. Quantification of immunoreaction intensity is an error filled process, since no linear relationship exists between amount of antigen and the expressed antigen-antibody reaction. A

35
more valid form of quantification is determining the percentage of estimated antigen-antibody specific cellular reaction. Identifying hotspot areas and estimating relative area with immunoreactions is a highly operator-dependent process, and other investigators have emphasized the advantage of using the methods described by Chalkey [234]. In Paper IV, variation in results obtained from different methods was shown, but nevertheless a correlation between observations could be noted. Thus, the established VEGF expression in cytoplasm as being predictor for survival led us to further examine the role of VEGF location in RCC. Cytoplasmic VEGF was demonstrated to be of prognostic signi-ficance only in smaller tumours. Further-more, in solid tumours, several other growth factors have been proposed as candidate to persuade the growth when tumours reached a certain volume [235].
Study V
Both RCC and kidney cortex samples expressed immunoreactive VEGF bands. Samples from kidney cortex expressed VEGF189 and VEGF165 isoforms, but not VEGF121. Both VEGF189 and VEGF165
isoforms were strongly expressed in kid-ney cortex, compared to RCC samples. In RCC samples, VEGF189 had highest immunoreactive expression and VEGF165 intermediate, while VEGF121 was rarely detected. VEGF121 expression was con-sidered positive when a band was visually detected on the Western blot. There was considerable variation in VEGF189 ex-pression between the different RCC types, in contrary to VEGF165 expression, which was usually low. No differences in VEGF isoforms were found according to gender, age, or due to storage time of tumour samples. CRCC had lower levels of both
VEGF189 and VEGF165 isoforms compared with kidney cortex. There were no corre-lations between isoforms and tumour stage or grade in CRCC, although VEGF189 was verified as low in tumours with renal capsule invasion (pT3a, N0-1, M0) compared with those without invasion (pT1-2, N0, M0). VEGF121 was more frequently detected in smaller (pT1) and localized tumours (stage I-II) compared with lager (pT2) and advanced tumour stages (stage III-IV). PRCC showed also lower levels of VEGF189 and VEGF165 compared with kidney cortex. In PRCC, protein VEGF165 level correlated inversely with tumour size, and VEGF189 levels correlated inversely with tumour stage and grade. In ChRCC, protein level of VEGF189 was comparable to that in kidney cortex, while VEGF165 level was significantly decreased. Also in ChRCC, the VEGF189 levels were lower in advanced tumour stages (stage III-IV) compared with tumours confined to kidney (stage I-II). VEGF121 was not detected in chromophobe RCC. In univariate analysis, VEGF165 showed no predictive information. However, low VEGF189 levels were associated with shorter survival in PRCC. In Cox multi-variate analysis, the VEGF189 expression remained an independent prognostic factor in patients with PRCC, while TNM stage was the last variable to be omitted after stepwise elimination analysis.
Comments on paper V
The WB-technique is based on protein extractions separated in SDS-PAGE gel. An equal loading of samples is crucial when WB is used for quantification. Assessment of protein extract concen-tration was reproduced at least twice, and the loaded protein was controlled by

36
Ponceau-Red staining, and sample protein concentration re-assured through actin detection by WB. Each sample was analysed in duplicate, and in this paper with different blocking solutions, which were used due to the EU import restrict-tion of hoarse serum from New Zealand in 1995. Any discordance in duplicate results led to re-analysis with available blocking solutions. Advantages with WB-technique are the option to evaluate different VEGF protein isoform by their molecular weight separated in SDS-PAGE gel, as well as the possibility to quantify expression of each VEGF iso-form. A weakness with the WB method is that protein extractions are based on expression from a mixture of cells of different origin including some other than tumour cells. Therefore imprints were made to verify all samples in fact con-tained tumour tissue. The findings in this
paper support the idea that VEGF189 may be the primary isoform reflecting tumour angiogenesis. Although mRNA VEGF189 was barely detectable in Paper III, the high VEGF189 protein expression in tumour tissue can be interpreted that VEGF189 protein is produced and most likely stored locally in tumour cells. Expression of mRNA VEGF189 has been demonstrated in tumours stage T3-T4, and furthermore is associated with higher microvessel density in contrast to other isoforms [236]. Angiogenesis is induced by released VEGF and enhanced by increased VEGF receptor density on endothelial cells. In addition, angio-genesis is thought to be promoted by release of bioactive different growth factors such as VEGF165, VEGF189, and bFGF during proteolytic degradation of ECM [143, 237].

37
FUTURE DIRECTIONS
Molecular biomarkers are expected to have an important impact on future diagnosis, prognostication and selection of therapeutic targets for RCC. Several potential drugs appear promising to in-hibit tumour angiogenesis in RCC. These include targeting VEGF with monoclonal antibody (Bevacizumb) [238, 239] and inhibition of VEGF-R2 and PDGF-R (BAY 43-9006 / Sorafenib) [240, 241], or multitargeted tyrosine kinase inhibitor of VEGF-R1, VEGF-R2, Flt-3 and PDGF-R (SU11248) [240, 241], whereas Thalidomide [242] and TPN-470 [243] have provided less promising clinical results. Novel targeted therapies such as matrix metalo-proteinase inhibitors (AG3340 / Primo-mastat), (BB2516 / Marimstat), AE-941 / Neovastat) have undergone clinical tes-ting with positive results [244]. Unique approaches with the aim of selectively inhibiting angiogenesis is provoking apoptosis in tumour stimulated endo-thelial cells in order to undermine growth and support of existing tumour vessels and cause tumour cell death from ische-mia (ZD6126 / Combretastatin) [245]. Considering the number of different genetic defects in RCC, it is unlikely that a single drug will be effective in treatment of advanced RCC. More likely, these new angiogenesis inhibitors can be expected to be most effective when used as adjuvant therapy to surgery where risk for re-
currences of RCC exist or in combination with drugs having different mechanisms of action. Currently no prognostic bio-markers are available to independently validate the therapeutic effect in indivi-duals with RCC. These evaluations still depend on traditional clinical surveillance. The complexities of multiple different growth factors are not completely under-stood, and their role and relationship re-mains important as a subject of future investigation in RCC.
There are several issues, which need to be further investigated, as whether cancer cells have receptors for VEGF and what significance this existence of auto-crine effect might have. Do different VEGF isoforms have an auxiliary effect on potential target cells i.e. causing vascular permeability to macromolecules and oedema, hemodynamic alteration by vasodilatation, and incitation of angio-genesis? Despite evidence of vascular dilatation and increased permeability the effects and regulation are not clear in pathological angiogenesis. Does VEGF independently correlate to increased microvessel density, or are there other factors that counteract or counterpart VEGF? Hypoxy inducible factor (HIF) affects transcription level of VEGF, however do there exist other factors that could be considered as regulatory of VEGF in RCC?

38
Figure 7. Antiangiogenic drugs currently under evaluation in cancer therapy.

39
CONCLUSIONS
Paper I
In RCC, S-VEGF level was significantly correlated to tumour stage and grade. Increased levels of S-VEGF were associated with shorter survival. Although, S-VEGF did not remain as an independent prognostic factor in multivariate analysis, the levels of S-VEGF were found useful for the identification of patients with potentially progressive disease especially those with vein invasion.
Paper II
S-VEGF showed a stronger association to prognosis than blood platelets and leukocyte counts. Blood platelet count seems to be affected in patients with intercurrent chronic disease and long-term medication, and in this large group of patients blood platelets counts are not associated with survival. Blood leukocyte count had no prognostic value.
Paper III
No correlation was found between VEGF165 mRNA and S-VEGF levels. Both VEGF121 and VEGF165 mRNA isoforms and Flt-1 mRNA (VEGF-R1) levels were higher in CRCC compared with PRCC. The trend of decreased levels of isoforms VEGF121 and VEGF165 mRNA in locally advanced RCC indicates that initiation of angiogenic activity by VEGF might be an early event and during tumour progression other factors are more relevant.
Paper IV
IHC VEGF staining was observed at cell membrane and cytoplasm in RCC cells. There was no difference in cytoplasm VEGF expression between the different RCC types. Associations between expression of cytoplasm VEGF and tumour size as well as prognosis indicate the significance of VEGF in growth and progression. The expression of IHC VEGF in cytoplasm might be useful for identification of potentially progressive RCC but was not an independent prognostic factor in multivariate analysis.
Paper V
Different VEGF isoform patterns were found between the specific RCC types. VEGF165 protein expression had no predictive value for survival and was observed in low levels in RCC samples. The protein VEGF189 expression was associated with tumour stage and grade and was an independent prognostic factor in PRCC.

40
ACKNOWLEDGEMENTS
I wish to express my sincere gratitude to all those who have supported me in this work. Especially I thank: Börje Ljungberg, (my supervisor, for never failing support and who introduced me to the field of RCC), Professor at the Department of Surgical and Perioperative Sciences, Urology and Andrology Kjell Grankvist, (my cosupervisor, for contribution to discussions and new ideas), Professor at the Department of Medical Biosciences, Clinical Chemistry Torgny Rasmuson, (my cosupervisor and for valuable help with manuscripts), Assistant Professor at the Department of Radiation Sciences, Oncology Anders Bergh, (my cosupervisor, for guiding me through immunohistochemical evaluation), Professor at the Department of Medical Biosciences, Pathology Göran Landberg, (for introducing TMA to this thesis), Professor at Division of Pathology, Department of Laboratory Medicine, Lund University, Malmö University Hospital Radisa Tomic, (for providing me with good working conditions and skilful guidance in surgery), Chief surgeon and former Head of the Department of Urology Stina Häggström-Rudolfssson, (for introducing the RT-PCR to this thesis) Ph.D., Assistant at the Department of Surgical and Perioperative Sciences, Urology and Andrology Gudrun Lindh, (for carrying out the RT-PCR technique), Assistant at the Department of Surgical and Perioperative Sciences, Urology and Andrology Ylva Hedberg, (for advice and positive attitude in my first contact with lab facilities), Ph.D., Assistant at the Department of Medical Biosciences, Pathology

41
Britta Lindgren, (for carrying out the numerous slides and TMA to this thesis), Assistant at the Department of Medical Biosciences, Pathology Michael Haney, (for effective help in revising the English text), Ph.D., M.D. at Department of Surgical and Perioperative Science, Anaesthesiology and Intensive Care Medicine Solveig Isberg, (for taking care of economy and secretarial services), Secretary at the Department of Surgical and Perioperative Sciences, Urology and Andrology Veronica Berglund, Kerstin Almroth and Britt-Inger Dahlin (for helping me with categorize blood and tissue samples), Reseach Assistants at the Department of Urology My colleagues, all staff and friends at the Department of Urology, Umeå University Hospital, Umeå, Sweden This work was supported by the Swedish Cancer Society (4431-B01-02XBB), the Maud and Birger Gustavsson Foundation, and the Lions Research Foundation, Umeå University, Umeå, Sweden.

42
REFERENCES
1. Socialstyrelsen: Cancer Incidence in Sweden 2003. The National Board of Health and Welfare, Centre for Epidemiology, 2004.
2. Tsui KH, Shvarts O, Smith RB et al.: Prognostic indicators for renal cell carcinoma: a multivariate analysis of 643 patients using the revised 1997 TNM staging criteria. J Urol. 163: 1090-5; quiz 1295, 2000.
3. Lang H JD: Prognostic factors in renal cell carcinoma. EAU, Update Series. 1: 215-219, 2003.
4. Erber R, Thurnher A, Katsen AD et al.: Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. Faseb J. 18: 338-40, 2004.
5. Hicklin DJ and Ellis LM: Role of the vascular endothelial growth factor pathway in tumor growth and angio-genesis. J Clin Oncol. 23: 1011-27, 2005.
6. Jacqmin D, van Poppel H, Kirkali Z et al.: Renal cancer. Eur Urol. 39: 361-9, 2001.
7. Parkin DM: The global burden of cancer. Semin Cancer Biol. 8: 219-35, 1998.
8. Chow WH, Devesa SS, Warren JL et al.: Rising incidence of renal cell cancer in the United States. Jama. 281: 1628-31, 1999.
9. Homma Y, Kawabe K, Kitamura T et al.: Increased incidental detection and reduced mortality in renal cancer--recent retrospective analysis at eight institutions. Int J Urol. 2: 77-80, 1995.
10. Pantuck AJ, Zisman A and Belldegrun AS: The changing natural history of renal cell carcinoma. J Urol. 166: 1611-23, 2001.
11. McLaughlin JK, Lindblad P, Mellemgaard
A et al.: International renal-cell cancer study. I. Tobacco use. Int J Cancer. 60: 194-8, 1995.
12. Dhote R, Pellicer-Coeuret M, Thiounn N et al.: Risk factors for adult renal cell carcinoma: a systematic review and implications for prevention. BJU Int. 86: 20-7, 2000.
13. Lindblad P: Epidemiology of renal cell carcinoma. Scand J Surg. 93: 88-96, 2004.
14. Gnarra JR: von Hippel-Lindau gene mutations in human and rodent renal tumors--association with clear cell phenotype. J Natl Cancer Inst. 90: 1685-7, 1998.
15. Shiao YH, Rice JM, Anderson LM et al.: von Hippel-Lindau gene mutations in N-nitrosodimethylamine-induced rat renal epithelial tumors. J Natl Cancer Inst. 90: 1720-3, 1998.
16. Semenza JC, Ziogas A, Largent J et al.: Gene-environment interactions in renal cell carcinoma. Am J Epidemiol. 153: 851-9, 2001.
17. Hu J, Mao Y and White K: Overweight and obesity in adults and risk of renal cell carcinoma in Canada. Soz Praventivmed. 48: 178-85, 2003.
18. Bergstrom A, Hsieh CC, Lindblad P et al.: Obesity and renal cell cancer--a quantitative review. Br J Cancer. 85: 984-90, 2001.
19. Prineas RJ, Folsom AR, Zhang ZM et al.: Nutrition and other risk factors for renal cell carcinoma in post-menopausal women. Epidemiology. 8: 31-6, 1997.
20. Mellemgaard A, Lindblad P, Schlehofer B et al.: International renal-cell cancer study. III. Role of weight, height, physical activity, and use of amphetamines. Int J Cancer. 60: 350-4, 1995.
21. Boeing H, Schlehofer B and

43
Wahrendorf J: Diet, obesity and risk for renal cell carcinoma: results from a case control-study in Germany. Z Ernahrungswiss. 36: 3-11, 1997.
22. McLaughlin JK, Chow WH, Mandel JS et al.: International renal-cell cancer study. VIII. Role of diuretics, other anti-hypertensive medications and hypertension. Int J Cancer. 63: 216-21, 1995.
23. Chow WH, McLaughlin JK, Mandel JS et al.: Risk of renal cell cancer in relation to diuretics, antihypertensive drugs, and hypertension. Cancer Epidemiol Biomarkers Prev. 4: 327-31, 1995.
24. Yuan JM, Castelao JE, Gago-Dominguez M et al.: Hypertension, obesity and their medications in relation to renal cell carcinoma. Br J Cancer. 77: 1508-13, 1998.
25. McCredie M, Pommer W, McLaughlin JK et al.: International renal-cell cancer study. II. Analgesics. Int J Cancer. 60: 345-9, 1995.
26. Lindblad P, Mellemgaard A, Schlehofer B et al.: International renal-cell cancer study. V. Reproductive factors, gynecologic operations and exogenous hormones. Int J Cancer. 61: 192-8, 1995.
27. Mandel JS, McLaughlin JK, Schlehofer B et al.: International renal-cell cancer study. IV. Occupation. Int J Cancer. 61: 601-5, 1995.
28. Yu MC and Ross RK: Obesity, hypertension, and renal cancer. N Engl J Med. 344: 531-2, 2001.
29. Lindblad P, McLaughlin JK, Mellemgaard A et al.: Risk of kidney cancer among patients using analgesics and diuretics: a population-based cohort study. Int J Cancer. 55: 5-9, 1993.
30. Mendelsohn J and Baselga J: Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol. 21: 2787-99, 2003.
31. Green DR: A Myc-induced apoptosis pathway surfaces. Science. 278: 1246-7, 1997.
32. Olivero M, Valente G, Bardelli A et al.: Novel mutation in the ATP-binding site of the MET oncogene tyrosine kinase in a HPRCC family. Int J Cancer. 82: 640-3, 1999.
33. Jeffers M, Schmidt L, Nakaigawa N et al.: Activating mutations for the met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci U S A. 94: 11445-50, 1997.
34. Xiao GH, Jeffers M, Bellacosa A et al.: Anti-apoptotic signaling by hepatocyte growth factor/Met via the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase pathways. Proc Natl Acad Sci U S A. 98: 247-52, 2001.
35. Fan S, Wang JA, Yuan RQ et al.: Scatter factor protects epithelial and carcinoma cells against apoptosis induced by DNA-damaging agents. Oncogene. 17: 131-41, 1998.
36. Giordano S, Maffe A, Williams TA et al.: Different point mutations in the met oncogene elicit distinct biological properties. Faseb J. 14: 399-406, 2000.
37. Hedberg Y, Davoodi E, Roos G et al.: Cyclin-D1 expression in human renal-cell carcinoma. Int J Cancer. 84: 268-72, 1999.
38. Hedberg Y, Ljungberg B, Roos G et al.: Expression of cyclin D1, D3, E, and p27 in human renal cell carci-noma analysed by tissue microarray. Br J Cancer. 88: 1417-23, 2003.
39. Hedberg Y, Roos G, Ljungberg B et al.: Cyclin D3 protein content in human renal cell carcinoma in relation to cyclin D1 and clinico-pathological parameters. Acta Oncol. 41: 175-81, 2002.
40. Hedberg Y, Davoodi E, Ljungberg B et al.: Cyclin E and p27 protein content in human renal cell carcinoma: clinical outcome and associations with cyclin D. Int J

44
Cancer. 102: 601-7, 2002. 41. Rioux-Leclercq N, Turlin B, Bansard
J et al.: Value of immunohistochemical Ki-67 and p53 determinations as predictive factors of outcome in renal cell carcinoma. Urology. 55: 501-5, 2000.
42. Ljungberg B, Bozoky B, Kovacs G et al.: p53 expression in correlation to clinical outcome in patients with renal cell carcinoma. Scand J Urol Nephrol. 35: 15-20, 2001.
43. Wiesener MS, Munchenhagen PM, Berger I et al.: Constitutive activation of hypoxia-inducible genes related to overexpression of hypoxia-inducible factor-1alpha in clear cell renal carcinomas. Cancer Res. 61: 5215-22, 2001.
44. Devarajan P, De Leon M, Talasazan F et al.: The von Hippel-Lindau gene product inhibits renal cell apoptosis via Bcl-2-dependent pathways. J Biol Chem. 276: 40599-605, 2001.
45. Kovacs G, Akhtar M, Beckwith BJ et al.: The Heidelberg classification of renal cell tumours. J Pathol. 183: 131-3, 1997.
46. Linehan WM, Walther MM and Zbar B: The genetic basis of cancer of the kidney. J Urol. 170: 2163-72, 2003.
47. Zbar B, Klausner R and Linehan WM: Studying cancer families to identify kidney cancer genes. Annu Rev Med. 54: 217-33, 2003.
48. Turner KJ: Inherited renal cancer. BJU Int. 86: 155-64;quiz i, 2000.
49. Hemminki K, Li X and Czene K: Familial risk of urological cancers: data for clinical counseling. World J Urol. 21: 377-81, 2004.
50. Kiuru M, Kujala M and Aittomaki K: Inherited forms of renal cell carcinoma. Scand J Surg. 93: 103-11, 2004.
51. Storkel S, Eble JN, Adlakha K et al.: Classification of renal cell carcinoma: Workgroup No. 1. Union Internationale Contre le Cancer (UICC) and the
American Joint Committee on Cancer (AJCC). Cancer. 80: 987-9, 1997.
52. Maranchie JK and Linehan WM: Genetic disorders and renal cell carcinoma. Urol Clin North Am. 30: 133-41, 2003.
53. Polascik TJ, Bostwick DG and Cairns P: Molecular genetics and histo-pathologic features of adult distal nephron tumors. Urology. 60: 941-6, 2002.
54. Wu SQ, Hafez GR, Xing W et al.: The correlation between the loss of chromosome 14q with histologic tumor grade, pathologic stage, and outcome of patients with nonpapillary renal cell carcinoma. Cancer. 77: 1154-60, 1996.
55. Junker K, Weirich G, Amin MB et al.: Genetic subtyping of renal cell carcinoma by comparative genomic hybridization. Recent Results Cancer Res. 162: 169-75, 2003.
56. Cussenot O and Fournier G: [Genetics and urology]. Prog Urol. 10: 681-1097, 2000.
57. Hemminki K and Li X: Age-specific familial risks for renal cell carcinoma with evidence on recessive heritable effects. Kidney Int. 65: 2298-302, 2004.
58. Woodward ER, Clifford SC, Astuti D et al.: Familial clear cell renal cell carcinoma (FCRC): clinical features and mutation analysis of the VHL, MET, and CUL2 candidate genes. J Med Genet. 37: 348-53, 2000.
59. Teh BT, Giraud S, Sari NF et al.: Familial non-VHL non-papillary clear-cell renal cancer. Lancet. 349: 848-9, 1997.
60. Takahashi M, Kahnoski R, Gross D et al.: Familial adult renal neoplasia. J Med Genet. 39: 1-5, 2002.
61. Lonser RR, Glenn GM, Walther M et al.: von Hippel-Lindau disease. Lancet. 361: 2059-67, 2003.
62. Maher ER, Iselius L, Yates JR et al.: Von Hippel-Lindau disease: a genetic

45
study. J Med Genet. 28: 443-7, 1991. 63. Grignon DJ and Eble JN: Papillary
and metanephric adenomas of the kidney. Semin Diagn Pathol. 15: 41-53, 1998.
64. Zbar B, Glenn G, Lubensky I et al.: Hereditary papillary renal cell carcinoma: clinical studies in 10 families. J Urol. 153: 907-12, 1995.
65. Zbar B, Tory K, Merino M et al.: Hereditary papillary renal cell carcinoma. J Urol. 151: 561-6, 1994.
66. Tomlinson IP, Alam NA, Rowan AJ et al.: Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 30: 406-10, 2002.
67. Kiuru M, Launonen V, Hietala M et al.: Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 159: 825-9, 2001.
68. Delahunt B, Eble JN, McCredie MR et al.: Morphologic typing of papillary renal cell carcinoma: comparison of growth kinetics and patient survival in 66 cases. Hum Pathol. 32: 590-5, 2001.
69. Allory Y, Ouazana D, Boucher E et al.: Papillary renal cell carcinoma. Prognostic value of morphological subtypes in a clinicopathologic study of 43 cases. Virchows Arch. 442: 336-42, 2003.
70. Renshaw AA, Henske EP, Loughlin KR et al.: Aggressive variants of chromophobe renal cell carcinoma. Cancer. 78: 1756-61, 1996.
71. Kuroda N, Toi M, Hiroi M et al.: Review of chromophobe renal cell carcinoma with focus on clinical and pathobiological aspects. Histol Histopathol. 18: 165-71, 2003.
72. Pavlovich CP, Walther MM, Eyler RA et al.: Renal tumors in the Birt-Hogg-Dube syndrome. Am J Surg Pathol. 26: 1542-52, 2002.
73. Schmidt LS, Warren MB, Nickerson ML et al.: Birt-Hogg-Dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 69: 876-82, 2001.
74. Nickerson ML, Warren MB, Toro JR et al.: Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2: 157-64, 2002.
75. Ljungberg B, Grankvist K and Rasmuson T: Serum acute phase reactants and prognosis in renal cell carcinoma. Cancer. 76: 1435-9, 1995.
76. Ljungberg B, Landberg G and Alamdari FI: Factors of importance for prediction of survival in patients with metastatic renal cell carcinoma, treated with or without nephrectomy. Scand J Urol Nephrol. 34: 246-51, 2000.
77. Mejean A, Oudard S and Thiounn N: Prognostic factors of renal cell carcinoma. J Urol. 169: 821-7, 2003.
78. Kirkali Z and Lekili M: Renal cell carcinoma: new prognostic factors? Curr Opin Urol. 13: 433-8, 2003.
79. Guinan P, Sobin LH, Algaba F et al.: TNM staging of renal cell carcinoma: Workgroup No. 3. Union International Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer. 80: 992-3, 1997.
80. Frank I, Blute ML, Cheville JC et al.: An outcome prediction model for patients with clear cell renal cell carcinoma treated with radical nephrectomy based on tumor stage, size, grade and necrosis: the SSIGN score. J Urol. 168: 2395-400, 2002.
81. Gettman MT, Blute ML, Spotts B et al.: Pathologic staging of renal cell carcinoma: significance of tumor classification with the 1997 TNM staging system. Cancer. 91: 354-61, 2001.

46
82. Ficarra V, Righetti R, Pilloni S et al.: Prognostic factors in patients with renal cell carcinoma: retrospective analysis of 675 cases. Eur Urol. 41: 190-8, 2002.
83. Ljungberg B, Alamdari FI, Rasmuson T et al.: Follow-up guidelines for nonmetastatic renal cell carcinoma based on the occurrence of metastases after radical nephrectomy. BJU Int. 84: 405-11, 1999.
84. Dinney CP, Awad SA, Gajewski JB et al.: Analysis of imaging modalities, staging systems, and prognostic indicators for renal cell carcinoma. Urology. 39: 122-9, 1992.
85. Sobin LHaWC: TNM Classification of Malignant Tumours. Union International Contre le Cancer (UICC). Sixth Edition, 2002.
86. Kinouchi T, Saiki S, Meguro N et al.: Impact of tumor size on the clinical outcomes of patients with Robson State I renal cell carcinoma. Cancer. 85: 689-95, 1999.
87. Belldegrun A, Tsui KH, deKernion JB et al.: Efficacy of nephron-sparing surgery for renal cell carcinoma: analysis based on the new 1997 tumor-node-metastasis staging system. J Clin Oncol. 17: 2868-75, 1999.
88. Hafez KS, Fergany AF and Novick AC: Nephron sparing surgery for localized renal cell carcinoma: impact of tumor size on patient survival, tumor recurrence and TNM staging. J Urol. 162: 1930-3, 1999.
89. Weiss LM, Gelb AB and Medeiros LJ: Adult renal epithelial neoplasms. Am J Clin Pathol. 103: 624-35, 1995.
90. Amin MB, Corless CL, Renshaw AA et al.: Papillary (chromophil) renal cell carcinoma: histomorphologic characteristics and evaluation of conventional pathologic prognostic parameters in 62 cases. Am J Surg Pathol. 21: 621-35, 1997.
91. Fuhrman SA, Lasky LC and Limas C: Prognostic significance of morpho-
logic parameters in renal cell carcinoma. Am J Surg Pathol. 6: 655-63, 1982.
92. Lohse CM, Blute ML, Zincke H et al.: Comparison of standardized and nonstandardized nuclear grade of renal cell carcinoma to predict outcome among 2,042 patients. Am J Clin Pathol. 118: 877-86, 2002.
93. Ficarra V, Righetti R, Martignoni G et al.: Prognostic value of renal cell carcinoma nuclear grading: multi-variate analysis of 333 cases. Urol Int. 67: 130-4, 2001.
94. Bretheau D, Lechevallier E, de Fromont M et al.: Prognostic value of nuclear grade of renal cell carcinoma. Cancer. 76: 2543-9, 1995.
95. Fiori E, De Cesare A, Galati G et al.: Prognostic significance of primary-tumor extension, stage and grade of nuclear differentiation in patients with renal cell carcinoma. J Exp Clin Cancer Res. 21: 229-32, 2002.
96. Skinner DG, Colvin RB, Vermillion CD et al.: Diagnosis and management of renal cell carcinoma. A clinical and pathologic study of 309 cases. Cancer. 28: 1165-77, 1971.
97. Ficarra V, Martignoni G, Maffei N et al.: Original and reviewed nuclear grading according to the Fuhrman system: a multivariate analysis of 388 patients with conventional renal cell carcinoma. Cancer. 103: 68-75, 2005.
98. Goldstein NS: The current state of renal cell carcinoma grading. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer. 80: 977-80, 1997.
99. Goldstein NS: Grading of renal cell carcinoma. Urol Clin North Am. 26: 637-42, vii, 1999.
100. True LD: The time for accurate Fuhrman grading of renal cell carcinomas has arrived. Am J Clin Pathol. 118: 827-9, 2002.
101. Zambrano NR, Lubensky IA, Merino

47
MJ et al.: Histopathology and molecular genetics of renal tumors toward unification of a classification system. J Urol. 162: 1246-58, 1999.
102. Saito S, Orikasa S, Ohyama C et al.: Changes in glycolipids in human renal-cell carcinoma and their clinical significance. Int J Cancer. 49: 329-34, 1991.
103. Steinberg P, Storkel S, Oesch F et al.: Carbohydrate metabolism in human renal clear cell carcinomas. Lab Invest. 67: 506-11, 1992.
104. Farrow GM, Harrison EG, Jr. and Utz DC: Sarcomas and sarcomatoid and mixed malignant tumors of the kidney in adults. 3. Cancer. 22: 556-63, 1968.
105. Eble JN: Recommendations for exa-mining and reporting tumor-bearing kidney specimens from adults. Semin Diagn Pathol. 15: 77-82, 1998.
106. Storkel S, Keymer R, Steinbach F and Thoenes W: Reaction patterns of tumor infiltrating lymphocytes in different renal cell carcinomas and oncocytomas. Prog Clin Biol Res. 378: 217-23, 1992.
107. Renshaw AA and Corless CL: Papillary renal cell carcinoma. Histology and immunohistochemistry. Am J Surg Pathol. 19: 842-9, 1995.
108. Matsumoto T, Iijima T, Kuwabara N et al.: Papillary renal cell carcinoma. Report of two cases. Acta Pathol Jpn. 42: 298-303, 1992.
109. Delahunt B and Eble JN: Papillary renal cell carcinoma: a clinico-pathologic and immunohistochemical study of 105 tumors. Mod Pathol. 10: 537-44, 1997.
110. Thoenes W, Storkel S, Rumpelt HJ et al.: Chromophobe cell renal car-cinoma and its variants--a report on 32 cases. J Pathol. 155: 277-87, 1988.
111. Thoenes W, Storkel S and Rumpelt HJ: Human chromophobe cell renal carcinoma. Virchows Arch B Cell Pathol Incl Mol Pathol. 48: 207-17,
1985. 112. Baer SC, Ro JY, Ordonez NG et al.:
Sarcomatoid collecting duct carcinoma: a clinicopathologic and immuno-histochemical study of five cases. Hum Pathol. 24: 1017-22, 1993.
113. Srigley JR and Eble JN: Collecting duct carcinoma of kidney. Semin Diagn Pathol. 15: 54-67, 1998.
114. Cheville JC, Lohse CM, Zincke H et al.: Comparisons of outcome and prognostic features among histologic subtypes of renal cell carcinoma. Am J Surg Pathol. 27: 612-24, 2003.
115. Beck SD, Patel MI, Snyder ME et al.: Effect of papillary and chromophobe cell type on disease-free survival after nephrectomy for renal cell carcinoma. Ann Surg Oncol. 11: 71-7, 2004.
116. Ljungberg B, Alamdari FI, Stenling R et al.: Prognostic significance of the Heidelberg classification of renal cell carcinoma. Eur Urol. 36: 565-9, 1999.
117. Delahunt B: Histopathologic prog-nostic indicators for renal cell carcinoma. Semin Diagn Pathol. 15: 68-76, 1998.
118. Gelb AB: Renal cell carcinoma: current prognostic factors. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer. 80: 981-6, 1997.
119. Amin MB, Tamboli P, Javidan J et al.: Prognostic impact of histologic subtyping of adult renal epithelial neoplasms: an experience of 405 cases. Am J Surg Pathol. 26: 281-91, 2002.
120. Moch H, Gasser T, Amin MB et al.: Prognostic utility of the recently recommended histologic classification and revised TNM staging system of renal cell carcinoma: a Swiss experience with 588 tumors. Cancer. 89: 604-14, 2000.
121. Upton MP, Parker RA, Youmans A et al.: Histologic predictors of renal cell carcinoma response to interleukin-2-

48
based therapy. J Immunother. 28: 488-95, 2005.
122. Folkman J: Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 175: 409-16, 1972.
123. Carmeliet P and Jain RK: Angio-genesis in cancer and other diseases. Nature. 407: 249-57, 2000.
124. Liotta LA, Steeg PS and Stetler-Stevenson WG: Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell. 64: 327-36, 1991.
125. Fleming S: Renal cancer genetics: von Hippel Lindau and other syndromes. Int J Dev Biol. 43: 469-71, 1999.
126. Ferrara N: Role of vascular endo-thelial growth factor in the regulation of angiogenesis. Kidney Int. 56: 794-814, 1999.
127. Yoshida A, Anand-Apte B and Zetter BR: Differential endothelial migration and proliferation to basic fibroblast growth factor and vascular endothelial growth factor. Growth Factors. 13: 57-64, 1996.
128. Ferrara N and Henzel WJ: Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 161: 851-8, 1989.
129. Senger DR, Galli SJ, Dvorak AM et al.: Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 219: 983-5, 1983.
130. Senger DR, Connolly DT, Van de Water L et al.: Purification and NH2-terminal amino acid sequence of guinea pig tumor-secreted vascular permeability factor. Cancer Res. 50: 1774-8, 1990.
131. Ferrara N: Molecular and biological properties of vascular endothelial growth factor. J Mol Med. 77: 527-43, 1999.
132. Maglione D, Guerriero V, Viglietto G
et al.: Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci U S A. 88: 9267-71, 1991.
133. Leung DW, Cachianes G, Kuang WJ et al.: Vascular endothelial growth factor is a secreted angiogenic mito-gen. Science. 246: 1306-9, 1989.
134. Keck PJ, Hauser SD, Krivi G et al.: Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 246: 1309-12, 1989.
135. Tischer E, Mitchell R, Hartman T et al.: The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 266: 11947-54, 1991.
136. Poltorak Z, Cohen T, Sivan R et al.: VEGF145, a secreted vascular endo-thelial growth factor isoform that binds to extracellular matrix. J Biol Chem. 272: 7151-8, 1997.
137. Ferrara N: Vascular endothelial growth factor. Eur J Cancer. 32A: 2413-22, 1996.
138. Bates DO and Harper SJ: Regulation of vascular permeability by vascular endothelial growth factors. Vascul Pharmacol. 39: 225-37, 2002.
139. Lei J, Jiang A and Pei D: Identification and characterization of a new splicing variant of vascular endothelial growth factor: VEGF183. Biochim Biophys Acta. 1443: 400-6, 1998.
140. Vincenti V, Cassano C, Rocchi M et al.: Assignment of the vascular endo-thelial growth factor gene to human chromosome 6p21.3. Circulation. 93: 1493-5, 1996.
141. Bates DO, Cui TG, Doughty JM et al.: VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 62: 4123-31, 2002.
142. Potgens AJ, Lubsen NH, van Altena

49
MC et al.: Covalent dimerization of vascular permeability factor/vascular endothelial growth factor is essential for its biological activity. Evidence from Cys to Ser mutations. J Biol Chem. 269: 32879-85, 1994.
143. Houck KA, Leung DW, Rowland AM et al.: Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 267: 26031-7, 1992.
144. Soker S, Takashima S, Miao HQ et al.: Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 92: 735-45, 1998.
145. Keyt BA, Berleau LT, Nguyen HV et al.: The carboxyl-terminal domain (111-165) of vascular endothelial growth factor is critical for its mitogenic potency. J Biol Chem. 271: 7788-95, 1996.
146. Shweiki D, Itin A, Soffer D et al.: Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 359: 843-5, 1992.
147. Dantz D, Bewersdorf J, Fruehwald-Schultes B et al.: Vascular endo-thelial growth factor: a novel endocrine defensive response to hypoglycemia. J Clin Endocrinol Metab. 87: 835-40, 2002.
148. Jackson JR, Minton JA, Ho ML et al.: Expression of vascular endothelial growth factor in synovial fibroblasts is induced by hypoxia and interleukin 1beta. J Rheumatol. 24: 1253-9, 1997.
149. Tsai JC, Goldman CK and Gillespie GY: Vascular endothelial growth factor in human glioma cell lines: induced secretion by EGF, PDGF-BB, and bFGF. J Neurosurg. 82: 864-73, 1995.
150. Bos R, van Diest PJ, de Jong JS et al.: Hypoxia-inducible factor-1alpha is associated with angiogenesis, and
expression of bFGF, PDGF-BB, and EGFR in invasive breast cancer. Histopathology. 46: 31-6, 2005.
151. Beckner ME: Factors promoting tumor angiogenesis. Cancer Invest. 17: 594-623, 1999.
152. Punglia RS, Lu M, Hsu J et al.: Regulation of vascular endothelial growth factor expression by insulin-like growth factor I. Diabetes. 46: 1619-26, 1997.
153. Parker A, Cheville JC, Lohse C et al.: Expression of insulin-like growth factor I receptor and survival in patients with clear cell renal cell carcinoma. J Urol. 170: 420-4, 2003.
154. Rak J, Filmus J, Finkenzeller G et al.: Oncogenes as inducers of tumor angiogenesis. Cancer Metastasis Rev. 14: 263-77, 1995.
155. Kieser A, Weich HA, Brandner G et al.: Mutant p53 potentiates protein kinase C induction of vascular endothelial growth factor expression. Oncogene. 9: 963-9, 1994.
156. Aaltomaa S, Lipponen P, Ala-Opas M et al.: Expression of cyclins A and D and p21(waf1/cip1) proteins in renal cell cancer and their relation to clinicopathological variables and patient survival. Br J Cancer. 80: 2001-7, 1999.
157. Hedberg Y, Ljungberg B, Roos G et al.: Retinoblastoma protein in human renal cell carcinoma in relation to alterations in G1/S regulatory proteins. Int J Cancer. 109: 189-93, 2004.
158. Vasavada SP, Novick AC and Williams BR: P53, bcl-2, and Bax expression in renal cell carcinoma. Urology. 51: 1057-61, 1998.
159. Tomasino RM, Morello V, Tralongo V et al.: p53 expression in human renal cell carcinoma: an immuno-histochemical study and a literature outline of the cytogenetic character-rization. Pathologica. 86: 227-33, 1994.

50
160. Levy AP, Levy NS, Iliopoulos O et al.: Regulation of vascular endothelial growth factor by hypoxia and its modulation by the von Hippel-Lindau tumor suppressor gene. Kidney Int. 51: 575-8, 1997.
161. Lazar-Molnar E, Hegyesi H, Toth S et al.: Autocrine and paracrine regulation by cytokines and growth factors in melanoma. Cytokine. 12: 547-54, 2000.
162. de Vries C, Escobedo JA, Ueno H et al.: The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 255: 989-91, 1992.
163. Ferrara N, Carver-Moore K, Chen H et al.: Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 380: 439-42, 1996.
164. Terman BI, Dougher-Vermazen M, Carrion ME et al.: Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun. 187: 1579-86, 1992.
165. Quinn TP, Peters KG, De Vries C et al.: Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium. Proc Natl Acad Sci U S A. 90: 7533-7, 1993.
166. Joukov V, Kaipainen A, Jeltsch M et al.: Vascular endothelial growth factors VEGF-B and VEGF-C. J Cell Physiol. 173: 211-5, 1997.
167. Millauer B, Wizigmann-Voos S, Schnurch H et al.: High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell. 72: 835-46, 1993.
168. Eriksson U and Alitalo K: Structure, expression and receptor-binding pro-perties of novel vascular endothelial growth factors. Curr Top Microbiol Immunol. 237: 41-57, 1999.
169. Barleon B, Sozzani S, Zhou D et al.:
Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 87: 3336-43, 1996.
170. Waltenberger J, Claesson-Welsh L, Siegbahn A et al.: Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 269: 26988-95, 1994.
171. Park JE, Chen HH, Winer J et al.: Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem. 269: 25646-54, 1994.
172. Olofsson B, Pajusola K, Kaipainen A et al.: Vascular endothelial growth factor B, a novel growth factor for endothelial cells. Proc Natl Acad Sci U S A. 93: 2576-81, 1996.
173. Achen MG, Jeltsch M, Kukk E et al.: Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci U S A. 95: 548-53, 1998.
174. Polverini PJ: How the extracellular matrix and macrophages contribute to angiogenesis-dependent diseases. Eur J Cancer. 32A: 2430-7, 1996.
175. Cirri L, Donnini S, Morbidelli L et al.: Endostatin: a promising drug for antiangiogenic therapy. Int J Biol Markers. 14: 263-7, 1999.
176. Brooks PC, Stromblad S, Sanders LC et al.: Localization of matrix metallo-proteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 85: 683-93, 1996.
177. Kandel J, Bossy-Wetzel E, Radvanyi F et al.: Neovascularization is asso-ciated with a switch to the export of bFGF in the multistep development of fibrosarcoma. Cell. 66: 1095-104, 1991.

51
178. Fang J, Yan L, Shing Y et al.: HIF-1alpha-mediated up-regulation of vascular endothelial growth factor, independent of basic fibroblast growth factor, is important in the switch to the angiogenic phenotype during early tumorigenesis. Cancer Res. 61: 5731-5, 2001.
179. Bussolino F, Albini A, Camussi G et al.: Role of soluble mediators in angiogenesis. Eur J Cancer. 32A: 2401-12, 1996.
180. O'Brien TS and Harris AL: Angio-genesis in urological malignancy. Br J Urol. 76: 675-82, 1995.
181. Folkman J, Klagsbrun M, Sasse J et al.: A heparin-binding angiogenic protein--basic fibroblast growth factor--is stored within basement membrane. Am J Pathol. 130: 393-400, 1988.
182. Pepper MS, Ferrara N, Orci L et al.: Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem Biophys Res Commun. 189: 824-31, 1992.
183. Duensing S, Grosse J and Atzpodien J: Increased serum levels of basic fibroblast growth factor (bFGF) are associated with progressive lung metastases in advanced renal cell carcinoma patients. Anticancer Res. 15: 2331-3, 1995.
184. Wechsel HW, Bichler KH, Feil G et al.: Renal cell carcinoma: relevance of angiogenetic factors. Anticancer Res. 19: 1537-40, 1999.
185. Fujimoto K, Ichimori Y, Kakizoe T et al.: Increased serum levels of basic fibroblast growth factor in patients with renal cell carcinoma. Biochem Biophys Res Commun. 180: 386-92, 1991.
186. Fujimoto K, Ichimori Y, Yamaguchi H et al.: Basic fibroblast growth factor as a candidate tumor marker for renal cell carcinoma. Jpn J Cancer
Res. 86: 182-6, 1995. 187. Takahashi A, Sasaki H, Kim SJ et al.:
Markedly increased amounts of messenger RNAs for vascular endo-thelial growth factor and placenta growth factor in renal cell carcinoma associated with angiogenesis. Cancer Res. 54: 4233-7, 1994.
188. Singh RK, Llansa N, Bucana CD et al.: Cell density-dependent regulation of basic fibroblast growth factor expression in human renal cell carcinoma cells. Cell Growth Differ. 7: 397-404, 1996.
189. Nanus DM, Schmitz-Drager BJ, Motzer RJ et al.: Expression of basic fibroblast growth factor in primary human renal tumors: correlation with poor survival. J Natl Cancer Inst. 85: 1597-9, 1993.
190. Heldin CH: Structural and functional studies on platelet-derived growth factor. Embo J. 11: 4251-9, 1992.
191. Ostman A and Heldin CH: Involve-ment of platelet-derived growth factor in disease: development of specific antagonists. Adv Cancer Res. 80: 1-38, 2001.
192. Betsholtz C and Raines EW: Platelet-derived growth factor: a key regulator of connective tissue cells in embryo-genesis and pathogenesis. Kidney Int. 51: 1361-9, 1997.
193. Kovacs G: Papillary renal cell carcinoma. A morphologic and cyto-genetic study of 11 cases. Am J Pathol. 134: 27-34, 1989.
194. Kovacs G: Molecular differential pathology of renal cell tumours. Histopathology. 22: 1-8, 1993.
195. Besa EC: Physiological changes in blood volume. CRC Crit Rev Clin Lab Sci. 6: 67-79, 1975.
196. Ljungberg B, Stenling R and Roos G: DNA content in renal cell carcinoma with reference to tumor hetero-geneity. Cancer. 56: 503-8, 1985.
197. Häggström S. Vascular endothelial growth factor and prostate cancer.

52
Umeå University Medical Disser-tations. New Series No. 666, ISSN 0346-6612, ISBN 91-7191-832-9. Umeå 2000.
198. Houck KA, Ferrara N, Winer J et al.: The vascular endothelial growth factor family: identification of a fourth molecular species and characte-rization of alternative splicing of RNA. Mol Endocrinol. 5: 1806-14, 1991.
199. Hormbrey E, Gillespie P, Turner K et al.: A critical review of vascular endothelial growth factor (VEGF) analysis in peripheral blood: is the current literature meaningful? Clin Exp Metastasis. 19: 651-63, 2002.
200. Banks RE, Forbes MA, Kinsey SE et al.: Release of the angiogenic cytokine vascular endothelial growth factor (VEGF) from platelets: significance for VEGF measurements and cancer biology. Br J Cancer. 77: 956-64, 1998.
201. Jelkmann W: Pitfalls in the measure-ment of circulating vascular endo-thelial growth factor. Clin Chem. 47: 617-23, 2001.
202. Salven P, Orpana A and Joensuu H: Leukocytes and platelets of patients with cancer contain high levels of vascular endothelial growth factor. Clin Cancer Res. 5: 487-91, 1999.
203. Webb NJ, Bottomley MJ, Watson CJ et al.: Vascular endothelial growth factor (VEGF) is released from platelets during blood clotting: implications for measurement of circulating VEGF levels in clinical disease. Clin Sci (Lond). 94: 395-404, 1998.
204. Maloney JP, Silliman CC, Ambruso DR et al.: In vitro release of vascular endothelial growth factor during platelet aggregation. Am J Physiol. 275: H1054-61, 1998.
205. Lee JK, Hong YJ, Han CJ et al.: Clinical usefulness of serum and plasma vascular endothelial growth
factor in cancer patients: which is the optimal specimen? Int J Oncol. 17: 149-52, 2000.
206. Adams J, Carder PJ, Downey S et al.: Vascular endothelial growth factor (VEGF) in breast cancer: comparison of plasma, serum, and tissue VEGF and microvessel density and effects of tamoxifen. Cancer Res. 60: 2898-905, 2000.
207. Gunsilius E, Petzer A, Stockhammer G et al.: Thrombocytes are the major source for soluble vascular endo-thelial growth factor in peripheral blood. Oncology. 58: 169-74, 2000.
208. Salgado R, Vermeulen PB, Benoy I et al.: Platelet number and interleukin-6 correlate with VEGF but not with bFGF serum levels of advanced cancer patients. Br J Cancer. 80: 892-7, 1999.
209. Blay JY, Favrot M, Rossi JF et al.: Role of interleukin-6 in para-neoplastic thrombocytosis. Blood. 82: 2261-2, 1993.
210. Harker LA and Slichter SJ: Platelet and fibrinogen consumption in man. N Engl J Med. 287: 999-1005, 1972.
211. Verheul HM, Hoekman K, Lupu F et al.: Platelet and coagulation acti-vation with vascular endothelial growth factor generation in soft tissue sarcomas. Clin Cancer Res. 6: 166-71, 2000.
212. Dvorak HF, Nagy JA, Berse B et al.: Vascular permeability factor, fibrin, and the pathogenesis of tumor stroma formation. Ann N Y Acad Sci. 667: 101-11, 1992.
213. Dvorak HF, Brown LF, Detmar M et al.: Vascular permeability factor/vascular endothelial growth factor, micro-vascular hyperpermeability, and angiogenesis. Am J Pathol. 146: 1029-39, 1995.
214. Verheul HM and Pinedo HM: Tumor Growth: A Putative Role for Platelets? Oncologist. 3: II, 1998.
215. Verheul HM, Hoekman K, Luykx-de

53
Bakker S et al.: Platelet: transporter of vascular endothelial growth factor. Clin Cancer Res. 3: 2187-90, 1997.
216. Pinedo HM, Verheul HM, D'Amato RJ et al.: Involvement of platelets in tumour angiogenesis? Lancet. 352: 1775-7, 1998.
217. Sulzbacher I, Birner P, Traxler M et al.: Expression of platelet-derived growth factor-alpha alpha receptor is associated with tumor progression in clear cell renal cell carcinoma. Am J Clin Pathol. 120: 107-12, 2003.
218. Yamamoto Y, Toi M, Kondo S et al.: Concentrations of vascular endo-thelial growth factor in the sera of normal controls and cancer patients. Clin Cancer Res. 2: 821-6, 1996.
219. Ferrara N, Houck KA, Jakeman LB et al.: The vascular endothelial growth factor family of polypeptides. J Cell Biochem. 47: 211-8, 1991.
220. Symbas NP, Townsend MF, El-Galley R et al.: Poor prognosis asso-ciated with thrombocytosis in patients with renal cell carcinoma. BJU Int. 86: 203-7, 2000.
221. George JN and Shattil SJ: The clinical importance of acquired ab-normalities of platelet function. N Engl J Med. 324: 27-39, 1991.
222. Dworsky R, Paganini-Hill A, Ducey B et al.: Lymphocyte immuno-phenotyping in an elderly population: age, sex and medication effects--a flow cytometry study. Mech Ageing Dev. 48: 255-66, 1989.
223. Allan RN and Alexander MK: A sex difference in the leucocyte count. J Clin Pathol. 21: 691-4, 1968.
224. Polednak AP: Age changes in diffe-rential leukocyte count among female adults. Hum Biol. 50: 301-11, 1978.
225. Tavani A and La Vecchia C: Epi-demiology of renal-cell carcinoma. J Nephrol. 10: 93-106, 1997.
226. Kreiger N, Marrett LD, Dodds L et al.: Risk factors for renal cell car-cinoma: results of a population-based
case-control study. Cancer Causes Control. 4: 101-10, 1993.
227. Kantor AL, Meigs JW, Heston JF et al.: Epidemiology of renal cell car-cinoma in Connecticut, 1935-1973. J Natl Cancer Inst. 57: 495-500, 1976.
228. Folkman J: The role of angiogenesis in tumor growth. Semin Cancer Biol. 3: 65-71, 1992.
229. Sato K, Tsuchiya N, Sasaki R et al.: Increased serum levels of vascular endothelial growth factor in patients with renal cell carcinoma. Jpn J Cancer Res. 90: 874-9, 1999.
230. Baccala AA, Zhong H, Clift SM et al.: Serum vascular endothelial growth factor is a candidate bio-marker of metastatic tumor response to ex vivo gene therapy of renal cell cancer. Urology. 51: 327-32, 1998.
231. Kanellis J, Mudge SJ, Fraser S et al.: Redistribution of cytoplasmic VEGF to the basolateral aspect of renal tubular cells in ischemia-reperfusion injury. Kidney Int. 57: 2445-56, 2000.
232. Leong AS: Pitfalls in diagnostic immunohistology. Adv Anat Pathol. 11: 86-93, 2004.
233. Leong AS and Gilham PN: The effects of progressive formaldehyde fixation on the preservation of tissue antigens. Pathology. 21: 266-8, 1989.
234. Vermeulen PB, Gasparini G, Fox SB et al.: Second international consensus on the methodology and criteria of evaluation of angiogenesis quanti-fication in solid human tumours. Eur J Cancer. 38: 1564-79, 2002.
235. Yoshiji H, Harris SR and Thorgeirsson UP: Vascular endo-thelial growth factor is essential for initial but not continued in vivo growth of human breast carcinoma cells. Cancer Res. 57: 3924-8, 1997.
236. Tomisawa M, Tokunaga T, Oshika Y et al.: Expression pattern of vascular endothelial growth factor isoform is closely correlated with tumour stage and vascularisation in renal cell

54
carcinoma. Eur J Cancer. 35: 133-7, 1999.
237. Park JE, Keller GA and Ferrara N: The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 4: 1317-26, 1993.
238. Yang JC, Haworth L, Sherry RM et al.: A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 349: 427-34, 2003.
239. Jermann M, Stahel RA, Salzberg M et al.: A phase II, open-label study of gefitinib (IRESSA) in patients with locally advanced, metastatic, or relapsed renal-cell carcinoma. Cancer Chemother Pharmacol: 1-7, 2005.
240. Bergers G, Song S, Meyer-Morse N et al.: Benefits of targeting both peri-cytes and endothelial cells in the tumor vasculature with kinase
inhibitors. J Clin Invest. 111: 1287-95, 2003.
241. Motzer RJ, Michaelson MD, Redman BG et al.: Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol. 24: 16-24, 2006.
242. Escudier B, Lassau N, Couanet D et al.: Phase II trial of thalidomide in renal-cell carcinoma. Ann Oncol. 13: 1029-35, 2002.
243. Stadler WM, Kuzel T, Shapiro C et al.: Multi-institutional study of the angiogenesis inhibitor TNP-470 in metastatic renal carcinoma. J Clin Oncol. 17: 2541-5, 1999.
244. Gordon MS: Novel antiangiogenic therapies for renal cell cancer. Clin Cancer Res. 10: 6377S-81S, 2004.
245. Thorpe PE: Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 10: 415-27, 2004.